
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol., 04 November 2024
Sec. Virus and Host
Volume 14 - 2024 | https://doi.org/10.3389/fcimb.2024.1449423
 Chunmei Nong1,2,3
Chunmei Nong1,2,3 Zhenzhen Wu2
Zhenzhen Wu2 Chan Yang4
Chan Yang4 Wei Xu4Linyi Luo5
Wei Xu4Linyi Luo5 Jianping Zhou6
Jianping Zhou6 Lihan Shen5
Lihan Shen5 Yinghua Chen7,8*Yaoqin Yuan5*Guodong Hu1,2,3*
Yinghua Chen7,8*Yaoqin Yuan5*Guodong Hu1,2,3*Introduction: SARS-CoV-2 infection drove senescent cells and the senescence-associated phenotypes were reported playing roles in disease progression, which contributes to severe COVID-19 and related sequelae. Cdc42 is involved in the regulation of cellular senescence. This study, aimed to investigate the mechanism of the SARS-CoV-2 spike protein regulating cellular senescence through Cdc42.
Methods: K18-hACE2 mice were infected with SARS-CoV-2 Omicron BA.4 or stimulated with spike protein through the airway, the senescent cells and Cdc42 expression in lung tissue were detected. Overexpression of spike protein or exogenous incubation of spike protein was used to simulate the induction of cellular senescence by spike protein. Mechanistic insights into the role of Cdc42 were mainly explored using Western Blot and qRT-PCR.
Results: Spike protein, SARS-CoV-2 infection related, accelerates cell aging by upregulating Cdc42 expression, which furtherly activated the Wnt/β-catenin signaling pathway. Conversely, treatment with ML141 in animal models, a Cdc42 inhibitor, reduced cellular senescence and ameliorated lung injury and inflammation. These results suggest that the upregulation of Cdc42 by the SARS-CoV-2 spike protein induces cellular senescence and enhances β-catenin nuclear translocation.
Discussion: This study provides insights into the mechanisms underlying cellular senescence induced by the SARS-CoV-2 spike protein, offering potential strategies to mitigate the inflammatory response and complications associated with COVID-19 in both the acute and long-term phases.
Since its emergence as a global pandemic in December 2019, SARS-CoV-2 has infected over 700 million people worldwide, leading to more than 7 million deaths as of February 2024 (World Health Organization, 2024). Notably, a significant proportion of patients continue to experience persistent symptoms or related sequelae even after the virus has cleared, including systemic fatigue, chest pain, respiratory dysfunction, nervous system, and cognitive impairments (Ballering et al., 2022; Ceban et al., 2022; Davis et al., 2023; Mandal et al., 2021; Mo et al., 2020). Consequently, a deeper understanding of the biological responses to SARS-CoV-2 infection, particularly the persistent manifestations, is urgently required.
Aging is recognized as a significant risk factor for severe COVID-19, with older individuals being more susceptible to severe disease manifestations. This heightened susceptibility is primarily attributed to age-related impairments in pulmonary function and immune responses within the aging lung (Bartleson et al., 2021; Schneider et al., 2021). Cellular senescence, initially observed in normal diploid cells, ceasing proliferation after a limited number of replications and divisions (Hayflick and Moorhead, 1961), is considered a response to various stressors, including exposure to toxic substances, hypoxia, mitochondrial dysfunction, activation of oncogenes, and viral infections (Gorgoulis et al., 2019). Cellular senescence represents an irreversible state of cell-cycle arrest accompanied by alterations in transcriptional, epigenetic, morphological, secretory properties, and metabolic capacity (Wiley and Campisi, 2021). While senescence serves as a beneficial mechanism for tumor suppression and wound healing (Campisi, 2001; Demaria et al., 2014; Munoz-Espin and Serrano, 2014), the aberrant accumulation of senescent cells can contribute to an inflammatory microenvironment, chronic tissue damage, and the onset and progression of chronic diseases such as chronic obstructive pulmonary disease and pulmonary fibrosis (Araya et al., 2019; Barnes et al., 2019; Guan et al., 2022). A principal hazard associated with these cells is their ability to secrete a range of bioactive substances, including proinflammatory factors, chemokines, growth factors, and matrix metalloproteinases, collectively referred to as senescence-associated secretory phenotypes (SASP) (Gorgoulis et al., 2019; Rodier and Campisi, 2011). The release of these factors contributes to the development of acute or chronic inflammation and potentially regulates immune responses. Furthermore, SASP can induce secondary senescence in neighboring healthy and proliferative cells, through paracrine signaling while activating immune surveillance (Acosta et al., 2013; Coppe et al., 2008).
SARS-CoV-2 induces cellular senescence through various mechanisms, a phenomenon known as virus-induced senescence (VIS) (Meyer et al., 2021; Tripathi et al., 2021). Bulk and single-cell transcriptomic analyses further support the concept that SARS-CoV-2 triggered senescence drives pathology in COVID-19 and the lungs of patients with severe COVID-19 showed higher levels of p16INK4a positive cells compared with that in individuals with other lung diseases (Schmitt et al., 2022). Senescence is a universal host cell response to SARS-CoV-2 stress. VIS cells aggravate the inflammatory response by secreting a plethora of SASP factors, many of which are bonafide NF-κB targets. Besides, only VIS cells show activation of the cGAS–STING pathway and are involved in driving SASP-mediated interferon responses, which indicates that virus-induced senescence is a driver and therapeutic target in COVID-19 (Lee et al., 2021). Given that older individuals already have a high prevalence of senescent cells, exposure to SARS-CoV-2 may increase this burden, potentially leading to an abnormal release of SASP and resulting in a cascade of inflammatory factors that culminate in an inflammatory storm. This inflammatory storm may be a primary reason why older individuals are more susceptible to developing severe illness from COVID-19.
SARS-CoV-2 S1 protein exacerbates the SASP of human senescent cells, leading to an active inflammatory response in severe patients. Furthermore, anti-aging drugs targeting senescent cells have been found to reduce mortality rates in infected mice (Weiss and Murdoch, 2020). Moreover, SARS-CoV-2 triggers senescence in infected cells, upregulating the expression of ACE2 receptors, thereby increasing the likelihood of infection and establishing a vicious cycle (Camell et al., 2021; Duarte et al., 2021). Even after viral clearance, the persistence of senescent cells in lung tissue, evidenced by the increased number of cells expressing p16 and p21, may contribute to post-acute COVID-19 syndrome (Lipskaia et al., 2022). These findings suggest that senescent cells play a role in the pathogenesis of COVID-19, highlighting the importance of reducing cellular senescence for the treatment of COVID-19 and the prevention of its long-term effects.
Cell division cycle protein 42 (Cdc42) is a small GTPase belonging to the Rho family, which plays a pivotal role in various fundamental cellular processes, including the reorganization of the actin cytoskeleton, cell polarity, and growth (Cerione, 2004; Fu et al., 2022; Mosaddeghzadeh and Ahmadian, 2021). Aberrant activation of Cdc42 has been implicated in several pathological conditions, including carcinogenesis and neurodegenerative diseases (Sinha and Yang, 2008). In addition to its involvement in these pathological states, recent research has highlighted the critical role of Cdc42 in cellular senescence. Inhibiting Cdc42 activity can restore the regenerative potential of senescent intestinal stem cells. Furthermore, Cdc42 is implicated in the senescence process of hematopoietic stem cells and mesenchymal stem cells (Florian et al., 2013; Nalapareddy et al., 2021; Umbayev et al., 2018). In studies involving aged mice, a specific inhibitor of Cdc42 called Casin has been utilized to reduce chronic inflammation levels and extend the average lifespan(Florian et al., 2020).
The removal of senescent cells, either through genetic or pharmacological means, can reportedly delay the onset of age-related inflammatory diseases in aged mice (Baker et al., 2016; Di Micco et al., 2021). Building upon the observed relationship between Cdc42 and cellular senescence, our study aimed to explore the potential of targeting Cdc42 as a therapeutic and preventive approach for COVID-19 and its associated complications.
All animal experiments and protocols conducted in this study were approved by the National Institutional Animal Care and Medical Ethics Committee of Southern Medical University. Male-specific pathogen-free (SPF) K18-hACE2 mice, aged 8−10 weeks, were obtained from Gempharmatech company (Jiangsu, China). Prior to the experiment, all mice were acclimated in cages for five days. The animal model was generated using two approaches: infection with SARS-CoV-2 Omicron BA.4 and stimulation with SARS-CoV-2 spike protein via the trachea.
The mice were allocated randomly into control and BA.4 groups (n=6 each). Mice in the control group were intranasally inoculated with 50 μL of PBS. Conversely, mice in the BA.4 group were intranasally inoculated with 105 plaque-forming units (PFU) of SARS-CoV-2 Omicron BA.4, prediluted in 50 μL of PBS. On the fourth day following inoculation, all mice were euthanized, and organ tissues were collected for histopathological analyses. The experimental procedures were conducted in accordance with approved guidelines under Biosafety Level 3 (BSL-3) conditions.
The mice were segregated randomly into control, SARS-CoV-2 ancestral spike, and SARS-CoV-2 omicron spike groups (n=6 each). K18-hACE2 mice were stimulated via the trachea with 5 μg spike protein, prediluted in 40 μL of PBS. The control group received an equivalent volume of PBS similarly. For mice undergoing ML141 treatment, ML141 (8 mg/kg) was administered intraperitoneally 1 h prior to SARS-CoV-2 spike protein stimulation. This regimen was administered once daily for five consecutive days during the same time period. On the sixth day, the mice were euthanized for further analysis.
Lung tissues from experimental mice were fixed in 4% paraformaldehyde for 48 h and embedded in paraffin. Sections (5μm thick) were stained with H&E and examined under a light microscope (Nikon, Japan) to assess histopathological alterations.
Hydrated sections were separately incubated in citrate buffer (pH 6.0), followed by exposure to 3% H2O2. A blocking solution with 10% goat serum was applied at room temperature for 1 h. The sections were then incubated overnight at 4°C with primary antibodies, including anti-p16 (1:200, Affinity Biosciences, OH, USA), anti-p21 (1:200, ABMART, China), and anti-Cdc42 (1:250, Abcam, UK). Subsequently, the sections were incubated with HRP-labeled goat anti-rabbit IgG or anti-mouse IgG secondary antibodies (both from ZSGB-BIO, Beijing, China) for 1 h at 37°C, and immunoreactivity was detected using DAB staining (Solarbio, Beijing, China). Quantitative analysis of related indicators was performed using ImageJ 1.8.0.
Total RNA was extracted using the FastPure Cell/Tissue Total RNA Isolation Kit V2 (RC112-01, Vazyme, Nanjing, China), and reverse transcription was performed using the PrimeScript™ RT Master Mix (RR036A, Takara Biomedical Technology, Beijing, China) according to the manufacturer’s protocol. For quantitative real-time polymerase chain reaction (qRT-PCR), SYBR Green Real-time PCR Master Mix (Q711-02, Vazyme, Nanjing, China) was used. The sequences used are shown in Table 1.
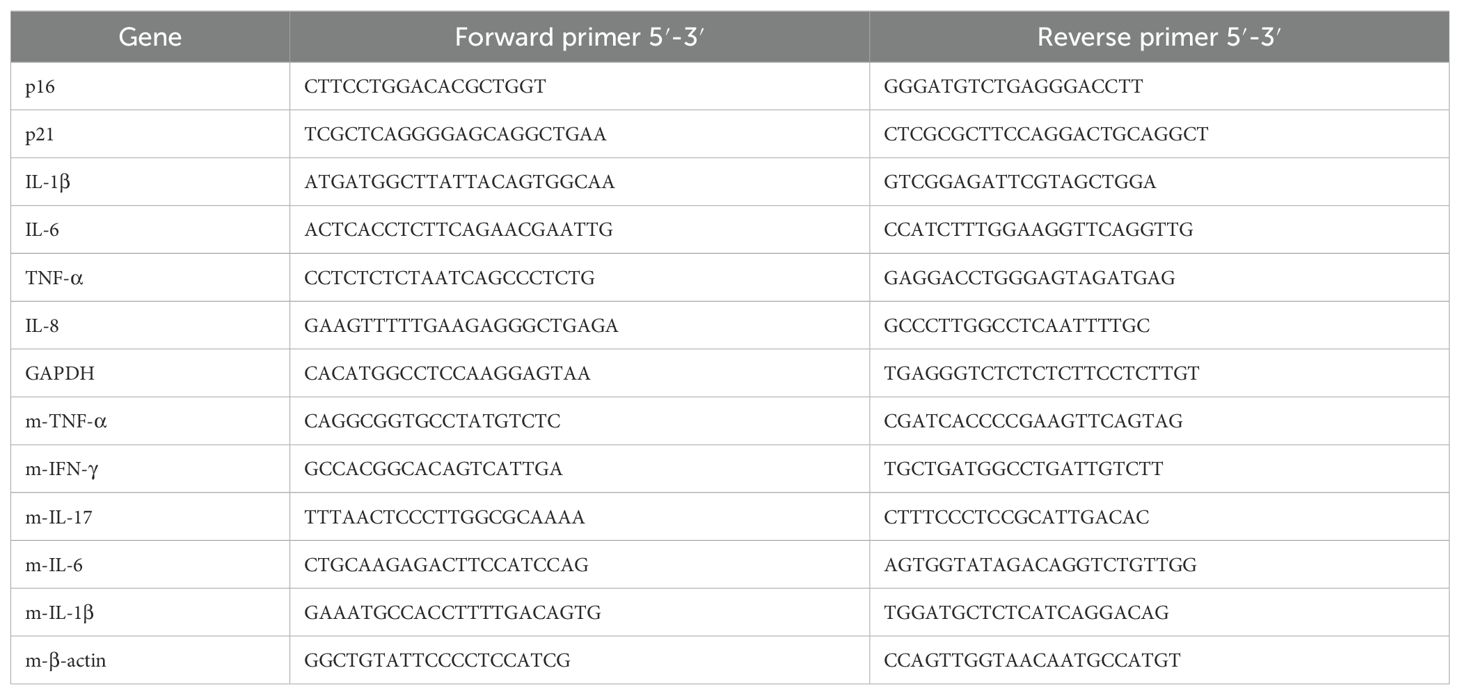
Table 1. Primer sequences for qRT-PCR.
According to the provided proportions, RIPA Lysis Buffer (ES-8148-100ml, ECOTOP SCIENTIFIC, Guangzhou, China), Protease Inhibitor Cocktail (20124ES03, Yeasen, Shanghai, China), and Phosphatase Inhibitor Cocktail (20109ES05, Yeasen, Shanghai, China) were prepared to extract the protein. Approximately 20 μg of proteins were separated by SDS-PAGE and then transferred to polyvinyl difluoride (PVDF) membranes (ISEQ00010, Merck Millipore, Darmstadt, Germany). Primary antibodies, including anti-p16 (AF0228-50μL, Affinity Biosciences, OH, USA), anti-p21 (TD6423S, Abmart, China), anti-Cdc42 (10155-1-AP, Proteintech, USA), anti-β-catenin (51067-2-AP, Proteintech, USA), and anti-GAPDH (FD0063, Hangzhou Fude Biological Technology, Hangzhou, China) were used. The next day, Goat Anti-Rabbit HRP IgG (FDR007, Hangzhou Fude Biological Technology, Hangzhou, China) and Goat Anti-Mouse HRP IgG (Hangzhou Fude Biological Technology, Hangzhou, China) were used for room-temperature incubation.
HEK-293T cells stably expressing human ACE2 (ACE2/293T) were previously described (Yang et al., 2021). Similarly, we generated A549 cells that stably express human ACE2 receptors using the same method. The cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) or RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. The cell cultures were maintained in a humidified atmosphere containing 5% CO2 at 37°C. The plasmid encoding the SARS-CoV-2 S protein (pcDNA3.1-SARS-CoV-2 S) was described in a previous study (Yang et al., 2021). Additionally, our laboratory constructed the pcDNA3.1-Spike-omicron plasmid and maintained an empty vector, pcDNA3.1. To perform transfections, the cells were cultured overnight in 6-well plates and transfected with the desired plasmid (pcDNA-SARS-CoV-2 S, pcDNA3.1-Spike-omicron) or the empty vector (2.5μg/well). Liofectamine 3000 (L3000001, ThermoFisher, USA) was used for transfection following the manufacturer’s instructions. The corresponding assays or experiments were conducted 72 hours after transfection.
The senescence-associated β-galactosidase (SA-β-Gal) activity in ACE2/A549 cells overexpressing spike proteins was assessed using a cellular senescence staining kit (C0602, Beyotime Biotechnology, China) following the manufacturer’s instructions. The staining procedure allows the visualization of blue-colored cells indicative of senescent cells.
Data are presented as mean ± SD. Statistical comparisons were performed using one-way analysis of variance (ANOVA) and unpaired Student’s t-tests, utilizing GraphPad Prism 9. Significance thresholds were set at *p<0.05, **p<0.01, and ***p<0.001.
Cellular senescence has been implicated in the poor clinical outcomes of patients with COVID-19 (Paschalaki et al., 2021). In our study, we induced lung injury in mice using SARS-CoV-2 Omicron BA.4 (Figure 1A) and identified senescent cells through immunohistochemical staining. Our results demonstrated an increase in the expression of senescence-related markers, p16, and p21, in the lungs of K18-hACE2 mice four days after infection with SARS-CoV-2 Omicron BA.4 (Figures 1B, C), indicating a potential role of novel coronaviruses in triggering cellular senescence.
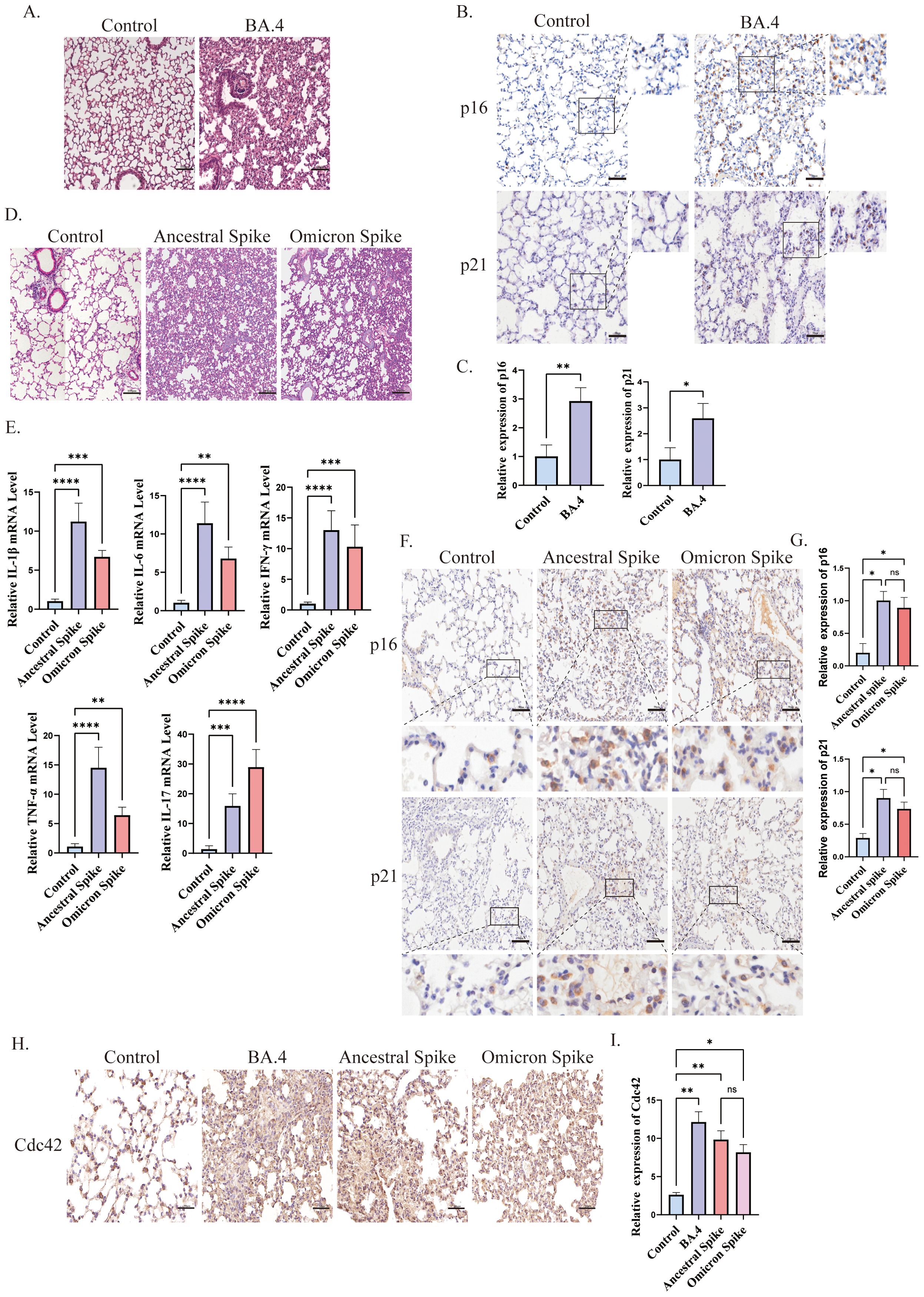
Figure 1. Spike protein accelerates the senescence of alveolar cells. (A) H&E staining of lung injury in mice induced by SARS-CoV-2 Omicron BA.4, bar = 100μm; (B) IHC staining of p16 and p21 and (C) quantitative analysis of p16 and p21 in lung tissue after SARS-CoV-2 Omicron BA.4 infection, bar = 50μm; (D) H&E staining of lung injury in mice induced by spike protein, bar = 100μm; (E) mRNA expression of IL-1β, IL-6, TNF-α, INF-γ, and IL-17 in lung tissues of K18-hACE2 mice stimulated by spike protein; (F) IHC staining and (G) quantitative analysis of p16 and p21 in lung tissue of mice stimulated by spike protein, bar = 50μm; (H) IHC staining and (I) quantitative analysis of Cdc42 in lung tissue of mice under different treatments, bar = 50μm. Data represent the mean ± SD (n=6). *p<0.05, **p<0.01, ***p<0.001.
To further investigate the role of the SARS-CoV-2 spike protein in promoting cellular senescence, we administered the spike protein to K18-hACE2 mice via the trachea. Subsequently, we observed lung injury and evaluated the senescence of alveolar epithelial cells. Given the emergence of several SARS-CoV-2 variants with heightened transmissibility and immune evasion, such as the Omicron variants, these have become dominant globally and have led to increased morbidity (He et al., 2023). Therefore, in this study, we focused on the potential mechanisms of cellular senescence induced by both ancestral and omicron spike variants. Post-treatment with the spike protein, the lungs of mice displayed signs of damage and elevated inflammation levels. Consistent with other reports (Sievers et al., 2022; Wolter et al., 2022), histological examination of ancestral spike-stimulated mice lungs revealed prominent pathological changes in the alveoli, including alveolar wall collapse and infiltration of inflammatory cells. Conversely, Omicron spike-stimulated mice exhibited less severe pathological changes and inflammation (Figures 1D, E). Additionally, an increased senescence phenotype was observed compared with that in the control group (Figures 1F, G).
The relationship between Cdc42 and aging is well-established, thus, we also examined its expression. We observed upregulation of Cdc42 expression post-infection with SARS-CoV-2 Omicron BA.4 or following spike protein stimulation (Figures 1H, I), indicating that the spike protein induces senescence in alveolar epithelial cells in mice, and Cdc42 may be involved in this process.
To investigate the role of Cdc42 in initiating cellular senescence triggered by the SARS-CoV-2 spike protein, we employed the Cdc42 inhibitor ML141 and assessed senescent cells status in mouse lung tissues post-spike protein stimulation. The experimental flow chart is depicted in Figure 2A.
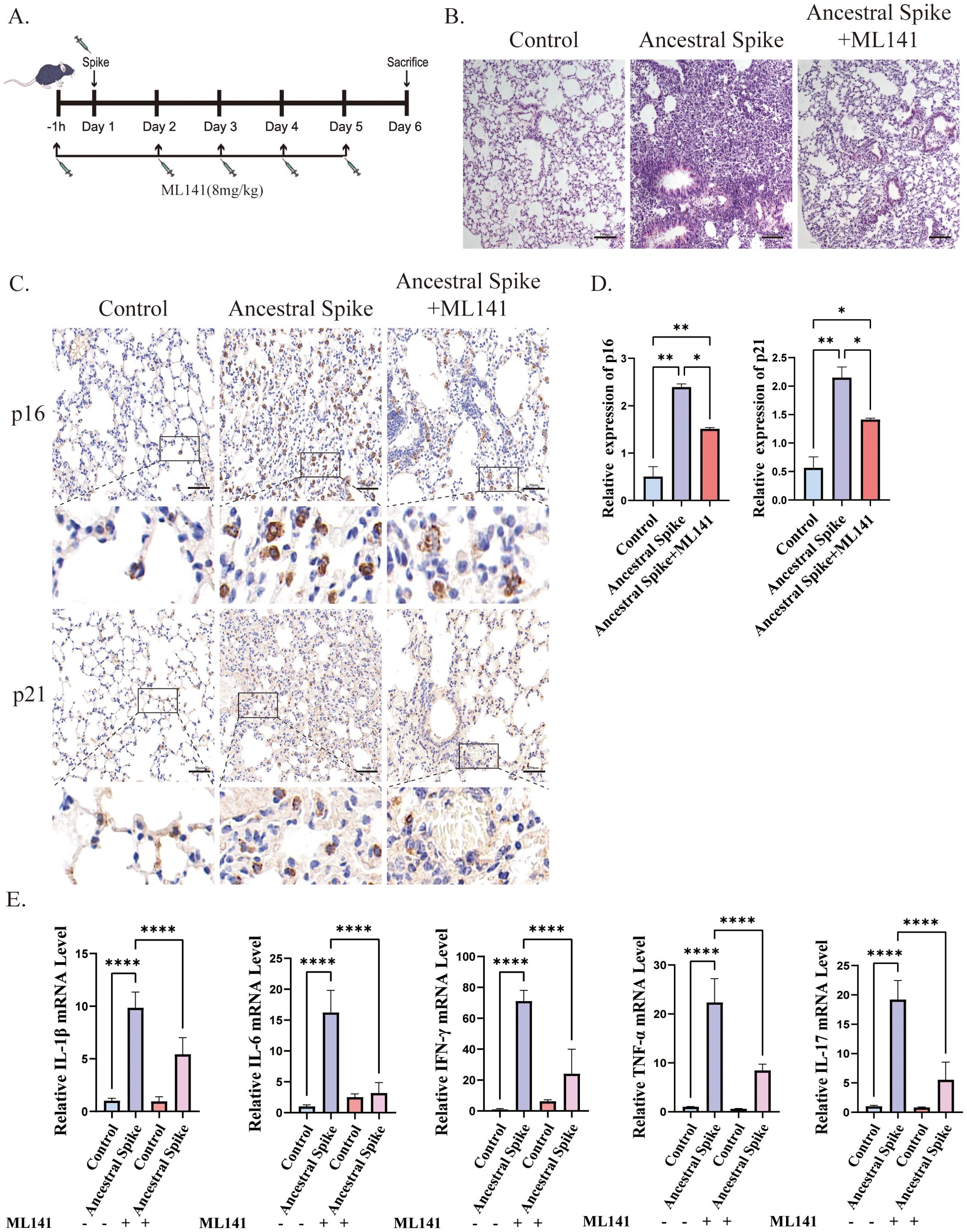
Figure 2. ML141 attenuates spike protein-induced senescence-related phenotypes in mice lung tissues. (A) Diagram showing the experimental design for animal treatment; (B) H&E staining of mice lung injury induced by spike protein was alleviated by ML141, bar = 100 μm; (C) IHC staining of p16 and p21 in mouse lung tissue and (D) quantitative analysis, bar = 50μm; (E) mRNA expression of inflammatory factors in mouse lung. Data represent the mean ± SD (n=6). *p<0.05, **p<0.01 and ***p<0.001.
Immunohistochemical analysis revealed that ML141 prevented the spike protein-induced increase in the number of senescent cells in the lungs, primarily manifested by decreased expression of p16 and p21, however, the number of senescent cells remained higher than that in the control group after ML141 treatment (Figures 2C, D). Additionally, ML141 administration resulted in a reduction in the levels of inflammatory factors and contributed to the alleviation of lung injury in mice, but conditions did not return to normal compared with that in the control group (Figures 2B, E).
In our in vivo experiments, we observed that the spike protein promotes lung aging in mice. To further explore the role of the spike protein in aging, we incubated spike protein exogenously and transfected ACE2/A549 cells with different plasmids: pcDNA-SARS-CoV-2 S (Ancestral), pcDNA3.1-Spike-Omicron, and pcDNA3.1 empty vector as a control. Subsequently 72h post-transfection, we confirmed the expression of cellular spike protein (Figure 3A).
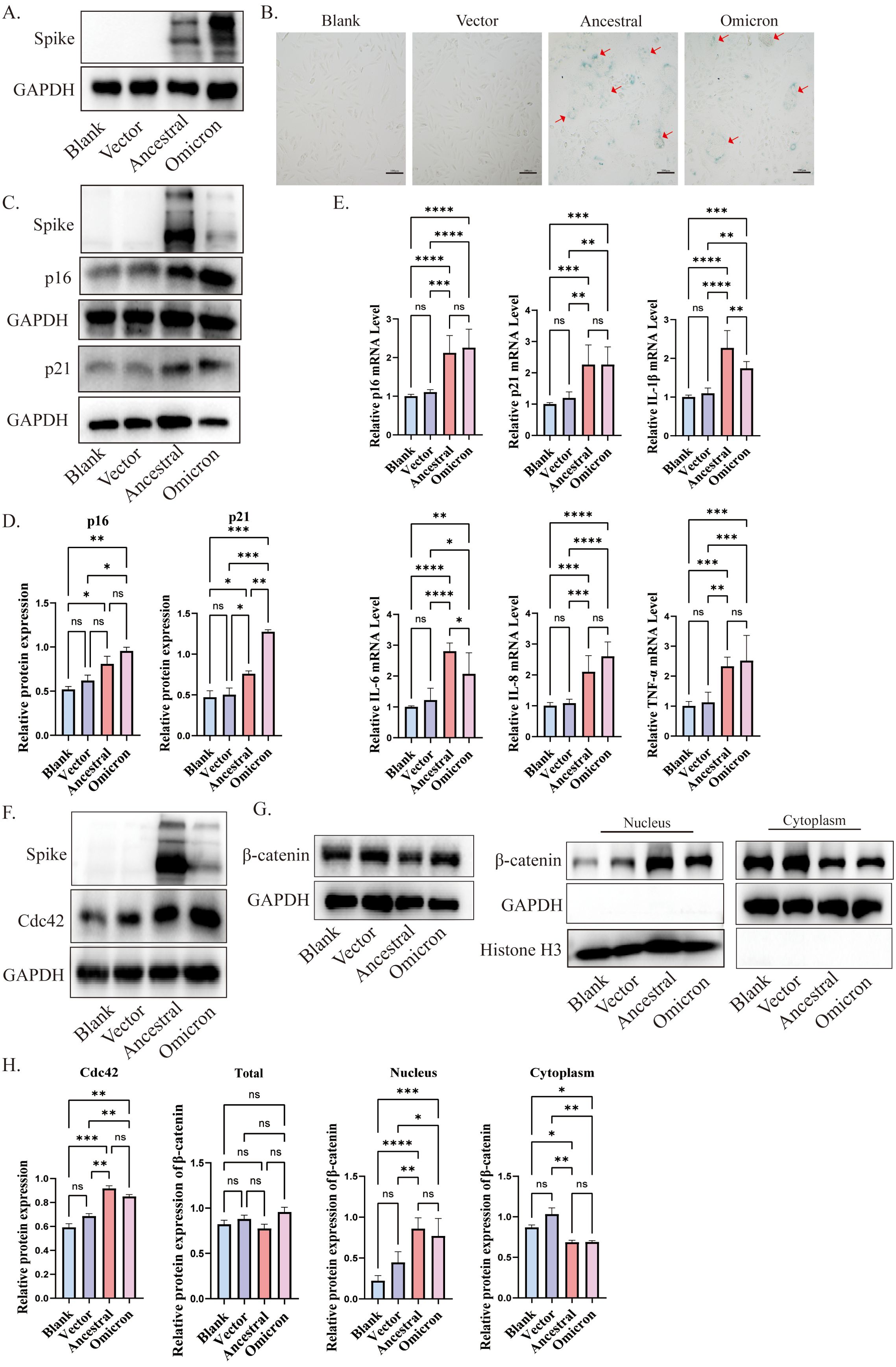
Figure 3. Cdc42 expression increases in senescent cells induced by spike protein. (A) Expression of spike protein in ACE2/A549 cells transfected with plasmids for 72 h; (B) SA-β-Gal staining of ACE2/A549 cells in different treatment groups and the red arrow indicates spike protein-mediated membrane fusion, bar =100μm; (C) Western blot analysis of senescence indicators p16, p21 in ACE2/A549 cells under different treatment factors and (D) quantitative analysis; (E) mRNA levels of p16, p21, and SASP in ACE2/A549 cells in different groups; (F) Western blot analysis of Cdc42 expression and level of β-catenin in the whole cells under different treatments; (G) Western blot analysis of β-catenin in cytoplasm and nucleus and (H) quantitative analysis. Data represent the mean ± SD (n=3). *p<0.05, ** p<0.01, ***p<0.001.
SA-β-Gal, a lysosomal hydrolase and classic marker of cellular senescence (Esposito et al., 2024). Following transfection with SARS-CoV-2 ancestral-spike and omicron-spike plasmids, we observed an increase in the number of SA-β-Gal-positive cells, along with the formation of giant fusion cells induced by the spike protein (Figure 3B). Consistent with the experimental results in vivo, both p16 and p21 were elevated under the stimulation of spike protein, and the expression of p16 and p21 induced by omicron spike seems to be higher than that induced by ancestral (Figures 3C, D), and the expression of some SASP factors was increased compared to the control group (Figure 3E). Cdc42 expression, consistent with the in vivo results, increased after spike transfection (Figures 3F, H). Moreover, treatment of ACE2/A549 cells with purified spike protein for 96 hours enhanced protein levels of p16, p21, and Cdc42, indicating that spike protein incubation could accelerate cellular senescence, potentially mediated by Cdc42 (Supplementary Figure 1B).
β-Catenin, a downstream effector of WNT signals and closely related to cellular senescence (Zhou et al., 2015). Previous research has shown that WNT/β-catenin activity is increased in the type II alveolar epithelial cells of aged mice compared to young mice, and prolonged activation of WNT/β-catenin signaling accelerates cellular senescence (Lehmann et al., 2020). β-catenin is a substrate of GSK3β, in the resting state, GSK3β and CKI could phosphorylate β-catenin, triggering its destabilization and degradation to maintain the balance of β-catenin in the cytosol/nucleus. We observed increased phosphorylation of GSK3β, suggesting decreased GSK3β activity (Supplementary Figures 1A, C). Compared with control cells, following spike protein stimulation or overexpression, β-catenin translocated to the nuclear compartment, indicating activation of the WNT/β-catenin pathway (Figures 3G, H, Supplementary Figure 1C). Collectively, our findings suggest that spike protein induces cellular senescence, potentially mediated by regulation of Cdc42 expression and β-catenin translocation to the nucleus.
To investigate the contribution of Cdc42 upregulation to spike-induced senescence, we pretreated ACE2/A549 cells, which overexpressed or incubated with spike protein, with the Cdc42 inhibitor ML141. SA-β-Gal staining assessed cellular senescence, revealing that ML141 treatment effectively blocked the spike-mediated increase in the number of senescent cells (Figure 4A). Furthermore, the upregulation of p16, p21, and SASP, induced by spike protein, was abolished by ML141 treatment, and for the other inflammatory factors, there were no significant differences between ancestral and omicron, but it seems to be a higher TNF-α level of omicron induced than that in ancestral, while there was no significant change in Cdc42 (Figures 4B, C, F). Besides, similar results were obtained in the ACE2/A549 cells groups with exogenous spike protein incubation (Supplementary Figure 2B). Compared with the untreated ML141 groups, ML141 reduced the phosphorylation of GSK3β and restored its activity, correspondingly, ML141 significantly inhibited the spike-induced localization of β-catenin to the nucleus (Figures 4D, E, Supplementary Figure 2A, C). In order to further clarify the role of Cdc42 in regulating cellular senescence, we knockdown Cdc42 by siRNA. Consistent with the effect of using ML141, Cdc42 knockdown resulted in down-regulation of both p16 and p21 expression (Supplementary Figure 3A). Meanwhile, the transfecting cells with Cdc42 siRNA abolished GSK3β phosphorylation and reversed the spike protein incubation-induced increase in β-catenin nuclear translocation (Supplementary Figures 3B, C).
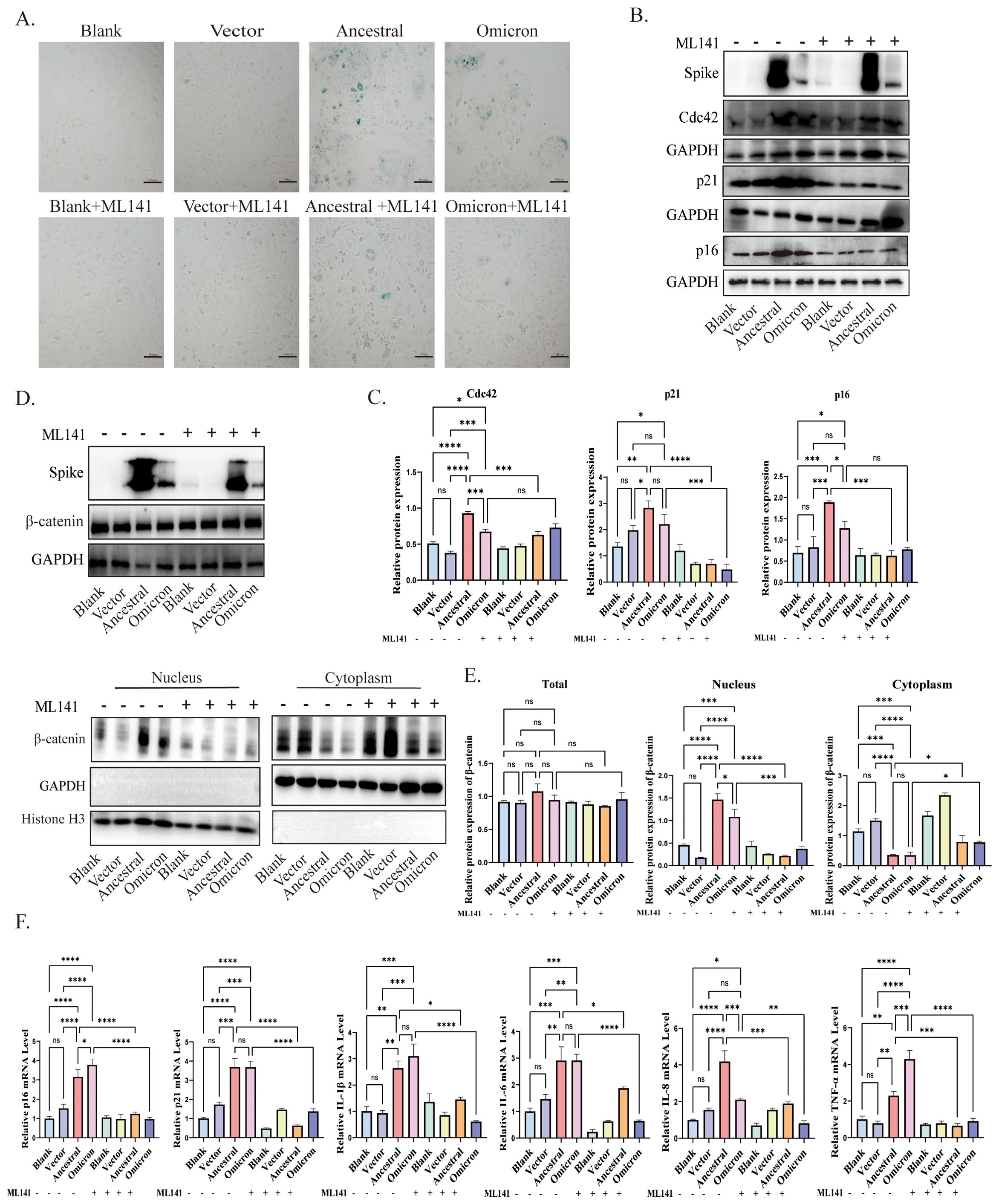
Figure 4. Cdc42 inhibition attenuates spike protein-induced senescence phenotype. (A) SA-β-Gal staining of ACE2/A549 cells in different treatment groups, bar = 100μm; (B) Western blot detection of Cdc42, p16, and p21 expression in ACE2/A549 cells under different treatments and (C) quantitative analysis; (D) Western blot detection of the whole cell, cytoplasmic and nuclear expression levels of β-catenin and (E) quantitative analysis; (F) mRNA levels of p16, p21, and SASP in different groups. Data represent the mean ± SD (n=3). *p<0.05, ** p<0.01, ***p<0.001.
These findings provide evidence that Cdc42 contributes to cellular senescence triggered by the spike protein. The inhibitory effects of Cdc42 are mainly manifested as the reduction of age-related phenotypes and the inhibition of β-catenin nuclear translocation, highlighting the therapeutic potential of targeting Cdc42 in mitigating spike protein-induced senescence.
Our results confirm the central role of Cdc42 in spike protein-mediated cellular senescence. To investigate whether this effect is mediated through the activation of the WNT/β-catenin signaling pathway, we treated ACE2/A549 cells with the WNT/β-catenin pathway inhibitor, KYA1797K, followed by transfection with plasmids.
The application of KYA1797K significantly influenced cellular senescence, as evidenced by the decreased number of SA-β-Gal-positive cells compared to groups expressing spike protein alone (Figure 5A). Furthermore, upon treatment with KYA1797K, western blot analysis revealed a downregulation of p16 and p21 (Figures 5B, C). We also examined the WNT/β-catenin pathway, spike protein overexpression resulted in reduced β-catenin translocation into the nucleus (Figures 5D, E). Additionally, the mRNA levels of SASP factors decreased in response to KYA1797K (Figure 5F). These findings highlight the impact of the interaction between Cdc42 and the WNT/β-catenin pathway in spike protein-induced cellular senescence.
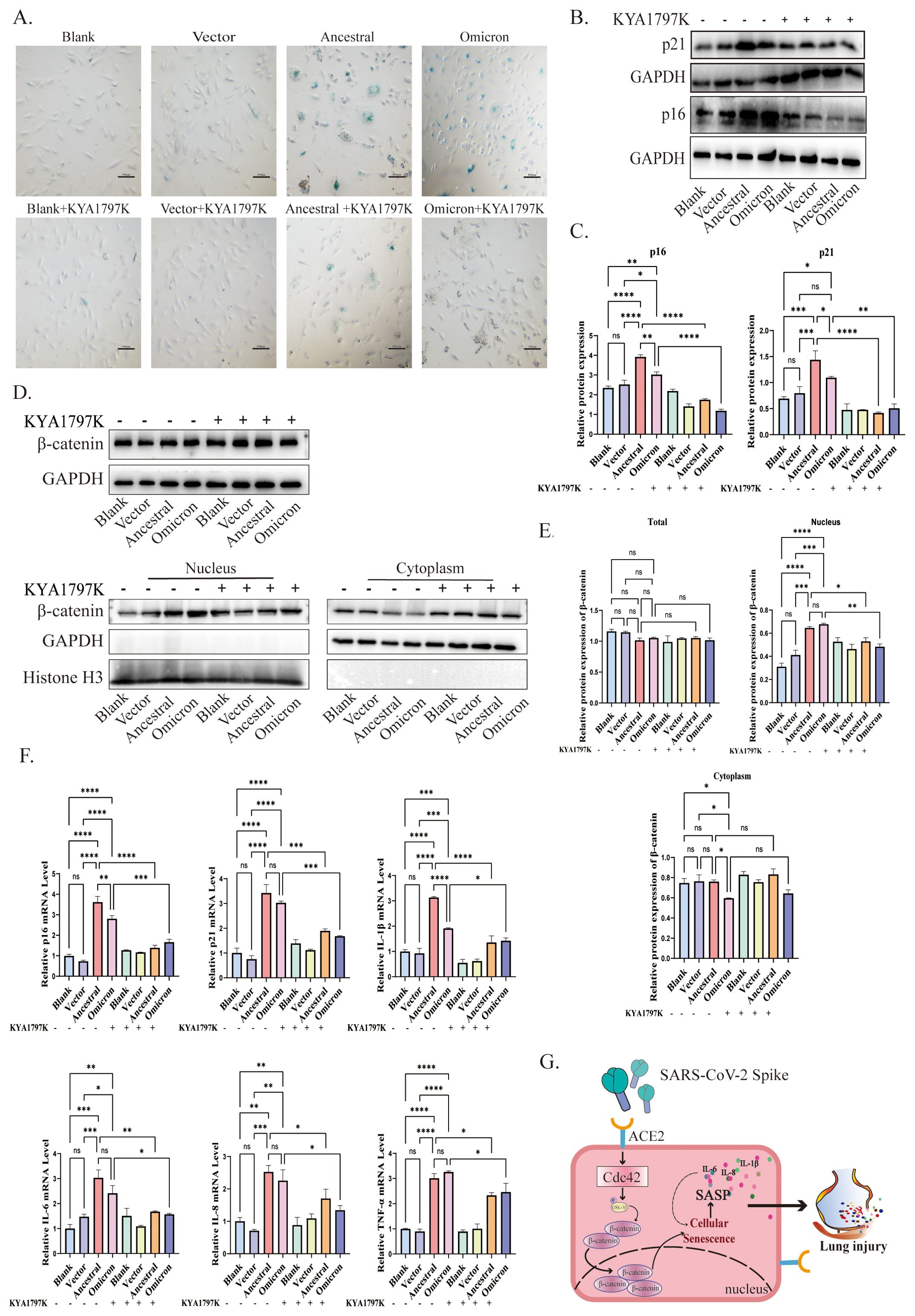
Figure 5. Cdc42 drives senescence by promoting β-catenin translocation to the nucleus. (A) SA-β-Gal staining of ACE2/A549 cells in different groups, bar = 100μm; (B) Protein levels of p16, p21, and (C) quantitative analysis in different treatments; (D) Protein levels of β-catenin and (E) quantitative analysis in different treatment groups; (F) mRNA levels of SASP in different groups; (G) Schematic of Cdc42 involved in the spike-induced cellular senescence by promoting β-catenin translocation to the nucleus. Data represent the mean ± SD (n=3). *p<0.05, ** p<0.01, ***p<0.001.
Aging is a major risk factor for severe COVID-19, with older individuals being more susceptible to worsened symptoms and outcomes (D’Agnillo et al., 2021; Santesmasses et al., 2020). This association may be attributed to the accumulation of senescent cells during aging. Alongside naturally occurring senescent cells, SARS-CoV-2 infection can induce cellular senescence, further exacerbating the accumulation of these cells. When the burden of senescent cells surpasses the capacity of the immune system to eliminate them, they release SASPs, provoking an inflammatory response. This chronic inflammation hampers tissue proliferation and repair, exacerbating disease progression. Several studies have highlighted the potential role of senescent cells in intensifying the immune response against SARS-CoV-2 (Blagosklonny, 2020; Kirkland and Tchkonia, 2020; Malavolta et al., 2020; Nehme et al., 2020). Thus, comprehending the connection between cellular senescence and COVID-19 pathogenesis is crucial for developing therapeutic and preventive strategies against its sequelae.
Senescence is a cellular response that occurs in the presence of various internal and external stress signals. In this study, we investigated the role of the spike protein in triggering cellular senescence. In mice lung tissues exposed to the spike protein, we observed an increase in the number of cells positive for the senescence markers p16 and p21, with ancestral spike-induced cell aging appearing more severe than omicron. Moreover, compared with the ancestral spike, omicron induced substantially attenuated lung pathology with downregulation of proinflammatory cytokines, consistent with the decreased pathogenicity of omicron that has been reported (Chan et al., 2022; Shuai et al., 2022; Tamura et al., 2023), and may even relate to the weaker ability of omicron to induce cellular senescence. Notably, we also noticed an upregulation in the expression of Cdc42, a protein associated with cellular processes.
Cdc42 is an evolutionarily ancient protein widely expressed and involved in fundamental cellular functions. Numerous studies have highlighted its role in the development and progression of age-related pathologies, including neurodegenerative diseases (Stankiewicz and Linseman, 2014), cardiovascular disease (Florian et al., 2017; Hirano et al., 2008), and degenerative joint diseases (Suzuki et al., 2015). For example, in Alzheimer’s disease, there is a significant upregulation of Cdc42 activity (Chen-Plotkin et al., 2009; Zhu et al., 2000). Additionally, while elevated Cdc42 activity has been associated with the senescence of intestinal stem cells, its inhibition enhances the regenerative capacity of the stem cells (Castillo-Azofeifa et al., 2023). Senolytics, which selectively eliminate senescent cells by inducing apoptosis, have shown promise in delaying, preventing, and even reversing the aging process, thereby extending lifespan (Lagoumtzi and Chondrogianni, 2021). Studies focusing on Cdc42 inhibition have demonstrated significant therapeutic and anti-aging effects in aged mice and models of aging-related diseases (Amoah et al., 2022; Florian et al., 2020). Based on these findings, we infer that Cdc42 plays a crucial role in spike protein-induced cellular senescence. For inflammation, inhibition of Cdc42 reduced cytokine secretion during TNF-α-induced inflammation, and also had an effect on cytokine gene transcription. Additionally, other reports showed that Rho proteins are needed for cytokine activation of NF-κB for proinflammatory signaling and it was reported that Cdc42 could promote the release of IL-1β by activating IQGAP1 (Bahia et al., 2020; Rowayna and Gary, 2024; Yun et al., 2022). Notably, however, Takashi K et al. suggest that inhibition of Cdc42 signaling had a much weaker influence on acute inflammation than chronic inflammation, which supports the notion that the Cdc42-dependent proinflammatory pathway is specifically activated by senescence-associated stimuli (Takashi K et al., 2014). Inhibition of CDC42, particularly using Cdc42 inhibitors as senolytic-associated drugs could potentially block spike protein-induced cellular senescence, alleviating the inflammatory response and mitigating the disease progression associated with senescent cells.
β-catenin is a crucial molecule involved in the classical WNT signaling pathway. Under normal conditions, the WNT pathway is inactive. However, abnormal activation of the pathway leads to the inhibition of β-catenin phosphorylation and ubiquitination, leading to an increase in free cytoplasmic β-catenin levels. This elevated β-catenin translocates into the nucleus, activating downstream WNT target genes (Angers and Moon, 2009; Vallee et al., 2021). Recent evidence suggests that classical WNT/β-catenin pathway activation is associated with inflammation and cytokine storms in COVID-19 patients (Choi et al., 2020; Vallee et al., 2021). Additionally, the WNT/β-catenin signaling pathway has been implicated in cellular senescence processes such as intervertebral disc degeneration, senescence of type II alveolar epithelial cells promoting pulmonary fibrosis, and renal tubular senescence (Chen et al., 2019; Gong et al., 2021; Lehmann et al., 2020; Yang et al., 2022). Furthermore, the relationship between Cdc42 and β-catenin has been established, with Cdc42 being involved in cell proliferation, migration, and differentiation through the activation of WNT/β-catenin signaling (Han et al., 2017; Wu et al., 2006). Cdc42 is known to initiate p-PKCζ/pGSK3β signaling, which is involved in renal fibrosis by inhibiting p-β-catenin and upregulating β-catenin (Hu et al., 2024). Consistently, we observed that Cdc42 regulates β-catenin, with Cdc42 inhibition increasing its degradation and reducing its entry into the nucleus. It has been reported that the increase of β-catenin nuclear translocation can induce cellular senescence by activating the p53/p21 signaling pathway, and in our study, spike protein was found to promote the up-regulation of p21 expression (Gu et al., 2014). Furthermore, p16 was found to colocalize with β-catenin to regulate the cellular aging process, indicating that these signals are closely related (Meng et al., 2022).
In our study, we observed that the spike protein induces increased translocation of β-catenin into the nucleus, activating the WNT/β-catenin pathway. However, Cdc42 knockdown and the use of ML141, a Cdc42 inhibitor, and KYA1797K, a WNT/β-catenin pathway inhibitor, can block the translocation of cytoplasmic β-catenin into the nucleus, thus alleviating the senescence-associated phenotype induced by SARS-CoV-2 infection.
In conclusion, our study findings confirm that the spike protein of SARS-CoV-2 promotes β-catenin translocation into the nucleus by upregulating Cdc42. This activates the WNT/β-catenin pathway, inducing cellular senescence (Figure 5G). By inhibiting Cdc42 activity, we observed decreased spike-induced cellular senescence and lung injury alleviation in mice lung tissues. These findings suggest targeting Cdc42 as a therapeutic strategy may mitigate the detrimental effects of spike protein-induced cellular senescence associated with SARS-CoV-2 infection and the lack of validation of possible pathways by which Cdc42 regulates related inflammatory factors is a limitation of this study. Further study is warranted to explore the precise mechanisms underlying the interplay between Cdc42, β-catenin, spike protein-mediated cellular senescence, and inflammatory response, and evaluate the therapeutic implications of targeting Cdc42 in the context of COVID-19 and other age-related diseases.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was approved by Southern Medical University Experimental Animal Ethics Committee (SMUL202404042). The study was conducted in accordance with the local legislation and institutional requirements.
CN: Conceptualization, Data curation, Methodology, Validation, Writing – original draft, Writing – review & editing. ZW: Data curation, Formal analysis, Methodology, Validation, Writing – original draft. CY: Validation, Visualization, Writing – review & editing. WX: Formal analysis, Writing – review & editing. LL: Formal analysis, Writing – review & editing. JZ: Investigation, Writing – review & editing. LS: Visualization, Writing – review & editing. YC: Investigation, Writing – review & editing, Funding acquisition, Project administration. YY: Conceptualization, Investigation, Writing – review & editing, Project administration. GH: Conceptualization, Funding acquisition, Writing – review & editing, Supervision.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This project was supported by the following grants: the National Natural Science Funding of China (82070083), the Natural Science Funding of Guangdong Province (2024A1515012874, 2022A1515012312), and Dongguan Social Development Technology Projects Science and Technology Planning Project (20231800940822, 20231800940792).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2024.1449423/full#supplementary-material
Acosta, J. C., Banito, A., Wuestefeld, T., Georgilis, A., Janich, P., Morton, J. P., et al. (2013). A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978–U221. doi: 10.1038/ncb2784
Amoah, A., Keller, A., Emini, R., Hoenicka, M., Liebold, A., Vollmer, A., et al. (2022). Aging of human hematopoietic stem cells is linked to changes in Cdc42 activity. Haematologica 107, 393–402. doi: 10.3324/haematol.2020.269670
Angers, S., Moon, R. T. (2009). Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 10, 468–477. doi: 10.1038/nrm2717
Araya, J., Tsubouchi, K., Sato, N., Ito, S., Minagawa, S., Hara, H., et al. (2019). PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy 15, 510–526. doi: 10.1080/15548627.2018.1532259
Bahia, B., Aurore, T., Yamini, R., Rachida, T., Laurence, A., Michael, D., et al. (2020). A toxic palmitoylation of Cdc42 enhances NF-κB signaling and drives a severe autoinflammatory syndrome. J. Allergy Clin. Immunol. 146 (5), 1201-1204. doi: 10.1016/j.jaci.2020.03.020
Baker, D. J., Childs, B. G., Durik, M., Wijers, M. E., Sieben, C. J., Zhong, J., et al. (2016). Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 530, 184–18+. doi: 10.1038/nature16932
Ballering, A. V., van Zon, S. K. R., Hartman, T. C., Rosmalen, J. G. M., Lifelines Corona Res, I (2022). Persistence of somatic symptoms after COVID-19 in the Netherlands: an observational cohort study. Lancet 400, 452–461. doi: 10.1016/S0140-6736(22)01214-4
Barnes, P. J., Baker, J., Donnelly, L. E. (2019). Cellular senescence as a mechanism and target in chronic lung diseases. Am. J. Respir. Crit. Care Med. 200, 556–564. doi: 10.1164/rccm.201810-1975TR
Bartleson, J. M., Radenkovic, D., Covarrubias, A. J., Furman, D., Winer, D. A., Verdin, E. (2021). SARS-CoV-2, COVID-19 and the aging immune system. Nat. Aging 1, 769–782. doi: 10.1038/s43587-021-00114-7
Blagosklonny, M. V. (2020). From causes of aging to death from COVID-19. Aging-Us 12, 10004–10021. doi: 10.18632/aging.103493
Camell, C. D., Yousefzadeh, M. J., Zhu, Y., Prata, L. G. P. L., Huggins, M. A., Pierson, M., et al. (2021). Senolytics reduce coronavirus-related mortality in old mice. Science 373, eabe4832. doi: 10.1126/science.abe4832
Campisi, J. (2001). Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 11, S27–S31. doi: 10.1016/s0962-8924(01)82148-6
Castillo-Azofeifa, D., Wald, T., Reyes, E. A., Gallagher, A., Schanin, J., Vlachos, S., et al. (2023). A DLG1-ARHGAP31-CDC42 axis is essential for the intestinal stem cell response to fluctuating niche Wnt signaling. Cell Stem Cell 30, 188–206. doi: 10.1016/j.stem.2022.12.008
Ceban, F., Ling, S., Lui, L. M. W., Lee, Y., Gill, H., Teopiz, K. M., et al. (2022). Fatigue and cognitive impairment in Post-COVID-19 Syndrome: A systematic review and meta-analysis. Brain Behav. Immun. 101, 93–135. doi: 10.1016/j.bbi.2021.12.020
Cerione, R. A. (2004). Cdc42: new roads to travel. Trends Cell Biol. 14, 127–132. doi: 10.1016/j.tcb.2004.01.008
Chan, J., Hu, B., Chai, Y., Shuai, H., Liu, H., Shi, J., et al. (2022). Virological features and pathogenicity of SARS-CoV-2 Omicron BA.2. Cell Rep. Med. 3, 100743. doi: 10.1016/j.xcrm.2022.100743
Chen, J., Mei, Z., Huang, B., Zhang, X., Liu, J., Shan, Z., et al. (2019). IL-6/YAP1/β-catenin signaling is involved in intervertebral disc degeneration. J. Cell. Physiol. 234, 5964–5971. doi: 10.1002/jcp.27065
Chen-Plotkin, A., Geser, F., Plotkin, J., Clark, C., Kwong, L., Yuan, W., et al. (2009). Variations in the progranulin gene affect global gene expression in frontotemporal lobar degeneration. Neurology 72, A256–A256. doi: 10.1093/hmg/ddn023
Choi, E. Y., Park, H. H., Kim, H., Kim, H. N., Kim, I., Jeon, S., et al. (2020). Wnt5a and Wnt11 as acute respiratory distress syndrome biomarkers for severe acute respiratory syndrome coronavirus 2 patients. Eur. Respir. J. 56, 2001531. doi: 10.1183/13993003.01531-2020
Coppe, J.-P., Patil, C. K., Rodier, F., Sun, Y., Munoz, D. P., Goldstein, J., et al. (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PloS Biol. 6, 2853–2868. doi: 10.1371/journal.pbio.0060301
D’Agnillo, F., Walters, K.-A., Xiao, Y., Sheng, Z.-M., Scherler, K., Park, J., et al. (2021). Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci. Trans. Med. 13, eabj7790. doi: 10.1126/scitranslmed.abj7790
Davis, H. E., McCorkell, L., Vogel, J. M., Topol, E. J. (2023). Long COVID: major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 21, 133–146. doi: 10.1038/s41579-022-00846-2
Demaria, M., Ohtani, N., Youssef, S. A., Rodier, F., Toussaint, W., Mitchell, J. R., et al. (2014). An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733. doi: 10.1016/j.devcel.2014.11.012
Di Micco, R., Krizhanovsky, V., Baker, D., di Fagagna, F. (2021). Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 22, 75–95. doi: 10.1038/s41580-020-00314-w
Duarte, C., Akkaoui, J., Ho, A., Garcia, C., Yamada, C., Movila, A. (2021). Age-dependent effects of the recombinant spike protein/SARS-CoV-2 on the M-CSF- and IL-34-differentiated macrophages in vitro. Biochem. Biophys. Res. Commun. 546, 97–102. doi: 10.1016/j.bbrc.2021.01.104
Esposito, P., Picciotto, D., Verzola, D., Garibotto, G., Parodi, E. L., Sofia, A., et al. (2024). SA-β-gal in kidney tubules as a predictor of renal outcome in patients with chronic kidney disease. J. Clin. Med. 13, 322. doi: 10.3390/jcm13020322
Florian, C., Doerr, K., Scharffetter-Kochanek, K., Zheng, Y., Geiger, H. (2013). CDC42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell 10, 520–530. doi: 10.1016/j.stem.2012.04.007
Florian, M. C., Klenk, J., Marka, G., Soller, K., Kiryakos, H., Peter, R., et al. (2017). Expression and activity of the small rhoGTPase cdc42 in blood cells of older adults are associated with age and cardiovascular disease. J. Gerontol. Ser. a-Biol. Sci. Med. Sci. 72, 1196–1200. doi: 10.1093/gerona/glx091
Florian, M. C., Leins, H., Gobs, M., Han, Y., Marka, G., Soller, K., et al. (2020). Inhibition of Cdc42 activity extends lifespan and decreases circulating inflammatory cytokines in aged female C57BL/6 mice. Aging Cell 19, e13208. doi: 10.1111/acel.13208
Fu, J., Liu, B., Zhang, H., Fu, F., Yang, X., Fan, L., et al. (2022). The role of cell division control protein 42 in tumor and non-tumor diseases: A systematic review. J. Cancer 13, 800–814. doi: 10.7150/jca.65415
Gong, W., Luo, C., Peng, F., Xiao, J., Zeng, Y., Yin, B., et al. (2021). Brahma-related gene-1 promotes tubular senescence and renal fibrosis through Wnt/β-catenin/autophagy axis. Clin. Sci. 135, 1873–1895. doi: 10.1042/cs20210447
Gorgoulis, V., Adams, P. D., Alimonti, A., Bennett, D. C., Bischof, O., Bishop, C., et al. (2019). Cellular senescence: defining a path forward. Cell 179, 813–827. doi: 10.1016/j.cell.2019.10.005
Gu, Z., Tan, W., Feng, G., Meng, Y., Shen, B., Liu, H., et al. (2014). Wnt/β-catenin signaling mediates the senescence of bone marrow-mesenchymal stem cells from systemic lupus erythematosus patients through the p53/p21 pathway. Mol. Cell. Biochem. 387, 27–37. doi: 10.1007/s11010-013-1866-5
Guan, R., Yuan, L., Li, J., Wang, J., Li, Z., Cai, Z., et al. (2022). Bone morphogenetic protein 4 inhibits pulmonary fibrosis by modulating cellular senescence and mitophagy in lung fibroblasts. Eur. Respir. J. 60, 2102307. doi: 10.1183/13993003.02307-2021
Han, B., Zhao, J.-y., Wang, W.-t., Li, Z.-w., He, A.-p., Song, X.-y. (2017). Cdc42 promotes schwann cell proliferation and migration through wnt/β-catenin and p38 MAPK signaling pathway after sciatic nerve injury. Neurochem. Res. 42, 1317–1324. doi: 10.1007/s11064-017-2175-2
Hayflick, L., Moorhead, P. S. (1961). The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621. doi: 10.1016/0014-4827(61)90192-6
He, X., He, C., Hong, W., Yang, J., Wei, X. (2023). Research progress in spike mutations of SARStions0 variants and vaccine development. Med. Res. Rev. 43, 932–971. doi: 10.1002/med.21941
Hirano, K.-i., Ikegami, C., Zhang, Z. (2008). Contribution of Cdc42 to cholesterol efflux in fibroblasts from Tangier disease and Werner syndrome. Methods Enzymol. 439, 159–169. doi: 10.1016/s0076-6879(07)00412-0
Hu, X., Gan, L., Tang, Z., Lin, R., Liang, Z., Li, F., et al. (2024). A natural small molecule mitigates kidney fibrosis by targeting cdc42-mediated GSK-3β/β-catenin signaling. Adv. Sci. 11, e2307850. doi: 10.1002/advs.202307850
Kirkland, J. L., Tchkonia, T. (2020). Senolytic drugs: from discovery to translation. J. Internal Med. 288, 518–536. doi: 10.1111/joim.13141
Lagoumtzi, S. M., Chondrogianni, N. (2021). Senolytics and senomorphics: Natural and synthetic therapeutics in the treatment of aging and chronic diseases. Free Radical Biol. Med. 171, 169–190. doi: 10.1016/j.freeradbiomed.2021.05.003
Lee, S., Yu, Y., Trimpert, J., Benthani, F., Mairhofer, M., Richter-Pechanska, P., et al. (2021). Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 599, 283–289. doi: 10.1038/s41586-021-03995-1
Lehmann, M., Hu, Q., Hu, Y., Hafner, K., Costa, R., van den Berg, A., et al. (2020). Chronic WNT/β-catenin signaling induces cellular senescence in lung epithelial cells. Cell. Signalling. 70, 109588. doi: 10.1016/j.cellsig.2020.109588
Lipskaia, L., Maisonnasse, P., Fouillade, C., Sencio, V., Pascal, Q., Flaman, J.-M., et al. (2022). Evidence that SARS-coV-2 induces lung cell senescence: potential impact on COVID-19 lung disease. Am. J. Respir. Cell Mol. Biol. 66, 107–111. doi: 10.1165/rcmb.2021-0205LE
Malavolta, M., Giacconi, R., Brunetti, D., Provinciali, M., Maggi, F. (2020). Exploring the relevance of senotherapeutics for the current SARS-coV-2 emergency and similar future global health threats. Cells 9, 909. doi: 10.3390/cells9040909
Mandal, S., Barnett, J., Brill, S. E., Brown, J. S., Denneny, E. K., Hare, S. S., et al. (2021). [amp]]lsquo;Long-COVID’: a cross-sectional study of persisting symptoms, biomarker and imaging abnormalities following hospitalisation for COVID-19. Thorax 76, 396–398. doi: 10.1136/thoraxjnl-2020-215818
Meng, P., Huang, J., Ling, X., Zhou, S., Wei, J., Zhu, M., et al. (2022). CXC chemokine receptor 2 accelerates tubular cell senescence and renal fibrosis via β-catenin-induced mitochondrial dysfunction. Front. Cell Dev. Biol. 10, 862675. doi: 10.3389/fcell.2022.862675
Meyer, K., Patra, T., VijayamahanteshRay, R. (2021). SARS-coV-2 spike protein induces paracrine senescence and leukocyte adhesion in endothelial cells. J. Virol. 95, e0079421. doi: 10.1128/jvi.00794-21
Mo, X., Jian, W., Su, Z., Chen, M., Peng, H., Peng, P., et al. (2020). Abnormal pulmonary function in COVID-19 patients at time of hospital discharge. Eur. Respir. J. 55, 2001217. doi: 10.1183/13993003.01217-2020
Mosaddeghzadeh, N., Ahmadian, M. R. (2021). The RHO family GTPases: mechanisms of regulation and signaling. Cells 10, 1831. doi: 10.3390/cells10071831
Munoz-Espin, D., Serrano, M. (2014). Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 15, 482–496. doi: 10.1038/nrm3823
Nalapareddy, K., Hassan, A., Sampson, L. L., Zheng, Y., Geiger, H. (2021). Suppression of elevated Cdc42 activity promotes the regenerative potential of aged intestinal stem cells. Iscience 24, 102362. doi: 10.1016/j.isci.2021.102362
Nehme, J., Borghesan, M., Mackedenski, S., Bird, T. G., Demaria, M. (2020). Cellular senescence as a potential mediator of COVID-19 severity in the elderly. Aging Cell 19, e13237. doi: 10.1111/acel.13237
Paschalaki, K., Evangelou, K., Veroutis, D., Foukas, P., Kittas, C., Tzioufas, A., et al. (2021). Alveolar type II cells harbouring SARS-CoV-2 show senescence with a proinflammatory phenotype. Eur. Respir. J. 60, 2102951. doi: 10.1183/13993003.congress-2021.OA4312
Rodier, F., Campisi, J. (2011). Four faces of cellular senescence. J. Cell Biol. 192, 547–556. doi: 10.1083/jcb.201009094
Rowayna, S., Gary, E. (2024). Inflammatory gene regulation by Cdc42 in airway epithelial cells. Cell Signal 122, 111321. doi: 10.1016/j.cellsig.2024.111321
Santesmasses, D., Castro, J. P., Zenin, A. A., Shindyapina, A. V., Gerashchenko, M. V., Zhang, B., et al. (2020). COVID-19 is an emergent disease of aging. Aging Cell 19, e13230. doi: 10.1111/acel.13230
Schmitt, C. A., Tchkonia, T., Niedernhofer, L. J., Robbins, P. D., Kirkland, J. L., Lee, S. (2022). COVID-19 and cellular senescence. Nat. Rev. Immunol. 23, 251–263. doi: 10.1038/s41577-022-00785-2
Schneider, J. L., Rowe, J. H., Garcia-de-Alba, C., Kim, C. F., Sharpe, A. H., Haigis, M. C. (2021). The aging lung: Physiology, disease, and immunity. Cell 184, 1990–2019. doi: 10.1016/j.cell.2021.03.005
Shuai, H., Chan, J., Hu, B., Chai, Y., Yuen, T., Yin, F., et al. (2022). Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 603, 693–699. doi: 10.1038/s41586-022-04442-5
Sievers, C., Zacher, B., Ullrich, A., Huska, M., Fuchs, S., Buda, S., et al. (2022). SARS-CoV-2 Omicron variants BA.1 and BA.2 both show similarly reduced disease severity of COVID-19 compared to Delta, Germany 2021 to 2022. Eurosurveillance 27 (22), 2200396. doi: 10.2807/1560-7917.Es.2022.27.22.2200396
Sinha, S., Yang, W. (2008). Cellular signaling for activation of Rho GTPase Cdc42. Cell. Signalling. 20, 1927–1934. doi: 10.1016/j.cellsig.2008.05.002
Stankiewicz, T. R., Linseman, D. A. (2014). Rho family GTPases: key players in neuronal development neuronal survival, and neurodegeneration. Front. Cell. Neurosci. 8. doi: 10.3389/fncel.2014.00314
Suzuki, W., Yamada, A., Aizawa, R., Suzuki, D., Kassai, H., Harada, T., et al. (2015). Cdc42 is critical for cartilage development during endochondral ossification. Endocrinology 156, 314–322. doi: 10.1210/en.2014-1032
Takashi K, I., Masataka, Y., Yohko, Y., Aika, N., Hidetoshi, K., Kengo, O., et al. (2014). A crucial role for CDC42 in senescence-associated inflammation and atherosclerosis. PloS One 9 (7), e102186. doi: 10.1371/journal.pone.0102186
Tamura, T., Yamasoba, D., Oda, Y., Ito, J., Kamasaki, T., Nao, N., et al. (2023). Comparative pathogenicity of SARS-CoV-2 Omicron subvariants including BA.1, BA.2, and BA.5. Commun. Biol. 6, 772. doi: 10.1038/s42003-023-05081-w
Tripathi, U., Nchioua, R., Prata, L. G. P. L., Zhu, Y., Gerdes, E. O. W., Giorgadze, N., et al. (2021). SARS-CoV-2 causes senescence in human cells and exacerbates the senescence-associated secretory phenotype through TLR-3. Aging-Us 13, 21838–21854. doi: 10.18632/aging.203560
Umbayev, B., Masoud, A.-R., Tsoy, A., Alimbetov, D., Olzhayev, F., Shramko, A., et al. (2018). Elevated levels of the small GTPase Cdc42 induces senescence in male rat mesenchymal stem cells. Biogerontology 19, 287–301. doi: 10.1007/s10522-018-9757-5
Vallee, A., Lecarpentier, Y., Vallee, J.-N. (2021). Interplay of opposing effects of the WNT/β-catenin pathway and PPARγ and implications for SARS-coV2 treatment. Front. Immunol. 12. doi: 10.3389/fimmu.2021.666693
Weiss, P., Murdoch, D. R. (2020). Clinical course and mortality risk of severe COVID-19. Lancet 395, 1014–1015. doi: 10.1016/s0140-6736(20)30633-4
World Health Organization. (2024). WHO COVID-19 dashboard. Available online at: https://data.who.int/dashboards/covid19/cases.
Wiley, C. D., Campisi, J. (2021). The metabolic roots of senescence: mechanisms and opportunities for intervention. Nat. Metab. 3, 1290–1301. doi: 10.1038/s42255-021-00483-8
Wolter, N., Jassat, W., Walaza, S., Welch, R., Moultrie, H., Groome, M., et al. (2022). Early assessment of the clinical severity of the SARS-CoV-2 omicron variant in South Africa: a data linkage study. Lancet 399, 437–446. doi: 10.1016/s0140-6736(22)00017-4
Wu, X. W., Quondamatteo, F., Lefever, T., Czuchra, A., Meyer, H., Chrostek, A., et al. (2006). Cdc42 controls progenitor cell differentiation and β-catenin turnover in skin. Genes Dev. 20, 571–585. doi: 10.1101/gad.361406
Yang, C., Pan, X., Huang, Y., Cheng, C., Xu, X., Wu, Y., et al. (2021). Drug repurposing of itraconazole and estradiol benzoate against COVID-19 by blocking SARS-coV-2 spike protein-mediated membrane fusion. Adv. Ther. 4, 2000224. doi: 10.1002/adtp.202000224
Yang, X., Sun, Y., Li, X., Zhang, W. (2022). Rac1 regulates nucleus pulposus cell degeneration by activating the Wnt/β-catenin signaling pathway and promotes the progression of intervertebral disc degeneration. Am. J. Physiology-Cell. Physiol. 322, C496–C507. doi: 10.1152/ajpcell.00355.2021
Yun, L., Xing, C., William, M.-L., Han, W., Belinda, W., Katarzyna, B., et al. (2022). The Ras GTPase-activating-like protein IQGAP1 bridges Gasdermin D to the ESCRT system to promote IL-1β release via exosomes. EMBO J. 42 (1), e110780. doi: 10.15252/embj.2022110780
Zhou, L., Li, Y., Hao, S., Zhou, D., Tan, R. J., Nie, J., et al. (2015). Multiple genes of the renin-angiotensin system are novel targets of wnt/β-catenin signaling. J. Am. Soc. Nephrol. 26, 107–120. doi: 10.1681/asn.2014010085
Keywords: SARS-CoV-2, spike, senescence, Cdc42, β-catenin
Citation: Nong C, Wu Z, Yang C, Xu W, Luo L, Zhou J, Shen L, Chen Y, Yuan Y and Hu G (2024) Cdc42 improve SARS-CoV-2 spike protein-induced cellular senescence through activating of Wnt/β-Catenin signaling pathway. Front. Cell. Infect. Microbiol. 14:1449423. doi: 10.3389/fcimb.2024.1449423
Received: 15 June 2024; Accepted: 11 October 2024;
Published: 04 November 2024.
Edited by:
Moises Leon Juarez, Instituto Nacional de Perinatología (INPER), MexicoReviewed by:
Daniel Humphreys, The University of Sheffield, United KingdomCopyright © 2024 Nong, Wu, Yang, Xu, Luo, Zhou, Shen, Chen, Yuan and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yinghua Chen, bXJjaGNoQDEyNi5jb20=; Yaoqin Yuan, eXVhbnlxOTM4QHNtdS5lZHUuY24=; Guodong Hu, aHVndW9kb25nQHNtdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.