
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 26 March 2025
Sec. Cellular Biochemistry
Volume 13 - 2025 | https://doi.org/10.3389/fcell.2025.1521336
This article is part of the Research Topic Redox Signaling in Metabolism, Development, and Aging View all articles
 Satya Srirama Karthik Divvela1
Satya Srirama Karthik Divvela1 Marialucia Gallorini2
Marialucia Gallorini2 Morris Gellisch1
Morris Gellisch1 Gaurav Deepak Patel1
Gaurav Deepak Patel1 Luciano Saso3*
Luciano Saso3* Beate Brand-Saberi1*
Beate Brand-Saberi1*Embryonic development is a complex process of concurrent events comprising cell proliferation, differentiation, morphogenesis, migration, and tissue remodeling. To cope with the demands arising from these developmental processes, cells increase their nutrient uptake, which subsequently increases their metabolic activity. Mitochondria play a key role in the maintenance of metabolism and production of reactive oxygen species (ROS) as a natural byproduct. Regulation of ROS by antioxidants is critical and tightly regulated during embryonic development, as dysregulation results in oxidative stress that damages essential cellular components such as DNA, proteins, and lipids, which are crucial for cellular maintenance and in extension development. However, during development, exposure to certain exogenous factors or damage to cellular components can result in an imbalance between ROS production and its neutralization by antioxidants, leading to detrimental effects on the developmental process. In this review article, we highlight the crucial role of redox homeostasis in normal development and how disruptions in redox balance may result in developmental defects.
Embryonic development is a highly coordinated process that culminates in the formation of a fully functional organism governed by a complex interplay of genetic, cellular, and environmental factors. During this pivotal phase, the developing embryo undergoes a series of precise and tightly regulated remodeling events involving cell proliferation, rearrangement, migration, and morphogenetic movements. All of these features are accompanied by dynamic changes in gene activity, ultimately giving rise to a diverse array of tissues and organs that constitute the adult organism. Interestingly, each of these steps is characterized by different oxygen levels, ranging from 1% to 5% during the formation of the placenta and up to 21% upon fetal development, where tissue differentiation becomes the predominant process (Michiels, 2004; Hitchler and Domann, 2007; Ortega et al., 2016). These dynamic remodeling steps, summarized as morphogenesis, are accompanied by a variety of challenges experienced by cells and tissues during embryonic development. One critical stage is the generation of oxidative stress within the developing embryo (Danielsson et al., 2023). Oxidation–reduction (redox) homeostasis, like pH control, is central to life. Redox processes pervade almost all fundamental life processes, from bioenergetics to metabolism and life functions (Laforgia et al., 2018).
Oxidative stress arises from an imbalance between the production of reactive oxygen species (ROS), that is, the production of free radicals such as hydrogen peroxide (H2O2), superoxide anions (O2●−), and hydroxyl radicals (●OH) (Figure 1) as well as the ability of the embryo’s antioxidant defense mechanisms to neutralize these potentially harmful molecules (Fathollahipour et al., 2019). In other words, oxidative stress disrupts mitochondrial redox signaling and control. High-flux pathways, such as the electron transport chain involved in ATP production, generate ROS as a byproduct. These ROS molecules feed into low-flux pathways, which also engage antioxidants to neutralize free radicals and oxidized macromolecules. Dysfunction in the high-flux pathways results in higher ROS production, disrupting cellular homeostasis, whereas dysfunction in the low-flux pathways compromises cell signaling in metabolism, growth, and apoptosis (Jones, 2006).

Figure 1. Prominent species of reactive oxygen.
In the context of embryonic development, a delicate balance between ROS generation and regulation becomes paramount, as oxidative stress can have profound consequences on the normal course of development (DeFreitas et al., 2022). In mammals, the origin of oxidative stress during embryonic development is multifaceted, encompassing a wide range of factors. This review aims to comprehensively explore the causes underlying oxidative stress development during embryogenesis, shedding light on intracellular sources of ROS, cell mechanisms involved, and the potential consequences of oxidative stress during this critical developmental period. By deepening our knowledge regarding the intracellular burst of oxidative stress during embryogenesis, valuable insights into the intricate processes governing the formation of life and the potential implications for developmental disorders and birth defects can be gained (Danielsson et al., 2023). Additionally, oxidative stress generated during morphogenesis may contribute to long-term health complications that manifest later in life (Ducsay et al., 2018; Suzuki, 2018).
The origin of oxidative stress, including both endogenous and exogenous sources, is described in the following sections. Intracellular molecular responses activated by oxidative stress during embryonic development have been presented in parallel. Additionally, this review explores the consequences of oxidative stress on morphogenesis and how an imbalanced redox equilibrium may have detrimental effects on normal mammalian embryonic development. The potential impact of oxidative stress is analyzed in terms of developmental disorders. Finally, the protective role of antioxidant defense mechanisms is considered, and potential avenues for future research in this vital field of study are discussed.
Embryogenesis requires specific signaling pathways to regulate cell proliferation and differentiation. Oxidative stress, due to an imbalance between the production of ROS and antioxidant defenses, disrupts signaling pathways and plays a causative role in birth defects. ROS generation within embryos can arise from various endogenous and exogenous sources (Jomova et al., 2023). Understanding these sources is essential for unraveling the complex mechanisms driving embryonic development and vulnerability to oxidative stress.
One of the primary endogenous sources of ROS in embryonic tissues is the normal metabolic activity of the developing cells. Mitochondria, the powerhouses of cells, produce ROS as byproducts of oxidative phosphorylation (Zorov et al., 2014). During embryogenesis, as cells rapidly divide and differentiate, there is an increased demand for energy production, leading to a higher rate of mitochondrial respiration and, consequently, elevated ROS production. In the early embryonic stages, mitochondria exhibit distinct characteristics (small and immature), which align with reduced oxidative phosphorylation and greater reliance on glycolysis. However, as cells differentiate, oxidative phosphorylation becomes a major source of ATP (Blerkom, 2004; Facucho-Oliveira and John, 2009). Given this crucial role, any mutations in mitochondria or dysfunction of mitochondria, as observed in maternal metabolic disorders such as diabetes and obesity, have been shown to be detrimental to organogenesis, compromising mechanisms such as proliferation, differentiation, and apoptosis, all of which affect organ maturation and development (Lu et al., 2009; Steffann et al., 2015).
Life processes rely on enzymatic reactions. Enzymes involved in various cellular processes, such as those responsible for DNA replication, repair, and cellular signaling, can generate ROS as part of their normal function (Magnani and Mattevi, 2019). More than 40 enzymes generate O2●−/H2O2, including the NOX family of multi-subunit NADPH oxidases, the transmembrane components of which are responsible for electron transport across biological membranes. Members of the NADPH oxidase family (Nox1-3) are involved in the catalysis of superoxides by transferring electrons from NADPH to molecular oxygen, whereas Nox4, Duox1, and Duox2 are involved in the catalysis of H2O2 from molecular oxygen (Geiszt et al., 2003; Joshi et al., 2013; Nisimoto et al., 2014; Lam et al., 2015; Szanto et al., 2019). In addition to NOX, found principally on the plasma membrane, nuclear, and endoplasmic reticulum (ER) membranes, peroxisomes are major generators of ROS. Peroxisomes contain various oxidases that produce H2O2 as a byproduct of fatty acid β-oxidation. They also contain antioxidants like catalase and superoxide dismutase to neutralize ROS (Antonenkov et al., 2010; Zhang et al., 2015). Another enzyme known to produce ROS is xanthine oxidase, which catalyzes the oxidation of hypoxanthine to uric acid, generating superoxide and hydrogen peroxide as byproducts (Cantu-Medellin and Kelley, 2013). These enzymatic reactions play a physiological role and are essential for proper development (Dutta et al., 2020). However, they can contribute to oxidative stress if not properly controlled (Cobbaut and Lint, 2018). During embryonic development, reactive oxygen species (ROS) are also produced in response to growth factors and cytokines. Growth factors, such as vascular endothelial growth factor (VEGF), and cytokines, such as TNF-α and IL-1, have been shown to stimulate ROS production in different contexts (Gurjar et al., 2001; Kim et al., 2010; Maraldi et al., 2010).
A special scenario is represented by the mammalian maternal–fetal interface. During pregnancy, the maternal–fetal interface is a critical site for oxidative stress. As a matter of fact, the placenta plays a crucial role in nutrient transport and gas exchange between the maternal and fetal circulations. Changes in oxygen levels (hypoxia or hyperoxia) during development and fluctuations in blood flow and tissue perfusion can lead to oxidative stress (Torres-Cuevas et al., 2017). These changes can occur naturally as part of the embryonic developmental program but may also result from various pathological conditions (Danielsson et al., 2023).
In addition, exposure to maternal agents, such as infections and inflammation, or maternal lifestyle choices, such as smoking or alcohol consumption, can elevate ROS levels in embryonic tissues (Jiang et al., 2012). It has been recently reported that cigarette smoke is associated with the upregulation of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) protein expression and activity in granulosa cells of women undergoing in vitro fertilization (Budani et al., 2022). Additionally, placental dysfunction or inadequate blood supply may compromise oxygen delivery to the developing embryo, leading to hypoxia–reperfusion injury that can trigger ROS production (Leslie et al., 2015). Moreover, infections and inflammatory responses in the maternal system can lead to the release of cytokines and immune cells, which generate ROS (Yu et al., 2022). These inflammatory mediators can potentially reach the developing embryo and initiate oxidative stress. In the case of maternal infections, such as those caused by viruses or bacteria, the development of fragile embryo immune defenses may be less effective in combating infection-triggered oxidative stress (Hussain et al., 2021).
Maternal diet and nutrition play crucial roles in mammalian embryonic development. A diet lacking essential nutrients and antioxidants can lead to oxidative stress in both the mother and the developing embryo (Diniz et al., 2023). Conversely, excessive intake of certain nutrients, such as iron or vitamin A, can also contribute to oxidative stress and depletion of the intracellular pool of glutathione through mechanisms such as the Fenton reaction or excessive production of ROS (Morales et al., 2022).
Exogenous sources of oxidative stress are equally significant contributors to embryonic oxidative stress. Environmental factors, including exposure to pollutants, radiation, and toxins, can affect embryo development (Al-Gubory, 2014). Some pollutants and chemicals, such as heavy metals and pesticides, can induce oxidative stress by promoting ROS production or interfering with antioxidant defenses (Ruder et al., 2008). Recently, micro- and nanoplastic (MNP) accumulation has been observed in the human placenta, raising important questions regarding the biological effects of these contaminants on the health of pregnant women and offspring (Zurub et al., 2024). In both rat and mouse models, oral exposure to MNPs results in the accumulation of these particles within the uterine tissue and in various ovarian compartments, including growing follicles (Wei et al., 2022). A recent study showed the accumulation of MNPs in the placenta has also been shown to increase apoptosis and induce endoplasmic reticulum stress, accompanied by elevated ROS levels, resulting in placental dysfunction and growth retardation (Bai et al., 2024).
In summary, the sources of oxidative stress in embryonic tissues are diverse and multifaceted and arise from both endogenous and exogenous factors (Figure 2). Developing embryos must navigate these challenges while maintaining a delicate balance between unavoidable ROS generation and antioxidant defense mechanisms. Understanding the origin of oxidative stress during embryonic development is essential for understanding its impact on normal development and its potential contribution to developmental disorders and birth defects.
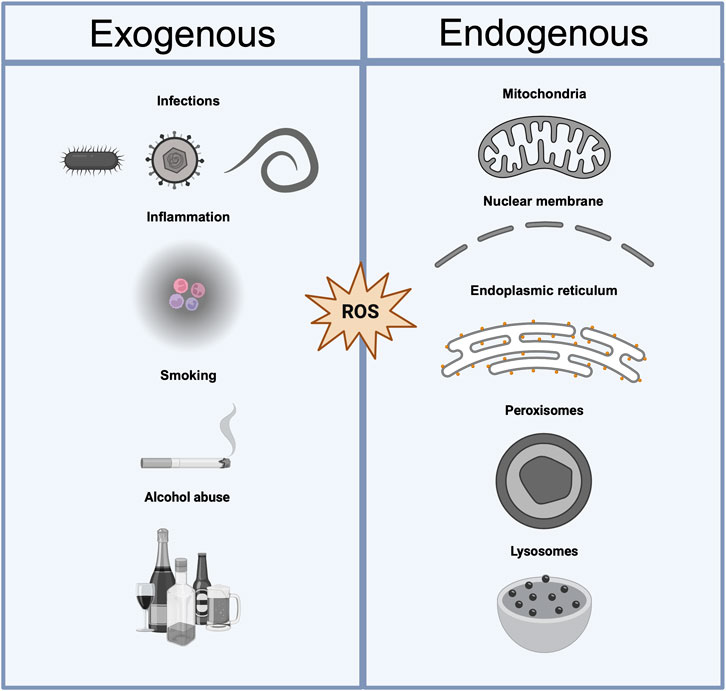
Figure 2. Exogenous and endogenous sources of ROS. Sources of oxidative stress can be categorized as exogenous or endogenous. Exogenous sources include external factors such as infections (e.g., Mycobacterium tuberculosis, Pseudomonas aeruginosa, HIV, influenza virus, and Plasmodium falciparum), inflammation, smoking, and alcohol abuse, which contribute to the production of reactive oxygen species (ROS). Endogenous sources arise from internal cellular processes, including mitochondrial activity, disruptions in the nuclear membrane, protein misfolding in the endoplasmic reticulum, and lysosomal dysfunction. Both categories lead to an imbalance in ROS homeostasis, potentially resulting in oxidative damage to biomolecules and cellular structures.
Oxidative stress, which is characterized by an imbalance between the production of reactive oxygen species (ROS) and the capacity of antioxidant defense mechanisms to neutralize them, can exert profound effects on embryonic development. During this critical period, the developing embryo undergoes the intricate processes of cell division, differentiation, and tissue formation (Dennery, 2007). When oxidative stress disrupts these processes, it can lead to growth retardation and malformations in the embryo and fetus (Dennery, 2010).
In response to the aforementioned stressors that result in free radicals (FR), cells activate antioxidants to limit the damage caused by the free radicals. Antioxidants can be classified into two categories: enzymatic and non-enzymatic. The enzymatic group comprises superoxide dismutases, catalase, glutathione peroxidases, glutathione reductase, peroxiredoxins, and thioredoxin. The non-enzymatic group includes glutathione, vitamin C, vitamin E, beta-carotene, and ubiquinone. Superoxide dismutases (SODs) catalyze the dismutation of superoxide radicals into hydrogen peroxide (H2O2) and molecular oxygen (O2). Intracellular H2O2 normally oxidizes cysteine residues in proteins to initiate redox biology, or it may be converted to H2O by cellular antioxidant proteins, such as peroxiredoxins (PRx), glutathione peroxidase (GPx), and catalase (CAT). Glutathione reductase (GR) catalyzes the reduction of glutathione disulfide (GSSG) to glutathione, which is crucial for redox homeostasis. Thioredoxin (Txn) catalyzes the reduction of disulfide bonds in proteins and acts as an electron donor for peroxiredoxins.
The effect of free radicals on embryonic development is complex, as these molecules have diverse effects, such as deterioration of cell promotion depending on the number of free radicals, starting from the first stages of development (fertilization, cleavage state, compaction, and blastocyst formation). Under normal conditions, a balance of ROS is observed prior to fertilization in both sperm and oocytes. However, it has been reported that disproportionate production and neutralization of ROS affects sperm maturation (Barati et al., 2020), motility, and capacitation (Takeshima et al., 2021). During fertilization, ROS also play a crucial role in the regulation of calcium levels and ATP synthesis in oocytes (Lewis et al., 2014). Dysregulation of ROS has been shown to have profound effects on development, resulting from spindle instability and chromosomal abnormalities, which directly affect the developmental competence of oocytes (Sasaki et al., 2019). In parallel, ROS have been observed to increase during the very first cell divisions post-fertilization, in the time of blastula formation, and during hatching prior to implantation in murine and bovine embryos (Lopes et al., 2010; Deluao et al., 2022). Recent evidence has shown that increased ROS in early embryonic stages alters embryonic programming and enhances the risk of diseases later in life, with transgenerational effects (Figure 3) (Loeken, 2004; Ornoy, 2007; Morrison, 2008).
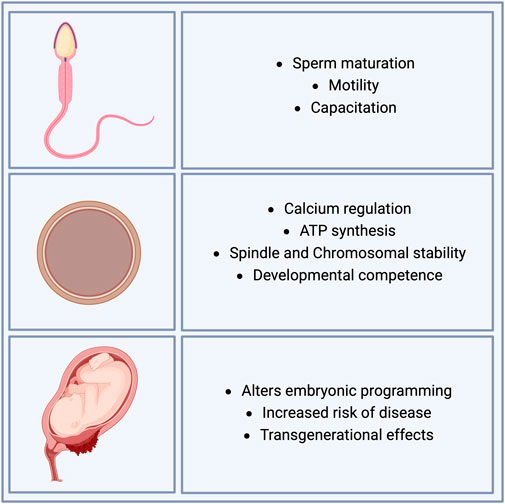
Figure 3. Effects of ROS on sperm, oocyte, and developmental competence. Oxidative stress plays a crucial role in reproductive processes, impacting both male and female gametes and embryonic development. In sperm, oxidative stress impairs maturation, motility, and capacitation, key processes essential for successful fertilization. In oocytes, oxidative stress influences calcium regulation, ATP synthesis, and the stability of the spindle and chromosomes, all of which are critical for ensuring developmental competence. During embryonic development, oxidative stress affects embryonic programming, increases the risk of disease, and may have transgenerational effects, potentially influencing the health of future generations.
Several pro-and antioxidant genes have been studied for loss-of-function mutations to better understand the impact of oxidative stress on embryonic development. In mouse models, loss-of-function mutations of pro-oxidant genes, such as Nox1, Nox2, Nox3, Nox4, Duox1, and Duox2, did not reveal any embryonic phenotype. However, adult mice showed different pathological conditions. In contrast, loss-of-function mutations of key antioxidant genes have been shown to be detrimental to embryonic development. For instance, thioredoxin (Txn) has been shown to be indispensable for early embryonic development, as its loss resulted in embryonic lethality after implantation (Matsui et al., 1996). Another antioxidant, glutathione peroxidase 4 (GPx4), is a highly evolutionarily conserved enzyme that acts as a phospholipid hydroperoxidase, utilizing reduced glutathione (GSH) to convert phospholipid hydroperoxides (PL─OOH) to phospholipid alcohols (PL─OH), which serves to regulate lipid peroxide levels. The depletion of GSH or inactivation of GPx4 in cells leads to excessive reactive oxygen species (ROS)-induced lipid peroxides and compromises redox homeostasis. Gpx4-null mice show embryonic lethality due to intrauterine resorption around embryonic day 7.5 during gastrulation (Yant et al., 2003). Similarly, studies have shown that the absence of two key antioxidant enzymes leads to impaired gastrulation and embryonic death in mouse models. These enzymes include glutathione synthetase (Gss), which is crucial for glutathione (Gsh) production, and thioredoxin reductase 1 (Txnrd1), which maintains thioredoxin in its reduced state (Bondareva et al., 2007; Winkler et al., 2011). Similarly, knockout models of the glutamate–cysteine ligase catalytic subunit (Gclc), thioredoxin reductase 2 (Txnrd2), and glutaredoxin 3 (Glrx3), which play crucial roles as antioxidants, also showed embryonic lethality during mid-gestation (E12.5–E13.5) (Dalton et al., 2000; Conrad et al., 2004; Cheng et al., 2011).
Loss of the other key antioxidant enzyme, superoxide dismutase (Sod2), was shown to be peri- or postnatally lethal in mice. Embryos exhibited severe oxidative stress during development, with pathologies such as dilated cardiomyopathy, neurodegeneration, metabolic acidosis, and lipid accumulation in the liver and skeletal muscles (Li et al., 1995; Huang et al., 2001; Ikegami et al., 2002). Additionally, the loss of function of genes such as isocitrate dehydrogenase 1 (Idh1), ferritin heavy chain 1 (Fth1), and glucose-6-phosphate dehydrogenase (G6pd), which play antioxidant roles, also resulted in embryonic lethality (Ferreira et al., 2000; Longo et al., 2002; Sasaki et al., 2012). We curated a list of well-known pro-and antioxidant knockout mouse models by briefly showing the phenotype during embryonic development (Table 1).
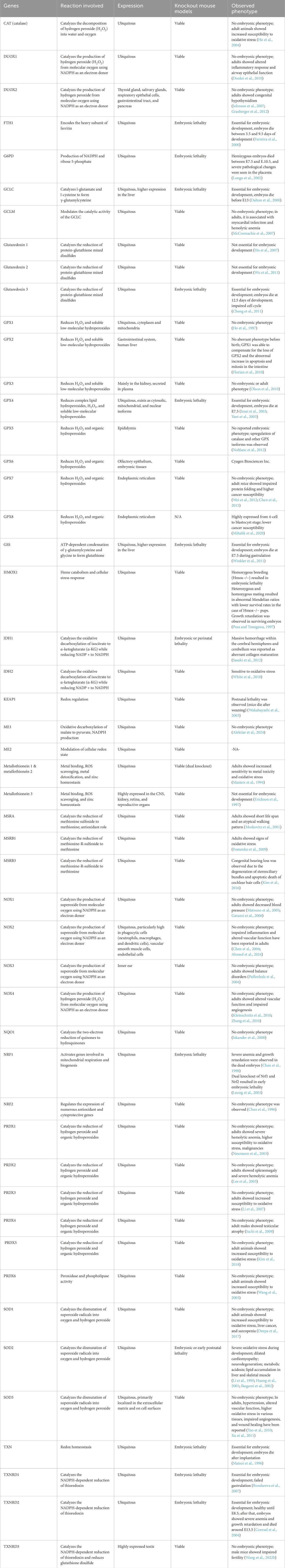
Table 1. List of phenotypes observed following ablation of pro- and antioxidant genes.
In parallel with studies on developing embryos, stem cells and stem cell-derived systems, such as organoids, have been studied regarding their oxygen consumption, dependence, and the impact thereof, revealing a tightly regulated network between oxygen metabolism and free radical (FR) production (Mohyeldin et al., 2010; Närvä et al., 2013). These in vitro systems allow researchers to study the impact of oxygen concentrations and oxidative stress under more standardized conditions than in living embryos and even enable them to analyze the molecular components and culture materials that alleviate cellular stress conditions to a certain extent (Harrison et al., 2007; Khattak et al., 2007; Gholipourmalekabadi et al., 2016; Oyefeso et al., 2021). During early embryonic development, embryonic stem cells (ESCs) reside in a hypoxic microenvironment, where cells use glycolysis to quickly produce very low levels of ATP. However, during differentiation, ATP production increases via oxidative phosphorylation (OxPhos), which in turn generates ROS (Cho et al., 2006). Metabolic shifts between glycolysis and OxPhos are accompanied by the differentiation of pluripotent stem cells (PSCs). The enhancement of glycolysis via hypoxia and the suppression of OxPhos leads to concomitantly decreased ROS levels, promoting the maintenance and proliferation of PSCs and thereby repressing differentiation (Mandal et al., 2011). Endogenous ROS levels are increased by sirtuin 1 (SIRT1)-mediated inhibition of p53 antioxidant function. SIRT1, a longevity-promoting NAD+-dependent class III histone deacetylase, is also involved in PSC function by regulating the p53-dependent expression of the pluripotency marker Nanog. SIRT1 is precisely suppressed during human PSC differentiation, resulting in the reactivation of developmental genes, such as the neuroretinal morphogenesis regulators DLL4, TBX3, and PAX6. Similarly, superoxide dismutase 1 (Sod1) is modulated by Oct4, Sox2, and Nanog, suggesting a core relationship between redox homeostasis and pluripotency in PSCs (Lee et al., 2018).
In addition to PSCs, mesenchymal stem cells (MSCs) have low levels of intracellular ROS and high levels of glutathione, a key antioxidant. They also constitutively express high levels of the enzymes required to manage oxidative stress. In terms of redox regulation, numerous recent reports have described the importance of oxidants in MSC differentiation into adipocytes, osteocytes, chondrocytes, and myocytes through the activation of signaling cascades involved in differentiation. In contrast, elevated levels of ROS lead to cell cycle arrest and apoptosis in MSCs (Atashi et al., 2015). Thus, the ability to respond to environmental oxidative damage is a universal property of MSC, but the biological mechanisms employed by fetal and placental MSCs in response to oxidative stress might be compromised under pathophysiological conditions such as preeclampsia (Kusuma et al., 2022).
ROS primarily act as secondary messengers by regulating key transcription factors, thereby influencing cellular signaling. The rapid turnover of ROS through enzymatic reactions is a major contributor to this process. However, during embryonic development, ROS are produced locally, acting as primary messengers that affect specific signaling pathways (Hansen, 2006; Schieber and Chandel, 2014). In addition, different ROS concentrations have been observed to influence various cellular mechanisms. Studies have shown that a reduction in ROS promotes cell proliferation, moderate ROS levels promote the differentiation of stem cells, and highly elevated ROS results in apoptosis or necrosis (Milkovic et al., 2019).
During development, ROS have been shown to alter vital pathways by regulating transcription factors such as activator protein (Ap1), hypoxia-inducible factor (HIF1), nuclear factor κB (NF-κB), nuclear factor (NF)-E2 related factor 1 and 2 (Nrf1, Nrf2), and redox-sensitive factors such as redox effector factor-1 (Ref-1) and wingless-related integration site (Wnt), which play crucial roles in proliferation, differentiation, and apoptosis (Dennery, 2007). In addition to enzymatic and non-enzymatic cellular responses to oxidative stress, living tissues can combat the consequences of oxidative stress by activating redox signaling pathways by regulating transcription factors. Nuclear factor (NF)-E2-related factor 2 (Nrf2) is the master regulator of antioxidant cell responses and orchestrates the expression of antioxidant genes, enabling cells to mount a defense against oxidative damage (Ma, 2013). Being located at the intersection of crucial signaling pathways, Nrf2 can influence a number of critical cellular functions, which extend beyond the maintenance of redox balance but include cellular metabolism, proteostasis, mitochondrial function, inflammation, and cell differentiation during development. Therefore, Nrf2 exhibits biological dualism by being involved in many pathological conditions, such as cancer (Gallorini et al., 2024). It has been reported that prolonged or excessive activation of Nrf2 can disrupt embryonic development by perturbing the balance of redox-regulated genes (Harris and Hansen, 2012). Recently, the role of Nrf2 during the blastocyst stage was described, and its mRNA expression was found to be attenuated in porcine embryos cultured under metabolically stressful conditions (Glanzner et al., 2024).
Nrf2 is regulated by Kelch-like ECH-associated protein 1 (KEAP1), an important sensor of oxidative stress (Motohashi and Yamamoto, 2004). KEAP1 inhibits Nrf2 by promoting its ubiquitination. However, upon conformational changes in KEAP1 resulting from the oxidation of cysteine residues, Nrf2 is stabilized and moves into the nucleus, binding the antioxidant-responsive element (ARE) in the promoter region (Figure 4) (Yamamoto et al., 2018). Despite its crucial role, Nrf2 null mice did not show any phenotype during embryonic development and are viable (Chan et al., 1996). Interestingly, a dual knockout of Nrf1 and Nrf2 was embryonically lethal. Nrf1 is known to regulate proteostasis and mitochondrial biogenesis (Leung et al., 2003).
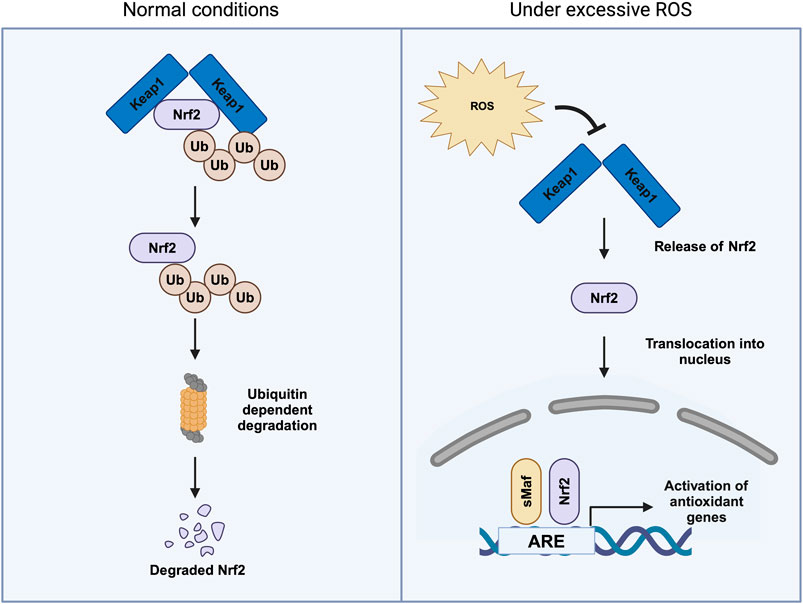
Figure 4. Regulation of Nrf2 under both normal conditions and in the presence of excessive reactive oxygen species (ROS). Under normal conditions, Nrf2 is bound to its cytoplasmic inhibitor, Keap1 (Kelch-like ECH-associated protein 1). This interaction promotes the ubiquitination (Ub) of Nrf2, marking it for proteasomal degradation through the ubiquitin–proteasome pathway, thereby maintaining low intracellular levels of Nrf2. During oxidative stress, characterized by elevated ROS, the interaction between Nrf2 and Keap1 is disrupted. This results in the release of Nrf2, which then translocates into the nucleus. Once in the nucleus, Nrf2 binds to antioxidant response elements (ARE) in the promoter regions of target genes, initiating the transcription of antioxidant and cytoprotective genes that help mitigate oxidative damage.
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), a master regulator of inflammation, has also been identified as being regulated by ROS. ROS have been shown to activate and inhibit NF-κB function depending on the context. Cytoplasmic ROS (H2O2) have been observed to activate NF-κB via oxidation and activation of IKK, whereas nuclear ROS (H2O2) have been observed to inhibit the binding of NF-κB to DNA via oxidation of cysteine residues, thereby decreasing its transcriptional activity (Nakajima and Kitamura, 2013). Knocking out the proteins involved in NF-κB signaling also resulted in embryonic lethality, compromising not only redox homeostasis but also various mechanisms (Pasparakis et al., 2006). Other vital transcription factors that have been observed to be activated by ROS are hypoxia-inducible factors (HIF-1α and HIF-2α), in which oxidants help stabilize HIF factors and initiate the hypoxic response (Pagé et al., 2008). The knockout mouse model of HIF-1α showed embryonic lethality after E8.0 with multiple developmental defects (Ryan et al., 1998). However, HIF-2α null mice were viable but showed multiple organ pathologies (Scortegagna et al., 2003). Activator protein (Ap1) is a transcription factor complex consisting of Jun and Fos family proteins that regulate the oxidative stress response via modulation of gene expression. Knockout models of c-Fos, FosB, and JunD have suggested that these proteins are indispensable for embryonic development. In contrast, c-Jun, JunB, and Fra-1 are essential for both the embryonic and adult stages (Jochum et al., 2001).
Intrinsic ROS generation is tightly regulated to prevent overproduction and subsequent oxidative damage. However, imbalances can occur, mainly during critical developmental stages, which may lead to significant damage to macromolecules such as DNA, proteins, and lipids.
It is well known that ROS can cause DNA damage in developing cells. The activation of DNA repair mechanisms, such as the base excision repair (BER) and nucleotide excision repair (NER) pathways, aims to restore genomic integrity (Musson et al., 2022). However, persistent DNA damage can lead to mutations and chromosomal abnormalities, contributing to developmental defects (Ribeiro et al., 2023).
ROS have been reported to cause a variety of lesions in DNA (such as base and/or sugar alterations, sugar-base cyclization, DNA-protein cross-links, and intra- and inter-strand cross-links), which, in turn, can result in DNA strand breaks. The consensus is that cell cycle checkpoints, including G1/S, intra-S, and G2/M, are involved in DNA damage response reactions. In addition, γH2AX, a marker of DNA damage, is an early indicator of DNA double-strand breaks and plays an important role in the DNA damage response. It has been reported that mouse embryos fertilized with H2O2-treated sperm show the appearance of γH2AX and a delay in first cleavage (Wang et al., 2013). In addition, checkpoint proteins ATM, Chk1, and Cdc25 are phosphorylated and activated in zygotes fertilized with H2O2-treated sperm (Song et al., 2014), which indicates that embryos fertilized with treated sperm might be arrested at the G2/M checkpoint through the ATM → Chk1 → Cdc25B/Cdc25C pathway.
ROS can induce structural and functional changes in proteins and disrupt critical developmental processes. ROS can oxidize thiol (-SH) groups in cysteine residues, resulting in the formation of disulfide bonds between proteins, which in turn alters their conformation and aggregation states. This affects crucial processes, such as fertilization, cell division, and morphogenesis, where damage to the extracellular matrix and cytoskeletal proteins has been observed (Hernebring et al., 2006; Wong and Wessel, 2008). In addition, several other amino acids have been shown to be prone to side-chain modifications by free radicals (Ahmad et al., 2016). For instance, oxidation of methionine to its oxidized form, methionine sulfoxide, can significantly affect S-adenosylmethionine (SAM), a universal methyl donor, resulting in hypomethylation in hESCs (Shiraki et al., 2014; Winkle and Ryznar, 2019). Similarly, the tyrosine residue in superoxide dismutase, a crucial antioxidant, is prone to nitration by peroxynitrite, rendering SOD inactive (Demicheli et al., 2018). Iron-sulfur (Fe-S) clusters in DNA repair enzymes are also prone to oxidation, which compromises the base-excision repair pathway (Musson et al., 2022). Free radicals have also been shown to result in the carbonylation of chaperone proteins such as HSP90, which plays a crucial role in protein folding during embryonic development. HSP90 has been shown to be associated with OCT4 and NANOG and to prevent their ubiquitin-dependent degradation. Therefore, it can be inferred that HSP90 dysfunction due to carbonylation can directly affect the stability of OCT4 and NANOG, directly influencing pluripotency and early embryonic differentiation (Bradley et al., 2012).
Advanced oxidation protein products (AOPPs) are markers of oxidation-mediated protein damage and are usually carried by plasma proteins. As a key product of oxidative reactions, AOPPs and their effects on the female reproductive system have received increasing attention. A high level of AOPPs in the follicular fluid has been reported to have adverse effects on oocytes and early embryonic development. High AOPP concentrations in the follicular fluid are also associated with poor IVF outcomes (Song et al., 2009). AOPPs have also been observed to result in cellular senescence in placental trophoblast cells, disrupting trophoblast cell invasion and placental development and contributing to preeclampsia (PE). Similarly, their accumulation is correlated with infertility, congenital malformations, and pregnancy-related complications (Li et al., 2022).
Lipid peroxidation is a chain reaction induced by free radicals (FR) that impair cell membrane integrity. Free radicals, such as hydroxyl radicals (●OH) or superoxide anions (O2●−), remove a hydrogen atom from polyunsaturated fatty acids (PUFAs) in cell membranes, forming lipid radicals. These lipid radicals react with molecular oxygen to form peroxyl radicals (ROO●). Thus, the formed peroxyl radical removes hydrogen atoms from adjacent PUFAs, generating new radicals and lipid hydroperoxides, further damaging cellular membranes. The reaction continues until the antioxidant interrupts and neutralizes the ROS. Lipid radicals also react with each other to form non-reactive products such as malondialdehyde (MDA) or 4-hydroxynonenal (4-HNE) (Ayala et al., 2014). Overall, lipid peroxidation compromises the membrane integrity, impairs enzyme function, and triggers apoptosis and necrosis. It has also been implicated in several congenital abnormalities.
Excessive lipid peroxidation in the uterine epithelium has been reported to cause implantation failure and pregnancy loss (Lu et al., 2024). Post-implantation, lipid peroxidation has also been observed to interfere with key developmental processes, resulting in several congenital anomalies such as esophageal atresia and autosomal dominant polycystic kidney disease.
Oxidative stress, characterized by excess reactive oxygen species (ROS) in cells, has been extensively studied for its detrimental effects on embryonic and fetal development in mammals (Takahashi, 2012). The most obvious effect of oxidative stress is growth impairment. Intrauterine growth restriction (IUGR) is a common consequence of oxidative stress that occurs during pregnancy. Maternal exposure to high levels of ROS also resulted in reduced fetal growth in mice (Xu et al., 2006). Researchers have attributed this to impaired placental function and reduced nutrient transport. This phenomenon manifests itself as impaired skeletal development. Schoppa et al. (2022) showed that ROS could inhibit osteoblast differentiation in mouse embryos, leading to skeletal abnormalities.
Oxidative stress-induced growth retardation in embryos often results from the disruption of critical cellular processes. Impaired cell proliferation, altered cell cycle regulation, and reduced nutrient uptake due to oxidative damage can all contribute to reduced fetal growth (Joo et al., 2021). In contrast, intrauterine growth restriction (IUGR) increases the risk of preterm births and long-term health issues in offspring. Organogenesis critically depends on a precise orchestration of events: embryonic tissues must proliferate sufficiently to interact by direct fusion, migration, or generation of permissive and instructive signals. Hence, growth retardation and inhibition of proliferation inevitably result in structural malformations. These malformations may affect various organs and systems, including the cardiovascular, nervous, and musculoskeletal systems. For example, oxidative stress-induced damage to neural crest cells can result in congenital heart defects, neural tube defects, and craniofacial abnormalities (Carmichael et al., 2023) (Table 2).
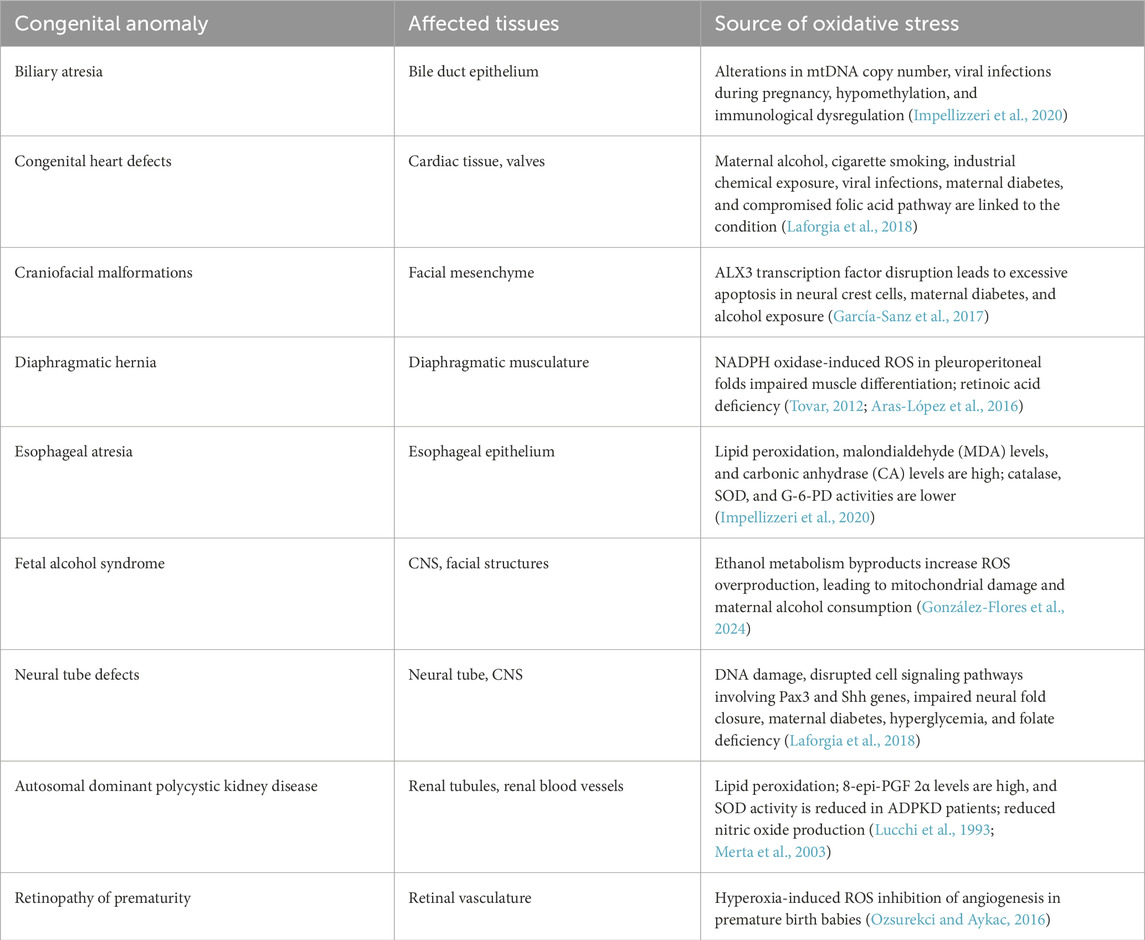
Table 2. List of congenital anomalies associated with oxidative stress.
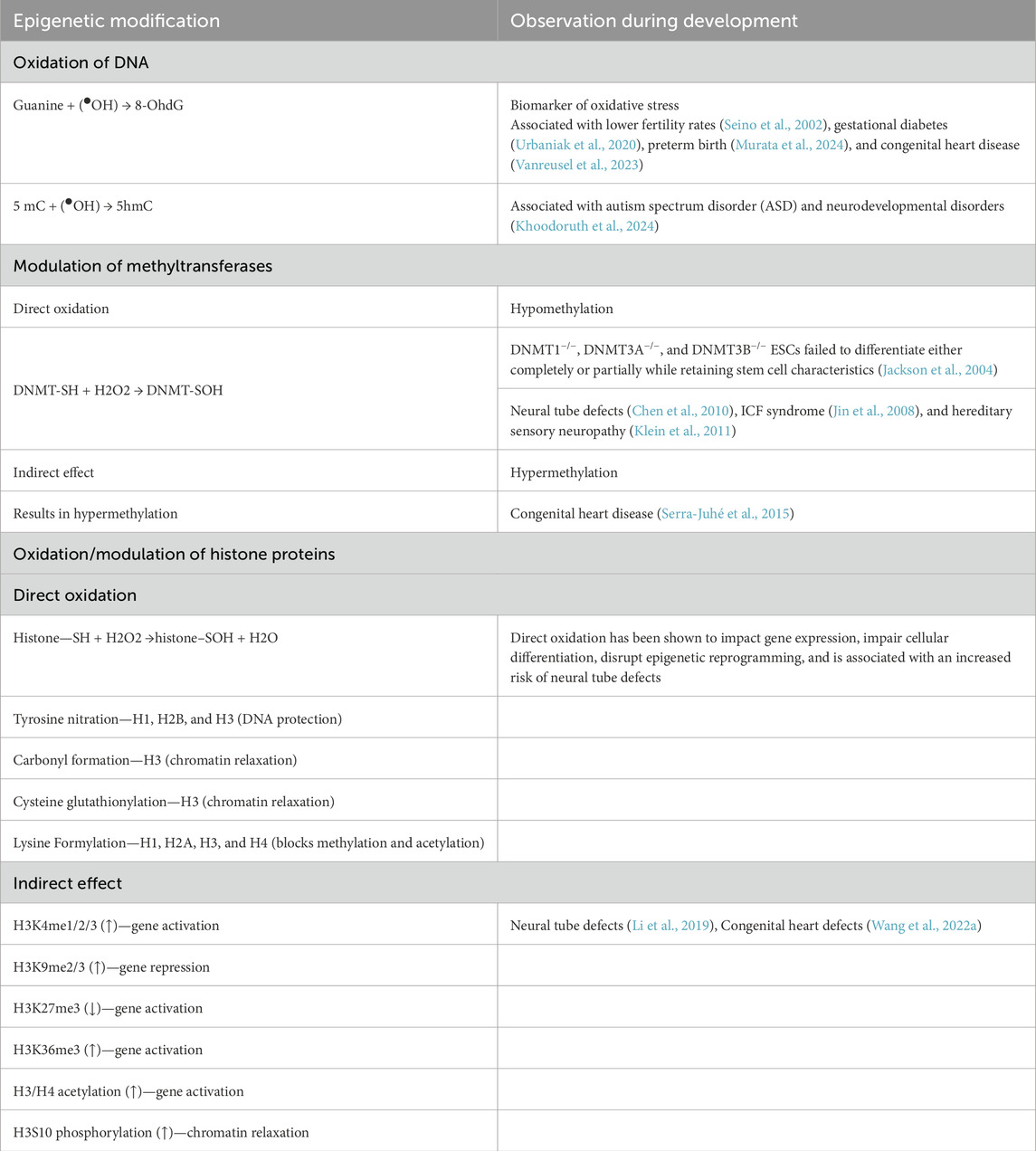
Table 3. ROS alter epigenetic landscape during embryonic development.
Oxidative stress during embryogenesis can affect neurodevelopment (Rains et al., 2021). Studies in rats have demonstrated that oxidative stress induced by prenatal alcohol exposure leads to impaired neuronal migration and neurogenesis in the fetal brain (Sogut et al., 2015). The developing fetal brain is particularly vulnerable to oxidative stress because of its high metabolic rate and low antioxidant defense system. Oxidative damage can disrupt essential processes, such as neurogenesis, neuronal migration, and synaptogenesis, leading to long-lasting changes in brain structure and function (Chen et al., 2012; Derme et al., 2024).
In the absence of fully functional compensatory mechanisms, mutations in key transcription factors, such as those in the HIF transcription complex, result in stage- and gene-specific effects on organogenesis, including placental formation and heart morphogenesis [reviewed by Dunwoodie (2009)]. Recently, attention has shifted to include epigenetic modifications. Oxidative stress can induce epigenetic modifications such as DNA methylation and histone modifications in developing tissues (Scarpato et al., 2020). These modifications can alter gene expression patterns; although primarily a coping strategy related to “developmental plasticity” (Ducsay et al., 2018), it may result in developmental abnormalities. Epigenetic changes induced by oxidative stress may persist into adulthood and influence an individual’s susceptibility to chronic diseases (Menezo et al., 2016).
Oxidative stress has also been reported to impair apoptosis regulation. Cells experiencing severe oxidative stress may undergo apoptosis or enter a state of cellular senescence (Redza-Dutordoir and Averill-Bates, 2016). Although apoptosis is essential for eliminating damaged cells, excessive apoptosis can impair proper tissue development. In the last few decades, many researchers have attempted to decipher the molecular mechanisms that initiate and execute apoptosis during development. Two distinct but ultimately converging pathways initiate apoptosis: the mitochondrial, intrinsic, or B-cell lymphoma 2 (BCL-2)-regulated pathway and the extrinsic or death receptor pathway. Mice lacking individual apoptotic regulators provided evidence for the requirement of specific regulators and suggested that developmental apoptosis is essential for mammalian development. In particular, it has become clear that reducing apoptosis typically causes webbed digits, vaginal septa, and lymphadenopathy, which commonly cause exencephaly, cleft face or palate, and occasionally omphalocele (Voss and Strasser, 2020). Cellular senescence can disrupt tissue homeostasis by altering the secretory profile of affected cells, influencing nearby cells, and contributing to developmental abnormalities (Lorda-Diez et al., 2015).
Epigenetic regulators are susceptible to free radicals. Free radicals can directly oxidize DNA. Guanine was found to react with the hydroxyl radical (●OH) to form 8-hydroxy-2′-deoxyguanosine (8-OHdG), which inhibits DNA methylation at the nearest cytosine bases, resulting in local hypomethylation. Research has shown that the presence of 8-OHdG correlates with reduced fertilization rates and low-quality embryos during in vitro fertilization (Seino et al., 2002). It has also been demonstrated as a potential biomarker for oxidative stress-induced hyperglycemia prior to the development of gestational diabetes mellitus (Urbaniak et al., 2020). Similarly, 5-methylcytosine (5 mC) is directly oxidized to 5-hydroxymethylcytosine (5-hmC), rendering it unrecognizable by DNA methyltransferases and leading to changes in the overall methylation pattern. This modification has also been associated with neurodevelopmental and autism spectrum disorders (Khoodoruth et al., 2024).
Another group of epigenetic modifiers is DNA methyltransferases (DNMTs), a family of enzymes that transfer methyl groups from S-adenosylmethionine (SAM) to cytosine residues in DNA, predominantly at CpG dinucleotides. DNMT1 primarily maintains methylation by copying the methylation patterns during DNA replication, whereas DNMT3A and DNMT3B are involved in de novo DNA methylation. ROS can directly oxidize DNMTs, resulting in global hypomethylation, and can indirectly increase the expression of DNMTs, resulting in hypermethylation (Kietzmann et al., 2017). ROS-induced DNA hypomethylation and hypermethylation have been linked to hereditary sensory neuropathy and congenital heart disease, respectively (Klein et al., 2011; Serra-Juhé et al., 2015).
In addition to DNA, histone proteins are subjected to direct oxidation by reactive oxygen species (ROS). Modifications to core histone proteins H3 and H4, due to their accessible tails around the nucleosome, alter chromatin organization and gene expression patterns. Similar to many other proteins, cysteine residues in histone proteins are liable to direct oxidation (Kietzmann et al., 2017). Apart from cysteine residues, histone proteins can also be oxidized at different amino acid residues. For instance, histones H1, H2B, and H3 undergo nitration and oxidation by reacting with peroxynitrite, resulting in alterations to chromatin structure and genome stability (Khan et al., 2016).
ROS can also affect the histone proteins indirectly. ROS reduce SAM levels. Since SAM is a cofactor for histone methyltransferases (HMTs), its reduction leads to decreased histone methylation (Mentch et al., 2015). Moreover, ROS can modulate the expression of histone demethylases (HDMs); for example, the expression of JmjC KDMs is reduced by the decreased availability of cofactors, such as Fe(II) and ascorbate, whereas ROS have been observed to increase the expression of KDM6B via STAT6 signaling (Monfort and Wutz, 2013; He et al., 2015). Overall, it can be concluded that ROS play a crucial role in the modulation of the expression, activity, and localization of histone proteins that affect the epigenetic landscape both during development and disease (Table 3).
The prenatal period is a crucial phase during which the developing brain is highly vulnerable to environmental influences, and there is ongoing discussion about a causal relationship between oxidative stress and the development of psychological disorders, as these influences significantly shape an individual’s psychological and cognitive wellbeing (Salim, 2014). Recent research indicates that oxidative stress during pregnancy can significantly increase the risk of developing psychological disorders later in life (Figure 5) (Pham et al., 2023). Research using high oxygen tension (hyperoxia) in neonatal mice demonstrated that oxidative stress induces reactive oxygen species, cell death, and disruptions in hippocampal circuits (Abbah et al., 2022). While such studies tend to focus on learning and memory processes, other studies have suggested that oxidative stress may also play a crucial role in the etiology of autism spectrum disorders (ASD) (Bjørklund et al., 2020). Oxidative stress and genetic polymorphisms in antioxidant enzymes such as glutathione transferases (GSTs) are significant contributors to the development of ASD (Mandic-Maravic et al., 2019). Perinatal complications such as prematurity, neonatal jaundice, and respiratory distress syndrome significantly increase the risk of ASD. Moreover, the GSTM1 genotype interacts with prenatal factors, including medication use, and influences ASD risk, particularly in patients homozygous for GSTM1-null. These findings underscore the importance of oxidative stress and genetic factors in ASD etiology and potential therapeutic approaches (Mandic-Maravic et al., 2019). Additionally, high levels of ROS and immune system dysfunction in ASD patients suggest that oxidative stress and inflammation may contribute to the pathogenesis and severity of ASD (Pangrazzi et al., 2020).

Figure 5. Effect of oxidative stress on embryonic development.
Attention-deficit/hyperactivity disorder (ADHD) is a neurodevelopmental disorder in children that is linked to abnormalities in particular circumscribed brain regions and disturbances in the catecholaminergic pathway. Its pathophysiology, although not fully understood, involves multiple factors, including increased oxidative stress and neuroinflammation (Corona, 2020). Oxidative damage during critical periods of brain development can affect the maturation of neural circuits involved in executive function and attention regulation. Urinary concentrations of oxidative stress biomarkers linked to inflammation, such as PGF2α, correlate with increased behavioral problems, indicative of ADHD (Rommel et al., 2020). In addition, early risk factors for ADHD, such as maternal infections, exposure to pollutants, alcohol, tobacco, and obesity, elevate maternal inflammation levels (Costenbader and Karlson, 2006; Rommel et al., 2020). This suggests that prenatal oxidative stress driven by these inflammatory conditions may play a critical role in the development of ADHD. The interplay between oxidative stress and inflammation during prenatal development is crucial for understanding the pathophysiology of ADHD and highlights the need for strategies to mitigate oxidative stress during pregnancy to reduce ADHD risk.
These conditions demonstrate a complex interplay between oxidative stress, genetic factors, and environmental factors. Understanding this interplay is essential for developing effective strategies against oxidative stress during pregnancy that may reduce the risk of various psychological disorders.
Oxidative stress represents a significant challenge to embryonic development that is capable of disrupting crucial cellular processes and leading to growth retardation and malformations in the embryo and fetus. A deeper understanding of the intricacies of cellular responses to oxidative stress and their impact on development is essential for advancing our knowledge of developmental biology and improving the outcomes of pregnancies at risk of oxidative stress-related complications. Further research is needed to uncover specific molecular targets and pathways that can be therapeutically manipulated to protect the developing embryo from the detrimental effects of oxidative stress.
Understanding the cellular mechanisms and signaling pathways activated by oxidative stress during embryonic development is crucial for developing strategies to mitigate its adverse effects. Researchers are exploring potential therapeutic interventions to counteract oxidative stress during pregnancy, including the administration of antioxidants (Mistry and Williams, 2011). However, the timing, dosage, and safety of such interventions should be carefully considered to avoid unintended consequences.
Published research has highlighted the significant impact of oxidative stress on the growth and organogenesis of mammalian embryos and fetuses. Oxidative stress-induced intrauterine growth restriction (IUGR), skeletal abnormalities, neurodevelopmental consequences, cardiovascular defects, gastrointestinal malformations, and limb abnormalities underscore the vulnerability of prenatal development to ROS imbalance. Therefore, extensive research is required to understand the impact of ROS on both pre- and post-implantation embryonic development. Understanding these effects is critical for developing strategies to mitigate oxidative stress-related developmental disorders and to improve the outcomes of pregnancies exposed to oxidative stress-inducing factors. Further research is needed to uncover the intricate mechanisms underlying these effects and explore potential interventions to protect embryonic and fetal development from the adverse consequences of oxidative stress.
Interdisciplinary collaboration is indispensable for gaining a comprehensive understanding of the relationship between oxidative stress and prenatal development. The intricate nature of this relationship demands expertise from various fields, including molecular biology, toxicology, epidemiology, clinical medicine, and neuroscience. By combining insights from these disciplines and employing cutting-edge techniques and technologies, researchers can uncover the mechanisms, risk factors, and potential interventions that contribute to the understanding of oxidative stress during prenatal development. These collaborative efforts hold the promise of improving maternal and fetal health outcomes and addressing the long-term consequences of oxidative stress in offspring.
SD: Writing–original draft, Writing–review and editing. MG: Writing–original draft, Writing–review and editing. MG: Writing–original draft, Writing–review and editing. GP: Writing–original draft, writing–review and editing. LS: Conceptualization, Writing–original draft, writing–review and editing. BB-S: Conceptualization, Writing–original draft, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
All the figures presented in the article were created using Biorender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbah, J., Vacher, C.-M., Goldstein, E. Z., Li, Z., Kundu, S., Talbot, B., et al. (2022). Oxidative stress-induced damage to the developing Hippocampus is mediated by GSK3β. J. Neurosci. 42, 4812–4827. doi:10.1523/jneurosci.2389-21.2022
Ahmad, S., Khan, H., Shahab, U., Rehman, S., Rafi, Z., Khan, M. Y., et al. (2016). Protein oxidation: an overview of metabolism of sulphur containing amino acid, cysteine. Front. Biosci. (Sch. Ed.) 9, 71–87. doi:10.2741/s474
Ahmed, A., Patil, G., Sonkar, V. K., Jensen, M., Streeter, J., and Dayal, S. (2024). Loss of endogenous Nox2-NADPH oxidase does not prevent age-induced platelet activation and arterial thrombosis in mice. Res. Pr. Thromb. Haemost. 8, 102597. doi:10.1016/j.rpth.2024.102597
Alektiar, J. M., Shan, M., Radyk, M. D., Zhang, L., Halbrook, C. J., Lin, L., et al. (2024). Malic enzyme 1 knockout has no deleterious phenotype and is favored in the male germline under standard laboratory conditions. PLOS ONE 19, e0303577. doi:10.1371/journal.pone.0303577
Al-Gubory, K. H. (2014). Environmental pollutants and lifestyle factors induce oxidative stress and poor prenatal development. Reprod. Biomed. Online 29, 17–31. doi:10.1016/j.rbmo.2014.03.002
Antonenkov, V. D., Grunau, S., Ohlmeier, S., and Hiltunen, J. K. (2010). Peroxisomes are oxidative organelles. Antioxid. Redox Signal. 13, 525–537. doi:10.1089/ars.2009.2996
Aras-López, R., Tovar, J. A., and Martínez, L. (2016). Possible role of increased oxidative stress in pulmonary hypertension in experimental diaphragmatic hernia. Pediatr. Surg. Int. 32, 141–145. doi:10.1007/s00383-015-3826-5
Atashi, F., Modarressi, A., and Pepper, M. S. (2015). The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: a review. Stem Cells Dev. 24, 1150–1163. doi:10.1089/scd.2014.0484
Ayala, A., Muñoz, M. F., and Argüelles, S. (2014). Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell. Longev. 2014, 360438. doi:10.1155/2014/360438
Bai, J., Wang, Y., Deng, S., Yang, Y., Chen, S., and Wu, Z. (2024). Microplastics caused embryonic growth retardation and placental dysfunction in pregnant mice by activating GRP78/IRE1α/JNK axis induced apoptosis and endoplasmic reticulum stress. Part. Fibre Toxicol. 21, 36. doi:10.1186/s12989-024-00595-5
Barati, E., Nikzad, H., and Karimian, M. (2020). Oxidative stress and male infertility: current knowledge of pathophysiology and role of antioxidant therapy in disease management. Cell. Mol. Life Sci. 77, 93–113. doi:10.1007/s00018-019-03253-8
Bjørklund, G., Meguid, N. A., El-Bana, M. A., Tinkov, A. A., Saad, K., Dadar, M., et al. (2020). Oxidative stress in autism spectrum disorder. Mol. Neurobiol. 57, 2314–2332. doi:10.1007/s12035-019-01742-2
Blerkom, J. V. (2004). Mitochondria in human oogenesis and preimplantation embryogenesis: engines of metabolism, ionic regulation and developmental competence. Reproduction 128, 269–280. doi:10.1530/rep.1.00240
Bondareva, A. A., Capecchi, M. R., Iverson, S. V., Li, Y., Lopez, N. I., Lucas, O., et al. (2007). Effects of thioredoxin reductase-1 deletion on embryogenesis and transcriptome. Free Radic. Biol. Med. 43, 911–923. doi:10.1016/j.freeradbiomed.2007.05.026
Bradley, E., Bieberich, E., Mivechi, N. F., Tangpisuthipongsa, D., and Wang, G. (2012). Regulation of embryonic stem cell pluripotency by heat shock protein 90. Stem Cells 30, 1624–1633. doi:10.1002/stem.1143
Budani, M. C., Gallorini, M., Elsallabi, O., Pino, V., Fratta, I. L., Pesce, M., et al. (2022). Cigarette smoke is associated with up-regulation of inducible NOS and COX-2 protein expression and activity in granulosa cells of women undergoing in vitro fertilization. Reprod. Toxicol. 113, 128–135. doi:10.1016/j.reprotox.2022.08.013
Cantu-Medellin, N., and Kelley, E. E. (2013). Xanthine oxidoreductase-catalyzed reactive species generation: a process in critical need of reevaluation. Redox Biol. 1, 353–358. doi:10.1016/j.redox.2013.05.002
Carmichael, S. L., Yang, W., Ma, C., Desrosiers, T. A., Weber, K., Collins, R. T., et al. (2023). Oxidative balance scores and neural crest cell-related congenital anomalies. Birth Defects Res. 115, 1151–1162. doi:10.1002/bdr2.2211
Chan, J. Y., Kwong, M., Lu, R., Chang, J., Wang, B., Yen, T. S. B., et al. (1998). Targeted disruption of the ubiquitous CNC-bZIP transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO J. 17, 1779–1787. doi:10.1093/emboj/17.6.1779
Chan, K., Lu, R., Chang, J. C., and Kan, Y. W. (1996). NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. 93, 13943–13948. doi:10.1073/pnas.93.24.13943
Chen, G., Ke, Z., Xu, M., Liao, M., Wang, X., Qi, Y., et al. (2012). Autophagy is a protective response to ethanol neurotoxicity. Autophagy 8, 1577–1589. doi:10.4161/auto.21376
Chen, X., Guo, J., Lei, Y., Zou, J., Lu, X., Bao, Y., et al. (2010). Global DNA hypomethylation is associated with NTD-affected pregnancy: a case-control study. Birth Defects Res. Part A Clin. Mol. Teratol. 88, 575–581. doi:10.1002/bdra.20670
Chen, Y.-I., Wei, P.-C., Hsu, J.-L., Su, F.-Y., and Lee, W.-H. (2015). NPGPx (GPx7): a novel oxidative stress sensor/transmitter with multiple roles in redox homeostasis. Am. J. Transl. Res. 8, 1626–1640.
Chen, Z., Keaney, J. F., Schulz, E., Levison, B., Shan, L., Sakuma, M., et al. (2004). Decreased neointimal formation in Nox2-deficient mice reveals a direct role for NADPH oxidase in the response to arterial injury. Proc. Natl. Acad. Sci. 101, 13014–13019. doi:10.1073/pnas.0405389101
Cheng, N., Zhang, W., Chen, W., Jin, J., Cui, X., Butte, N. F., et al. (2011). A mammalian monothiol glutaredoxin, Grx3, is critical for cell cycle progression during embryogenesis. FEBS J. 278, 2525–2539. doi:10.1111/j.1742-4658.2011.08178.x
Cho, Y. M., Kwon, S., Pak, Y. K., Seol, H. W., Choi, Y. M., Park, D. J., et al. (2006). Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem. Biophys. Res. Commun. 348, 1472–1478. doi:10.1016/j.bbrc.2006.08.020
Cobbaut, M., and Lint, J. V. (2018). Function and regulation of protein kinase D in oxidative stress: a tale of isoforms. Oxidative Med. Cell. Longev. 2018, 2138502. doi:10.1155/2018/2138502
Conrad, M., Jakupoglu, C., Moreno, S. G., Lippl, S., Banjac, A., Schneider, M., et al. (2004). Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol. Cell. Biol. 24, 9414–9423. doi:10.1128/mcb.24.21.9414-9423.2004
Corona, J. C. (2020). Role of oxidative stress and neuroinflammation in attention-deficit/hyperactivity disorder. Antioxidants 9, 1039. doi:10.3390/antiox9111039
Costenbader, K. H., and Karlson, E. W. (2006). Cigarette smoking and autoimmune disease: what can we learn from epidemiology? Lupus 15, 737–745. doi:10.1177/0961203306069344
Dalton, T. P., Dieter, M. Z., Yang, Y., Shertzer, H. G., and Nebert, D. W. (2000). Knockout of the mouse glutamate cysteine ligase catalytic subunit (gclc) gene: embryonic lethal when homozygous, and proposed model for moderate glutathione deficiency when heterozygous. Biochem. Biophys. Res. Commun. 279, 324–329. doi:10.1006/bbrc.2000.3930
Danielsson, B., Vargesson, N., and Danielsson, C. (2023). Teratogenicity and Reactive Oxygen Species after transient embryonic hypoxia: experimental and clinical evidence with focus on drugs causing failed abortion in humans. Reprod. Toxicol. 122, 108488. doi:10.1016/j.reprotox.2023.108488
Deepa, S. S., Bhaskaran, S., Espinoza, S., Brooks, S. V., McArdle, A., Jackson, M. J., et al. (2017). A new mouse model of frailty: the Cu/Zn superoxide dismutase knockout mouse. GeroScience 39, 187–198. doi:10.1007/s11357-017-9975-9
DeFreitas, M. J., Katsoufis, C. P., Benny, M., Young, K., Kulandavelu, S., Ahn, H., et al. (2022). Educational review: the impact of perinatal oxidative stress on the developing kidney. Front. Pediatr. 10, 853722. doi:10.3389/fped.2022.853722
Deluao, J. C., Winstanley, Y., Robker, R. L., Pacella-Ince, L., Gonzalez, M. B., and McPherson, N. O. (2022). Oxidative Stress and Reproductive Function: reactive oxygen species in the mammalian pre-implantation embryo. Reproduction 164, F95–F108. doi:10.1530/rep-22-0121
Demicheli, V., Moreno, D. M., and Radi, R. (2018). Human Mn-superoxide dismutase inactivation by peroxynitrite: a paradigm of metal-catalyzed tyrosine nitration in vitro and in vivo. Metallomics 10, 679–695. doi:10.1039/c7mt00348j
Dennery, P. A. (2007). Effects of oxidative stress on embryonic development. Birth Defects Res. Part C. Embryo Today Rev. 81, 155–162. doi:10.1002/bdrc.20098
Dennery, P. A. (2010). Oxidative stress in development: nature or nurture? Free Radic. Biol. Med. 49, 1147–1151. doi:10.1016/j.freeradbiomed.2010.07.011
Derme, M., Briante, M., Ceccanti, M., Giannini, G., Vitali, M., Messina, M. P., et al. (2024). Prenatal alcohol exposure and metabolic disorders in pediatrics: the role of the oxidative stress—a review of the literature. Children 11, 269. doi:10.3390/children11030269
Diniz, M. S., Magalhães, C. C., Tocantins, C., Grilo, L. F., Teixeira, J., and Pereira, S. P. (2023). Nurturing through nutrition: exploring the role of antioxidants in maternal diet during pregnancy to mitigate developmental programming of chronic diseases. Nutrients 15, 4623. doi:10.3390/nu15214623
Donkó, Á., Ruisanchez, É., Orient, A., Enyedi, B., Kapui, R., Péterfi, Z., et al. (2010). Urothelial cells produce hydrogen peroxide through the activation of Duox1. Free Radic. Biol. Med. 49, 2040–2048. doi:10.1016/j.freeradbiomed.2010.09.027
Ducsay, C. A., Goyal, R., Pearce, W. J., Wilson, S., Hu, X.-Q., and Zhang, L. (2018). Gestational hypoxia and developmental plasticity. Physiol. Rev. 98, 1241–1334. doi:10.1152/physrev.00043.2017
Dunwoodie, S. L. (2009). The role of hypoxia in development of the mammalian embryo. Dev. Cell. 17, 755–773. doi:10.1016/j.devcel.2009.11.008
Dutta, S., Henkel, R., Sengupta, P., and Agarwal, A. (2020). Physiological role of ROS in sperm function. Androl. ART Antioxidants, 337–345. doi:10.1007/978-3-030-32300-4_27
Erickson, J. C., Hollopeter, G., Thomas, S. A., Froelick, G. J., and Palmiter, R. D. (1997). Disruption of the metallothionein-III gene in mice: analysis of brain zinc, behavior, and neuron vulnerability to metals, aging, and seizures. J. Neurosci. 17, 1271–1281. doi:10.1523/jneurosci.17-04-01271.1997
Facucho-Oliveira, J. M., and John, J. C.St. (2009). The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell. Rev. Rep. 5, 140–158. doi:10.1007/s12015-009-9058-0
Fathollahipour, S., Patil, P. S., and Leipzig, N. D. (2019). Oxygen regulation in development: lessons from embryogenesis towards tissue engineering. Cells Tissues Organs 205, 350–371. doi:10.1159/000493162
Ferreira, C., Bucchini, D., Martin, M.-E., Levi, S., Arosio, P., Grandchamp, B., et al. (2000). Early embryonic lethality of H Ferritin gene deletion in mice. J. Biol. Chem. 275, 3021–3024. doi:10.1074/jbc.275.5.3021
Florian, S., Krehl, S., Loewinger, M., Kipp, A., Banning, A., Esworthy, S., et al. (2010). Loss of GPx2 increases apoptosis, mitosis, and GPx1 expression in the intestine of mice. Free Radic. Biol. Med. 49, 1694–1702. doi:10.1016/j.freeradbiomed.2010.08.029
Fomenko, D. E., Novoselov, S. V., Natarajan, S. K., Lee, B. C., Koc, A., Carlson, B. A., et al. (2009). MsrB1 (Methionine-R-sulfoxide reductase 1) knock-out mice roles of Msrb1 in redox regulation and identification of A novel selenoprotein form. J. Biol. Chem. 284, 5986–5993. doi:10.1074/jbc.m805770200
Gallorini, M., Carradori, S., Panieri, E., Sova, M., and Saso, L. (2024). Modulation of NRF2: biological dualism in cancer, targets and possible therapeutic applications. Antioxid. Redox Signal. 40, 636–662. doi:10.1089/ars.2022.0213
García-Sanz, P., Mirasierra, M., Moratalla, R., and Vallejo, M. (2017). Embryonic defence mechanisms against glucose-dependent oxidative stress require enhanced expression of Alx3 to prevent malformations during diabetic pregnancy. Sci. Rep. 7, 389. doi:10.1038/s41598-017-00334-1
Gavazzi, G., Banfi, B., Deffert, C., Fiette, L., Schappi, M., Herrmann, F., et al. (2006). Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 580, 497–504. doi:10.1016/j.febslet.2005.12.049
Geiszt, M., Witta, J., Baff, J., Lekstrom, K., and Leto, T. L. (2003). Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 17, 1–14. doi:10.1096/fj.02-1104fje
Gholipourmalekabadi, M., Zhao, S., Harrison, B. S., Mozafari, M., and Seifalian, A. M. (2016). Oxygen-generating biomaterials: a new, viable paradigm for tissue engineering? Trends Biotechnol. 34, 1010–1021. doi:10.1016/j.tibtech.2016.05.012
Glanzner, W. G., Sousa, L. R., Gutierrez, K., Macedo, M. P., Currin, L., Perecin, F., et al. (2024). NRF2 attenuation aggravates detrimental consequences of metabolic stress on cultured porcine parthenote embryos. Sci. Rep. 14, 2973. doi:10.1038/s41598-024-53480-8
González-Flores, D., Márquez, A., and Casimiro, I. (2024). Oxidative effects in early stages of embryo development due to alcohol consumption. Int. J. Mol. Sci. 25, 4100. doi:10.3390/ijms25074100
Grasberger, H., Deken, X. D., Mayo, O. B., Raad, H., Weiss, M., Liao, X.-H., et al. (2012). Mice deficient in dual oxidase maturation factors are severely hypothyroid. Mol. Endocrinol. 26, 481–492. doi:10.1210/me.2011-1320
Gurjar, M. V., Deleon, J., Sharma, R. V., and Bhalla, R. C. (2001). Role of reactive oxygen species in IL-1 beta-stimulated sustained ERK activation and MMP-9 induction. Am. J. Physiol.-Hear. Circ. Physiol. 281, H2568–H2574. doi:10.1152/ajpheart.2001.281.6.h2568
Hansen, J. M. (2006). Oxidative stress as a mechanism of teratogenesis. Birth Defects Res. Part C. Embryo Today Rev. 78, 293–307. doi:10.1002/bdrc.20085
Harris, C., and Hansen, J. M. (2012). Nrf2-Mediated resistance to oxidant-induced redox disruption in embryos. Birth Defects Res. Part B Dev. Reprod. Toxicol. 95, 213–218. doi:10.1002/bdrb.21005
Harrison, B. S., Eberli, D., Lee, S. J., Atala, A., and Yoo, J. J. (2007). Oxygen producing biomaterials for tissue regeneration. Biomaterials 28, 4628–4634. doi:10.1016/j.biomaterials.2007.07.003
He, C., Larson-Casey, J. L., Gu, L., Ryan, A. J., Murthy, S., and Carter, A. B. (2015). Cu,Zn–Superoxide dismutase–mediated redox regulation of jumonji domain containing 3 modulates macrophage polarization and pulmonary fibrosis. Am. J. Respir. Cell. Mol. Biol. 55, 58–71. doi:10.1165/rcmb.2015-0183oc
Hernebring, M., Brolén, G., Aguilaniu, H., Semb, H., and Nyström, T. (2006). Elimination of damaged proteins during differentiation of embryonic stem cells. Proc. Natl. Acad. Sci. 103, 7700–7705. doi:10.1073/pnas.0510944103
Hitchler, M. J., and Domann, F. E. (2007). An epigenetic perspective on the free radical theory of development. Free Radic. Biol. Med. 43, 1023–1036. doi:10.1016/j.freeradbiomed.2007.06.027
Ho, Y.-S., Magnenat, J.-L., Bronson, R. T., Cao, J., Gargano, M., Sugawara, M., et al. (1997). Mice deficient in cellular glutathione peroxidase develop normally and show No increased sensitivity to hyperoxia. J. Biol. Chem. 272, 16644–16651. doi:10.1074/jbc.272.26.16644
Ho, Y.-S., Xiong, Y., Ho, D. S., Gao, J., Chua, B. H. L., Pai, H., et al. (2007). Targeted disruption of the glutaredoxin 1 gene does not sensitize adult mice to tissue injury induced by ischemia/reperfusion and hyperoxia. Free Radic. Biol. Med. 43, 1299–1312. doi:10.1016/j.freeradbiomed.2007.07.025
Ho, Y.-S., Xiong, Y., Ma, W., Spector, A., and Ho, D. S. (2004). Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J. Biol. Chem. 279, 32804–32812. doi:10.1074/jbc.m404800200
Huang, T.-T., Carlson, E. J., Kozy, H. M., Mantha, S., Goodman, S. I., Ursell, P. C., et al. (2001). Genetic modification of prenatal lethality and dilated cardiomyopathy in Mn superoxide dismutase mutant mice. Free Radic. Biol. Med. 31, 1101–1110. doi:10.1016/s0891-5849(01)00694-3
Hussain, T., Murtaza, G., Metwally, E., Kalhoro, D. H., Kalhoro, M. S., Rahu, B. A., et al. (2021). The role of oxidative stress and antioxidant balance in pregnancy. Mediat. Inflamm. 2021, 9962860. doi:10.1155/2021/9962860
Ikegami, T., Suzuki, Y., Shimizu, T., Isono, K., Koseki, H., and Shirasawa, T. (2002). Model mice for tissue-specific deletion of the manganese superoxide dismutase (MnSOD) gene. Biochem. Biophys. Res. Commun. 296, 729–736. doi:10.1016/s0006-291x(02)00933-6
Imai, H., Hirao, F., Sakamoto, T., Sekine, K., Mizukura, Y., Saito, M., et al. (2003). Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem. Biophys. Res. Commun. 305, 278–286. doi:10.1016/s0006-291x(03)00734-4
Impellizzeri, P., Nascimben, F., Fabrizio, D. D., Antonuccio, P., Antonelli, E., Peri, F. M., et al. (2020). Pathogenesis of congenital malformations: possible role of oxidative stress. Am. J. Perinatol. 39, 816–823. doi:10.1055/s-0040-1721081
Iskander, K., Barrios, R. J., and Jaiswal, A. K. (2008). Disruption of NAD(P)H:quinone oxidoreductase 1 gene in mice leads to radiation-induced myeloproliferative disease. Cancer Res. 68, 7915–7922. doi:10.1158/0008-5472.can-08-0766
Iuchi, Y., Okada, F., Tsunoda, S., Kibe, N., Shirasawa, N., Ikawa, M., et al. (2009). Peroxiredoxin 4 knockout results in elevated spermatogenic cell death via oxidative stress. Biochem. J. 419, 149–158. doi:10.1042/bj20081526
Jackson, M., Krassowska, A., Gilbert, N., Chevassut, T., Forrester, L., Ansell, J., et al. (2004). Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Mol. Cell. Biol. 24, 8862–8871. doi:10.1128/mcb.24.20.8862-8871.2004
Jiang, X., Bar, H. Y., Yan, J., West, A. A., Perry, C. A., Malysheva, O. V., et al. (2012). Pregnancy induces transcriptional activation of the peripheral innate immune system and increases oxidative DNA damage among healthy third trimester pregnant women. PLoS One 7, e46736. doi:10.1371/journal.pone.0046736
Jin, B., Tao, Q., Peng, J., Soo, H. M., Wu, W., Ying, J., et al. (2008). DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum. Mol. Genet. 17, 690–709. doi:10.1093/hmg/ddm341
Jochum, W., Passegué, E., and Wagner, E. F. (2001). AP-1 in mouse development and tumorigenesis. Oncogene 20, 2401–2412. doi:10.1038/sj.onc.1204389
Johnson, K. R., Marden, C. C., Ward-Bailey, P., Gagnon, L. H., Bronson, R. T., and Donahue, L. R. (2007). Congenital hypothyroidism, dwarfism, and hearing impairment caused by a missense mutation in the mouse dual oxidase 2 gene, Duox2. Mol. Endocrinol. 21, 1593–1602. doi:10.1210/me.2007-0085
Jomova, K., Raptova, R., Alomar, S. Y., Alwasel, S. H., Nepovimova, E., Kuca, K., et al. (2023). Reactive oxygen species, toxicity, oxidative stress, and antioxidants: chronic diseases and aging. Arch. Toxicol. 97, 2499–2574. doi:10.1007/s00204-023-03562-9
Jones, D. P. (2006). Disruption of mitochondrial redox circuitry in oxidative stress. Chem.-Biol. Interact. 163, 38–53. doi:10.1016/j.cbi.2006.07.008
Joo, E. H., Kim, Y. R., Kim, N., Jung, J. E., Han, S. H., and Cho, H. Y. (2021). Effect of endogenic and exogenic oxidative stress triggers on adverse pregnancy outcomes: preeclampsia, fetal growth restriction, gestational diabetes mellitus and preterm birth. Int. J. Mol. Sci. 22, 10122. doi:10.3390/ijms221810122
Joshi, S., Peck, A. B., and Khan, S. R. (2013). NADPH oxidase as a therapeutic target for oxalate induced injury in kidneys. Oxidative Med. Cell. Longev. 2013, 462361. doi:10.1155/2013/462361
Khan, M. A., Alam, K., Dixit, K., and Rizvi, M. M. A. (2016). Role of peroxynitrite induced structural changes on H2B histone by physicochemical method. Int. J. Biol. Macromol. 82, 31–38. doi:10.1016/j.ijbiomac.2015.10.085
Khattak, S. F., Chin, K., Bhatia, S. R., and Roberts, S. C. (2007). Enhancing oxygen tension and cellular function in alginate cell encapsulation devices through the use of perfluorocarbons. Biotechnol. Bioeng. 96, 156–166. doi:10.1002/bit.21151
Khoodoruth, M. A. S., Khoodoruth, W. N. C., and Alwani, R. A. (2024). Exploring the epigenetic landscape: the role of 5-hydroxymethylcytosine in neurodevelopmental disorders. Camb. Prism. Precis. Med. 2, e5. doi:10.1017/pcm.2024.2
Kietzmann, T., Petry, A., Shvetsova, A., Gerhold, J. M., and Görlach, A. (2017). The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 174, 1533–1554. doi:10.1111/bph.13792
Kim, J. J., Lee, S. B., Park, J. K., and Yoo, Y. D. (2010). TNF-alpha-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-X(L). Cell. Death Differ. 17, 1420–1434. doi:10.1038/cdd.2010.19
Kim, M.-A., Cho, H.-J., Bae, S.-H., Lee, B., Oh, S.-K., Kwon, T.-J., et al. (2016). Methionine sulfoxide reductase B3-targeted in utero gene therapy rescues hearing function in a mouse model of congenital sensorineural hearing loss. Antioxid. Redox Signal. 24, 590–602. doi:10.1089/ars.2015.6442
Kim, M. H., Park, S.-J., Kim, J.-H., Seong, J. B., Kim, K.-M., Woo, H. A., et al. (2018). Peroxiredoxin 5 regulates adipogenesis-attenuating oxidative stress in obese mouse models induced by a high-fat diet. Free Radic. Biol. Med. 123, 27–38. doi:10.1016/j.freeradbiomed.2018.05.061
Klein, C. J., Botuyan, M.-V., Wu, Y., Ward, C. J., Nicholson, G. A., Hammans, S., et al. (2011). Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet. 43, 595–600. doi:10.1038/ng.830
Kleinschnitz, C., Grund, H., Wingler, K., Armitage, M. E., Jones, E., Mittal, M., et al. (2010). Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 8, e1000479. doi:10.1371/journal.pbio.1000479
Kusuma, G. D., Georgiou, H. M., Perkins, A. V., Abumaree, M. H., Brennecke, S. P., and Kalionis, B. (2022). Mesenchymal stem/stromal cells and their role in oxidative stress associated with preeclampsia. Yale J. Biol. Med. 95, 115–127.
Laforgia, N., Mauro, A. D., Guarnieri, G. F., Varvara, D., Cosmo, L. D., Panza, R., et al. (2018). The role of oxidative stress in the pathomechanism of congenital malformations. Oxidative Med. Cell. Longev. 2018, 7404082. doi:10.1155/2018/7404082
Lam, G., Apostolopoulos, V., Zulli, A., and Nurgali, K. (2015). NADPH oxidases and inflammatory bowel disease. Curr. Med. Chem. 22, 2100–2109. doi:10.2174/0929867322666150416095114
Lee, J., Cho, Y. S., Jung, H., and Choi, I. (2018). Pharmacological regulation of oxidative stress in stem cells. Oxidative Med. Cell. Longev. 2018, 4081890. doi:10.1155/2018/4081890
Lee, T.-H., Kim, S.-U., Yu, S.-L., Kim, S. H., Park, D. S., Moon, H.-B., et al. (2003). Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood 101, 5033–5038. doi:10.1182/blood-2002-08-2548
Leslie, K., Whitley, G. S. J., Herse, F., Dechend, R., Ashton, S. V., Laing, K., et al. (2015). Increased apoptosis, altered oxygen signaling, and antioxidant defenses in first-trimester pregnancies with high-resistance uterine artery blood flow. Am. J. Pathol. 185, 2731–2741. doi:10.1016/j.ajpath.2015.06.020
Leung, L., Kwong, M., Hou, S., Lee, C., and Chan, J. Y. (2003). Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J. Biol. Chem. 278, 48021–48029. doi:10.1074/jbc.m308439200
Lewis, A., Hayashi, T., Su, T.-P., and Betenbaugh, M. J. (2014). Bcl-2 family in inter-organelle modulation of calcium signaling; roles in bioenergetics and cell survival. J. Bioenerg. Biomembr. 46, 1–15. doi:10.1007/s10863-013-9527-7
Li, D., Wan, C., Bai, B., Cao, H., Liu, C., and Zhang, Q. (2019). Identification of histone acetylation markers in human fetal brains and increased H4K5ac expression in neural tube defects. Mol. Genet. Genom. Med. 7, e1002. doi:10.1002/mgg3.1002
Li, L., Shoji, W., Takano, H., Nishimura, N., Aoki, Y., Takahashi, R., et al. (2007). Increased susceptibility of MER5 (peroxiredoxins III) knockout mice to LPS-induced oxidative stress. Biochem. Biophys. Res. Commun. 355, 715–721. doi:10.1016/j.bbrc.2007.02.022
Li, Y., Huang, T.-T., Carlson, E. J., Melov, S., Ursell, P. C., Olson, J. L., et al. (1995). Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 11, 376–381. doi:10.1038/ng1295-376
Li, Z., Wang, S., and Li, L. (2022). Advanced oxidative protein products drive trophoblast cells into senescence by inhibiting the autophagy: the potential implication of preeclampsia. Front. Cell. Dev. Biol. 10, 810282. doi:10.3389/fcell.2022.810282
Loeken, M. R. (2004). Free radicals and birth defects. J. Matern.-Fetal Neonatal Med. 15, 6–14. doi:10.1080/14767050310001650662
Longo, L., Vanegas, O. C., Patel, M., Rosti, V., Li, H., Waka, J., et al. (2002). Maternally transmitted severe glucose 6-phosphate dehydrogenase deficiency is an embryonic lethal. EMBO J. 21, 4229–4239. doi:10.1093/emboj/cdf426
Lopes, A. S., Lane, M., and Thompson, J. G. (2010). Oxygen consumption and ROS production are increased at the time of fertilization and cell cleavage in bovine zygotes. Hum. Reprod. 25, 2762–2773. doi:10.1093/humrep/deq221
Lorda-Diez, C. I., Garcia-Riart, B., Montero, J. A., Rodriguez-León, J., Garcia-Porrero, J. A., and Hurle, J. M. (2015). Apoptosis during embryonic tissue remodeling is accompanied by cell senescence. Aging (Albany NY) 7, 974–985. doi:10.18632/aging.100844
Lu, J., Sharma, L. K., and Bai, Y. (2009). Implications of mitochondrial DNA mutations and mitochondrial dysfunction in tumorigenesis. Cell. Res. 19, 802–815. doi:10.1038/cr.2009.69
Lu, Y., Shao, Y., Cui, W., Jia, Z., Zhang, Q., Zhao, Q., et al. (2024). Excessive lipid peroxidation in uterine epithelium causes implantation failure and pregnancy loss. Adv. Sci. 11, 2302887. doi:10.1002/advs.202302887
Lucchi, L., Banni, S., Botti, B., Cappelli, G., Medici, G., Melis, M. P., et al. (1993). Conjugated diene fatty acids in patients with chronic renal failure: evidence of increased lipid peroxidation? Nephron 65, 401–409. doi:10.1159/000187520
Ma, Q. (2013). Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 53, 401–426. doi:10.1146/annurev-pharmtox-011112-140320
Magnani, F., and Mattevi, A. (2019). Structure and mechanisms of ROS generation by NADPH oxidases. Curr. Opin. Struct. Biol. 59, 91–97. doi:10.1016/j.sbi.2019.03.001
Mandal, S., Lindgren, A. G., Srivastava, A. S., Clark, A. T., and Banerjee, U. (2011). Mitochondrial function controls proliferation and early differentiation potential of embryonic stem cells. Stem Cells 29, 486–495. doi:10.1002/stem.590
Mandic-Maravic, V., Mitkovic-Voncina, M., Pljesa-Ercegovac, M., Savic-Radojevic, A., Djordjevic, M., Pekmezovic, T., et al. (2019). Autism spectrum disorders and perinatal complications—is oxidative stress the connection? Front. Psychiatry 10, 675. doi:10.3389/fpsyt.2019.00675
Maraldi, T., Prata, C., Caliceti, C., Sega, F. V. D., Zambonin, L., Fiorentini, D., et al. (2010). VEGF-induced ROS generation from NAD(P)H oxidases protects human leukemic cells from apoptosis. Int. J. Oncol. 36, 1581–1589. doi:10.3892/ijo_00000645
Masters, B. A., Kelly, E. J., Quaife, C. J., Brinster, R. L., and Palmiter, R. D. (1994). Targeted disruption of metallothionein I and II genes increases sensitivity to cadmium. Proc. Natl. Acad. Sci. 91, 584–588. doi:10.1073/pnas.91.2.584
Matsui, M., Oshima, M., Oshima, H., Takaku, K., Maruyama, T., Yodoi, J., et al. (1996). Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev. Biol. 178, 179–185. doi:10.1006/dbio.1996.0208
Matsuno, K., Yamada, H., Iwata, K., Jin, D., Katsuyama, M., Matsuki, M., et al. (2005). Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation 112, 2677–2685. doi:10.1161/circulationaha.105.573709
McConnachie, L. A., Mohar, I., Hudson, F. N., Ware, C. B., Ladiges, W. C., Fernandez, C., et al. (2007). Glutamate cysteine ligase modifier subunit deficiency and gender as determinants of acetaminophen-induced hepatotoxicity in mice. Toxicol. Sci. 99, 628–636. doi:10.1093/toxsci/kfm165
Menezo, Y. J. R., Silvestris, E., Dale, B., and Elder, K. (2016). Oxidative stress and alterations in DNA methylation: two sides of the same coin in reproduction. Reprod. Biomed. Online 33, 668–683. doi:10.1016/j.rbmo.2016.09.006
Mentch, S. J., Mehrmohamadi, M., Huang, L., Liu, X., Gupta, D., Mattocks, D., et al. (2015). Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell. Metab. 22, 861–873. doi:10.1016/j.cmet.2015.08.024
Merta, M., Reiterová, J., Rysavá, R., Tesar, V., Závada, J., Jáchymová, M., et al. (2003). Role of endothelin and nitric oxide in the pathogenesis of arterial hypertension in autosomal dominant polycystic kidney disease. Physiol. Res. 52, 433–437. doi:10.33549/physiolres.930324
Michiels, C. (2004). Physiological and pathological responses to hypoxia. Am. J. Pathol. 164, 1875–1882. doi:10.1016/s0002-9440(10)63747-9
Mihalik, J., Kreheľová, A., Kovaříková, V., Solár, P., Domoráková, I., Pavliuk-Karachevtseva, A., et al. (2020). GPx8 expression in rat oocytes, embryos, and female genital organs during preimplantation period of pregnancy. Int. J. Mol. Sci. 21, 6313. doi:10.3390/ijms21176313
Milkovic, L., Gasparovic, A. C., Cindric, M., Mouthuy, P.-A., and Zarkovic, N. (2019). Short overview of ROS as cell function regulators and their implications in therapy concepts. Cells 8, 793. doi:10.3390/cells8080793
Mistry, H. D., and Williams, P. J. (2011). The importance of antioxidant micronutrients in pregnancy. Oxidative Med. Cell. Longev. 2011, 841749. doi:10.1155/2011/841749
Mohyeldin, A., Garzón-Muvdi, T., and Quiñones-Hinojosa, A. (2010). Oxygen in stem cell biology: a critical component of the stem cell niche. Cell. Stem Cell. 7, 150–161. doi:10.1016/j.stem.2010.07.007
Monfort, A., and Wutz, A. (2013). Breathing-in epigenetic change with vitamin C. EMBO Rep. 14, 337–346. doi:10.1038/embor.2013.29
Morales, E., García-Serna, A. M., Larqué, E., Sánchez-Campillo, M., Serrano-Munera, A., Martinez-Graciá, C., et al. (2022). Dietary patterns in pregnancy and biomarkers of oxidative stress in mothers and offspring: the NELA birth cohort. Front. Nutr. 9, 869357. doi:10.3389/fnut.2022.869357
Morrison, J. L. (2008). Sheep models of intrauterine growth restriction: fetal adaptations and consequences. Clin. Exp. Pharmacol. Physiol. 35, 730–743. doi:10.1111/j.1440-1681.2008.04975.x
Moskovitz, J., Bar-Noy, S., Williams, W. M., Requena, J., Berlett, B. S., and Stadtman, E. R. (2001). Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc. Natl. Acad. Sci. 98, 12920–12925. doi:10.1073/pnas.231472998
Motohashi, H., and Yamamoto, M. (2004). Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 10, 549–557. doi:10.1016/j.molmed.2004.09.003
Murata, T., Kyozuka, H., Fukuda, T., Imaizumi, K., Isogami, H., Kanno, A., et al. (2024). Urinary 8-hydroxy-2′-deoxyguanosine levels and preterm births: a prospective cohort study from the Japan Environment and Children’s Study. BMJ Open 14, e063619. doi:10.1136/bmjopen-2022-063619
Musson, R., Gąsior, Ł., Bisogno, S., and Ptak, G. E. (2022). DNA damage in preimplantation embryos and gametes: specification, clinical relevance and repair strategies. Hum. Reprod. Updat. 28, 376–399. doi:10.1093/humupd/dmab046
Nakajima, S., and Kitamura, M.(2013). Bidirectional regulation of NF-κB by reactive oxygen species: A role of unfolded protein response. Free Radic. Biol. Med. 65, 162–174. doi:10.1016/j.freeradbiomed.2013.06.020
Närvä, E., Pursiheimo, J.-P., Laiho, A., Rahkonen, N., Emani, M. R., Viitala, M., et al. (2013). Continuous hypoxic culturing of human embryonic stem cells enhances SSEA-3 and MYC levels. PLoS ONE 8, e78847. doi:10.1371/journal.pone.0078847
Neumann, C. A., Krause, D. S., Carman, C. V., Das, S., Dubey, D. P., Abraham, J. L., et al. (2003). Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 424, 561–565. doi:10.1038/nature01819
Nisimoto, Y., Diebold, B. A., Cosentino-Gomes, D., Constentino-Gomes, D., and Lambeth, J. D. (2014). Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry 53, 5111–5120. doi:10.1021/bi500331y
Noblanc, A., Peltier, M., Damon-Soubeyrand, C., Kerchkove, N., Chabory, E., Vernet, P., et al. (2012). Epididymis response partly compensates for spermatozoa oxidative defects in snGPx4 and GPx5 double mutant mice. PLoS ONE 7, e38565. doi:10.1371/journal.pone.0038565
Olson, G. E., Whitin, J. C., Hill, K. E., Winfrey, V. P., Motley, A. K., Austin, L. M., et al. (2010). Extracellular glutathione peroxidase (Gpx3) binds specifically to basement membranes of mouse renal cortex tubule cells. Am. J. Physiol.-Ren. Physiol. 298, F1244–F1253. doi:10.1152/ajprenal.00662.2009
Ornoy, A. (2007). Embryonic oxidative stress as a mechanism of teratogenesis with special emphasis on diabetic embryopathy. Reprod. Toxicol. 24, 31–41. doi:10.1016/j.reprotox.2007.04.004
Ortega, J. A., Sirois, C. L., Memi, F., Glidden, N., and Zecevic, N. (2016). Oxygen levels regulate the development of human cortical radial glia cells. Cereb. Cortex 27, 3736–3751. doi:10.1093/cercor/bhw194
Oyefeso, F. A., Muotri, A. R., Wilson, C. G., and Pecaut, M. J. (2021). Brain organoids: a promising model to assess oxidative stress-induced central nervous system damage. Dev. Neurobiol. 81, 653–670. doi:10.1002/dneu.22828
Ozsurekci, Y., and Aykac, K. (2016). Oxidative stress related diseases in newborns. Oxidative Med. Cell. Longev. 2016, 2768365. doi:10.1155/2016/2768365
Paffenholz, R., Bergstrom, R. A., Pasutto, F., Wabnitz, P., Munroe, R. J., Jagla, W., et al. (2004). Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes. Dev. 18, 486–491. doi:10.1101/gad.1172504
Pagé, E. L., Chan, D. A., Giaccia, A. J., Levine, M., and Richard, D. E. (2008). Hypoxia-inducible Factor-1α Stabilization in Nonhypoxic Conditions: Role of Oxidation and Intracellular Ascorbate Depletion. Mol. Biol. Cell 19, 86–491. doi:10.1091/mbc.e07-06-0612
Pangrazzi, L., Balasco, L., and Bozzi, Y.(2020). Oxidative stress and immune system dysfunction in autism spectrum disorders. Int. J. Mol. Sci. 21, 3293. doi:10.3390/ijms21093293
Pasparakis, M., Luedde, T., and Schmidt-Supprian, M. (2006). Dissection of the NF-kappaB signalling cascade in transgenic and knockout mice. Cell. Death Differ. 13, 861–872. doi:10.1038/sj.cdd.4401870
Pham, C., Thomson, S., Chin, S.-T., Vuillermin, P., O’Hely, M., Burgner, D., et al. (2023). Maternal oxidative stress during pregnancy associated with emotional and behavioural problems in early childhood: implications for foetal programming. Mol. Psychiatry 28, 3760–3768. doi:10.1038/s41380-023-02284-9
Poss, K. D., and Tonegawa, S. (1997). Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. 94, 10919–10924. doi:10.1073/pnas.94.20.10919
Rains, M. E., Muncie, C. B., Pang, Y., Fan, L.-W., Tien, L.-T., and Ojeda, N. B. (2021). Oxidative stress and neurodevelopmental outcomes in rat offspring with intrauterine growth restriction induced by reduced uterine perfusion. Brain Sci. 11, 78. doi:10.3390/brainsci11010078
Redza-Dutordoir, M., and Averill-Bates, D. A. (2016). Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta (BBA) - Mol. Cell. Res. 1863, 2977–2992. doi:10.1016/j.bbamcr.2016.09.012
Ribeiro, J. H., Altinisik, N., Rajan, N., Verslegers, M., Baatout, S., Gopalakrishnan, J., et al. (2023). DNA damage and repair: underlying mechanisms leading to microcephaly. Front. Cell. Dev. Biol. 11, 1268565. doi:10.3389/fcell.2023.1268565
Rommel, A.-S., Milne, G. L., Barrett, E. S., Bush, N. R., Nguyen, R., Sathyanarayana, S., et al. (2020). Associations between urinary biomarkers of oxidative stress in the third trimester of pregnancy and behavioral outcomes in the child at 4 years of age. Brain, Behav. Immun. 90, 272–278. doi:10.1016/j.bbi.2020.08.029
Ruder, E. H., Hartman, T. J., Blumberg, J., and Goldman, M. B. (2008). Oxidative stress and antioxidants: exposure and impact on female fertility. Hum. Reprod. Updat. 14, 345–357. doi:10.1093/humupd/dmn011
Ryan, H. E., Lo, J., and Johnson, R. S. (1998). HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 17, 3005–3015. doi:10.1093/emboj/17.11.3005
Salim, S. (2014). Oxidative stress and psychological disorders. Curr. Neuropharmacol. 12, 140–147. doi:10.2174/1570159x11666131120230309
Sasaki, H., Hamatani, T., Kamijo, S., Iwai, M., Kobanawa, M., Ogawa, S., et al. (2019). Impact of oxidative stress on age-associated decline in oocyte developmental competence. Front. Endocrinol. 10, 811. doi:10.3389/fendo.2019.00811
Sasaki, M., Knobbe, C. B., Itsumi, M., Elia, A. J., Harris, I. S., Chio, I. I. C., et al. (2012). D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes. Dev. 26, 2038–2049. doi:10.1101/gad.198200.112
Scarpato, R., Testi, S., Colosimo, V., Crespo, C. G., Micheli, C., Azzarà, A., et al. (2020). Role of oxidative stress, genome damage and DNA methylation as determinants of pathological conditions in the newborn: an overview from conception to early neonatal stage. Mutat. Res.Rev. Mutat. Res. 783, 108295. doi:10.1016/j.mrrev.2019.108295
Schieber, M., and Chandel, N. S. (2014). ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–R462. doi:10.1016/j.cub.2014.03.034
Schoppa, A. M., Chen, X., Ramge, J.-M., Vikman, A., Fischer, V., Haffner-Luntzer, M., et al. (2022). Osteoblast lineage Sod2 deficiency leads to an osteoporosis-like phenotype in mice. Dis. Model. Mech. 15, dmm049392. doi:10.1242/dmm.049392
Scortegagna, M., Ding, K., Oktay, Y., Gaur, A., Thurmond, F., Yan, L.-J., et al. (2003). Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat. Genet. 35, 331–340. doi:10.1038/ng1266
Seino, T., Saito, H., Kaneko, T., Takahashi, T., Kawachiya, S., and Kurachi, H. (2002). Eight-hydroxy-2′-deoxyguanosine in granulosa cells is correlated with the quality of oocytes and embryos in an in vitro fertilization-embryo transfer program. Fertil. Steril. 77, 1184–1190. doi:10.1016/s0015-0282(02)03103-5
Serra-Juhé, C., Cuscó, I., Homs, A., Flores, R., Torán, N., and Pérez-Jurado, L. A. (2015). DNA methylation abnormalities in congenital heart disease. Epigenetics 10, 167–177. doi:10.1080/15592294.2014.998536
Shiraki, N., Shiraki, Y., Tsuyama, T., Obata, F., Miura, M., Nagae, G., et al. (2014). Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell. Metab. 19, 780–794. doi:10.1016/j.cmet.2014.03.017
Sogut, I., Oglakci, A., Kartkaya, K., Ol, K. K., Sogut, M. S., Kanbak, G., et al. (2015). Effect of boric acid on oxidative stress in rats with fetal alcohol syndrome. Exp. Ther. Med. 9, 1023–1027. doi:10.3892/etm.2014.2164
Song, Y., Li, Z., Wang, B., Xiao, J., Wang, X., and Huang, J. (2014). Phospho-Cdc25 correlates with activating G2/M checkpoint in mouse zygotes fertilized with hydrogen peroxide-treated mouse sperm. Mol. Cell. Biochem. 396, 41–48. doi:10.1007/s11010-014-2140-1
Song, Y., Quan, S., Tian, J., Li, H., Chen, S., and Xing, F. (2009). Relationship between protein oxidation levels in the follicular fluid and the outcome parameters of in vitro fertilization-embryo transplantation. Nan fang yi ke xue xue bao South. Méd. Univ. 29, 160–163.
Steffann, J., Monnot, S., and Bonnefont, J.-P. (2015). mtDNA mutations variously impact mtDNA maintenance throughout the human embryofetal development. Clin. Genet. 88, 416–424. doi:10.1111/cge.12557
Suzuki, K. (2018). The developing world of DOHaD. J. Dev. Orig. Heal. Dis. 9, 266–269. doi:10.1017/s2040174417000691
Szanto, I., Pusztaszeri, M., and Mavromati, M. (2019). H2O2 metabolism in normal thyroid cells and in thyroid tumorigenesis: focus on NADPH oxidases. Antioxidants 8, 126. doi:10.3390/antiox8050126
Takahashi, M. (2012). Oxidative stress and redox regulation on in vitro development of mammalian embryos. J. Reprod. Dev. 58, 1–9. doi:10.1262/jrd.11-138n
Takeshima, T., Usui, K., Mori, K., Asai, T., Yasuda, K., Kuroda, S., et al. (2021). Oxidative stress and male infertility. Reprod. Med. Biol. 20, 41–52. doi:10.1002/rmb2.12353
Torres-Cuevas, I., Parra-Llorca, A., Sánchez-Illana, A., Nuñez-Ramiro, A., Kuligowski, J., Cháfer-Pericás, C., et al. (2017). Oxygen and oxidative stress in the perinatal period. Redox Biol. 12, 674–681. doi:10.1016/j.redox.2017.03.011
Tovar, J. A. (2012). Congenital diaphragmatic hernia. Orphanet J. Rare Dis. 7, 1. doi:10.1186/1750-1172-7-1
Urbaniak, S. K., Boguszewska, K., Szewczuk, M., Kaźmierczak-Barańska, J., and Karwowski, B. T. (2020). 8-Oxo-7,8-Dihydro-2′-Deoxyguanosine (8-oxodG) and 8-hydroxy-2′-deoxyguanosine (8-OHdG) as a potential biomarker for gestational diabetes mellitus (GDM) development. Molecules 25, 202. doi:10.3390/molecules25010202
Vanreusel, I., Taeymans, J., Craenenbroeck, E. V., Segers, V. F. M., Berendoncks, A. V., Briedé, J. J., et al. (2023). Elevated oxidative stress in patients with congenital heart disease and the effect of cyanosis: a meta-analysis. Free Radic. Res. 57, 470–486. doi:10.1080/10715762.2023.2284639
Voss, A. K., and Strasser, A. (2020). The essentials of developmental apoptosis. F1000Research 9. doi:10.12688/f1000research.21571.1
Wakabayashi, N., Itoh, K., Wakabayashi, J., Motohashi, H., Noda, S., Takahashi, S., et al. (2003). Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 35, 238–245. doi:10.1038/ng1248
Wang, B., Li, Z., Wang, C., Chen, M., Xiao, J., Wu, X., et al. (2013). Zygotic G2/M cell cycle arrest induced by ATM/Chk1 activation and DNA repair in mouse embryos fertilized with hydrogen peroxide-treated epididymal mouse sperm. PLoS One 8, e73987. doi:10.1371/journal.pone.0073987
Wang, G., Wang, B., and Yang, P. (2022a). Epigenetics in congenital heart disease. J. Am. Hear. Assoc. 11, e025163. doi:10.1161/jaha.121.025163
Wang, H., Dou, Q., Jeong, K. J., Choi, J., Gladyshev, V. N., and Chung, J.-J. (2022b). Redox regulation by TXNRD3 during epididymal maturation underlies capacitation-associated mitochondrial activity and sperm motility in mice. J. Biol. Chem. 298, 102077. doi:10.1016/j.jbc.2022.102077
Wang, X., Phelan, S. A., Forsman-Semb, K., Taylor, E. F., Petros, C., Brown, A., et al. (2003). Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J. Biol. Chem. 278, 25179–25190. doi:10.1074/jbc.m302706200
Wei, P.-C., Hsieh, Y.-H., Su, M.-I., Jiang, X., Hsu, P.-H., Lo, W.-T., et al. (2012). Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol. Cell. 48, 747–759. doi:10.1016/j.molcel.2012.10.007
Wei, Z., Wang, Y., Wang, S., Xie, J., Han, Q., and Chen, M. (2022). Comparing the effects of polystyrene microplastics exposure on reproduction and fertility in male and female mice. Toxicology 465, 153059. doi:10.1016/j.tox.2021.153059
White, K., Kim, M.-J., Han, C., Park, H.-J., Ding, D., Boyd, K., et al. (2018). Loss of IDH2 accelerates age-related hearing loss in male mice. Sci. Rep. 8, 5039. doi:10.1038/s41598-018-23436-w
Winkle, L. J. V., and Ryznar, R. (2019). One-carbon metabolism regulates embryonic stem cell fate through epigenetic DNA and histone modifications: implications for transgenerational metabolic disorders in adults. Front. Cell. Dev. Biol. 7, 300. doi:10.3389/fcell.2019.00300
Winkler, A., Njålsson, R., Carlsson, K., Elgadi, A., Rozell, B., Abraham, L., et al. (2011). Glutathione is essential for early embryogenesis – analysis of a glutathione synthetase knockout mouse. Biochem. Biophys. Res. Commun. 412, 121–126. doi:10.1016/j.bbrc.2011.07.056
Wong, J. L., and Wessel, G. M. (2008). Free-radical crosslinking of specific proteins alters the function of the egg extracellular matrix at fertilization. Development 135, 431–440. doi:10.1242/dev.015503
Wu, H., Lin, L., Giblin, F., Ho, Y.-S., and Lou, M. F. (2011). Glutaredoxin 2 knockout increases sensitivity to oxidative stress in mouse lens epithelial cells. Free Radic. Biol. Med. 51, 2108–2117. doi:10.1016/j.freeradbiomed.2011.09.011
Xu, D., Guo, H., Xu, X., Lu, Z., Fassett, J., Hu, X., et al. (2011). Exacerbated pulmonary arterial hypertension and right ventricular hypertrophy in animals with loss of function of extracellular superoxide dismutase. Hypertension 58, 303–309. doi:10.1161/hypertensionaha.110.166819
Xu, D.-X., Chen, Y.-H., Zhao, L., Wang, H., and Wei, W. (2006). Reactive oxygen species are involved in lipopolysaccharide-induced intrauterine growth restriction and skeletal development retardation in mice. Am. J. Obstet. Gynecol. 195, 1707–1714. doi:10.1016/j.ajog.2006.03.047
Yamamoto, M., Kensler, T. W., and Motohashi, H. (2018). The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 98, 1169–1203. doi:10.1152/physrev.00023.2017
Yant, L. J., Ran, Q., Rao, L., Remmen, H. V., Shibatani, T., Belter, J. G., et al. (2003). The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 34, 496–502. doi:10.1016/s0891-5849(02)01360-6
Yao, H., Arunachalam, G., Hwang, J., Chung, S., Sundar, I. K., Kinnula, V. L., et al. (2010). Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc. Natl. Acad. Sci. 107, 15571–15576. doi:10.1073/pnas.1007625107
Yu, W., Tu, Y., Long, Z., Liu, J., Kong, D., Peng, J., et al. (2022). Reactive oxygen species bridge the gap between chronic inflammation and tumor development. Oxidative Med. Cell. Longev. 2022, 2606928. doi:10.1155/2022/2606928
Zhang, J., Tripathi, D. N., Jing, J., Alexander, A., Kim, J., Powell, R. T., et al. (2015). ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell. Biol. 17, 1259–1269. doi:10.1038/ncb3230
Zhang, M., Brewer, A. C., Schröder, K., Santos, C. X. C., Grieve, D. J., Wang, M., et al. (2010). NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc. Natl. Acad. Sci. 107, 18121–18126. doi:10.1073/pnas.1009700107
Zorov, D. B., Juhaszova, M., and Sollott, S. J. (2014). Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 94, 909–950. doi:10.1152/physrev.00026.2013
Keywords: ADHD, antioxidants, ASD, embryo, malformations, morphogenesis, ROS, oxidative stress
Citation: Divvela SSK, Gallorini M, Gellisch M, Patel GD, Saso L and Brand-Saberi B (2025) Navigating redox imbalance: the role of oxidative stress in embryonic development and long-term health outcomes. Front. Cell Dev. Biol. 13:1521336. doi: 10.3389/fcell.2025.1521336
Received: 01 November 2024; Accepted: 19 February 2025;
Published: 26 March 2025.
Edited by:
Fernando Antunes, University of Lisbon, PortugalReviewed by:
Nicolas Santander, Universidad de O’Higgins, ChileCopyright © 2025 Divvela, Gallorini, Gellisch, Patel, Saso and Brand-Saberi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luciano Saso, THVjaWFuby5zYXNvQHVuaXJvbWExLml0; Beate Brand-Saberi, QmVhdGUuQnJhbmQtU2FiZXJpQHJ1aHItdW5pLWJvY2h1bS5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.