
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 21 June 2023
Sec. Clinical Microbiology
Volume 13 - 2023 | https://doi.org/10.3389/fcimb.2023.1146571
This article is part of the Research Topic Finding Potential Approaches for Tuberculosis Prevention and Treatment View all 9 articles
 Alex Kayongo1,2,3
Alex Kayongo1,2,3 Brian Nyiro1,2
Brian Nyiro1,2 Trishul Siddharthan4Bruce Kirenga3William Checkley5,6Moses Lutaakome Joloba3
Trishul Siddharthan4Bruce Kirenga3William Checkley5,6Moses Lutaakome Joloba3 Jerrold Ellner1
Jerrold Ellner1 Padmini Salgame1*
Padmini Salgame1*Pulmonary tuberculosis is increasingly recognized as a risk factor for COPD. Severe lung function impairment has been reported in post-TB patients. Despite increasing evidence to support the association between TB and COPD, only a few studies describe the immunological basis of COPD among TB patients following successful treatment completion. In this review, we draw on well-elaborated Mycobacterium tuberculosis-induced immune mechanisms in the lungs to highlight shared mechanisms for COPD pathogenesis in the setting of tuberculosis disease. We further examine how such mechanisms could be exploited to guide COPD therapeutics.
Chronic obstructive pulmonary disease (COPD) refers to chronic lung diseases characterized by slowly progressive irreversible airflow obstruction. Individuals diagnosed with COPD have varying degrees of chronic bronchitis, small airway obstruction, and emphysema (Cruz et al., 2007; Joint United Nations Programme on HA, 2011; Organization WH, 2015; Venkatesan, 2022). COPD is the third leading cause of death worldwide (Salvi, 2015; Venkatesan, 2022), with over 65 million people having moderate to severe COPD (Organization WH, 2015). It is one of the most common non-communicable diseases (NCDs), affecting over 329 million (Salvi, 2015). In 2012, COPD contributed to 6% of all total deaths globally, with more than 90% of mortalities occurring in low- and middle-income countries (LMICs) where effective strategies for prevention and control are not consistently implemented or accessible (Cruz et al., 2007). The prevalence of COPD, defined by spirometry, is 11.7% worldwide, and mortality shows an increasing trend (Adeloye et al., 2015).
Globally, over ten million people fell ill with TB disease in 2021, with over 1.4 million TB-related mortalities among HIV-negative individuals (Bagcchi, 2023). Men account for more TB cases than women (Organization WH, 2020; Bagcchi, 2023). Thirty high-TB-burden countries account for almost 90% of those who fall sick with the disease annually, with South-East Asia and Africa contributing to the most significant TB burden (Harding, 2020). Although cigarette smoking, exposure to pollutants, and HIV infection are predominant risk factors for COPD, pulmonary tuberculosis remains an under-recognized risk factor for developing COPD (Chakrabarti et al., 2007; Caballero et al., 2008; Allwood et al., 2014; Pefura-Yone et al., 2014; Byrne et al., 2015; Ngahane et al., 2016; Sarkar et al., 2017; Siddharthan et al., 2019; Kayongo et al., 2020; Byanova et al., 2021; Kamenar et al., 2021). Respiratory function is impaired in TB-induced COPD, characterized by significantly reduced forced vital capacity (FVC) and post-bronchodilator expiratory volume in 1 second (FEV1) compared to those with smoke-induced COPD (Anno and Tomashefski, 1955; Lee and Chang, 2003; Kamenar et al., 2021). Furthermore, the post-bronchodilator response is significantly reduced in tuberculosis-induced COPD compared to smoke-induced COPD, indicating the irreversible nature of airflow obstruction (Lee and Chang, 2003; Menezes et al., 2007). Tuberculosis-induced COPD risk is higher in males than females, with adjusted odds ratios of 4.0 and 1.7, respectively (Menezes et al., 2007). History of prior tuberculosis has been strongly associated with severe forms of COPD (Menezes et al., 2007; Kamenar et al., 2021). According to the Burden of Obstructive Lung Diseases (BOLD) study, a history of tuberculosis increases the risk of developing airflow obstruction in later life with an adjusted odds ratio of 2.5 (Amaral et al., 2015). The frequency and severity of airflow obstruction in pulmonary tuberculosis positively correlate with the number of episodes of tuberculosis (Hnizdo et al., 2000). Structural damage of the lungs increases with an increasing number of tuberculosis episodes and persists in many patients despite anti‐tuberculosis treatment (Plit et al., 1998; Hnizdo et al., 2000).
Several predictors of COPD severity among tuberculosis patients have been reported, including smear‐positive disease, extensive pulmonary involvement before anti‐tuberculosis treatment, reduced radiographic improvement post‐treatment, and delay in initiating tuberculosis treatment (Chung et al., 2011). These factors imply that host-tuberculosis immune interactions in the lung microenvironment during active tuberculosis disease predominantly drive COPD pathogenesis. In this Review, we draw on well-described tuberculosis immune responses in the lung microenvironment to discuss immunological processes that could underlie the pathogenesis of COPD in the setting of tuberculosis disease (Figure 1). We further examine the extent to which such immunological processes could be exploited to guide tuberculosis-associated COPD therapeutics (Figure 2).
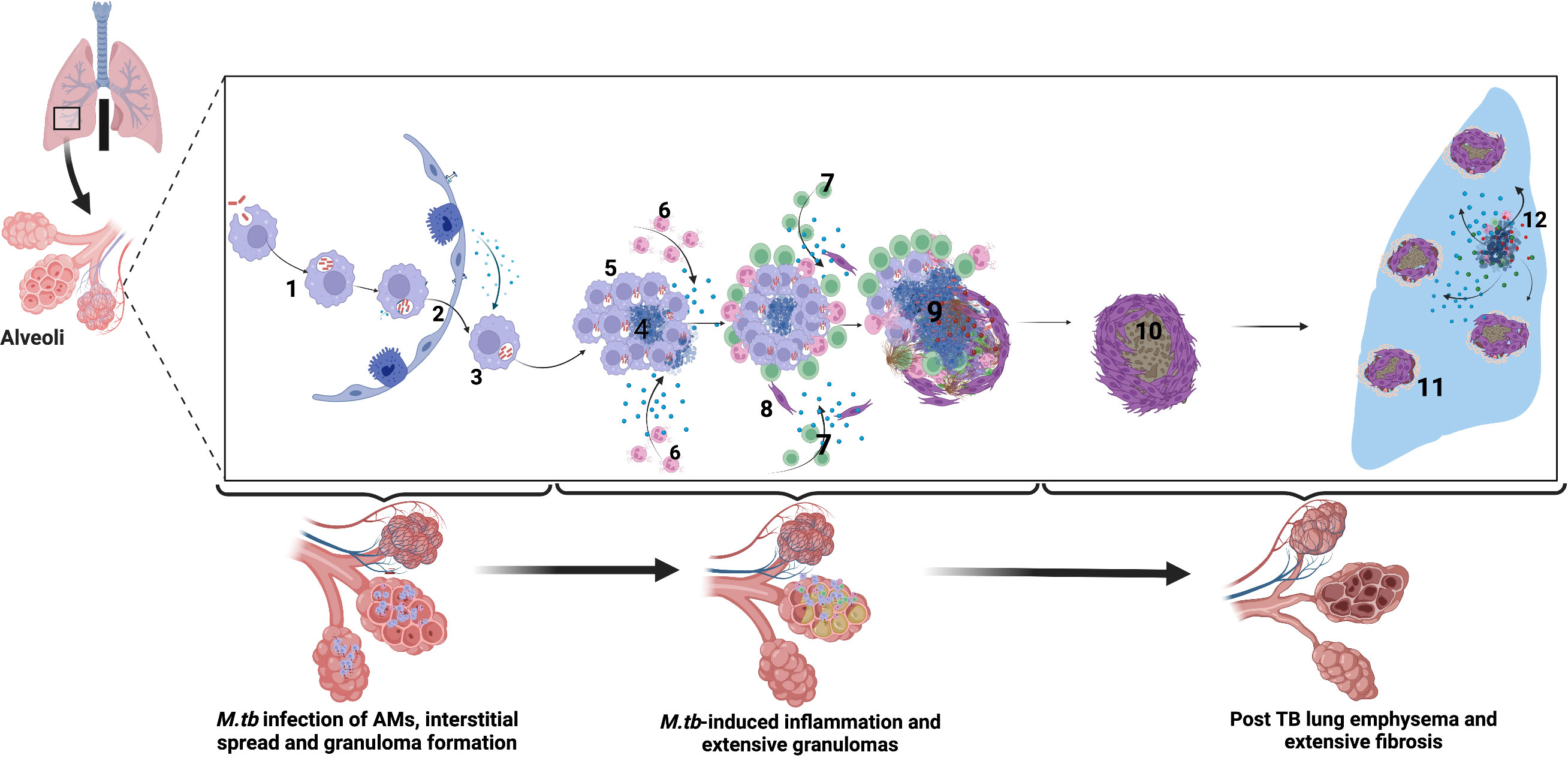
Figure 1 Immune-mediated mechanisms of TB-associated COPD. Alveolar macrophages engulf M.tb in the alveolar space. 2. Infected alveolar macrophages migrate from the alveolar space into the interstitium in an IL-1R-dependent manner. 3. M.tb replicate within alveolar macrophages. 4. M.tb induce infected macrophage apoptosis and expression of host lytic proteins in an ESX-1-dependent manner. 5. Newly recruited alveolar macrophages engulf infected cell debris. 6. Lung infiltrating neutrophils move by chemotaxis towards the growing granuloma, engulfing dying infected cells and killing bacteria through NETosis and release of lytic enzymes. 7. M.tb-specific T cells arrive at the granuloma and produce IFN-γ to enhance the microbicidal activity of alveolar macrophages. However, activated T cells are walled off from accessing the inner core of the granuloma, and their effect is dampened by the cytokine TGFβ. 8. Alveolar macrophage necrosis leads to granuloma rupture and release of M.tb into the extracellular space. Subsequent induction of lytic proteins causes granuloma cavitation and release of Mtb into the airways. 9. In the post-TB stage following treatment, extensive lung fibrosis and emphysema reduces lung compliance and are observed as reduced lung function. 10-11. Extensive fibrosis and calcification further reduce lung compliance and worsens COPD. 12. Periodic insults such as bacterial, viral, and fungal infections and air pollution or smoking trigger periodic COPD exacerbations after that. Created with Biorender.com.
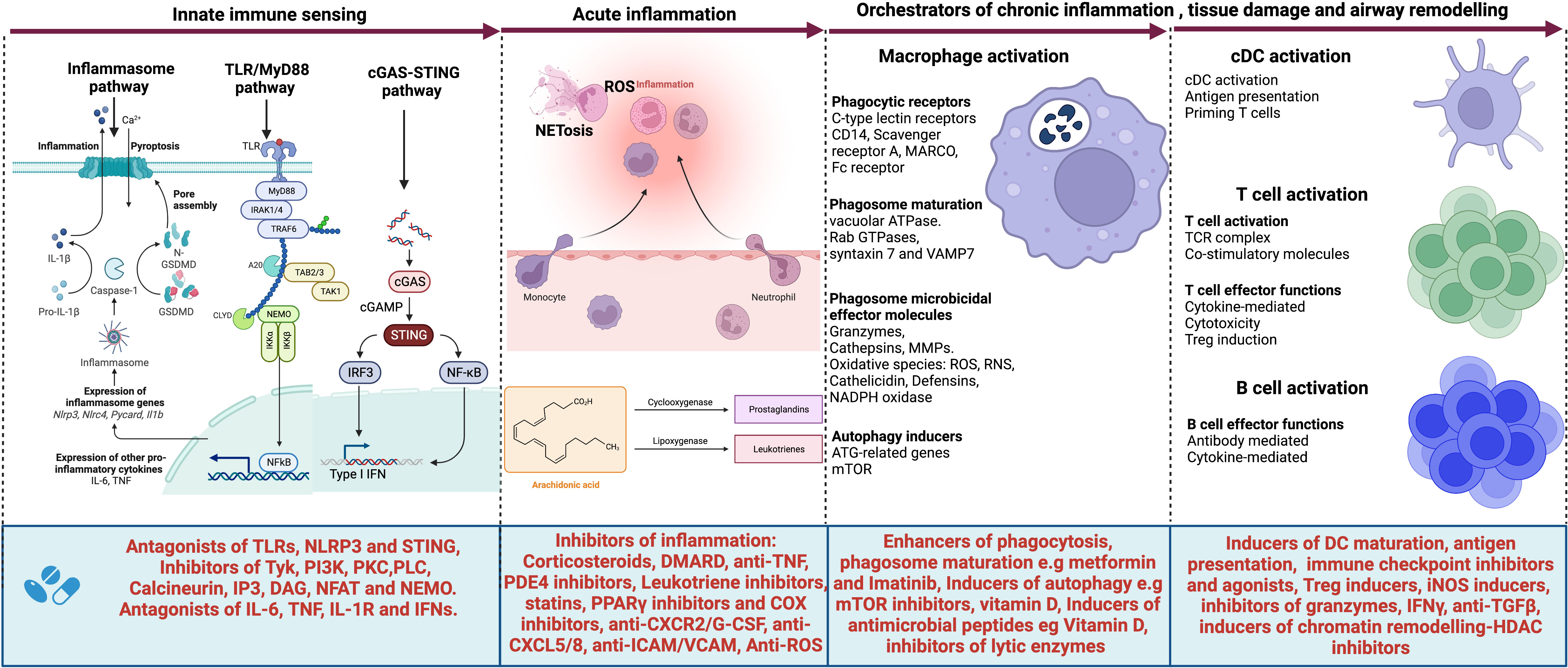
Figure 2 Targets for host-directed therapy (HDTs) for TB-associated COPD. A select list of immune-based therapies could offer benefits for patients with TB-associated COPD. Several agents have been developed or are under development that target (i) the innate immune sensors such as Toll-like receptors (TLRs) and their signaling pathway; (ii) the inflammasome activation pathway, their executioner Gasdermin D and pro-inflammatory cytokines IL-1β and IL-18; the cGAS-STING pathway and their effector cytokines, type I interferons as well as (iv) pro-inflammatory cytokines such as IL-6, TNF, IL1β and type I and II interferons. Such agents include antagonists of TLRs, NLRP3, and STING; inhibitors of the signaling molecules and enzymes such as tyrosine kinase (Tyk), PI3 kinase (PI3K), Phosphokinase C (PKCθ), phospholipase C (PLCγ), calcineurin, IP3, DAG, NFAT, and NEMO. Cytokine antagonists include anti-IL6, anti-TNF, IL-1R inhibitors, and inhibitors of type I interferons. Other agents target acute inflammation, such as corticosteroids, disease-modifying antirheumatic drugs (DMARDs), anti-TNF, Phosphodiesterase 4 (PDE4) inhibitors, Leukotriene BLT1-receptor antagonists, anti-TNF- therapies, TNFα-converting enzyme inhibitors, statins, PPARγ inhibitors, COX selective and non-selective inhibitors, TGFβ-1 receptor kinase antagonists, anti-IL-8 neutralizing antibody, CXCR2 inhibitors, CXCR3 antagonists, anti-Reactive Oxygen Species (ROS), anti-ICAM/VCAM and anti-CXCL5/8 and inhibitors. Other molecules target macrophage phagocytic receptors, autophagy, executioner machinery, and phagosome maturation. These include metformin, Imatinib as enhancers of phagosome maturation and autophagy induction, mammalian target of rapamycin (mTOR) inhibitors, Vitamin D, and inhibitors of lytic proteins and enzymes in phagolysosomes. cDCs, T and B cell activation also contribute molecules that orchestrate tissue damage. Such molecules can be targeted for therapeutic purposes. These include inducers of DC maturation like FLT3L, immune checkpoint inhibitors, inducers of Treg cells, inducers of iNOS, inhibitors of granzymes, anti-TGFβ, and finally, inhibitors of chromatin remodeling such as HDAC inhibitors. Created with Biorender.com.
Distinct immune cell populations reside in the respiratory system, reflecting specialization along the tract to suit differing intensities of exposure to airborne antigens and airway microbiome in the transition between the upper and lower respiratory tract (Holt et al., 2008; Huffnagle et al., 2017). In conducting airways, the mucosal epithelial lining comprises ciliated cells interspersed with goblet cells, providing mucociliary clearance of inhaled antigens and locally secreted immunoglobulin A (IgA) (Holt et al., 2008; Hewitt and Lloyd, 2021). The mucosa contains dense networks of macrophages and dendritic cells (DCs) (Kopf et al., 2015). Airway macrophages are the most predominant cells residing on the mucosa and continuously sample airborne antigens (Cohen et al., 2018). The airway DC population comprises both myeloid DCs (mDCs) and plasmacytoid DCs (pDCs), with a predominance of mDCs (Kopf et al., 2015). A specialized group of DCs known for immune surveillance is strategically located within and directly below the mucosa (Holt et al., 2008). These DCs extend their protrusions into the airway mucosa, routinely sampling microbial antigens (Jahnsen et al., 2006). Another population of cells referred to as innate lymphoid cells (ILCs) resides within lymphoid tissue at airway branch points (Sonnenberg and Hepworth, 2019). They co-localize with CD4+T cells, DCs, and specialized stromal cells, which provide activating cytokines (Sonnenberg and Hepworth, 2019). Three ILC subsets closely mirroring the transcriptional and functional biology of CD4+T helper (Th) cells (i.e., ILC1, ILC2, and ILC3) have been described elsewhere (Sonnenberg and Hepworth, 2019).
T cells are also found in relatively high numbers in the mucosa and within the underlying lamina propria (Holt et al., 2008). Most intra-epithelial T cells express CD8, whereas CD4+T cells are more frequently found in the lamina propria (Holt et al., 2008). Both cell subtypes have effector and memory cell phenotypes defined by the expression of CD45RO (Holt et al., 2008). The submucosa also harbors B, mast, and plasma cells, mainly producing polymeric IgA (Brandtzaeg, 2015). In addition to their role in antibody production, airway B cells contribute to local antigen presentation. As described elsewhere (Holt et al., 2008), the airway mucosa also contains bronchial-associated lymphoid tissue (BALT), comprising discrete lymphoid-cell aggregates underlying a specialized epithelium, similar to Peyer’s patches in the gut. Under homeostasis, several immune cells, including interstitial macrophages, DCs, T cells, B cells, and mast cells, populate the lung parenchyma, whereas large numbers of T cells sequester in the lung parenchymal vascular bed (Holt et al., 2008). In the setting of inflammation, other cells, such as neutrophils and monocytes, infiltrate and predominate the lung parenchyma (Cohen et al., 2018). The contribution of bronchial epithelial cells, fibroblasts, and the extracellular matrix in airway immune response has been described elsewhere (Kitamura et al., 2011; Gao et al., 2015; Hewitt and Lloyd, 2021).
Several authors have described the pathogenesis of COPD with emphasis on cigarette smoke, biomass exposure, and HIV infection (Hogg, 2004; Chung and Adcock, 2008; Churg et al., 2008; Rajendrasozhan et al., 2008; Atkinson et al., 2011; Milara et al., 2013; Pichavant et al., 2014; Agustí and Celli, 2017; Yoshida et al., 2019; Byanova et al., 2021). Other post-TB sequelae, such as bronchiectasis and bronchopulmonary aspergillosis, cannot be underestimated (Meghji et al., 2021). Whereas the role of innate and adaptive immune cells in COPD has been extensively described elsewhere (Barbu et al., 2011; Ezzati Givi et al., 2012; Craig et al., 2017; Le Rouzic et al., 2017; Ni and Dong, 2018), in this review, we describe how immune mechanisms in the setting of Mycobacterium tuberculosis infection intersect with known COPD-immune mechanisms and possibly contribute to immuno-pathology in TB-associated COPD. To date, literature describing the immunological relationship between tuberculosis and COPD remains limited (Radovic et al., 2011; Cao et al., 2012; Allwood et al., 2019; Chin et al., 2019; Singh et al., 2022). Recent evidence suggests that tuberculosis orchestrates chronic lung inflammation and tissue necrosis, with resultant airway fibrosis and remodeling observed in COPD (Radovic et al., 2011; Cao et al., 2012; Sarkar et al., 2017; Stek et al., 2018; Allwood et al., 2019; Chin et al., 2019; Singh et al., 2022). The immune response, particularly in the distal airways, is characterized by activation of alveolar macrophages, dendritic cells, innate lymphoid, and γδ-T cells, which promote recruitment and activation of neutrophils, monocytes, as well as cells of the adaptive immune response (B cells, Th1, Th17, and cytotoxic T-cells). Further interaction between immune cells and the airway epithelial cells, fibroblasts, and the extracellular matrix culminates into granuloma formation, which upon degeneration (caseation), orchestrates lung inflammation and tissue necrosis coupled with fibrosis and airway remodeling (Chung and Adcock, 2008). Although the immune mechanisms of lung parenchymal damage in tuberculosis have been extensively described and the role of host-directed therapy elucidated elsewhere (Ravimohan et al., 2018; Stek et al., 2018), this review focuses on the immunologic sequence of events in early versus late tuberculosis disease that orchestrate COPD immunopathology (Figure 1).
Following infection of a host with Mycobacterium tuberculosis (Mtb) via inhalation of viable bacilli in exhaled droplets, bacilli are internalized by alveolar macrophages (AM) via phagocytosis (Pieters, 2008). Based on in vitro studies with various macrophage types, including AMs, viable bacilli have been shown to modify phagosome activity, preventing its maturation and fusion with lysosomes via mechanisms well-described elsewhere (Sturgill-Koszycki et al., 1994; Ferrari et al., 1999; Gatfield and Pieters, 2000; Fratti et al., 2003; Rosenberger and Finlay, 2003; Walburger et al., 2004; Nguyen and Pieters, 2005; Vergne et al., 2005; Rohde et al., 2007; Warner and Mizrahi, 2007; Pieters, 2008; Vandal et al., 2008; Sun et al., 2010; Abramovitch et al., 2011; O’Leary et al., 2011; Wong et al., 2011; Sullivan et al., 2012; Jamwal et al., 2016; Awuh and Flo, 2017; Queval et al., 2017; Buter et al., 2019). Some internalized bacilli perforate the phagosome membrane using the ESX-1 type III secretion system, escaping into the cytosolic space (Manzanillo et al., 2012; Augenstreich et al., 2017; Conrad et al., 2017; Queval et al., 2017; Wong, 2017). Therefore, by engaging several immune evasion strategies such as those described above (Hmama et al., 2015), M.tb bacilli successfully establish infection in the AMs dividing exponentially until the immune pressure contains the pathogen (Russell et al., 2009). Consequently, either localized sterilization of M.tb bacilli and mineralization of lesions into Ghon foci or extensive caseation and tissue necrosis occurs with lung damage and eventual release of M.tb bacilli into the airways (Russell et al., 2009). Although several authors have described macrophage activation in the context of the M.tb control (Kornfeld et al., 1999; Kaufmann, 2002; Nau et al., 2002; Flynn, 2004; Lin et al., 2007; Blumenthal et al., 2009; Moreira-Teixeira et al., 2016; BoseDasgupta and Pieters, 2018; Marakalala et al., 2018; Khan et al., 2019; Li et al., 2021; Mata et al., 2021; Thakur and Muniyappa, 2023), in this section, we elaborate on events surrounding macrophage activation in early and late M.tb infection, which orchestrate lung damage as observed in COPD.
In the early stages of M.tb infection, airways initially show increased infiltration with innate immune cells beneath the lamina propria, without any obvious pathological lesions (North and Jung, 2004; Russell et al., 2009). Until recently, the precise composition, pattern, and mechanisms underlying this immune cell infiltration within the airways in the early stages of M.tb infection have been largely speculated. However, evidence from the murine model shows that M.tb infection exclusively occurs in the AMs during the first week of infection (Cohen et al., 2018). In this period, M.tb-activated AMs infiltrate the lung interstitium in a MyD88/IL-1R-dependent manner to establish a focus of infection in the interstitium (Cohen et al., 2018; Lovey et al., 2022). This precedes chemotaxis and infiltration of the lung interstitium and the airways with neutrophils, monocyte, and lymphocytes. Cytokines produced by AMs play a critical role in attracting other immune cells into the primary area of insult to contain M.tb growth. For instance, TNF has been reported as a critical cytokine in the initial stages of granuloma formation and M.tb control (Kindler et al., 1989; Roach et al., 2002; Algood et al., 2005). This cytokine maintains the concentration gradient required for sustained cellular recruitment and retention into the growing granuloma (Roach et al., 2002). In the tnfr knock-out murine model, granuloma cellular organization is severely disrupted, resulting in an amorphous and necrotic structure that is poor at containing M.tb growth (Gil et al., 2006). However, as the infection progresses, too much TNF drives airway pathology, where the granulomatous response becomes too aggressive and causes extensive tissue destruction (Casadevall and Pirofski, 2003). Although the intention is usually to contain M.tb growth, this excessive response orchestrates airway damage and drives structural changes that severely impair lung function (Casadevall and Pirofski, 2003; Russell et al., 2009). Besides TNF, other pro-inflammatory cytokines such as IFN-γ, IL-12, IL-1β, and IL-18 not only drive M.tb killing but also sustain macrophage activation (Flynn, 2004; Koo et al., 2008; Fan et al., 2013; Yang et al., 2013; Moreira-Teixeira et al., 2016; Agarwal et al., 2021; Li et al., 2021), which sustains airway damage. What is currently unknown is the exact timing when such damage becomes irreversible to portray the pathology observed in COPD. However, from the natural history of COPD, repetitive insults accumulate over time and cause structural abnormalities with impaired lung function (Tantucci and Modina, 2012; Vestbo and Lange, 2016; Lange et al., 2021). In the setting of M.tb, airway insults most likely occur during the acute phase of inflammation and become sustained in the chronic phase. Unfortunately, multiple attempts at healing result in extensive fibrosis and airway remodeling, altering lung anatomy and distal airway mechanics (Dheda et al., 2005). Performing serial lung function tests in a murine infection model in a carefully designed experimental setting could assist in determining the specific time when irreversible damage occurs. Furthermore, conducting in-depth omics analyses of such changes would provide critical information about TB-associated COPD immunopathology.
Upon activation, AMs demonstrate plasticity, differentiating into either classical (M1) phenotype in the presence of specific cytokines such as IFN-γ or alternative (M2) phenotype in the presence of specific cytokines such as TGF-β and IL-10 (Khan et al., 2019; Locati et al., 2020; Ge et al., 2021; Ahmad et al., 2022; Arish and Naz, 2022). M1 phenotype is pro-inflammatory, expresses the iNOS gene, induces reactive oxygen (ROS)/nitrogen (Ernst, 2012) species (Ernst, 2012), and is highly specialized in phagolysosomal killing and containment of M.tb bacilli, while M2 is anti-inflammatory and aids in the clearance of debris at the down or resolution of inflammation (Kasmi and Stenmark, 2015; Marino et al., 2015; Arora et al., 2018; Shapouri-Moghaddam et al., 2018). M2 also drives tissue repair and regeneration (Kasmi and Stenmark, 2015; Marino et al., 2015; Arora et al., 2018; Shapouri-Moghaddam et al., 2018). Since macrophage polarization states affect the growth of M.tb bacilli in either a restrictive or permissive manner, a fine balance between M1 and M2 phenotypes ensures containment of the M.tb bacilli with minimal tissue damage (Refai et al., 2018; Chen et al., 2022). In chronic inflammation, excessive M1 activity orchestrates tissue damage. Macrophages unleash ROS and RNS species, mount oxidative stress, and cause bystander damage to surrounding tissue (Bogdan, 2001; Guirado et al., 2013). Other molecules, such as cathelicidins (Herr et al., 2007; Tecle et al., 2010), defensins (Ganz, 2003; Doss et al., 2010), cathepsins (Zavašnik-Bergant and Turk, 2006; Lecaille et al., 2008; Patel et al., 2018), matrix metalloproteases (Page-McCaw et al., 2007; Salgame, 2011; Houghton, 2015; Grzela et al., 2016), and S100 proteins (Donato et al., 2013; Kessel et al., 2013; Pouwels et al., 2014; Singh and Ali, 2022), drive connective tissue damage, fibrosis, and angiogenesis, as described elsewhere (Barnes et al., 2003; Guirado et al., 2013; Whitsett and Alenghat, 2015). Of particular importance are collagenases (MMP1 and MMP13), elastases (MMP12), and gelatinases (MMP2 and MMP9), which degrade underlying connective lung tissue, promoting tissue necrosis (Guirado et al., 2013). MMP-1 and MMP-8 levels correlate with airway tissue damage in TB patients, whereas MMP-14 promotes collagen degradation and regulates monocyte migration (Sathyamoorthy et al., 2015). Overall, existing evidence supports the role of these molecules in driving lung damage in TB. Following treatment with anti-TB drugs for at least two months, findings show a significant reduction in the serum levels of MMP-8 in TB patients with severe lung tissue damage (de Melo et al., 2019). Similarly, serum MMP-1, -2, -3, -9, and -12 levels are higher in TB patients with severe structural lung damage. These molecules decrease following successful treatment (Kumar et al., 2018). In emphysema and chronic bronchitis (which are forms of COPD), similar molecules mediate lung inflammation and promote the release of fibrogenic growth factors (Churg et al., 2012), suggesting that these molecules, in a way, contribute to TB-associated COPD. As previously noted, we currently do not know the exact time in the course of the disease when these molecules induce irreversible airway damage and tissue remodeling, hence warrant further investigation.
Besides the collateral damage from activated macrophages, the genesis and evolution of the TB granuloma (Russell, 2007; Russell et al., 2009; Ramakrishnan, 2012; Nunes-Alves et al., 2014; Kiran et al., 2016; Marakalala et al., 2016; Cadena et al., 2017; Cohen et al., 2022; McCaffrey et al., 2022), from its nascent form through caseous, fibrocaseous, and resolved forms as the infection progresses, plays a direct role in distorting the lung anatomy, as observed on chest X-ray films and CT scans of most post-TB patients (Stek et al., 2018). Similarly, evidence of impaired respiratory mechanics has been documented by spirometry among post-TB individuals (Amaral et al., 2015; Meghji et al., 2020; Mpagama et al., 2021; Ivanova et al., 2023). During the process of granuloma formation, activated AMs, as previously described, invade subtending epithelium and attract mononuclear cells from neighboring blood vessels through chemotaxis and form the cellular matrix of the early granuloma (Russell et al., 2009). As M.tb infection progresses, the newly recruited monocyte-derived macrophages engulf infected cell debris and contribute to primary granuloma expansion (Pagán and Ramakrishnan, 2015). Some of these newly infected macrophages exit the primary granuloma and establish secondary granulomas in distal tissues, spreading the infection further (Pagán and Ramakrishnan, 2015). Infected macrophages in the primary granuloma undergo necrotic cell death (Behar et al., 2011), majorly driven by pathogen-induced subversion of eicosanoid synthesis from prostaglandin E2 to Lipoxin A4 (LXA4) (Chen et al., 2006). This promotes lung inflammation, tissue destruction, and remodeling, favoring chronic lung damage. Other immune cells, such as neutrophils and lymphocytes, infiltrate TB granuloma and lung tissue (Cohen et al., 2018). Consequently, by the 3rd week of infection, infected alveolar macrophage number plateaus while infected neutrophils and monocyte-derived macrophages increase drastically to become the most predominant cells in the granuloma (Russell et al., 2009; Cohen et al., 2018). In its nascent stage, the granuloma has a center of infected macrophages. As it evolves into its caseous form, these cells become enclosed by lipid-laden (foamy) macrophages, which drive caseous necrosis (Russell et al., 2009; Agarwal et al., 2021). In the fibrocaseous stage, the macrophages differentiate into multinucleated giant epithelioid cells surrounded by lymphocytes. A thick fibrous cuff forms outside the epithelioid cells at this stage, excluding lymphocytes from the granuloma core (Russell et al., 2009; Agarwal et al., 2021). In the resolved form, extensive fibrosis occurs, walling off this area. The bacillary load remains relatively constant in this period, establishing a state of latency in 90% of cases (Russell et al., 2009). In some scenarios, calcification occurs, giving rise to Ghon foci, where TB re-activation into active disease never occurs (Donald et al., 2021). However, in some situations, for instance, conditions associated with immunosuppression such as AIDS, M.tb re-activation occurs, driving active TB disease (Innes et al., 2009). For granulomas that show increasing accumulation of caseum in the core, extensive necrosis and breakdown occur, releasing viable bacilli into the airways to propagate the M.tb infection cycle (Russell et al., 2009). Despite a prolonged course of anti-tuberculosis therapy, the damage caused by extensive granulomatous disease and fibrosis in the airways is permanent (Pasipanodya et al., 2007). It persists and increases with future TB episodes (Plit et al., 1998; Hnizdo et al., 2000), resulting in an irreversible decline in pulmonary lung function and structural damage to the lung parenchyma observed among post-TB individuals (Hnizdo et al., 2000). Several studies have described this phenotype as pulmonary impairment after tuberculosis (PIAT) (Pasipanodya et al., 2007; Pasipanodya et al., 2010; Gandhi et al., 2016; Chushkin and Ots, 2017).
In a nutshell, airway macrophages play a critical role in orchestrating lung damage as a bystander effect in M.tb infection. Insults most likely occur during the acute phase of inflammation and become sustained in the chronic phase, as evidenced by increased uptake or intensity of fludeoxyglucose F18 on PET scans taken among TB patients a year after completion of anti-TB therapy compared to baseline (Malherbe et al., 2016). In such a setting, dysregulated healing results in extensive fibrosis and airway remodeling. Distorted lung anatomy and abnormal airway mechanics consequently ensue. Given the heterogeneity of immune cells infiltrating the airways during acute and chronic inflammation in M.tb infection, immunopathology is multifactorial. In the proceeding sections, we discuss how other immune cells drive lung damage in TB, highlighting similarities to COPD immunopathology.
Although earlier studies report neutrophilic infiltration into the sites of M.tb infection within hours of inoculation, the response depends on the route of M.tb exposure (Lowe et al., 2012). Several experimental infection animal models report neutrophilic infiltration into multiple perivascular sites an hour post-infection following intravenous M.tb inoculation (Long et al., 1931), while cutaneous infiltration in rabbit infection model following BCG inoculation occurs within 3 hours of infection, peaking at 12 hours (Shigenaga et al., 2001). In a murine model, neutrophils arrive at the site of dermal BCG inoculation within 4 hours, peaking at 24 hours post-intranasal or intrapleural BCG inoculation (Abadie et al., 2005). A sharp contrast exists when the exposure is aerosol or intratracheal. Following intratracheal BCG inoculation, a 1- to 2-week’ airway neutrophilic infiltration has been reported (Fulton et al., 2000). In a recently published murine infection model, airway neutrophilic infiltration occurs within the 1st week and a few days post-infection, following interstitial localization of alveolar macrophages (Cohen et al., 2018), and as the infection progresses through the 3rd week, neutrophils predominate the airways (Cohen et al., 2018). Few studies have investigated the timing of neutrophilic influx in early TB infection in humans. However, a massive influx of neutrophils into the airways occurs in individuals with established tuberculosis disease, associated with tissue necrosis (Ehlers and Schaible, 2013). Evidence shows that neutrophils accumulate in broncho-alveolar lavage fluid and sputum of individuals with active tuberculosis (Eum et al., 2010). Mechanistically, the inflammatory cytokine milieu in the airways activates the endothelium, increasing the expression of adhesion molecules, ICAM-1, E-selectin, and P-selectin, which results in a neutrophilic influx (Lowe et al., 2012). This influx is driven by several immune pathways. In the IL-23/Th17 axis, alveolar macrophages, upon activation, produce IL-23, which activates mucosal Th17 cells to produce IL-17, associated with neutrophil recruitment into the airways (Sergejeva et al., 2005; Lockhart et al., 2006; Guirado et al., 2013). In a second pathway, activation of bronchial epithelial cells via TLR2 signaling promotes the secretion of CXCL5, a known ligand for CXCR2 receptors expressed on neutrophils. Consequently, neutrophils move into the airways following the CXCL5 chemokine concentration gradient (Nouailles et al., 2014). Early neutrophilic infiltration into the airways depends exclusively on CXCR2 and primarily on CXCL5 as genetic ablation of CXCL5 results in impaired neutrophil recruitment and reduced lung inflammation (Nouailles et al., 2014).
Whereas early neutrophilic inflammation plays a critical role in protecting against tuberculosis disease progression (Kisich et al., 2002; Dallenga and Schaible, 2016; Kroon et al., 2018), sustained neutrophilic inflammation harms the host. Following phagocytosis, neutrophils fail to neutralize viable M.tb bacilli. Instead, the highly oxidative state generated in the setting of M.tb infection drives neutrophils into necrotic versus apoptotic cell death (Dallenga and Schaible, 2016). Mechanistically, M.tb induces ESX-1-dependent neutrophilic necrosis driven by reactive oxygen species (ROS). Consequently, massive accumulation of infected and dying neutrophils further unleashes highly lytic and oxidative molecules which cause extensive tissue necrosis (Dallenga et al., 2017). Unfortunately, impaired dead cell removal drives further inflammation, even after M.tb sterilization. This greatly hinders the resolution of inflammation (Malherbe et al., 2016).
Infected and dying neutrophils mediate tissue damage via a number of pathways, as briefly explained in this section. The production of reactive oxygen species (ROS) during the oxidative burst exceeds cellular antioxidant capacity, damaging cellular structures, including lipids, protein, and DNA (Thannickal and Fanburg, 2000). This oxidative stress drives M.tb-induced tissue necrosis, further promoting tissue inflammation and damage (Sun et al., 2015; Dallenga et al., 2017). Secondly, NETosis, which involves the expulsion of DNA complexed with antimicrobial proteins into extracellular space to form neutrophil extracellular traps (NETs), contains M.tb bacilli. However, high levels of NETosis coupled with ineffective clearance of bacteria in tuberculosis pose pathological consequences in the lungs. The antimicrobial histones and peptides coating the NET-DNA are directly cytotoxic to lung tissue, and inadequate support of NETs causes deleterious inflammation of host tissue (Xu et al., 2009). NETs, particularly extracellular histones, also cause epithelial and endothelial cell death (Xu et al., 2011). Such NETosis-induced cell necrosis orchestrates lung inflammation and damage (Ramos-Kichik et al., 2009). M.tb-induced NETosis has also been associated with macrophage activation, which drives lung inflammation and tissue damage in TB disease, as previously described (Braian et al., 2013). Several human studies have validated the role of NETosis in lung tissue damage. A study reported high citrullinated H3, a standard NET marker in serum samples from tuberculosis patients with extensive pulmonary damage (de Melo et al., 2019), suggesting that NET formation is associated with severe lung tissue damage in TB patients. Similarly, NETosis was markedly increased in the airways of stable COPD patients in another study (Pedersen et al., 2015; Pedersen et al., 2018; Uddin et al., 2019). Extracellular DNA correlated with absolute neutrophil numbers in sputum and airway obstruction (Pedersen et al., 2018; Uddin et al., 2019). Other studies have reported similar correlations between NETosis and COPD (Obermayer et al., 2014; Grabcanovic-Musija et al., 2015; Uddin et al., 2019). NETosis and ROS in TB disease could contribute to irreversible lung damage, as observed in COPD, although mechanistic studies are needed to underpin the role of NETosis in COPD pathogenesis. Thirdly, the production of lytic molecules such as matrix metalloproteases (MMPs), cathepsins, S100 proteins, cathelicidins, and beta-defensins, as is the case for activated macrophages, also drive airway pathology (Pugin et al., 1999; Ong et al., 2015; Muefong and Sutherland, 2020). Finally, there is increasing evidence that activated neutrophils mediate tissue damage via cytokine production (Mantovani et al., 2011; Etna et al., 2014). High levels of IL-8, TNFα, and IL-1β secreted from activated neutrophils in TB disease correlate with enhanced neutrophil migratory capacity, airway recruitment, and heightened pro-inflammatory response in the lung environment (Muefong and Sutherland, 2020), which could contribute to the observed tissue necrosis. Previous studies have supported IL-8 as a well-known factor in the pathogenesis of COPD (Larsson, 2008; Zhang et al., 2011). IL-8 has been reported to recruit neutrophils in airways and induce increased MUC5AC and MUC5B mRNA expression in bronchial epithelial cells (Bautista et al., 2009). In addition, previous studies have demonstrated higher baseline levels of IL-8 expression in airway epithelial cells from patients with COPD compared to healthy controls (Schneider et al., 2010). TNFα has also been reported to promote tissue necrosis, lung fibrosis, and weight loss in COPD (De Godoy et al., 1996; Takabatake et al., 2000). Similarly, IL-1β has been demonstrated to promote lung inflammation, emphysema, and airway remodeling (Schneider et al., 2010). As COPD progresses in severity, airway neutrophilia increases and is associated with more significant airflow obstruction and accelerated lung function decline (Stănescu et al., 1996; O’Donnell et al., 2004).
Besides CXCL5 production by epithelial cells, alarmins (IL-25, IL-33, TSLP) (Vannella et al., 2016; Gupta et al., 2017; Roan et al., 2019) as well as cytokine production (IL-1β, IL-8, and G-CSF) (Wickremasinghe et al., 1999; Roan et al., 2019) results in further mobilization of innate lymphoid cells (ILCs). As elaborated in this section, these cells promote the recruitment of other immune cells, driving airway inflammation and tissue damage. Following infection, ILC2/3 cells rapidly accumulate in the airways, promoting the recruitment of monocyte-derived macrophages and neutrophils (Ardain et al., 2019). In a murine model, ILC3s accumulate in the airways early in the infection, followed by ILC2 later. As the infection progresses, the ILC3 number increases drastically, mirroring the expansion of alveolar macrophages. These cells precede the accumulation of monocytes and monocyte-derived macrophages in the airways. Interestingly, mice that lacked ILC3s exhibited a significant reduction in early AMs (Ardain et al., 2019). During M.tb infection, CXCR5 expression on circulating ILC3s is upregulated, and parallel increases in plasma levels of its ligand, CXCL13, have been observed in human studies. This implies that ILC3s migrate in response to CXCL13 concentration gradient (Ardain et al., 2019). Moreover, IL-23-dependent expansion of ILC3s in mice and the production of the cytokines IL-17 and IL-22 are critical inducers of lung CXCL13, promoting early mobilization of lung ILC3 and macrophages to initiate inflammation in an attempt to control tuberculosis. This, however, promotes tissue necrosis, consequently (Ardain et al., 2019). Similarly, ILC3-driven immunopathology has been reported in COPD. In a recent report, the frequency of natural cytotoxicity receptor-expressing ILC3 cells was increased in COPD lungs (Marashian et al., 2015). Other studies have shown that the frequency of ILC1 cells in patients with COPD correlates with disease severity and susceptibility to COPD exacerbations (Silver et al., 2016). In contrast, the combination of micro-CT analysis, histology, and gene expression profiling indicated that ILC1 signatures were enriched in centrilobular emphysema, suggesting that the alveolar destruction observed in COPD may be driven by ILC1s (Suzuki et al., 2017).
In the airways, conventional dendritic cells (cDCs) exist as immature cells, residing beneath the mucosa for an extended period of time (Banchereau and Steinman, 1998). These cells continuously sample the mucosa via their dendrites’ extension in between the epithelial cells’ tight junctions (Artis, 2008). Immature cDCs partially mature to express CCR7, which allows them to home into draining lymphoid tissues, in which they interact with naive T cells (Geissmann et al., 2002). Upon contact with M.tb, immature DCs phagocytose the bacilli, become activated, and undergo differentiation from highly phagocytic into less phagocytic cells (Kapsenberg, 2003). At the onset of the inflammatory response, immature cDCs are highly represented at sites of M.tb infection and are specialized for antigen uptake (Sertl et al., 1986; Mihret, 2012). During differentiation, however, cDCs upregulate the expression of MHC class I and II molecules, CD40, CD54, CD58, and CD80, resulting in a cellular phenotype consistent with mature and activated DCs, excellent at antigen presentation (Mihret et al., 2011). Upon phagocytosis, the fate of antigen presentation to either CD4+ or CD8+ T cell depends on the intracellular M.tb routing (Burgdorf et al., 2007; Chatterjee et al., 2012; Cohn et al., 2013). Delivery of the pathogen cargo into the late endolysosomes results in a much quicker degradation and MHC class II antigenic loading, whereas delivery into early endosomes results in a slower degradation with prolonged MHC class I antigenic loading. Upon migration into draining lymphoid tissue, mature cDCs with class II antigen load prime naïve CD4+T cells, while those with class I antigen load prime CD8+T cells (Burgdorf et al., 2007; Chatterjee et al., 2012; Cohn et al., 2013). cDCs prime the adaptive immune system and contribute significantly to early, intermediate, and late cytokine production. During early M.tb-induced response, the release of early mediators of inflammation such as TNF-α, IL-1α, IL-1β, IFN-γ and chemokines such as CXCL5 and CXCL8 promotes the influx of additional monocytes, macrophages, and neutrophils into the airways (Etna et al., 2014). As previously described, these cells orchestrate inflammation, tissue necrosis, airway remodeling, and fibrosis. cDCs also phagocytose apoptotic bodies from dying neutrophils, and macrophages infected with M.tb bacilli, promoting cross-presentation via class I molecules. Notably, the sustained release of IL-12, IFN-γ, and IFN-β cytokines from activated DCs, particularly in the early stage of M.tb infection, drive early granuloma formation and containment of M.tb (Etna et al., 2014). However, excessive type I interferon production contributes to tissue damage in the chronic phase (LeibundGut-Landmann et al., 2007; Steinman and Banchereau, 2007). As elaborated in the next section, activated cDCs produce several molecules with Th-polarizing abilities. For instance, IL-12, IL-23, IL-27, and type I IFNs induce a Th1 phenotype, while MCP1 and OX40 ligand induce a Th2 phenotype (Kapsenberg, 2003). During chronic inflammation, DCs alternatively induce CD4+T cells to become suppressive by making IL-10 or differentiating into FOXP3+ CD4+T cells (Jonuleit et al., 2000; Luo et al., 2007). IL-10 counter-regulates inflammation and induces tissue healing. However, in chronic inflammation, it also drives fibrosis (Robb et al., 2016). Finally, activated cDCs also produce high levels of IL-4, IL-33, and TGF-β, which drive airway fibrosis (Mihret, 2012). Whereas the direct role of cDC in inducing airway damage in TB has not been described, the accumulation of mucosal cDCs in the small airways of COPD patients supports the role of such cells in the immunopathology of COPD (Demedts et al., 2007).
Activating the adaptive immune response to M.tb restricts bacterial growth but rarely eliminates the bacilli (Ernst, 2012). Evidence suggests that adaptive immune responses contribute to airway pathology (Ernst, 2012) in TB disease as observed in COPD. This response depends predominantly on cell-mediated immunity because M.tb lives within macrophages; thus, effector T-cell responses are required to contain or kill the bacteria (Flynn and Chan, 2001). The role of B cells in controlling M.tb infection was still unknown until recently when it was demonstrated that antigen-specific B cells nurture and direct follicular-like T cells into lymphoid follicles to mediate M.tb control (Swanson et al., 2023).
Within a week of M.tb infection, activated CD4+ and CD8+T cells migrate to the lung-draining lymph nodes (Feng et al., 1999; Ernst, 2012). By the end of a month, both cellular phenotypes have increased in the lung environment, demonstrating both effector and memory phenotypes (Feng et al., 1999). Half of these cells are CD69+ (Kornfeld et al., 1999; Refai et al., 2018; Chen et al., 2022), indicating that activated T cells migrate to the site of infection, interact with M.tb-infected macrophages and DCs through CD40L on CD4+ T cells and CD40 on macrophages or dendritic cells (Kalams and Walker, 1998; Clarke, 2000). The outcome was previously reported as enhanced antigen presentation and costimulatory activity resulting in the generation of robust CD4+ and CD8+T cells (Kalams and Walker, 1998; Clarke, 2000). However, recent studies show that IFNγ production by M.tb-specific CD4+T cells becomes locally restricted by the granuloma microenvironment despite ongoing antigen recognition through TGFβ dependent immune mechanisms (Gern et al., 2021). TNFα and type I IFNs orchestrate macrophage necrosis and release of M.tb into the extracellular space (Pagán and Ramakrishnan, 2015). Subsequent induction of several tissue proteases causes granuloma degeneration (caseation), orchestrating lung tissue inflammation, necrosis, fibrosis, and remodeling, resulting in cavitation and release of M.tb into the airways. Consequently, these changes gradually and irreversibly compromise lung function leading to obstructive airway pathology observed in COPD. In the presence of the necessary cytokine milieu (Cooper et al., 1995), various Th1 effector subtypes are produced, ranging from early activated cells producing only IL-2 to cells producing IFNγ and multifunctional cells expressing IL-2, IFNγ, and TNFα (Darrah et al., 2007; Cooper, 2009). The presence of these multifunctional cells is associated with protection and lung tissue inflammation and necrosis (Darrah et al., 2007; Cooper, 2009). In addition, cytolytic CD4+T cells secreting perforins and granulysin are produced, promoting necrosis of M.tb-infected macrophages. Multifunctional CD4+T cells have been reported frequently among tuberculosis patients (Winkler et al., 2005), individuals from endemic tuberculosis areas, and vaccinated infants (Scriba et al., 2008; Soares et al., 2008). The conditions optimal for multifunctional and cytolytic antigen-specific lymphocytes have yet to be fully defined in human studies; however, recent literature suggests the role of IL-12p70 in IFN-producing cells in maintaining this cellular profile (Cooper et al., 2007).
In addition to Th1 cells, tuberculosis-specific Th17 cells are induced following aerosol tuberculosis infection in mouse models; Th17 cellular immune response in the lung microenvironment depends on the secretion of IL-23 from activated macrophages (Khader et al., 2005). Also, gamma delta (γδ) T cells have been shown to produce IL-17 following a high-dose intranasal challenge with BCG in mouse models. A large portion of the IL-17 response in the mouse model is within the γδ T cell population (Lockhart et al., 2006). Interestingly, upon blockade of IL-17 secretion during a high-dose challenge, neutrophil recruitment into the lung microenvironment is significantly hindered, altering the subsequent inflammation and lung pathology (Umemura et al., 2007). The repeated challenge of tuberculosis-infected animals with tuberculosis antigen results in increased lung neutrophil infiltration and subsequent tissue necrosis (Taylor et al., 2003). Studies investigating the role of IL-17 in this enhanced pathology have found that increased tissue necrosis and neutrophilic infiltration is dependent on IL-23 and could be ablated by the delivery of anti-IL-17 antibody, suggesting that lung inflammation developing in response to chronic tuberculosis exposure depends on IL-23 and IL-17 (Darrah et al., 2007; Cooper, 2009). In human studies, CD4+ M.tb-specific IL-17- and IL-22-producing cells have been detected in individuals exposed to tuberculosis, although only IL-22 is seen in the lung (Scriba et al., 2008). Similarly, CD1- and MHC class I–restricted CD8+ T cells in the lungs, upon activation, produce cytotoxic granules which, upon degranulation on infected macrophages and dendritic cells, release perforins forming pores in cells and granulysin, killing both bacteria and infected macrophages (Ernst, 2012). In addition, CD8+T cells produce IFNγ, which potentiates macrophage function (Flynn and Chan, 2001). In summary, Th1 CD4+ and CD8+T cell activation in tuberculosis amplify macrophage activation and necrosis, while Th17 promotes neutrophil-mediated tissue necrosis, which drives lung inflammation, tissue damage, and remodeling.
In addition to T cell-mediated immune responses to M.tb, naïve B cells also develop into activated plasma cells that secrete M.tb-specific antibodies and produce cytokines (Rao et al., 2015; Loxton, 2019). Antibodies regulate effector functions, including opsonization, antibody-dependent cellular cytotoxicity (ADCC), and antigen neutralization. Furthermore, activated B cells are effective antigen-presenting cells that respond to whole or partial pathogens, presenting processed antigen peptides through their MHC-II to CD4+T cells (Abbas et al., 2019). As a result, B-cells contribute to the induction of CD4+ T-cells responses to tuberculosis, providing early protection against infection and driving antibody-mediated phagocytosis in which they modify macrophage behavior (Phuah et al., 2012). B-cells also respond in a non-humoral manner when stimulated by M.tb to produce pro- and anti-inflammatory cytokines, including TNFα, IL-10, IL-1β, IL-17, and IL-21 (Du Plessis et al., 2016b; Du Plessis et al., 2016a). Plasma (memory B) cells predominantly drive IL-10, IL-21, and TNFα production (Du Plessis et al., 2016b; Du Plessis et al., 2016a). Through the production of IL-12, IFNγ, and TNFα, effector B cells drive Th1 responses potentiating classical macrophage activation and, consequently, lung inflammation, tissue damage, and fibrosis (Chan et al., 2014). Production of IL-4, IL-33, and TGF-β by effector B cells disrupts Th1 response and favors alternative macrophage activation, promoting fibroblast activation, tissue remodeling, and fibrosis (Zhang et al., 2012). Preliminary COPD studies have suggested B cells’ role in COPD pathogenesis. Lymphoid follicles consisting of B-cells and follicular dendritic cells (DCs) with adjacent T-cells have been reported in both parenchyma and bronchial walls of patients with smoke-induced emphysema (van der Strate et al., 2006). Similarly, reported oligoclonal antigen-specific B-cell reactions among current smokers have been published (Brandsma et al., 2009). Plasma cells derived from B-cell maturation occur in more significant numbers in sub-epithelial and submucosal glands in patients with COPD. Most of these B cells express IL-4 and IL-5 (Zhu et al., 2007), which promote mucous hypersecretion and stimulation of fibroblasts to produce TGFβ, promoting tissue remodeling. More research is needed to characterize M.tb-specific B cell responses in the context of COPD pathogenesis.
Until recently, the role of bacterial genetics in M.tb clinical outcome was only associated with the host and environmental factors (Warner et al., 2015). With the advent of whole genome sequencing (WGS), seven human-adapted lineages were reported and associated with distinct virulence and transmissibility (Coscolla and Gagneux, 2014). The modern M.tb lineages (Lineages 2, 3, and 4) have been reported to be more virulent and transmissible than the ancient lineages (Lineages 1, 5, 6, and 7) (Wang et al., 2010; Sarkar et al., 2012). This heterogeneity in virulence and transmissibility has implications for the clinical outcome of the disease. For example, in a preclinical M.tb infection model, virulent Beijing strains cause higher bacillary loads, more lung damage, and earlier mortality compared to strains from other lineages (Ordway et al., 2007). Follow-up mechanistic studies have suggested that Beijing strains have enhanced capacity to inhibit protective immunity in the lungs through induction of higher levels of type-I interferons, lower levels of IL-12 and TNF-α, and reduced CD4/CD8+T-cell activation (Manca et al., 2001; Reed et al., 2004; Manca et al., 2005). However, other studies have indicated that the Beijing strains induce a more robust regulatory T-cell response than different strains, thereby down-regulating protective immunity (Ordway et al., 2007; Shang et al., 2011). Furthermore, a mouse infection model demonstrated that different M.tb transmission phenotypes are associated with distinct pulmonary pathologies (Verma et al., 2019; Lovey et al., 2022). High transmission M.tb strains (M.tb-HT) were related to necrotic caseating lung granulomas with great potential to cavitate, while low transmission strains (M.tb-LT) were associated with diffuse lung inflammation. Therefore, the distinct pulmonary pathologies related to the different M.tb genotypes could have implications for COPD. Literature examining the relationship between M.tb strains and COPD pathology is very limited. With recent evidence for pulmonary impairment after tuberculosis (PAIT) in the context of high versus low M.tb transmission strains, investigators are now examining the relationship between high vs. low transmission M.tb strains and COPD outcome phenotype. Follow-up human studies involving spirometry and radiological imaging in individuals diagnosed with high transmission versus low transmission strains will illuminate this relationship further.
Immune pathways orchestrating lung tissue damage and fibrosis in tuberculosis or post-tuberculosis disease warrant a careful search into possibilities of using a select group of immune-based therapies optimal for patients diagnosed with tuberculosis-associated COPD (Barnes and Stockley, 2005). This section discusses a select list of immune-based therapies for tuberculosis, traditionally referred to as host-directed therapies (HDTs). Host-directed immune therapies have been extensively reviewed as potential adjuvants to the standard M.tb treatment and have demonstrated the potential to shorten TB treatment duration, curb emerging antimicrobial drug resistance, and reduce airway injury and fibrosis in post-TB patients (Wallis and Hafner, 2015; O’Connor et al., 2016; Wallis et al., 2016; Kolloli and Subbian, 2017; Yang, 2017; Du Plessis et al., 2018; Kaufmann et al., 2018). Figure 2 summarizes the potential targets of host-directed therapy that could be used to treat TB-associated COPD. Below, we discuss a few host-directed therapies (HDTs) that have shown promise in clinical trials. Phosphodiesterase 4 (PDE4) inhibitor has shown promise in clinical investigations. In a 6-week trial, a PDE4 inhibitor improved lung function among individuals with moderate-to-severe COPD (Vestbo et al., 2009). In patients with advanced TB disease, PDE4 inhibitors improved FEV1 at six months post-treatment (Wallis et al., 2021). mTOR inhibitors have also been investigated as host-directed therapies in TB patients. In a similar study as described above, Everolimus, an mTOR inhibitor, enhanced the recovery of FEV1 at month 6 post-TB treatment (Wallis et al., 2021). Auranofin (oral gold salt), and ergocalciferol (vitamin D), tested in the same study, were reported ineffective (Wallis et al., 2021). Leukotriene BLT1-receptor antagonists had been developed to treat neutrophilic inflammation among COPD patients (Silbaugh et al., 2000; Beeh et al., 2003). The same antagonists also inhibit the neutrophil chemotactic activity of sputum obtained from COPD patients, indicating their potential clinical value (Silbaugh et al., 2000; Beeh et al., 2003). A human monoclonal anti-IL-8 neutralizing antibody has been tested in tuberculosis associated-COPD. Whereas it minimally reduced dyspnea scores, there was no statistically significant clinical improvement field (Mahler et al., 2004). CXCR2 inhibitors have proven particularly beneficial in COPD and are being tested in clinical trials (Widdowson et al., 2004). Also, CXCR3 antagonists, which inhibit the recruitment of CD8+ T-cells into airways, might be helpful (Saetta et al., 2002). Antioxidants such as N-acetyl cysteine, stable glutathione compounds, analogs of superoxide dismutase, and selenium-based drugs have also been developed for clinical use and selective inhibitors of iNOS (MacNee, 2000; Kharitonov and Barnes, 2003). Using non-selective signal transduction pathways inhibitors such as NF-kB and p38 MAPK inhibitors may result in immune suppression and impair host defense, worsening tuberculosis disease (Underwood et al., 2000; Castro et al., 2003). However, more selective PI3Kγ inhibitors may have relevant anti-inflammatory activity in COPD, and small molecule inhibitors of PI-3Kγ have been developed (Sasaki et al., 2000). In addition, Metformin and statins have been implicated as potential therapeutic agents for lung fibrosis (Wallis and Hafner, 2015; Sato et al., 2016; Tahir et al., 2020). Mechanistically, metformin enhances autophagy and improves mitochondrial bioenergetics (Bharath et al., 2020), whereas statins block 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase, the rate-limiting enzyme in the cholesterol biosynthesis pathway (Stancu and Sima, 2001).
In summary, whereas host-directed therapies have been extensively investigated as potential adjuvants to the standard M.tb treatment and have demonstrated the potential to shorten TB treatment duration, such evidence is lacking for COPD after tuberculosis. More importantly, the exact time when HDTs should be started in TB patients is unknown. Therefore, more studies are warranted testing the timing of administration and effectiveness of these molecules in the context of TB-associated COPD.
In summary, in this review article, we have described immune pathways that may drive the immunopathogenesis of tuberculosis-associated COPD and their implications in the management of tuberculosis associated-COPD. The immune responses are predominantly characterized by activation and migration of alveolar macrophages from alveolar spaces into the lung parenchyma, coupled with recruitment of neutrophils; monocytes derived macrophages, B-cells, innate lymphoid cells, Th1, Th17, and cytotoxic CD8+T-cells into lung microenvironment. Their interaction with airway epithelial cells, fibroblasts, and the extracellular matrix culminates in sustained lung inflammation, tissue necrosis, and airway remodeling. Therefore, these changes gradually compromise lung function. Using this knowledge, we can harness the latest developments in COPD immune therapies and utilize them to make host-directed therapeutic choices optimal for tuberculosis-associated COPD or prevent lung injury.
AK: Conceptualization, Methodology, Original draft writing, and editing; BK, TS, BN, and WC: draft review and editing; ML: funding acquisition, draft review, and editing; JE: funding acquisition, draft review, and editing and PS: Conceptualization, Mentorship, Design, funding acquisition and drafting the manuscript for important intellectual content. All authors contributed to the article and approved the submitted version.
This review article was funded by a grant supported by the US National Institutes of Health through theFogarty International Center, NIH award number D43TW009093, titled Training of Ugandans in Basic and Translational Research on TB and Emerging Infectious Diseases. Its contents are solely the authors’ responsibility and do not necessarily represent the official views of the supporting offices. AK is also funded by a grant supported by the US National Institutes of Health through the Fogarty International Center, NIH award number R21TW012354. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the supporting offices.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abadie, V., Badell, E., Douillard, P., Ensergueix, D., Leenen, P. J., Tanguy, M., et al. (2005). Neutrophils rapidly migrate via lymphatics after mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood 106 (5), 1843–1850. doi: 10.1182/blood-2005-03-1281
Abbas, A. K., Lichtman, A. H., Pillai, S. (2019). Basic immunology e-book: functions and disorders of the immune system (Amsterdam, Netherlands: Elsevier Health Sciences).
Abramovitch, R. B., Rohde, K. H., Hsu, F. F., Russell, D. G. (2011). aprABC: a mycobacterium tuberculosis complex-specific locus that modulates pH-driven adaptation to the macrophage phagosome. Mol. Microbiol. 80 (3), 678–694. doi: 10.1111/j.1365-2958.2011.07601.x
Adeloye, D., Chua, S., Lee, C., Basquill, C., Papana, A., Theodoratou, E., et al. (2015). Global and regional estimates of COPD prevalence: systematic review and meta–analysis. J. Global Health 5 (2), 020415–020415. doi: 10.7189/jogh.05.020415
Agarwal, P., Gordon, S., Martinez, F. O. (2021). Foam cell macrophages in tuberculosis. Front. Immunol. 12, 775326. doi: 10.3389/fimmu.2021.775326
Agustí, A., Celli, B. (2017). Natural history of COPD: gaps and opportunities. ERJ. Open Res. 3 (4). doi: 10.1183/23120541.00117-2017
Ahmad, F., Rani, A., Alam, A., Zarin, S., Pandey, S., Singh, H., et al. (2022). Macrophage: a cell with many faces and functions in tuberculosis. Front. Immunol. 13. doi: 10.3389/fimmu.2022.747799
Algood, H. M., Lin, P. L., Flynn, J. L. (2005). Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin. Infect. Dis. 41 Suppl 3, S189–S193. doi: 10.1086/429994
Allwood, B. W., Gillespie, R., Galperin-Aizenberg, M., Bateman, M., Olckers, H., Taborda-Barata, L., et al. (2014). Mechanism of airflow obstruction in tuberculosis-associated obstructive pulmonary disease (TOPD). D39 CONNECTING THE DOTS: DRAWING LINES BETWEEN COPD AND COMORBID CONDITIONS (New York, USA: American Thoracic Society), A5832–A583A.
Allwood, B., Rigby, J., Griffith-Richards, S., Kanarek, D., du Preez, L., Mathot, B., et al. (2019). Histologically confirmed tuberculosis-associated obstructive pulmonary disease. Int. J. Tuberculosis. Lung Dis. 23 (5), 552–554. doi: 10.5588/ijtld.18.0722
Amaral, A. F., Coton, S., Kato, B., Tan, W. C., Studnicka, M., Janson, C., et al. (2015). Tuberculosis associates with both airflow obstruction and low lung function: BOLD results. Eur. Respir. J. 46 (4), 1104–1112. doi: 10.1183/13993003.02325-2014
Anno, H., Tomashefski, J. F. (1955). Studies on the impairment of respiratory function in pulmonary tuberculosis. Am. Rev. Tuberculosis. Pulmonary. Dis. 71 (3-1), 333–348. doi: 10.1164/artpd.1955.71.3-1.333
Ardain, A., Domingo-Gonzalez, R., Das, S., Kazer, S. W., Howard, N. C., Singh, A., et al. (2019). Group 3 innate lymphoid cells mediate early protective immunity against tuberculosis. Nature 570 (7762), 528–532. doi: 10.1038/s41586-019-1276-2
Arish, M., Naz, F. (2022). Macrophage plasticity as a therapeutic target in tuberculosis. Eur. J. Immunol. 52 (5), 696–704. doi: 10.1002/eji.202149624
Arora, S., Dev, K., Agarwal, B., Das, P., Syed, M. A. (2018). Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology 223 (4-5), 383–396. doi: 10.1016/j.imbio.2017.11.001
Artis, D. (2008). Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 8 (6), 411–420. doi: 10.1038/nri2316
Atkinson, J. J., Lutey, B. A., Suzuki, Y., Toennies, H. M., Kelley, D. G., Kobayashi, D. K., et al. (2011). The role of matrix metalloproteinase-9 in cigarette smoke–induced emphysema. Am. J. Respir. Crit. Care Med. 183 (7), 876–884. doi: 10.1164/rccm.201005-0718OC
Augenstreich, J., Arbues, A., Simeone, R., Haanappel, E., Wegener, A., Sayes, F., et al. (2017). ESX-1 and phthiocerol dimycocerosates of mycobacterium tuberculosis act in concert to cause phagosomal rupture and host cell apoptosis. Cell. Microbiol. 19 (7), e12726. doi: 10.1111/cmi.12726
Awuh, J. A., Flo, T. H. (2017). Molecular basis of mycobacterial survival in macrophages. Cell. Mol. Life Sci. 74, 1625–1648. doi: 10.1007/s00018-016-2422-8
Bagcchi, S. (2023). WHO’s global tuberculosis report 2022. Lancet Microbe 4 (1), e20. doi: 10.1016/S2666-5247(22)00359-7
Banchereau, J., Steinman, R. M. (1998). Dendritic cells and the control of immunity. Nature 392 (6673), 245–252. doi: 10.1038/32588
Barbu, C., Iordache, M., Man, M. (2011). Inflammation in COPD: pathogenesis, local and systemic effects. Rom. J. Morphol. Embryol. 52 (1), 21–27. doi: 10.1136/thorax.57.12.1067
Barnes, P. J., Shapiro, S. D., Pauwels, R. (2003). Chronic obstructive pulmonary disease: molecular and cellularmechanisms. Eur. Respir. J. 22 (4), 672–688. doi: 10.1183/09031936.03.00040703
Barnes, P., Stockley, R. (2005). COPD: current therapeutic interventions and future approaches. Eur. Respir. J. 25 (6), 1084–1106. doi: 10.1183/09031936.05.00139104
Bautista, M. V., Chen, Y., Ivanova, V. S., Rahimi, M. K., Watson, A. M., Rose, M. C. (2009). IL-8 regulates mucin gene expression at the posttranscriptional level in lung epithelial cells. J. Immunol. 183 (3), 2159–2166. doi: 10.4049/jimmunol.0803022
Beeh, K. M., Kornmann, O., Buhl, R., Culpitt, S. V., Giembycz, M. A., Barnes, P. J. (2003). Neutrophil chemotactic activity of sputum from patients with COPD: role of interleukin 8 and leukotriene B4. Chest 123 (4), 1240–1247. doi: 10.1378/chest.123.4.1240
Behar, S., Martin, C., Booty, M., Nishimura, T., Zhao, X., Gan, H., et al. (2011). Apoptosis is an innate defense function of macrophages against mycobacterium tuberculosis. Mucosal Immunol. 4 (3), 279–287. doi: 10.1038/mi.2011.3
Bharath, L. P., Agrawal, M., McCambridge, G., Nicholas, D. A., Hasturk, H., Liu, J., et al. (2020). Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 32 (1), 44–55. e6. doi: 10.1016/j.cmet.2020.04.015
Blumenthal, A., Kobayashi, T., Pierini, L. M., Banaei, N., Ernst, J. D., Miyake, K., et al. (2009). RP105 facilitates macrophage activation by mycobacterium tuberculosis lipoproteins. Cell Host Microbe 5 (1), 35–46. doi: 10.1016/j.chom.2008.12.002
Bogdan, C. (2001). Nitric oxide and the immune response. Nat. Immunol. 2 (10), 907–916. doi: 10.1038/ni1001-907
BoseDasgupta, S., Pieters, J. (Eds.) (2018). “Macrophage-microbe interaction: lessons learned from the pathogen mycobacterium tuberculosis,” in Seminars in immunopathology (Germany: Springer Berlin Heidelberg).
Braian, C., Hogea, V., Stendahl, O. (2013). Mycobacterium tuberculosis-induced neutrophil extracellular traps activate human macrophages. J. Innate. Immunity. 5 (6), 591–602. doi: 10.1159/000348676
Brandsma, C.-A., Hylkema, M. N., Geerlings, M., van Geffen, W. H., Postma, D. S., Timens, W., et al. (2009). Increased levels of (class switched) memory b cells in peripheral blood of current smokers. Respir. Res. 10 (1), 108. doi: 10.1186/1465-9921-10-108
Brandtzaeg, P. (2015). The mucosal b cell system. Mucosal Immunol.: Elsevier; p, 623–681. doi: 10.1016/B978-0-12-415847-4.00031-8
Burgdorf, S., Kautz, A., Boühnert, V., Knolle, P. A., Kurts, C. (2007). Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. science 316 (5824), 612–616. doi: 10.1126/science.1137971
Buter, J., Cheng, T.-Y., Ghanem, M., Grootemaat, A. E., Raman, S., Feng, X., et al. (2019). Mycobacterium tuberculosis releases an antacid that remodels phagosomes. Nat. Chem. Biol. 15 (9), 889–899. doi: 10.1038/s41589-019-0336-0
Byanova, K., Kunisaki, K. M., Vasquez, J., Huang, L. (2021). Chronic obstructive pulmonary disease in HIV. Expert Rev. Respir. Med. 15 (1), 71–87. doi: 10.1080/17476348.2021.1848556
Byrne, A. L., Marais, B. J., Mitnick, C. D., Lecca, L., Marks, G. B. (2015). Tuberculosis and chronic respiratory disease: a systematic review. Int. J. Infect. Dis. 32, 138–146. doi: 10.1016/j.ijid.2014.12.016
Caballero, A., Torres-Duque, C. A., Jaramillo, C., Bolívar, F., Sanabria, F., Osorio, P., et al. (2008). Prevalence of COPD in five Colombian cities situated at low, medium, and high altitude (PREPOCOL study). Chest 133 (2), 343–349. doi: 10.1378/chest.07-1361
Cadena, A. M., Fortune, S. M., Flynn, J. L. (2017). Heterogeneity in tuberculosis. Nat. Rev. Immunol. 17 (11), 691–702. doi: 10.1038/nri.2017.69
Cao, J., Zhang, L., Li, D., Xu, F., Huang, S., Xiang, Y., et al. (2012). IL-27 is elevated in patients with COPD and patients with pulmonary TB and induces human bronchial epithelial cells to produce CXCL10. Chest 141 (1), 121–130. doi: 10.1378/chest.10-3297
Casadevall, A., Pirofski, L. A. (2003). The damage-response framework of microbial pathogenesis. Nat. Rev. Microbiol. 1 (1), 17–24. doi: 10.1038/nrmicro732
Castro, A. C., Dang, L. C., Soucy, F., Grenier, L., Mazdiyasni, H., Hottelet, M., et al. (2003). Novel IKK inhibitors: β-carbolines. Bioorg. Med. Chem. Lett. 13 (14), 2419–2422. doi: 10.1016/S0960-894X(03)00408-6
Chakrabarti, B., Calverley, P. M., Davies, P. D. (2007). Tuberculosis and its incidence, special nature, and relationship with chronic obstructive pulmonary disease. Int. J. Chronic. Obstructive. Pulmonary. Dis. 2 (3), 263.
Chan, J., Mehta, S., Bharrhan, S., Chen, Y., Achkar, J. M., Casadevall, A., et al. (2014). “The role of b cells and humoral immunity in mycobacterium tuberculosis infection,” in Seminars in immunology (Springer Berlin Heidelberg Germany: Elsevier).
Chatterjee, B., Smed-Sörensen, A., Cohn, L., Chalouni, C., Vandlen, R., Lee, B.-C., et al. (2012). Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood. J. Am. Soc. Hematol. 120 (10), 2011–2020. doi: 10.1182/blood-2012-01-402370
Chen, M., Gan, H., Remold, H. G. (2006). A mechanism of virulence: virulent mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J. Immunol. 176 (6), 3707–3716. doi: 10.4049/jimmunol.176.6.3707
Chen, Q., Hu, C., Lu, W., Hang, T., Shao, Y., Chen, C., et al. (2022). Characteristics of alveolar macrophages in bronchioalveolar lavage fluids from active tuberculosis patients identified by single-cell RNA sequencing. J. Biomed. Res. 36 (3), 167. doi: 10.7555/JBR.36.20220007
Chin, A., Rylance, J., Makumbirofa, S., Meffert, S., Vu, T., Clayton, J., et al. (2019). Chronic lung disease in adult recurrent tuberculosis survivors in Zimbabwe: a cohort study. Int. J. Tuberculosis. Lung Dis. 23 (2), 203–211. doi: 10.5588/ijtld.18.0313
Chung, K., Adcock, I. (2008). Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur. Respir. J. 31 (6), 1334–1356. doi: 10.1183/09031936.00018908
Chung, K.-P., Chen, J.-Y., Lee, C.-H., Wu, H.-D., Wang, J.-Y., Lee, L.-N., et al. (2011). Trends and predictors of changes in pulmonary function after treatment for pulmonary tuberculosis. Clinics 66 (4), 549–556. doi: 10.1590/S1807-59322011000400005
Churg, A., Cosio, M., Wright, J. L. (2008). Mechanisms of cigarette smoke-induced COPD: insights from animal models. Am. J. Physiology-Lung. Cell. Mol. Physiol. 294 (4), L612–LL31. doi: 10.1152/ajplung.00390.2007
Churg, A., Zhou, S., Wright, J. L. (2012). Matrix metalloproteinases in COPD. Eur. Respir. J. 39 (1), 197–209. doi: 10.1183/09031936.00121611
Chushkin, M. I., Ots, O. N. (2017). Impaired pulmonary function after treatment for tuberculosis: the end of the disease? J. Brasileiro. Pneumol. 43, 38–43. doi: 10.1590/s1806-37562016000000053
Clarke, S. R. M. (2000). The critical role of CD40/CD40L in the CD4-dependent generation of CD8+ T cell immunity. J. Leukocyte. Biol. 67 (5), 607–614. doi: 10.1002/jlb.67.5.607
Cohen, S. B., Gern, B. H., Delahaye, J. L., Adams, K. N., Plumlee, C. R., Winkler, J. K., et al. (2018). Alveolar macrophages provide an early mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe 24 (3), 439–46. e4. doi: 10.1016/j.chom.2018.08.001
Cohen, S. B., Gern, B. H., Urdahl, K. B. (2022). The tuberculous granuloma and preexisting immunity. Annu. Rev. Immunol. 40, 589–614. doi: 10.1146/annurev-immunol-093019-125148
Cohn, L., Chatterjee, B., Esselborn, F., Smed-Sörensen, A., Nakamura, N., Chalouni, C., et al. (2013). Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3+ dendritic cells at cross presentation. J. Exp. Med. 210 (5), 1049–1063. doi: 10.1084/jem.20121251
Conrad, W. H., Osman, M. M., Shanahan, J. K., Chu, F., Takaki, K. K., Cameron, J., et al. (2017). Mycobacterial ESX-1 secretion system mediates host cell lysis through bacterium contact-dependent gross membrane disruptions. Proc. Natl. Acad. Sci. 114 (6), 1371–1376. doi: 10.1073/pnas.1620133114
Cooper, A. M. (2009). Cell-mediated immune responses in tuberculosis. Annu. Rev. Immunol. 27, 393–422. doi: 10.1146/annurev.immunol.021908.132703
Cooper, A., Roberts, A., Rhoades, E., Callahan, J., Getzy, D., Orme, I. (1995). The role of interleukin-12 in acquired immunity to mycobacterium tuberculosis infection. Immunology 84 (3), 423.
Cooper, A. M., Solache, A., Khader, S. A. (2007). Interleukin-12 and tuberculosis: an old story revisited. Curr. Opin. Immunol. 19 (4), 441–447. doi: 10.1016/j.coi.2007.07.004
Coscolla, M., Gagneux, S. (2014). “Consequences of genomic diversity in mycobacterium tuberculosis,” in Seminars in immunology (Springer Berlin Heidelberg Germany: Elsevier).
Craig, J. M., Scott, A. L., Mitzner, W. (2017). Immune-mediated inflammation in the pathogenesis of emphysema: insights from mouse models. Cell Tissue Res. 367 (3), 591–605. doi: 10.1007/s00441-016-2567-7
Cruz, A. A., Bousquet, J., Khaltaev, N. (2007). Global surveillance, prevention and control of chronic respiratory diseases: a comprehensive approach (Elsevier, Kidlington, United Kingdom: World Health Organization).
Dallenga, T., Repnik, U., Corleis, B., Eich, J., Reimer, R., Griffiths, G. W., et al. (2017). M. tuberculosis-induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host Microbe 22 (4), 519–30.e3. doi: 10.1016/j.chom.2017.09.003
Dallenga, T., Schaible, U. E. (2016). Neutrophils in tuberculosis–first line of defence or booster of disease and targets for host-directed therapy? Pathog. Dis. 74 (3). doi: 10.1093/femspd/ftw012
Darrah, P. A., Patel, D. T., De Luca, P. M., Lindsay, R. W., Davey, D. F., Flynn, B. J., et al. (2007). Multifunctional TH 1 cells define a correlate of vaccine-mediated protection against leishmania major. Nat. Med. 13 (7), 843–850. doi: 10.1038/nm1592
De Godoy, I., Donahoe, M., Calhoun, W. J., Mancino, J., Rogers, R. M. (1996). Elevated TNF-alpha production by peripheral blood monocytes of weight-losing COPD patients. Am. J. Respir. Crit. Care Med. 153 (2), 633–637. doi: 10.1164/ajrccm.153.2.8564110
Demedts, I. K., Bracke, K. R., Van Pottelberge, G., Testelmans, D., Verleden, G. M., Vermassen, F. E., et al. (2007). Accumulation of dendritic cells and increased CCL20 levels in the airways of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 175 (10), 998–1005. doi: 10.1164/rccm.200608-1113OC
de Melo, M. G. M., Mesquita, E. D. D., Oliveira, M. M., Silva-Monteiro, Cd, Silveira, A. K., Malaquias, T. S., et al. (2019). Imbalance of NET and alpha-1-antitrypsin in tuberculosis patients is related with hyper inflammation and severe lung tissue damage. Front. Immunol. 9, 3147. doi: 10.3389/fimmu.2018.03147
Dheda, K., Booth, H., Huggett, J. F., Johnson, M. A., Zumla, A., Rook, G. A. (2005). Lung remodeling in pulmonary tuberculosis. J. Infect. Dis. 192 (7), 1201–1209. doi: 10.1086/444545
Donald, P. R., Diacon, A. H., Thee, S. (2021). Anton Ghon and his colleagues and their studies of the primary focus and complex of tuberculosis infection and their relevance for the twenty-first century. Respiration 100 (7), 557–567. doi: 10.1159/000509522
Donato, R., R cannon, B., Sorci, G., Riuzzi, F., Hsu, K., J Weber, D., et al. (2013). Functions of S100 proteins. Curr. Mol. Med. 13 (1), 24–57. doi: 10.2174/156652413804486214
Doss, M., White, M. R., Tecle, T., Hartshorn, K. L. (2010). Human defensins and LL-37 in mucosal immunity. J. Leukocyte. Biol. 87 (1), 79–92. doi: 10.1189/jlb.0609382
Du Plessis, W. J., Keyser, A., Walzl, G., Loxton, A. G. (2016a). Phenotypic analysis of peripheral b cell populations during mycobacterium tuberculosis infection and disease. J. Inflamm. 13 (1), 23. doi: 10.1186/s12950-016-0133-4
Du Plessis, W. J., Kleynhans, L., Du Plessis, N., Stanley, K., Malherbe, S. T., Maasdorp, E., et al. (2016b). The functional response of b cells to antigenic stimulation: a preliminary report of latent tuberculosis. PloS One 11 (4), e0152710. doi: 10.1371/journal.pone.0152710
Du Plessis, N., Kotze, L. A., Leukes, V, Walzl, G.. (2018). Tuberculosis: progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect. Dis. 18 (7), e183–ee98. doi: 10.3389/fcimb.2018.00332
Ehlers, S., Schaible, U. E. (2013). The granuloma in tuberculosis: dynamics of a host–pathogen collusion. Front. Immunol. 3, 411. doi: 10.3389/fimmu.2012.00411
Ernst, J. D. (2012). The immunological life cycle of tuberculosis. Nat. Rev. Immunol. 12 (8), 581–591. doi: 10.1038/nri3259
Etna, M. P., Giacomini, E., Severa, M., Coccia, M. E. (Eds.) (2014). “Pro-and anti-inflammatory cytokines in tuberculosis: a two-edged sword in TB pathogenesis,” in Seminars in immunology (Elsevier).
Eum, S. Y., Kong, J. H., Hong, M. S., Lee, Y. J., Kim, J. H., Hwang, S. H., et al. (2010). Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest 137 (1), 122–128. doi: 10.1378/chest.09-0903
Ezzati Givi, M., Redegeld F, A., Folkerts, G., Mortaz, E. (2012). Dendritic cells in pathogenesis of COPD. Curr. Pharm. Design. 18 (16), 2329–2335. doi: 10.2174/138161212800166068
Fan, Q., Lu, M., Xia, Z. Y., Bao, L. (2013). Mycobacterium tuberculosis MPT64 stimulates the activation of murine macrophage modulated by IFN-γ. Eur. Rev. Med. Pharmacol. Sci. 17 (24), 3296–3305. doi: 10.1007/s00330-012-2729-2
Feng, C. G., Bean, A. G., Hooi, H., Briscoe, H., Britton, W. J. (1999). Increase in gamma interferon-secreting CD8+, as well as CD4+, T cells in lungs following aerosol infection with mycobacterium tuberculosis. Infect. Immunity. 67 (7), 3242–3247. doi: 10.1128/IAI.67.7.3242-3247.1999
Ferrari, G., Langen, H., Naito, M., Pieters, J. (1999). A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell 97 (4), 435–447. doi: 10.1016/S0092-8674(00)80754-0
Flynn, J. L. (2004). Immunology of tuberculosis and implications in vaccine development. Tuberculosis. (Edinb). 84 (1-2), 93–101. doi: 10.1016/j.tube.2003.08.010
Flynn, J. L., Chan, J. (2001). Immunology of tuberculosis. Annu. Rev. Immunol. 19 (1), 93–129. doi: 10.1146/annurev.immunol.19.1.93
Fratti, R. A., Chua, J., Vergne, I., Deretic, V. (2003). Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc. Natl. Acad. Sci. 100 (9), 5437–5442. doi: 10.1073/pnas.0737613100
Fulton, S. A., Martin, T. D., Redline, R. W., Henry Boom, W. (2000). Pulmonary immune responses during primary mycobacterium bovis–Calmette-Guerin bacillus infection in C57Bl/6 mice. Am. J. Respir. Cell Mol. Biol. 22 (3), 333–343. doi: 10.1165/ajrcmb.22.3.3776
Gandhi, K., Gupta, S., Singla, R. (2016). Risk factors associated with development of pulmonary impairment after tuberculosis. Indian J. Tuberculosis. 63 (1), 34–38. doi: 10.1016/j.ijtb.2016.01.006
Ganz, T. (2003). Defensins: antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 3 (9), 710–720. doi: 10.1038/nri1180
Gao, W., Li, L., Wang, Y., Zhang, S., Adcock, I. M., Barnes, P. J., et al. (2015). Bronchial epithelial cells: the key effector cells in the pathogenesis of chronic obstructive pulmonary disease? Respirology 20 (5), 722–729. doi: 10.1111/resp.12542
Gatfield, J., Pieters, J. (2000). Essential role for cholesterol in entry of mycobacteria into macrophages. Science 288 (5471), 1647–1651. doi: 10.1126/science.288.5471.1647
Ge, G., Jiang, H., Xiong, J., Zhang, W., Shi, Y., Tao, C., et al. (2021). Progress of the art of macrophage polarization and different subtypes in mycobacterial infection. Front. Immunol. 4570. doi: 10.3389/fimmu.2021.752657
Geissmann, F., Dieu-Nosjean, M., Dezutter, C., Valladeau, J., Kayal, S., Leborgne, M., et al. (2002). Accumulation of immature langerhans cells in human lymph nodes draining chronically inflamed skin. J. Exp. Med. 196 (4), 417–430. doi: 10.1084/jem.20020018
Gern, B. H., Adams, K. N., Plumlee, C. R., Stoltzfus, C. R., Shehata, L., Moguche, A. O., et al. (2021). TGFβ restricts expansion, survival, and function of T cells within the tuberculous granuloma. Cell Host Microbe 29 (4), 594–606. e6. doi: 10.1016/j.chom.2021.02.005
Gil, O., Guirado, E., Gordillo, S., Díaz, J., Tapia, G., Vilaplana, C., et al. (2006). Intragranulomatous necrosis in lungs of mice infected by aerosol with mycobacterium tuberculosis is related to bacterial load rather than to any one cytokine or T cell type. Microbes Infect. 8 (3), 628–636. doi: 10.1016/j.micinf.2005.08.014
Grabcanovic-Musija, F., Obermayer, A., Stoiber, W., Krautgartner, W.-D., Steinbacher, P., Winterberg, N., et al. (2015). Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir. Res. 16 (1), 1–12. doi: 10.1186/s12931-015-0221-7
Grzela, K., Litwiniuk, M., Zagorska, W., Grzela, T. (2016). Airway remodeling in chronic obstructive pulmonary disease and asthma: the role of matrix metalloproteinase-9. Archivum. Immunol. Ther. Experimentalis. 64, 47–55. doi: 10.1007/s00005-015-0345-y
Guirado, E., Schlesinger, L. S., Kaplan, G. (Eds.) (2013). “Macrophages in tuberculosis: friend or foe,” in Seminars in immunopathology (Lausanne, Switzerland: Springer).
Gupta, R. K., Gupta, K., Dwivedi, P. D. (2017). Pathophysiology of IL-33 and IL-17 in allergic disorders. Cytokine Growth Factor. Rev. 38, 22–36. doi: 10.1016/j.cytogfr.2017.09.005
Harding, E. (2020). WHO global progress report on tuberculosis elimination. Lancet Respir. Med. 8 (1), 19. doi: 10.1016/S2213-2600(19)30418-7
Herr, C., Shaykhiev, R., Bals, R. (2007). The role of cathelicidin and defensins in pulmonary inflammatory diseases. Expert Opin. Biol. Ther. 7 (9), 1449–1461. doi: 10.1517/14712598.7.9.1449
Hewitt, R. J., Lloyd, C. M. (2021). Regulation of immune responses by the airway epithelial cell landscape. Nat. Rev. Immunol. 21 (6), 347–362. doi: 10.1038/s41577-020-00477-9
Hmama, Z., Peña-Díaz, S., Joseph, S., Av-Gay, Y. (2015). Immunoevasion and immunosuppression of the macrophage by m ycobacterium tuberculosis. Immunol. Rev. 264 (1), 220–232. doi: 10.1111/imr.12268
Hnizdo, E., Singh, T., Churchyard, G. (2000). Chronic pulmonary function impairment caused by initial and recurrent pulmonary tuberculosis following treatment. Thorax 55 (1), 32–38. doi: 10.1136/thorax.55.1.32
Hogg, J. C. (2004). Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet 364 (9435), 709–721. doi: 10.1056/NEJMoa032158
Holt, P. G., Strickland, D. H., Wikström, M. E., Jahnsen, F. L. (2008). Regulation of immunological homeostasis in the respiratory tract. Nat. Rev. Immunol. 8 (2), 142–152. doi: 10.1038/nri2236
Houghton, A. M. (2015). Matrix metalloproteinases in destructive lung disease. Matrix. Biol. 44, 167–174. doi: 10.1016/j.matbio.2015.02.002
Huffnagle, G., Dickson, R., Lukacs, N. (2017). The respiratory tract microbiome and lung inflammation: a two-way street. Mucosal Immunol. 10 (2), 299–306. doi: 10.1038/mi.2016.108
Innes, S., Schaaf, H., Cotton, M. (2009). Cavitation of the ghon focus in an hiv-infected infant who acquired tuberculosis after the initiation of haart. South. Afr. J. HIV Med. 10 (1). doi: 10.4102/sajhivmed.v10i1.1001
Ivanova, O., Hoffmann, V. S., Lange, C., Hoelscher, M., Rachow, A. (2023). Post-tuberculosis lung impairment: systematic review and meta-analysis of spirometry data from 14 621 people. Eur. Respir. Rev. 32 (168), 220221. doi: 10.1183/16000617.0221-2022
Jahnsen, F. L., Strickland, D. H., Thomas, J. A., Tobagus, I. T., Napoli, S., Zosky, G. R., et al. (2006). Accelerated antigen sampling and transport by airway mucosal dendritic cells following inhalation of a bacterial stimulus. J. Immunol. 177 (9), 5861–5867. doi: 10.4049/jimmunol.177.9.5861
Jamwal, S. V., Mehrotra, P., Singh, A., Siddiqui, Z., Basu, A., Rao, K. V. (2016). Mycobacterial escape from macrophage phagosomes to the cytoplasm represents an alternate adaptation mechanism. Sci. Rep. 6 (1), 23089. doi: 10.1038/srep23089
Joint United Nations Programme on HA (2011). Countdown to zero: global plan towards the elimination of new HIV infections among children by 2015 and keeping their mothers alive, 2011-2015 (Geneva: UNAIDS).
Jonuleit, H., Schmitt, E., Schuler, G., Knop, J., Enk, A. H. (2000). Induction of interleukin 10–producing, nonproliferating CD4+ T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J. Exp. Med. 192 (9), 1213–1222. doi: 10.1084/jem.192.9.1213
Kalams, S. A., Walker, B. D. (1998). The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J. Exp. Med. 188 (12), 2199–2204. doi: 10.1084/jem.188.12.2199
Kamenar, K., Hossen, S., Gupte, A. N., Siddharthan, T., Pollard, S., Chowdhury, M., et al. (2021). Previous tuberculosis disease as a risk factor for chronic obstructive pulmonary disease: a cross-sectional analysis of multicountry, population-based studies. Thorax 77 (11), 1088–1097. doi: 10.1136/thoraxjnl-2020-216500
Kapsenberg, M. L. (2003). Dendritic-cell control of pathogen-driven T-cell polarization. Nat. Rev. Immunol. 3 (12), 984–993. doi: 10.1038/nri1246
Kasmi, K. C. E., Stenmark, K. R. (Eds.) (2015). “Contribution of metabolic reprogramming to macrophage plasticity and function,” in Seminars in immunology (Springer Berlin Heidelberg, Germany: Elsevier).
Kaufmann, S. H. (2002). Protection against tuberculosis: cytokines, T cells, and macrophages. Ann. Rheumatic. Dis. 61 (suppl 2), ii54–iii8. doi: 10.1136/ard.61.suppl_2.ii54
Kaufmann, S. H., Dorhoi, A., Hotchkiss, R. S., Bartenschlager, R. (2018). Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discov. 17 (1), 35–56. doi: 10.1038/nrd.2017.162
Kayongo, A., Wosu, A. C., Naz, T., Nassali, F., Kalyesubula, R., Kirenga, B., et al. (2020). Chronic obstructive pulmonary disease prevalence and associated factors in a setting of well-controlled HIV, a cross-sectional study. COPD.: J. Chronic. Obstructive. Pulmonary. Dis. 17 (3), 297–305. doi: 10.1080/15412555.2020.1769583
Kessel, C., Holzinger, D., Foell, D. (2013). Phagocyte-derived S100 proteins in autoinflammation: putative role in pathogenesis and usefulness as biomarkers. Clin. Immunol. 147 (3), 229–241. doi: 10.1016/j.clim.2012.11.008
Khader, S. A., Pearl, J. E., Sakamoto, K., Gilmartin, L., Bell, G. K., Jelley-Gibbs, D. M., et al. (2005). IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-γ responses if IL-12p70 is available. J. Immunol. 175 (2), 788–795. doi: 10.4049/jimmunol.175.2.788
Khan, A., Singh, V. K., Hunter, R. L., Jagannath, C. (2019). Macrophage heterogeneity and plasticity in tuberculosis. J. Leukoc. Biol. 106 (2), 275–282. doi: 10.1002/JLB.MR0318-095RR
Kharitonov, S. A., Barnes, P. J. (2003). Nitric oxide, nitrotyrosine, and nitric oxide modulators in asthma and chronic obstructive pulmonary disease. Curr. Allergy Asthma Rep. 3 (2), 121–129. doi: 10.1007/s11882-003-0024-7
Kindler, V., Sappino, A. P., Grau, G. E., Piguet, P. F., Vassalli, P. (1989). The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell 56 (5), 731–740. doi: 10.1016/0092-8674(89)90676-4
Kiran, D., Podell, B. K., Chambers, M., Basaraba, R. J. (Eds.) (2016). “Host-directed therapy targeting the mycobacterium tuberculosis granuloma: a review,” in Seminars in immunopathology (Springer Berlin Heidelberg, Germany: Springer).
Kisich, K. O., Higgins, M., Diamond, G., Heifets, L. (2002). Tumor necrosis factor alpha stimulates killing of mycobacterium tuberculosis by human neutrophils. Infect. Immun. 70 (8), 4591–4599. doi: 10.1128/IAI.70.8.4591-4599.2002
Kitamura, H., Cambier, S., Somanath, S., Barker, T., Minagawa, S., Markovics, J., et al. (2011). Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin αvβ8–mediated activation of TGF-β. J. Clin. Invest. 121 (7), 2863–2875. doi: 10.1172/JCI45589
Kolloli, A., Subbian, S. (2017). Host-directed therapeutic strategies for tuberculosis. Front. Med. 4, 171. doi: 10.3389/fmed.2017.00171
Koo, I. C., Wang, C., Raghavan, S., Morisaki, J. H., Cox, J. S., Brown, E. J. (2008). ESX-1-dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell Microbiol. 10 (9), 1866–1878. doi: 10.1111/j.1462-5822.2008.01177.x
Kopf, M., Schneider, C., Nobs, S. P. (2015). The development and function of lung-resident macrophages and dendritic cells. Nat. Immunol. 16 (1), 36–44. doi: 10.1038/ni.3052
Kornfeld, H., Mancino, G., Colizzi, V. (1999). The role of macrophage cell death in tuberculosis. Cell Death Differ. 6 (1), 71–78. doi: 10.1038/sj.cdd.4400454
Kroon, E. E., Coussens, A. K., Kinnear, C., Orlova, M., Möller, M., Seeger, A., et al. (2018). Neutrophils: innate effectors of TB resistance? Front. Immunol. 9, 2637. doi: 10.3389/fimmu.2018.02637
Kumar, N. P., Moideen, K., Viswanathan, V., Shruthi, B. S., Sivakumar, S., Menon, P. A., et al. (2018). Elevated levels of matrix metalloproteinases reflect severity and extent of disease in tuberculosis-diabetes co-morbidity and are predominantly reversed following standard anti-tuberculosis or metformin treatment. BMC Infect. Dis. 18 (1), 345. doi: 10.1186/s12879-018-3246-y
Lange, P., Ahmed, E., Lahmar, Z. M., Martinez, F. J., Bourdin, A. (2021). Natural history and mechanisms of COPD. Respirology 26 (4), 298–321. doi: 10.1111/resp.14007
Larsson, K. (2008). Inflammatory markers in COPD. Clin. Respir. J. 2, 84–87. doi: 10.1111/j.1752-699X.2008.00089.x
Lecaille, F., Brömme, D., Lalmanach, G. (2008). Biochemical properties and regulation of cathepsin K activity. Biochimie 90 (2), 208–226. doi: 10.1016/j.biochi.2007.08.011
Lee, J., Chang, J. (2003). Lung function in patients with chronic airflow obstruction due to tuberculous destroyed lung. Respir. Med. 97 (11), 1237–1242. doi: 10.1016/S0954-6111(03)00255-5
LeibundGut-Landmann, S., Groß, O., Robinson, M. J., Osorio, F., Slack, E. C., Tsoni, S. V., et al. (2007). Syk-and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 8 (6), 630–638. doi: 10.1038/ni1460
Le Rouzic, O., Pichavant, M., Frealle, E., Guillon, A., Si-Tahar, M., Gosset, P. (2017). Th17 cytokines: novel potential therapeutic targets for COPD pathogenesis and exacerbations. Eur. Respir. J. 50 (4), 1602434–1602434. doi: 10.1183/13993003.02434-2016
Li, X., Wang, M., Ming, S., Liang, Z., Zhan, X., Cao, C., et al. (2021). TARM-1 is critical for macrophage activation and Th1 response in mycobacterium tuberculosis infection. J. Immunol. 207 (1), 234–243. doi: 10.4049/jimmunol.2001037
Lin, P. L., Plessner, H. L., Voitenok, N. N., Flynn, J. L. (Eds.) (2007). “Tumor necrosis factor and tuberculosis,” in Journal of investigative dermatology symposium proceedings (California, San Francisco: Elsevier).
Locati, M., Curtale, G., Mantovani, A. (2020). Diversity, mechanisms, and significance of macrophage plasticity. Annu. Rev. Pathol.: Mech. Dis. 15, 123–147. doi: 10.1146/annurev-pathmechdis-012418-012718
Lockhart, E., Green, A. M., Flynn, J. L. (2006). IL-17 production is dominated by γδ T cells rather than CD4 T cells during mycobacterium tuberculosis infection. J. Immunol. 177 (7), 4662–4669. doi: 10.4049/jimmunol.177.7.4662
Long, E., Vorwald, A., Donaldson, L. (1931). Early cellular reaction to tubercle bacilli. Arch. Path. 12 (Dec.), 956.
Lovey, A., Verma, S., Kaipilyawar, V., Ribeiro-Rodrigues, R., Husain, S., Palaci, M., et al. (2022). Early alveolar macrophage response and IL-1R-dependent T cell priming determine transmissibility of mycobacterium tuberculosis strains. Nat. Commun. 13 (1), 884. doi: 10.1038/s41467-022-28506-2
Lowe, D. M., Redford, P. S., Wilkinson, R. J., O’Garra, A., Martineau, A. R. (2012). Neutrophils in tuberculosis: friend or foe? Trends Immunol. 33 (1), 14–25.
Loxton, A. G. (2019). Bcells and their regulatory functions during tuberculosis: latency and active disease. Mol. Immunol. 111, 145–151. doi: 10.1016/j.molimm.2019.04.012
Luo, X., Tarbell, K. V., Yang, H., Pothoven, K., Bailey, S. L., Ding, R., et al. (2007). Dendritic cells with TGF-β1 differentiate naive CD4+ CD25– T cells into islet-protective Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. 104 (8), 2821–2826. doi: 10.1073/pnas.0611646104
MacNee, W. (2000). Oxidants/antioxidants and COPD. Chest 117 (5), 303S–317S. doi: 10.1378/chest.117.5_suppl_1.303S-a
Mahler, D. A., Huang, S., Tabrizi, M., Bell, G. M. (2004). Efficacy and safety of a monoclonal antibody recognizing interleukin-8 in COPD: a pilot study. Chest 126 (3), 926–934. doi: 10.1378/chest.126.3.926
Malherbe, S. T., Shenai, S., Ronacher, K., Loxton, A. G., Dolganov, G., Kriel, M., et al. (2016). Persisting positron emission tomography lesion activity and mycobacterium tuberculosis mRNA after tuberculosis cure. Nat. Med. 22 (10), 1094–1100. doi: 10.1038/nm.4177
Manca, C., Tsenova, L., Bergtold, A., Freeman, S., Tovey, M., Musser, J. M., et al. (2001). Virulence of a mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-α/β. Proc. Natl. Acad. Sci. 98 (10), 5752–5757. doi: 10.1073/pnas.091096998
Manca, C., Tsenova, L., Freeman, S., Barczak, A. K., Tovey, M., Murray, P. J., et al. (2005). Hypervirulent m. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the jak-stat pathway. J. Interferon Cytokine Res. 25 (11), 694–701. doi: 10.1089/jir.2005.25.694
Mantovani, A., Cassatella, M. A., Costantini, C., Jaillon, S. (2011). Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 11 (8), 519–531. doi: 10.1038/nri3024
Manzanillo, P. S., Shiloh, M. U., Portnoy, D. A., Cox, J. S. (2012). Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 11 (5), 469–480. doi: 10.1016/j.chom.2012.03.007
Marakalala, M. J., Martinez, F. O., Plüddemann, A., Gordon, S. (2018). Macrophage heterogeneity in the immunopathogenesis of tuberculosis. Front. Microbiol. 9, 1028. doi: 10.3389/fmicb.2018.01028
Marakalala, M. J., Raju, R. M., Sharma, K., Zhang, Y. J., Eugenin, E. A., Prideaux, B., et al. (2016). Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat. Med. 22 (5), 531–538. doi: 10.1038/nm.4073
Marashian, S. M., Mortaz, E., Jamaati, H. R., Alavi-Moghaddam, M., Kiani, A., Abedini, A., et al. (2015). Role of innate lymphoid cells in lung disease. Iran J. Allergy Asthma Immunol. 14 (4), 346–360.
Marino, S., Cilfone, N. A., Mattila, J. T., Linderman, J. J., Flynn, J. L., Kirschner, D. E. (2015). Macrophage polarization drives granuloma outcome during mycobacterium tuberculosis infection. Infect. Immunity. 83 (1), 324–338. doi: 10.1128/IAI.02494-14
Mata, E., Tarancon, R., Guerrero, C., Moreo, E., Moreau, F., Uranga, S., et al. (2021). Pulmonary BCG induces lung-resident macrophage activation and confers long-term protection against tuberculosis. Sci. Immunol. 6 (63), eabc2934.
McCaffrey, E. F., Donato, M., Keren, L., Chen, Z., Delmastro, A., Fitzpatrick, M. B., et al. (2022). The immunoregulatory landscape of human tuberculosis granulomas. Nat. Immunol. 23 (2), 318–329. doi: 10.1038/s41590-021-01121-x
Meghji, J., Lesosky, M., Joekes, E., Banda, P., Rylance, J., Gordon, S., et al. (2020). Patient outcomes associated with post-tuberculosis lung damage in Malawi: a prospective cohort study. Thorax 75 (3), 269–278. doi: 10.1136/thoraxjnl-2019-213808
Meghji, J., Mortimer, K., Agusti, A., Allwood, B. W., Asher, I., Bateman, E. D., et al. (2021). Improving lung health in low-income and middle-income countries: from challenges to solutions. Lancet 397 (10277), 928–940. doi: 10.1016/S0140-6736(21)00458-X
Menezes, A. M. B., Hallal, P. C., Perez-Padilla, R., Jardim, J., Muino, A., Lopez, M., et al. (2007). Tuberculosis and airflow obstruction: evidence from the PLATINO study in Latin America. Eur. Respir. J. 30 (6), 1180–1185. doi: 10.1183/09031936.00083507
Mihret, A. (2012). The role of dendritic cells in mycobacterium tuberculosis infection. Virulence 3 (7), 654–659. doi: 10.4161/viru.22586
Mihret, A., Mamo, G., Tafesse, M., Hailu, A., Parida, S. (2011). Dendritic cells activate and mature after infection with mycobacterium tuberculosis. BMC Res. notes. 4 (1), 247. doi: 10.1186/1756-0500-4-247
Milara, J., Peiró, T., Serrano, A., Cortijo, J. (2013). Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax 68 (5), 410–420. doi: 10.1136/thoraxjnl-2012-201761
Moreira-Teixeira, L., Sousa, J., McNab, F. W., Torrado, E., Cardoso, F., Machado, H., et al. (2016). Type I IFN inhibits alternative macrophage activation during mycobacterium tuberculosis infection and leads to enhanced protection in the absence of IFN-γ signaling. J. Immunol. 197 (12), 4714–4726. doi: 10.4049/jimmunol.1600584
Mpagama, S., Msaji, K., Kaswaga, O., Zurba, L., Mbelele, P., Allwood, B. W., et al. (2021). The burden and determinants of post-TB lung disease. Int. J. Tuberculosis. Lung Dis. 25 (10), 846–853. doi: 10.5588/ijtld.21.0278
Muefong, C. N., Sutherland, J. S. (2020). Neutrophils in tuberculosis-associated inflammation and lung pathology. Front. Immunol. 11, 962. doi: 10.3389/fimmu.2020.00962
Nau, G. J., Richmond, J. F., Schlesinger, A., Jennings, E. G., Lander, E. S., Young, R. A. (2002). Human macrophage activation programs induced by bacterial pathogens. Proc. Natl. Acad. Sci. U. S. A. 99 (3), 1503–1508. doi: 10.1073/pnas.022649799
Ngahane, B. H. M., Nouyep, J., Motto, M. N., Njankouo, Y. M., Wandji, A., Endale, M., et al. (2016). Post-tuberculous lung function impairment in a tuberculosis reference clinic in Cameroon. Respir. Med. 114, 67–71. doi: 10.1016/j.rmed.2016.03.007
Nguyen, L., Pieters, J. (2005). The Trojan horse: survival tactics of pathogenic mycobacteria in macrophages. Trends Cell Biol. 15 (5), 269–276. doi: 10.1016/j.tcb.2005.03.009
Ni, L., Dong, C. (2018). Roles of myeloid and lymphoid cells in the pathogenesis of chronic obstructive pulmonary disease. Front. Immunol. 9, 1431. doi: 10.3389/fimmu.2018.01431
North, R. J., Jung, Y. J. (2004). Immunity to tuberculosis. Annu. Rev. Immunol. 22, 599–623. doi: 10.1146/annurev.immunol.22.012703.104635
Nouailles, G., Dorhoi, A., Koch, M., Zerrahn, J., Weiner, J., Faé, K. C., et al. (2014). CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. J. Clin. Invest. 124 (3), 1268–1282. doi: 10.1172/JCI72030
Nunes-Alves, C., Booty, M. G., Carpenter, S. M., Jayaraman, P., Rothchild, A. C., Behar, S. M. (2014). In search of a new paradigm for protective immunity to TB. Nat. Rev. Microbiol. 12 (4), 289–299. doi: 10.1038/nrmicro3230
Obermayer, A., Stoiber, W., Krautgartner, W.-D., Klappacher, M., Kofler, B., Steinbacher, P., et al. (2014). New aspects on the structure of neutrophil extracellular traps from chronic obstructive pulmonary disease and in vitro generation. PloS One 9 (5), e97784. doi: 10.1371/journal.pone.0097784
O’Connor, G., Gleeson, L. E., Fagan-Murphy, A., Cryan, S.-A., O’Sullivan, M. P., Keane, J. (2016). Sharpening nature’s tools for efficient tuberculosis control: a review of the potential role and development of host-directed therapies and strategies for targeted respiratory delivery. Adv. Drug Deliv. Rev. 102, 33–54. doi: 10.1016/j.addr.2016.04.024
O’Donnell, R. A., Peebles, C., Ward, J. A., Daraker, A., Angco, G., Broberg, P., et al. (2004). Relationship between peripheral airway dysfunction, airway obstruction, and neutrophilic inflammation in COPD. Thorax 59 (10), 837–842. doi: 10.1136/thx.2003.019349
O’Leary, Sn, O’Sullivan, M. P., Keane, J. (2011). IL-10 blocks phagosome maturation in mycobacterium tuberculosis–infected human macrophages. Am. J. Respir. Cell Mol. Biol. 45 (1), 172–180. doi: 10.1165/rcmb.2010-0319OC
Ong, C. W., Elkington, P. T., Brilha, S., Ugarte-Gil, C., Tome-Esteban, M. T., Tezera, L. B., et al. (2015). Neutrophil-derived MMP-8 drives AMPK-dependent matrix destruction in human pulmonary tuberculosis. PloS Pathog. 11 (5), e1004917. doi: 10.1371/journal.ppat.1004917
Ordway, D., Henao-Tamayo, M., Harton, M., Palanisamy, G., Troudt, J., Shanley, C., et al. (2007). The hypervirulent mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J. Immunol. 179 (1), 522–531. doi: 10.4049/jimmunol.179.1.522
Organization WH (2015). World health statistics 2015 Geneva (Geneva, Switzerland: World Health Organization). Available at: http://public.eblib.com/choice/publicfullrecord.aspx?p=2070501.
Organization WH (2020). Global tuberculosis report 2020 (Geneva, Switzerland: Global tuberculosis report 2020).
Pagán, A. J., Ramakrishnan, L. (2015). Immunity and immunopathology in the tuberculous granuloma. Cold Spring Harbor Perspect. Med. 5 (9), a018499. doi: 10.1101/cshperspect.a018499
Page-McCaw, A., Ewald, A. J., Werb, Z. (2007). Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 8 (3), 221–233. doi: 10.1038/nrm2125
Pasipanodya, J. G., McNabb, S. J., Hilsenrath, P., Bae, S., Lykens, K., Vecino, E., et al. (2010). Pulmonary impairment after tuberculosis and its contribution to TB burden. BMC Public Health 10, 1–10. doi: 10.1186/1471-2458-10-259
Pasipanodya, J. G., Miller, T. L., Vecino, M., Munguia, G., Garmon, R., Bae, S., et al. (2007). Pulmonary impairment after tuberculosis. Chest 131 (6), 1817–1824. doi: 10.1378/chest.06-2949
Patel, S., Homaei, A., El-Seedi, H. R., Akhtar, N. (2018). Cathepsins: proteases that are vital for survival but can also be fatal. Biomed. Pharmacother. 105, 526–532. doi: 10.2147/COPD.S150576
Pedersen, F., Marwitz, S., Holz, O., Kirsten, A., Bahmer, T., Waschki, B., et al. (2015). Neutrophil extracellular trap formation and extracellular DNA in sputum of stable COPD patients. Respir. Med. 109 (10), 1360–1362. doi: 10.1016/j.rmed.2015.08.008
Pedersen, F., Waschki, B., Marwitz, S., Goldmann, T., Kirsten, A., Malmgren, A., et al. (2018). Neutrophil extracellular trap formation is regulated by CXCR2 in COPD neutrophils. Eur. Respir. J. 51 (4), 717–723. doi: 10.1183/13993003.00970-2017
Pefura-Yone, E. W., Kengne, A. P., Tagne-Kamdem, P. E., Afane-Ze, E. (2014). Clinical significance of low forced expiratory flow between 25% and 75% of vital capacity following treated pulmonary tuberculosis: a cross-sectional study. BMJ Open 4 (7), e005361–e005361. doi: 10.1136/bmjopen-2014-005361
Phuah, J. Y., Mattila, J. T., Lin, P. L., Flynn, J. L. (2012). Activated b cells in the granulomas of nonhuman primates infected with mycobacterium tuberculosis. Am. J. Pathol. 181 (2), 508–514. doi: 10.1016/j.ajpath.2012.05.009
Pichavant, M., Rémy, G., Bekaert, S., Le Rouzic, O., Kervoaze, G., Vilain, E., et al. (2014). Oxidative stress-mediated i NKT-cell activation is involved in COPD pathogenesis. Mucosal Immunol. 7 (3), 568–578. doi: 10.1038/mi.2013.75
Pieters, J. (2008). Mycobacterium tuberculosis and the macrophage: maintaining a balance. Cell Host Microbe 3 (6), 399–407. doi: 10.1016/j.chom.2008.05.006
Plit, M., Anderson, R., Van Rensburg, C., Page-Shipp, L., Blott, J., Fresen, J., et al. (1998). Influence of antimicrobial chemotherapy on spirometric parameters and pro-inflammatory indices in severe pulmonary tuberculosis. Eur. Respir. J. 12 (2), 351–356. doi: 10.1183/09031936.98.12020351
Pouwels, S. D., Heijink, I. H., Ten Hacken, N. H., Vandenabeele, P., Krysko, D. V., Nawijn, M. C., et al. (2014). DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol. 7 (2), 215–226. doi: 10.1038/mi.2013.77
Pugin, J., Widmer, M.-C., Kossodo, S., Liang, C.-M., Preas, Hn, Suffredini, A. F. (1999). Human neutrophils secrete gelatinase b in vitro and in vivo in response to endotoxin and proinflammatory mediators. Am. J. Respir. Cell Mol. Biol. 20 (3), 458–464. doi: 10.1165/ajrcmb.20.3.3311
Queval, C. J., Brosch, R., Simeone, R. (2017). The macrophage: a disputed fortress in the battle against mycobacterium tuberculosis. Front. Microbiol. 8, 2284. doi: 10.3389/fmicb.2017.02284
Radovic, M., Ristic, L., Stankovic, I., Pejcic, T., Rancic, M., Ciric, Z., et al. (2011). Chronic airflow obstruction syndrome due to pulmonary tuberculosis treated with directly observed therapy–a serious changes in lung function. Med. Arh. 65 (5), 265–269. doi: 10.5455/medarh.2011.65.265-
Rajendrasozhan, S., Yang, S.-R., Edirisinghe, I., Yao, H., Adenuga, D., Rahman, I. (2008). Deacetylases and NF-κ b in redox regulation of cigarette smoke-induced lung inflammation: epigenetics in pathogenesis of COPD. Antioxid. Redox Signaling 10 (4), 799–812. doi: 10.1089/ars.2007.1938
Ramakrishnan, L. (2012). Revisiting the role of the granuloma in tuberculosis. Nat. Rev. Immunol. 12 (5), 352–366. doi: 10.1038/nri3211
Ramos-Kichik, V., Mondragón-Flores, R., Mondragón-Castelán, M., Gonzalez-Pozos, S., Muñiz-Hernandez, S., Rojas-Espinosa, O., et al. (2009). Neutrophil extracellular traps are induced by mycobacterium tuberculosis. Tuberculosis 89 (1), 29–37. doi: 10.1016/j.tube.2008.09.009
Rao, M., Valentini, D., Poiret, T., Dodoo, E., Parida, S., Zumla, A., et al. (2015). B in TB: b cells as mediators of clinically relevant immune responses in tuberculosis. Clin. Infect. Dis. 61 (suppl_3), S225–SS34. doi: 10.1093/cid/civ614
Ravimohan, S., Kornfeld, H., Weissman, D., Bisson, G. P. (2018). Tuberculosis and lung damage: from epidemiology to pathophysiology. Eur. Respir. Rev. 27 (147), 170077. doi: 10.1183/16000617.0077-2017
Reed, M. B., Domenech, P., Manca, C., Su, H., Barczak, A. K., Kreiswirth, B. N., et al. (2004). A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature 431 (7004), 84–87. doi: 10.1038/nature02837
Refai, A., Gritli, S., Barbouche, M.-R., Essafi, M. (2018). Mycobacterium tuberculosis virulent factor ESAT-6 drives macrophage differentiation toward the pro-inflammatory M1 phenotype and subsequently switches it to the anti-inflammatory M2 phenotype. Front. Cell. Infect. Microbiol. 8, 327. doi: 10.3389/fcimb.2018.00327
Roach, D. R., Bean, A. G., Demangel, C., France, M. P., Briscoe, H., Britton, W. J. (2002). TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J. Immunol. 168 (9), 4620–4627. doi: 10.4049/jimmunol.168.9.4620
Roan, F., Obata-Ninomiya, K., Ziegler, S. F. (2019). Epithelial cell–derived cytokines: more than just signaling the alarm. J. Clin. Invest. 129 (4), 1441–1451. doi: 10.1172/JCI124606
Robb, C., Regan, K., Dorward, D., Rossi, A. (Eds.) (2016). “Key mechanisms governing resolution of lung inflammation,” in Seminars in immunopathology (Springer Berlin Heidelberg, Germany: Springer).
Rohde, K., Yates, R. M., Purdy, G. E., Russell, D. G. (2007). Mycobacterium tuberculosis and the environment within the phagosome. Immunol. Rev. 219 (1), 37–54. doi: 10.1111/j.1600-065X.2007.00547.x
Rosenberger, C. M., Finlay, B. B. (2003). Phagocyte sabotage: disruption of macrophage signalling by bacterial pathogens. Nat. Rev. Mol. Cell Biol. 4 (5), 385–396. doi: 10.1038/nrm1104
Russell, D. G. (2007). Who puts the tubercle in tuberculosis? Nat. Rev. Microbiol. 5 (1), 39–47. doi: 10.1038/nrmicro1538
Russell, D. G., Cardona, P.-J., Kim, M.-J., Allain, S., Altare, F. (2009). Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 10 (9), 943–948. doi: 10.1038/ni.1781
Saetta, M., Mariani, M., Panina-Bordignon, P., Turato, G., Buonsanti, C., Baraldo, S., et al. (2002). Increased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 165 (10), 1404–1409. doi: 10.1164/rccm.2107139
Salgame, P. (2011). MMPs in tuberculosis: granuloma creators and tissue destroyers. J. Clin. Invest. 121 (5), 1686–1688. doi: 10.1172/JCI57423
Salvi, S. (2015). The silent epidemic of COPD in Africa. Lancet Global Health 3 (1), e6–e7. doi: 10.1016/S2214-109X(14)70359-6
Sarkar, R., Lenders, L., Wilkinson, K. A., Wilkinson, R. J., Nicol, M. P. (2012). Modern lineages of mycobacterium tuberculosis exhibit lineage-specific patterns of growth and cytokine induction in human monocyte-derived macrophages. PloS one 7 (8), e43170–e43170. doi: 10.1371/journal.pone.0043170
Sarkar, M., Madabhavi, I., Kumar, K. (2017). Tuberculosis associated chronic obstructive pulmonary disease. Clin. Respir. J. 11 (3), 285–295. doi: 10.1111/crj.12621
Sasaki, T., Irie-Sasaki, J., Jones, R. G., Oliveira-dos-Santos, A. J., Stanford, W. L., Bolon, B., et al. (2000). Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science 287 (5455), 1040–1046. doi: 10.1126/science.287.5455.1040
Sathyamoorthy, T., Tezera, L. B., Walker, N. F., Brilha, S., Saraiva, L., Mauri, F. A., et al. (2015). Membrane type 1 matrix metalloproteinase regulates monocyte migration and collagen destruction in tuberculosis. J. Immunol. 195 (3), 882–891. doi: 10.4049/jimmunol.1403110
Sato, N., Takasaka, N., Yoshida, M., Tsubouchi, K., Minagawa, S., Araya, J., et al. (2016). Metformin attenuates lung fibrosis development via NOX4 suppression. Respir. Res. 17 (1), 1–12. doi: 10.1186/s12931-016-0420-x
Schneider, D., Ganesan, S., Comstock, A. T., Meldrum, C. A., Mahidhara, R., Goldsmith, A. M., et al. (2010). Increased cytokine response of rhinovirus-infected airway epithelial cells in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 182 (3), 332–340. doi: 10.1164/rccm.200911-1673OC
Scriba, T. J., Kalsdorf, B., Abrahams, D.-A., Isaacs, F., Hofmeister, J., Black, G., et al. (2008). Distinct, specific IL-17-and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. J. Immunol. 180 (3), 1962–1970. doi: 10.4049/jimmunol.180.3.1962
Sergejeva, S., Ivanov, S., Lotvall, J., Lindén, A. (2005). Interleukin-17 as a recruitment and survival factor for airway macrophages in allergic airway inflammation. Am. J. Respir. Cell Mol. Biol. 33 (3), 248–253. doi: 10.1165/rcmb.2004-0213OC
Sertl, K., Takemura, T., Tschachler, E., Ferrans, V., Kaliner, M., Shevach, E. (1986). Dendritic cells with antigen-presenting capability reside in airway epithelium, lung parenchyma, and visceral pleura. J. Exp. Med. 163 (2), 436–451. doi: 10.1084/jem.163.2.436
Shang, S., Harton, M., Tamayo, M. H., Shanley, C., Palanisamy, G. S., Caraway, M., et al. (2011). Increased Foxp3 expression in guinea pigs infected with W-Beijing strains of m. tuberculosis. Tuberculosis 91 (5), 378–385. doi: 10.1016/j.tube.2011.06.001
Shapouri-Moghaddam, A., Mohammadian, S., Vazini, H., Taghadosi, M., Esmaeili, S. A., Mardani, F., et al. (2018). Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 233 (9), 6425–6440. doi: 10.1002/jcp.26429
Shigenaga, T., Dannenberg, J., Lowrie, D., Said, W., Urist, M., Abbey, H., et al. (2001). Immune responses in tuberculosis: antibodies and CD4-CD8 lymphocytes with vascular adhesion molecules and cytokines (chemokines) cause a rapid antigen-specific cell infiltration at sites of bacillus calmette–guérin reinfection. Immunology 102 (4), 466–479. doi: 10.1046/j.1365-2567.2001.01195.x
Siddharthan, T., Grigsby, M., Morgan, B., Kalyesubula, R., Wise, R. A., Kirenga, B., et al. (2019). Prevalence of chronic respiratory disease in urban and rural Uganda. Bull. World Health Organ. 97 (5), 318–327. doi: 10.2471/BLT.18.216523
Silbaugh, S. A., Stengel, P. W., Cockerham, S. L., Froelich, L. L., Bendele, A. M., Spaethe, S. M., et al. (2000). Pharmacologic actions of the second generation leukotriene b 4 receptor antagonist LY293111: in vivo pulmonary studies. Naunyn-Schmiedeberg’s. Arch. Pharmacol. 361 (4), 397–404. doi: 10.1007/s002109900211
Silver, J. S., Kearley, J., Copenhaver, A. M., Sanden, C., Mori, M., Yu, L., et al. (2016). Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat. Immunol. 17 (6), 626–635. doi: 10.1038/ni.3443
Singh, P., Ali, S. A. (2022). Multifunctional role of S100 protein family in the immune system: an update. Cells 11 (15), 2274. doi: 10.3390/cells11152274
Singh, S., Allwood, B., Chiyaka, T., Kleyhans-Cornelissen, L., Naidoo, C., Moodley, S., et al. (2022). Immunologic and imaging signatures in post tuberculosis lung disease. Tuberculosis 102244. doi: 10.1016/j.tube.2022.102244
Soares, A. P., Scriba, T. J., Joseph, S., Harbacheuski, R., Murray, R. A., Gelderbloem, S. J., et al. (2008). Bacillus calmette-guerin vaccination of human newborns induces T cells with complex cytokine and phenotypic profiles. J. Immunol. 180 (5), 3569–3577. doi: 10.4049/jimmunol.180.5.3569
Sonnenberg, G. F., Hepworth, M. R. (2019). Functional interactions between innate lymphoid cells and adaptive immunity. Nat. Rev. Immunol. 19 (10), 599–613. doi: 10.1038/s41577-019-0194-8
Stancu, C., Sima, A. (2001). Statins: mechanism of action and effects. J. Cell. Mol. Med. 5 (4), 378–387. doi: 10.1111/j.1582-4934.2001.tb00172.x
Stănescu, D., Sanna, A., Veriter, C., Kostianev, S., Calcagni, P. G., Fabbri, L. M., et al. (1996). Airways obstruction, chronic expectoration, and rapid decline of FEV1 in smokers are associated with increased levels of sputum neutrophils. Thorax 51 (3), 267–271. doi: 10.1136/thx.51.3.267
Steinman, R. M., Banchereau, J. (2007). Taking dendritic cells into medicine. Nature 449 (7161), 419–426. doi: 10.1038/nature06175
Stek, C., Allwood, B., Walker, N. F., Wilkinson, R. J., Lynen, L., Meintjes, G. (2018). The immune mechanisms of lung parenchymal damage in tuberculosis and the role of host-directed therapy. Front. Microbiol. 9, 2603. doi: 10.3389/fmicb.2018.02603
Sturgill-Koszycki, S., Schlesinger, P. H., Chakraborty, P., Haddix, P. L., Collins, H. L., Fok, A. K., et al. (1994). Lack of acidification in mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science 263 (5147), 678–681. doi: 10.1126/science.8303277
Sullivan, J. T., Young, E. F., McCann, J. R., Braunstein, M. (2012). The mycobacterium tuberculosis SecA2 system subverts phagosome maturation to promote growth in macrophages. Infect. Immunity. 80 (3), 996–1006. doi: 10.1128/IAI.05987-11
Sun, B., Wang, X., Ji, Z., Wang, M., Liao, Y. P., Chang, C. H., et al. (2015). NADPH oxidase-dependent NLRP3 inflammasome activation and its important role in lung fibrosis by multiwalled carbon nanotubes. Small 11 (17), 2087–2097. doi: 10.1002/smll.201402859
Sun, J., Wang, X., Lau, A., Liao, T.-Y. A., Bucci, C., Hmama, Z. (2010). Mycobacterial nucleoside diphosphate kinase blocks phagosome maturation in murine RAW 264. 7. Macrophages. PloS One 5 (1), e8769.
Suzuki, M., Sze, M. A., Campbell, J. D., Brothers, J. F., 2nd, Lenburg, M. E., McDonough, J. E., et al. (2017). The cellular and molecular determinants of emphysematous destruction in COPD. Sci. Rep. 7 (1), 9562. doi: 10.1038/s41598-017-10126-2
Swanson, R. V., Gupta, A., Foreman, T. W., Lu, L., Choreno-Parra, J. A., Mbandi, S. K., et al. (2023). Antigen-specific b cells direct T follicular-like helper cells into lymphoid follicles to mediate mycobacterium tuberculosis control. Nat. Immunol., 1–14. doi: 10.1038/s41590-023-01476-3
Tahir, F., Bin Arif, T., Ahmed, J., Shah, S. R., Khalid, M. (2020). Anti-tuberculous effects of statin therapy: a review of literature. Cureus 12 (3), e7404. doi: 10.7759/cureus.7404
Takabatake, N., Nakamura, H., Abe, S., Inoue, S., Hino, T., Saito, H., et al. (2000). The relationship between chronic hypoxemia and activation of the tumor necrosis factor-α system in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 161 (4), 1179–1184. doi: 10.1164/ajrccm.161.4.9903022
Tantucci, C., Modina, D. (2012). Lung function decline in COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 7, 95–99. doi: 10.2147/COPD.S27480
Taylor, J. L., Turner, O. C., Basaraba, R. J., Belisle, J. T., Huygen, K., Orme, I. M. (2003). Pulmonary necrosis resulting from DNA vaccination against tuberculosis. Infect. Immunity. 71 (4), 2192–2198. doi: 10.1128/IAI.71.4.2192-2198.2003
Tecle, T., Tripathi, S., Hartshorn, K. L. (2010). Defensins and cathelicidins in lung immunity. Innate. Immunity. 16 (3), 151–159. doi: 10.1177/1753425910365734
Thakur, M., Muniyappa, K. (2023). Macrophage activation highlight an important role for NER proteins in the survival, latency and multiplication of mycobacterium tuberculosis. Tuberculosis. (Edinb). 138, 102284. doi: 10.1016/j.tube.2022.102284
Thannickal, V. J., Fanburg, B. L. (2000). Reactive oxygen species in cell signaling. Am. J. Physiology-Lung. Cell. Mol. Physiol. 279 (6), L1005–L1L28. doi: 10.1152/ajplung.2000.279.6.L1005
Uddin, M., Watz, H., Malmgren, A., Pedersen, F. (2019). NETopathic inflammation in chronic obstructive pulmonary disease and severe asthma. Front. Immunol. 10, 47. doi: 10.3389/fimmu.2019.00047
Umemura, M., Yahagi, A., Hamada, S., Begum, M. D., Watanabe, H., Kawakami, K., et al. (2007). IL-17-mediated regulation of innate and acquired immune response against pulmonary mycobacterium bovis bacille calmette-guerin infection. J. Immunol. 178 (6), 3786–3796. doi: 10.4049/jimmunol.178.6.3786
Underwood, D. C., Osborn, R. R., Bochnowicz, S., Webb, E. F., Rieman, D. J., Lee, J. C., et al. (2000). SB 239063, a p38 MAPK inhibitor, reduces neutrophilia, inflammatory cytokines, MMP-9, and fibrosis in lung. Am. J. Physiology-Lung Cell. Mol. Physiol. 279, 5, 895–902. doi: 10.1152/ajplung.2000.279.5.L895
Vandal, O. H., Pierini, L. M., Schnappinger, D., Nathan, C. F., Ehrt, S. (2008). A membrane protein preserves intrabacterial pH in intraphagosomal mycobacterium tuberculosis. Nat. Med. 14 (8), 849–854. doi: 10.1038/nm.1795
van der Strate, B. W., Postma, D. S., Brandsma, C.-A., Melgert, B. N., Luinge, M. A., Geerlings, M., et al. (2006). Cigarette smoke–induced emphysema: a role for the b cell? Am. J. Respir. Crit. Care Med. 173 (7), 751–758. doi: 10.1164/rccm.200504-594OC
Vannella, K. M., Ramalingam, T. R., Borthwick, L. A., Barron, L., Hart, K. M., Thompson, R. W., et al. (2016). Combinatorial targeting of TSLP, IL-25, and IL-33 in type 2 cytokine–driven inflammation and fibrosis. Sci. Trans. Med. 8 (337), 337ra65–ra65. doi: 10.1126/scitranslmed.aaf1938
Venkatesan, P. (2022). GOLD report: 2022 update. Lancet Respir. Med. 10 (2), e20. doi: 10.1016/S2213-2600(21)00561-0
Vergne, I., Chua, J., Lee, H.-H., Lucas, M., Belisle, J., Deretic, V. (2005). Mechanism of phagolysosome biogenesis block by viable mycobacterium tuberculosis. Proc. Natl. Acad. Sci. 102 (11), 4033–4038. doi: 10.1073/pnas.0409716102
Verma, S., Bhatt, K., Lovey, A., Ribeiro-Rodrigues, R., Durbin, J., Jones-López, E. C., et al. (2019). Transmission phenotype of mycobacterium tuberculosis strains is mechanistically linked to induction of distinct pulmonary pathology. PloS Pathogens. 15 (3), e1007613. doi: 10.1371/journal.ppat.1007613
Vestbo, J., Lange, P. (2016). Natural history of COPD: focusing on change in FEV1. Respirology 21 (1), 34–43. doi: 10.1111/resp.12589
Vestbo, J., Tan, L., Atkinson, G., Ward, J. (2009). A controlled trial of 6-weeks’ treatment with a novel inhaled phosphodiesterase type-4 inhibitor in COPD. Eur. Respir. J. 33 (5), 1039–1044. doi: 10.1183/09031936.00068908
Walburger, A., Koul, A., Ferrari, G., Nguyen, L., Prescianotto-Baschong, C., Huygen, K., et al. (2004). Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 304 (5678), 1800–1804. doi: 10.1126/science.1099384
Wallis, R. S., Ginindza, S., Beattie, T., Arjun, N., Likoti, M., Edward, V. A., et al. (2021). Adjunctive host-directed therapies for pulmonary tuberculosis: a prospective, open-label, phase 2, randomised controlled trial. Lancet Respir. Med. 9 (8), 897–908. doi: 10.1016/S2213-2600(20)30448-3
Wallis, R. S., Hafner, R. (2015). Advancing host-directed therapy for tuberculosis. Nat. Rev. Immunol. 15 (4), 255–263. doi: 10.1038/nri3813
Wallis, R. S., Maeurer, M., Mwaba, P., Chakaya, J., Rustomjee, R., Migliori, G. B., et al. (2016). Tuberculosis–advances in development of new drugs, treatment regimens, host-directed therapies, and biomarkers. Lancet Infect. Dis. 16 (4), e34–e46. doi: 10.1016/S1473-3099(16)00070-0
Wang, C., Peyron, P., Mestre, O., Kaplan, G., Van Soolingen, D., Gao, Q., et al. (2010). Innate immune response to mycobacterium tuberculosis Beijing and other genotypes. PloS One 5 (10), e13594. doi: 10.1371/journal.pone.0013594
Warner, D. F., Koch, A., Mizrahi, V. (2015). Diversity and disease pathogenesis in mycobacterium tuberculosis. Trends Microbiol. 23 (1), 14–21. doi: 10.1016/j.tim.2014.10.005
Warner, D. F., Mizrahi, V. (2007). The survival kit of mycobacterium tuberculosis. Nat. Med. 13 (3), 282–284. doi: 10.1038/nm0307-282
Whitsett, J. A., Alenghat, T. (2015). Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 16 (1), 27–35. doi: 10.1038/ni.3045
Wickremasinghe, M. I., Thomas, L. H., Friedland, J. S. (1999). Pulmonary epithelial cells are a source of IL-8 in the response to mycobacterium tuberculosis: essential role of IL-1 from infected monocytes in a NF-κB-dependent network. J. Immunol. 163 (7), 3936–3947. doi: 10.4049/jimmunol.163.7.3936
Widdowson, K. L., Elliott, J. D., Veber, D. F., Nie, H., Rutledge, M. C., McCleland, B. W., et al. (2004). Evaluation of potent and selective small-molecule antagonists for the CXCR2 chemokine receptor. J. Med. Chem. 47 (6), 1319–1321. doi: 10.1021/jm034248l
Winkler, S., Necek, M., Winkler, H., Adegnika, A. A., Perkmann, T., Ramharter, M., et al. (2005). Increased specific T cell cytokine responses in patients with active pulmonary tuberculosis from central Africa. Microbes Infect. 7 (9-10), 1161–1169. doi: 10.1016/j.micinf.2005.03.020
Wong, K.-W. (2017). The role of ESX-1 in mycobacterium tuberculosis pathogenesis. Microbiol. Spectrum. 5 (3), 5.3. 02. doi: 10.1128/microbiolspec.TBTB2-0001-2015
Wong, D., Bach, H., Sun, J., Hmama, Z., Av-Gay, Y. (2011). Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+–ATPase to inhibit phagosome acidification. Proc. Natl. Acad. Sci. 108 (48), 19371–19376. doi: 10.1073/pnas.1109201108
Xu, J., Zhang, X., Monestier, M., Esmon, N. L., Esmon, C. T. (2011). Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J. Immunol. 187 (5), 2626–2631. doi: 10.4049/jimmunol.1003930
Xu, J., Zhang, X., Pelayo, R., Monestier, M., Ammollo, C. T., Semeraro, F., et al. (2009). Extracellular histones are major mediators of death in sepsis. Nat. Med. 15 (11), 1318–1321. doi: 10.1038/nm.2053
Yang, C.-S. (2017). Advancing host-directed therapy for tuberculosis: new therapeutic insights from the toxoplasma gondii. Microbial. Cell. 4 (3), 105. doi: 10.15698/mic2017.03.565
Yang, Y., Zhou, X., Kouadir, M., Shi, F., Ding, T., Liu, C., et al. (2013). The AIM2 inflammasome is involved in macrophage activation during infection with virulent mycobacterium bovis strain. J. Infect. Dis. 208 (11), 1849–1858. doi: 10.1093/infdis/jit347
Yoshida, M., Minagawa, S., Araya, J., Sakamoto, T., Hara, H., Tsubouchi, K., et al. (2019). Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat. Commun. 10 (1), 1–14. doi: 10.1038/s41467-019-10991-7
Zavašnik-Bergant, T., Turk, B. (2006). Cysteine cathepsins in the immune response. Tissue Antigens 67 (5), 349–355. doi: 10.1111/j.1399-0039.2006.00585.x
Zhang, X., Zheng, H., Zhang, H., Ma, W., Wang, F., Liu, C., et al. (2011). Increased interleukin (IL)-8 and decreased IL-17 production in chronic obstructive pulmonary disease (COPD) provoked by cigarette smoke. Cytokine 56 (3), 717–725. doi: 10.1016/j.cyto.2011.09.010
Zhang, M., Zheng, X., Zhang, J., Zhu, Y., Zhu, X., Liu, H., et al. (2012). CD19+ CD1d+ CD5+ b cell frequencies are increased in patients with tuberculosis and suppress Th17 responses. Cell. Immunol. 274 (1-2), 89–97. doi: 10.1016/j.cellimm.2012.01.007
Keywords: Tuberculosis, COPD - chronic obstructive pulmonary disease, adaptive immunity, innate immunity, host-directed therapy (HDT)
Citation: Kayongo A, Nyiro B, Siddharthan T, Kirenga B, Checkley W, Lutaakome Joloba M, Ellner J and Salgame P (2023) Mechanisms of lung damage in tuberculosis: implications for chronic obstructive pulmonary disease. Front. Cell. Infect. Microbiol. 13:1146571. doi: 10.3389/fcimb.2023.1146571
Received: 17 January 2023; Accepted: 05 June 2023;
Published: 21 June 2023.
Edited by:
Bindu Singh, Texas Biomedical Research Institute, United StatesReviewed by:
Kyle Rohde, University of Central Florida, United StatesCopyright © 2023 Kayongo, Nyiro, Siddharthan, Kirenga, Checkley, Lutaakome Joloba, Ellner and Salgame. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Padmini Salgame, c2FsZ2FtcGFAbmptcy5ydXRnZXJzLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.