 Yohei Shinmyo
Yohei Shinmyo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol., 24 February 2025
Sec. Signaling
Volume 13 - 2025 | https://doi.org/10.3389/fcell.2025.1560940
This article is part of the Research TopicNeuronal Guidance Signaling in Health and Neurological DiseasesView all 7 articles
Axon guidance proteins not only play a role in the formation of proper neural circuits but also have other important functions, such as cell survival, migration, and proliferation in the brain. Therefore, mutations in the genes encoding these proteins frequently cause various types of neurological disorders, including psychiatric disorders and neurodegenerative diseases. We previously identified an axon guidance protein, draxin, that is essential for the development of several neural circuits and cell survival in the brain. Recently, the deletion of the draxin gene was identified in an inbred BTBR T+ Itpr3tf/J (BTBR/J) mouse, which is a widely used model of Autism Spectrum Disorder (ASD), suggesting that draxin deletion is a genetic factor for ASD-like characteristics in BTBR/J mice. In this review, I summarize the neuroanatomical abnormalities in draxin knockout mice by comparing them to BTBR/J mice and discuss the possible contributions of draxin to anatomical and behavioral phenotypes in BTBR/J mice.
Draxin was first identified as an axon guidance protein that regulates commissural axons in the spinal cord and the forebrain. It is a secreted protein that shares no homology with other known proteins (Islam et al., 2009; Miyake et al., 2009). Draxin has been shown to bind to netrin-1 and its receptors, including Deleted in colorectal cancer (Dcc) and Neogenin (Neo1) (Ahmed et al., 2011; Shinmyo et al., 2015). Previous studies have suggested that draxin regulates the outgrowth of axons originating from various types of neurons in vitro (Islam et al., 2009; Naser et al., 2009; Ahmed et al., 2010; Ahmed et al., 2011; Chen et al., 2013; Meli et al., 2015; Shinmyo et al., 2015). Draxin knockout (KO) mice show developmental abnormalities in various neural circuits, including the corpus callosum, the hippocampal commissure, the anterior commissure, the fornix, and the thalamocortical axons (Islam et al., 2009; Zhang et al., 2010; Shinmyo et al., 2015). Thus, draxin may control the development of neural circuits in the brain through the netrin-1 receptors or by modulating netrin-1-mediated axon guidance.
Previous human and animal studies have shown that axon guidance proteins are associated with structural changes in neuronal connections during neurological disorders (Nugent et al., 2012; Van Battum et al., 2015). In addition, because axon guidance cues have other important functions in the brain, such as cell survival, migration, and proliferation (Mehlen et al., 2011), mutations in the genes encoding axon guidance proteins can cause many neurological disorders. Indeed, draxin and/or netrin signaling has been shown to be associated with several neurological disorders, including psychiatric disorders, gliomas, and neurodegenerative diseases (Infante et al., 2015; Vosberg et al., 2020; Ahn et al., 2021; Jasmin et al., 2021; Cai et al., 2024). Recently, an 8-bp frameshift deletion of the draxin gene was identified in an inbred BTBR T+ Itpr3tf/J (BTBR/J) mouse, a widely used model of Autism Spectrum Disorder (ASD) (Morcom et al., 2021; Arslan et al., 2023). Furthermore, draxin deletion in BTBR/J mice was shown to contribute to the dysgenesis of the corpus callosum, which is a neuroanatomical abnormality characteristic of human ASD (Arslan et al., 2023). In this review, I summarized the neuroanatomical abnormalities in draxin KO mice by comparing them to BRBR/J mice.
ASD is a neurodevelopmental disorder defined by impairments in social interactions, communication deficits, and repetitive behaviors with restricted interests (Lai et al., 2014). Identifying abnormalities in brain structures in ASD is critical for developing more precise and objective diagnoses and for creating effective new treatments. One prominent mechanism that has been suggested to contribute to the underlying pathology of ASD is abnormal long-range neuronal connectivity. This is because numerous MRI studies have demonstrated reduced fractional anisotropy in major white matter tracts in individuals with ASD, including the cingulum, uncinate fasciculi, occipitotemporal tracts, and, most consistently, the corpus callosum (Barnea-Goraly et al., 2004; Alexander et al., 2007; Keller et al., 2007; Frazier and Hardan, 2009; Kumar et al., 2010; Weinstein et al., 2011).
The corpus callosum is a large bundle of nerve fibers that connects the left and right hemispheres of the brain. Variable corpus callosum abnormalities have been reported in the anterior, midbody, and posterior regions of the forebrain in ASD (Egaas et al., 1995; Saitoh et al., 1995; Haas et al., 1996; Piven et al., 1997; Manes et al., 1999; Hardan et al., 2000). These observations suggest that the abnormal development of the corpus callosum is associated with ASD. This is consistent with recent results from mega-analyses comparing white matter microstructural differences between healthy participants and those with psychiatric disorders, showing that patients with schizophrenia, bipolar disorder, or ASD disorder have common alterations in the corpus callosum (Koshiyama et al., 2020).
The corpus callosum plays a critical role in the transmission and integration of information between the left and the right hemispheres. The anterior corpus callosum connects regions of the prefrontal cortex and is associated with higher-order cognitive, emotional, and social functions. The midbody of the corpus callosum connects multiple regions, including the primary motor and sensory cortices, and is involved in sensory and motor processing. The posterior corpus callosum links the occipital lobes and is crucial for the processing and integration of visual information. Abnormal development in specific regions of the corpus callosum may be associated with the specific cognitive and behavioral characteristics of ASD. However, abnormalities in brain structures in patients with ASD have been observed not only in the corpus callosum but also in other regions. Therefore, to understand the causes of behavioral abnormalities in ASD accurately, it is important to analyze animal models of specific anatomical and functional abnormalities.
Characteristic behavioral phenotypes of ASD have been modeled in mice. One such model is the inbred BTBR/J mouse, which is the most extensively researched and the most commonly reproduced inbred strain (Nadler et al., 2006; Bolivar et al., 2007; Moy et al., 2007). BTBR/J mice exhibit impaired in social interactions and high levels of repetitive behaviors (Moy et al., 2007; McFarlane et al., 2008; Dodero et al., 2013). Furthermore, this strain is characterized by the absence of the corpus callosum and a smaller-to-absent hippocampal commissure (Wahlsten et al., 2003). A previous study identified several genomic regions in BTBR/J mice that distinctly influenced their ASD-like characteristics (Jones-Davis et al., 2013). Recently, an 8-bp frameshift deletion of the draxin gene, leading to the loss of draxin function, was identified in BTBR/J mice (Morcom et al., 2021; Arslan et al., 2023). The draxin gene is located in a genomic region that was previously identified as contributing to commissural abnormalities in BTBR/J mice (Jones-Davis et al., 2013). Since draxin KO mice display malformations of the corpus callosum and the hippocampal commissure, draxin is a promising candidate for explaining the defects in these commissures in BTBR/J mice. Consistently, abnormal development of the corpus callosum was partially restored in BTBR/J mice with a heterozygous knock-in that reverted the 8 bp draxin deletion to the wild-type, suggesting that the draxin deletion contributes to agenesis of the corpus callosum in BTBR/J mice (Arslan et al., 2023).
Since previous studies have suggested that BTBR/J mice are characterized by multiple genetic aberrations, it is important to clarify the contribution of draxin to the anatomical and behavioral phenotypes of BTBR/J mice. Draxin KO mice show various developmental abnormalities in the brain similar to those observed in BTBR/J mice. BTBR/J mice exhibit an absence of the corpus callosum, and reductions in the hippocampal and the anterior commissures (Table 1) (Wahlsten et al., 2003; Ellegood et al., 2015). Similar to BTBR/J mice, draxin KO mice show severe defects in all forebrain commissures, the corpus callosum, the hippocampal commissure, and the anterior commissure (Islam et al., 2009). Given that the abnormal development of the corpus callosum was partially rescued in BTBR/J mice with a heterozygous knock-in that reverted the 8 bp draxin deletion to the wild-type, the draxin deletion contributes to the absence of the corpus callosum in BTBR/J mice (Arslan et al., 2023). However, this observation suggests that additional genetic factors contribute to the absence of the corpus callosum in BTBR/J mice. Both draxin KO mice and BTBR mice with a C57Bl/6J genetic background display variable penetrance of the corpus callosum defect, suggesting that other genetic factors modify the corpus callosum phenotype driven by the draxin mutation (Morcom et al., 2021).
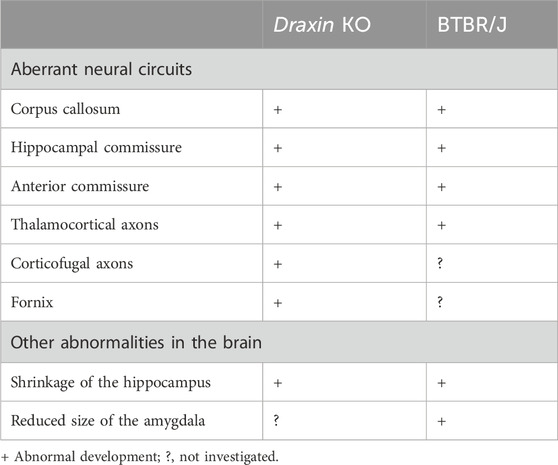
Table 1. Anatomical abnormalities in brains of draxin KO and BTBR mice.
Draxin KO mice also show severe defects in the thalamocortical and corticofugal projections (Shinmyo et al., 2015). During normal brain development, corticofugal and thalamocortical axons meet in the internal capsule and depend on each other for their guidance to the thalamus and neocortex, respectively (Lopez-Bendito and Molnar, 2003). Corticofugal axons grow from the cortex into the internal capsule in wild-type mice. In contrast, some corticofugal axons of draxin KO mice do not enter the internal capsule but instead grow toward the external capsule. Thalamocortical axons in draxin KO mice grow normally toward the internal capsule. However, some of them do not enter the cortex and instead either stall or turn laterally toward the external capsule, whereas others enter the cortex with an abnormal topographic organization. Visualization of the cortical sensory regions revealed disruptions in the spatial positions of thalamocortical axon terminals in draxin KO mice (Shinmyo et al., 2015). Thus, draxin is essential for guiding thalamocortical axons from the internal capsule to the cortex, as well as for their region-specific connections between the thalamus and cortex. Importantly, the topography of thalamocortical projections changes in BTBR/J mice, in which the primary somatosensory and visual cortical areas are medially shifted (Fenlon et al., 2015). Therefore, abnormalities in the topographic organization of thalamocortical projections are a common feature of draxin KO and BTBR/J mice, although this phenotype in draxin KO mice requires further investigation. Another similarity in the anatomical phenotype between draxin KO mice (Zhang et al., 2010) and BTBR/J mice (Mercier et al., 2012) is the shrinkage of the hippocampus. In addition to the hippocampus, the size of the amygdala nuclei is reduced in BTBR/J mice (Mercier et al., 2012). However, it remains unclear whether the anatomy of the amygdala is altered in draxin KO mice or not. Collectively, draxin deletion is likely to be the primary genetic factor underlying the neuroanatomical phenotypes in BTBR/J mice.
In this review, I have summarized the similarities in neuroanatomical phenotypes between draxin KO and BTBR/J mice. In addition to their phenotypical similarities, recent studies have suggested that draxin contributes to neuroanatomical phenotypes in BTBR/J mice (Morcom et al., 2021; Arslan et al., 2023). However, the contribution of draxin to the behavioral phenotypes of BTBR/J mice remains unclear. To address this issue, it is necessary to perform behavioral analyses in draxin KO mice and draxin knock-in BTBR mice.
It is important to determine the neuroanatomical abnormalities responsible for the behavioral phenotypes of ASD. Previous studies on humans with ASD and BTBR/J mice have suggested that dysgenesis of the corpus callosum is strongly associated with behavioral abnormalities in ASD. However, there is no direct evidence supporting this idea because dysgenesis of the corpus callosum is generally accompanied by other anomalies in brain structures in both humans and mice. For example, patients with corpus callosum anomalies frequently display dysgenesis of the hippocampal commissure (Hetts et al., 2006). Therefore, to examine whether the behavioral phenotypes characteristic of ASD are caused by anomalies in the corpus callosum, a mouse model with a specific defect in the corpus callosum is required. Surgical lesions of the corpus callosum at an early postnatal stage do not affect the juvenile play or adult social behaviors, nor do they increase repetitive self-grooming (Yang et al., 2009). This evidence does not support the hypothesis that disconnection of the corpus callosum is a causal factor for ASD-like behaviors in mice. However, experimental lesions at the postnatal stage may not replicate congenital corpus callosum anomalies. Both BTBR/J and draxin KO mice show corpus callosum agenesis with similar misprojections of the callosal axons. In these mice, callosal axons fail to cross the midline; instead, they form ipsilateral “Probst” bundles that run parallel to the midline (Islam et al., 2009; Fenlon et al., 2015). Since this aberrant neuronal circuitry is retained throughout adulthood, it may contribute to ASD-like behaviors in mice.
Furthermore, both draxin KO and BTBR/J mice have abnormalities in the topographic organization of connections between the thalamus and the cortex (Fenlon et al., 2015; Shinmyo et al., 2015). This suggests that the alteration in cortical area patterning caused by the deletion of the draxin gene contributes to the previously observed sensory and behavioral deficits in BTBR/J mice (Moy et al., 2007; McFarlane et al., 2008). It is critical to generate conditional draxin KO mice with specific neural structural abnormalities and perform behavioral analyses to investigate these possibilities. Recently, it was reported that BTBR TF/ArtRbrc (BTBR/R) mice, a sister strain of BTBR/J, show core symptoms of ASD despite having an intact draxin gene and preserved forebrain commissures (Lin et al., 2023). BTBR/R mice will be useful for understanding the draxin-independent mechanisms that cause ASD-like behaviors.
YS: Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), the Takeda Science Foundation, the Naito Foundation, and HUSM.
I am grateful to my lab members for their valuable support, critical discussions, and comments on this manuscript.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ahmed, G., Shinmyo, Y., Naser, I. B., Hossain, M., Song, X., and Tanaka, H. (2010). Olfactory bulb axonal outgrowth is inhibited by draxin. Biochem. Biophys. Res. Commun. 398, 730–734. doi:10.1016/j.bbrc.2010.07.010
Ahmed, G., Shinmyo, Y., Ohta, K., Islam, S. M., Hossain, M., Naser, I. B., et al. (2011). Draxin inhibits axonal outgrowth through the netrin receptor DCC. J. Neurosci. 31, 14018–14023. doi:10.1523/JNEUROSCI.0943-11.2011
Ahn, E. H., Kang, S. S., Liu, X., Cao, X., Choi, S. Y., Musazzi, L., et al. (2021). BDNF and Netrin-1 repression by C/EBPβ in the gut triggers Parkinson's disease pathologies, associated with constipation and motor dysfunctions. Prog. Neurobiol. 198, 101905. doi:10.1016/j.pneurobio.2020.101905
Alexander, A. L., Lee, J. E., Lazar, M., Boudos, R., Dubray, M. B., Oakes, T. R., et al. (2007). Diffusion tensor imaging of the corpus callosum in Autism. Neuroimage 34, 61–73. doi:10.1016/j.neuroimage.2006.08.032
Arslan, A., Fang, Z., Wang, M., Tan, Y., Cheng, Z., Chen, X., et al. (2023). Analysis of structural variation among inbred mouse strains. BMC Genomics 24, 97. doi:10.1186/s12864-023-09197-5
Barnea-Goraly, N., Kwon, H., Menon, V., Eliez, S., Lotspeich, L., and Reiss, A. L. (2004). White matter structure in autism: preliminary evidence from diffusion tensor imaging. Biol. Psychiatry 55, 323–326. doi:10.1016/j.biopsych.2003.10.022
Bolivar, V. J., Walters, S. R., and Phoenix, J. L. (2007). Assessing autism-like behavior in mice: variations in social interactions among inbred strains. Behav. Brain Res. 176, 21–26. doi:10.1016/j.bbr.2006.09.007
Cai, M., Zheng, Q., Chen, Y., Liu, S., Zhu, H., and Bai, B. (2024). Insights from the neural guidance factor Netrin-1 into neurodegeneration and other diseases. Front. Mol. Neurosci. 17, 1379726. doi:10.3389/fnmol.2024.1379726
Chen, Q., Sun, X., Zhou, X. H., Liu, J. H., Wu, J., Zhang, Y., et al. (2013). N-terminal horseshoe conformation of DCC is functionally required for axon guidance and might be shared by other neural receptors. J. Cell Sci. 126, 186–195. doi:10.1242/jcs.111278
Dodero, L., Damiano, M., Galbusera, A., Bifone, A., Tsaftsaris, S. A., Scattoni, M. L., et al. (2013). Neuroimaging evidence of major morpho-anatomical and functional abnormalities in the BTBR T+TF/J mouse model of autism. PLoS One 8, e76655. doi:10.1371/journal.pone.0076655
Egaas, B., Courchesne, E., and Saitoh, O. (1995). Reduced size of corpus callosum in autism. Arch. Neurol. 52, 794–801. doi:10.1001/archneur.1995.00540320070014
Ellegood, J., Anagnostou, E., Babineau, B. A., Crawley, J. N., Lin, L., Genestine, M., et al. (2015). Clustering autism: using neuroanatomical differences in 26 mouse models to gain insight into the heterogeneity. Mol. Psychiatry 20, 118–125. doi:10.1038/mp.2014.98
Fenlon, L. R., Liu, S., Gobius, I., Kurniawan, N. D., Murphy, S., Moldrich, R. X., et al. (2015). Formation of functional areas in the cerebral cortex is disrupted in a mouse model of autism spectrum disorder. Neural Dev. 10, 10. doi:10.1186/s13064-015-0033-y
Frazier, T. W., and Hardan, A. Y. (2009). A meta-analysis of the corpus callosum in autism. Biol. Psychiatry 66, 935–941. doi:10.1016/j.biopsych.2009.07.022
Haas, R. H., Townsend, J., Courchesne, E., Lincoln, A. J., Schreibman, L., and Yeung-Courchesne, R. (1996). Neurologic abnormalities in infantile autism. J. Child. Neurol. 11, 84–92. doi:10.1177/088307389601100204
Hardan, A. Y., Minshew, N. J., and Keshavan, M. S. (2000). Corpus callosum size in autism. Neurology 55, 1033–1036. doi:10.1212/wnl.55.7.1033
Hetts, S. W., Sherr, E. H., Chao, S., Gobuty, S., and Barkovich, A. J. (2006). Anomalies of the corpus callosum: an MR analysis of the phenotypic spectrum of associated malformations. AJR Am. J. Roentgenol. 187, 1343–1348. doi:10.2214/AJR.05.0146
Infante, J., Prieto, C., Sierra, M., Sanchez-Juan, P., Gonzalez-Aramburu, I., Sanchez-Quintana, C., et al. (2015). Identification of candidate genes for Parkinson's disease through blood transcriptome analysis in LRRK2-G2019S carriers, idiopathic cases, and controls. Neurobiol. Aging 36, 1105–1109. doi:10.1016/j.neurobiolaging.2014.10.039
Islam, S. M., Shinmyo, Y., Okafuji, T., Su, Y., Naser, I. B., Ahmed, G., et al. (2009). Draxin, a repulsive guidance protein for spinal cord and forebrain commissures. Science 323, 388–393. doi:10.1126/science.1165187
Jasmin, M., Ahn, E. H., Voutilainen, M. H., Fombonne, J., Guix, C., Viljakainen, T., et al. (2021). Netrin-1 and its receptor DCC modulate survival and death of dopamine neurons and Parkinson's disease features. EMBO J. 40, e105537. doi:10.15252/embj.2020105537
Jones-Davis, D. M., Yang, M., Rider, E., Osbun, N. C., Da Gente, G. J., Li, J., et al. (2013). Quantitative trait loci for interhemispheric commissure development and social behaviors in the BTBR T⁺ tf/J mouse model of autism. PLoS One 8, e61829. doi:10.1371/journal.pone.0061829
Keller, T. A., Kana, R. K., and Just, M. A. (2007). A developmental study of the structural integrity of white matter in autism. Neuroreport 18, 23–27. doi:10.1097/01.wnr.0000239965.21685.99
Koshiyama, D., Fukunaga, M., Okada, N., Morita, K., Nemoto, K., Usui, K., et al. (2020). White matter microstructural alterations across four major psychiatric disorders: mega-analysis study in 2937 individuals. Mol. Psychiatry 25, 883–895. doi:10.1038/s41380-019-0553-7
Kumar, A., Sundaram, S. K., Sivaswamy, L., Behen, M. E., Makki, M. I., Ager, J., et al. (2010). Alterations in frontal lobe tracts and corpus callosum in young children with autism spectrum disorder. Cereb. Cortex 20, 2103–2113. doi:10.1093/cercor/bhp278
Lai, M. C., Lombardo, M. V., and Baron-Cohen, S. (2014). Autism. Lancet 383, 896–910. doi:10.1016/S0140-6736(13)61539-1
Lin, C. W., Ellegood, J., Tamada, K., Miura, I., Konda, M., Takeshita, K., et al. (2023). An old model with new insights: endogenous retroviruses drive the evolvement toward ASD susceptibility and hijack transcription machinery during development. Mol. Psychiatry 28, 1932–1945. doi:10.1038/s41380-023-01999-z
Lopez-Bendito, G., and Molnar, Z. (2003). Thalamocortical development: how are we going to get there? Nat. Rev. Neurosci. 4, 276–289. doi:10.1038/nrn1075
Manes, F., Piven, J., Vrancic, D., Nanclares, V., Plebst, C., and Starkstein, S. E. (1999). An MRI study of the corpus callosum and cerebellum in mentally retarded autistic individuals. J. Neuropsychiatry Clin. Neurosci. 11, 470–474. doi:10.1176/jnp.11.4.470
Mcfarlane, H. G., Kusek, G. K., Yang, M., Phoenix, J. L., Bolivar, V. J., and Crawley, J. N. (2008). Autism-like behavioral phenotypes in BTBR T+tf/J mice. Genes Brain Behav. 7, 152–163. doi:10.1111/j.1601-183X.2007.00330.x
Mehlen, P., Delloye-Bourgeois, C., and Chedotal, A. (2011). Novel roles for Slits and netrins: axon guidance cues as anticancer targets? Nat. Rev. Cancer 11, 188–197. doi:10.1038/nrc3005
Meli, R., Weisova, P., and Propst, F. (2015). Repulsive axon guidance by Draxin is mediated by protein Kinase B (Akt), glycogen synthase kinase-3β (GSK-3β) and microtubule-associated protein 1B. PLoS One 10, e0119524. doi:10.1371/journal.pone.0119524
Mercier, F., Kwon, Y. C., and Douet, V. (2012). Hippocampus/amygdala alterations, loss of heparan sulfates, fractones and ventricle wall reduction in adult BTBR T+ tf/J mice, animal model for autism. Neurosci. Lett. 506, 208–213. doi:10.1016/j.neulet.2011.11.007
Miyake, A., Takahashi, Y., Miwa, H., Shimada, A., Konishi, M., and Itoh, N. (2009). Neucrin is a novel neural-specific secreted antagonist to canonical Wnt signaling. Biochem. Biophys. Res. Commun. 390, 1051–1055. doi:10.1016/j.bbrc.2009.10.113
Morcom, L., Edwards, T. J., Rider, E., Jones-Davis, D., Lim, J. W., Chen, K. S., et al. (2021). DRAXIN regulates interhemispheric fissure remodelling to influence the extent of corpus callosum formation. Elife 10, e61618. doi:10.7554/eLife.61618
Moy, S. S., Nadler, J. J., Young, N. B., Perez, A., Holloway, L. P., Barbaro, R. P., et al. (2007). Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behav. Brain Res. 176, 4–20. doi:10.1016/j.bbr.2006.07.030
Nadler, J. J., Zou, F., Huang, H., Moy, S. S., Lauder, J., Crawley, J. N., et al. (2006). Large-scale gene expression differences across brain regions and inbred strains correlate with a behavioral phenotype. Genetics 174, 1229–1236. doi:10.1534/genetics.106.061481
Naser, I. B., Su, Y., Islam, S. M., Shinmyo, Y., Zhang, S., Ahmed, G., et al. (2009). Analysis of a repulsive axon guidance molecule, draxin, on ventrally directed axon projection in chick early embryonic midbrain. Dev. Biol. 332, 351–359. doi:10.1016/j.ydbio.2009.06.004
Nugent, A. A., Kolpak, A. L., and Engle, E. C. (2012). Human disorders of axon guidance. Curr. Opin. Neurobiol. 22, 837–843. doi:10.1016/j.conb.2012.02.006
Piven, J., Bailey, J., Ranson, B. J., and Arndt, S. (1997). An MRI study of the corpus callosum in autism. Am. J. Psychiatry 154, 1051–1056. doi:10.1176/ajp.154.8.1051
Saitoh, O., Courchesne, E., Egaas, B., Lincoln, A. J., and Schreibman, L. (1995). Cross-sectional area of the posterior hippocampus in autistic patients with cerebellar and corpus callosum abnormalities. Neurology 45, 317–324. doi:10.1212/wnl.45.2.317
Shinmyo, Y., Asrafuzzaman Riyadh, M., Ahmed, G., Bin Naser, I., Hossain, M., Takebayashi, H., et al. (2015). Draxin from neocortical neurons controls the guidance of thalamocortical projections into the neocortex. Nat. Commun. 6, 10232. doi:10.1038/ncomms10232
Van Battum, E. Y., Brignani, S., and Pasterkamp, R. J. (2015). Axon guidance proteins in neurological disorders. Lancet Neurol. 14, 532–546. doi:10.1016/S1474-4422(14)70257-1
Vosberg, D. E., Leyton, M., and Flores, C. (2020). The Netrin-1/DCC guidance system: dopamine pathway maturation and psychiatric disorders emerging in adolescence. Mol. Psychiatry 25, 297–307. doi:10.1038/s41380-019-0561-7
Wahlsten, D., Metten, P., and Crabbe, J. C. (2003). Survey of 21 inbred mouse strains in two laboratories reveals that BTBR T/+ tf/tf has severely reduced hippocampal commissure and absent corpus callosum. Brain Res. 971, 47–54. doi:10.1016/s0006-8993(03)02354-0
Weinstein, M., Ben-Sira, L., Levy, Y., Zachor, D. A., Ben Itzhak, E., Artzi, M., et al. (2011). Abnormal white matter integrity in young children with autism. Hum. Brain Mapp. 32, 534–543. doi:10.1002/hbm.21042
Yang, M., Clarke, A. M., and Crawley, J. N. (2009). Postnatal lesion evidence against a primary role for the corpus callosum in mouse sociability. Eur. J. Neurosci. 29, 1663–1677. doi:10.1111/j.1460-9568.2009.06714.x
Keywords: axon guidance, draxin, BTBR mouse, ASD, corpus callosum
Citation: Shinmyo Y (2025) Implications of draxin in neurological disorders. Front. Cell Dev. Biol. 13:1560940. doi: 10.3389/fcell.2025.1560940
Received: 15 January 2025; Accepted: 11 February 2025;
Published: 24 February 2025.
Edited by:
Junichi Yuasa-Kawada, Juntendo University, JapanReviewed by:
Mitsuharu Hattori, Nagoya City University, JapanCopyright © 2025 Shinmyo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yohei Shinmyo, c2hpbm15b0BoYW1hLW1lZC5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.