
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci. , 11 March 2025
Sec. Cellular Neuropathology
Volume 19 - 2025 | https://doi.org/10.3389/fncel.2025.1523773
 Cecilia Mei1,2
Cecilia Mei1,2 Valentina Magliocca2
Valentina Magliocca2 Xin Chen3
Xin Chen3 Keith Massey4
Keith Massey4 Anai Gonzalez-Cordero5
Anai Gonzalez-Cordero5 Steven J. Gray3
Steven J. Gray3 Marco Tartaglia2
Marco Tartaglia2 Enrico Silvio Bertini6
Enrico Silvio Bertini6 Stefania Corti1*
Stefania Corti1* Claudia Compagnucci2*
Claudia Compagnucci2*Riboflavin transporter deficiency syndrome (RTD) is a rare childhood-onset neurodegenerative disorder caused by mutations in SLC52A2 and SLC52A3 genes, encoding the riboflavin (RF) transporters hRFVT2 and hRFVT3. In the present study we focused on RTD Type 2, which is due to variants in SLC52A2 gene. There is no cure for RTD patients and, although studies have reported clinical improvements with administration of RF, an effective treatment is still unavailable. Here we tested gene augmentation therapy on RTD type 2 patient-derived motoneurons using an adeno-associated viral vector 2/9 (AAV9) carrying the human codon optimized SLC52A2 cDNA. We optimized the in vitro transduction of motoneurons using sialidase treatment. Treated RTD motoneurons showed a significant increase in neurite’s length when compared to untreated samples demonstrating that AAV9-SLC52A2 gene therapy can rescue RTD motoneurons. This leads the path towards in vivo studies offering a potential treatment for RTD patients.
Riboflavin transporter deficiency (RTD), formerly known as Brown-Vialetto Van Laere syndrome, is a rare recessive neurologic condition. The disorder is a motor neuron disease characterized by defective motoneurons controlling speech, walking, swallowing, breathing and general body movements (Amir et al., 2020; O’Callaghan et al., 2019).
The syndrome is characterized by a phenotypic spectrum of motor, sensory, and cranial nerve neuropathy, resulting in muscle weakness, respiratory compromise, vision loss, sensorineural hearing loss, and sensory ataxia (Bosch et al., 2012; Foley et al., 2014; Manole et al., 2017). RTD type 2 specifically is caused by biallelic pathogenic variants in SLC52A2 gene (Jaeger and Bosch, 2016; Johnson et al., 2012), encoding the riboflavin transporters, hRFVT2 (Foley et al., 2014; Haack et al., 2012). Riboflavin (RF) is a precursor of flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) and reduction of its intracellular availability, through defective transporters, compromises several vital processes. RF cannot be synthesized de novo and is taken from the diet through riboflavin transporters hRFVT1, 2, 3, which have different tissue distribution. Specifically, hRFVT1 is preferentially expressed in the intestinal epithelium and in placenta, hRFVT2 is localized in the central and peripheral nervous system, while hRFVT3 is mainly localized in the testis, small intestine, kidney, and placenta (Jin and Yonezawa, 2022). Albeit empirical studies reported clinical improvement with the administration of RF, an effective cure is still lacking (Marioli et al., 2020; Rizzo et al., 2017).
Over the last two decades, AAV gene therapy has showed substantial improvements and benefits to patients. AAV vectors have emerged as one of the safest and most used vectors for gene replacement (Kantor et al., 2014). Specific targeting capabilities conferring a variety of capsid choice have made recombinant AAV the ideal vector used for gene delivery to many tissues and organs, including the central nervous system (CNS) (Hocquemiller et al., 2016; Ling et al., 2023). Furthermore, AAV exhibit a stable transgene expression in post-mitotic cells, neuronal tropism, low immunogenicity (Murlidharan et al., 2014). The serotype 9 (AAV9) is able to cross the blood–brain barrier with high transduction efficacy, representing a good vector for intravascular administration (Saraiva et al., 2016). The FDA approved Onasemnogene abeparvovec (Zolgensma) as a AAV9 gene therapy for infants with spinal muscular atrophy (SMA) (Ogbonmide et al., 2023).
Recombinant AAV9 vectors display widespread transduction in animals, from mice to larger animal models (Choudhury et al., 2017; Karumuthil-Melethil et al., 2016; Federici et al., 2012; Gray et al., 2013; Snyder et al., 2011; Masamizu et al., 2011; Bucher et al., 2013; Haurigot et al., 2013; Samaranch et al., 2013), but often present low transduction efficiency in cells in vitro (Gao et al., 2004; Zincarelli et al., 2008).
We previously generated and characterized several induced pluripotent stem cells (iPSCs) lines generated from RTD patients with different variants in the SLC52A2 gene. Specifically, we assessed their molecular (i.e., antioxidant response), morphological (i.e., neurite’s length) and functional (i.e., calcium metabolism) features (Niceforo et al., 2021; Colasuonno et al., 2020). Data collected in previous studies suggest that RF supplementation partially rescues the RTD phenotype, and the combined treatment of RF plus antioxidants provides further improvements of these biomarkers.
In this study we confirmed the morphological phenotype of RTD iPSC-derived motoneurons showing a shorter neurite’s length. In parallel we demonstrated that sialidase treatment increased in vitro transduction of AAVs. Finally, we demonstrated the potential of gene therapy to rescue RTD neurodegeneration. Since all aspects of RTD type 2 disease stem from the loss of SLC52A2 gene function, gene replacement therapy represents a reasonable and promising approach to provide a meaningful benefit for RTD patients.
Clinical features of RTD P1 (carrying the variants c.155C > T (p.Ser52Phe) and c.935 T > C (p.Leu312Pro)) had been previously reported (Niceforo et al., 2021). In particular, thanks to the timely treatment with riboflavin (75 mg/kg QID) and antioxidant therapy at 2.5 years of age she has remained neurologically stable.
RTD P2 (carrying the variants c.505C > T (p.Arg169Cys) and c.1030_1031del (p.Leu344Alafs*100)) is a girl presenting the first symptoms (arm weakness and dysphagia) at 8 months of age. At 12 months old she was diagnosed reporting severe optic atrophy, bilateral sensorineural deafness, sensory neuropathy and complete diaphragm paralysis (Magliocca et al., 2024).
The studies were conducted in compliance with the Code of Ethics of the World Medical Association (Declaration of Helsinki), and with national legislation and institutional guidelines (local institutional ethical committee, Ref. 1410_OPBG_2021, date of approval 11 February 2019). Informed consent was obtained from the subjects involved in the study. Patient skin fibroblasts were cultured in Dulbecco’s Modified Eagle Medium (Sigma Aldrich, Cod D5671), supplemented with 10% of Fetal Bovine Serum (Gibco Cod 10082-147) and penicillin/streptomycin (Gibco, Cod 15140148) at 37°C, 5% CO2 and 21% O2. When the cells reached 80% of confluence, they were reprogrammed as described (Okita et al., 2011).
Pluripotency characterization of RTD P1 and RTD P2 iPSC lines had been previously reported (Rizzo et al., 2017; Okita et al., 2011). Clinical features and characterization of RTD P1 and P2 iPSC lines had previously been reported (Rizzo et al., 2017; Okita et al., 2011). Control iPSCs (CTRL iPSCs) were obtained from healthy individual from System Biosciences Coriell (GM23338 and AG28869) and were derived from fibroblasts of a healthy individual using non-integrating episomal technology. Both control lines were used for experiments shown in Figures 1, 2, while for the transduction experiments only one control line (GM23338) was used.
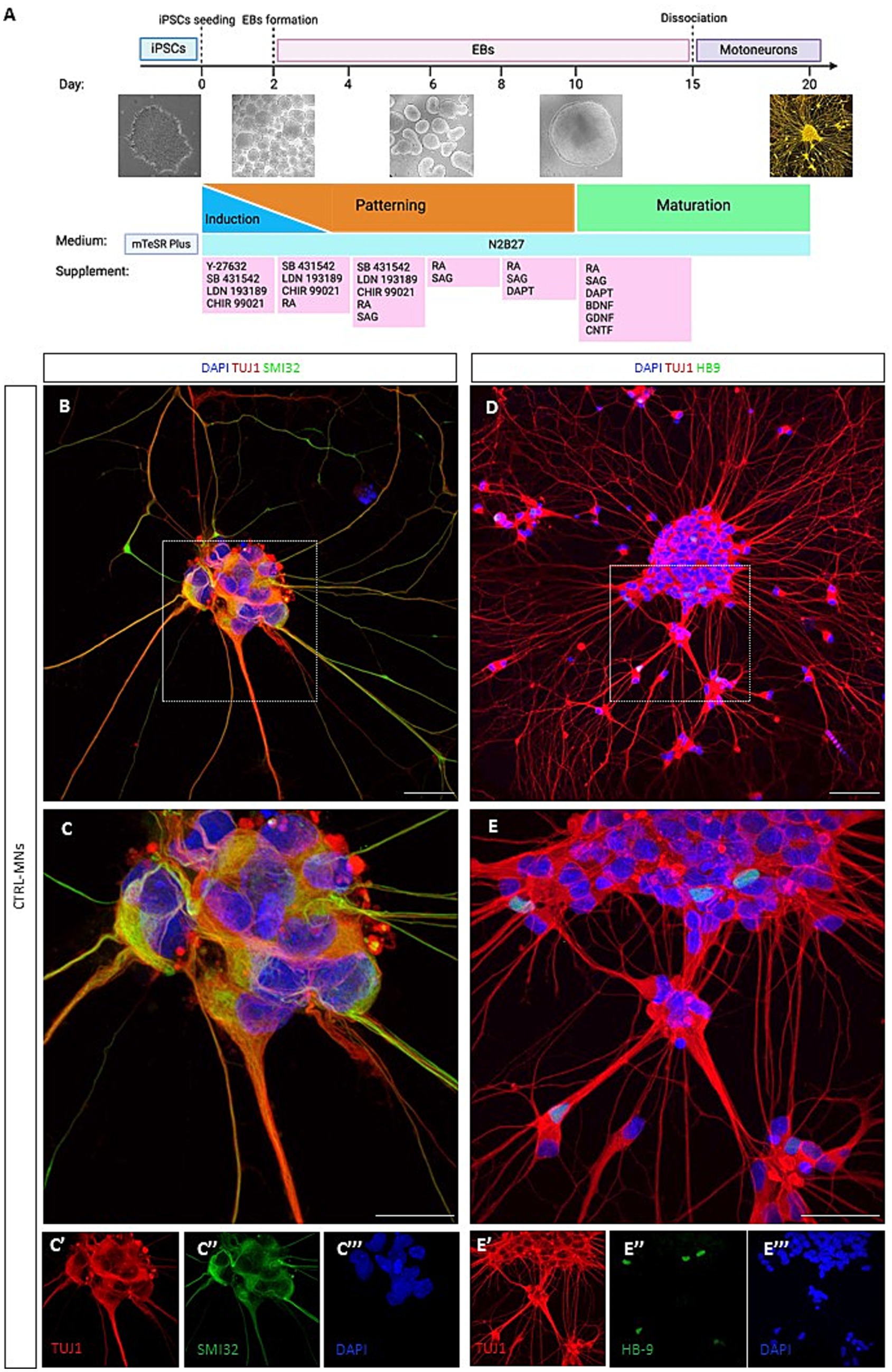
Figure 1. 3D/2D neural differentiation and characterization of iPSC-derived motoneurons. (A) Schematic showing a stepwise differentiation of motoneurons using the optimized 3D/2D protocol, showing EBs formation and the mature 2D motoneurons. Created with Biorender.com. (B) Day 20 immunofluorescence confocal image analysis for the neural marker TUJ1 (in red) and SMI-32 (in green). (C) High-magnification confocal image of control motoneurons co-stained for TUJ1 and SMI32 with images showing separate color channels (C′, C″, C″’). (D) Representative image of neurons positive for TUJ1 (in red) and HB9 (in green). (E) High-magnification confocal image of TUJ1 and HB9positive control motoneurons with images showing separate color channels (E’, E,” E”’). Nuclei counterstained with DAPI. Scale bar = 50um.
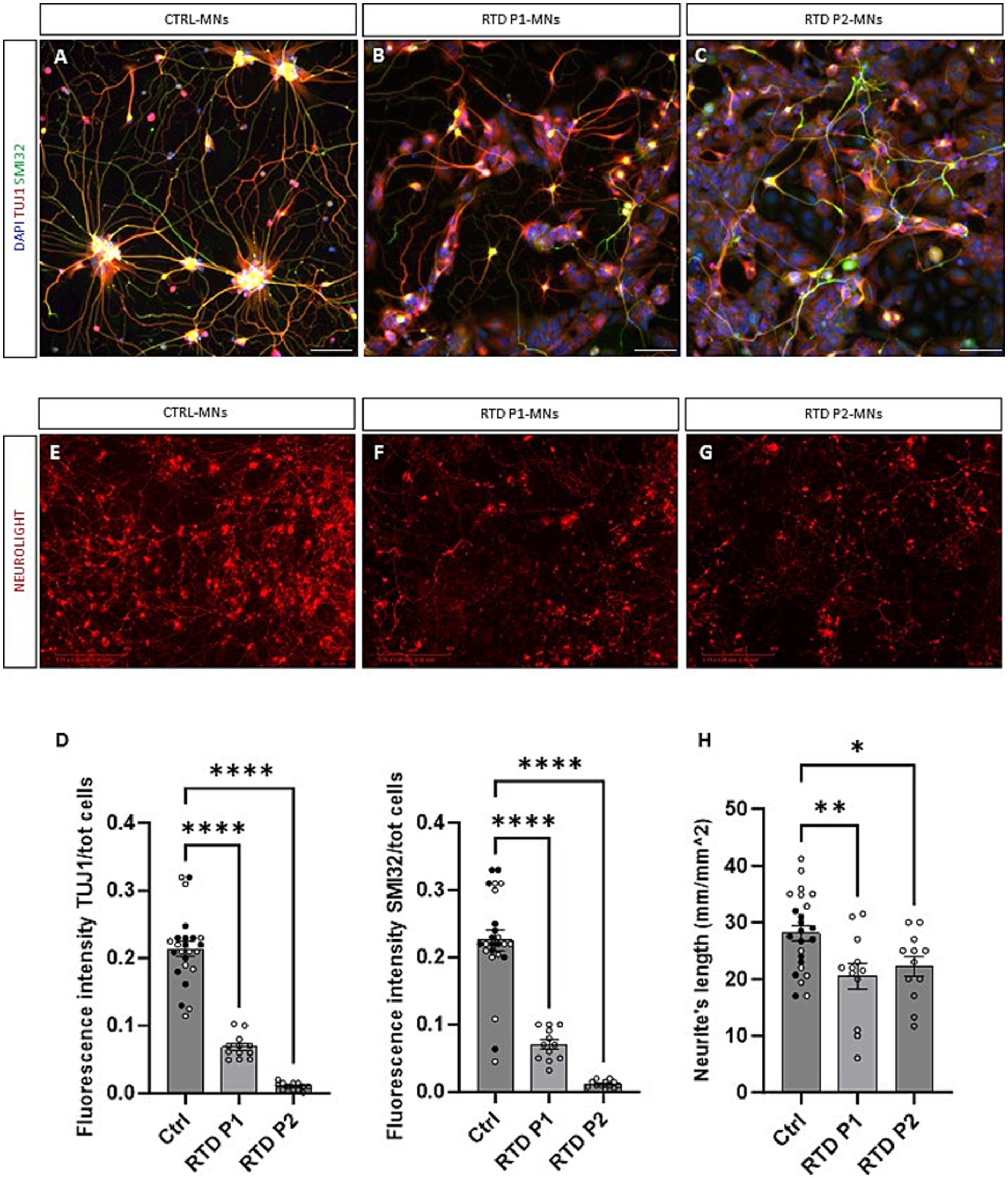
Figure 2. Morphological characterization of RTD motoneurons. (A–C) Representative images of control and RTD motoneurons positive for TUJ1 (in red) and SMI32 (in green). (D) Quantification of TUJ1 and SMI32 fluorescent signals demonstrated a significant decrease in fluorescence intensity in RTD iPSC-derived motoneurons (****p < 0.0001, one-way ANOVA; mean ± SEM; N = 3 independent experiments). (E–G) Neurites’ length measurement of RTD and control motoneurons positive for Neurolight red lentivirus in the Incucyte system at day 28. (H) Quantification confirmed a significant reduction of neurite extension in both lines of RTD motoneurons (*p < 0.05, **p < 0.01, one-way ANOVA; mean ± SEM; N = 3 independent experiments). Nuclei co-stained with DAPI in (A–C). The CTRL bar includes data points from two CTRL cell lines (one represented by black dots, the other by white dots). Scale bar = 50 um in (A–C). Scale bar = 400 um in (E–G).
RTD iPSCs and CTRL iPSCs (DIV 6–8) were detached with Accutase (Sigma-Aldrich, Cod SCR005) for 5 min at 37°C. Then, single cells were cultured on ultra-low attachment dishes, using the Basal Medium N2/B27 consisting of: DMEM/F12 (Sigma Aldrich, Cod D0697), Neurobasal (Gibco, Cod 21103049), Glutamax (Gibco, Cod 35050061), Pen-strep (Gibco, Cod 15140122), B27 supplement minus Vit A, (Life technologies, Cod 12587010), N2 supplement (Life technologies, Cod 17502-048) and 2-ME (Gibco, Cod 21985-023). At Day 0, the medium was supplemented with Y27632 (10 uM) (Cell Signaling, Cod 13624) LDN193189 (0.1 uM) (StemCell, Cod 72149), SB431542 (20uM) (StemCell, Cod S4317) and CHIR 99021 (3 uM) (Sigma-Aldrich, Cod SML1046). At Day 2, the EBs were resuspended in the previous medium plus RA (100 nM) (Sigma Aldrich, Cod R2625) and no Y27632 was added. Then, at day 4, SAG (500 nM) (Sigma-Aldrich, Cod 566660) was added; at day 7, the EBs were resuspended in N2B27 plus RA and SAG. At day 9, DAPT (10 uM) (StemCell, Cod 72082) was added and at day 11 the EBs were resuspended in N2B27 plus RA, SAG, DAPT, BDNF (10 ng/mL), GDNF (10 ng/mL), and CNTF (10 ng/mL) (Sigma-Aldrich, Cod C3710). At day 15, the EBs were collected in a tube, washed in PBS and rinsed with Trypsin–EDTA 1X (Euroclone, Cod ECB3052) plus DNAse (20 ug/mL) (Thermo Fisher Scientific, Cod AM2235). Tubes were then placed in warm bath for 15 min and then EBs were dissociated to become a single cell suspension and FBS was added to neutralize the trypsin. Cells were then centrifuged for 5 min at 300 g and, after the removal of the supernatant, they were passed through a 70-um strainer. After the dissociation process, MNs were plated for further analyses.
The AAV9-GFP vector design has been previously described (Gray et al., 2011). The AAV9-SLC52A2 vector was developed similarly, but with a moderate strong UsP promoter (Chen et al., 2023) and bovine growth hormone (BGH) polyadenylation signal to drive expression of a codon-optimized human SLC52A2 sequence. Codon optimization was carried out by ATUM (Menlo Park, CA, USA). Both AAV vectors used a self-complementary genome configuration. The AAV vectors were manufactured by the University of North Carolina Vector core, according to published methods (Grieger et al., 2016).
To increase the infectivity of the virus, Sialidase (Roche, cod. 10269611001) was used. After the EBs dissociation, motoneurons were left to adhere on a precoated plates with poly-O-ornithine (50 ug/mL) and laminin (20 ug/mL) for two days. Then, cells were washed 3 times with PBS and pretreated with Sialidase at 2 different concentrations: 0.125 U/mL and 1.25 U/mL for 3 h at 37°C. Following washing with PBS (three times), cells were transduced with AAV9-GFP or AAV9-SLC52A2 in a concentration of 107 m.o.i for 24 h. The following day, the cell media was completely removed and replaced with the fresh media. Then, cell media was changed every other day until day 40 (Figure 3).
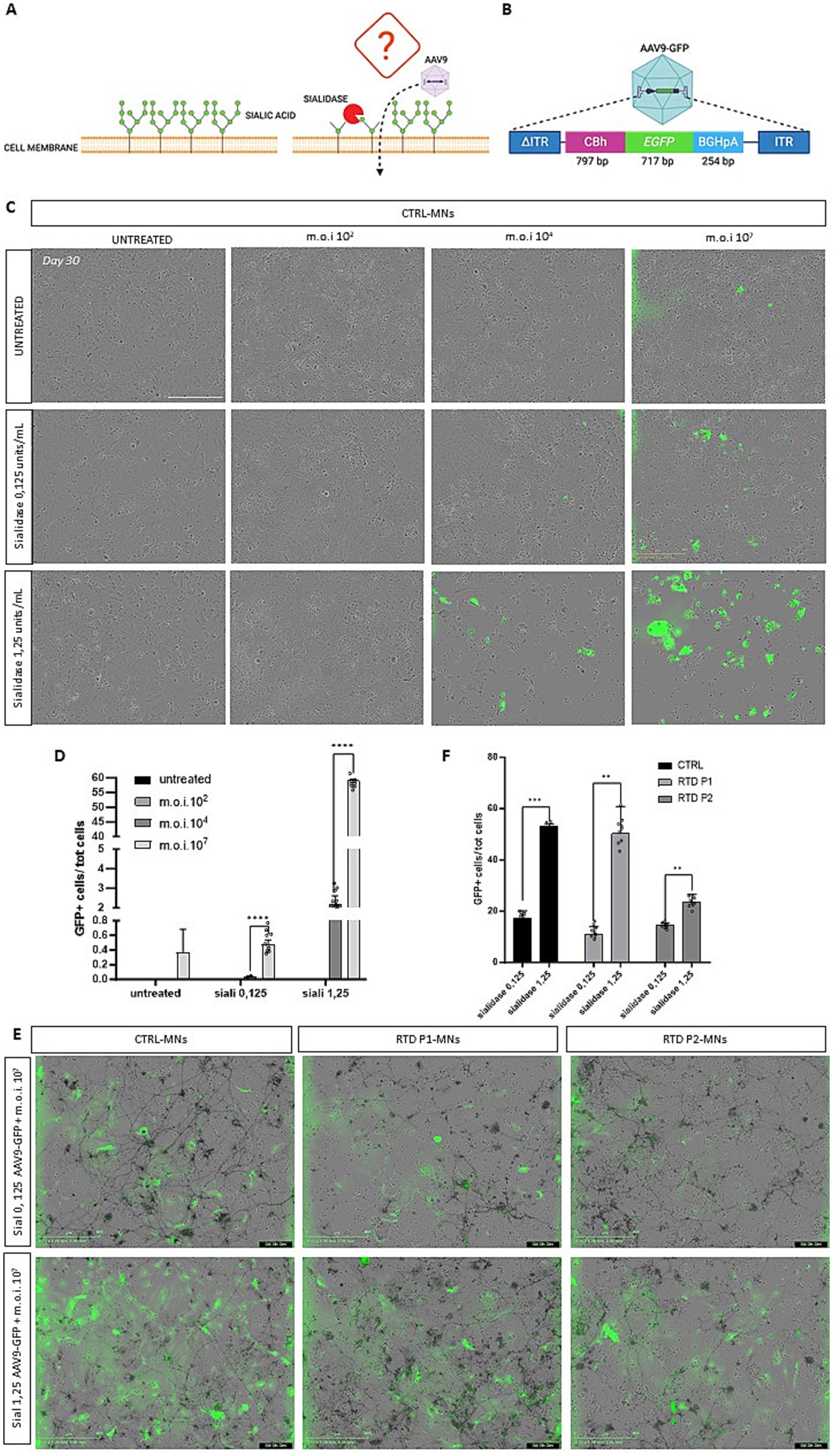
Figure 3. Sialidase treatment and the effect on AAV9-GFP vector transduction. (A) Schematic of sialidase mechanism. Removal of siliac acids from the cells surface, might increase the infectivity of the virus. (B) Schematic drawing of AAV9-CBh-GFP viral vector construct. (A,B) Created with Biorender.com. (C) Representative images of day 30 motoneurons after sialidase treatment and after transduction with 10 (O’Callaghan et al., 2019); 104 and 107 vg/cell MOI GFP expression shows cells that have been transduced. (D) Graph showing the percentage of GFP transduced motoneurons (****p < 0.0318, Kruskal-Wallis; mean ± SEM; N = 3 independent experiments). (E) Representative images of motoneurons pre-treated with sialidase and then transduced with the AAV9-GFP vector. (F) Graphs showing the percentage of GFP-positive cells demonstrate a significant increase in transduced cells with 1.25 U/mL sialidase treatment (*** p < 0.0001, **p < 0.001, unpaired t test; mean ± SEM, N = 3 independent experiments). Scale bar 400 um in (C,E).
Mature RTD and control MNs were plated on coverslips placed in a pre-coated 24-well plate with poly-O-ornithine (50 ug/mL) and laminin (20 ug/mL). After 5 days, MNs were fixed with 4% paraformaldehyde for 10 min at RT. BSA 5% blocking solution was used and a 0.1% Triton X-100 was added (Sigma) for permeabilization. Incubation was performed with the primary antibody against TUJ1 (Sigma Aldrich, Cod 8578) diluted 1:500 and maintained at RT for 2 h, anti-SMI32 (Sigma Aldrich, Cod N4142) diluted 1:200 O/N at 4°C and anti HB-9 (Invitrogen, Cod PA5-23407). AlexaFluor 555 (Thermo Fisher Scientific, Cod A21425) and AlexaFluor 488 (Thermo Fisher Scientific, Cod A11070) were used as secondary antibodies, diluted 1:500 and incubated at RT for 1 h. Nuclei were counterstained using DAPI (Thermo Fisher Scientific, Cod. D1306). Finally, the cells were observed with a confocal Leica Dmi8 fluorescence microscope (Leica Microsystems, Germany) and acquired images were digitally elaborated with a modular image processing and analysis software (LasX Software, Leica).
Motoneurons were plated at a density of 5′000 cells/well in a Matrigel precoated 96-well plate (TPP, Cod 92696) and neurites’ length was measured using the IncuCyte System (Sartorius, Essen BioScience) with the Neurite Analysis application for Neurolight labeled cells. Cells were transduced with a lentiviral-based vector encoding the Incucyte Neurolight Lentivirus (Sartorius, Essen BioScience, Cod 4807) following manufacturer’s instructions (Figure 4). Live imaging experiments (in Figure 5) were performed by acquiring every 4 h for 15 days, from the 25th to the 40th day of the neural differentiation process. Phase-contrast and fluorescent images were acquired for every experiment. Analysis parameters for NeuroTrack software module-processing definitions were optimized individually for each experiment according to the workflow outlined in the manufacturer’s manual. Microplate graphs were generated using the time plot feature in the graph/export menu of the Incucyte SX5 software. Raw data of neurites’ lengths were exported to Microsoft Excel and GraphPad Prism to calculate mean values ± SEM.
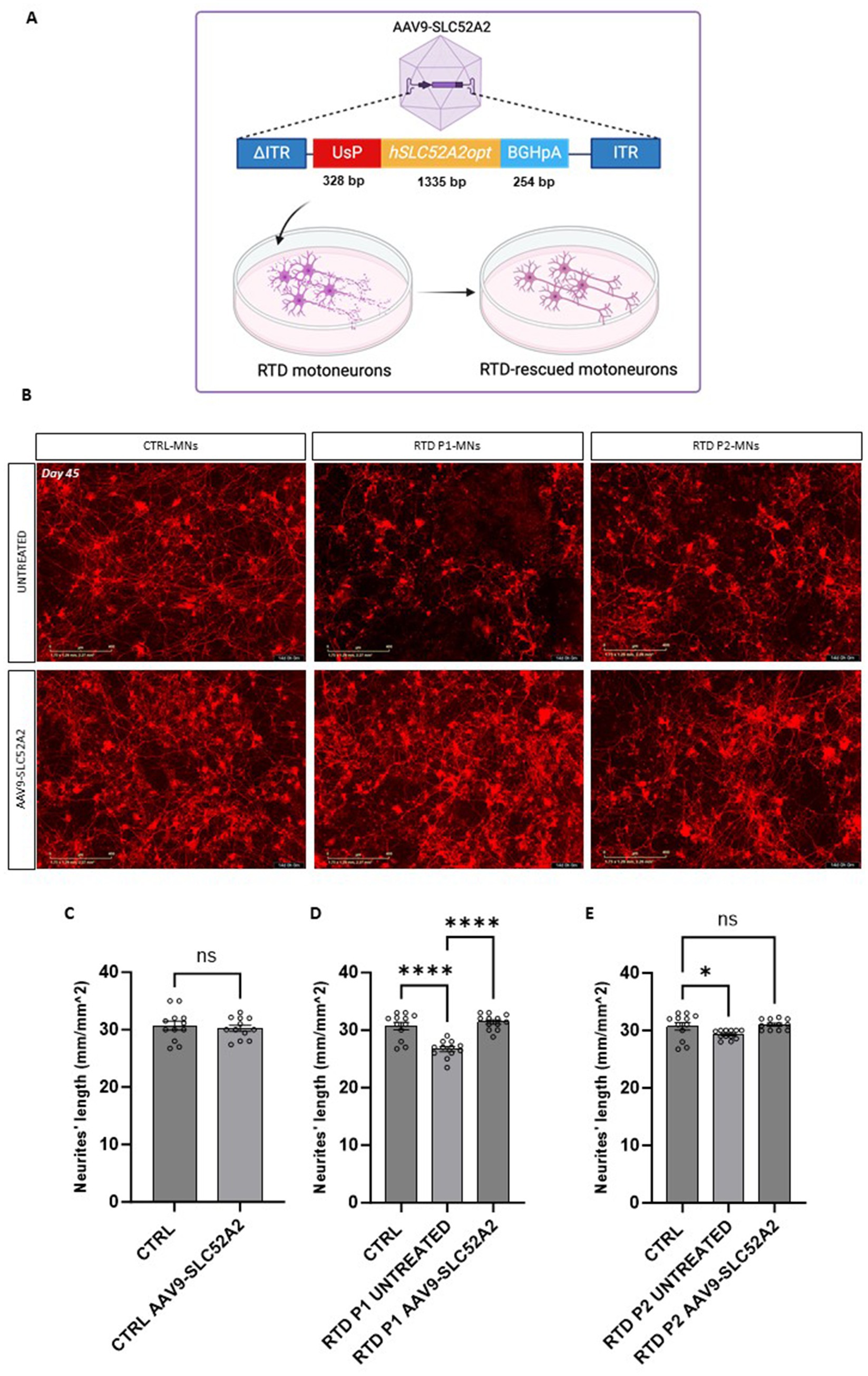
Figure 4. SLC52A2 gene therapy rescues RTD motoneurons neurites morphology. (A) Schematic of gene therapy construct. Created with Biorender.com. (B) Representative Incucyte image of the neural network formation of CTRL and RTD motoneurons (positive for Neurolight lentivirus in red) following gene therapy. (C–E) Graphs depicting the quantification of neurites’ length at day 45 of differentiation in treated and untreated motoneurons (****p < 0.0001, *p < 0.01, one-way ANOVA; mean ± SEM; N = 3 independent experiments). Scale bar 400 um.
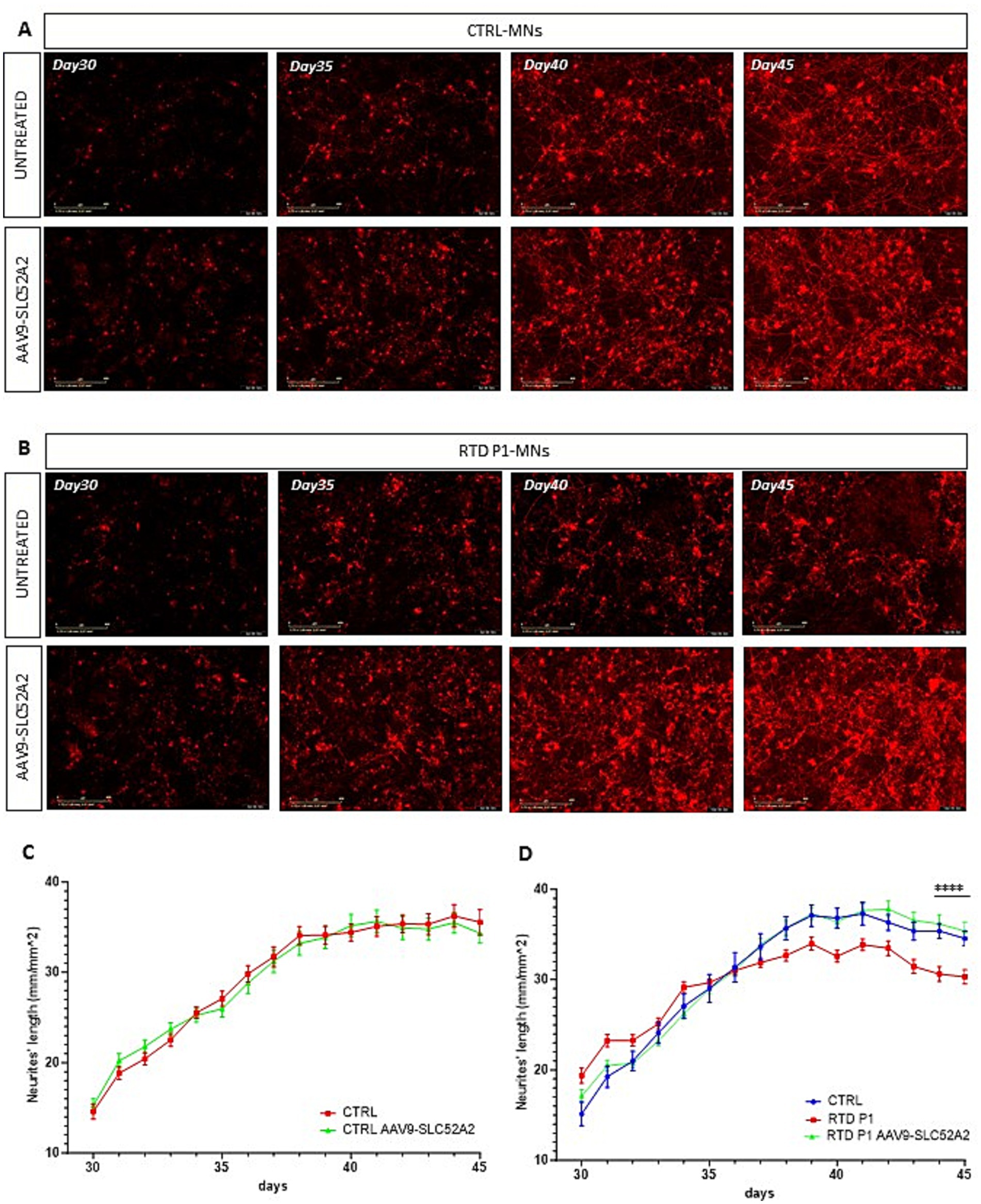
Figure 5. Time-laps imaging of the neurites’ length in untreated and treated motoneurons with AAV9-SLC52A2 vector. (A,B) Representative fluorescent images of Neurolight red positive neurons of CTRL and RTD P1 at different days of neuronal differentiation, showing that at the end of in vitro neurogenesis (day 45), motoneurons of RTD patient showed longer neurites compared to those untreated. (C,D) Graphs showing neurites’ length of CTRL and RTD P1 over in vitro neurogenesis (**p = 0.001, one-way ANOVA; mean ± SEM, N = 3 independent experiments, N = 4 images per independent experiment per sample). Scale bar 400 um.
Raw data were collected from each individual experiment of three independent biological replicates. The data were analyzed as the mean ± the standard deviation (SD) or the standard error of the mean (SEM). Significance was tested using ordinary unpaired t test (when two sample groups were compared) or one-way ANOVA (parametric test) (when more than two sample groups were compared) for normally distributed data. For data that do not passed the normality test, Kruskal Wallis (non parametric) test has been used to calculate statistical significance. GraphPad-Prism software (v9.3.1, GraphPad Software) was used to statistically analyze the data.
We first tested two protocols for the generation of mature motoneurons using a control iPSC line (Corti et al., 2012). Using a 2D adherent differentiation protocol neurons expressed the pan-neural marker TUJ1 but failed to show the mature motoneuron marker HB9 (Supplementary Figures S1C,D) even at day 30 in culture. iPSCs were successfully differentiated into motoneurons using an adapted 3D/2D protocol (Figure 1A; Maury et al., 2014). This directed differentiation protocol is based on the generation of embryoid bodies (EBs), which at later stages are dissociated to obtain pure cultures of motoneurons (90 ± 1.49%) (Supplementary Figure S1E).
At day 20 in vitro neural differentiation was confirmed by immunopositivity of the pan-neural marker TUJ1 and SMI32 (Figures 1B,C). We further assessed for motoneuron maturation using the post-mitotic motoneuronal marker HB9 (Figures 1D,E). Results confirmed the ability of the 3D/2D protocol to generate mature motoneurons being positive for the markers TUJ1, SMI32 and HB9.
Next, we differentiated two iPSC lines from healthy donors (CTRL) and two iPSC lines from RTD patients, carrying different mutations (RTD P1 155C > T;935 T > C and RTD P2 1030_31 del;505C > T) in the SLC52A2 gene. RTD motoneurons expressed TUJ1 and SMI32 proteins, however formation of neuronal networks was less evident (Figures 2A–C). A significant decrease in fluorescence intensity was observed in RTD iPSC-derived motoneurons (Figure 2D, **** p < 0.0001, one-way ANOVA; mean ± SEM; N = 3 independent experiments, N = 3 images per independent experiment per sample), confirming the impaired neuronal differentiation demonstrated using a different methodology (see Conclusions). Next, we sought to confirm if neuronal networks were impaired in diseased motoneurons. Day 20 motoneurons were cultured on 96-well plates for live imaging and transduced using a Neurolight Red Lentivirus (Incucyte SX5 system) which enabled highly efficient and non-disruptive labeling of iPSC-derived neurons (Figures 2E–G). Fourteen days following transduction RTD motoneurons showed a significant reduction in neurite extension (Figure 2H, ****p = <0.0001, one-way ANOVA; mean ± SEM; N = 3 independent experiments, N = 4 for each independent experiment per sample). These data demonstrate that using 3D/2D neural differentiation RTD motoneurons exhibit disrupted neurite extension starting to elucidate characteristics of disease in vitro.
Efficient gene transfer by AAV9 vectors requires an atypical interaction with non-sialylated cell surface glycans (Shen et al., 2011; Bell et al., 2011). We sought to develop an effective AAV gene therapy for RTD by testing whether sialidase treatment, which removes the siliac acid from the cell surface increases AAV9 transduction (Figure 3A). Different multiplicity of infection (MOI) of a control AAV9-CBh-GFP vector (Figure 3B) was used to assess transduction efficiency in motoneurons following sialidase treatment at various concentrations (Figure 3C). Transduction efficiency was analyzed 5 days post-transduction at day 30 in culture. MOI 102 was too low to detect any GFP positive cells even in both untreated and sialidase treated cells. However, treatment at both 0.125 and 1.25 units/mL significantly increase transduction when compared to untreated control at moi 104 (****p < 0.0001, Kruskal-Wallis; mean ± SEM; N = 3 independent experiments, N = 4 images per independent experiment per sample). AAV9 vector at 107 moi significantly outperformed all other treatments (Figure 3D, ****p < 0.0318, Kruskal-Wallis; mean ± SEM; n = 3 images, N = 3 independent experiments). Finally, to evaluate if transduction varies between control and RTD motoneurons we tested the transduction efficiency of the AAV9-CBh-GFP vector in control and RTD motoneurons pre-treated with 0.125 and 1.25 u/mL sialidase. A significant increase in the percentage of GFP motoneurons in control, RTD P1 and P2 was observed in the 1.25 u/mL sialidase treated motoneurons (Figures 3E,F, *** p < 0.0001, ** p = 0.0033 for RTD P1, **p = 0.0078 for RTD P2, unpaired t-test; mean ± SEM; N = 3 independent experiments, N = 3 images per independent experiment per sample).
Next, to treat RTD motoneurons, we designed a AAV9-UsP-SCL52A2 vector carrying the human codon optimized SLC52A2 cDNA (Figure 4A).
We transduced RTD P1 and P2 motoneurons, with the AAV9-UsP-SLC52A2 at day 25 in culture using the optimized 1.25 u/mL sialidase treatment and m.o.i of 107 and evaluated motoneurons at day 45 in culture. To establish the effects of the gene therapy on the neurites’ length disease phenotype we also transduced motoneurons with the Neurolight red lentivirus (Figure 4B; Supplementary Video S1) for the automated measurement of the neurite’s length (Supplementary Figure S2). Importantly, following AAV treatment, we found that neurites’ length of RTD P1 and P2 motoneurons were significantly longer than untreated (Figures 4C–E, p = 0.5011 for CTRL, **p = 0.0050 for RTD P1, *p = 0.0192 for RTD P2, ANOVA; mean ± SEM; N = 3 independent experiments, N = 4 images per independent experiment per sample). Furthermore, length was restored to levels similar to control MNs (Figures 4D,E). A more pronounced rescue was observed in treated RTD P1 motoneurons, therefore to evaluate the effect of the gene therapy on neurogenesis, we imaged untreated and treated control and RTD P1 motoneurons at several time points (days 30, 35, 40 and 45 of neuronal differentiation) (Figures 5A,B). We showed that in untreated RTD motoneurons, a presumptive neural network was able to form, but over time, motoneurons neurites were lost and cells underwent neurodegeneration (Figures 5A,B; Supplementary Video S1). Meanwhile, following gene therapy with the AAV9-SLC52A2 vector the neural network in RTD patient was preserved over time, showing a progressive amelioration of the neurites’ length in transduced RTD motoneurons (Figure 5C, **p = 0.001, ANOVA; mean ± SEM; N = 3 independent experiments, N = 4 images per independent experiment per sample). No changes in terms of neurites’ length nor network formation/degeneration were observed in control motoneurons treated with AAV9- UsP-SLC52A2 indicating the overexpression of SLC52A2 is not detrimental to neurons in vitro (Figure 5C).
In vitro stem cell-derived motoneurons are a valuable tool to analyse the pathomechanisms underlying motoneuron diseases. In this study, we demonstrated the efficient differentiation of iPSCs into motoneurons by adapting a previously described protocol (Maury et al., 2014). Various types of neuronal cultures have been described to date, and although this model has been broadly used, the majority of the models generate immature motoneurons (Bucchia et al., 2018). Here we showed that our iPSC-derived motoneurons matured and expressed the post-mitotic marker HB9 enabling proper RTD modelling in vitro. Despite the improved methodology used in this study to obtain iPSC-derived motoneurons, the fluorescent intensities of TUJ1 and SMI32 are significantly decreased in RTD motoneurons (Figure 2D), thus confirming the impaired neuronal differentiation of RTD cells as previously demonstrated using different neuronal differentiation methodology (Marioli et al., 2020; Rizzo et al., 2017; Niceforo et al., 2021). Additionally, we do not exclude that the decreased TUJ1 and SMI32 levels in RTD motoneurons are a consequence of impaired neuronal survival of RTD motoneurons, since neuronal apoptosis has been recently demonstrated as a contributing event in RTD pathogenesis (Marioli et al., 2024).
One of the most compromised features in RTD motoneurons is the cytoskeleton (Marioli et al., 2020; Rizzo et al., 2017; Niceforo et al., 2021). Our morphometric analyses of RTD iPSC-derived motoneurons confirmed that RTD disease leads to neurites that are significantly shorter than those of the healthy motoneurons (Marioli et al., 2020; Rizzo et al., 2017; Niceforo et al., 2021).
Previous studies have reported that gene therapy testing in iPSC-derived motoneurons is limited due to low transduction efficiency by AAV vectors. Here, we described that treatment with Sialidase significantly increased cell surface binding and infectivity of adeno-associated virus (AAV) serotype 9 (Shen et al., 2011). We also established the optimal experimental condition to obtain the maximal rate of infection. Various combinations of sialidase and MOI vector allowed to obtain more than the 50% of transduction efficiency.
The majority of RTD patients are responsive to RF supplementation, however, it remains unclear if this supplementation will be effective in preventing symptoms for life in responsive patients, or simply delay the occurrence of symptoms (Jaeger and Bosch, 2016; Koy et al., 2012). It is therefore necessary to look for a strategy that can alleviate the sufferings of all RTD patients. Considering the recent advances in the field of gene therapy, we designed and tested a AAV9-SLC52A2 gene supplementation therapy which would be efficacious on all RTD patients independently to the carried mutations and/or their responsiveness to RF treatment.
In the present study, we aimed at promoting the expression of the correct SLC52A2 protein by introducing it by AAV9-SLC52A2 transduction. Promisingly, gene therapy was able to restore neurites length of RTD motoneurons generating long neurites maintaining a robust neural network during the neurogenesis compared to the untreated RTD motoneurons, which resulted in a breakable and fragile neuronal network undergoing degradation over time.
Collectively, our results indicate that AAV9-SLC52A2 vector rescues the neural phenotype in motoneurons derived from RTD Type 2 patient iPSCs, which warrants further in vitro as well as in vivo studies to develop gene therapy as a potential clinical treatment for these patients. In fact, we observed recovery of the morphological abnormalities of the RTD neurites and we are currently working on the evaluation of the functional aspects of AAV9-SLC52A2 transduced RTD neuronal cells. Recently, several in vivo viral-vector gene therapies are currently on the market and the field of gene therapy for neurological conditions is continuously evolving (Ling et al., 2023). For many years, rare neurodegenerative disorders as amyotrophic lateral sclerosis (SLA), spinal muscular atrophy (SMA) and RTD were considered incurable, but now, gene therapy might offer an effective treatment. Currently, many research efforts are focused on finding new therapeutic strategies that would treat RTD independently from patients’ variants. These findings, offer the first insights into gene therapy efficacy for RTD patients worldwide.
The data presented in this study have been deposited in Zenodo with the following doi: 10.5281/zenodo.14916787.
The studies involving humans were approved by Ospedale Pediatrico Bambino Gesù. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired as part of a previous study for which ethical approval was obtained. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. No potentially identifiable images or data are presented in this study.
CM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft. VM: Formal analysis, Writing – review & editing. XC: Data curation, Formal analysis, Investigation, Writing – review & editing. KM: Data curation, Formal analysis, Writing – review & editing. AG-C: Data curation, Writing – review & editing. SG: Data curation, Formal analysis, Writing – review & editing. MT: Data curation, Formal analysis, Writing – review & editing. EB: Formal analysis, Resources, Writing – review & editing. SC: Data curation, Formal analysis, Funding acquisition, Resources, Writing – review & editing. CC: Conceptualization, Data curation, Formal analysis, Funding acquisition, Project administration, Resources, Supervision, Writing – original draft.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Italian Ministry of Health with Current Research funds, RCR-2022-23682289 and SG-2021-12373714; from MNESYS (to CC and EB), the Cure RTD Foundation, Cure RTD Italia Odv, Fondazione Bambino Gesù (Vite Coraggiose).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2025.1523773/full#supplementary-material
Amir, F., Atzinger, C., Massey, K., Greinwald, J., Hunter, L. L., Ulm, E., et al. (2020). The clinical journey of patients with riboflavin transporter deficiency type 2. J. Child Neurol. 35, 283–290. doi: 10.1177/0883073819893159
Bell, C. L., Vandenberghe, L. H., Bell, P., Limberis, M. P., Gao, G. P., Van Vliet, K., et al. (2011). The AAV9 receptor and its modification to improve in vivo lung gene transfer in mice. J. Clin. Invest. 121, 2427–2435. doi: 10.1172/JCI57367
Bosch, A. M., Stroek, K., Abeling, N. G., Waterham, H. R., Ijlst, L., and Wanders, R. J. A. (2012). The Brown-Vialetto-Van Laere and Fazio Londe syndrome revisited: natural history, genetics, treatment and future perspectives. Orphanet J. Rare Dis. 7:83. doi: 10.1186/1750-1172-7-83
Bucchia, M., Merwin, S. J., Re, D. B., and Kariya, S. (2018). Limitations and challenges in modeling diseases involving spinal motor neuron degeneration in vitro. Front. Cell. Neurosci. 12:2018. doi: 10.3389/fncel.2018.00061
Bucher, T., Colle, M. A., Wakeling, E., Dubreil, L., Fyfe, J., Briot-Nivard, D., et al. (2013). ScAAV9 intracisternal delivery results in efficient gene transfer to the central nervous system of a feline model of motor neuron disease. Hum. Gene Ther. 24, 670–682. doi: 10.1089/hum.2012.218
Chen, X., Dong, T., Hu, Y., De Pace, R., Mattera, R., Eberhardt, K., et al. (2023). Intrathecal AAV9/AP4M1 gene therapy for hereditary spastic paraplegia 50 shows safety and efficacy in preclinical studies. J. Clin. Invest. 133:e164575. doi: 10.1172/JCI164575
Choudhury, S. R., Hudry, E., Maguire, C. A., Sena-Esteves, M., Breakefield, X. O., and Grandi, P. (2017). Viral vectors for therapy of neurologic diseases. Neuropharmacology 120, 63–80. doi: 10.1016/j.neuropharm.2016.02.013
Colasuonno, F., Bertini, E., Tartaglia, M., Compagnucci, C., and Moreno, S. (2020). Mitochondrial abnormalities in induced pluripotent stem cells-derived motor neurons from patients with riboflavin transporter deficiency. Antioxidants 9:1252. doi: 10.3390/antiox9121252
Corti, S., Nizzardo, M., Simone, C., Falcone, M., Nardini, M., Ronchi, D., et al. (2012). Genetic correction of human induced pluripotent stem cells from patients with spinal muscular atrophy. Sci. Transl. Med. 4:165ra162. doi: 10.1126/scitranslmed.3004108
Federici, T., Taub, J. S., Baum, G. R., Gray, S. J., Grieger, J. C., Matthews, K. A., et al. (2012). Robust spinal motor neuron transduction following intrathecal delivery of AAV9 in pigs. Gene Ther. 19, 852–859. doi: 10.1038/gt.2011.130
Foley, A. R., Menezes, M. P., Pandraud, A., Gonzalez, M. A., Al-Odaib, A., Abrams, A. J., et al. (2014). Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain 137, 44–56. doi: 10.1093/brain/awt315
Gao, G., Vandenberghe, L. H., Alvira, M. R., Lu, Y., Calcedo, R., Zhou, X., et al. (2004). Clades of adeno-associated viruses are widely disseminated in human tissues. J. Virol. 78, 6381–6388. doi: 10.1128/jvi.78.12.6381-6388.2004
Gray, S. J., Foti, S. B., Schwartz, J. W., Bachaboina, L., Taylor-Blake, B., Coleman, J., et al. (2011). Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum. Gene Ther. 22, 1143–1153. doi: 10.1089/hum.2010.245
Gray, S. J., Nagabhushan Kalburgi, S., McCown, T. J., and Jude Samulski, R. (2013). Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 20, 450–459. doi: 10.1038/gt.2012.101
Grieger, J. C., Soltys, S. M., and Samulski, R. J. (2016). Production of recombinant adeno-associated virus vectors using suspension HEK293 cells and continuous harvest of vector from the culture media for GMP FIX and FLT1 clinical vector. Mol. Ther. 24, 287–297. doi: 10.1038/mt.2015.187
Haack, T. B., Makowski, C., Yao, Y., Graf, E., Hempel, M., Wieland, T., et al. (2012). Impaired riboflavin transport due to missense mutations in SLC52A2 causes Brown-Vialetto-Van Laere syndrome. J. Inherit. Metab. Dis. 35, 943–948. doi: 10.1007/s10545-012-9513-y
Haurigot, V., Marcó, S., Ribera, A., Garcia, M., Ruzo, A., Villacampa, P., et al. (2013). Whole body correction of mucopolysaccharidosis IIIA by intracerebrospinal fluid gene therapy. J. Clin. Invest. 123, 3254–3271. doi: 10.1172/JCI66778
Hocquemiller, M., Giersch, L., Audrain, M., Parker, S., and Cartier, N. (2016). Adeno-associated virus-based gene therapy for CNS diseases. Hum. Gene Ther. 27, 478–496. doi: 10.1089/hum.2016.087
Jaeger, B., and Bosch, A. M. (2016). Clinical presentation and outcome of riboflavin transporter deficiency: mini review after five years of experience. J. Inherit. Metab. Dis. 39, 559–564. doi: 10.1007/s10545-016-9924-2
Jin, C., and Yonezawa, A. (2022). Recent advances in riboflavin transporter RFVT and its genetic disease. Pharmacol. Ther. 233:108023. doi: 10.1016/j.pharmthera.2021.108023
Johnson, J. O., Gibbs, J. R., Megarbane, A., Urtizberea, J. A., Hernandez, D. G., Foley, A. R., et al. (2012). Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain 135, 2875–2882. doi: 10.1093/brain/aws161
Kantor, B., McCown, T., Leone, P., and Gray, S. J. (2014). Clinical applications involving CNS gene transfer. Adv. Genet. 87, 71–124. doi: 10.1016/B978-0-12-800149-3.00002-0
Karumuthil-Melethil, S., Marshall, M. S., Heindel, C., Jakubauskas, B., Bongarzone, E. R., and Gray, S. J. (2016). Intrathecal administration of AAV/GALC vectors in 10–11-day-old twitcher mice improves survival and is enhanced by bone marrow transplant. J. Neurosci. Res. 94, 1138–1151. doi: 10.1002/jnr.23882
Koy, A., Pillekamp, F., Hoehn, T., Waterham, H., Klee, D., Mayatepek, E., et al. (2012). Brown-Vialetto-Van Laere syndrome: a riboflavin-unresponsive patient with a novel mutation in the C20orf54 gene. Pediatr. Neurol. 46, 407–409. doi: 10.1016/j.pediatrneurol.2012.03.008
Ling, Q., Herstine, J. A., Bradbury, A., and Gray, S. J. (2023). AAV-based in vivo gene therapy for neurological disorders. Nat. Rev. Drug Discov. 22, 789–806. doi: 10.1038/s41573-023-00766-7
Magliocca, V., Lanciotti, A., Ambrosini, E., Travaglini, L., D'Ezio, V., D'Oria, V., et al. (2024). Modeling riboflavin transporter deficiency type 2: from iPSC-derived motoneurons to iPSC-derived astrocytes. Front. Cell. Neurosci. 18:1440555. doi: 10.3389/fncel.2024.1440555
Manole, A., Jaunmuktane, Z., Hargreaves, I., Ludtmann, M. H. R., Salpietro, V., Bello, O. D., et al. (2017). Clinical, pathological and functional characterization of riboflavin-responsive neuropathy. Brain 140, 2820–2837. doi: 10.1093/brain/awx231
Marioli, C., Magliocca, V., Petrini, S., Niceforo, A., Borghi, R., Petrillo, S., et al. (2020). Antioxidant amelioration of riboflavin transporter deficiency in motoneurons derived from patient-specific induced pluripotent stem cells. Int. J. Mol. Sci. 21:7402. doi: 10.3390/ijms21197402
Marioli, C., Muzzi, M., Colasuonno, F., Fiorucci, C., Cicolani, N., Petrini, S., et al. (2024). Caspase-dependent apoptosis in riboflavin transporter deficiency iPSCs and derived motor neurons. Cell Death Discovery 10:54. doi: 10.1038/s41420-024-01812-y
Masamizu, Y., Okada, T., Kawasaki, K., Ishibashi, H., Yuasa, S., Takeda, S., et al. (2011). Local and retrograde gene transfer into primate neuronal pathways via adeno-associated virus serotype 8 and 9. Neuroscience 193, 249–258. doi: 10.1016/j.neuroscience.2011.06.080
Maury, Y., Come, J., Piskorowski, R. A., Salah-Mohellibi, N., Chevaleyre, V., Peschanski, M., et al. (2014). Combinatorial analysis of developmental cues efficiently converts human pliripotent stem cells into multiple neuronal subtypes. Nat Biothecnol. 33, 89–96. doi: 10.1038/nbt.3049
Murlidharan, G., Samulski, R. J., and Asokan, A. (2014). Biology of adeno-associated viral vectors in the central nervous system. Front. Mol. Neurosci. 7:2014. doi: 10.3389/fnmol.2014.00076
Niceforo, A., Marioli, C., Colasuonno, F., Petrini, S., Massey, K., Tartaglia, M., et al. (2021). Altered cytoskeletal arrangement in induced pluripotent stem cells and motor neurons from patients with riboflavin transporter deficiency. DMM Dis. Model. Mech. 14:46391. doi: 10.1242/DMM.046391
O’Callaghan, B., Bosch, A. M., and Houlden, H. (2019). An update on the genetics, clinical presentation, and pathomechanisms of human riboflavin transporter deficiency. J. Inherit. Metab. Dis. 42, 598–607. doi: 10.1002/jimd.12053
Ogbonmide, T., Rathore, R., Rangrej, S. B., Hutchinson, S., Lewis, M., Ojilere, S., et al. (2023). Gene therapy for spinal muscular atrophy (SMA): a review of current challenges and safety considerations for Onasemnogene Abeparvovec (Zolgensma). Cureus. 15:e36197. doi: 10.7759/cureus.36197
Okita, K., Matsumura, Y., Sato, Y., Okada, A., Morizane, A., Okamoto, S., et al. (2011). A more efficient method to generate integration-free human iPS cells. Nat. Methods 8, 409–412. doi: 10.1038/nmeth.1591
Rizzo, F., Ramirez, A., Compagnucci, C., Salani, S., Melzi, V., Bordoni, A., et al. (2017). Genome-wide RNA-seq of iPSC-derived motor neurons indicates selective cytoskeletal perturbation in Brown-Vialetto disease that is partially rescued by riboflavin. Sci. Rep. 7:271. doi: 10.1038/srep46271
Samaranch, L., Salegio, E. A., San Sebastian, W., Kells, A. P., Bringas, J. R., Forsayeth, J., et al. (2013). Strong cortical and spinal cord transduction after AAV7 and AAV9 delivery into the cerebrospinal fluid of nonhuman primates. Hum. Gene Ther. 24, 526–532. doi: 10.1089/hum.2013.005
Saraiva, J., Nobre, R. J., and Pereira de Almeida, L. (2016). Gene therapy for the CNS using AAVs: the impact of systemic delivery by AAV9. J. Control. Release 241, 94–109. doi: 10.1016/j.jconrel.2016.09.011
Shen, S., Bryant, K. D., Brown, S. M., Randell, S. H., and Asokan, A. (2011). Terminal n-linked galactose is the primary receptor for adeno-associated virus. J. Biol. Chem. 286, 13532–13540. doi: 10.1074/jbc.M110.210922
Snyder, B. R., Gray, S. J., Quach, E. T., Huang, J. W., Leung, C. H., Samulski, R. J., et al. (2011). Comparison of adeno-associated viral vector serotypes for spinal cord and motor neuron gene delivery. Hum. Gene Ther. 22, 1129–1135. doi: 10.1089/hum.2011.008
Keywords: human pluripotent stem cells, motoneuronal differentiation, neurodegenerative autosomal recessive disease, morphological neuronal phenotyping, gene therapy
Citation: Mei C, Magliocca V, Chen X, Massey K, Gonzalez-Cordero A, Gray SJ, Tartaglia M, Bertini ES, Corti S and Compagnucci C (2025) Riboflavin transporter deficiency: AAV9-SLC52A2 gene therapy as a new therapeutic strategy. Front. Cell. Neurosci. 19:1523773. doi: 10.3389/fncel.2025.1523773
Edited by:
Manoj Menezes, The Children’s Hospital at Westmead, AustraliaReviewed by:
Maria do Carmo Costa, University of Michigan, United StatesCopyright © 2025 Mei, Magliocca, Chen, Massey, Gonzalez-Cordero, Gray, Tartaglia, Bertini, Corti and Compagnucci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefania Corti, c3RlZmFuaWEuY29ydGlAdW5pbWkuaXQ=; Claudia Compagnucci, Y2xhdWRpYS5jb21wYWdudWNjaUBvcGJnLm5ldA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.