
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 14 July 2023
Sec. Clinical Microbiology
Volume 13 - 2023 | https://doi.org/10.3389/fcimb.2023.1208473
 Yingmiao Zhang1†
Yingmiao Zhang1† Yue Fan1†Yu Zhan1Hao Wang2Xun Li1Hui Wang1Tian Feng1Lifeng Shi1Jing Wang1
Yue Fan1†Yu Zhan1Hao Wang2Xun Li1Hui Wang1Tian Feng1Lifeng Shi1Jing Wang1 Hui Wang1
Hui Wang1 Zhongxin Lu1,3*
Zhongxin Lu1,3*Introduction: Pantoea anthophila (P. anthophila) is a Gram-negative bacterium initially isolated from Impatiens balsamina in India. P. anthophila has been characterized with low pathogenicity, and no human infections caused by this organism have been reported yet. We report the first case of urinary tract infection caused by P. anthophila in a 73-year-old man after bladder cancer surgery.
Methods: The bacterial isolate gained from urine was named UI705 and identified as P. anthophila by MALDI-TOF mass spectrometry. The genome sequencing and analysis were performed to further characterize the pathogenesis of the clinical isolate.
Result and discussion: To the best of our knowledge, this is the first report of human infection caused by P. anthophila in China. The draft genome sequence of P. anthophila UI705 provides a fundamental resource for subsequent investigation of its virulence factors, antibiotic resistance, host–pathogen interactions, and comparative genomics of genus Pantoea.
The genus Pantoea are Gram-negative, facultatively anaerobic, motile, and non-spore-forming bacteria that are generally isolated from agricultural or clinical sources, such as eucalyptus, Impatiens balsamina, maize, and human feces (Brady et al., 2008). At present, the genus comprises 18 species with validly published names in the List of Prokaryotic names with Standing in Nomenclature (LPSN) database (Meier-Kolthoff et al., 2022). Among those species, only a few species have been reported to be associated with human infectious diseases, including Pantoea agglomerans, Pantoea ananatis, and Pantoea dispersa (Cruz et al., 2007; Manoharan et al., 2012; Mehar et al., 2013). P. anthophila is a member of the genus Pantoea, which was originally isolated from I. balsamina in India in the 20th century. P. anthophila was once assigned to protein profile group VII in the “Erwinia herbicola–Enterobacter agglomerans complex” (Beji et al., 1988). Studies by Brady et al. placed this strain into a separate cluster and proposed its current name as P. anthophila by using multilocus sequence analyses (MLSAs), amplified fragment length polymorphism (AFLP) analysis, and DNA–DNA hybridization in 2009 (Brady et al., 2009). Upon searching in PubMed, only one study reported that P. anthophila is pathogenic to plants and naturally causes soft rot disease and cracking on Clausena lansium (wampee) in China (Zhou et al., 2015).
The study on pathogen genome is conducive to a comprehensive understanding of pathogenicity and molecular epidemiological characteristics of pathogens. However, there is currently a small amount of genomic information available on P. anthophila, mainly due to its rarity. He et al. have recently reported a complete genome sequence of P. anthophila CL1 that was initially isolated from symptomatic tissues of wampee, using both next- and third-generation sequencing technologies (He et al., 2022). The genome sequence of P. anthophila UI705 in this study represents the first genome of clinical isolate of this microorganism and may provide a better understanding of its pathogenicity in medical practice.
A 73-year-old male patient with frequent urination after bladder cancer surgery was admitted to our hospital on 4 July 2022. The patient underwent bladder tumor resection and bilateral ureteral stent placement for bladder tumor in the trigone 40 days ago, and postoperative pathology showed high-grade non-invasive urothelial carcinoma. The laboratory tests revealed increased neutrophils and fasting blood glucose, but decreased erythrocyte, hemoglobin, and albumin. Urinalysis revealed a WBC count of 523/μl (normal 0–6/μl), an erythrocyte count of 9/μl, urine protein of 1+, and urine glucose of 2+. After isolation of the purified bacterial strain, the patient was diagnosed with urinary tract infection. The details of the case report are presented in the Supplementary Material.
After admission, the clean-catch urine of the patient was collected prior to antibiotic therapy and plated onto Columbia blood agar and MacConkey agar plates (Guangzhou Dijing Microbial Technology Co., Ltd., Guangzhou, China) at 35°C in the presence of 5% CO2. After incubation overnight, bacterial colonies with homogeneous morphology were observed and counted on both agar plates, and the bacterial titer was higher than 105 colony-forming units per milliliter (CFUs/ml).
The isolated strain UI705 was identified on matrix-assisted laser desorption ionization/time of flight mass spectrometry (MALDI-TOF MS, Bruker Daltonik GmbH, Germany) platform by direct smear of fresh bacterial colony on the target plate in accordance with the manufacturer’s instructions. The acquired spectra data from tested bacteria were compared with the existing known spectrum by using the MALDI Biotyper 3.1 software (Bruker Daltonik GmbH, Germany).
All the reagents used for biochemical characterization of strain UI705 were purchased from Hangzhou Microbial Reagent, Co., Ltd (Hangzhou, China). The catalytic activity of catalase was detected using 3% (v/v) H2O2. The oxidase activity was measured by filter paper soaked with the reagent tetramethyl-p-phenylenediamine.
Enzymatic activity and acid production from various carbohydrates of the strain were determined by bacterial microbiochemical reaction tubes according to the manufacturer’s instructions. Antimicrobial susceptibility test (AST) was performed by the Kirby-Bauer disc diffusion method with antibiotics (Thermo Fisher Scientific, Waltham, MA, USA).
The genomic DNA of purified bacteria was extracted using the TIANamp Bacteria DNA Kit (TIANGEN Biotech, Co., Ltd, Beijing, China) according to the instructions of the manufacturer. 16S rRNA gene sequencing was performed with universal primers (27F: 5′;-AGTTTGATCMTGGCTCAG-3′;; 1492R: 5′;-GGTTACCTTGTTACGACTT-3′;). The amplification was performed on C1000 Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, USA) with the following procedure: denaturation at 95°C for 3 min, 30 cycles of denaturation at 95°C for 30 s, annealing at 58°C for 30 s and elongation at 72°C for 1 min, and a further 10 min of elongation at 72°C. The complete 16S rRNA sequence of the strain UI705 was analyzed with the EzBioCloud Database (Yoon et al., 2017a). A phylogenetic tree was constructed using the neighbor-joining method by MEGA software version 11 (Tamura et al., 2021).
The draft genome sequencing of the strain UI705 was performed using the Illumina HiSeq platform by generating paired-end libraries (PE 150 bp). Fragmented genomic DNA with an average size of 300 bp was selected for sequencing. The raw data of sequencing were evaluated by FastQC v0.11.2 and cut by Trimmomatic v0.36 (Bolger et al., 2014) to obtain relatively accurate and effective data. The filtered reads were assembled into contigs using SPAdes v3.5.0 (Bankevich et al., 2012), and GapFiller v1.11 (Boetzer and Pirovano, 2012) is used to fill the gap between contigs. The genetic elements were predicted by Prokka v1.10 (Seemann, 2014). The average nucleotide identity was calculated using the OrthoANIu algorithm (Yoon et al., 2017b). The DNA–DNA hybridization (DDH) value between UI705 and its related type strain was analyzed by Genome-to-Genome Distance Calculator 3.0 (Meier-Kolthoff et al., 2022). The prediction of antibiotic resistance, virulence factors, determinants of pathogen–host interaction, and carbohydrate-active enzymes was performed by means of the Comprehensive Antibiotic Resistance Database (CARD) (McArthur et al., 2013), the Virulence Factors of Pathogenic Bacteria (VFDB) (Chen et al., 2016), the Pathogen–Host Interactions (PHI) database (Urban et al., 2015), and the Carbohydrate-Active enZYmes (CAZy) Database (Lombard et al., 2014), respectively. The automatic annotation server of protein sequence such as NCBI non-redundant protein sequences (NR) (https://www.ncbi.nlm.nih.gov/), Protein family (PFAM) (Finn et al., 2016), Swiss-Prot (UniProt-Consortium, 2015), Clusters of Orthologous Groups of proteins (COG) (Tatusov et al., 2000), Conserved Domain Database (CDD) (Marchler-Bauer et al., 2013), and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000) was applied under the right conditions.
The urine sample developed bacterial colonies on Columbia blood agar after being cultured overnight. The colonies appeared beige to yellow, smooth and circular without apparent hemolytic zones and had diameters between 1 and 2 mm (Figure 1A). A spectrogram with protein molecular mass of the strain was acquired by MALDI-TOF MS (Figure 1B) and compared with that of known bacterial species in the database provided by the manufacturer. According to the spectra comparison, the strain UI705 was matched to P. anthophila DSM 23080T with a high confidence level (Figure 1C). Biochemical tests revealed that the strain UI705 is catalase-positive and oxidase-negative. β-galactosidase is produced. Indole, H2S, arginine dihydrolase, and urease are not produced. Acid is produced from glucose, mannose, arabinose, rhamnose, and xylose (Table 1). The AST via the Kirby–Bauer method showed that the strain is susceptible to almost all tested antibiotics except for ampicillin and cefazolin, which belong to penicillin and first-generation cephalosporin antibiotics, respectively (Table 2).
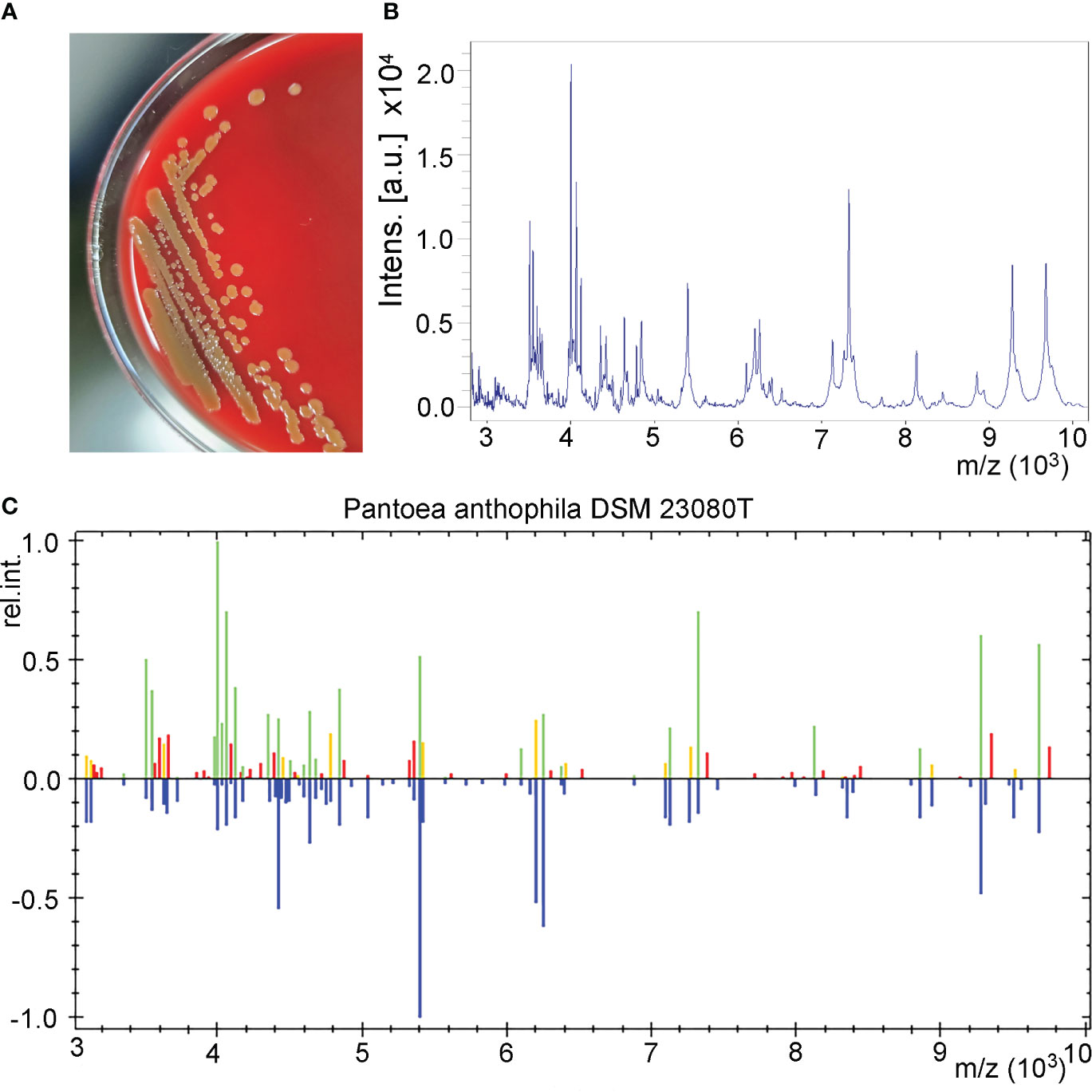
Figure 1 Isolation and identification of P. anthophila strain UI705. (A) Bacterial colonies on Columbia blood agar after being cultured at 37°C in the presence of 5% CO2 for 24 h. (B) The spectrogram with protein molecular mass of the strain acquired by MALDI-TOF MS. (C) Spectra comparison of the strain UI705 with that of known strains in a database matched to P. anthophila DSM 23080T with a high confidence level.
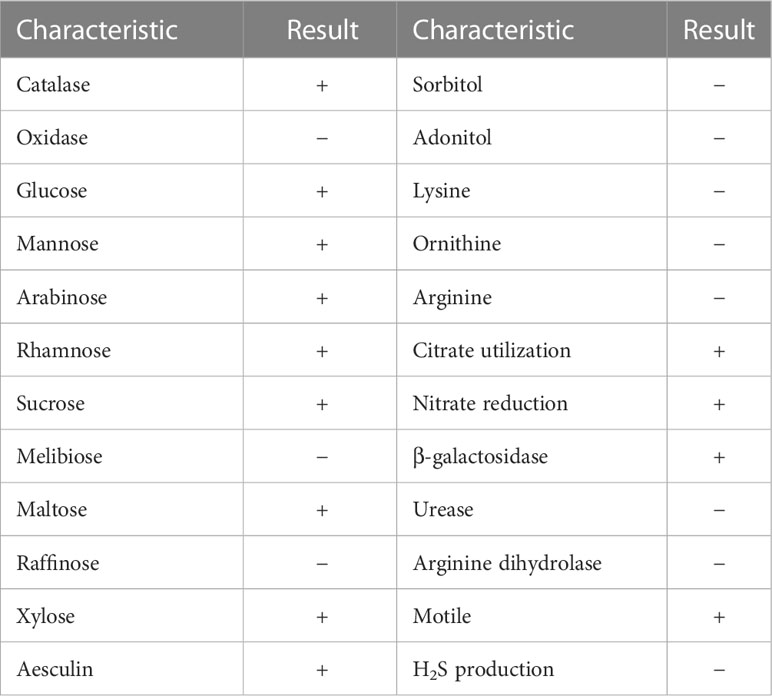
Table 1 Biochemical characteristics of strain P. anthophila UI705.

Table 2 Antimicrobial susceptibility of P. anthophila UI705.
To further investigate the phylogenetic features of the isolated strain UI705 in this study, 16S rRNA gene sequencing was performed. The complete 16S rRNA sequence of the strain UI705 was analyzed with the EzBioCloud Database (Yoon et al., 2017a). The strain UI705 exhibited highest (99.64%) 16S rRNA gene sequence similarity with the type strain of P. anthophila LMG 2558T (GenBank accession no. EF688010). A total of 1,387 contiguous nucleotides of the strain UI705 were determined and compared with that of P. anthophila LMG 2558T with a completeness of 98.5%. Among the 1,387 bases, there was only a five-base difference from strain LMG 2558T. The 16S rRNA sequencing results were submitted to GenBank (accession no. OQ704296). Multiple alignments with sequences of the related Pantoea and the calculations of the levels of sequence similarity were carried out using the MUSCLE algorithm (Edgar, 2004). A phylogenetic tree was constructed using the neighbor-joining method by MEGA software version 11 (Tamura et al., 2021). The topology of the phylogenetic tree was evaluated by using the bootstrap resampling method with 1,000 replicates. The phylogenetic tree showed that strain UI705 was clustered with the type strain LMG 2558T, and this cluster was strongly supported with a bootstrap value of 82% (Figure 2). The results of the comparative 16S rRNA gene sequence analysis demonstrated that the isolated strain UI705 belongs to the P. anthophila species.

Figure 2 Phylogenetic tree based on the 16S rRNA gene sequences showing the relationship of isolated strain UI705 (black arrow) and members within genus Pantoea. The tree was reconstructed by the neighbor-joining method, and Enterobacter huaxiensis 90008T (MK049964) was used as an outgroup. Bootstrap values (>50%) based on 1,000 replicates are shown at branch nodes. T, type strain.
The draft genome sequence of P. anthophila UI705 revealed 4,542,010 bp in size and assembled into 15 contigs, with an N50 value of 564,400 bp and 243.23 × coverage. The GC content of the assembled genome is 56.95% (Figure 3), which is consistent with that of other reports available. Automatic annotation with the NCBI PGAP revealed 4,288 protein coding genes. The details of gene element prediction were listed in Table 3. Genome-based taxonomy classification using OrthoANIu algorithm validated that UI705 belongs to P. anthophila as it revealed a high ANI value of 99.88% between UI705 and the P. anthophila type strain LMG 2558T. Moreover, the DDH value between UI705 and its related type strain was analyzed by GGDC 3.0 with an estimated value of 99.30% (Formula 2). The gene function annotation of the UI705 was performed in a database that is based on NCBI Blast+ and KEGG Automatic Annotation Server, including CDD, COG, NR, and PFAM, and the number of annotated genes is the highest in the NR database (Supplement Figure S1). The assembled sequences of P. anthophila UI705 were subjected to GO for further annotation and description of the properties of genes and gene products that were divided into molecular function, cellular component, and biological process (Figure 4A). The result from KEGG revealed 100 predictive genes involved in human diseases, mainly those related to infectious diseases and drug resistance (Figure 4B), indicating that P. anthophila may emerge as a human pathogen.
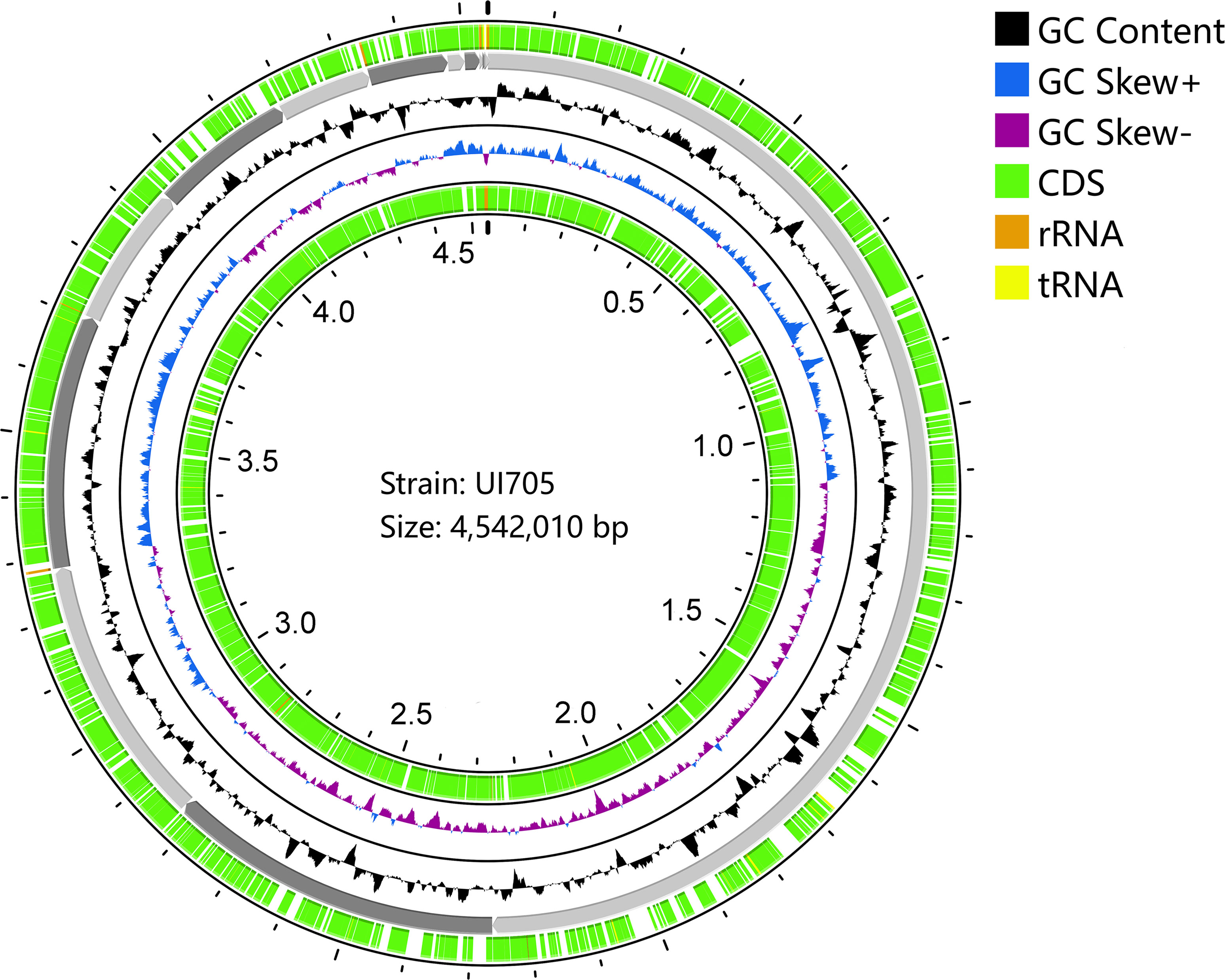
Figure 3 Circular display of the draft genome of P. anthophila UI705. The circle from outside to inside represent the following: coding sequences (CDS) on the forward strand (green), the contigs (gray), GC content (black), GC-skew (blue and purple indicate values higher and lower than the mean value, respectively), and CDS on the reverse strand (green). The tRNA (yellow) and rRNA (brown) on both forward and reverse strands are indicated.
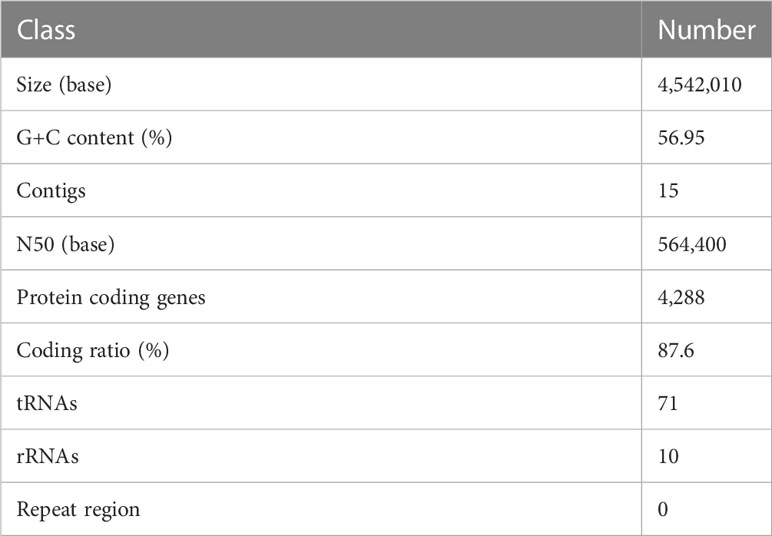
Table 3 Genome features of P. anthophila UI705.
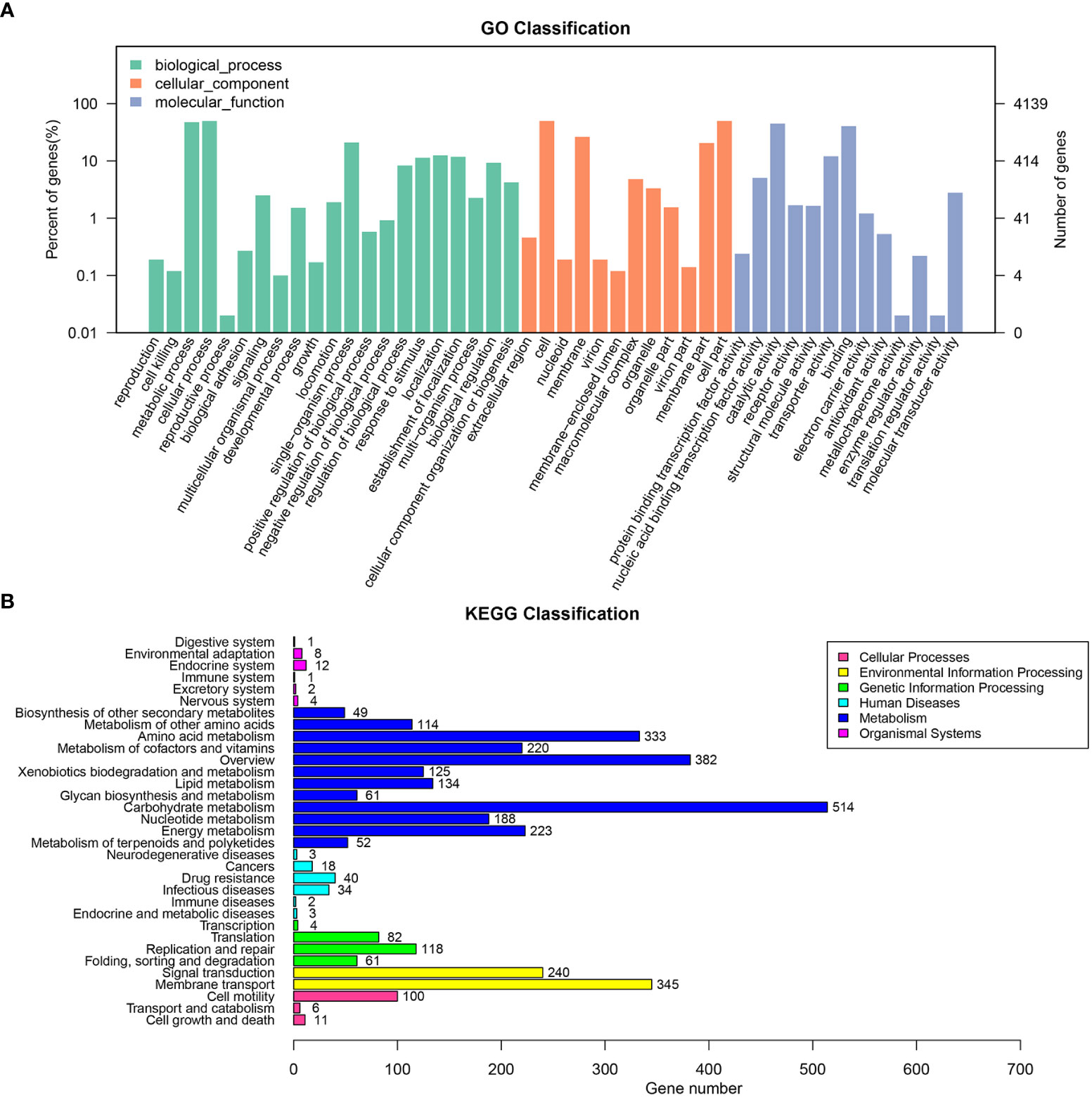
Figure 4 The gene function annotations of the P. anthophila strain UI705. (A) GO classification describes the properties of genes and gene products as three terms: molecular function (light blue), cellular component (brownish red), and biological process (cyan). The left and right axis indicate percentage of genes and gene numbers, respectively. (B) KEGG classification reveals the predicted genes involved in metabolic pathways and human diseases. The numbers on the right of the bar graphs represent predicted genes that are related to correspondent activities.
The antibiotic resistance of the isolate U1705 was predicted by the CARD and 81 proteins were predicted, such as those related to antibiotic efflux, antibiotic inactivation, and reduced permeability to antibiotic (Figure 5A). The antibiotic resistance ontology adeF, rsmA, and CRP were annotated with high percentage identity of matching region (61.44%–89.66%), and the length ratio of reference sequence ranges from 99.24% to 100%. These AROs belong to the resistance-nodulation-cell division (RND) antibiotic efflux pump family and mediate antibiotic resistance in plenty of Gram-negative bacteria (Blair and Piddock, 2009). In addition, a total of 126 virulence factor terms were predicted through the core dataset of the VFDB, which contains 368 predicted proteins (Figure 5B). The database predicted the most immune modulation-related proteins, followed by an effector delivery system that includes Type III and VI Secretion Systems (T3SS and T6SS). The T3SS is a needle-like protein complex found in several species of plant or human pathogenic organisms within genus Pantoea, contributing to the pathogenicity to their hosts (Correa et al., 2012; Kirzinger et al., 2015). Based on PHI database, the U1705 exhibited pathogenicity to both animals and plants, with a total of 198 PHI-related proteins predicted (Figure 5C). There are 11 annotated genes that are related to reduced virulence or loss of pathogenicity in human host. The carbohydrate active enzyme annotation was carried out by CAZy with e-value < 1e−5 and glycosyl transferases remain the highest (Figure 5D). The lipopolysaccharide core heptosyltransferase RfaQ and RfaG, and capsular polysaccharide synthesis protein that belong to glycosyl transferases play important roles in the pathogenicity of many bacteria (Aguiniga et al., 2016; St John et al., 2023).
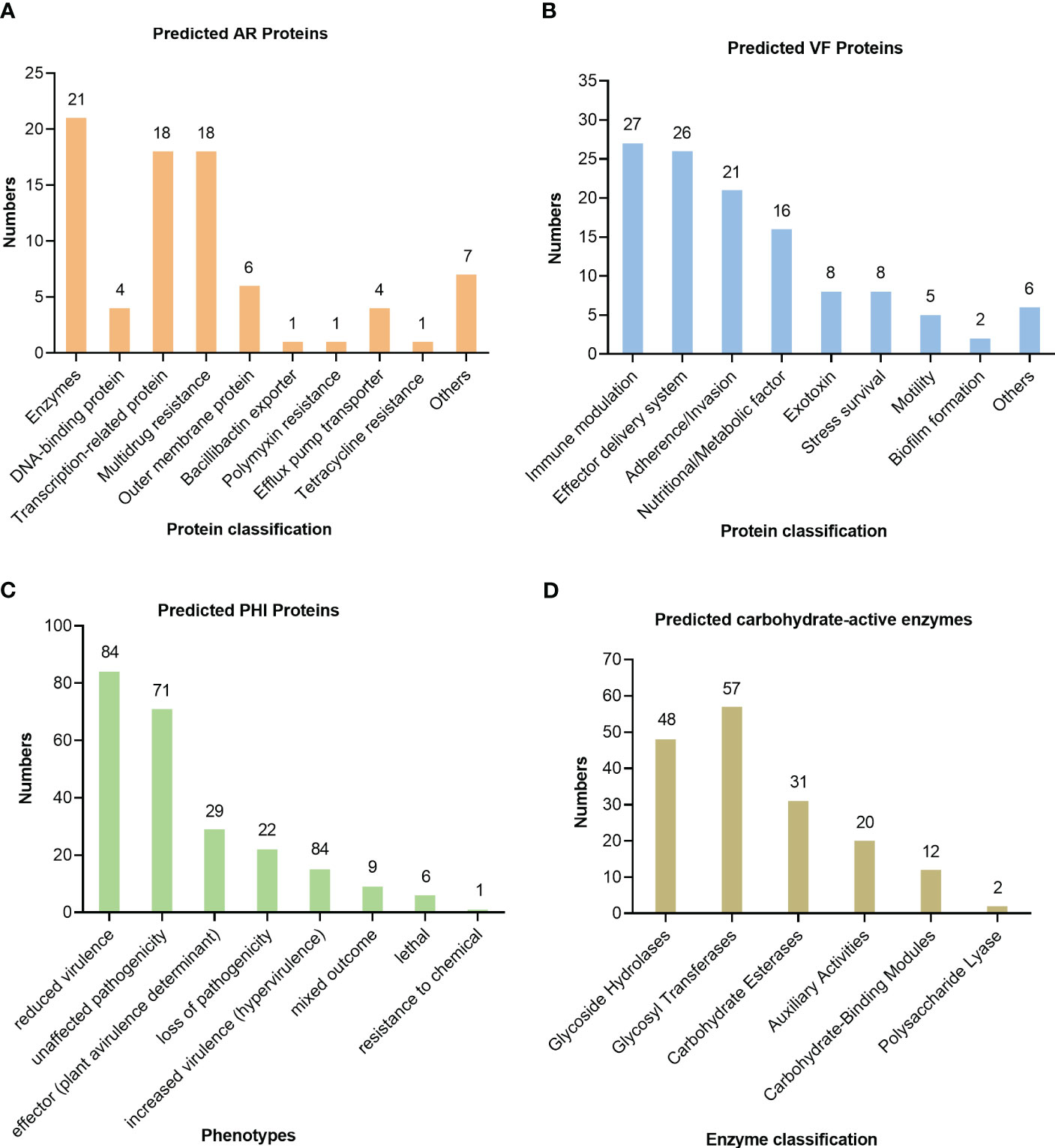
Figure 5 Prediction of pathogenicity of the strain UI705 through the database. (A) The antibiotic resistance was predicted by CARD. (B) The virulence factors were predicted through the core dataset of the VFDB. (C) The pathogen–host interactions were predicted by the PHI database. (D) The carbohydrate active enzyme annotation was carried out through the CAZy database. The numbers on the top of the bar graphs represent the predicted proteins in each channel.
Pantoea is a genus of Gram-negative bacteria derived from Erwiniaceae family that was recently proposed as a novel family on the basis of comprehensive and genome-scale taxonomic analysis by Adeolu et al. (2016). The majority of strains within the genus are isolated from plants and the environment, and it is generally considered as a plant pathogen. Among the members of the genus Pantoea, only a limited number of species are reported to be associated with human infectious diseases. Currently, the most common causative agent of opportunistic human infections within the genus Pantoea is P. agglomerans, which can cause infections in different parts of the body, such as bloodstream infections, arthritis or synovitis, periostitis, and endocarditis (Dutkiewicz et al., 2016). Moreover, P. agglomerans has caused several nosocomial outbreaks of septicemia since the great epidemic occurred in 1970 and resulted in 152 cases by this organism alone (Dutkiewicz et al., 2016). Infections caused by P. agglomerans are mainly from wound infection with plant material and hospital-acquired infection in immunocompromised individuals. Recently, a couple of studies report that P. dispersa can cause bacteremia and neonatal sepsis, especially in immunocompromised patients (Mehar et al., 2013; Hagiya and Otsuka, 2014; Asai et al., 2019). Rare cases of bacteriuria are described in P. ananatis and P. stewartii (De Baere et al., 2004; Cobo et al., 2022). An increasing number of Pantoea species related to human infections are described, but the pathogenic mechanisms underlying these species remain unclear.
P. anthophila has been characterized with non-pathogenic organism and initially isolated from I. balsamina and Tagetes erecta, showing no external sign of infection or any negative effect on its hosts (Brady et al., 2009). Wan et al. reported the draft genome sequence of P. anthophila strain 11-2 isolated from hypersaline water from the lake on Laysan, Hawaii and numerous virulence-related components were identified based on bioinformatic analysis (Wan et al., 2015). Only one study by Zhou et al. reported for the first time that P. anthophila naturally caused soft rot disease and cracking on C. lansium (Zhou et al., 2015), indicating that P. anthophila is pathogenic to plant.
We encountered the first case of urinary tract infection caused by P. anthophila in a 73-year-old man with bladder cancer. The patient denied having been in contact with or being injured by plants or other materials, so the exact source of the isolate from our patient remains unknown. The elderly patient has undergone chemotherapy after surgical treatment of bladder cancer and belongs to the immunocompromised population, which may contribute to the infection of P. anthophila. Infections by Pantoea species may not present with the conventional symptoms such as fever and leukocytosis, which mainly result from the low virulence of the bacteria. The main symptom of the patient in this study is frequent urination, without fever or other clinical manifestations, and the leukocyte count remained normal with a high percentage of neutrophils (85.6%). Therefore, it is difficult to determine the presence of infection through hematological indicators, and the isolation of pathogenic bacteria from urine confirms the existence of infection. The isolate was susceptible to all antibiotics tested, except for ampicillin and cefazolin, which is consistent with the reported drug sensitivity of P. stewartia (Cobo et al., 2022). The patient’s condition improved rapidly due to appropriate antibiotic treatment.
The isolated strain was identified as P. anthophila by MALDI-TOF MS, an emerging technology developed in recent years for rapid identification of bacterial pathogens. MALDI-TOF MS can accurately identify commonly isolated micro-organisms due to high coverage rate in the comparison database, but it has limited ability in identification of novel or rare pathogenic species (Ha et al., 2019). Mass spectrometry may be misidentified in the identification of some Pantoea species, such as P. dispersa, which was initially mistaken as Klebsiella ozaenae (Asai et al., 2019). 16S rRNA gene sequencing has been frequently used for molecular classification and phylogeny of bacterial isolates. The 16S rRNA sequence of strain UI705 was found to have a five-base difference from the type strain LMG 2588T within a total of 1,387 nucleotides. A phylogenetic tree also revealed a cluster within the previously reported P. anthophila strains and our isolate UI705, indicating that our strain is most closely related to the type strain. Owing to the diversity of microbial species, multi-dimensional analysis, including MALDI-TOF MS and 16S rRNA gene sequencing, will be valuable in accurately distinguishing the large amounts of species from their closest phylogenetic neighbors.
The draft genome sequencing of the clinical isolate UI705 reveals plenty of virulence factors that are involved in human diseases, such as adherence/invasion, toxin, and stress survival. The chaperone protein fimbriae, Myf/pH6 antigen, outer membrane protein OmpU, and multivalent adhesion molecule MAM7 are important adhesins or invasins that mediate the initiation of host–pathogen interactions (Krachler et al., 2011; Liu et al., 2015; Pakharukova et al., 2016). Moreover, there are a number of predicted proteins related to host–pathogen interactions, such as TyrR, yfeAB, Crp, and RelA, and most of the genes are annotated from the reservoirs of Yersinia pestis and Vibrio cholerae. These results indicate that P. anthophila is a potential pathogen in both animal and plant hosts, and further experimental investigations are necessary to determine its pathogenicity. In this case, the P. anthophila caused urinary tract infection, which was cured by appropriate treatment with fosfomycin. The antibiotic resistance of the isolate UI705 was characterized by analysis through the CARD database based on the genome with kinds of multidrug resistance proteins. However, the AST results of this study show that the UI705 is only resistant to first-generation cephalosporin antibiotics, which may be due to the poor concordance between bacterial resistance genotypes and phenotypes.
In conclusion, we report the first case of urinary tract infection caused by P. anthophila in an elder man with bladder cancer. The clinical isolate was identified and analyzed by means of MALDI-TOF MS, 16S rRNA gene sequencing, and genome sequence. It should be noted that P. anthophila may lead to asymptomatic infection in an immunocompromised patient, which deserves the attention of clinicians. Although the pathogenicity of P. anthophila is unclear, the present case may indicate an emerging pathogen in medical practice. More cases of infections by this organism should be collected and integrated to explore the potential pathogenicity as well as epidemiology of P. anthophila infections.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by Medical Ethics Committee of the Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology. The patients/participants provided their written informed consent to participate in this study.
YZhang, YF, and ZL designed the research. YZhang, YF, YZhan, and XL performed the experiments. HaW, TF, LS, HaW and JW analyzed the data. YZhang, YF, and HuW wrote the initial draft of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by a grant from the Wuhan Municipal Health Commission (Project no. WX21Q42) and by one local grant from The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology to YMZ.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1208473/full#supplementary-material
Adeolu, M., Alnajar, S., Naushad, S., Gupta, R. S. (2016). Genome-based phylogeny and taxonomy of the 'Enterobacteriales': proposal for enterobacterales ord. nov. divided into the families enterobacteriaceae, erwiniaceae fam. nov., pectobacteriaceae fam. nov., yersiniaceae fam. nov., hafniaceae fam. nov., morganellaceae fam. nov., and budviciaceae fam. nov. Int. J. Syst. Evol. Microbiol. 66 (12), 5575–5599. doi: 10.1099/ijsem.0.001485
Aguiniga, L. M., Yaggie, R. E., Schaeffer, A. J., Klumpp, D. J. (2016). Lipopolysaccharide domains modulate urovirulence. Infect. Immun. 84 (11), 3131–3140. doi: 10.1128/iai.00315-16
Asai, N., Koizumi, Y., Yamada, A., Sakanashi, D., Watanabe, H., Kato, H., et al. (2019). Pantoea dispersa bacteremia in an immunocompetent patient: a case report and review of the literature. J. Med. Case Rep. 13 (1), 33. doi: 10.1186/s13256-019-1969-z
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19 (5), 455–477. doi: 10.1089/cmb.2012.0021
Beji, A., Mergaert, J., Gavini, F., Izard, D., Kersters, K., Leclerc, H., et al. (1988). Subjective synonymy of Erwinia herbicola, Erwinia milletiae, and Enterobacter agglomerans and redefinition of the taxon by genotypic and phenotypic data. Int. J. Syst. Evol. Microbiol. 38, (1), 77–88. doi: 10.1099/00207713-38-1-77
Blair, J. M., Piddock, L. J. (2009). Structure, function and inhibition of RND efflux pumps in gram-negative bacteria: an update. Curr. Opin. Microbiol. 12 (5), 512–519. doi: 10.1016/j.mib.2009.07.003
Boetzer, M., Pirovano, W. (2012). Toward almost closed genomes with GapFiller. Genome Biol. 13 (6), R56. doi: 10.1186/gb-2012-13-6-r56
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Brady, C., Cleenwerck, I., Venter, S., Vancanneyt, M., Swings, J., Coutinho, T. (2008). Phylogeny and identification of pantoea species associated with plants, humans and the natural environment based on multilocus sequence analysis (MLSA). Syst. Appl. Microbiol. 31 (6-8), 447–460. doi: 10.1016/j.syapm.2008.09.004
Brady, C. L., Venter, S. N., Cleenwerck, I., Engelbeen, K., Vancanneyt, M., Swings, J., et al. (2009). Pantoea vagans sp. nov., pantoea eucalypti sp. nov., Pantoea deleyi sp. nov. and Pantoea anthophila sp. nov. Int. J. Syst. Evol. Microbiol. 59 (Pt 9), 2339–2345. doi: 10.1099/ijs.0.009241-0
Chen, L., Zheng, D., Liu, B., Yang, J., Jin, Q. (2016). VFDB 2016: hierarchical and refined dataset for big data analysis–10 years on. Nucleic Acids Res. 44 (D1), D694–D697. doi: 10.1093/nar/gkv1239
Cobo, F., Gonzalez, A., Perez-Carrasco, V., Garcia-Salcedo, J. A. (2022). Pantoea stewartii: a new pathogen as a cause of bacteremia? Enferm Infecc Microbiol. Clin. (Engl Ed) 40 (5), 278–280. doi: 10.1016/j.eimce.2021.03.005
Correa, V. R., Majerczak, D. R., Ammar el, D., Merighi, M., Pratt, R. C., Hogenhout, S. A., et al. (2012). The bacterium Pantoea stewartii uses two different type III secretion systems to colonize its plant host and insect vector. Appl. Environ. Microbiol. 78 (17), 6327–6336. doi: 10.1128/aem.00892-12
Cruz, A. T., Cazacu, A. C., Allen, C. H. (2007). Pantoea agglomerans, a plant pathogen causing human disease. J. Clin. Microbiol. 45 (6), 1989–1992. doi: 10.1128/JCM.00632-07
De Baere, T., Verhelst, R., Labit, C., Verschraegen, G., Wauters, G., Claeys, G., et al. (2004). Bacteremic infection with Pantoea ananatis. J. Clin. Microbiol. 42 (9), 4393–4395. doi: 10.1128/JCM.42.9.4393-4395.2004
Dutkiewicz, J., Mackiewicz, B., Kinga Lemieszek, M., Golec, M., Milanowski, J. (2016). Pantoea agglomerans: a mysterious bacterium of evil and good. part III. deleterious effects: infections of humans, animals and plants. Ann. Agric. Environ. Med. 23 (2), 197–205. doi: 10.5604/12321966.1203878
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 5, 113. doi: 10.1186/1471-2105-5-113
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44 (D1), D279–D285. doi: 10.1093/nar/gkv1344
Ha, S. M., Kim, C. K., Roh, J., Byun, J. H., Yang, S. J., Choi, S. B., et al. (2019). Application of the whole genome-based bacterial identification system, TrueBac ID, using clinical isolates that were not identified with three matrix-assisted laser Desorption/Ionization time-of-Flight mass spectrometry (MALDI-TOF MS) systems. Ann. Lab. Med. 39 (6), 530–536. doi: 10.3343/alm.2019.39.6.530
Hagiya, H., Otsuka, F. (2014). Pantoea dispersa bacteremia caused by central line-associated bloodstream infection. Braz. J. Infect. Dis. 18 (6), 696–697. doi: 10.1016/j.bjid.2014.06.006
He, Q., Li, J., Liu, Q., Li, Y., Liao, X., Zhou, J., et al. (2022). Complete genome sequence resource of Pantoea anthophila CL1 causing soft rot disease in clausena lansium (Wampee) in China. Plant Dis. 107 (5), 1613–1616. doi: 10.1094/pdis-10-22-2504-a
Kanehisa, M., Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30. doi: 10.1093/nar/28.1.27
Kirzinger, M. W., Butz, C. J., Stavrinides, J. (2015). Inheritance of Pantoea type III secretion systems through both vertical and horizontal transfer. Mol. Genet. Genomics 290 (6), 2075–2088. doi: 10.1007/s00438-015-1062-2
Krachler, A. M., Ham, H., Orth, K. (2011). Outer membrane adhesion factor multivalent adhesion molecule 7 initiates host cell binding during infection by gram-negative pathogens. Proc. Natl. Acad. Sci. U.S.A. 108 (28), 11614–11619. doi: 10.1073/pnas.1102360108
Liu, X., Gao, H., Xiao, N., Liu, Y., Li, J., Li, L. (2015). Outer membrane protein U (OmpU) mediates adhesion of Vibrio mimicus to host cells via two novel n-terminal motifs. PloS One 10 (3), e0119026. doi: 10.1371/journal.pone.0119026
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42 (Database issue), D490–D495. doi: 10.1093/nar/gkt1178
Manoharan, G., Lalitha, P., Jeganathan, L. P., Dsilva, S. S., Prajna, N. V. (2012). Pantoea ananatis as a cause of corneal infiltrate after rice husk injury. J. Clin. Microbiol. 50 (6), 2163–2164. doi: 10.1128/JCM.06743-11
Marchler-Bauer, A., Zheng, C., Chitsaz, F., Derbyshire, M. K., Geer, L. Y., Geer, R. C., et al. (2013). CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 41 (Database issue), D348–D352. doi: 10.1093/nar/gks1243
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57 (7), 3348–3357. doi: 10.1128/aac.00419-13
Mehar, V., Yadav, D., Sanghvi, J., Gupta, N., Singh, K. (2013). Pantoea dispersa: an unusual cause of neonatal sepsis. Braz. J. Infect. Dis. 17 (6), 726–728. doi: 10.1016/j.bjid.2013.05.013
Meier-Kolthoff, J. P., Carbasse, J. S., Peinado-Olarte, R. L., Göker, M. (2022). TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 50 (D1), D801–d807. doi: 10.1093/nar/gkab902
Pakharukova, N., Roy, S., Tuittila, M., Rahman, M. M., Paavilainen, S., Ingars, A. K., et al. (2016). Structural basis for myf and psa fimbriae-mediated tropism of pathogenic strains of Yersinia for host tissues. Mol. Microbiol. 102 (4), 593–610. doi: 10.1111/mmi.13481
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30 (14), 2068–2069. doi: 10.1093/bioinformatics/btu153
St John, A., Perault, A. I., Giacometti, S. I., Sommerfield, A. G., DuMont, A. L., Lacey, K. A., et al. (2023). Capsular polysaccharide is essential for the virulence of the antimicrobial-resistant pathogen Enterobacter hormaechei. mBio 14 (2), e0259022. doi: 10.1128/mbio.02590-22
Tamura, K., Stecher, G., Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38 (7), 3022–3027. doi: 10.1093/molbev/msab120
Tatusov, R. L., Galperin, M. Y., Natale, D. A., Koonin, E. V. (2000). The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28 (1), 33–36. doi: 10.1093/nar/28.1.33
UniProt-Consortium (2015). UniProt: a hub for protein information. Nucleic Acids Res. 43 (Database issue), D204–D212. doi: 10.1093/nar/gku989
Urban, M., Pant, R., Raghunath, A., Irvine, A. G., Pedro, H., Hammond-Kosack, K. E. (2015). The pathogen-host interactions database (PHI-base): additions and future developments. Nucleic Acids Res. 43 (Database issue), D645–D655. doi: 10.1093/nar/gku1165
Wan, X., Hou, S., Phan, N., Malone Moss, J. S., Donachie, S. P., Alam, M. (2015). Draft genome sequence of Pantoea anthophila strain 11-2 from hypersaline lake laysan, Hawaii. Genome Announc. 3 (3), e00321-15. doi: 10.1128/genomeA.00321-15
Yoon, S. H., Ha, S. M., Kwon, S., Lim, J., Kim, Y., Seo, H., et al. (2017a). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67 (5), 1613–1617. doi: 10.1099/ijsem.0.001755
Yoon, S. H., Ha, S. M., Lim, J., Kwon, S., Chun, J. (2017b). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110 (10), 1281–1286. doi: 10.1007/s10482-017-0844-4
Keywords: Pantoea anthophila, urinary tract infection, 16S rRNA, MALDI-TOF MS, whole genome sequencing (WGS)
Citation: Zhang Y, Fan Y, Zhan Y, Wang H, Li X, Wang H, Feng T, Shi L, Wang J, Wang H and Lu Z (2023) Genomic characterization of Pantoea anthophila strain UI705 causing urinary tract infections in China. Front. Cell. Infect. Microbiol. 13:1208473. doi: 10.3389/fcimb.2023.1208473
Received: 19 April 2023; Accepted: 26 June 2023;
Published: 14 July 2023.
Edited by:
Michael Marceau, Université Lille Nord de France, FranceReviewed by:
Grzegorz Czerwonka, Jan Kochanowski University, PolandCopyright © 2023 Zhang, Fan, Zhan, Wang, Li, Wang, Feng, Shi, Wang, Wang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongxin Lu, bHV6aG9uZ3hpbkB6eGhvc3BpdGFsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.