
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Infect. Microbiol., 15 September 2023
Sec. Extra-intestinal Microbiome
Volume 13 - 2023 | https://doi.org/10.3389/fcimb.2023.1271117
This article is part of the Research TopicReviews in Microbiome in Health & DiseaseView all 13 articles
 Florian Lussac-Sorton1,2*
Florian Lussac-Sorton1,2* Éléna Charpentier1,2
Éléna Charpentier1,2 Sébastien Imbert1,2,3Maxime Lefranc3
Sébastien Imbert1,2,3Maxime Lefranc3 Stéphanie Bui1,2,3
Stéphanie Bui1,2,3 Michael Fayon1,2,3Patrick Berger1,2,3
Michael Fayon1,2,3Patrick Berger1,2,3 Raphaël Enaud1,2,3Laurence Delhaes1,2,3*
Raphaël Enaud1,2,3Laurence Delhaes1,2,3*The advent of CFTR modulators represents a turning point in the history of cystic fibrosis (CF) management, changing profoundly the disease’s clinical course by improving mucosal hydration. Assessing changes in airway and digestive tract microbiomes is of great interest to better understand the mechanisms and to predict disease evolution. Bacterial and fungal dysbiosis have been well documented in patients with CF; yet the impact of CFTR modulators on microbial communities has only been partially deciphered to date. In this review, we aim to summarize the current state of knowledge regarding the impact of CFTR modulators on both pulmonary and digestive microbiomes. Our analysis also covers the inter-organ connections between lung and gut communities, in order to highlight the gut-lung axis involvement in CF pathophysiology and its evolution in the era of novel modulators therapies.
Cystic fibrosis (CF) is the most common severe life-limiting genetic disease in Caucasian populations. It affects more than 80,000 people worldwide, with a prevalence at birth of about 1 in 3,500 in Europe (Brown et al., 2017) but with a wide heterogeneity in its distribution. While it is diagnosed equally in men and women, its clinical expression appears to be more severe in women, which may be linked to estrogen involvement in the disease pathophysiology (Lam et al., 2021). CF is a monogenic disease with an autosomal recessive inheritance, caused by mutations in the gene coding the cystic fibrosis transmembrane conductance regulator (CFTR) protein, responsible for impaired chloride and bicarbonate secretions across epithelial cell apical membranes. More than 2,000 cftr gene mutations have been reported, which are classified into 6 groups according to the subsequent functional defect. These dysfunctions in the CFTR chloride channel cause an accumulation of viscous and dehydrated mucus in exocrine glands epithelia and airway lumen. While pulmonary complications remain the main cause of morbidity-mortality in CF, the disease also affects other organs in particular the gastro-intestinal tract (Karb and Cummings, 2021). The mucus accumulation may lead to an obstruction of the intestinal lumen as well as pancreatic and bile ducts. People with CF (pwCF) also exhibit an increased risk of gastro-intestinal cancers compared to general population (Yamada et al., 2018). Hence, CF represents a multi-organ pathology responsible for a wide variety of symptoms across patients’ lives (Figure 1), with respiratory and digestive manifestations playing key roles in disease progression.
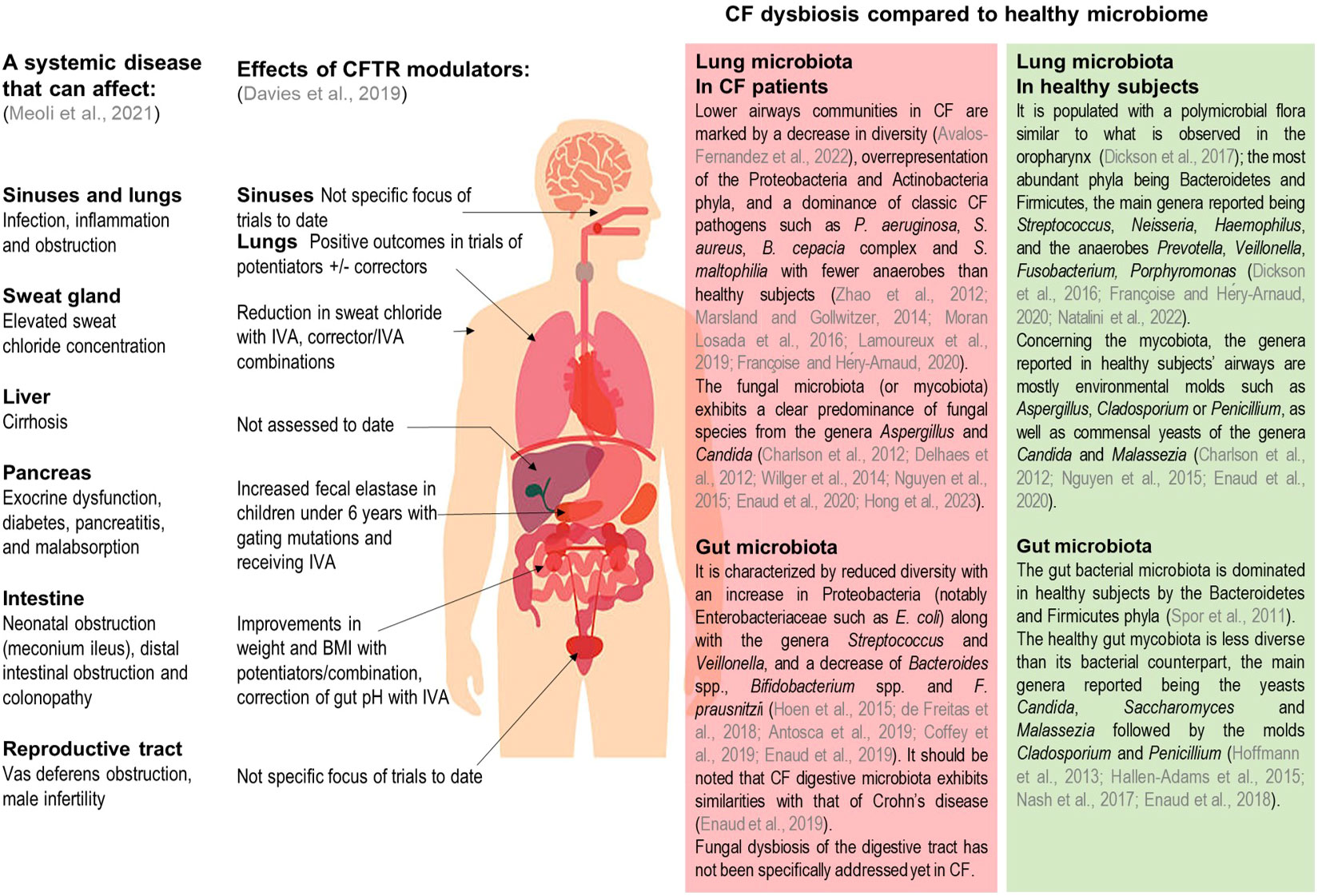
Figure 1 Summary of clinical manifestations, documented CFTR modulator effects, and of known lung and gut dysbiosis in pwCF. Adapted from Meoli et al., Pharmaceuticals 2021, and Davies et al., Kendig's Disorders of the Respiratory Tract in Children 2019. IVA, Ivacaftor; BMI, body mass index.
In both the airways and gastro-intestinal tracts altered mucus composition leads to an imbalance in microbial communities. This dysbiosis also contributes to the establishment of chronic inflammation associated with functional lung and digestive decline (Enaud et al., 2019). Metagenomic approaches based on next generation sequencing have clearly facilitated the investigation of the CF microbiome (Françoise and Héry-Arnaud, 2020) which is of great importance to the understanding of the underlying mechanisms and to predict disease evolution. While assessment of both bacterial and fungal communities is well documented using metabarcoding approaches, the viral component of the microbiome requires shotgun metagenomics methods and remains largely unknown (Billard et al., 2017).
Pulmonary bacterial and to a lesser degree fungal microbiotas have been well documented in pwCF and healthy subjects (Figure 1) (Charlson et al., 2012; Delhaes et al., 2012; Zhao et al., 2012; Marsland and Gollwitzer, 2014; Willger et al., 2014; Nguyen et al., 2015; Dickson et al., 2016; Moran Losada et al., 2016; Dickson et al., 2017; Lamoureux et al., 2019; Enaud et al., 2020; Françoise and Héry-Arnaud, 2020; Avalos-Fernandez et al., 2022; Natalini et al., 2022; Hong et al., 2023). In the gut, microbial communities have also been well characterized (Figure 1) (Spor et al., 2011; Hoffmann et al., 2013; Hallen-Adams et al., 2015; Hoen et al., 2015; Nash et al., 2017; de Freitas et al., 2018; Enaud et al., 2018; Antosca et al., 2019; Coffey et al., 2019; Enaud et al., 2019); however it should be noted that fungal microbiota (also called mycobiota) has not been specifically addressed yet in pwCF digestive tract. Distant microbial florae display inter-organ connections involving a bidirectional crosstalk. As such, the gut-lung axis is an emerging concept describing how pulmonary and intestinal communities influence each other and are linked to clinical outcomes. In CF, this gut-lung axis is supported by studies results showing how gut bacterial microbiome is correlated with its pulmonary counterpart or the occurrence of respiratory complications (Enaud et al., 2020).
Before the 2010s, CF therapies were solely able to alleviate the disease symptoms. Over the last decade the development of CFTR modulators which directly address the underlying defects in the impaired protein has represented a turning point in the history of CF management (Davies et al., 2019). This has dramatically changed the disease’s clinical course (Figure 1) leading to meaningful improvements in the daily lives of a large CF population and enabling to reach an average life expectancy of nearly 50 years (Karb and Cummings, 2021). In this review we will focus on CFTR modulators, their impact on pulmonary and intestinal microbiotas and discuss how these novel therapies may modify profoundly the gut-lung axis in CF and which long-term clinical benefit CF patients may expect.
Current advances in CFTR modulators have been reviewed recently (Meoli et al., 2021). Here, we will focus on CFTR modulators for which microbiota data have been published. Briefly, Ivacaftor (IVA) (Kalydeco®, Vertex pharmaceuticals, Boston, Massachusetts) was the first modulator to obtain approval by the FDA (2012); it belongs to the “potentiator” class, which increases CFTR gating at the apical surface of epithelial cells allowing for extended channel opening time. IVA was first approved in adult patients with at least one G551D gating mutation, representing only 2% of CF patients (McKone et al., 2003). Its use has been secondarily extended to other rare CFTR mutations with gating defects and nowadays from the age of 4 months upwards. Its clinical efficacy was demonstrated in children and adult cohorts, with an improvement in lung function (i.e., increase of percent predicted forced expiratory volume in 1 second (ppFEV1)), a decrease in the rate of pulmonary exacerbations, an improvement in nutritional status (i.e., gain of weight and increase in body mass index (BMI)), an improvement in pancreatic function, and a strong reduction of the sweat chloride concentration (Ramsey et al., 2011; Davies et al., 2013). IVA displayed an acceptable safety profile (the most common adverse effects being headaches, oropharyngeal pain, upper respiratory tract infections and nasal congestion) (Ramsey et al., 2011; Davies et al., 2013). A risk of increased blood liver enzymes has been reported, justifying regular monitoring in all treated patients. No positive effects were observed when tested in patients with the more frequent F508del mutation (Flume et al., 2012), suggesting that a combination with a corrector is required to rectify the activity defect.
Several CFTR correctors have been developed to treat the protein misfolding and improve its trafficking to the apical surface, lumacaftor (LUM) being the first molecule of this class. Though LUM did not show any significant clinical effects as monotherapy (Clancy et al., 2012), it proved beneficial when associated with the potentiator IVA, and as such, has been FDA-approved as a combination since 2015 for patients aged 2 years and older, with homozygous F508del mutations (about 40% of pwCF (Wainwright et al., 2015)). The combination of lumacaftor-ivacaftor (LUM/IVA) (Orkambi®,Vertex pharmaceuticals) was evaluated in two randomized controlled trials, which showed modest but significant benefits on lung function at 24 weeks and on nutritional status (increase in BMI) (Wainwright et al., 2015). These benefits continued to be observed in the long term at week 96 (Konstan et al., 2017). LUM/IVA combination displayed an acceptable safety profile in the pivotal trials. However, clinical responses may also vary significantly amongst patients with the same genotype, and acute respiratory events (chest tightness, dyspnea and drop of ppFEV1) were reported later on in real life studies, responsible for treatment discontinuation in approximately 20% of patients (Hubert et al., 2017; Jennings et al., 2017; Labaste et al., 2017; Burgel et al., 2020).
The second dual-combination of a CFTR corrector and potentiator was tezacaftor-ivacaftor (TEZ/IVA) (Symdeko® in the USA and Symkevi® in Europe, Vertex pharmaceuticals), FDA-approved in 2018 for patients aged 6 years and older, with homozygous F508del mutations or heterozygous in association with a residual-function mutation. TEZ/IVA combination showed significant clinical efficacy similar to that of LUM/IVA, but a better side-effect profile with less occurrence of acute respiratory events (Rowe et al., 2017; Taylor-Cousar et al., 2017; Lommatzsch and Taylor-Cousar, 2019). Thereafter, a triple-combination approach was developed by adding elexacaftor (ELX), a new CFTR corrector, to the former TEZ/IVA regimen. ELX and TEZ bind to different sites of the CFTR protein, displaying an additive effect to improve the protein processing. The triple-combination elexacaftor-tezacaftor-ivacaftor (ELX/TEZ/IVA) (Trikafta® in USA and Kaftrio® in Europe, Vertex pharmaceuticals) was approved in 2019 for patients aged 6 years and older having at least one F508del mutation (regardless of the other mutation on the second allele), representing nearly 90% of pwCF (Cystic Fibrosis Foundation, 2022; Vaincre la Mucoviscidose, 2022). ELX/TEZ/IVA has been assessed in several randomized clinical trials (Heijerman et al., 2019; Middleton et al., 2019; Zemanick et al., 2021) which demonstrated an unprecedented clinical improvement with an acceptable safety profile leading to a significant decrease in the number of lung transplantations (Burgel et al., 2021).
CFTR modulators therapies lead to improved mucus hydration by correcting chloride channel activity, which improves mucociliary clearance and, in turn, induces changes in airway microbial communities. The impact of modulators on the pulmonary microbiome has been more extensively studied during IVA monotherapy (as the first developed molecule) in patients bearing at least one G551D mutation. Results of these studies are summarized in Table 1. Briefly, a first study assessing IVA effects reported no significant changes in bacterial alpha-diversity, but a trend towards decreased abundance of pathogens as well as a significant increase in the anaerobe Prevotella (Rowe et al., 2014). A subsequent study performed on 3 patients did not find any significant differences in total bacterial load and global community composition, but described a notable decrease in Streptococcus mitis abundance and increase in the anaerobe Porphyromonas (Bernarde et al., 2015). A third study reported a significant increase of bacterial diversity indices during the first year of treatment in patients chronically infected with P. aeruginosa, as well as a decrease in the relative abundance of P. aeruginosa with reciprocal increase of oropharyngeal bacteria such as Streptococcus and Prevotella (Hisert et al., 2017). Nonetheless, these changes were not sustained with a notable rebound observed during the second year of IVA monotherapy (Burgel et al., 2021). A more recent study reported increased richness and diversity of the pulmonary bacterial microbiome, correlated with lower levels of circulating inflammatory markers (Einarsson et al., 2021). By contrast, other studies did not find any significant changes in pulmonary bacterial microbiome related to IVA in patients with G551D or S1251N mutations (Peleg et al., 2018; Harris et al., 2020; Kristensen et al., 2021), in spite of limitations due to small sample sizes, short follow-up periods and differences in antibiotic exposures. Overall, these results suggest a trend towards increased bacterial diversity in the airways on IVA monotherapy, accompanied with a decrease but no eradication of P. aeruginosa and reciprocal increase in commensal anaerobes.
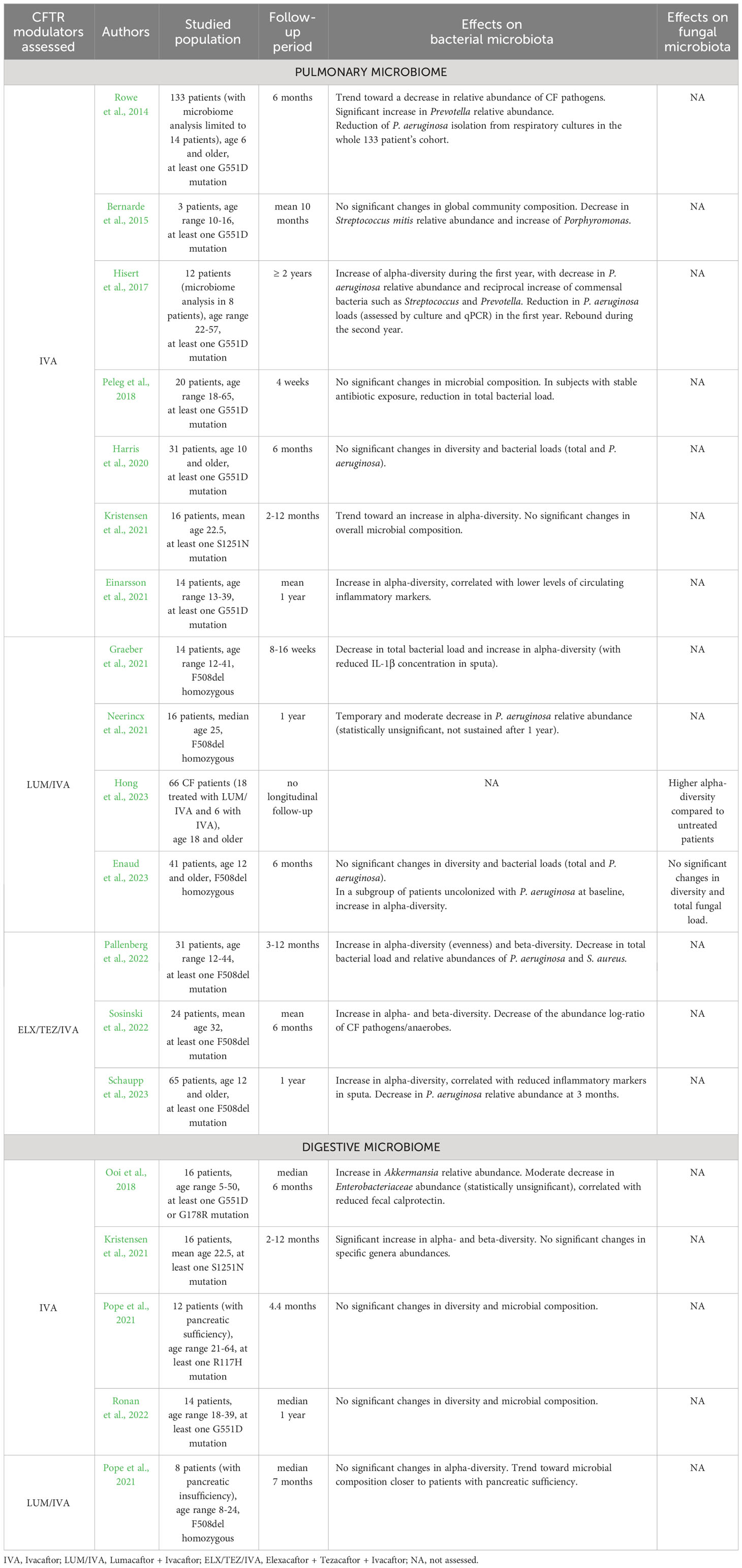
Table 1 Impact of CFTR modulators on pulmonary and digestive microbiomes.
Regarding the impact of the LUM/IVA dual-combination on the airway microbiome, a first study reported similarly a moderate decrease in P. aeruginosa relative abundance that did not reach statistical significance and was not sustained after 12 months of treatment (Neerincx et al., 2021). A second study described a significant decrease in total bacterial load and an increase in bacterial alpha-diversity calculated with the Shannon index, along with a reduced concentration of the proinflammatory cytokine IL-1β in pwCF sputa (Graeber et al., 2021). Regarding the mycobiome, one recent cross-sectional study showed that adult patients treated with modulators (the majority of whom were receiving LUM/IVA) had significantly higher fungal alpha-diversity compared to untreated patients (Hong et al., 2023). A last study did not find any significant differences in both airway bacterial and fungal microbiota and pulmonary inflammation assessed by calprotectin measurement (Enaud et al., 2023). However, when focusing on a subgroup of patients uncolonized with P. aeruginosa at LUM/IVA initiation, calprotectin levels were lower and a significant increase in bacterial alpha-diversity was observed after 6 months of therapy, suggesting that microbiome changes on LUM/IVA are dependent on P. aeruginosa colonization status.
Only three studies have addressed the effect of the most recent and promising ELX/TEZ/IVA triple-combination on pulmonary microbiome in pwCF wearing at least one F508del mutation. One study performed on 24 patients (Sosinski et al., 2022) demonstrated a significant increase in bacterial alpha- and beta-diversity in the sputa after treatment, as well as a trend towards a reduced bacterial load. Although the relative abundances of specific bacterial taxa were unchanged, the authors described a significant decrease in the CF pathogens to anaerobes log-ratio. These findings were partially confirmed by a subsequent study reporting a significant increase in the evenness of distribution of bacterial taxa, along with a decrease in total bacterial load and relative abundances of major CF pathogens P. aeruginosa and Staphylococcus aureus (Pallenberg et al., 2022). Similarly, the authors of a recent study described an increase in bacterial alpha-diversity after 1, 3 and 12 months on ELX/TEZ/IVA (Schaupp et al., 2023) while the decrease in P. aeruginosa relative abundance was only significant at 3 months. In addition, the authors also reported a significant reduction in inflammatory markers (IL-1β, IL-8 and neutrophil elastase) in the sputa. All these findings appear to be consistent with those reported with previous generations of CFTR modulators.
To date the impact of CFTR modulators on the intestinal microbiome has been less extensively studied compared to its pulmonary counterpart. Several studies suggest that IVA monotherapy may have an impact on the intestinal microbiome in pwCF with gating defects; such results are summarized in Table 1. A first study showed that IVA provokes a significant increase in the relative abundance of the bacterial genus Akkermansia (Ooi et al., 2018) which is known for its anti-inflammatory effects and as a biomarker of healthy gut mucosa (Derrien et al., 2017; Rodrigues et al., 2022). This finding was associated with a significant decrease in intestinal inflammation assessed by fecal calprotectin measurement, while bacterial alpha- and beta-diversities were unchanged. Conversely, another study reported a significant increase in both alpha- and beta-diversity indices after IVA treatment, whereas no significant changes were found in specific bacterial genera abundances (Kristensen et al., 2021). Two other studies did not show any significant effect related to IVA monotherapy on the gut microbiome (Pope et al., 2021; Ronan et al., 2022). Finally, trends toward a bacterial microbiota composition closer to subjects with normal exocrine pancreatic function were reported with LUM/IVA dual-combination (Pope et al., 2021). These limited findings need to be further validated by larger studies, especially with next-generation modulators like the ELX/TEZ/IVA triple-combination. Moreover, it must be noted that the intestinal mycobiota has not been assessed to date in patients treated with CFTR modulators. By extrapolation based on similarities regarding the bacterial component (Enaud et al., 2019) it may display similarities with patterns reported in inflammatory bowel diseases, which are characterized by a notable increase in the Basidiomycota/Ascomycota ratio during flares episodes and a decrease during remission (Sokol et al., 2017). In addition, although the intestinal mycobiome has been reported to influence airway outcomes (Zhang et al., 2017) no studies have currently examined this hypothesis or the role of the gut in the context of CF.
In this review we have summarized the published data regarding the impact of CFTR modulators on pulmonary and digestive microbiomes respectively. However, these two microbial florae are not strictly independent as indicated by growing evidence describing inter-organ connections (Enaud et al., 2020; Françoise and Héry-Arnaud, 2020), and supporting the concept of a gut-lung axis. Due to the anatomical links the microbial communities of both tracts have direct interactions through gastroesophageal content inhalations and sputum swallowing (Enaud et al., 2020). Long-reaching interactions between the airways and intestine are also involved, mediated via the mesenteric lymphatic system through which microbial metabolites are exchanged (Bingula et al., 2017; Anand and Mande, 2018; Enaud et al., 2020). Among them, short-chain fatty acids synthesized by intestinal bacteria are of particular importance given their well-known immunomodulatory properties. Such fatty acids have been shown to mitigate the airway inflammatory response (Trompette et al., 2014; Halnes et al., 2017).
In pwCF, several studies suggest interactions between bacterial communities at the pulmonary and digestive levels. In infants with CF, a high degree of concordance was observed between bacterial genera evolution over time at both sites, with some genera colonizing the gut prior to their appearance in the respiratory tract (Madan et al., 2012). Furthermore, the gut microbiome is closely related to CF respiratory complications. Indeed, some gut microbiota alterations (such as a decrease of Parabacteroides) precede the onset of P. aeruginosa colonization, while its composition is associated with CF exacerbation in early life (Hoen et al., 2015). Regarding dietary exposures, breastfeeding was associated with microbial diversity of the respiratory tract as well as a prolonged time to first CF exacerbation (Madan et al., 2012; Hoen et al., 2015), the role of short-chain fatty acids being discussed but not demonstrated yet. Finally, the oral administration of probiotics (supposed to modulate the gut microbiota) showed a significant reduction in the number of pulmonary exacerbations, although the size of effect is unclear (Anderson et al., 2017).
The gut-lung axis involvement in pwCF receiving CFTR modulators has been far less extensively investigated. To date, only one study has assessed bacterial communities in both the airways and gut in 16 patients with the rare S1251N mutation treated with IVA monotherapy (Kristensen et al., 2021). The authors reported significant improvements in the gut microbiota diversity that were not observed in the respiratory samples (including sputum, nasopharynx and oropharynx samples). Nonetheless, these findings are limited by the small sample size (n=16, among which only 8 patients were followed during 12 months). In addition, the authors suggested a follow-up time in excess of 12 months to detect significant improvements in lung microbiota on CFTR modulators, since the development of respiratory tract microbiota appeared to be presaged by gut colonization (Madan et al., 2012).
Further studies are thus required to fully decipher the gut-lung axis evolution on modulator therapies, involving a joint analysis of pulmonary and fecal samples. Moreover, microbiome analyses have so far been almost exclusively limited to the bacterial kingdom, the fungal kingdom being neglected in the assessment of pwCF microbiotas on modulators, especially in the digestive tract analysis. In addition, the use of shotgun metagenomics approaches will allow to investigate the viral component of the microbiome, providing an exhaustive inter-kingdom assessment of CF microbial communities. The entire microbiome data should also be correlated with clinical parameters and surrogate inflammatory biomarkers for a comprehensive evaluation of the CF disease landscape in a personalized medicine approach that next generation of CFTR modulators will enable (Yi et al., 2021; Enaud et al., 2023).
Regarding the populations studied, it should be noted that the patients included in the quoted studies were over 5 years of age, including a majority of adults with already established chronic colonization and infection (Table 1). The modulators’ impact should also be assessed in future studies in large pediatric populations including children under 5 years of age, since CFTR modulator treatment will be initiated earlier, before the patients are chronically colonized and the gut-lung axis mucosa irreversibly damaged (Enaud et al., 2023). Moreover, other features such as pwCF gender should be considered, since differential outcomes between men and women treated with IVA have been described (Secunda et al., 2020). Finally, early published studies being focused on the assessment of patients treated with IVA monotherapy, future studies are needed to investigate the gut-lung axis in patients receiving combinations of modulators such as LUM/IVA, TEZ/IVA and ELX/TEZ/IVA.
In conclusion, the advent of CFTR modulators marks a turning point in the history of CF management and outcome, with unprecedented clinical improvements and favorable safety profiles, even if data about long-term adverse effects are not yet available. The impact of such molecules on the resident microbial communities of the respiratory and digestive tracts has only been partially deciphered, mostly with first-generation therapies such as IVA. While the results of available studies are not totally consistent, they suggest that CFTR modulators may induce an increase in bacterial diversity, associated with a decrease in conventional culture-based CF pathogens in the airways and an increase in anti-inflammatory bacteria in the gut. The impact of next-generation modulators on the gut-lung axis will have to be more extensively investigated in future studies, with additional data regarding the mycobiota and virobiota, especially in younger pwCF who will benefit even more from these innovative therapies.
FL-S: Conceptualization, Writing – original draft, Writing – review & editing. ÉC: Conceptualization, Writing – review & editing. SI: Conceptualization, Writing – review & editing. ML: Writing – review & editing. SB: Writing – review & editing. MF: Writing – review & editing. PB: Writing – review & editing. RE: Conceptualization, Writing – review & editing. LD: Conceptualization, Writing – original draft, Writing – review & editing.
The authors declare financial support was received for the research, authorship, and/or publication of this article. LD team had annual grants from the Bordeaux University Hospital, University of Bordeaux and INSERM U1045; they also benefited from Vaincre la Mucoviscidose’s grant (RCI20210502769). All the funders had no role in article design, data analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Anand, S., Mande, S. S. (2018). Diet, microbiota and gut-lung connection. Front. Microbiol. 9, 2147. doi: 10.3389/fmicb.2018.02147
Anderson, J. L., Miles, C., Tierney, A. C. (2017). Effect of probiotics on respiratory, gastrointestinal and nutritional outcomes in patients with cystic fibrosis: A systematic review. J. Cyst Fibros 16 (2), 186–197. doi: 10.1016/j.jcf.2016.09.004
Antosca, K. M., Chernikova, D. A., Price, C. E., Ruoff, K. L., Li, K., Guill, M. F., et al. (2019). Altered stool microbiota of infants with cystic fibrosis shows a reduction in genera associated with immune programming from birth. J. Bacteriol. 201 (16), e00274-19. doi: 10.1128/JB.00274-19
Avalos-Fernandez, M., Alin, T., Métayer, C., Thiébaut, R., Enaud, R., Delhaes, L. (2022). The respiratory microbiota alpha-diversity in chronic lung diseases: first systematic review and meta-analysis. Respir. Res. 23 (1), 214. doi: 10.1186/s12931-022-02132-4
Bernarde, C., Keravec, M., Mounier, J., Gouriou, S., Rault, G., Férec, C., et al. (2015). Impact of the CFTR-potentiator ivacaftor on airway microbiota in cystic fibrosis patients carrying A G551D mutation. PloS One 10 (4), e0124124. doi: 10.1371/journal.pone.0124124
Billard, L., Le Berre, R., Pilorgé, L., Payan, C., Héry-Arnaud, G., Vallet, S. (2017). Viruses in cystic fibrosis patients’ airways. Crit. Rev. Microbiol. 43 (6), 690–708. doi: 10.1080/1040841X.2017.1297763
Bingula, R., Filaire, M., Radosevic-Robin, N., Bey, M., Berthon, J. Y., Bernalier-Donadille, A., et al. (2017). Desired turbulence? Gut-lung axis, immunity, and lung cancer. J. Oncol. 2017, 5035371. doi: 10.1155/2017/5035371
Brown, S. D., White, R., Tobin, P. (2017). Keep them breathing: Cystic fibrosis pathophysiology, diagnosis, and treatment. J. Am. Acad. Physician Assistants 30 (5), 23–27. doi: 10.1097/01.JAA.0000515540.36581.92
Burgel, P. R., Durieu, I., Chiron, R., Ramel, S., Danner-Boucher, I., Prevotat, A., et al. (2021). Rapid improvement after starting elexacaftor-tezacaftor-ivacaftor in patients with cystic fibrosis and advanced pulmonary disease. Am. J. Respir. Crit. Care Med. 204 (1), 64–73. doi: 10.1164/rccm.202011-4153OC
Burgel, P. R., Munck, A., Durieu, I., Chiron, R., Mely, L., Prevotat, A., et al. (2020). Real-life safety and effectiveness of lumacaftor–ivacaftor in patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 201 (2), 188–197. doi: 10.1164/rccm.201906-1227OC
Charlson, E. S., Diamond, J. M., Bittinger, K., Fitzgerald, A. S., Yadav, A., Haas, A. R., et al. (2012). Lung-enriched organisms and aberrant bacterial and fungal respiratory microbiota after lung transplant. Am. J. Respir. Crit. Care Med. 186 (6), 536–545. doi: 10.1164/rccm.201204-0693OC
Clancy, J. P., Rowe, S. M., Accurso, F. J., Aitken, M. L., Amin, R. S., Ashlock, M. A., et al. (2012). Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 67 (1), 12–18. doi: 10.1136/thoraxjnl-2011-200393
Coffey, M. J., Nielsen, S., Wemheuer, B., Kaakoush, N. O., Garg, M., Needham, B., et al. (2019). Gut microbiota in children with cystic fibrosis: A taxonomic and functional dysbiosis. Sci. Rep. 9 (1), 18593. doi: 10.1038/s41598-019-55028-7
Cystic Fibrosis Foundation (2022). Cystic fibrosis foundation patient registry 2021 annual data report (Bethesda, Maryland). Available at: https://www.cff.org/sites/default/files/2021-11/Patient-Registry-Annual-Data-Report.pdf.
Davies, G., Griesenbach, U., Alton, E., Davies, J. C. (2019). Molecular therapies for cystic fibrosis. In: Kendig’s Disord. Respir. Tract Children Elsevier;, 800–811.e3. doi: 10.1016/B978-0-323-44887-1.00053-5
Davies, J. C., Wainwright, C. E., Canny, G. J., Chilvers, M. A., Howenstine, M. S., Munck, A., et al. (2013). Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 187 (11), 1219–1225. doi: 10.1164/rccm.201301-0153OC
de Freitas, M. B., Moreira, E. A. M., Tomio, C., Moreno, Y. M. F., Daltoe, F. P., Barbosa, E., et al. (2018). Altered intestinal microbiota composition, antibiotic therapy and intestinal inflammation in children and adolescents with cystic fibrosis. PloS One 13 (6), e0198457. doi: 10.1371/journal.pone.0198457
Delhaes, L., Monchy, S., Fréalle, E., Hubans, C., Salleron, J., Leroy, S., et al. (2012). The airway microbiota in cystic fibrosis: a complex fungal and bacterial community–implications for therapeutic management. PloS One 7 (4), e36313. doi: 10.1371/journal.pone.0036313
Derrien, M., Belzer, C., de Vos, W. M. (2017). Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 106, 171–181. doi: 10.1016/j.micpath.2016.02.005
Dickson, R. P., Erb-Downward, J. R., Freeman, C. M., McCloskey, L., Falkowski, N. R., Huffnagle, G. B., et al. (2017). Bacterial topography of the healthy human lower respiratory tract. mBio. 8 (1), e02287–e02216. doi: 10.1128/mBio.02287-16
Dickson, R. P., Erb-Downward, J. R., Martinez, F. J., Huffnagle, G. B. (2016). The microbiome and the respiratory tract. Annu. Rev. Physiol. 78, 481–504. doi: 10.1146/annurev-physiol-021115-105238
Einarsson, G. G., Ronan, N. J., Mooney, D., McGettigan, C., Mullane, D., NiChroinin, M., et al. (2021). Extended-culture and culture-independent molecular analysis of the airway microbiota in cystic fibrosis following CFTR modulation with ivacaftor. J. Cystic Fibrosis 20 (5), 747–753. doi: 10.1016/j.jcf.2020.12.023
Enaud, R., Hooks, K. B., Barre, A., Barnetche, T., Hubert, C., Massot, M., et al. (2019). Intestinal inflammation in children with cystic fibrosis is associated with crohn’s-like microbiota disturbances. J. Clin. Med. 8 (5), 645. doi: 10.3390/jcm8050645
Enaud, R., Lussac-Sorton, F., Charpentier, E., Velo-Suárez, L., Guiraud, J., Bui, S., et al. (2023). Effects of lumacaftor-ivacaftor on airway microbiota-mycobiota and inflammation in patients with cystic fibrosis appear to be linked to pseudomonas aeruginosa chronic colonization. Microbiol. Spectr. 11 (2), e0225122. doi: 10.1128/spectrum.02251-22
Enaud, R., Prevel, R., Ciarlo, E., Beaufils, F., Wieërs, G., Guery, B., et al. (2020). The gut-lung axis in health and respiratory diseases: A place for inter-organ and inter-kingdom crosstalks. Front. Cell Infect. Microbiol. 10, 9. doi: 10.3389/fcimb.2020.00009
Enaud, R., Vandenborght, L. E., Coron, N., Bazin, T., Prevel, R., Schaeverbeke, T., et al. (2018). The mycobiome: A neglected component in the microbiota-gut-brain axis. Microorganisms 6 (1), 22. doi: 10.3390/microorganisms6010022
Flume, P. A., Liou, T. G., Borowitz, D. S., Li, H., Yen, K., Ordoñez, C. L., et al. (2012). Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 142 (3), 718–724. doi: 10.1378/chest.11-2672
Françoise, A., Héry-Arnaud, G. (2020). The microbiome in cystic fibrosis pulmonary disease. Genes 11 (5), 536. doi: 10.3390/genes11050536
Graeber, S. Y., Boutin, S., Wielpütz, M. O., Joachim, C., Frey, D. L., Wege, S., et al. (2021). Effects of lumacaftor-ivacaftor on lung clearance index, magnetic resonance imaging and airway microbiome in phe508del homozygous patients with cystic fibrosis. Ann. Am. Thorac. Soc. 18 (6), 971–980. doi: 10.1513/AnnalsATS.202008-1054OC
Hallen-Adams, H. E., Kachman, S. D., Kim, J., Legge, R. M., Martínez, I. (2015). Fungi inhabiting the healthy human gastrointestinal tract: a diverse and dynamic community. Fungal Ecol. 15, 9–17. doi: 10.1016/j.funeco.2015.01.006
Halnes, I., Baines, K. J., Berthon, B. S., MacDonald-Wicks, L. K., Gibson, P. G., Wood, L. G. (2017). Soluble fibre meal challenge reduces airway inflammation and expression of GPR43 and GPR41 in asthma. Nutrients 9 (1), 57. doi: 10.3390/nu9010057
Harris, J. K., Wagner, B. D., Zemanick, E. T., Robertson, C. E., Stevens, M. J., Heltshe, S. L., et al. (2020). Changes in airway microbiome and inflammation with ivacaftor treatment in patients with cystic fibrosis and the G551D mutation. Ann. ATS 17 (2), 212–220. doi: 10.1513/AnnalsATS.201907-493OC
Heijerman, H. G. M., McKone, E. F., Downey, D. G., Van Braeckel, E., Rowe, S. M., Tullis, E., et al. (2019). Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 394 (10212), 1940–1948. doi: 10.1016/S0140-6736(19)32597-8
Hisert, K. B., Heltshe, S. L., Pope, C., Jorth, P., Wu, X., Edwards, R. M., et al. (2017). Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am. J. Respir. Crit. Care Med. 195 (12), 1617–1628. doi: 10.1164/rccm.201609-1954OC
Hoen, A. G., Li, J., Moulton, L. A., O’Toole, G. A., Housman, M. L., Koestler, D. C., et al. (2015). Associations between gut microbial colonization in early life and respiratory outcomes in cystic fibrosis. J. Pediatr. 167 (1), 138–147.e1-3. doi: 10.1016/j.jpeds.2015.02.049
Hoffmann, C., Dollive, S., Grunberg, S., Chen, J., Li, H., Wu, G. D., et al. (2013). Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PloS One 8 (6), e66019. doi: 10.1371/journal.pone.0066019
Hong, G., Daniel, S. G., Lee, J. J., Bittinger, K., Glaser, L., Mattei, L. M., et al. (2023). Distinct community structures of the fungal microbiome and respiratory health in adults with cystic fibrosis. J. Cystic Fibrosis. 22 (4), 636–643. doi: 10.1016/j.jcf.2023.02.003
Hubert, D., Chiron, R., Camara, B., Grenet, D., Prévotat, A., Bassinet, L., et al. (2017). Real-life initiation of lumacaftor/ivacaftor combination in adults with cystic fibrosis homozygous for the Phe508del CFTR mutation and severe lung disease. J. Cyst Fibros 16 (3), 388–391. doi: 10.1016/j.jcf.2017.03.003
Jennings, M. T., Dezube, R., Paranjape, S., West, N. E., Hong, G., Braun, A., et al. (2017). An observational study of outcomes and tolerances in patients with cystic fibrosis initiated on lumacaftor/ivacaftor. Ann. Am. Thorac. Soc 14 (11), 1662–1666. doi: 10.1513/AnnalsATS.201701-058OC
Karb, D. B., Cummings, L. C. (2021). The intestinal microbiome and cystic fibrosis transmembrane conductance regulator modulators: emerging themes in the management of gastrointestinal manifestations of cystic fibrosis. Curr. Gastroenterol. Rep. 23 (10), 17. doi: 10.1007/s11894-021-00817-2
Konstan, M. W., McKone, E. F., Moss, R. B., Marigowda, G., Tian, S., Waltz, D., et al. (2017). Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study. Lancet Respir. Med. 5 (2), 107–118. doi: 10.1016/S2213-2600(16)30427-1
Kristensen, M. I., de Winter-de Groot, K. M., Berkers, G., Chu, M. L. J. N., Arp, K., Ghijsen, S., et al. (2021). Individual and group response of treatment with ivacaftor on airway and gut microbiota in people with CF and a S1251N mutation. JPM 11 (5), 350. doi: 10.3390/jpm11050350
Labaste, A., Ohlmann, C., Mainguy, C., Jubin, V., Perceval, M., Coutier, L., et al. (2017). Real-life acute lung function changes after lumacaftor/ivacaftor first administration in pediatric patients with cystic fibrosis. J. Cyst Fibros 16 (6), 709–712. doi: 10.1016/j.jcf.2017.05.002
Lam, G. Y., Goodwin, J., Wilcox, P. G., Quon, B. S. (2021). Sex disparities in cystic fibrosis: review on the effect of female sex hormones on lung pathophysiology and outcomes. ERJ Open Res. 7 (1), 00475–02020. doi: 10.1183/23120541.00475-2020
Lamoureux, C., Guilloux, C. A., Beauruelle, C., Jolivet-Gougeon, A., Héry-Arnaud, G. (2019). Anaerobes in cystic fibrosis patients’ airways. Crit. Rev. Microbiol. 45 (1), 103–117. doi: 10.1080/1040841X.2018.1549019
Lommatzsch, S. T., Taylor-Cousar, J. L. (2019). The combination of tezacaftor and ivacaftor in the treatment of patients with cystic fibrosis: clinical evidence and future prospects in cystic fibrosis therapy. Ther. Adv. Respir. Dis. 13, 1753466619844424. doi: 10.1177/1753466619844424
Madan, J. C., Koestler, D. C., Stanton, B. A., Davidson, L., Moulton, L. A., Housman, M. L., et al. (2012). Serial analysis of the gut and respiratory microbiome in cystic fibrosis in infancy: interaction between intestinal and respiratory tracts and impact of nutritional exposures. Ausubel FM editor mBio. 3 (4), e00251–e00212. doi: 10.1128/mBio.00251-12
Marsland, B. J., Gollwitzer, E. S. (2014). Host-microorganism interactions in lung diseases. Nat. Rev. Immunol. 14 (12), 827–835. doi: 10.1038/nri3769
McKone, E. F., Emerson, S. S., Edwards, K. L., Aitken, M. L. (2003). Effect of genotype on phenotype and mortality in cystic fibrosis: a retrospective cohort study. Lancet 361 (9370), 1671–1676. doi: 10.1016/S0140-6736(03)13368-5
Meoli, A., Fainardi, V., Deolmi, M., Chiopris, G., Marinelli, F., Caminiti, C., et al. (2021). State of the art on approved cystic fibrosis transmembrane conductance regulator (CFTR) modulators and triple-combination therapy. Pharmaceuticals 14 (9), 928. doi: 10.3390/ph14090928
Middleton, P. G., Mall, M. A., Dřevínek, P., Lands, L. C., McKone, E. F., Polineni, D., et al. (2019). Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single phe508del allele. N Engl. J. Med. 381 (19), 1809–1819. doi: 10.1056/NEJMoa1908639
Moran Losada, P., Chouvarine, P., Dorda, M., Hedtfeld, S., Mielke, S., Schulz, A., et al. (2016). The cystic fibrosis lower airways microbial metagenome. ERJ Open Res. 2 (2), 00096–02015. doi: 10.1016/S1569-1993(16)30337-X
Nash, A. K., Auchtung, T. A., Wong, M. C., Smith, D. P., Gesell, J. R., Ross, M. C., et al. (2017). The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 5 (1), 153. doi: 10.1186/s40168-017-0373-4
Natalini, J. G., Singh, S., Segal, L. N. (2022). The dynamic lung microbiome in health and disease. Nat. Rev. Microbiol. 16, 1–14. doi: 10.1038/s41579-022-00821-x
Neerincx, A. H., Whiteson, K., Phan, J. L., Brinkman, P., Abdel-Aziz, M. I., Weersink, E. J. M., et al. (2021). Lumacaftor/ivacaftor changes the lung microbiome and metabolome in cystic fibrosis patients. ERJ Open Res. 7 (2), 00731–02020. doi: 10.1183/23120541.00731-2020
Nguyen, L. D. N., Viscogliosi, E., Delhaes, L. (2015). The lung mycobiome: an emerging field of the human respiratory microbiome. Front. Microbiol. 6, 89. doi: 10.3389/fmicb.2015.00089
Ooi, C. Y., Syed, S. A., Rossi, L., Garg, M., Needham, B., Avolio, J., et al. (2018). Impact of CFTR modulation with ivacaftor on gut microbiota and intestinal inflammation. Sci. Rep. 8 (1), 17834. doi: 10.1038/s41598-018-36364-6
Pallenberg, S. T., Pust, M. M., Rosenboom, I., Hansen, G., Wiehlmann, L., Dittrich, A. M., et al. (2022). Impact of elexacaftor/tezacaftor/ivacaftor therapy on the cystic fibrosis airway microbial metagenome. Goldberg JB editor Microbiol. Spectr. 10 (5), e01454–e01422. doi: 10.1128/spectrum.01454-22
Peleg, A. Y., Choo, J. M., Langan, K. M., Edgeworth, D., Keating, D., Wilson, J., et al. (2018). Antibiotic exposure and interpersonal variance mask the effect of ivacaftor on respiratory microbiota composition. J. Cyst Fibros 17 (1), 50–56. doi: 10.1016/j.jcf.2017.08.002
Pope, C., Vo, A., Hayden, H., Weiss, E., Durfey, S., McNamara, S., et al. (2021). Changes in fecal microbiota with CFTR modulator therapy: A pilot study. J. Cystic Fibrosis 20 (5), 742–746. doi: 10.1016/j.jcf.2020.12.002
Ramsey, B. W., Davies, J., McElvaney, N. G., Tullis, E., Bell, S. C., Dřevínek, P., et al. (2011). A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl. J. Med. 365 (18), 1663–1672. doi: 10.1056/NEJMoa1105185
Rodrigues, V. F., Elias-Oliveira, J., Pereira, ÍS, Pereira, J. A., Barbosa, S. C., MaChado, M. S. G., et al. (2022). Akkermansia muciniphila and gut immune system: A good friendship that attenuates inflammatory bowel disease, obesity, and diabetes. Front. Immunol. 13, 934695. doi: 10.3389/fimmu.2022.934695
Ronan, N. J., Einarsson, G. G., Deane, J., Fouhy, F., Rea, M., Hill, C., et al. (2022). Modulation, microbiota and inflammation in the adult CF gut: A prospective study. J. Cyst Fibros 21 (5), 837–843. doi: 10.1016/j.jcf.2022.06.002
Rowe, S. M., Daines, C., Ringshausen, F. C., Kerem, E., Wilson, J., Tullis, E., et al. (2017). Tezacaftor-ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl. J. Med. 377 (21), 2024–2035. doi: 10.1056/NEJMoa1709847
Rowe, S. M., Heltshe, S. L., Gonska, T., Donaldson, S. H., Borowitz, D., Gelfond, D., et al. (2014). Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am. J. Respir. Crit. Care Med. 190 (2), 175–184. doi: 10.1164/rccm.201404-0703OC
Schaupp, L., Addante, A., Völler, M., Fentker, K., Kuppe, A., Bardua, M., et al. (2023). Longitudinal effects of elexacaftor/tezacaftor/ivacaftor on sputum viscoelastic properties, airway infection and inflammation in patients with cystic fibrosis. Eur. Respir. J. 62 (2), 2202153. doi: 10.1183/13993003.02153-2022
Secunda, K. E., Guimbellot, J. S., Jovanovic, B., Heltshe, S. L., Sagel, S. D., Rowe, S. M., et al. (2020). Females with cystic fibrosis demonstrate a differential response profile to ivacaftor compared with males. Am. J. Respir. Crit. Care Med. 201 (8), 996–998. doi: 10.1164/rccm.201909-1845LE
Sokol, H., Leducq, V., Aschard, H., Pham, H. P., Jegou, S., Landman, C., et al. (2017). Fungal microbiota dysbiosis in IBD. Gut 66 (6), 1039–1048. doi: 10.1136/gutjnl-2015-310746
Sosinski, L. M., CM, H., Neugebauer, K. A., Ghuneim, L. A. J., Guzior, D. V., Castillo-Bahena, A., et al. (2022). A restructuring of microbiome niche space is associated with Elexacaftor-Tezacaftor-Ivacaftor therapy in the cystic fibrosis lung. J. Cystic Fibrosis 21 (6), 996–1005. doi: 10.1016/j.jcf.2021.11.003
Spor, A., Koren, O., Ley, R. (2011). Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 9 (4), 279–290. doi: 10.1038/nrmicro2540
Taylor-Cousar, J. L., Munck, A., McKone, E. F., van der Ent, C. K., Moeller, A., Simard, C., et al. (2017). Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for phe508del. N Engl. J. Med. 377 (21), 2013–2023. doi: 10.1056/NEJMoa1709846
Trompette, A., Gollwitzer, E. S., Yadava, K., Sichelstiel, A. K., Sprenger, N., Ngom-Bru, C., et al. (2014). Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 20 (2), 159–166. doi: 10.1038/nm.3444
Vaincre la Mucoviscidose (2022). Registre français de la mucoviscidose – Bilan des données 2021 (Paris). Available at: https://www.vaincrelamuco.org/sites/default/files/registre_francais_de_la_mucoviscidose_bilan_2021_0.pdf.
Wainwright, C. E., Elborn, J. S., Ramsey, B. W., Marigowda, G., Huang, X., Cipolli, M., et al. (2015). Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for phe508del CFTR. N Engl. J. Med. 373 (3), 220–231. doi: 10.1056/NEJMoa1409547
Willger, S. D., Grim, S. L., Dolben, E. L., Shipunova, A., Hampton, T. H., Morrison, H. G., et al. (2014). Characterization and quantification of the fungal microbiome in serial samples from individuals with cystic fibrosis. Microbiome 2, 40. doi: 10.1186/2049-2618-2-40
Yamada, A., Komaki, Y., Komaki, F., Micic, D., Zullow, S., Sakuraba, A. (2018). Risk of gastrointestinal cancers in patients with cystic fibrosis: a systematic review and meta-analysis. Lancet Oncol. 19 (6), 758–767. doi: 10.1016/S1470-2045(18)30188-8
Yi, B., Dalpke, A. H., Boutin, S. (2021). Changes in the cystic fibrosis airway microbiome in response to CFTR modulator therapy. Front. Cell Infect. Microbiol. 11, 548613. doi: 10.3389/fcimb.2021.548613
Zemanick, E. T., Taylor-Cousar, J. L., Davies, J., Gibson, R. L., Mall, M. A., McKone, E. F., et al. (2021). A Phase 3 Open-Label Study of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 through 11 Years of Age with Cystic Fibrosis and at Least One F508del Allele. Am. J. Respir. Crit. Care Med. 203 (12), 1522–1532. doi: 10.1164/rccm.202102-0509OC
Zhang, I., Pletcher, S. D., Goldberg, A. N., Barker, B. M., Cope, E. K. (2017). Fungal microbiota in chronic airway inflammatory disease and emerging relationships with the host immune response. Front. Microbiol. 8, 2477. doi: 10.3389/fmicb.2017.02477
Keywords: gut-lung axis, microbiota, mycobiota, cystic fibrosis, CFTR modulators
Citation: Lussac-Sorton F, Charpentier É, Imbert S, Lefranc M, Bui S, Fayon M, Berger P, Enaud R and Delhaes L (2023) The gut-lung axis in the CFTR modulator era. Front. Cell. Infect. Microbiol. 13:1271117. doi: 10.3389/fcimb.2023.1271117
Received: 01 August 2023; Accepted: 30 August 2023;
Published: 15 September 2023.
Edited by:
Suhana Chattopadhyay, College Park, United StatesReviewed by:
Vedita Anand Singh, The Scripps Research Institute, United StatesCopyright © 2023 Lussac-Sorton, Charpentier, Imbert, Lefranc, Bui, Fayon, Berger, Enaud and Delhaes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Florian Lussac-Sorton, Zmxvcmlhbi5sdXNzYWMtc29ydG9uQHUtYm9yZGVhdXguZnI=; Laurence Delhaes, bGF1cmVuY2UuZGVsaGFlc0B1LWJvcmRlYXV4LmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.