 Marta Camici
Marta Camici Giulia Del Duca
Giulia Del Duca Anna Clelia Brita
Anna Clelia Brita Andrea Antinori
Andrea Antinori- 1Clinical and Research Infectious Diseases Department, National Institute for Infectious Diseases Lazzaro Spallanzani Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Rome, Italy
- 2Department of Clinical Psychology, National Institute for Infectious Diseases Lazzaro Spallanzani Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Rome, Italy
The pathogenesis of long COVID (LC) still presents many areas of uncertainty. This leads to difficulties in finding an effective specific therapy. We hypothesize that the key to LC pathogenesis lies in the presence of chronic functional damage to the main anti-inflammatory mechanisms of our body: the three reflexes mediated by the vagus nerve, the hypothalamic-pituitary-adrenal (HPA) hormonal axis, and the mitochondrial redox status. We will illustrate that this neuro-endocrine-metabolic axis is closely interconnected and how the SARS-CoV-2 can damage it at all stages through direct, immune-inflammatory, epigenetic damage mechanisms, as well as through the reactivation of neurotropic viruses. According to our theory, the direct mitochondrial damage carried out by the virus, which replicates within these organelles, and the cellular oxidative imbalance, cannot be countered in patients who develop LC. This is because their anti-inflammatory mechanisms are inconsistent due to reduced vagal tone and direct damage to the endocrine glands of the HPA axis. We will illustrate how acetylcholine (ACh) and cortisol, with its cytoplasmatic and cellular receptors respectively, are fundamental players in the LC process. Both Ach and cortisol play multifaceted and synergistic roles in reducing inflammation. They achieve this by modulating the activity of innate and cell-mediated immunity, attenuating endothelial and platelet activation, and modulating mitochondrial function, which is crucial for cellular energy production and anti-inflammatory mechanisms. In our opinion, it is essential to study the sensitivity of the glucocorticoids receptor in people who develop LC and whether SARS-CoV-2 can cause long-term epigenetic variations in its expression and function.
Introduction
Long COVID (LC) was redefined by the US National Academies of Sciences, Engineering, and Medicine as “an infection-associated chronic condition that occurs after severe acute respiratory syndrome Coronavirus 2 (SARS-CoV-2) infection, present for at least three months, manifesting as continuous, relapsing, or progressive disease affecting one or more organ systems” (A Long COVID Definition: A Chronic, Systemic Disease State with Profound Consequences, 2024). This consensus classified LC among chronic health issues following infections of any kind, such as myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and Lyme-associated chronic illness, without needing laboratory confirmation of the initial infection (A Long COVID Definition: A Chronic, Systemic Disease State with Profound Consequences, 2024). LC can worsen preexisting conditions or emerge as new issues, impacting individuals’ ability to work, attend school, care for families, and manage self-care. A recent meta-analysis found that nearly half of Coronavirus disease 2019 (COVID-19) survivors reported lingering symptoms around 120 days post-recovery, with about almost 7.3% developing high-morbidity LC, predominantly affecting young, productive individuals (Mastrorosa et al., 2023). In minors, LC is linked to a more severe acute illness requiring hospitalization, whereas in adults, it often occurs in those with mild to moderate COVID (Su et al., 2022). Risk factors for developing LC include female sex (Bai et al., 2022), multimorbidity, unvaccinated status (Robertson et al., 2023), EBV reactivation, autoimmunity disorders, asthma, type 2 diabetes (Su et al., 2022), attention deficit hyperactivity disorder (ADHD), chronic urticaria and allergic rhinitis (Merzon et al., 2022), although a third of people with LC have no identified pre-existing conditions (Davis et al., 2023). Unfortunately, no biological marker for LC has been found, and no cures exist, with spontaneous recovery being rare (Ely et al., 2024). Actually, various pathogenetic mechanisms of LC have been proposed, including persistent infection, autoimmunity, antigenic mimicry, mitochondrial damage, vagus nerve (VN) injury (Woo et al., 2023), hypercoagulability, microbiota alterations (Alvarez-Santacruz et al., 2024), neurotropic virus reactivation, hypothalamic-pituitary-adrenal (HPA) gland axis dysfunction, and epigenetic modification in gene cell expression (Cheong et al., 2023). Different clinical form of LC have been identified, based on symptoms, but it remains unclear if they reflect distinct pathogenetic pathways. SARS-CoV-2 infection likely has a pleiotropic effect, with multiple causative pathways present concurrently. Notably, LC symptoms fluctuate unpredictably, and the hierarchy and relationships between these mechanisms are still not fully understood, nor is it clear if a common thread connects them.
Vagus nerve damage may play a central role in long COVID pathogenesis, potentially reducing the body’s anti-inflammatory response and mitochondrial cell function
Our hypothesis proposes that VN damage is a primary contributor to LC development, leading to dysautonomia and disruption of key anti-inflammatory pathways, and mitochondrial function (VanElzakker, 2013). SARS-CoV-2 has been shown to induce both direct and indirect VN damage (Andersson and Tracey, 2024). Woo et al. conducted a post-mortem analysis demonstrating direct SARS-CoV-2 infection of the VN, accompanied by significant neuroinflammation (Woo et al., 2023). Specifically, by studying cells gene expression, they observed increased interferon (IFN) signaling in activated monocytes, glial cells, endothelial cells, and Schwann cells within brain tissue, irrespective of viral load (Woo et al., 2023). Conversely, SARS-CoV-2 RNA load in vulnerable neurons showed a positive correlation with upregulation of stress response mechanisms (e.g., autophagy, proteasomal breakdown). However, these higher levels of intracellular SARS-CoV-2 RNA were also associated with reduced activity in genes responsible for neurotransmitter signaling and neuronal transport, showing a dose-dependent direct VN damage and a dose independent neuroinflammatory response (Woo et al., 2023). Moreover, autoantibodies targeting receptors involved in the vagal anti-inflammatory reflex have been found in convalescent COVID-19 patients, indicating potential functional impairment beyond nerve damage (Dobrowolska et al., 2023). In addition to the VN impairment mechanisms already mentioned, alterations in coagulation and the reactivation of herpes viruses can collaborate to neurotoxicity (VanElzakker, 2013). Interestingly, VN dysfunction triggered by SARS-CoV-2 infection occurs early on during the infection and contributes to the virus’s virulence. In the acute phase, the impairment of the anti-inflammatory reflex may sustain the development of severe cytokine storms, leading to conditions like Acute Respiratory Distress Syndrome (ARDS) and micro embolisms, which increase organ damage and worsen prognosis severity (Johnston and Webster, 2009; Song et al., 2020). Accordingly, Woo et al. found that COVID-19 patients who died exhibited a lower respiratory rate compared to survivors, even in the presence of elevated blood carbon dioxide and severe respiratory insufficiency. This finding suggests that damage to the VN, resulting in autonomic dysfunction, may contribute to respiratory failure in severe COVID-19 cases (Woo et al., 2023).
In the post-COVID phase, impaired vagal signaling could have a pivotal role in preventing the body from restoring inflammatory balance, ultimately perpetrating the LC syndrome (Llados et al., 2024). To strengthen our position, this mechanism was already postulated in a cohort of patients affected by irritable bowel syndrome (IBS) and Crohn’s disease (CD) (Pellissier et al., 2014). In fact, this relevant research found that healthy individuals with higher vagal tone in the morning, defined as an high heart rate variability (HRV), tended to have lower cortisol levels in the evening (Pellissier et al., 2014). This inverse relationship proves the clear neuroendocrinal correlation between VN and axis and suggests that greater vagal tone is associated with better regulation of the stress hormone cortisol (Pellissier et al., 2014). Interestingly, this correlation was not observed in patients with IBS, suggesting an impairment in the vagal anti-inflammatory mechanisms. Further supporting this finding, lower vagal tone in CD patients correlated with higher levels of the inflammatory marker tumor necrosis factor-alpha (TNF-α) (Pellissier et al., 2014). The same study underlined that IBS patients with low vagal tone display high epinephrine levels, indicating heightened and potentially maladaptive sympathetic nervous system activity (Pellissier et al., 2014). Accordingly, HRV was found to be reduced in LC patients compared to a control group, thus reflecting a reduced VN tone (da Silva et al., 2023).
The VN plays a crucial role in regulating the anti-inflammatory response and maintaining balance within the neuro-endocrine-immune system. This is achieved by stimulating the nucleus of the solitary tract (NTS) in the medulla oblongata. The NTS, activated by afferent VN fibers, detects and responds to inflammatory and immune signals from body tissues, initiating three separate pathways (Figure 1) (Kaniusas et al., 2020).
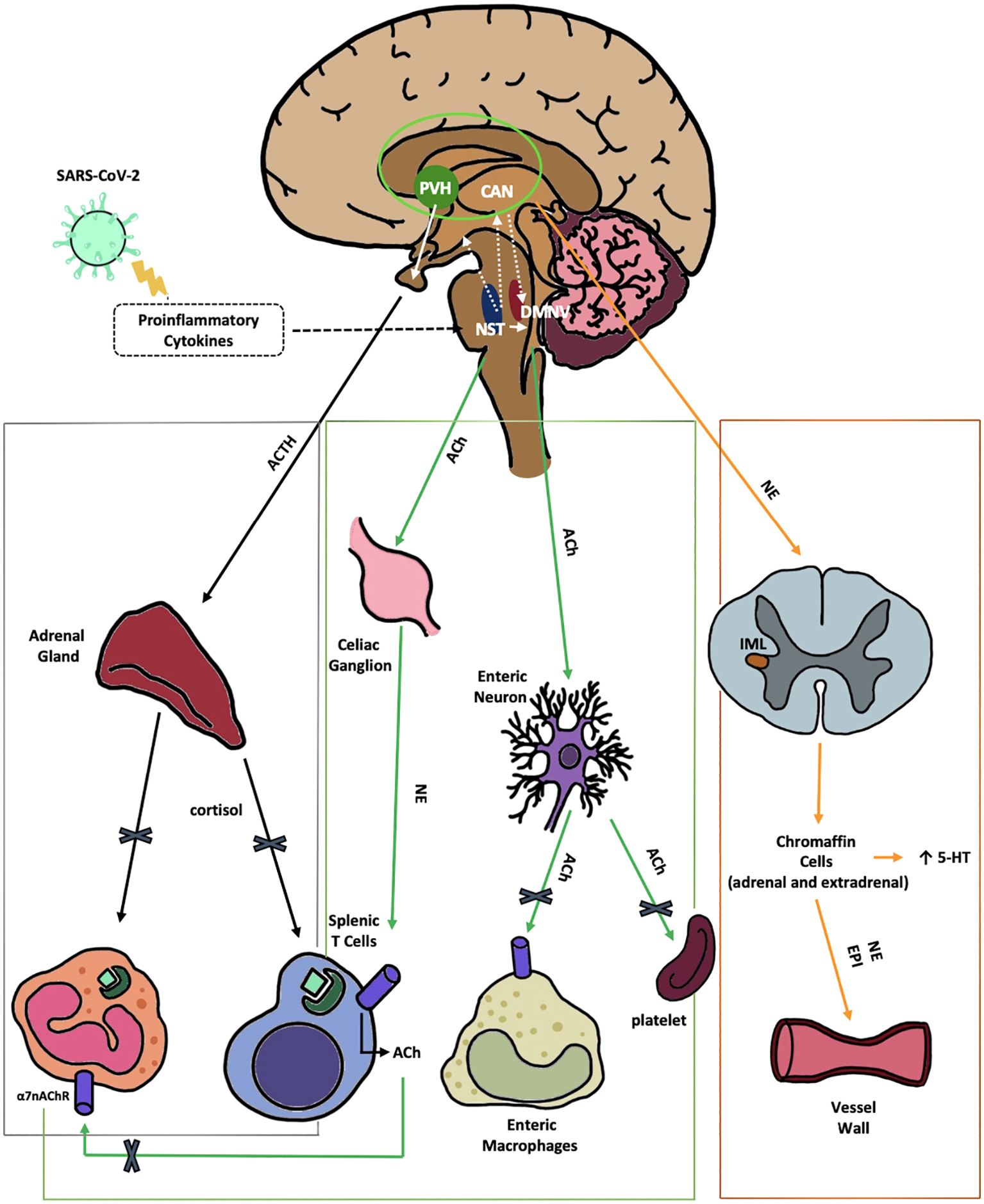
Figure 1. Anti-inflammatory pathways of the VN disrupted by SARS-CoV-2. 1) The HPA axis reflex (black) depicts the suppression of the hypothalamic-pituitary-adrenal axis reflex, leading to a reduction in cortisol production by SARS-CoV-2. This reduction in cortisol impairs the regulation of both innate and adaptive immune responses. Nerve fibers from the NTS stimulate the release of CRH by neurons in the PVH, a hypothalamic nucleus within the central autonomic network. The CAN, comprised of the thalamus, amygdala, hypothalamus, and brainstem nuclei, integrates emotional, sensory, and cognitive stimuli to produce autonomic and endocrine responses. 2) The cholinergic anti-inflammatory reflex (green) illustrates the VN projection to the gastrointestinal tract. This reflex is initiated by afferent fibers from the NTS, which relay peripheral visceral sensory information to the DMNV. ACh, released from DMNV efferent fibers, finally inhibits cytokine release from intestinal macrophages, thereby mitigating local inflammation. Moreover ACh suppress platelets activation. 3) The vago-sympathetic pathway (orange) is activated by vagal efferent stimuli originating from the NTS. This pathway regulates sympathetic outflow, thought the CAN. It targets preganglionic sympathetic neurons in the IML of the spinal cord and chromaffin cells. This complex network modulates peripheral blood tone, vasoconstriction, and immune responses. Enterochromaffin cells are involved in the gut metabolism of 5-HT. α7-nAChR, αlpha7-nicotinicACh-Receptor; ACh, acetylcholine; ACTH, adrenocorticotropic hormone; CAN, central autonomic network; DMNV, dorsal motor nucleus of vagus nerve; EPI, epinephrine; HPA, hypothalamic–pituitary–adrenal; IML, intermediolateral nucleus; NE, norepinephrine; NTS, nucleus tractus solitarius; PVH, parvo-cellular nucleus; VN, vagus nerve; 5-HT, serotonin.
The hypothalamic-pituitary-adrenal axis reflex: the neuroendocrine anti-inflammatory pathway
The VN carries out its anti-inflammatory function by activating the axis reflex, leading to increased cortisol production. Therefore, it plays a crucial role in the neuro-immuno-endocrine axis (Figure 1, Black line).
The efferent A2 noradrenergic group of VN fibers from the NTS directly activate the secretion of corticotrophin-releasing hormone (CRH) by the hypothalamic neurons located in the parvo-cellular PVH nucleus (Kaniusas et al., 2020). This nucleus plays a role within the central autonomic network (CAN), an intricate network connecting various nervous centers such as the thalamus, amygdala, hypothalamus, and brain stem nuclei. Together, these centers integrate emotional, sensory, and cognitive stimuli to generate autonomic, behavioral, and endocrine responses. Upon CRH stimulation, adrenocorticotropic hormone (ACTH) is released from the pituitary gland. ACTH then travels to the adrenal glands where it prompts the production of cortisol. Cortisol, in turn, hinders the activation of both innate immunity (splenic and tissue macrophages) and adaptive immunity (spleen T-lymphocytes) via interaction with the glucocorticoid receptor (GR). Glucocorticoids (GCs) bind to the cytoplasmic GR, facilitating its translocation into the nucleus as a transcription factor (Scheinman et al., 1995). A neurovisceral integration model suggests that impaired VN tone, frequently seen in LC patients (Acanfora et al., 2022), contributes to chronic changes in cortisol production and elevated levels of proinflammatory cytokines and acute-phase proteins (Thayer and Sternberg, 2006).
GCs reduce inflammation in several ways. One key mechanism is increasing the production of IKBα protein, which blocks the activity of nuclear factor kappa B (NF-KB), a factor that activates immune response genes (Auphan et al., 1995). GCs also impact immune cells like monocytes, macrophages, and T helper (Th) lymphocytes, influencing their movement, function, and survival (Quatrini and Ugolini, 2021). For example, GCs can suppress inflammation in asthma by altering the balance of Th1 and Th2 cells (Hu et al., 2018). Additionally, GCs can increase CXCR4 expression, affecting the migration of B cells (Cain et al., 2020). Recently, it was discovered that GCs can also activate immune checkpoints in cytotoxic lymphocytes, further suppressing immune responses (Quatrini et al., 2018).
During SARS-CoV-2 infection, both vagal signaling and axis function appear to be impaired, potentially hindering this reflex at various stages. A comparison of autopsies from individuals who died from COVID-19 and those who died from influenza revealed that all COVID-19 patients, but only a small number of influenza patients, had adrenalitis with significantly more severe damage to the structure of the adrenal cortex. Additionally, COVID-19 patients with intensive care unit (ICU) stays exceeding one week showed widespread fibrosis and degeneration of their adrenal glands (Paul et al., 2022). The same authors demonstrated that the SARS-CoV-2 virus has a strong affinity for and replicates effectively in vivo within adrenal cortical cells, which express angiotensin converting enzyme 2 (ACE2) and transmembrane protease serine 2 (TMPRSS2) membrane receptors (Paul et al., 2022). Intriguingly, a UK study on 353 hospitalized patients with suspected COVID-19 found that those with confirmed infections had significantly higher cortisol levels early in their illness compared to those without COVID-19 (Tan et al., 2020). This suggests potential adrenal injury and a strong stress response in COVID-19 patients. Additionally, the high cortisol levels was a reliable severity disease marker during the acute infection (Tan et al., 2020). The authors propose that these patients may develop adrenal insufficiency later in their illness, similar to what has been seen in patients with prolonged ICU stays (Tan et al., 2020). Accordingly, impairment of the VN neuro-endocrine reflex, coupled with potential direct damage to endocrine glands by SARS-CoV-2, can lead to reduced cortisol secretion, that was an hallmark of LC (Klein et al., 2023). Notably, a US study identified low morning blood cortisol as a strong predictor of LC development (Wallukat et al., 2021; Klein et al., 2023). This study also observed a blunted stress-induced increase in ACTH and a flat diurnal rhythm of GCs in LC patients, suggesting impaired axis feedback regulation (Jacobson et al., 1988) as previously observed for SARS-CoV (Leow et al., 2005). Indeed, after the acute phase of SARS-CoV, many survivors develop central hypocortisolism, which is characterized by low or inappropriately normal ACTH levels (Leow et al., 2005).
It is also fascinating that molecular mimicry between ACTH and the SARS-CoV viruses has been shown (Wheatland, 2004). The presence of antibodies against SARS-CoV-2 could collaborate to impair the body’s stress response and potentially impact the cortisol response during LC (Wheatland, 2004). Even more interesting is the fact that cortisol seems to directly inhibit the binding of the Spike S1 protein to its intracellular ACE2 receptor, a mechanism that has been postulated to underlie asymptomatic infections (Sarker et al., 2022). The central role of adrenal dysfunction in both LC and ME/CFS is further underscored by the significant overlap of symptoms with adrenal fatigue (AF). AF, a stress-related disorder, typically arises when the adrenal glands struggle to meet heightened cellular energy demands following prolonged stress or trauma (Wilson, 2014). While the similarities in clinical presentation are striking, unlike LC, AF can often be reversed through significant lifestyle modifications, dietary adjustments, and supplements, although recovery may take up to two years (Wilson, 2014).
The cholinergic anti-inflammatory reflex is fundamental in maintaining anti-inflammatory and coagulative homeostasis
The second anti-inflammatory reflex mediated by the VN, the cholinergic anti-inflammatory reflex (Figure 1, green lines), involves efferent fibers from the dorsal motor nucleus of the VN (DMNV). This reflex is initiated by afferent fibers from the NTS, which relay peripheral visceral sensory information to the DMNV (Kaniusas et al., 2020). The DMNV, through its efferent cholinergic pathway, modulates immune responses in the spleen (via the celiac ganglion), liver, and gastrointestinal (GI) tract by the enteric neurons suppressing pro-inflammatory cytokines. In the spleen, acetylcholine (ACh) stimulates celiac neurons, leading to norepinephrine (NE) release, which suppresses cytokines from macrophages, both directly and via splenic T-cell activation. Similarly, ACh stimulates cholinergic enteric neurons in the GI tract, locally inhibiting the innate immune response (Pavlov and Tracey, 2012). Research in mice shows that absence of this reflex enhances innate immune responses and cytokine-related toxicity (Auphan et al., 1995). Moreover, animal models inducing sepsis, ischemia-reperfusion, and pancreatitis, have indicated that VN nerve stimulation (VNS) reduces TNF-α synthesis, prevent acute inflammation, and ultimately enhance overall health outcomes (Borovikova et al., 2000). This reflex involves ACh receptors (AChR) in the celiac ganglion, α7-nicotinic acetylcholine receptors (α7-nAChR) on macrophages, and both NE receptors and β2-adrenergic receptors (β2AR) on T-lymphocytes and macrophages in the spleen. In α7-subunit knockout mice, TNF-α levels fail to decrease after endotoxin exposure, along with a less significant reduction in interleukin(IL)-1β and IL-6 production (Wang et al., 2003), highlighting the receptor’s role in reducing inflammation. T lymphocytes, central to both effector and regulatory immune functions, play a critical role in inflammation and autoimmunity (Hu et al., 2018). Notably, distinct splenic and intestinal T cell subsets express functional choline acetyltransferase (ChAT) and synthesize ACh, constituting a population termed ChAT+ T cells. These ChAT+ T cell subsets are predominantly localized near catecholaminergic splenic nerve fibers, establishing a non-neuronal cholinergic reservoir. The ACh released within this microenvironment activates α7-nAChR on T cells, thereby facilitating their activation and proliferation (Halder and Lal, 2021). Nicotine activation of α7-nAChR in a mouse model of autoimmune encephalomyelitis alleviated symptoms, shifting CD4+ T cells tow an anti-inflammatory IL-4-producing Th2 phenotype while reducing Th1 and Th17 cytokines (Yamakawa et al., 2020). Intriguingly, a sequence in the SARS-CoV-2 spike glycoprotein receptor-binding domain (RBD), termed SARS-CoV-2 glycoprotein peptide (SCoV2P), shares homology with the snake venom neurotoxin NL1, that interact with nAChRs (Farsalinos et al., 2020). This peptide, SCoV2P, modulates α7-nAChRs, enhancing ACh-mediated currents at low concentrations and inhibiting them at higher concentrations (Farsalinos et al., 2020). Other immune cells like mast cells, microglia cells, Kupffer cells may also express α7-nAChRs and could potentially be sensitive to ACh’s anti-inflammatory effects (Wang et al., 2003). Moreover, α7-nAChRs on platelets form functional Ca2+ channels, suggesting that ACh acts as a natural inhibitor of platelet activation (Schedel et al., 2011), with implications for thrombotic complications in COVID-19. Li et al. demonstrated that chronic VN stimulation in mice can suppress endothelial activation during inflammation (Li et al., 2016). Further research by the same group revealed that Ach, by binding nAChRs, inhibits the expression of adhesion molecules like VCAM-1, ICAM-1, and E-selectin on endothelial cells. This binding also reduces cytokine production, providing new insights into the anti-inflammatory and anti-thrombotic effects associated with the cholinergic anti-inflammatory reflex (Li et al., 2022). Supporting this, some LC patients experienced improvement with nicotine patches, which act as competitive agonists for nAChRs, potentially restoring cholinergic function (Leitzke, 2023).
The vagus-sympathetic pathway influences the anti-inflammatory reflex and regulates serotonin production and reabsorption in the gut
The vagus-sympathetic pathway is the third autonomic route potentially impaired in LC. Vagal efferent stimuli from the NTS activate five brain nuclei within the CAN, which regulates sympathetic outflow and targeting preganglionic sympathetic neurons located in the intermediolateral nucleus (IML) of the spinal cord (Woo et al., 2023) (Figure 1, orange line). This network modulates blood tone, vasoconstriction, and immune responses through postganglionic sympathetic neurons targeting adrenal chromaffin cells, enterochromaffin cells (ECs), the celiac ganglion, and the spleen (Andersson and Tracey, 2024). Challenging the traditional view of the VN as purely parasympathetic, experimental studies has shown that in many species the VN stimulation has a double role in both increasing and reducing heart rate (Chiang and Leaders, 1966), and allow to retain endogenous NE in heart (Jellinek et al., 1965). Finally, an animal histochemical study identified a component of adrenergic fibers in the VN, which are characterized as small and unmyelinated (Muryobayashi et al., 1968). This suggests the VN may influence the anti-inflammatory reflex also by modulating sympathetic activity.
Interestingly, a recent study found that LC is associated with decreased enteral reabsorption of serotonin (5-HT), potentially due to gut inflammation and impaired vagal signaling, both mediated by SARS-CoV-2 (Wong et al., 2023). While primarily stored in ECs, 5-HT is also present in gut nerve terminals and mast cells. The vagal-adrenergic pathway is essential for 5-HT release from ECs into portal circulation and the gut lumen, mediated by sympathetic fibers (Linan-Rico et al., 2016). Furthermore, gut 5-HT has been shown to upregulate VN activity, demonstrating a bidirectional relationship. Intact vagal signaling between the gut and brain is essential for mediating the behavioral effects of orally administered selective 5-HT reuptake inhibitors (SSRIs) (McVey Neufeld et al., 2019). This suggests that SSRIs may function, in part, by restoring VN function, highlighting its crucial in 5-HT regulation (McVey Neufeld et al., 2019). According to this theory, while intestinal inflammation may contribute to reduced 5-HT absorption, the inability to restore efficient VN signaling could be a key factor in the 5-HT depletion observed in LC. Peripheral 5-HT deficiency, in turn, impairs cognitive function via reduced vagal signaling (Manta et al., 2009; McVey Neufeld et al., 2019). Given this understanding, therapeutic strategies targeting the VN hold promise for LC treatment. Preliminary studies indicate that non-invasive VNS may improve cognitive function, mood, sleep, and fatigue in LC patients (Yap et al., 2020; Zheng et al., 2024).
Autoantibodies targeting G-protein-coupled receptors may play a pivotal role in altering neurological, immunological, and cellular functions in long COVID
The impairment of the three identified anti-inflammatory reflexes appears central to the pathogenesis of LC. This impairment may stem from initial direct VN damage, perpetuated by a dysregulated host response to SARS-CoV-2 remnants, particularly in the gut. This sustained immune response could lead to the production of autoantibodies targeting receptors crucial to these reflexes and vasal nerve tone. Intriguingly, a recent preprint demonstrated that transferring IgG from specific LC patient subgroups, stratified by symptom patterns, to healthy mice induced similar LC symptoms. This finding aligns with previous research in ME/CFS and fibromyalgia (Blomberg et al., 2018; Goebel et al., 2021; Chen et al., 2024).
This theory is supported by the observation of elevated circulating autoantibodies in LC patients compared to healthy individuals. Many of these autoantibodies target GPCR muscarinic (mAChR) or nAChR and adrenergic receptors (Wallukat et al., 2021). While the precise pathogenic mechanisms of anti-GPCR autoantibodies in LC warrant further investigation, these autoantibodies could contribute to LC pathology by causing damage to nerve signal transmission, reducing microvascular blood flow, directly impacting cardiac tissue, and/or triggering mast cell activation. Given the established roles of anti-GPCR autoantibodies in diseases like glaucoma and cardiomyopathy, and the promising efficacy of GPCR-targeting molecules in preclinical studies (Campbell and Smrcka, 2018), further research is warranted to elucidate the interplay between autoimmunity, GPCR dysfunction, and the development of LC.
Mitochondrial dysfunction in SARS-CoV-2 acute infection and long COVID
Mitochondria generate adenosine triphosphate (ATP), the primary energy currency of cells, through cellular respiration and are crucial for cells function. Additionally, mitochondria contribute to various cellular processes, including cell differentiation, autophagy, apoptosis, calcium 2+ (Ca2+) signaling, and thermoregulation (Andrieux et al., 2021). This double-membrane organelles contain multiple copies of their own DNA (mtDNA), that encode subunits of the respiratory-chain complexes, as well as ribosomal and transfer RNAs needed for their synthesis (Wallace, 2005). However, complex II subunit and other mitochondrial proteins, are encoded by the nuclear genome and transported into the mitochondria, where they fold into their functional forms (Walker and Moraes, 2022). Under stressful events, if the mechanisms ensuring mitochondrial integrity and protein folding fail, misfolded proteins can accumulate within mitochondria, leading to dysfunction and cellular apoptosis (Tatsuta, 2009). The dynamic nature of mitochondria allows them to adapt their mass based on cellular energy demands and external stimuli (Scarpulla et al., 2012). They constantly undergo fission and fusion to maintain cellular homeostasis and eliminate damaged organelles through mitophagy (Twig et al., 2008). Notably, mitochondria play a vital role in the innate immune and inflammatory responses. In fact, upon detecting infection, pattern-recognition receptors (PRRs) starts mitochondrial antiviral signaling proteins, leading to increased production of mitochondrial reactive oxygen species (mtROS) (Andrieux et al., 2021). This process activates the Nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin domain containing 3 (NLRP3) inflammasome, induces pro-inflammatory gene expression, and triggers the release of pro-inflammatory cytokines, ultimately leading to the elimination of infected cells and viral clearance (Mantle et al., 2024).
Although mitochondrial damage is a common virulence mechanism for many pathogens to evade the immune response (Mehrzadi et al., 2021; Singh et al., 2021), animal models have confirmed that coronaviruses (CoVs) are particularly pernicious against them, directly invading mitochondria and relying on them for its replication (Zhang et al., 2020). A study, comparing gene expression in lung cells infected with Influenza A virus, SARS-CoV-2, and Middle East respiratory syndrome (MERS)-CoV, revealed that all three viruses caused an increase in IFN signaling genes. However, only the two CoVs caused increment of mtROS production and perturbation in autophagy. Besides, only SARS-CoV-2 infection led to an increase in inflammatory and cytokine signaling genes (Singh et al., 2021). SARS-CoV-2 has been shown to manipulate host cell mitochondria for its benefit, inducing mitochondrial damage at multiple levels of biological function. The mitochondrial mechanisms damage, during SARS-CoV-2 infection, are listed in Table 1. For instance, the spike protein can reduce normal mitochondrial respiration and ATP production while simultaneously increasing glucose-induced glycolysis to promote its metabolic path (Lei et al., 2021; Shang et al., 2021). This effectively hijacks the host’s metabolic processes, as evidenced by elevated lactate and glucose levels in infected patients (Moolamalla et al., 2021). Studies show that the virus disrupts the organization and function of the electron transport chain, as evidenced by the mis-localization of key proteins in infected cells (Soria-Castro et al., 2021). This disruption, along with impaired expression of mitochondrial and antioxidant genes, compromises the mitochondrial membrane and the oxidative phosphorylation (OXPHOS), ultimately increasing harmful mtROS (Soria-Castro et al., 2021). Furthermore, to improve its replication, SARS-CoV-2 directly downregulate the expression of genes crucial for mitochondrial ribosomes and Complex I, thereby impairing the mitochondrial respiratory chain, increasing cell oxidative stress causing the loss of mitochondrial integrity and cell death (Singh et al., 2021). Another mitochondrial injury mechanism, is mediated by the CoVs non-structural proteins Open Reading Frame (ORF), such as ORF-7a, ORF-8a and ORF-9b, which are located in mitochondria and inhibit the retinoic acid-inducible gene I-mitochondrial antiviral signaling protein (RIG1-MAVS)-dependent IFN signaling, evading host cell immunity and promoting viral replication (Singh et al., 2020). Additionally, ORFs can directly trigger the mitochondrial apoptotic pathways (Ren et al., 2020; Mehrzadi et al., 2021). Specifically, the SARS-CoV-2 ORF3a protein can dimerize and generate a non-selective Ca2+ permeable cation channels on the mitochondrial membrane, that upregulates Ca2+ signaling from endoplasmic reticulum (Davies et al., 2020). This activates e cascade starting with Ca2+-dependent caspases, that leads to programmed cell death (Ren et al., 2020), contributing to the reduced lymphocyte count observed in COVID-19 (Ren et al., 2016; Mehrzadi et al., 2021). SARS-CoV-2 proteins can disrupt cellular Ca2+ balance also by direct blocking L-type Ca2+ channels, perturbing cardiac energetic and ultimately cause cardiomyocyte death (Ramachandran et al., 2022). Moreover, SARS-CoV-2 may induce the NLRP3 inflammasome, and upregulate the expression of the inflammatory cytokine genes such as IL-1β and IL-18 and contributing to the proinflammatory storm. This can trigger pyro-ptosis, a highly inflammatory form of programmed cell death, particularly in lymphocytes and macrophages (Yang, 2020). Additionally, CoVs can antagonize the unfolded protein response and organelle fission, resulting in hyperfused, non-active mitochondria (Tondera et al., 2009; Shi et al., 2014). Accordingly, a study on mitochondrial morphological changes during acute infection found mitochondrial swelling and vacuolization after one day (Shang et al., 2021). Notably, a recent study has shown that the degrading mitochondrial material eliminated by the cells stolen in extracellular vesicles can activate the innate immune response and autoantibodies cross reaction against mitochondrial antigens (Di Florio et al., 2024). In addition to that, SARS-CoV-2 has been shown to disrupt mitophagy. While the virus initially upregulates this process by activating the Pink1-Parkin-P62 pathway, it subsequently inhibits its completion by obstructing the binding of P62 and LC3, which is essential for the selective engulfment of targeted components into autophagosomes (Shang et al., 2021). Lastly, SARS-CoV-2 may indirectly affect mitochondrial function by impacting cortisol and Ach metabolism, as discussed later.
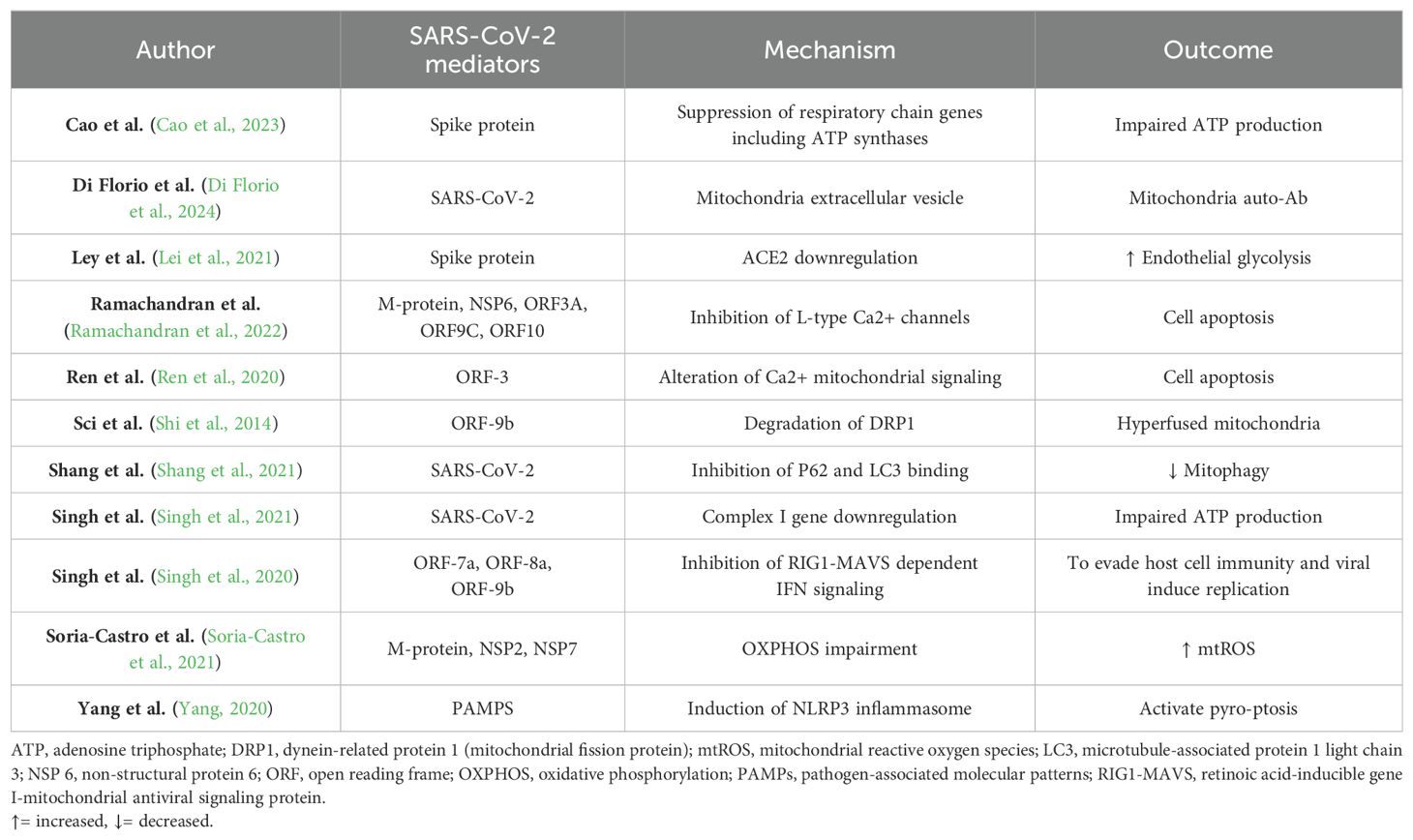
Table 1. Mitochondrial Damage Mechanisms Induced by SARS-CoV-2.
Interestingly, mitochondria dysfunction seems last long in those patients developing LC, although the precise mechanisms require further investigation (Chen et al., 2023; Molnar et al., 2024). For example, a study conducted on mice infected by a lentivirus vector encoding sequence of Spike gene demonstrated that Spike protein can induce transcriptional suppression of metabolic genes, determining an impairment in energy production and redox status, hesitating in morphologic cells change, and cardiac fibrosis (Cao et al., 2023). Similarly, neuropsychiatric manifestation of LC resulted correlated with the increment of mitochondrial proteins and SARS-CoV-2 proteins in neuronal and astrocytes-derived patients exosomes (Peluso et al., 2022). Further studies are needed to understand the evolution of mitochondrial damage over time and its specific impact on the pathogenesis of LC. It will also be important to determine the mechanisms by which those who do not develop LC restore basal mitochondrial function and what prevents patients from returning to the previous equilibrium.
The altered cortisol signaling can affect both cellular gene expression and mitochondrial function
Cortisol, produced in the zona fasciculata cells of the adrenal cortex, is synthesized from cholesterol in mitochondria-rich cells, regulated by two families of mitochondrial enzymes: cytochrome P450 and the hydroxysteroid dehydrogenase/ketosteroid reductase (HSD/KSR) (Midzak and Papadopoulos, 2016).
Although primarily produced in the adrenal gland, extra-adrenal cortisol is also synthesized in the brain, thymus (Talaber et al., 2015), blood vessels, and epithelial cells (Noti et al., 2009), contributing to local hormonal regulation (Taves et al., 2011). Transported in the blood by cortisol-binding protein (Henley et al., 2016), cortisol becomes active inside cells after conversion by 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1), an enzyme with tissue-specific regulation (Timmermans et al., 2019). Biologically active cortisol then binds to its intracellular receptors and mediates a wide range of cell-specific effects (Rhen and Cidlowski, 2005). The human GR gene is highly variable, producing multiple receptor isoforms, with 13 exon-1 variants that regulate transcription, reflecting the complexity of GR gene expression (Timmermans et al., 2019; Quatrini and Ugolini, 2021). The binding of transcription factors to gene variants can significantly affect receptor protein availability in target tissues (Vandevyver et al., 2014), while alternative splicing of the GR gene generates multiple protein isoforms, with intracellular factors further regulating ligand sensitivity. This intricate regulation enables the GR to function in a cell-specific manner, influencing responses to cortisol and GC treatments (Nagy et al., 2016). Epigenetic modifications like DNA methylation and histone acetylation further regulate GR gene expression, which can profoundly influence their function (Timmermans et al., 2019). Interestingly, the presence or absence of specific epigenetic variations has been linked to cancer development and mental health issues (Radtke et al., 2015). Classically, GC/GR controls the expression of around 1,500 genes involved to vital functions including metabolism, cardiac function, immune response, mood, and cognition (Wilson, 2014). However, recent research has uncovered a non-genomic role for GC/GR in mitochondrial function. GC/GR can activate mitochondrial transcription, enhancing cell energy production and regulating electron transport chain function and mtRNA expression in response to stressors (Lapp et al., 2019). While acute corticosteroid stimulation appears to boost mitochondrial oxidation, chronic or excessive stimulation can have the opposite effect (Picard et al., 2018). This is crucial since mitochondria synthesize all steroid hormones, including cortisol, indicating a self-regulating feedback loop (Bose et al., 2002). Progesterone may also act as a stress hormone (Musto and Camici, 2022) and its potential role in SARS-CoV-2 pathogenesis and treatment has to be assessed. As a result, an altered cortisol signaling affects both gene expression and mitochondrial function. Besides, GR and adrenal receptor (AR) are transcriptional activators of ACE2 and the TMPRSS2 both fundamental for the SARS-CoV-2 cells adhesion and penetration. A molecule modulating, through allostatic inhibition its activity, has shown to reduce the severity of SARS-CoV-2 infection in animal models (Rocha et al., 2022). Considering what has already been stated, we believe it is appropriate to evaluate whether the cortisol sensitivity of receptors in patients affected by LC differs compared from those who do not develop it. Additionally, it is important to study the existence of possible epigenetic alterations in the synthesis of this receptor, which is crucial for cellular function, induced by the interaction with the virus.
Consequences of detrimental cholinergic signaling in long COVID: from neurocognitive impairment to mitochondrial dysfunction
Adding another layer of complexity, a VN dysfunction and the consequent decline in cholinergic signaling can locally contribute to neurodegenerative processes (Chen et al., 2022). As is known, the cholinergic system regulates neurogenesis, synaptic plasticity, neuronal differentiation, and neuroprotection through nAChR and mAChR (Frinchi et al., 2018). Cholinergic neurons in the basal forebrain, rich in ACh-producing fibers, are particularly important for learning, memory, and cognitive function due to their influence on cortical and hippocampal regions (Latina et al., 2018). Experimental models using scopolamine, a muscarinic antagonist, have shown that blocking these receptors can induce Alzheimer Disease (AD)-like pathology by impairing mitochondrial antioxidant systems and increasing reactive oxygen species (Bujan et al., 2019). Conversely, xanomeline, a mAChR (M1/M4) agonist, has shown promise in treating AD and schizophrenia (Montani et al., 2021). Intriguingly, studies have found elevated levels of anti-muscarinic antibodies in the serum of LC and ME/CFS patients compared to controls, further implicating cholinergic dysfunction in these conditions (Wallukat et al., 2021). Notably, ACh synthesis, like cortisol production, depends on mitochondrial function, as ACh is synthesized from acetyl-CoA a byproduct of mitochondrial glycolysis (Halder and Lal, 2021). Further solidifying this link, a study investigating the cardioprotective effects of ACh found that it significantly increased mitochondrial density, mass, and mtDNA copy number, increasing ATP synthesis and mitochondrial activity (Sun et al., 2013). These suggests a feedback loop where ACh both relies on and boosts mitochondrial function, which can be highly impaired in LC.
In summary, these findings highlights the strong biochemical link between vagal and adrenal functions, both disrupted by SARS-CoV-2, and their modulation of mitochondrial activity. Indeed, in a scenario where SARS-CoV-2 infection significantly impairs mitochondria, vagal nerve dysfunction, leading to reduced cholinergic and cortisol signaling, could further hinder mitochondrial function. This could create a vicious cycle, perpetuating oxidative stress, inflammation, cell damage, and ultimately cell death.
Conclusions
In conclusion, the model of a dysfunctional VN nerve-HPA-mitochondrial axis can provide a comprehensive explanation for the various alterations observed in LC. Disruptions to more components of this intricate system, whether through direct damage, immune dysregulation, autoantibody interference or epigenetic gene modification in predisposed patients may determine the development and the persistence of this complex syndrome. As a consequence, individual susceptibility to LC likely can depends on baseline vagal tone, cortisol sensitivity, basal mitochondrial cell functioning and immunity factors (Dedoncker et al., 2021). According to this theory, individuals with mild pre-existing VN impairment may be more vulnerable to developing LC, particularly when faced with cortisol reductions and epigenetic modifications in GR signaling pathway and mitochondrial genes. Specifically, investigations into the long-term effects of chronically low morning cortisol levels and their impact on the epigenetic regulation of mtDNA will be instrumental, in our opinion, in unraveling the contributions of mitochondria to LC pathogenesis. For example, it has been hypothesized that dysregulation of cellular microRNAs (miRNAs), which are small non-coding RNAs that modulate gene expression by binding to messenger RNA, may contribute to the upregulation of the IL-6/signal transducers and activators of transcription 3 (STAT3) proinflammatory axis, leading to pain in LC patients (Reyes-Long et al., 2023).
With this paper, we aim to highlight the potential fundamental connection between VN dysfunction, the resulting deficit of Ach and cortisol, which directly leads to mitochondrial dysfunction, perpetuating the process and contributing to the exacerbation of LC symptoms. This theory could also partly explain why SARS-CoV infection accelerates aging (Strong, 2023) and facilitates the development of autoimmune diseases (Sharma and Bayry, 2023).
Given this complexity, an integrated and multidimensional approach to patient evaluation is paramount. This evaluation should encompass assessments of VN basal tone, comprehensive autoantibody panels, mitochondrial function, and potentially even genetic typing of relevant receptors and their epigenetic regulation. A thorough understanding of how stress affects mitochondrial physiology requires a multifaceted evaluation that includes measuring mitochondrial copy number, function, and methylation status. An integrated, multidimensional evaluation could enable personalized treatments by targeting simultaneously specific pathways, offering the best hope for addressing LC challenges.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
MC: Conceptualization, Writing – original draft, Writing – review & editing. GD: Writing – original draft, Writing – review & editing. AB: Writing – original draft, Writing – review & editing. AA: Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Italian Ministry of Health ‘Ricerca Corrente - Linea 1 - INMI L. Spallanzani IRCCS.
Acknowledgments
The authors gratefully acknowledge Dr. Enrica Cipriano for creating the image.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Acanfora, D., Nolano, M., Acanfora, C., Colella, C., Provitera, V., Caporaso, G., et al. (2022). Impaired vagal activity in long-COVID-19 patients. Viruses 14 (5), 1035. doi: 10.3390/v14051035
Alvarez-Santacruz, C., Tyrkalska, S. D., Candel, S. (2024). The microbiota in long COVID. Int. J. Mol. Sci. 25 (2), 1330. doi: 10.3390/ijms25021330
Andersson, U., Tracey, K. J. (2024). Vagus nerve SARS-CoV-2 infection and inflammatory reflex dysfunction: Is there a causal relationship? J. Intern. Med. 295, 91–102. doi: 10.1111/joim.13746
Andrieux, P., Chevillard, C., Cunha-Neto, E., Nunes, J. P. S. (2021). Mitochondria as a cellular hub in infection and inflammation. Int. J. Mol. Sci. 22(21), 11338. doi: 10.3390/ijms222111338
Auphan, N., DiDonato, J. A., Rosette, C., Helmberg, A., Karin, M. (1995). Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science 270, 286–290. doi: 10.1126/science.270.5234.286
Bai, F., Tomasoni, D., Falcinella, C., Barbanotti, D., Castoldi, R., Mulè, G., et al. (2022). Female gender is associated with long COVID syndrome: a prospective cohort study. Clin. Microbiol. Infect. 28, 611.e9–611.e16. doi: 10.1016/j.cmi.2021.11.002
Blomberg, J., Gottfries, C. G., Elfaitouri, A., Rizwan, M., Rosen, A. (2018). Infection elicited autoimmunity and myalgic encephalomyelitis/chronic fatigue syndrome: an explanatory model. Front. Immunol. 9. doi: 10.3389/fimmu.2018.00229
Borovikova, L. V., Ivanova, S., Zhang, M., Yang, H., Botchkina, G. I., Watkins, L., et al. (2000). Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405, 458–462. doi: 10.1038/35013070
Bose, H. S., Lingappa, V. R., Miller, W. L. (2002). Rapid regulation of steroidogenesis by mitochondrial protein import. Nature 417, 87–91. doi: 10.1038/417087a
Bujan, A., Lister, J. J., O’Brien, J. L., Edwards, J. D. (2019). Cortical auditory evoked potentials in mild cognitive impairment: Evidence from a temporal-spatial principal component analysis. Psychophysiology 56, e13466. doi: 10.1111/psyp.13466
Cain, D. W., Bortner, C. D., Diaz-Jimenez, D., Petrillo, M. G., Gruver-Yates, A., Cidlowski, J. A. (2020). Murine glucocorticoid receptors orchestrate B cell migration selectively between bone marrow and blood. J. Immunol. 205, 619–629. doi: 10.4049/jimmunol.1901135
Campbell, A. P., Smrcka, A. V. (2018). Targeting G protein-coupled receptor signalling by blocking G proteins. Nat. Rev. Drug Discovery 17, 789–803. doi: 10.1038/nrd.2018.135
Cao, X., Nguyen, V., Tsai, J., Gao, C., Tian, Y., Zhang, Y., et al. (2023). The SARS-CoV-2 spike protein induces long-term transcriptional perturbations of mitochondrial metabolic genes, causes cardiac fibrosis, and reduces myocardial contractile in obese mice. Mol. Metab. 74, 101756. doi: 10.1016/j.molmet.2023.101756
Chen, H.-J., Appelman, B., Willemen, H., Bos, A., Prado, J., Geyer, C. E., et al. (2024). Transfer of IgG from Long COVID patients induces symptomology in mice. bioRxiv. doi: 10.1101/2024.05.30.596590
Chen, T. H., Chang, C. J., Hung, P. H. (2023). Possible pathogenesis and prevention of long COVID: SARS-CoV-2-induced mitochondrial disorder. Int. J. Mol. Sci. 24 (9), 8034. doi: 10.3390/ijms24098034
Chen, Z. R., Huang, J. B., Yang, S. L., Hong, F. F. (2022). Role of cholinergic signaling in alzheimer’s disease. Molecules 27 (6), 1816. doi: 10.3390/molecules27061816
Cheong, J. G., Ravishankar, A., Sharma, S., Parkhurst, C. N., Grassmann, S. A., Wingert, C. K., et al. (2023). Epigenetic memory of coronavirus infection in innate immune cells and their progenitors. Cell 186, 3882–3902.e24. doi: 10.1016/j.cell.2023.07.019
Chiang, T. S., Leaders, F. E. (1966). Cardiostimulatory responses to vagal stimulation, nicotine, and tyramine. Am. J. Physiol. 211, 1443–1446. doi: 10.1152/ajplegacy.1966.211.6.1443
da Silva, A. L. G., Vieira, L. D. P., Dias, L. S., Prestes, C. V., Back, G. D., Goulart, C. D. L., et al. (2023). Impact of long COVID on the heart rate variability at rest and during deep breathing maneuver. Sci. Rep. 13, 22695. doi: 10.1038/s41598-023-50276-0
Davies, J. P., Almasy, K. M., McDonald, E. F., Plate, L. (2020). Comparative multiplexed interactomics of SARS-CoV-2 and homologous coronavirus nonstructural proteins identifies unique and shared host-cell dependencies. ACS Infect. Dis. 6, 3174–3189. doi: 10.1021/acsinfecdis.0c00500
Davis, H. E., McCorkell, L., Vogel, J. M., Topol, E. J. (2023). Long COVID: major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 21, 133–146. doi: 10.1038/s41579-022-00846-2
Dedoncker, J., Vanderhasselt, M. A., Ottaviani, C., Slavich, G. M. (2021). Mental health during the COVID-19 pandemic and beyond: The importance of the vagus nerve for biopsychosocial resilience. Neurosci. Biobehav. Rev. 125, 1–10. doi: 10.1016/j.neubiorev.2021.02.010
Di Florio, D. N., Beetler, D. J., McCabe, E. J., Sin, J., Ikezu, T., Fairweather, D. (2024). Mitochondrial extracellular vesicles, autoimmunity and myocarditis. Front. Immunol. 15. doi: 10.3389/fimmu.2024.1374796
Dobrowolska, K., Zarebska-Michaluk, D., Poniedzialek, B., Jaroszewicz, J., Flisiak, R., Rzymski, P. (2023). Overview of autoantibodies in COVID-19 convalescents. J. Med. Virol. 95, e28864. doi: 10.1002/jmv.28864
Ely, E. W., Brown, L. M., Fineberg, H. V., National Academies of Sciences E, Medicine Committee on Examining the Working Definition for Long C (2024). Long covid defined. N Engl. J. Med. 391. doi: 10.1056/NEJMsb2408466
Farsalinos, K., Niaura, R., Le Houezec, J., Barbouni, A., Tsatsakis, A., Kouretas, D., et al. (2020). Editorial: Nicotine and SARS-CoV-2: COVID-19 may be a disease of the nicotinic cholinergic system. Toxicol. Rep. 7, 658–663. doi: 10.1016/j.toxrep.2020.04.012
Frinchi, M., Scaduto, P., Cappello, F., Belluardo, N., Mudo, G. (2018). Heat shock protein (Hsp) regulation by muscarinic acetylcholine receptor (mAChR) activation in the rat hippocampus. J. Cell Physiol. 233, 6107–6116. doi: 10.1002/jcp.26454
Goebel, A., Krock, E., Gentry, C., Israel, M. R., Jurczak, A., Urbina, C. M., et al. (2021). Passive transfer of fibromyalgia symptoms from patients to mice. J. Clin. Invest. 131. doi: 10.1172/JCI144201
Halder, N., Lal, G. (2021). Cholinergic system and its therapeutic importance in inflammation and autoimmunity. Front. Immunol. 12. doi: 10.3389/fimmu.2021.660342
Henley, D., Lightman, S., Carrell, R. (2016). Cortisol and CBG - Getting cortisol to the right place at the right time. Pharmacol. Ther. 166, 128–135. doi: 10.1016/j.pharmthera.2016.06.020
Hu, C., Li, Z., Feng, J., Tang, Y., Qin, L., Hu, X., et al. (2018). Glucocorticoids modulate th1 and th2 responses in asthmatic mouse models by inhibition of notch1 signaling. Int. Arch. Allergy Immunol. 175, 44–52. doi: 10.1159/000485890
Jacobson, L., Akana, S. F., Cascio, C. S., Shinsako, J., Dallman, M. F. (1988). Circadian variations in plasma corticosterone permit normal termination of adrenocorticotropin responses to stress. Endocrinology 122, 1343–1348. doi: 10.1210/endo-122-4-1343
Jellinek, M., Kaye, M. P., Kaiser, G. C., Cooper, T. (1965). Effect of cervical vagosympathectomy on myocardial catecholamine concentration. Am. J. Physiol. 209, 951–954. doi: 10.1152/ajplegacy.1965.209.5.951
Johnston, G. R., Webster, N. R. (2009). Cytokines and the immunomodulatory function of the vagus nerve. Br. J. Anaesth 102, 453–462. doi: 10.1093/bja/aep037
Kaniusas, E., Szeles, J. C., Kampusch, S., Alfageme-Lopez, N., Yucuma-Conde, D., Li, X., et al. (2020). Non-invasive auricular vagus nerve stimulation as a potential treatment for covid19-originated acute respiratory distress syndrome. Front. Physiol. 11. doi: 10.3389/fphys.2020.00890
Klein, J., Wood, J., Jaycox, J., Dhodapkar, R. M., Lu, P., Gehlhausen, J. R., et al. (2023). Distinguishing features of Long COVID identified through immune profiling. Nature. 623, 139–148. doi: 10.1038/s41586-023-06651-y
Lapp, H. E., Bartlett, A. A., Hunter, R. G. (2019). Stress and glucocorticoid receptor regulation of mitochondrial gene expression. J. Mol. Endocrinol. 62, R121–R128. doi: 10.1530/JME-18-0152
Latina, V., Caioli, S., Zona, C., Ciotti, M. T., Borreca, A., Calissano, P., et al. (2018). NGF-dependent changes in ubiquitin homeostasis trigger early cholinergic degeneration in cellular and animal AD-model. Front. Cell Neurosci. 12. doi: 10.3389/fncel.2018.00487
Lei, Y., Zhang, J., Schiavon, C. R., He, M., Chen, L., Shen, H., et al. (2021). SARS-CoV-2 spike protein impairs endothelial function via downregulation of ACE 2. Circ. Res. 128, 1323–1326. doi: 10.1161/CIRCRESAHA.121.318902
Leitzke, M. (2023). Is the post-COVID-19 syndrome a severe impairment of acetylcholine-orchestrated neuromodulation that responds to nicotine administration? Bioelectron Med. 9, 2. doi: 10.1186/s42234-023-00104-7
Leow, M. K., Kwek, D. S., Ng, A. W., Ong, K. C., Kaw, G. J., Lee, L. S. (2005). Hypocortisolism in survivors of severe acute respiratory syndrome (SARS). Clin. Endocrinol. (Oxf) 63, 197–202. doi: 10.1111/j.1365-2265.2005.02325.x
Li, P., Liu, H., Sun, P., Chen, Y., Zhang, G., Yang, J., et al. (2016). Chronic vagus nerve stimulation attenuates vascular endothelial impairments and reduces the inflammatory profile via inhibition of the NF-kappaB signaling pathway in ovariectomized rats. Exp. Gerontol 74, 43–55. doi: 10.1016/j.exger.2015.12.005
Li, P., Zhou, K., Li, J., Xu, X., Wang, L., Wang, T. (2022). Acetylcholine suppresses LPS-induced endothelial cell activation by inhibiting the MAPK and NF-kappaB pathways. Eur. Cytokine Netw. 33, 79–89. doi: 10.1684/ecn.2023.0481
Linan-Rico, A., Ochoa-Cortes, F., Beyder, A., Soghomonyan, S., Zuleta-Alarcon, A., Coppola, V., et al. (2016). Mechanosensory signaling in enterochromaffin cells and 5-HT release: potential implications for gut inflammation. Front. Neurosci. 10. doi: 10.3389/fnins.2016.00564
Llados, G., Massanella, M., Coll-Fernandez, R., Rodriguez, R., Hernandez, E., Lucente, G., et al. (2024). Vagus nerve dysfunction in the post-COVID-19 condition: a pilot cross-sectional study. Clin. Microbiol. Infect. 30, 515–521. doi: 10.1016/j.cmi.2023.11.007
Manta, S., Dong, J., Debonnel, G., Blier, P. (2009). Enhancement of the function of rat serotonin and norepinephrine neurons by sustained vagus nerve stimulation. J. Psychiatry Neurosci. 34, 272–280.
Mantle, D., Hargreaves, I. P., Domingo, J. C., Castro-Marrero, J. (2024). Mitochondrial dysfunction and coenzyme Q10 supplementation in post-viral fatigue syndrome: an overview. Int. J. Mol. Sci. 25 (1), 574. doi: 10.3390/ijms25010574
Mastrorosa, I., Del Duca, G., Pinnetti, C., Lorenzini, P., Vergori, A., Brita, A. C., et al. (2023). What is the impact of post-COVID-19 syndrome on health-related quality of life and associated factors: a cross-sectional analysis. Health Qual Life Outcomes 21, 28. doi: 10.1186/s12955-023-02107-z
McVey Neufeld, K. A., Bienenstock, J., Bharwani, A., Champagne-Jorgensen, K., Mao, Y., West, C., et al. (2019). Oral selective serotonin reuptake inhibitors activate vagus nerve dependent gut-brain signalling. Sci. Rep. 9, 14290. doi: 10.1038/s41598-019-50807-8
Mehrzadi, S., Karimi, M. Y., Fatemi, A., Reiter, R. J., Hosseinzadeh, A. (2021). SARS-CoV-2 and other coronaviruses negatively influence mitochondrial quality control: beneficial effects of melatonin. Pharmacol. Ther. 224, 107825. doi: 10.1016/j.pharmthera.2021.107825
Merzon, E., Weiss, M., Krone, B., Cohen, S., Ilani, G., Vinker, S., et al. (2022). Clinical and socio-demographic variables associated with the diagnosis of long COVID syndrome in youth: A population-based study. Int. J. Environ. Res. Public Health 19(10), 5993. doi: 10.3390/ijerph19105993
Midzak, A., Papadopoulos, V. (2016). Adrenal mitochondria and steroidogenesis: from individual proteins to functional protein assemblies. Front. Endocrinol. (Lausanne) 7. doi: 10.3389/fendo.2016.00106
Molnar, T., Lehoczki, A., Fekete, M., Varnai, R., Zavori, L., Erdo-Bonyar, S., et al. (2024). Mitochondrial dysfunction in long COVID: mechanisms, consequences, and potential therapeutic approaches. Geroscience 46, 5267–5286. doi: 10.1007/s11357-024-01165-5
Montani, C., Canella, C., Schwarz, A. J., Li, J., Gilmour, G., Galbusera, A., et al. (2021). The M1/M4 preferring muscarinic agonist xanomeline modulates functional connectivity and NMDAR antagonist-induced changes in the mouse brain. Neuropsychopharmacology 46, 1194–1206. doi: 10.1038/s41386-020-00916-0
Moolamalla, S. T. R., Balasubramanian, R., Chauhan, R., Priyakumar, U. D., Vinod, P. K. (2021). Host metabolic reprogramming in response to SARS-CoV-2 infection: A systems biology approach. Microb. Pathog. 158, 105114. doi: 10.1016/j.micpath.2021.105114
Muryobayashi, T., Mori, J., Fujiwara, M., Shimamoto, K. (1968). Fluorescence histochemical demonstration of adrenergic nerve fibers in the vagus nerve of cats and dogs. Jpn J. Pharmacol. 18, 285–293. doi: 10.1254/jjp.18.285
Musto, A., Camici, M. (2022). Can Hormonal Contraceptive Therapy be a useful treatment in Fibromyalgia? A Case Report. Arch. Clin. Med. Case Rep. doi: 10.26502/acmcr.96550484
Nagy, Z., Acs, B., Butz, H., Feldman, K., Marta, A., Szabo, P. M., et al. (2016). Overexpression of GRss in colonic mucosal cell line partly reflects altered gene expression in colonic mucosa of patients with inflammatory bowel disease. J. Steroid Biochem. Mol. Biol. 155, 76–84. doi: 10.1016/j.jsbmb.2015.10.006
National Academies of Sciences, Engineering, and Medicine. (2024). A Long COVID Definition: A Chronic, Systemic Disease State with Profound Consequences (Washington, DC: The National Academies Press). doi: 10.17226/27768
Noti, M., Sidler, D., Brunner, T. (2009). Extra-adrenal glucocorticoid synthesis in the intestinal epithelium: more than a drop in the ocean? Semin. Immunopathol. 31, 237–248. doi: 10.1007/s00281-009-0159-2
Paul, T., Ledderose, S., Bartsch, H., Sun, N., Soliman, S., Markl, B., et al. (2022). Adrenal tropism of SARS-CoV-2 and adrenal findings in a post-mortem case series of patients with severe fatal COVID-19. Nat. Commun. 13, 1589. doi: 10.1038/s41467-022-29145-3
Pavlov, V. A., Tracey, K. J. (2012). The vagus nerve and the inflammatory reflex–linking immunity and metabolism. Nat. Rev. Endocrinol. 8, 743–754. doi: 10.1038/nrendo.2012.189
Pellissier, S., Dantzer, C., Mondillon, L., Trocme, C., Gauchez, A. S., Ducros, V., et al. (2014). Relationship between vagal tone, cortisol, TNF-alpha, epinephrine and negative affects in Crohn’s disease and irritable bowel syndrome. PloS One 9, e105328. doi: 10.1371/journal.pone.0105328
Peluso, M. J., Deeks, S. G., Mustapic, M., Kapogiannis, D., Henrich, T. J., Lu, S., et al. (2022). SARS-CoV-2 and mitochondrial proteins in neural-derived exosomes of COVID-19. Ann. Neurol. 91, 772–781. doi: 10.1002/ana.26350
Picard, M., Prather, A. A., Puterman, E., Cuillerier, A., Coccia, M., Aschbacher, K., et al. (2018). A mitochondrial health index sensitive to mood and caregiving stress. Biol. Psychiatry 84, 9–17. doi: 10.1016/j.biopsych.2018.01.012
Quatrini, L., Ugolini, S. (2021). New insights into the cell- and tissue-specificity of glucocorticoid actions. Cell Mol. Immunol. 18, 269–278. doi: 10.1038/s41423-020-00526-2
Quatrini, L., Wieduwild, E., Escaliere, B., Filtjens, J., Chasson, L., Laprie, C., et al. (2018). Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat. Immunol. 19, 954–962. doi: 10.1038/s41590-018-0185-0
Radtke, K. M., Schauer, M., Gunter, H. M., Ruf-Leuschner, M., Sill, J., Meyer, A., et al. (2015). Epigenetic modifications of the glucocorticoid receptor gene are associated with the vulnerability to psychopathology in childhood maltreatment. Transl. Psychiatry 5, e571. doi: 10.1038/tp.2015.63
Ramachandran, K., Maity, S., Muthukumar, A. R., Kandala, S., Tomar, D., Abd El-Aziz, T. M., et al. (2022). SARS-CoV-2 infection enhances mitochondrial PTP complex activity to perturb cardiac energetics. iScience 25, 103722. doi: 10.1016/j.isci.2021.103722
Ren, Y., Shu, T., Wu, D., Mu, J., Wang, C., Huang, M., et al. (2020). The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol. Immunol. 17, 881–883. doi: 10.1038/s41423-020-0485-9
Ren, F., Zhang, L., Zhang, X., Shi, H., Wen, T., Bai, L., et al. (2016). Inhibition of glycogen synthase kinase 3beta promotes autophagy to protect mice from acute liver failure mediated by peroxisome proliferator-activated receptor alpha. Cell Death Dis. 7, e2151. doi: 10.1038/cddis.2016.56
Reyes-Long, S., Cortes-Altamirano, J. L., Bandala, C., Avendano-Ortiz, K., Bonilla-Jaime, H., Bueno-Nav, A., et al. (2023). Role of the microRNAs in the pathogenic mechanism of painful symptoms in long COVID: systematic review. Int. J. Mol. Sci. 24(4), 3574. doi: 10.3390/ijms24043574
Rhen, T., Cidlowski, J. A. (2005). Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N Engl. J. Med. 353, 1711–1723. doi: 10.1056/NEJMra050541
Robertson, M. M., Qasmieh, S. A., Kulkarni, S. G., Teasdale, C. A., Jones, H. E., McNairy, M., et al. (2023). The epidemiology of long coronavirus disease in US adults. Clin. Infect. Dis. 76, 1636–1645. doi: 10.1093/cid/ciac961
Rocha, S. M., Fagre, A. C., Latham, A. S., Cummings, J. E., Aboellail, T. A., Reigan, P., et al. (2022). A novel glucocorticoid and androgen receptor modulator reduces viral entry and innate immune inflammatory responses in the Syrian hamster model of SARS-CoV-2 infection. Front. Immunol. 13. doi: 10.3389/fimmu.2022.811430
Sarker, H., Panigrahi, R., Hardy, E., Glover, J. N. M., Elahi, S., Fernandez-Patron, C. (2022). Glucocorticoids bind to SARS-CoV-2 S1 at multiple sites causing cooperative inhibition of SARS-CoV-2 S1 interaction with ACE2. Front. Immunol. 13. doi: 10.3389/fimmu.2022.906687
Scarpulla, R. C., Vega, R. B., Kelly, D. P. (2012). Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 23, 459–466. doi: 10.1016/j.tem.2012.06.006
Schedel, A., Thornton, S., Schloss, P., Kluter, H., Bugert, P. (2011). Human platelets express functional alpha7-nicotinic acetylcholine receptors. Arterioscler. Thromb. Vasc. Biol. 31, 928–934. doi: 10.1161/ATVBAHA.110.218297
Scheinman, R. I., Cogswell, P. C., Lofquist, A. K., Baldwin, A. S., Jr. (1995). Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science 270, 283–286. doi: 10.1126/science.270.5234.283
Shang, C., Liu, Z., Zhu, Y., Lu, J., Ge, C., Zhang, C., et al. (2021). SARS-CoV-2 causes mitochondrial dysfunction and mitophagy impairment. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.780768
Sharma, C., Bayry, J. (2023). High risk of autoimmune diseases after COVID-19. Nat. Rev. Rheumatol 19, 399–400. doi: 10.1038/s41584-023-00964-y
Shi, C. S., Qi, H. Y., Boularan, C., Huang, N. N., Abu-Asab, M., Shelhamer, J. H., et al. (2014). SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 193, 3080–3089. doi: 10.4049/jimmunol.1303196
Singh, K. K., Chaubey, G., Chen, J. Y., Suravajhala, P. (2020). Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am. J. Physiol. Cell Physiol. 319, C258–C267. doi: 10.1152/ajpcell.00224.2020
Singh, K., Chen, Y. C., Hassanzadeh, S., Han, K., Judy, J. T., Seifuddin, F., et al. (2021). Network analysis and transcriptome profiling identify autophagic and mitochondrial dysfunctions in SARS-CoV-2 infection. Front. Genet. 12. doi: 10.3389/fgene.2021.599261
Song, P., Li, W., Xie, J., Hou, Y., You, C. (2020). Cytokine storm induced by SARS-CoV-2. Clin. Chim. Acta 509, 280–287. doi: 10.1016/j.cca.2020.06.017
Soria-Castro, E., Soto, M. E., Guarner-Lans, V., Rojas, G., Perezpena-Diazconti, M., Criales-Vera, S. A., et al. (2021). The kidnapping of mitochondrial function associated with the SARS-CoV-2 infection. Histol Histopathol 36, 947–965. doi: 10.14670/HH-18-354
Strong, M. J. (2023). SARS-CoV-2, aging, and Post-COVID-19 neurodegeneration. J. Neurochem. 165, 115–130. doi: 10.1111/jnc.15736
Su, Y., Yuan, D., Chen, D. G., Ng, R. H., Wang, K., Choi, J., et al. (2022). Multiple early factors anticipate post-acute COVID-19 sequelae. Cell 185, 881–895.e20. doi: 10.1016/j.cell.2022.01.014
Sun, L., Zhao, M., Yu, X. J., Wang, H., He, X., Liu, J. K., et al. (2013). Cardioprotection by acetylcholine: a novel mechanism via mitochondrial biogenesis and function involving the PGC-1alpha pathway. J. Cell Physiol. 228, 1238–1248. doi: 10.1002/jcp.24277
Talaber, G., Jondal, M., Okret, S. (2015). Local glucocorticoid production in the thymus. Steroids. Nov 103, 58–63. doi: 10.1016/j.steroids.2015.06.010
Tan, T., Khoo, B., Mills, E. G., Phylactou, M., Patel, B., Eng, P. C., et al. (2020). Association between high serum total cortisol concentrations and mortality from COVID-19. Lancet Diabetes Endocrinol. 8, 659–660. doi: 10.1016/S2213-8587(20)30216-3
Tatsuta, T. (2009). Protein quality control in mitochondria. J. Biochem. 146, 455–461. doi: 10.1093/jb/mvp122
Taves, M. D., Gomez-Sanchez, C. E., Soma, K. K. (2011). Extra-adrenal glucocorticoids and mineralocorticoids: evidence for local synthesis, regulation, and function. Am. J. Physiol. Endocrinol. Metab. 301, E11–E24. doi: 10.1152/ajpendo.00100.2011
Thayer, J. F., Sternberg, E. (2006). Beyond heart rate variability: vagal regulation of allostatic systems. Ann. N Y Acad. Sci. 1088, 361–372. doi: 10.1196/annals.1366.014
Timmermans, S., Souffriau, J., Libert, C. (2019). A general introduction to glucocorticoid biology. Front. Immunol. 10. doi: 10.3389/fimmu.2019.01545
Tondera, D., Grandemange, S., Jourdain, A., Karbowski, M., Mattenberger, Y., Herzig, S., et al. (2009). SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 28, 1589–1600. doi: 10.1038/emboj.2009.89
Twig, G., Hyde, B., Shirihai, O. S. (2008). Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim. Biophys. Acta 1777, 1092–1097. doi: 10.1016/j.bbabio.2008.05.001
Vandevyver, S., Dejager, L., Libert, C. (2014). Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr. Rev. 35, 671–693. doi: 10.1210/er.2014-1010
VanElzakker, M. B. (2013). Chronic fatigue syndrome from vagus nerve infection: a psychoneuroimmunological hypothesis. Med. Hypotheses 81, 414–423. doi: 10.1016/j.mehy.2013.05.034
Walker, B. R., Moraes, C. T. (2022). Nuclear-mitochondrial interactions. Biomolecules 12 (3), 427. doi: 10.3390/biom12030427
Wallace, D. C. (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 39, 359–407. doi: 10.1146/annurev.genet.39.110304.095751
Wallukat, G., Hohberger, B., Wenzel, K., Furst, J., Schulze-Rothe, S., Wallukat, A., et al. (2021). Functional autoantibodies against G-protein coupled receptors in patients with persistent Long-COVID-19 symptoms. J. Transl. Autoimmun 4, 100100. doi: 10.1016/j.jtauto.2021.100100
Wang, H., Yu, M., Ochani, M., Amella, C. A., Tanovic, M., Susarla, S., et al. (2003). Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421, 384–388. doi: 10.1038/nature01339
Wheatland, R. (2004). Molecular mimicry of ACTH in SARS - implications for corticosteroid treatment and prophylaxis. Med. Hypotheses 63, 855–862. doi: 10.1016/j.mehy.2004.04.009
Wilson, J. L. (2014). Clinical perspective on stress, cortisol and adrenal fatigue. Adv. Integr. Med. 1, 93–96. doi: 10.1016/j.aimed.2014.05.002
Wong, A. C., Devason, A. S., Umana, I. C., Cox, T. O., Dohnalova, L., Litichevskiy, L., et al. (2023). Serotonin reduction in post-acute sequelae of viral infection. Cell 186, 4851–4867 e20. doi: 10.1016/j.cell.2023.09.013
Woo, M. S., Shafiq, M., Fitzek, A., Dottermusch, M., Altmeppen, H., Mohammadi, B., et al. (2023). Vagus nerve inflammation contributes to dysautonomia in COVID-19. Acta Neuropathol. 146, 387–394. doi: 10.1007/s00401-023-02612-x
Yamakawa, M., Mukaino, A., Kimura, A., Nagasako, Y., Kitazaki, Y., Maeda, Y., et al. (2020). Antibodies to the alpha3 subunit of the ganglionic-type nicotinic acetylcholine receptors in patients with autoimmune encephalitis. J. Neuroimmunol 349, 577399. doi: 10.1016/j.jneuroim.2020.577399
Yang, M. (2020). Cell Pyroptosis, a Potential Pathogenic Mechanism of 2019-nCoV Infection. doi: 10.2139/ssrn.3527420
Yap, J. Y. Y., Keatch, C., Lambert, E., Woods, W., Stoddart, P. R., Kameneva, T. (2020). Critical review of transcutaneous vagus nerve stimulation: challenges for translation to clinical practice. Front. Neurosci. 14. doi: 10.3389/fnins.2020.00284
Zhang, J., Han, Y., Shi, H., Chen, J., Zhang, X., et al. (2020). Swine acute diarrhea syndrome coronavirus-induced apoptosis is caspase- and cyclophilin D- dependent. Emerg. Microbes Infect. 9, 439–456. doi: 10.1080/22221751.2020.1722758
Keywords: long COVID, vagus nerve dysfunction, hypothalamic-pituitary-adrenal axis reflex, cholinergic anti-inflammatory reflex, adrenergic anti-inflammatory reflex, cortisol, glucocorticoids receptor, mitochondrial dysfunction
Citation: Camici M, Del Duca G, Brita AC and Antinori A (2024) Connecting dots of long COVID-19 pathogenesis: a vagus nerve- hypothalamic-pituitary- adrenal-mitochondrial axis dysfunction. Front. Cell. Infect. Microbiol. 14:1501949. doi: 10.3389/fcimb.2024.1501949
Received: 25 September 2024; Accepted: 25 November 2024;
Published: 13 December 2024.
Edited by:
Chun Kiat Lee, National University Health System, SingaporeReviewed by:
Ana Rosa Pérez, National Scientific and Technical Research Council (CONICET), ArgentinaCopyright © 2024 Camici, Del Duca, Brita and Antinori. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Camici, bWFydGEuY2FtaWNpQGlubWkuaXQ=