
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 23 October 2024
Sec. Veterinary and Zoonotic Infection
Volume 14 - 2024 | https://doi.org/10.3389/fcimb.2024.1474676
This article is part of the Research Topic Viral Diseases in Swine View all 7 articles
 Yiwan Song1,2
Yiwan Song1,2 Yiqi Fang1,2Shuaiqi Zhu1,2Weijun Wang1Lianxiang Wang3Wenxian Chen1,2Yintao He1,2Lin Yi1,2Hongxing Ding1,2Mingqiu Zhao1Shuangqi Fan1,2Zhaoyao Li1,3*Jinding Chen1,2*
Yiqi Fang1,2Shuaiqi Zhu1,2Weijun Wang1Lianxiang Wang3Wenxian Chen1,2Yintao He1,2Lin Yi1,2Hongxing Ding1,2Mingqiu Zhao1Shuangqi Fan1,2Zhaoyao Li1,3*Jinding Chen1,2*Background: Senecavirus A (SVA) is a newly pathogenic virus correlated with the acute death of piglets and vesicular lesions in pigs. The further prevalence of SVA will cause considerable economic damage to the global pig farming industry. Therefore, rapid and accurate diagnostic tools for SVA are crucial for preventing and controlling the disease.
Methods: We designed multiple primer pairs targeting the most conserved region of the SVA 3D gene and selected those with the highest specificity. Nfo-probes were subsequently developed based on the highest specificity primer pairs. Subsequently, the recombinase-assisted amplification (RAA) reaction was completed, and the reaction temperature and duration were optimized. The RAA amplicons were detected using a lateral flow device (LFD). Finally, a rapid and intuitive RAA-LFD assay was established against SVA.
Results: The SVA RAA-LFD assay can be performed under reaction conditions of 35°C within 17 minutes, with results observable to the naked eye. We then evaluated the performance of this method. It exhibited high specificity and no cross-reaction with the other common swine pathogens. The lowest detectable limits of this method for the plasmid of pMD18-SVA-3D, DNA amplification product, and viral were 3.86×101 copies/µL, 8.76×10-7 ng/µL, and 1×100.25 TCID50/mL, respectively. A total of 44 clinical samples were then tested using the RAA-LFD, PCR, and RT-qPCR methods. The results demonstrated a consistent detection rate between the RAA-LFD and RT-qPCR assays.
Conclusion: The SVA RAA-LFD assay developed in our study exhibits excellent specificity, sensitivity, and time-saving attributes, making it ideally suited for utilization in lack-instrumented laboratory and field settings.
Senecavirus A (SVA) is a single-stranded, positive RNA virus belonging to the genus Senecavirus within the family Picornaviridae (Adams et al., 2015; King et al., 2012). SVA is among the pathogens associated with vesicular diseases in pigs, characterized primarily by acute mortality in neonatal piglets and the development of vesicular ulcers in the nostrils, hoof coronets, and coronary arteries of adult pigs (Joshi et al., 2016; Vannucci et al., 2015). Consequently, distinguishing disease infected by SVA from other swine vesicular diseases such as foot-and-mouth disease (FMD), swine vesicular disease (SVD), and vesicular stomatitis (VS) based solely on clinical symptoms has been a challenging task. Furthermore, this disease lacks distinct seasonal epidemic trends, posing additional challenges for prevention and control efforts (Hales et al., 2008). The SVA genome is approximately 7300 bp, comprising a 5’ untranslated region (5′ UTR), an open reading frame (ORF), and a 3’ untranslated region (3′ UTR) (Lin et al., 2009). The ORF consisted of a leader protein (L) and three precursor proteins (P1, P2, and P3) (Adams et al., 2016; Hales et al., 2008; Venkataraman et al., 2008). Subsequently, these regions are further processed by the virus-encoded cysteine protease (3Cpro) to form mature functional viral proteins (L-VP4-VP2-VP3-VP1-2A-2B-2C-3A-3B-3C-3D) (Hales et al., 2008; Leme et al., 2015).
In 2002, SVA was identified in cultivating transformed embryonic retinal cell lines (C6 cells) grown at a U.S. research company (Knowles et al., 2006). Following isolation and purification, the virus was named SVV 001 (Hales et al., 2008). Since the identification of SVV001, several studies have shown that it is closely associated with subsequent sporadic cases of porcine idiopathic vesicular disease (PIVD) in the USA (Bracht et al., 2016; K. et al., 2012) and Canada (Pasma et al., 2008). Interestingly, a series of outbreaks of PIVD occurred in Brazil between 2014 and 2015, subsequently confirmed to be caused by SVA infection. This confirmed that SVA is the pathogen responsible for swine vesicular disease (Vannucci et al., 2015). Since then, two outbreaks of SVA infections have been reported in the Brazilian region in 2018 and 2020 (Leme et al., 2019; Vieira et al., 2022). Additionally, several countries within the areas of Europe, Southeast Asia, and Asia have published reports of SVA infections (Arzt et al., 2019; Hause et al., 2016; Saeng-chuto et al., 2017; Sun et al., 2017; Wu et al., 2017; Xu et al., 2017). The above situation implies that SVA is gradually prevalent, which will pose a severe economic threat to the swine industry worldwide. Therefore, the prevention and control of SVA is crucial to avoid further dissemination.
Currently, a number of detection methods for SVA have been developed in the laboratory, including PCR (Pinheiro-de-Oliveira et al., 2019; Zhang et al., 2019), qPCR (Mu et al., 2020; Zhang et al., 2019), and ELISA (Dvorak et al., 2017; Yang et al., 2012). These methods are designed to prevent SVA infections from affecting the economy of the pig farming industry. However, these detection methods require a thermocycler for target gene amplification or proficient laboratory techniques, rendering them unsuitable for laboratories with limited infrastructure or field inspections.
Isothermal nucleic acid amplification is a technique developed in recent years that uses specific enzymes to amplify target genes under constant temperature conditions, with methods including methods such as Loop-mediated isothermal amplification (LAMP) (Peng et al., 2024; Wang et al., 2024) and recombinase-aided amplification (RAA) (Feng et al., 2024; Zhou et al., 2024). These methods require only a constant-temperature heating block or even body heat to achieve target gene amplification, significantly reducing the reaction time and complexity of nucleic acid amplification (Kolm et al., 2019). However, the design of the LAMP primer is more complex and technically challenging. In contrast, the primer design for RAA is straightforward, requiring only the control of the target gene length within 500 bp and the design of two primers with lengths of 30~35 bp. The RAA reaction primarily utilizes recombinase, single-stranded binding proteins (SSBs), and DNA polymerase with strand displacement activity to initiate the reaction. It can complete nucleic acid amplification in less than 40 minutes. This method has been used to detect a variety of pathogens (Hai, 2010; He et al., 2021; Li et al., 2023; Wu et al., 2022; Zhao et al., 2024b).
Lateral flow dipstick (LFD) is a technique for rapidly detecting nucleic acid samples based on capillary chromatography, molecular hybridization, and colloidal gold (Jaroenram et al., 2009). The method is utilized to detect biomarkers in nucleic acid samples, requiring neither specialized equipment nor operational expertise, thus rendering it accessible to a broad range of users (Jaroenram and Owens, 2014). The combination of RAA and LFD is based on the principle that the RAA reaction forms amplified products labeled with biotin and specific fluorescent antibodies, which then interact with the colloidal gold-labeled antibodies in the LFD, further cross-linking with biotin on the test (T)-line, ultimately achieving the visual detection of the target product (Chow et al., 2008). The RAA-LFD assay has been successfully used for the detection of a variety of pathogens (Bienes et al., 2022; Homklinkaew et al., 2023; Hou et al., 2022; Li et al., 2022).
In this study, the most optimal RAA primers and probes were designed for the most conserved region of the SVA 3D gene. The RAA products were detected using LFD, and a highly sensitive and specific SVA RAA-LFD rapid visualization assay was established. This method overcomes the difficulties of ordinary PCR and qPCR, which require expensive nucleic acid amplifiers. It provides a reliable method for the initial screening and detection of SVA in ordinary laboratories and the field.
SVA, foot and mouth disease virus (FMDV), african swine fever (ASFV), porcine circovirus type 2 (PVC2), japanese encephalitis virus (JEV), classical swine fever virus (CSFV), and porcine parvovirus (PPV) were obtained from the Laboratory of Veterinary Microbiology and Immunology, South China Agricultural University.
In 2018, forty-four clinical swine samples suspected of SVA infection were obtained from multiple pig farms in Guangzhou and were stored at -80°C for further utilization. RNA was extracted from each clinical sample using the E.Z.N.A.® FFPE RNA Kit (Omega Bio-Tek, Inc., Connecticut, USA) and stored at -80°C in our laboratory.
According to previous studies conducted in our laboratory and the research conducted by Li et al (Li et al., 2019), the most conserved region of SVA is located within the 3D gene, which can be a suitable target for RAA amplification. Thirty-seven SVA sequences were obtained from the National Center for Biotechnology Information (NCBI) (GenBank accession numbers: MF615501.1, MF615506.1, MF615507.1, MF615508.1, MF615509.1, MF189001.1, MF189000.1, KR063107.1, MF460448.1, MH885100.1, MH885099.1, MH634510.1, MH634508.1, MH634506.1, MH634518.1, MH634522.1, MH634516.1, MH716015.1, MHS88717.1, MH779611.1, MH817445.1, MH316117.1, MH316116.1, MK357117.1, MN781984.1, MN781983.1, MN781981.1, KRO63107.1, ON369394.1, ON868377.1, KR063109.1, MZ818785.1, MZ375462.1, KY486166.1, KY486165.1, MZ395819.1, MZ733980.1). The 3D gene regions of the above sequences were aligned using MegAlign 7.1 (DNAStar, USA) to confirm the most conserved region. Subsequently, six primer pairs were designed within this region using Oligo 7.0 (Molecular Biology Insights, Inc, USA) and Primer Premier 5 (Premier Biosoft International, Canada) (Table 1). The specificity of the six pairs of primers for SVA was verified by the RAA reaction provided by the RAA nucleic acid amplification kit based on agarose gel electrophoresis (AGE) (Jiangsu Qitian Gene Biotechnology Co., Ltd., Jiangsu, China), and the most specific primers were screened. Components including 25 μL rehydration buffer V, 2.0 μL forward primer (10 μM), 2.0 μL reverse primer (10 μM), and 17.5 μL nuclease-free water were sequentially added to the lyophilized enzyme base reaction unit in the kit. Then, the walls of the tubes were flicked by hand and centrifuged transiently for 10 seconds. To stimulate the reaction, 2.5 μL magnesium acetate was introduced into the inside of each tube cap, and 1μL of genomic DNA was added as a template. Additionally, the ddH2O and positive samples were added to the separately formulated reaction system as negative and positive controls, respectively. After 40 minutes incubation period at 37°C, 50 μL of chloroform and phenol mixture (1:1) was added to purify the amplicons. Finally, the supernatant was extracted for analysis using 2% AGE.
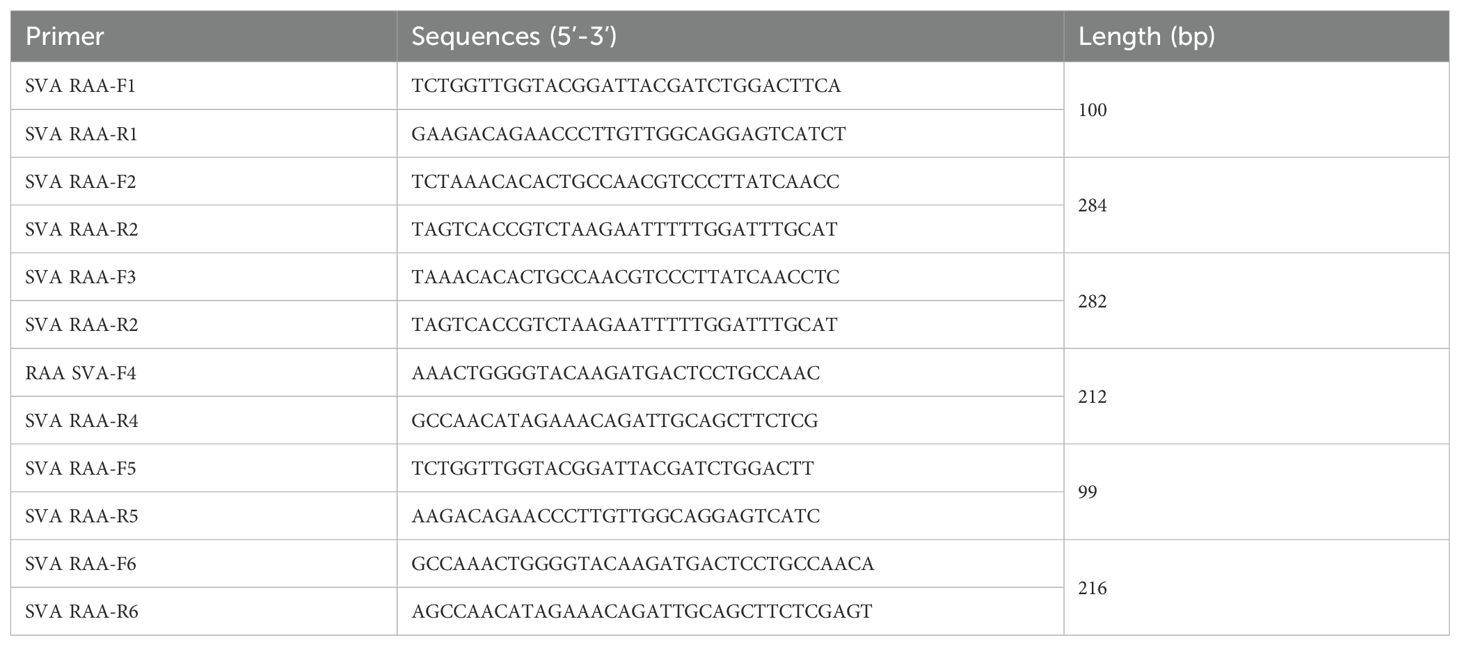
Table 1. The primers of the 3D-based RAA basic reaction for SVA were used in this study.
Following the primer screening, a specific probe was developed and situated within the middle of the primer pairs. This probe was labeled with 6-carboxy-fluorescein (FAM) at the 5’ end, modified with a polymerase extension blocking group (C3 spacer) at the 3’ end, and integrated with a tetrahydrofuran abasic-site mimic (THF) moiety positioned 30 nucleotides away from the 5’ end. Additionally, the reverse primer’s 5’ end was biotin-labeled. All primers were synthesized by Sangon Biotech (Sangon Biotech (Shanghai) Co., Ltd, Shanghai, China). The RAA basic reaction for LFD was performed according to instructions provided in the nfo-based RAA nucleic acid amplification kit (Jiangsu Qitian Gene Biotechnology Co., Ltd., Jiangsu, China). The nfo-based RAA reaction system was based on the AGE-based RAA reaction volume with the addition of 0.6 μL of fluorescent probe (10 μM) and an adjusted volume of nuclease-free water (16.9 μL). The reaction was carried out under the same time and temperature conditions, following 10 μL of nfo-based RAA amplicons were taken and diluted with 50 μL of PBS and later inserted into the LFD strips (Nanjing Wobo Biotechnology Co., Ltd., Nanjing, China) for 2 minutes. The design of RAA primers, Nfo-probe, and the principle of the RAA-LFD assay are illustrated in Figure 1.
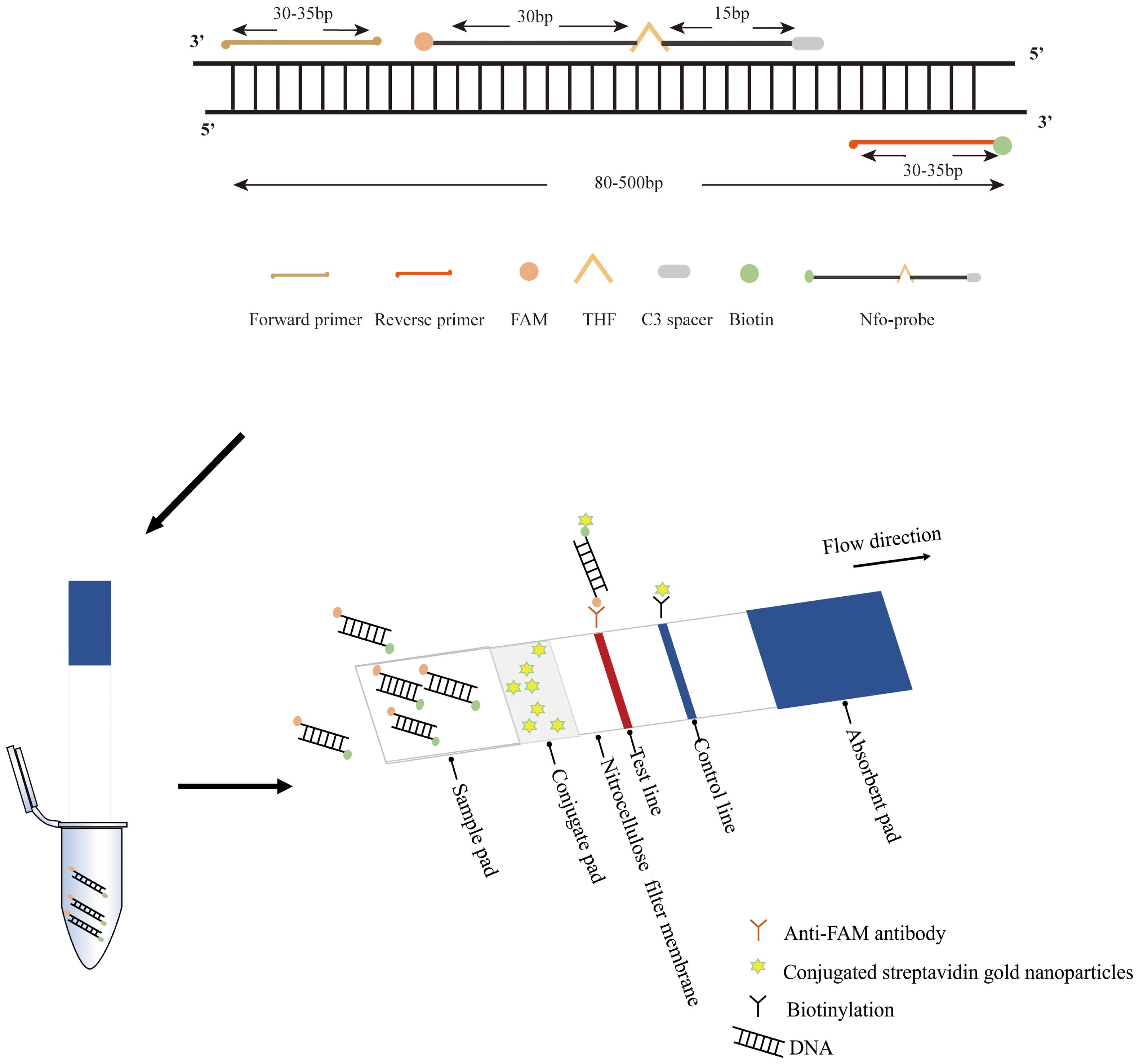
Figure 1. The principle of RAA primers, nfo-probe, and the RAA-LFD assay.
Initially, the RAA basic reaction was conducted with a fixed reaction time of 40 minutes at a gradient temperature from 34°C to 40°C. The results were visualized using a nucleic acid test strip. The test strips were positioned against a white backdrop, and the T-lines were digitally outlined using ImageJ 2 software (National Institutes of Health; Bethesda, MD, USA) to quantify color intensity. This process was conducted three times, and the collected data were subsequently inputted into GraphPad 8.0 software (Graph Pad Software, La Jolla, CA, USA) to generate a line graph illustrating the grayscale values of the T-lines at the different reaction temperatures. Subsequently, the RAA basic reaction was executed at the optimal temperature determined in the preliminary step, with reaction times varying from 5 to 40 minutes. The results were visualized using a nucleic acid test strip. Applying the methodology above, a line graph was formulated to assess the grayscale values of the T-line at various reaction durations.
The SVA 3D gene was amplified by PCR with designed primers (SVA 3D-F: 5’-TGATGACTGAGCTAGAGCCTGG-3’, SVA 3D-R: 5’-TCGAACAAGGCCCTCCATCTTG-3’). The amplicon was ligated to pMD18-T Vector (Takara Biomedical Technology Co., Ltd., Japan) and transformed into Escherichia coli DH5α. The pMD18-SVA-3D plasmid was subsequently confirmed in LB solid medium containing 1‰ ampicillin and expanded in LB broth for overnight incubation at 37°C. Finally, the plasmid was extracted using the Plasmid Mini Kit II (Omega Bio-Tek, Inc., Connecticut, USA) according to the manufacturer’s instructions. A NanoDrop™ 1000 Spectrophotometer (Thermo Fisher Scientific, WymanStreet, USA) was used to measure the concentration of the extracted plasmid and calculate copies using the following equation: Copy number per unit (copies/µL) = (concentration of DNA (ng) × 6.022 × 1023)/(entire length of template × 109 × 650) (dsDNA copy number calculator (uri.edu).
SVA and other laboratory-preserved viruses were used as templates to validate the specificity of the developed assay. The lowest detection limit of SVA RAA-LFD was determined using ten-fold serial dilutions of pMD18-SVA-3D plasmid, DNA amplification products, and viral fluid at a concentration of 108.25 TCID50/mL as templates. PCR and RT-qPCR methods were employed as parallel experiments for comparison.
The reaction system for PCR (Nanjing Vazyme Biotech Co., Ltd., China) is as follows: 25 µL of 2×Phanta Max Buffer, 1µL of dNTP Mix (10 mM each), 1µL of Phanta Max Super-Fidelity DNA Polymerase, 2 µL of forward primer (10 µM), 2 µL of reverse primer(10 µM), 1 µL of DNA template, and 19 µL of ddH2O. SVA specific forward primer: SVA PCR-F (5’-CAAGGAATTTGAATATGACATGG-3’) and reverse primer: SVA PCR-R (5’-GCAGCTTCTCGAGTAGTGTTCC-3’) were described previously (Qian et al., 2016). Amplicon product size was 298 bp. The thermal cycling conditions: one cycle at 95°C for 2 min; 35 cycles at 95°C for 15 s, 57°C for 30 s, and 72°C for 30 s, and one cycle at 72°C for 5 min. Amplicon products were analyzed by 2% AGE.
The reagent HiScript II Q Select RT SuperMix for qPCR (Nanjing Vazyme Biotech Co., Ltd., China) was used to reverse transcribe RNA into cDNA. 9 µL 2 × ChamQ SYBR qPCR Master Mix. The reaction system for qPCR is as follows: 5 µL 2 × ChamQ SYBR qPCR Master Mix (Nanjing Vazyme Biotech Co., Ltd., China), 0.2 µL forward primer (10 µM), 0.2 µL reverse primer (10 µM), 1 µL of DNA template, and 3.6 µL of ddH2O. SVA specific forward primer(SVA qPCR-F: 5’-GGGTAACACTGACACCGATTT-3’) and reverse primer (SVA qPCR-R: 5’-TCGAGATCGATCAAACAGGAAC-3’) were designed based on the VP1 gene region with a length of 87 bp (Bracht et al., 2016). The thermal cycling conditions: 48°C for 5 min, 95°C for 10 min, 40 cycles of 95°C for 5 sec, and 60°C for 1 min.
Several concentrations of the pMD18-SVA-3D plasmid were used as templates to assess the reproducibility of the established assay, including 3.86 × 104 copies/µL (strongly positive), 3.86 × 102 copies/µL (moderately positive) and 3.86 × 101 copies/µL (weak positive). The experiment was repeated three times.
The six candidate primers were evaluated using the RAA basic assay, conducted at 37°C for 40 minutes, with the GD-SVA2-2018 strain as the template. The 2%AGE results (Figure 2) demonstrated that the primer pair SVA RAA-F2/R2 produced the brightest specific bands compared to the other groups. These findings indicate that the SVA RAA-F2/R2 primer pair exhibits the highest specificity. Consequently, the SVA RAA-F2/R2 primer pair was selected for further research in the RAA-LFD assay.
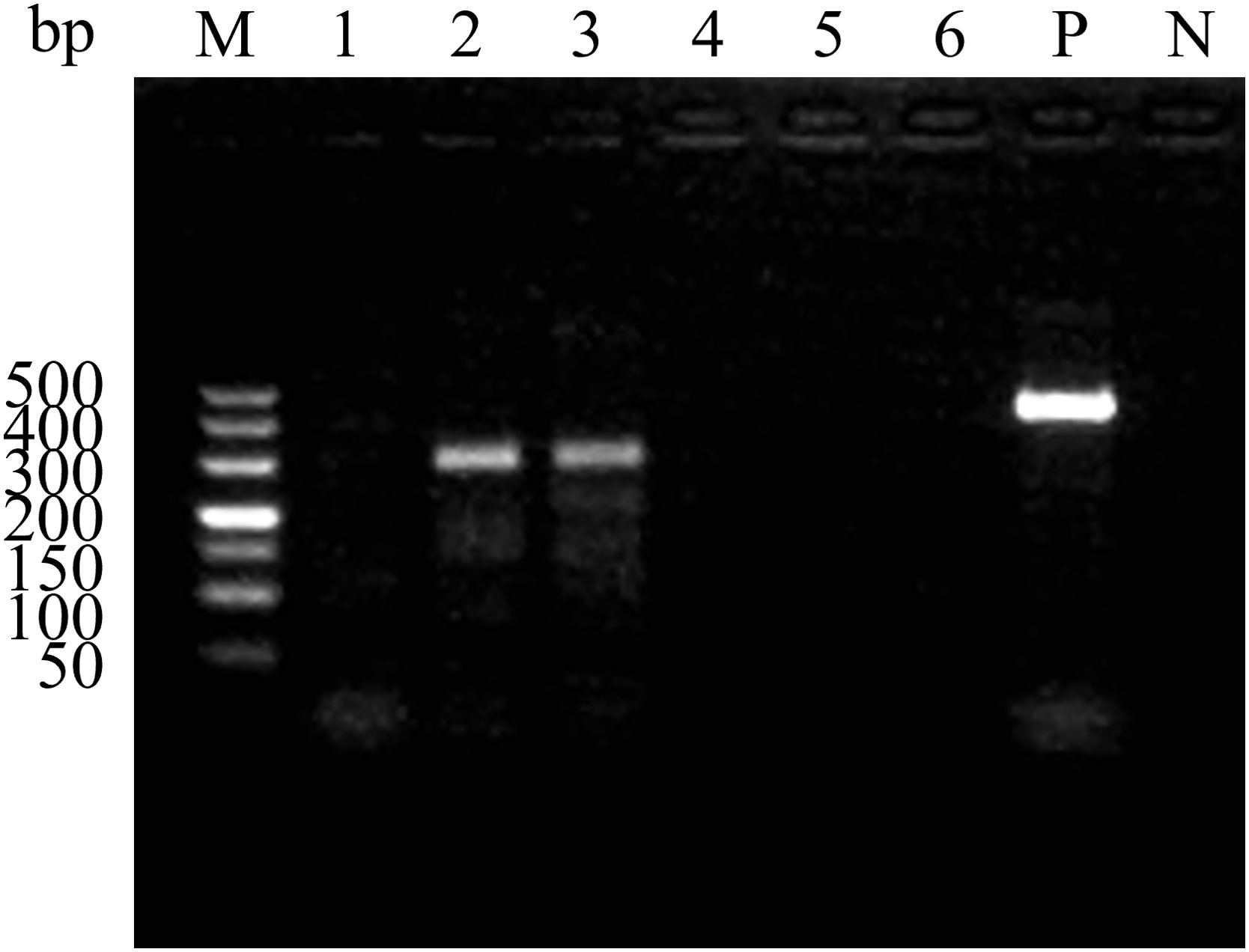
Figure 2. Amplification results of the RAA primer pairs. Serial number 1 to 6 are SVA RAA-F1/R1, SVA RAA-F2/R2, SVA RAA-F3/R2, SVA RAA-F4/R4, SVA RAA-F5/R5, and SVA RAA-F6/R6. M, 500 bp DNA marker; P, Positive control; N, Negative control.
To establish the SVA RAA-LFD assay, we have successfully designed an ideal fluorescent probe according to the gene sequence amplified by the SVA F2/R2 primer pair, nfo-probe (5’-6-FAM-TTTGTTCTACACATACATGTCAGAGTACGC-THF-CATCGGGTTTTCTCC-C3 spacer-3’) (Figure 3A). This design fully complies with the criteria for a good nfo-probe. The 5’ end of the reserve primer SAV RAA-R1 was also modified with biotin as RAA-FB: 5’-(Biotin) TAGTCACCGTCTAAGAATTTTTGGATTTGCAT-3’. Subsequently, the nfo-probe and modified and screened primers were used in the nfo-based RAA basic reaction, and LFD detected the amplicons. The results (Figure 3B) showed that the control (C)-line in both the negative control and the positive test groups was blue, indicating that the test strip was valid. The T-line in the negative control group showed no color, and the T-line in the SVA experimental group was red, indicating that the test result was positive and that the SVA RAA-LFD assay was initially established.
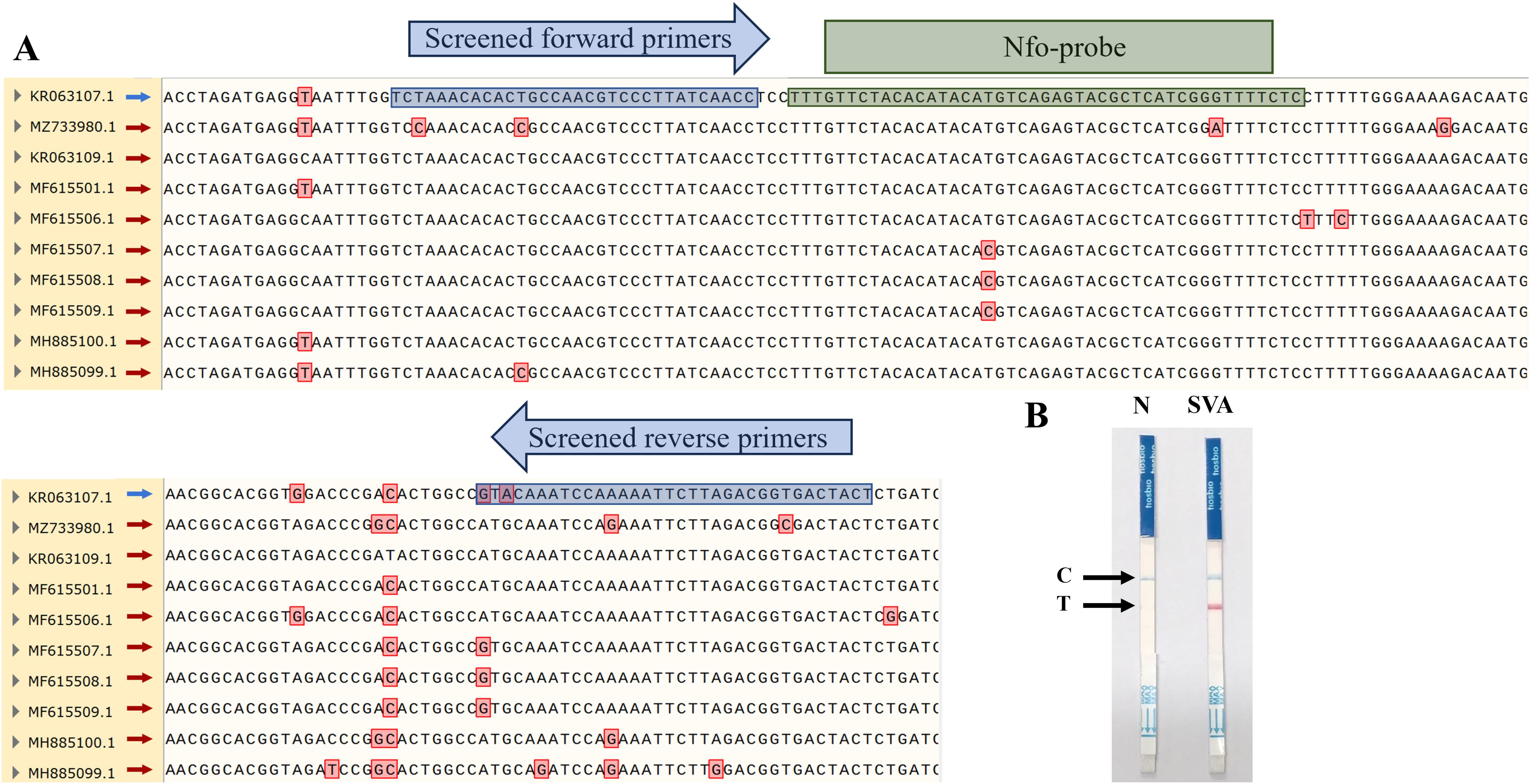
Figure 3. Partial results of multiple sequence alignment for SVA 3D gene and establishment of SVA RAA-LFD assay. (A) Red boxes represent nucleotide mutation sites in the alignment results. Blue boxes represent the binding sites of screened primers. Green boxes represent the binding sites of the nfo-probe. (B) Preliminary results of RAA-LFD for SVA detection.
To achieve the optimal reactivity performance of the SVA RAA-LFD assay, the preferred temperature and time for the RAA basic reaction were evaluated. RAA basic reactions were carried out at seven different temperatures from 34°C to 40°C (34°C, 35°C, 36°C, 37°C, 38°C, 39°C, and 40°C) for 40 minutes in a thermostatic. Subsequently, the RAA basic reaction target amplicon was detected by LFD under different temperature gradients. The results indicate that the T-lines of all test strips appeared red, demonstrating the effectiveness of the test strips within the temperature gradients. The result of the gray value analysis of the T-line (Figure 4A) showed that 35°C was the optimal reaction temperature for the SVA RAA-LFD assay. Furthermore, the highest value gray value analysis of the T-line was observed at 15 min (Figure 4B), indicating that 15 minutes was the optimal reaction time for the RAA basic reaction.
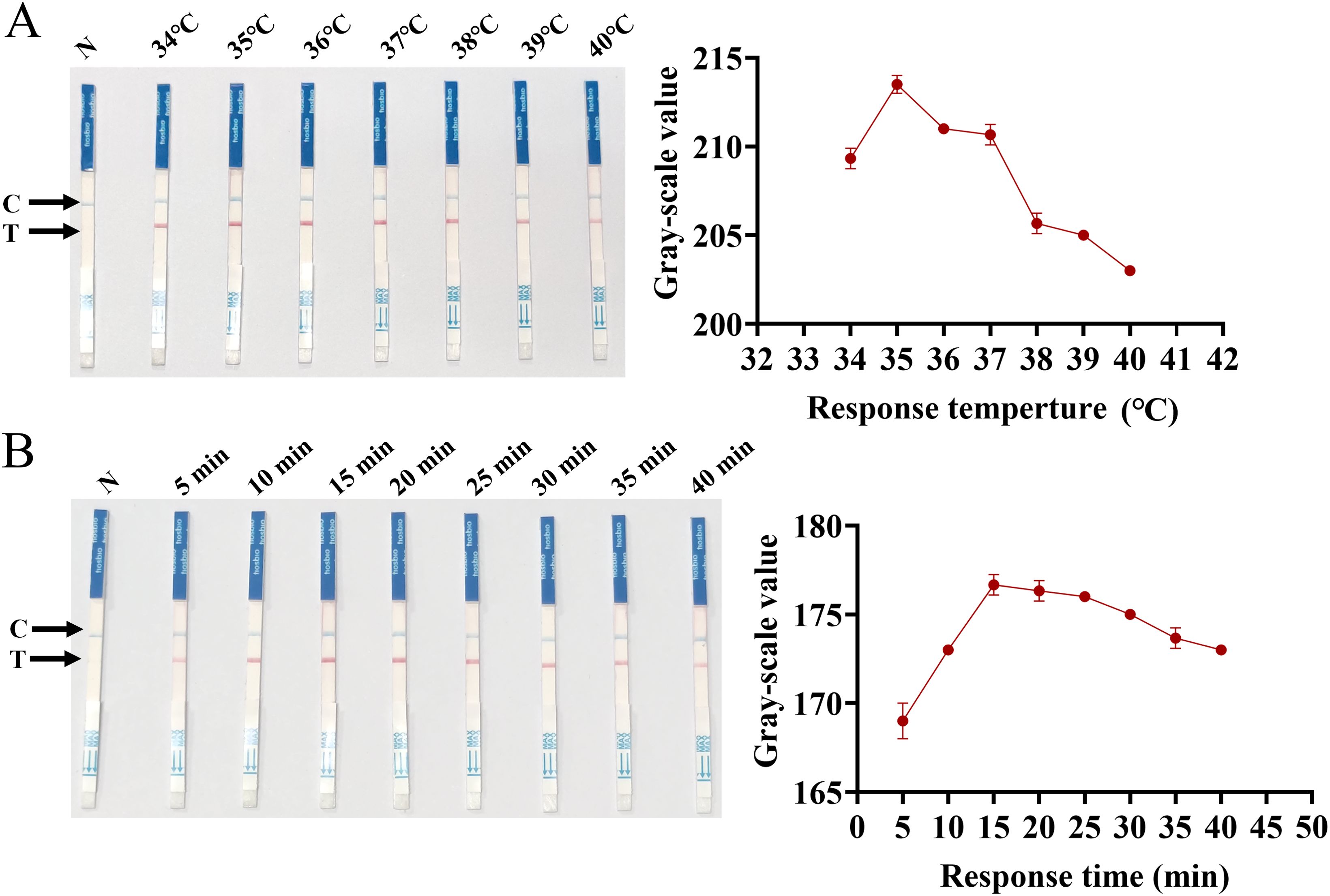
Figure 4. Optimization of SVA RAA-LFD reaction conditions. (A) Optimization of temperature for the RAA basic reaction. (B) Optimization of time for the RAA basic reaction.
As shown in Figure 5, in the analytical specificity analysis, the SVA RAA-LFD only produced visible red bands on the T-line for SVA. None of the other clinically important porcine pathogens, such as FMDV, ASFV, PCV2, JEV, CSFV, and PPV, produced visible bands on the T-line. The specificity of the SVA RAA-LFD assay was further validated by three independent technicians, confirming its high specificity.
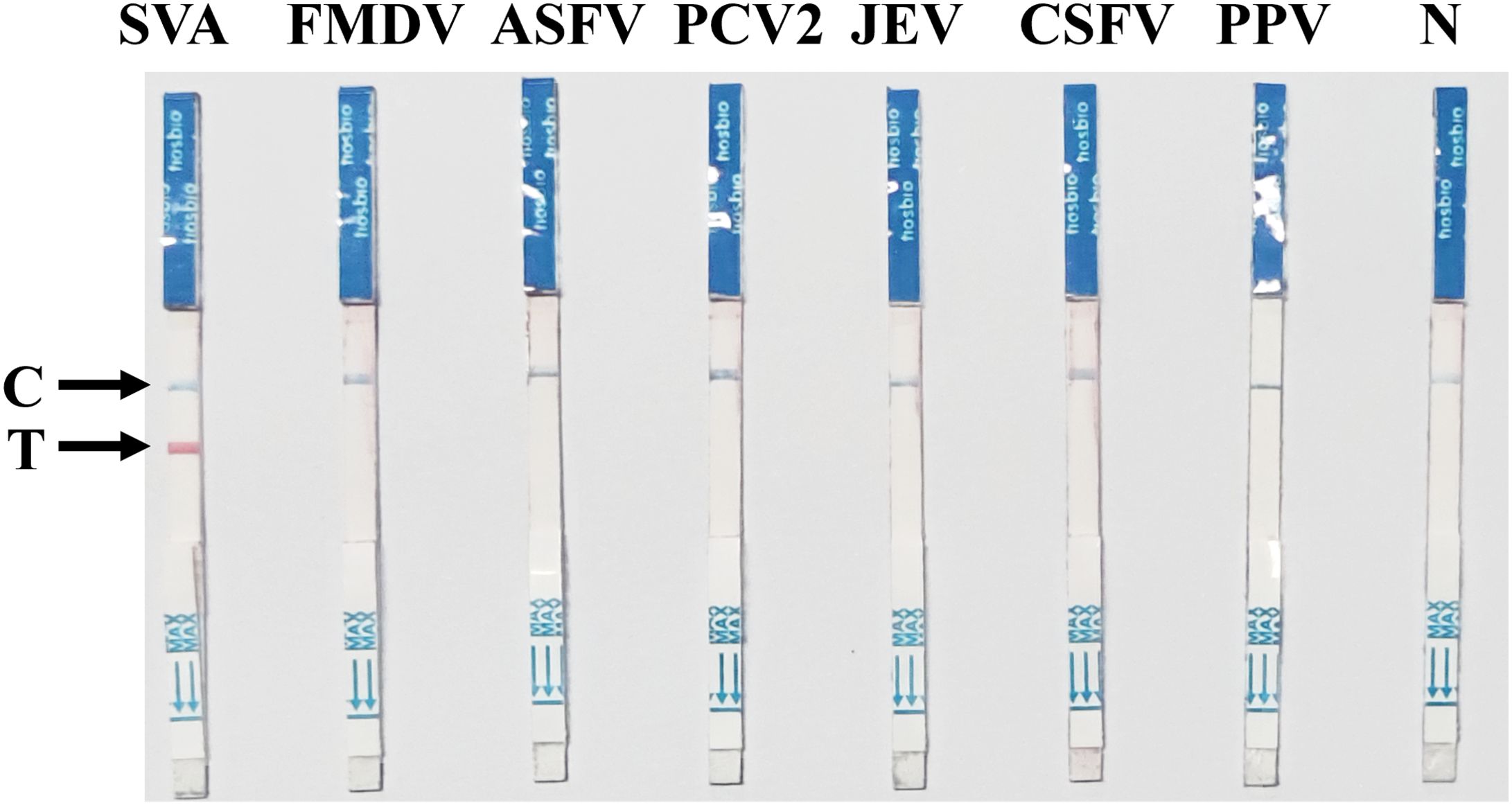
Figure 5. Specificity of the SVA RAA-LFD assay. The nucleic acids of a batch of pathogens, including FMDV, ASFV, PCV2, JEV, CSFV, and PPV, were used to evaluate the specificity of the SVA RAA-LFD assay.
To analyze the sensitivity of SVA RAA-LFD detection, this method was used to detect SVA pMD18-SVA-3D plasmids, DNA amplification products, and viral diluted tenfold (3.86×1010~3.86×100 copies/µL, 8.76×101~8.76×10-9 ng/µL, and 1×108.25~1×10-1.75 TCID50/mL, respectively). PCR and RT-qPCR were performed as parallel experiments. The results (Figure 6) showed that the lowest concentration of template detection of RAA-LFD for SVA plasmids (Figure 6A), DNA amplification product (Figure 6B), and viral fluid (Figure 6C) were 3.86×101 copies/µL, 8.76×10-7 ng/µL, and1×100.25 TCID50/mL, respectively. The sensitivity of the SVA RAA-LFD assay was significantly higher than that of the PCR assay, yet slightly less than 10 times that of the RT-qPCR assay. However, the sensitivity of this method in detecting the plasmid concentration remained consistent with that of the RT-qPCR assay.
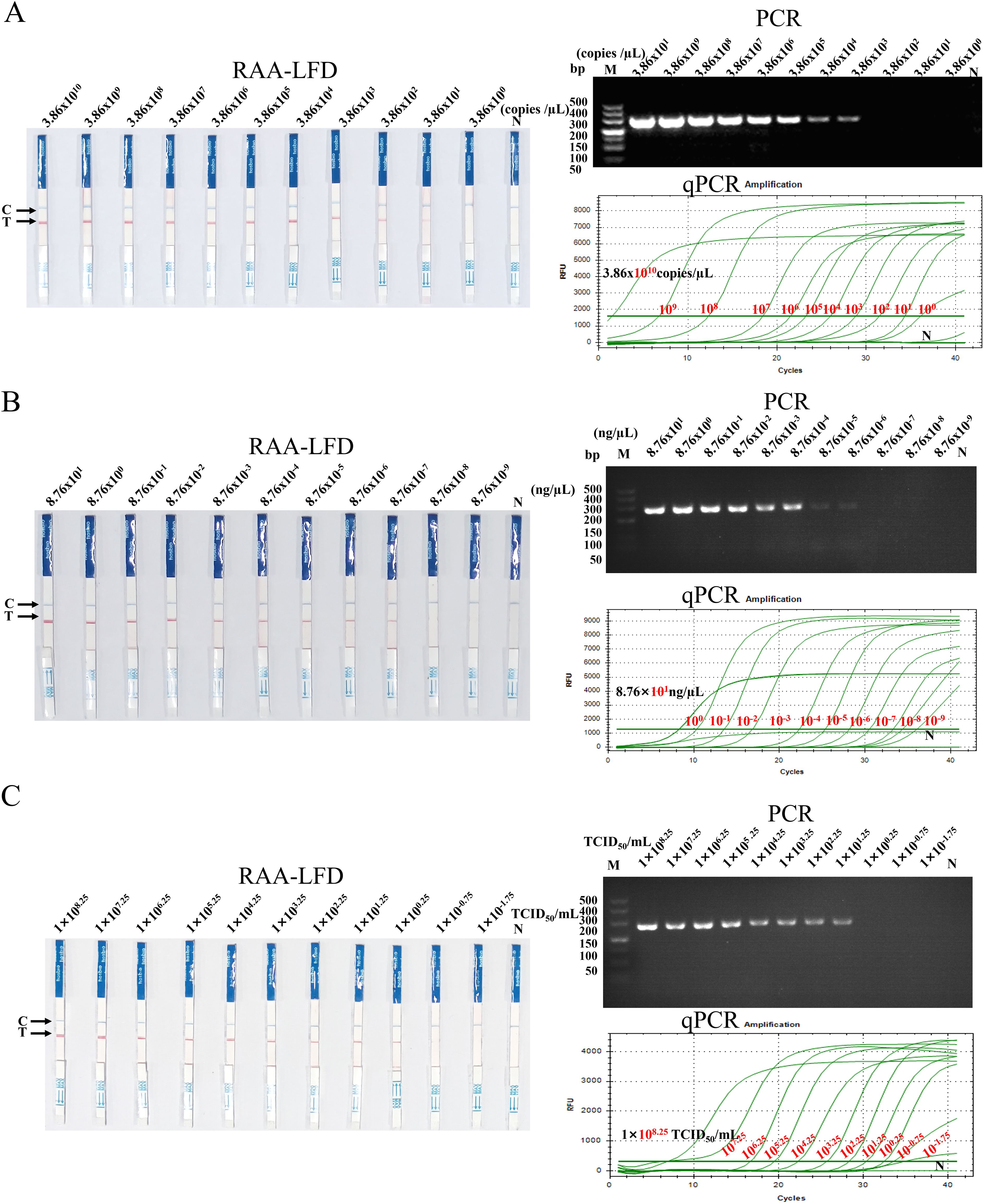
Figure 6. Sensitivity of the SVA RAA-LFD assay. (A) The results of detecting serially diluted pMD18-SVA-3D plasmids using the three methods. (B) The results of detecting serially diluted SVA DNA amplification product using the three methods. (C) The results of detecting serially diluted SVA viral fluid using the three methods. M: 500 bp DNA marker, N: Negative control, Cycles>35: Negative.
The SVA RAA-LFD assay for three different concentrations of the pMD18-SVA-3D plasmid showed all positive reaction signals at the T-line, with good reproducibility across three replicates. There was a slight difference in the intensity of the detection line positive response signal for the lowest detection concentration repeatability experiments, but all three replicates resulted in positive results (Figure 7).
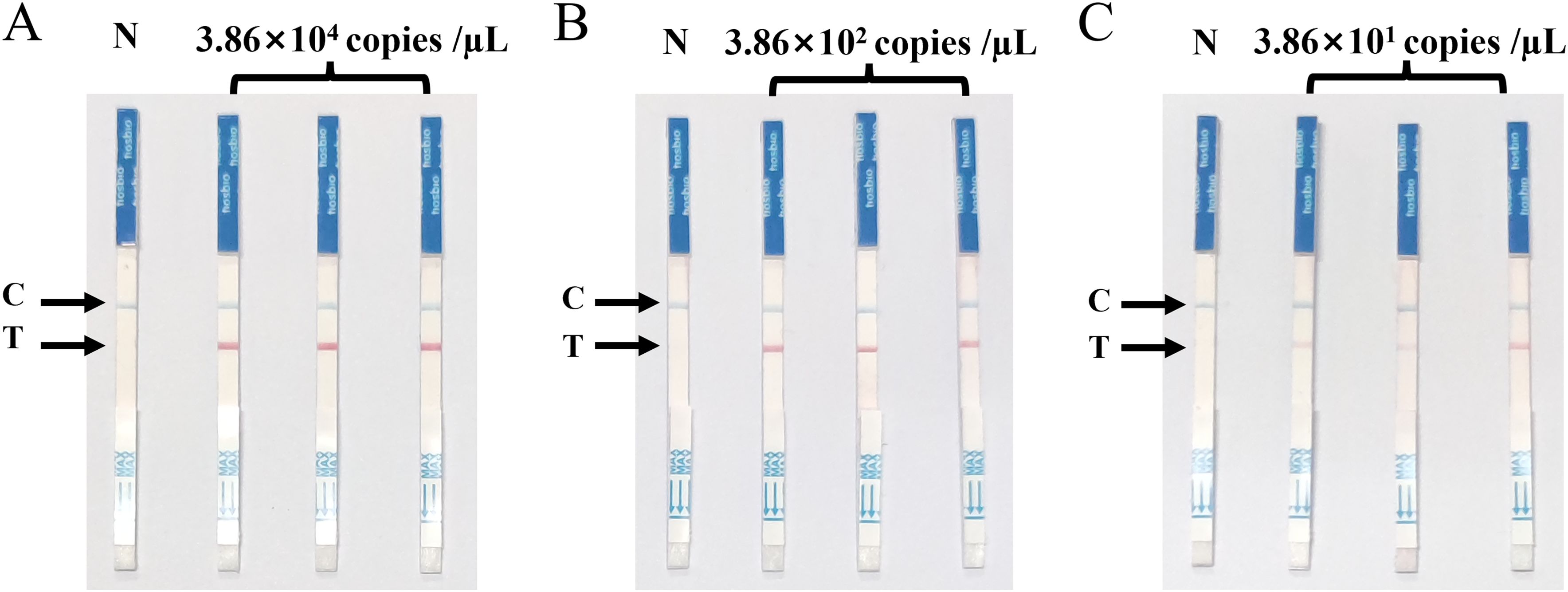
Figure 7. Repeatability of the SVA RAA-LFD assay. (A) Strong positive duplicate test of the SVA RAA-LFD assay for plasmid concentration of 3.86×104 copies/µL. (B) Moderate positive duplicate test of the SVA RAA-LFD assay for plasmid concentration of 3.86×102 copies/µL. (C) Weak positive duplicate test of the SVA RAA-LFD assay for plasmid concentration of 3.86×101 copies/µL.
To further assess the clinical performance of the SVA RAA-LFD assay, forty-four clinical samples were tested using the assay. Parallel experiments were conducted using PCR and RT-qPCR methods. The results of the PCR and RAA-LFD methods are presented in Figure 8, while the comparative outcomes of these three methods are displayed in Table 2. Specifically, the RAA-LFD method successfully detected 27 positive samples, the RT-qPCR method identified 27 samples, and the PCR method identified 24 samples. Notably, the RAA-LFD and RT-qPCR methods exhibited consistent detection rates, demonstrating the clinical feasibility of the SVA RAA-LFD assay in this study.
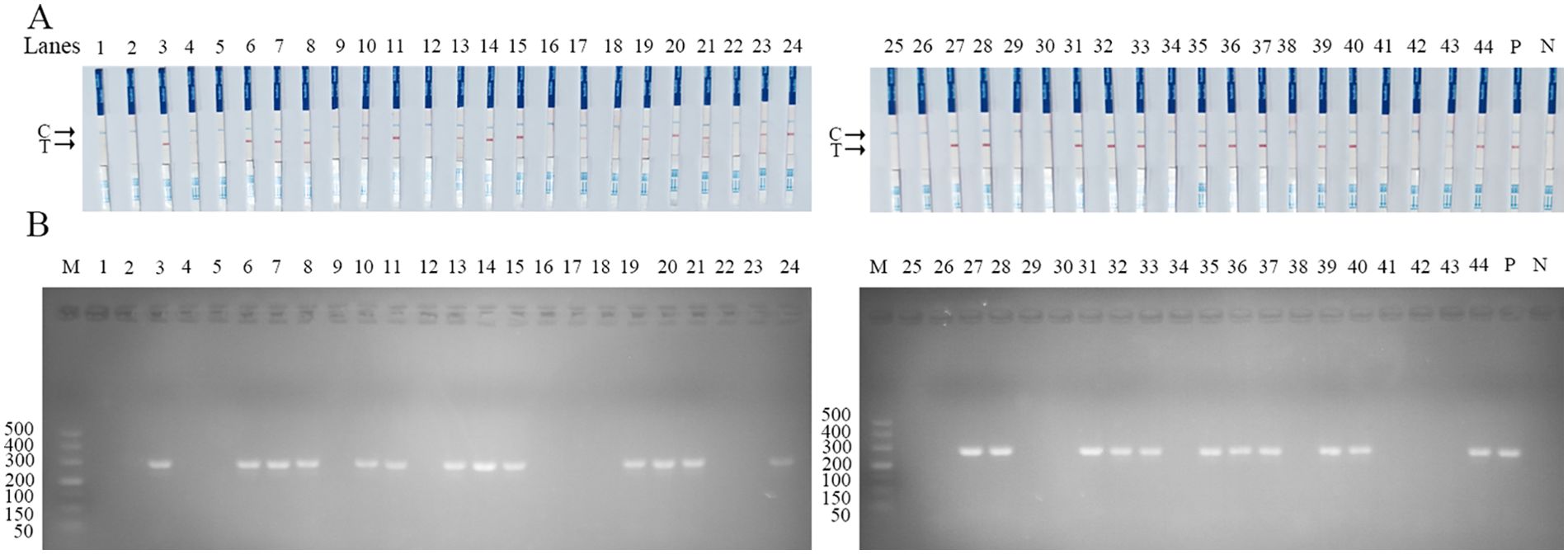
Figure 8. Performance of SVA RAA-LFD on clinical swine samples. (A) The result of clinical swine samples detection by the SVA RAA-LFD assay. (B) The result of clinical swine samples detection by conventional PCR method. Lanes1–44 Different clinical swine samples were preserved in our laboratory. N, Negative control; P, Positive control; M, 500 bp DNA marker.
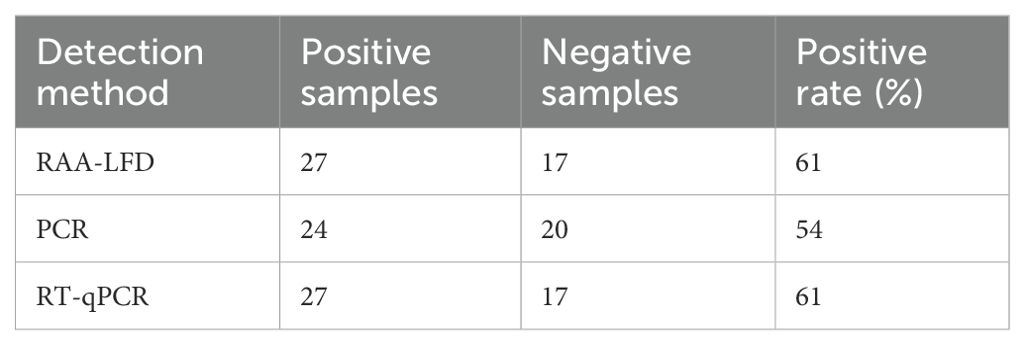
Table 2. Clinical application of the SVA RAA-LFD assay.
SVA is a recently emerged pathogen of pigs that is both pathogenic and infectious, and it is clinically indistinguishable from other pathogens causing vesicular lesions in pigs, including FMDV. Furthermore, contemporary SVA isolates exhibit significant genetic divergence and evolution compared to historical strains, demonstrating a trend toward greater pathogenicity (Chen et al., 2019; Fernandes et al., 2018; Jiang et al., 2021; Joshi et al., 2016, 2020; Li et al., 2024; Wang et al., 2019; Zhang et al., 2020). Early diagnosis of SVA is critical not only for providing valuable epidemiological information and for the rapid initiation of prevention and control strategies but also for facilitating the accurate diagnosis, prevention, and control of other porcine vesicular epidemics, particularly FMD. Therefore, developing a highly specific and sensitive rapid diagnostic method for SVA is essential to provide effective technical support for these needs.
RAA is a novel molecular biology assay that allows for the amplification of large amounts of target nucleic acids in a short period, which has been widely used in the detection of a variety of pathogens that infect livestock or humans (Chen et al., 2022; Fan et al., 2020; Liu et al., 2023; Tian et al., 2023; Zhao et al., 2024a). In this study, we first established a rapid visualization assay specific to SVA by combining RAA with LFD. Initially, to ensure optimal sensitivity and specificity during the development of the SVA RAA-LFD assay, we identified the conserved region of the SVA 3D gene as the target sequence by aligning the nucleotides of multiple sequences obtained from GenBank. We successfully screened for the optimal primer pair and designed the nfo-probe with the highest match.
Furthermore, building upon the preliminary SVA RAA-LFD assay, we fine-tuned the reaction time and temperature parameters. This adjustment allows the assay to be completed in just 17 minutes at 35 °C, comprising 15 minutes for the RAA basic reaction and 2 minutes for LFD processing. These modifications enhance the accuracy and efficiency of the developed assay. Subsequently, we further evaluated the specificity and sensitivity of the optimized assay. The final detection data showed that the lowest detection limits of the established assay for pMD18-SVA-3D plasmid, DNA amplification product, and viral were 3.86×101cpoies/µL, 8.76×10-7 ng/µL, and 1×100.25 TCID50/mL, respectively. The SVA RAA-LFD assay we developed is 100 times more sensitive to plasmids than the SVA 3D-based PCR method studied by Qian et al. (Qian et al., 2016) and nearly consistent with the VP1-based RT-qPCR studied by Bracht et al (Bracht et al., 2016).
To assess clinical applicability, we tested forty-four clinical samples using the RAA-LFD assay, which showed a positive detection rate consistent with RT-qPCR. In comparison with the other two detection methodologies, the PCR assay detected three fewer positive samples. This discrepancy may arise from the low viral load present in the clinical samples, which could be beneath the PCR assay’s limit of detection, or it may be attributable to the primers’ insufficient specificity for the identification of SVA within the clinical samples. The RAA-LFD assay established in this study produced T-line specific positive reaction signals only for SVA, with no cross-reactivity to other common swine pathogens, including FMD, and demonstrated good reproducibility. In summary, the developed SVA RAA-LFD assay exhibits excellent sensitivity, specificity, and clinical applicability, making it a promising method for detecting SVA.
It is well known that the primary advantage of RAA is that it does not require an expensive thermal cycler to amplify the target gene. Compared to standard laboratory molecular biology detection techniques such as PCR and qPCR, RAA is more convenient and time-efficient, requiring only a water bath or a portable suitcase lab for operation, thereby enabling on-site diagnosis of the target gene (El Wahed et al., 2021). In recent years, other isothermal amplification techniques, such as LAMP, have been used for SVA detection (Armson et al., 2019; Zeng et al., 2018). However, compared to LAMP, RAA offers two distinct advantages. First, while LAMP necessitates a reaction temperature range of 60~65°C, RAA operates efficiently at a lower temperature, approximately 37°C. In our study, target gene amplification occurred within a temperature range of 34~40°C, with 35°C being the most favorable; notably, the reaction can even proceed at body temperature. Second, LAMP requires 4~6 primers to support the amplification reaction. In contrast, the RAA reaction can be accomplished with just two conventional primers or by adding a specific probe.
The combined application of RAA and LFD integrates the high sensitivity of polymerase chain reaction with immunochromatographic techniques, enabling rapid visualization of detection results (Homklinkaew et al., 2023; Li et al., 2022; Yu et al., 2019). In brief, this method involves the combination of target genes labeled with specific probes with antibodies conjugated to colloidal gold encapsulated on the test strip, ultimately translating the detection results into visible color signals discernible to the naked eye. The simplicity of operation and lack of requirement for expensive instrumentation make this method suitable for application in remote areas and field settings.
Regrettably, the method established in this study was not validated with clinical samples from multiple countries. However, previous studies have shown that the RAA reaction can accommodate mismatches of 5-9 nucleotide bases (Abd El. Wahed et al., 2013). Upon aligning multiple SVA sequences from various countries, including China, the USA, and Brazil, we found that the individual base differences in the selected amplification sequences were less than 5 nucleotides, indicating the potential applicability of the assay used in this study. Testing a large variety of clinical swine samples would provide more robust evidence for the feasibility of the developed method. Therefore, we intend to seek out multiple clinical samples for further validation in subsequent investigations.
In summary, this study has established a convenient and sensitive RAA-LFD assay based on the conserved region of the SVA3D gene. The assay provides visually detectable results in under 17 minutes using an instrument-free technique. This makes it suitable for point-of-care diagnosis for SVA in the field or remote areas lacking instrumentation.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The animal study was approved by Ethics Committee and the Laboratory Animal Care and Use Committee of South China Agricultural University (protocol code No.44007200110300). The study was conducted in accordance with the local legislation and institutional requirements.
YS: Conceptualization, Data curation, Formal analysis, Investigation, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. YF: Formal analysis, Investigation, Methodology, Writing – review & editing. SZ: Data curation, Supervision, Writing – review & editing, Visualization. WW: Formal analysis, Methodology, Software, Writing – review & editing. LW: Conceptualization, Methodology, Supervision, Writing – review & editing. WC: Methodology, Project administration, Supervision, Writing – review & editing. YH: Formal analysis, Supervision, Writing – review & editing, Software. LY: Investigation, Resources, Writing – review & editing, Visualization. HD: Data curation, Supervision, Validation, Writing – review & editing. MZ: Formal analysis, Methodology, Visualization, Writing – review & editing. SF: Investigation, Methodology, Supervision, Writing – review & editing. ZL: Conceptualization, Investigation, Project administration, Supervision, Writing – review & editing. JC: Conceptualization, Project administration, Resources, Supervision, Validation, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Funding for this work was provided by the National Key Research and Development Program of China (No.2021YFD1800300), the Program of National Natural Science Foundation of China (No.32172824 and No.32102643), the Science and Technology Program of Guangzhou, China (No. 202206010161), and South China Agricultural University and Wen’s group Science and Technology Innovation Centre results incubation project (WS-HN-JKYZ-202404-118).
Authors ZL and LW were employed by the company Wen’s Foodstuffs Group Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2024.1474676/full#supplementary-material
Abd El. Wahed, A., El-Deeb, A., El-Tholoth, M., Abd El Kader, H., Ahmed, A., Hassan, S., et al. (2013). A portable reverse transcription recombinase polymerase amplification assay for rapid detection of foot-and-mouth disease virus. PloS One 8, e71642. doi: 10.1371/journal.pone.0071642
Adams, M. J., Lefkowitz, E. J., King, A. M. Q., Bamford, D. H., Breitbart, M., Davison, A. J., et al. (2015). Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses. Arch. Virol. 160, 1837–1850. doi: 10.1007/s00705-015-2425-z
Adams, M. J., Lefkowitz, E. J., King, A. M., Harrach, B., Harrison, R. L., Knowles, N. J., et al. (2016). Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses. Arch. Virol. 161, 2921–2949. doi: 10.1007/s00705-016-2977-6
Armson, B., Walsh, C., Morant, N., Fowler, V. L., Knowles, N. J., Clark, D. (2019). The development of two field-ready reverse transcription loop-mediated isothermal amplification assays for the rapid detection of Seneca Valley virus 1. Transbound Emerg. Dis. 66, 497–504. doi: 10.1111/tbed.13051
Arzt, J., Bertram, M. R., Vu, L. T., Pauszek, S. J., Hartwig, E. J., Smoliga, G. R., et al. (2019). First detection and genome sequence of senecavirus A in Vietnam. Microbiol. Resour. Announc. 8, e01247–e01218. doi: 10.1128/MRA.01247-18
Bienes, K. M., Mao, L., Selekon, B., Gonofio, E., Nakoune, E., Wong, G., et al. (2022). Rapid detection of the varicella-zoster virus using a recombinase-aided amplification-lateral flow system. Diagnostics (Basel) 12, 2957. doi: 10.3390/diagnostics12122957
Bracht, A. J., O’Hearn, E. S., Fabian, A. W., Barrette, R. W., Sayed, A. (2016). Real-time reverse transcription PCR assay for detection of senecavirus A in swine vesicular diagnostic specimens. PloS One 11, e0146211. doi: 10.1371/journal.pone.0146211
Chen, L., Zhang, J., Wang, M., Pan, S., Mou, C., Chen, Z. (2019). Pathogenicity of two Chinese Seneca Valley virus (SVV) strains in pigs. Microbial pathogenesis. 136, 103695. doi: 10.1016/j.micpath.2019.103695
Chen, Y., Zong, N., Ye, F., Mei, Y., Qu, J., Jiang, X. (2022). Dual-CRISPR/cas12a-assisted RT-RAA for ultrasensitive SARS-coV-2 detection on automated centrifugal microfluidics. Analytical Chem. 94, 9603–9609. doi: 10.1021/acs.analchem.2c00638
Chow, W. H., McCloskey, C., Tong, Y., Hu, L., You, Q., Kelly, C. P., et al. (2008). Application of isothermal helicase-dependent amplification with a disposable detection device in a simple sensitive stool test for toxigenic Clostridium difficile. J. Mol. Diagn. 10, 452–458. doi: 10.2353/jmoldx.2008.080008
Dvorak, C. M., Akkutay-Yoldar, Z., Stone, S. R., Tousignant, S. J., Vannucci, F. A., Murtaugh, M. P. (2017). An indirect enzyme-linked immunosorbent assay for the identification of antibodies to Senecavirus A in swine. BMC veterinary Res. 13, 50. doi: 10.1186/s12917-017-0967-x
El Wahed, A. A., Patel, P., Maier, M., Pietsch, C., Rüster, D., Böhlken-Fascher, S., et al. (2021). Suitcase lab for rapid detection of SARS-coV-2 based on recombinase polymerase amplification assay. Anal. Chem. 93, 2627–2634. doi: 10.1021/acs.analchem.0c04779
Fan, X., Li, L., Zhao, Y., Liu, Y., Liu, C., Wang, Q., et al. (2020). Clinical validation of two recombinase-based isothermal amplification assays (RPA/RAA) for the rapid detection of African swine fever virus. Front. Microbiol. 11, 1696. doi: 10.3389/fmicb.2020.01696
Feng, J., Chen, J., Du, B., Cui, X., Xia, Y., Xue, G., et al. (2024). Development of a recombinase-aided amplification assay for the rapid detection of candida auris. Anal. Chem. 96, 9424–9429. doi: 10.1021/acs.analchem.4c00450
Fernandes, M. H. V., Maggioli, M. F., Joshi, L. R., Clement, T., Faccin, T. C., Rauh, R., et al. (2018). Pathogenicity and cross-reactive immune responses of a historical and a contemporary Senecavirus A strains in pigs. Virology. 522, 147–157. doi: 10.1016/j.virol.2018.06.003
Hai, C. (2010). Recombinase-aid amplification:a novel technology of in vitro rapid nucleic acid amplification. Scientia Sinica (Vitae). 40, 983–988.
Hales, L. M., Knowles, N. J., Reddy, P. S., Xu, L., Hay, C., Hallenbeck, P. L. (2008). Complete genome sequence analysis of Seneca Valley virus-001, a novel oncolytic picornavirus. J. Gen. Virol. 89, 1265–1275. doi: 10.1099/vir.0.83570-0
Hause, B. M., Myers, O., Duff, J., Hesse, R. A. (2016). Senecavirus A in pigs, United States 2015. Emerg. Infect. Dis. 22, 1323–1325. doi: 10.3201/eid2207.151591
He, Y., Chen, W., Fan, J., Fan, S., Ding, H., Chen, J., et al. (2021). Recombinase-aided amplification coupled with lateral flow dipstick for efficient and accurate detection of porcine parvovirus. Life (Basel) 11, 762. doi: 10.3390/life11080762
Homklinkaew, P., Phatthanakunanan, S., Jala, S., Boonsoongnern, A., Lertwatcharasarakul, P. (2023). Development of a recombinase-aided amplification method combined with lateral flow dipstick assay to detect Porcine circovirus type 2. Vet. World 16, 2313–2320. doi: 10.14202/vetworld.
Hou, L., Li, D., Zhang, N., Zhao, J., Zhao, Y., Sun, X. (2022). Development of an isothermal recombinase-aided amplification assay for the rapid and visualized detection of Klebsiella pneumoniae. J. Sci. Food Agric. 102, 3879–3886. doi: 10.1002/jsfa.v102.9
Jaroenram, W., Kiatpathomchai, W., Flegel, T. W. (2009). Rapid and sensitive detection of white spot syndrome virus by loop-mediated isothermal amplification combined with a lateral flow dipstick. Mol. Cell Probes 23, 65–70. doi: 10.1016/j.mcp.2008.12.003
Jaroenram, W., Owens, L. (2014). Recombinase polymerase amplification combined with a lateral flow dipstick for discriminating between infectious Penaeus stylirostris densovirus and virus-related sequences in shrimp genome. J. Virol. Methods 208, 144–151. doi: 10.1016/j.jviromet.2014.08.006
Jiang, J., Zha, Y., Liu, J., Xing, C., Mi, S., Yu, J., et al. (2021). Isolation and evolutionary analysis of Senecavirus A isolates from Guangdong province, China. Infect. Genet. Evol. 91, 104819. doi: 10.1016/j.meegid.2021.104819
Joshi, L. R., Fernandes, M. H. V., Clement, T., Lawson, S., Pillatzki, A., Resende, T. P., et al. (2016). Pathogenesis of Senecavirus A infection in finishing pigs. J. Gen. Virol. 97, 3267–3279. doi: 10.1099/jgv.0.000631
Joshi, L. R., Mohr, K. A., Gava, D., Kutish, G., Buysse, A. S., Vannucci, F. A., et al. (2020). Genetic diversity and evolution of the emerging picornavirus Senecavirus A. J. Gen. Virol. 101, 175–187. doi: 10.1099/jgv.0.001360
K., S., S., C., G., C. S., G., S., R., F. (2012). Seneca valley virus and vesicular lesions in a pig with idiopathic vesicular disease. J. Vet. Sci. 03, 123. doi: 10.4172/2157-7579.1000123
King, A. M. Q., Adams, M. J., Carstens, E. B., Lefkowitz, E. J. (2012). Virus taxonomy: ninth report of the international committee on taxonomy of viruses. Arch. Virol. 140, 1221–1234. doi: 10.1007/BF01309873
Knowles, N. J., Hales, L. M., Jones, B. H., Landgraf, J. G., Hallenbeck, P. L. (2006). Epidemiology of seneca valley virus: identification and characterization of isolates from pigs in the United States.
Kolm, C., Martzy, R., Führer, M., Mach, R. L., Krska, R., Baumgartner, S., et al. (2019). Detection of a microbial source tracking marker by isothermal helicase-dependent amplification and a nucleic acid lateral-flow strip test. Sci. Rep. 9, 393. doi: 10.1038/s41598-018-36749-7
Leme, R. A., Miyabe, F. M., Dall Agnol, A. M., Alfieri, A. F., Alfieri, A. A. (2019). A new wave of Seneca Valley virus outbreaks in Brazil. Transbound Emerg. Dis. 66, 1101–1104. doi: 10.1111/tbed.2019.66.issue-3
Leme, R. A., Zotti, E., Alcântara, B. K., Oliveira, M. V., Freitas, L. A., Alfieri, A. F., et al. (2015). Senecavirus A: an emerging vesicular infection in Brazilian pig herds. Transbound Emerg. Dis. 62, 603–611. doi: 10.1111/tbed.2015.62.issue-6
Li, J. S., Hao, Y. Z., Hou, M. L., Zhang, X., Zhang, X. G., Cao, Y. X., et al. (2022). Development of a recombinase-aided amplification combined with lateral flow dipstick assay for the rapid detection of the African swine fever virus. BioMed. Environ. Sci. 35, 133–140. doi: 10.3967/bes2022.018
Li, J., Liang, W., Xu, S., Shi, J., Zhou, X., Liu, B., et al. (2019). Rapid and sensitive detection of Senecavirus A by reverse transcription loop-mediated isothermal amplification combined with a lateral flow dipstick method. PloS One 14, e0216245. doi: 10.1371/journal.pone.0216245
Li, Y., Liu, T., Zhang, Y., Duan, X., Liu, F. (2024). RNA recombination: non-negligible factor for preventing emergence or reemergence of Senecavirus A. Front. Vet. Sci. 11, 1357179. doi: 10.3389/fvets.2024.1357179
Li, Y., Shang, J., Wang, Y., Luo, J., Jiang, W., Yin, X., et al. (2023). Establishment of two assays based on reverse transcription recombinase-aided amplification technology for rapid detection of H5 subtype avian influenza virus. Microbiol. Spectr. 11, e0218623. doi: 10.1128/spectrum.02186-23
Lin, J. Y., Chen, T. C., Weng, K. F., Chang, S. C., Chen, L. L., Shih, S. R. (2009). Viral and host proteins involved in picornavirus life cycle. J. Biomed. Sci. 16, 103. doi: 10.1186/1423-0127-16-103
Liu, W., Zhang, G., Xu, D., Ye, J., Lu, Y. (2023). A novel RAA combined test strip method based on dual gene targets for pathogenic vibrio vulnificus in aquatic products. Foods 12, 3605. doi: 10.3390/foods12193605
Mu, S., Abdullah, S. W., Zhang, Y., Han, S., Guo, H., Li, M., et al. (2020). Development of a novel SYBR green I-based quantitative RT-PCR assay for Senecavirus A detection in clinical samples of pigs. Mol. Cell Probes. 53, 101643. doi: 10.1016/j.mcp.2020.101643
Pasma, T., Davidson, S., Shaw, S. L. (2008). Idiopathic vesicular disease in swine in Manitoba. Can. Vet. J. 49, 84–85.
Peng, Y., Hu, R., Xue, S., He, Y., Tian, L., Pang, Z., et al. (2024). Rapid and highly sensitive colorimetric LAMP assay and integrated device for visual detection of monkeypox virus. Anal. Chim. Acta 1311, 342720. doi: 10.1016/j.aca.2024.342720
Pinheiro-de-Oliveira, T. F., Fonseca-Júnior, A. A., Camargos, M. F., Laguardia-Nascimento, M., Giannattasio-Ferraz, S., Cottorello, A. C. P., et al. (2019). Reverse transcriptase droplet digital PCR to identify the emerging vesicular virus Senecavirus A in biological samples. Transbound Emerg. Dis. 66, 1360–1369. doi: 10.1111/tbed.13168
Qian, S., Fan, W., Qian, P., Chen, H., Li, X. (2016). Isolation and full-genome sequencing of Seneca Valley virus in piglets from China 2016. Virol. J. 13, 173. doi: 10.1186/s12985-016-0631-2
Saeng-chuto, K., Rodtian, P., Temeeyasen, G., Wegner, M., Nilubo, D. (2017). The first detection of Senecavirus A in pigs in Thailand 2016. Transbound Emerg. Dis. 65, 285–288. doi: 10.1111/tbed.12654
Sun, D., Vannucci, F., Knutson, T. P., Corzo, C., Marthaler, D. G. (2017). Emergence and whole-genome sequence of Senecavirus A in Colombia. Transbound Emerg. Dis. 64, 1346–1349. doi: 10.1111/tbed.2017.64.issue-5
Tian, Y., Fan, Z., Xu, L., Cao, Y., Chen, S., Pan, Z., et al. (2023). CRISPR/Cas13a-assisted rapid and portable HBV DNA detection for low-level viremia patients. Emerg. Microbes Infect. 12, e2177088. doi: 10.1080/22221751.2023.2177088
Vannucci, F. A., Linhares, D. C., Barcellos, D. E., Lam, H. C., Collins, J., Marthaler, D. (2015). Identification and complete genome of seneca valley virus in vesicular fluid and sera of pigs affected with idiopathic vesicular disease, Brazil. Transbound Emerg. Dis. 62, 589–593. doi: 10.1111/tbed.2015.62.issue-6
Venkataraman, S., Reddy, S. P., Loo, J., Idamakanti, N., Hallenbeck, P. L., Reddy, V. S. (2008). Structure of Seneca Valley Virus-001: an oncolytic picornavirus representing a new genus. Structure 16, 1555–1561. doi: 10.1016/j.str.2008.07.013
Vieira, M. V., Yasumitsu, C. Y., Dall Agnol, A. M., Leme, R. A., Alfieri, A. F., Alfieri, A. A. (2022). The third wave of Seneca Valley virus outbreaks in pig herds in southern Brazil. Braz. J. Microbiol. 53, 1701–1706. doi: 10.1007/s42770-022-00767-5
Wang, M., Chen, L., Pan, S., Mou, C., Shi, K., Chen, Z. (2019). Molecular evolution and characterization of novel Seneca Valley virus (SVV) strains in South China. Infect. Genet. Evol. 69, 1–7. doi: 10.1016/j.meegid.2019.01.004
Wang, Q., Heo, W., Choi, S., Jang, W., Lim, C. S., Jung, H. I. (2024). Hand-held all-in-one (HAO) self-test kit for rapid and on-site detection of SARS-CoV-2 with colorimetric LAMP. Lab. Chip 24, 3265–3275. doi: 10.1039/D4LC00199K
Wu, X., Liu, Y., Gao, L., Yan, Z., Zhao, Q., Chen, F., et al. (2022). Development and application of a reverse-transcription recombinase-aided amplification assay for porcine epidemic diarrhea virus. Viruses 14, 591. doi: 10.3390/v14030591
Wu, Q., Zhao, X., Bai, Y., Sun, B., Xie, Q., Ma, J. (2017). The first identification and complete genome of senecavirus A affecting pig with idiopathic vesicular disease in China. Transbound Emerg. Dis. 64, 1633–1640. doi: 10.1111/tbed.2017.64.issue-5
Xu, W., Hole, K., Goolia, M., Pickering, B., Nfon, C. (2017). Genome wide analysis of the evolution of Senecavirus A from swine clinical material and assembly yard environmental samples. PloS One 12, e0176964. doi: 10.1371/journal.pone.0176964
Yang, M., van Bruggen, R., Xu, W. (2012). Generation and diagnostic application of monoclonal antibodies against Seneca Valley virus. J. Vet. Diagn. Invest. 24, 42–50. doi: 10.1177/1040638711426323
Yu, J., Shen, D., Dai, T., Lu, X., Xu, H., Dou, D. (2019). Rapid and equipment-free detection of Phytophthora capsici using lateral flow strip-based recombinase polymerase amplification assay. Lett. Appl. Microbiol. 69, 64–70. doi: 10.1111/lam.13166
Zeng, F., Cong, F., Liu, X., Lian, Y., Wu, M., Xiao, L., et al. (2018). Development of a real time loop-mediated isothermal amplification method for detection of Senecavirus A. J. Virol. Methods 261, 98–103. doi: 10.1016/j.jviromet.2018.08.005
Zhang, H., Chen, P., Hao, G., Liu, W., Chen, H., Qian, P., et al. (2020). Comparison of the pathogenicity of two different branches of senecavirus a strain in China. Pathogens 9, 39. doi: 10.3390/pathogens9010039
Zhang, Z., Zhang, Y., Lin, X., Chen, Z., Wu, S. (2019). Development of a novel reverse transcription droplet digital PCR assay for the sensitive detection of Senecavirus A. Transbound Emerg. Dis. 66, 517–525. doi: 10.1111/tbed.2019.66.issue-1
Zhao, Y., Yang, M., Zhou, C., Guo, B., Wang, K., Song, C., et al. (2024a). Establishment of a simple, sensitive, and specific ASFV detection method based on Pyrococcus furiosus argonaute. Biosens. Bioelectron. 254, 116230. doi: 10.1016/j.bios.2024.116230
Zhao, Y., Zhou, C., Guo, B., Yang, X., Wang, H. (2024b). Pyrococcus furiosus Argonaute-mediated porcine epidemic diarrhea virus detection. Appl. Microbiol. Biotechnol. 108, 137. doi: 10.1007/s00253-023-12919-0
Keywords: Senecavirus A, recombinase-aided amplification, lateral flow dipstick, sensitivity, specificity, visual detection
Citation: Song Y, Fang Y, Zhu S, Wang W, Wang L, Chen W, He Y, Yi L, Ding H, Zhao M, Fan S, Li Z and Chen J (2024) A rapid and visual detection assay for Senecavirus A based on recombinase-aided amplification and lateral flow dipstick. Front. Cell. Infect. Microbiol. 14:1474676. doi: 10.3389/fcimb.2024.1474676
Received: 02 August 2024; Accepted: 01 October 2024;
Published: 23 October 2024.
Edited by:
Ke Liu, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Zhanbo Zhu, Heilongjiang Bayi Agricultural University, ChinaCopyright © 2024 Song, Fang, Zhu, Wang, Wang, Chen, He, Yi, Ding, Zhao, Fan, Li and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhaoyao Li, bGl6aGFveWFvMTIzQHdlbnMuY29tLmNu; Jinding Chen, amRjaGVuQHNjYXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.