
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 22 July 2022
Sec. Cancer Cell Biology
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.942500
This article is part of the Research TopicProtein Kinase and Pseudokinase in Cancer BiologyView all 7 articles
 Kristina Riegel1†
Kristina Riegel1† Parthiban Vijayarangakannan2†
Parthiban Vijayarangakannan2† Petros Kechagioglou1
Petros Kechagioglou1 Katarzyna Bogucka1
Katarzyna Bogucka1 Krishnaraj Rajalingam1*
Krishnaraj Rajalingam1*Kinases still remain the most favorable members of the druggable genome, and there are an increasing number of kinase inhibitors approved by the FDA to treat a variety of cancers. Here, we summarize recent developments in targeting kinases and pseudokinases with some examples. Targeting the cell cycle machinery garnered significant clinical success, however, a large section of the kinome remains understudied. We also review recent developments in the understanding of pseudokinases and discuss approaches on how to effectively target in cancer.
Kinases are a key class of enzymes, which catalyze the covalent attachment of the gamma-phosphate of ATP to their various targets including proteins, lipids, and nucleotides. Protein kinases represent one of the largest gene families in eukaryotes with more than 518 protein kinases forming the human kinome (Manning et al., 2002). Among them, protein kinases phosphorylating either tyrosine (tyrosine-specific protein kinases) or serine/threonine (Ser-/Thr-specific protein kinases) are the predominant ones (Martin et al., 2010). Phosphorylation of a protein can affect its function in a variety of ways, such as enhancing or inhibiting its biological activity in terms of enzymatic reactions, transcription, or translation, or affecting the stability, complex formation, or cellular localization of a protein (Roskoski, 2015). Thus, protein phosphorylation is a powerful tool for regulating almost all cellular functions. To name just a few examples, protein kinases play a central role in controlling cell division, cell movement, cell death, transcription, and cell metabolism (Manning et al., 2002; Roskoski, 2015). Further, dysregulation or mutation of protein kinases are linked to many human diseases. The extent of the correlation between kinase dysfunction and diseases was underlined, by the study of Manning et al. (2002), in which they were mapping all kinase genes to chromosomal loci revealing that 164 kinases map to amplicons seen in tumors and 80 kinases map to amplicons associated with other major diseases (Manning et al., 2002). Melnikova and Golden (2004) even suggest that protein kinases are directly or indirectly linked to 400 human diseases making them to attractive targets for therapeutic strategies (Melnikova and Golden, 2004). It took until 2001, after the first description of enzymatic phosphorylation in 1954 by Burnett and Kennedy (1954); Roskoski (2015) and the identification of Rous sarcoma virus (v-Src) as the first transforming factor in 1978 (Collett and Erikson, 1978), for imatinib to be approved as the first small molecule kinase inhibitor by the FDA, which in turn opened the way for further targeted therapeutics in clinical oncology (Cohen, 2002; Roskoski, 2015; Wu et al., 2016; Cohen et al., 2021). By 2021, the number of FDA-approved kinase inhibitors against neoplasm has increased to a total of 58 (Roskoski, 2022), with the majority targeting receptor protein-tyrosine kinases. Newer inhibitors approved by the FDA in 2021 include the MET kinase inhibitor tepotinib (Mathieu et al., 2022), the VEGFR kinase inhibitor tivozanib (Chang et al., 2022), and the EGFR kinase inhibitor mobocertinib (Markham, 2021). Furthermore, the inhibitor trilaciclib was approved to reduce chemotherapy-induced myelosuppression. Trilaciclib targets the serine/threonine kinases CDK4/6 (Powell and Prasad, 2021), which we will discuss in more detail later. Also worth mentioning are the latest developments in BTK inhibitors. All three FDA-approved BTK inhibitors are covalent inhibitors that bind to the nucleophilic Cys481 and are used to treat B-cell-associated malignancies (Guo et al., 2019; Gabizon and London, 2020). However, because there is a need for more selective BTK inhibitors, alternative binding modes of BTK have been exploited. This in turn has led to the development of CGI1746, which reversibly binds BTK in an inactive conformation (Di Paolo et al., 2011). Based on this compound, optimized inhibitors such as the reversible inhibitor fenebrutinib and the covalent inhibitor remibrutinib have been developed and are currently under clinical investigation (Angst et al., 2020; Gabizon and London, 2020).
To develop effective targeting strategies and rational kinase inhibitors, it is important to know the basic structure of kinase domains and how structural features in turn affect function. A typical protein kinase domain comprises of a small, mostly β-stranded N-lobe and a larger, α-helical C-lobe, which are connected by a short hinge region. As reviewed by Fabbro et al. (2015), the N-lobe harbors the glycine-rich loop (P-loop) that coordinates the phosphates of ATP and a single α-helix (C-helix) that can occupy different positions, contributing to the formation of an active or inactive kinase state (Fabbro et al., 2015). More precisely, the ‘α-helix-in’ position facilitates the interaction between the active site Lys and the Glu from the α-helix required for efficient catalysis (Cowan-Jacob, 2006; Fabbro et al., 2015). Deep in the ATP pocket is an additional residue, called “gatekeeper”, which restricts access to a pre-existing pocket and whose mutation leads to drug resistance (Fabbro et al., 2015). The C-lobe contains most of the catalytic machinery including the Y/HRD motif of the catalytic loop and the DFG motif. While the Y/HRD motif correctly positions the acceptor group to allow the contacts important for an efficient catalysis, the DFG motif brings the Asp in the correct position. In many instances, the DFG motif can take an “in” position where Phe rotates out of the ATP binding pocket and brings Asp to the site to coordinate Mg2+. In the DFG “out” position, however, Phe occupies the pocket (Fabbro et al., 2015). A generally accepted kinase activation model has been proposed in which kinases possess a series of highly conserved residues that form two parallel columns upon activation, termed regulatory and catalytic spines (Kornev et al., 2006).
As the example of RAF kinases shows, the activation of kinases can also be allosterically linked to the dimerization of the kinase. Here, the leucine residue of the α-helix involved in the regulatory spine formation is adjacent to an arginine residue, that is, part of the conserved RKTR motif at the dimer interface and thus plays a role in RAF dimer formation (Durrant and Morrison, 2018). Consequently, RAF dimerization pushes the α-helix to the “in” position, which contributes to the alignment of the regulatory spine and to the global stabilization of the active kinase conformation (Durrant and Morrison, 2018).
Protein kinase inhibitors have been classified as type I, II, and III inhibitors. Type I inhibitors bind reversibly at the ATP-binding pocket and extend into proximal regions to achieve greater selectivity. Another common feature is that these inhibitors bind the kinase in a DFG “in,” active state. A typical example of a type I inhibitor is the EGFR inhibitor gefinitib (Roskoski, 2016). In addition to type I inhibitors, there are type 1.5 inhibitors such as the RAF inhibitor vemurafenib, which are ATP-competitive inhibitors, that bind the kinase with a DFG “in” but α-helix “out” conformation (Tsai et al., 2008; Fabbro et al., 2015). In contrast to type I inhibitors, type II inhibitors bind to the DFG “out” conformation. Here, the ATP binding site as well as an additional hydrophobic pocket adjacent to the ATP pocket, which is present in the DFG “out” conformation, are preferentially occupied by type II inhibitors (Fabbro et al., 2015). An example for a type II inhibitor is imatinib (Nagar et al., 2002), the first FDA-approved kinase inhibitor, which inhibits BCR-Abl tyrosine kinase and is used for the treatment of CML (Roskoski, 2015). Inhibitors that do not bind the ATP binding site but an allosteric site are called type III or allosteric inhibitors. The advantage of type III inhibitors is that they do not compete with ATP binding and that they show a higher degree of selectivity since binding sites and regulatory mechanisms are targeted, which are unique for the respective kinase (Roskoski, 2016). A typical example for a type III inhibitor is trametinib, a MEK inhibitor approved for the treatment of melanoma.
Targeting strategies can be adapted dependent on what kind of problems are occurring with the developed inhibitors. A good example is the development of RAF inhibitors. There are three RAF isoforms (ARAF, BRAF and CRAF), which are part of the classical MAPK (mitogen-activated protein kinase) pathway RAF-MEK1/2-ERK1/2. MAPKs are a class of ubiquitously expressed serine/threonine-specific protein kinases. The mammalian family of MAPKs is represented by 14 members and organized into seven signaling pathways with the archetypal, RAS-dependent RAF-MEK1/2-ERK1/2 module at the forefront of the drug development research in cancer (Cargnello and Roux, 2011; Degirmenci et al., 2020).
Upon growth factor stimulation, RAS is activating this pathway, which regulates fundamental cellular processes including migration, cell survival and cell division. The classical MAPK pathway gained enormous interest since this pathway is often deregulated in human cancer with RAS and BRAF being among the most frequently mutated oncogenes. Therefore, much time has been invested in the development of drugs targeting this signaling pathway. Although the initial success of the RAF inhibitor vemurafenib has been impressive, rapidly developing drug resistance and the occurrence of secondary malignancies were of concern. Further studies revealed that the first-generation RAF inhibitors such as vemurafenib induced paradoxical MAPK activation in a BRAF mutation-free context by triggering RAF dimerization. To overcome this effect, next generation inhibitors such as pan-RAF inhibitors have been developed, which bind RAF kinases in a DFG “out”/α-helix “in” state thus inhibiting monomeric and dimeric RAF with equal efficiency (Durrant and Morrison, 2018). Drugs targeting the RAF dimer interface such as the PLX8394 represent an alternative approach to prevent the drug-induced paradoxical MAPK activation (Yao et al., 2019). As we found in our own study, a change in the targeting strategy could lead to different side effects than first-generation inhibitors, such as impairment of the function of certain immune cells (Riegel et al., 2019), which needs to be carefully considered in further inhibitor development.
Instead of aiming at inhibition of the kinase activity, new therapeutics induce degradation of the target by recruiting an E3 ligase. These so-called PROteolysis TArgeting Chimeras (PROTACs) are heterobifunctional molecules that bring the target and the E3 ligase in proximity, causing ubiquitination and subsequent degradation by the ubiquitin-proteasome system (Khan S. et al., 2020). This strategy would also be useful to target the kinase-independent functions.
In addition to the development of novel drugs, researchers studying the kinome now increasingly rely on its bioinformatics visualization, which can be used in different ways. For example, pathways in which kinases are involved may be visualized using the Reactome database. Moreover, using Kinome trees researchers can analyze the range of highly expressed kinases in diseases such as cancer (Figure 1). For advanced research, tumor samples could be also classified according to molecular subtypes (e.g., TCGA-COAD subtypes) to identify biomarkers or kinase drug targets of interest against specific cancer types or subtypes (Guinney et al., 2015). While it is useful to identify a single kinase target, that is highly expressed in a number of molecular subtypes or cancers, it is possible that a particular kinase is only highly expressed in one of the molecular subtypes, so a corresponding drug target for that subtype may be promising.
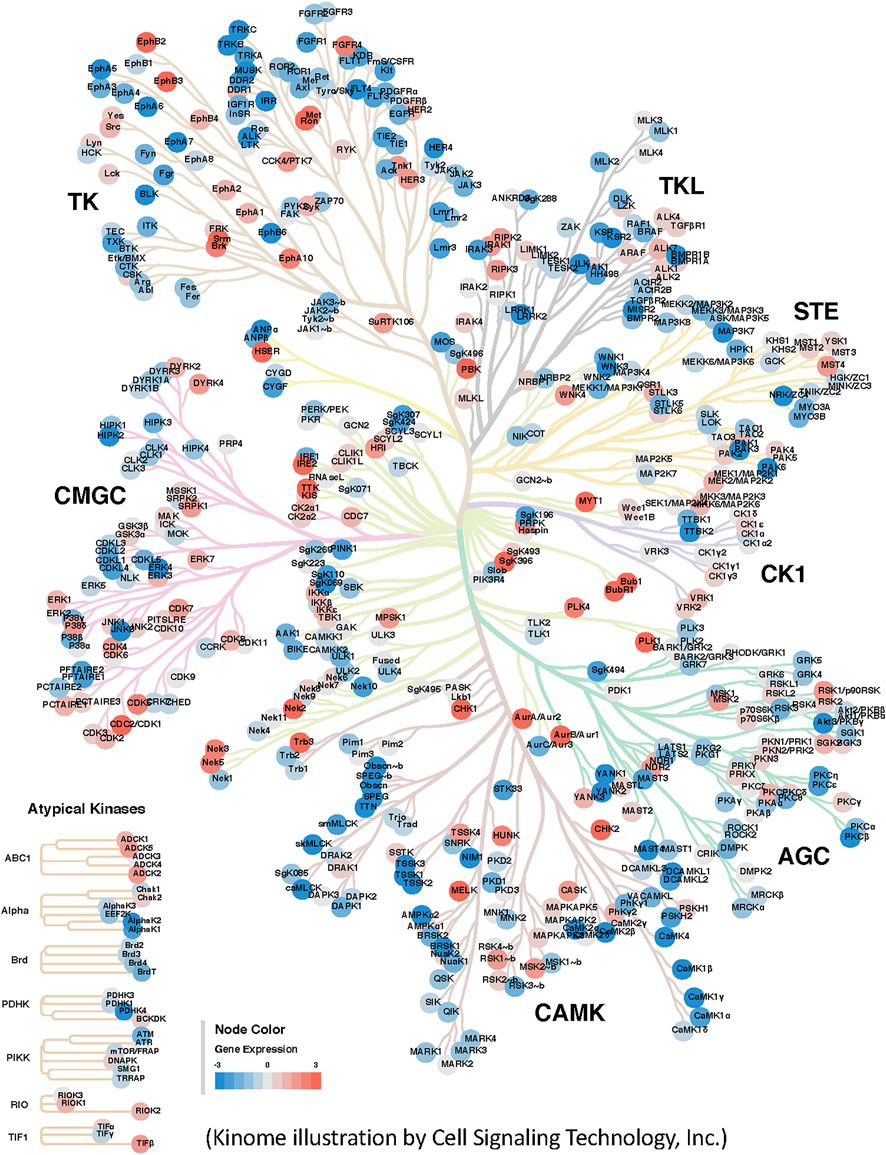
FIGURE 1. Visualization of differentially expressed kinases in cancer. Differential expression of kinases in Colon Adenocarcinoma was estimated using 100 TCGA-COAD RNA-Seq tumor samples (cases) and 165 normal samples (controls) derived from various human tissues by (Suntsova et al., 2019). Raw gene counts of both samples were downloaded from the Genomic Data Commons and Sequence Read Archive (SRA ID: SRP163252) respectively. DEseq2 R package was used to normalize the data and estimate the differential expression of genes. The log2 ratio of differentially expressed kinases were then extracted and used as quantitative input data within the CORAL web application (Metz et al., 2018). A few prominent differentially expressed kinases such as MET and RON as one of the highly expressed kinases in the results.
Since hyperactivation of cell cycle proteins and uncontrolled proliferation are hallmarks of cancer, one of the strategies is to target the cell cycle itself. The cyclin-dependent kinases (CDKs) are serine/threonine kinases that regulate cell cycle progression (Malumbres and Barbacid, 2001). CDK4/6 are crucial regulators of the G1/S transition, and their activity is activated by the D-type cyclins D1, D2, and D3 (cyclin D) (Morgan, 1997; Sherr and Roberts, 2004). For cells to enter S phase from G1 phase, CDK4 cyclin D1 and subsequently CDK2 cyclin E must hyperphosphorylate and thereby inactivate the retinoblastoma-associated protein (Rb) (Weinberg, 1995). Inactivation of pRb leads to the release of the transcription factor E2F from the Rb-E2F complex that actively represses transcription of cell cycle genes (Chellappan et al., 1991). Therefore, CDK4/6 enzymes are considered promising targets in cancer therapy (Hanahan and Weinberg, 2011; Otto and Sicinski, 2017), and the anti-tumor effect of small molecule CDK4/6 inhibitors is based on blocking the phosphorylation of the tumor suppressor retinoblastoma (Rb), and induction of G1/S arrest in tumor cells (Knudsen et al., 2019) (Luo et al., 1998).
Developing compounds with selectivity for CDK4/6 over other CDKs has been challenging, but Pyrido(2,3-d)pyrimidinone finally showed high selectivity for CDK4 compared to other CDKs (Barvian et al., 2000) and served as a precursor compound for the development of the FDA-approved ATP-competitive inhibitors palbociclib (Ibrance, PD0332991), ribociclib (Kisqali, LEE011), abemaciclib (Verzenio, LY2835219), and trilaciclib (G1 therapeutics, G1T28-1) (Beaver et al., 2015; Walker et al., 2016; Kim, 2017; Syed, 2017; Powell and Prasad, 2021). Palbociclib and ribociclib are both approved for the treatment of hormone receptor-positive (HR+), human epidermal growth factor receptor 2 (HER2)-negative advanced breast cancer patients in combination with endocrine therapy. Abemaciclib is the only one that can be used as monotherapy in adult patients with disease progression following endocrine therapy and prior chemotherapy in the advanced breast cancer, or in combination with fulvestrant in case of disease progression after endocrine therapy. CDK4/6 inhibitors have changed the treatment strategy for HR+/HER2− advanced breast cancer patients, and more than 17 CDK4/6 inhibitors in combination with various drugs, are now being tested or have been tested in clinical trials.
Several studies have suggested that the CDK4/6 inhibitors bind to the ATP-binding pocket of CDK4 and CDK6 (Mariaule and Belmont, 2014) and block the cell proliferation in a wide range of tumors and reduce tumor growth in cancer xenograft models (Fry et al., 2004; Marzec et al., 2006; Logan et al., 2013; Tate et al., 2014; Gong et al., 2017; Kim et al., 2018). Another potential mechanism, after prolonged treatment with CDK4/6 inhibitors, may be the induction of RB1 and FOXM1-dependent senescence (Dean et al., 2010; Anders et al., 2011; Vijayaraghavan et al., 2017). Senescence has been demonstrated as a desired mechanism of cell growth inhibition by blocking the cell cycle progression (Braig et al., 2005). However, one of the chemotoxicity-induced mechanisms of chemotherapy and radiotherapy is the excessive induction of senescence in non-malignant cells (Baar et al., 2017; Murali et al., 2018; Yao et al., 2020). The main regulator of these detrimental properties is the transcription factor NF-κB, which triggers the “senescence-associated secretory phenotype” in conventional cancer treatments (Rodier et al., 2009; Chien et al., 2011). Meanwhile, a new study has found that CDK4/6 inhibitors induced senescence in non-malignant cells without toxicity, which was dependent on the transcriptional activity of the tumor suppressor p53 rather than NF-κB and therefore lacked most of the common pro-inflammatory senescence-associated secretory phenotype factors responsible for several adverse reactions (Wang et al., 2022).
The discovery of CDK4/6 inhibitors is considered a game-changer in cancer treatment. However, intrinsic or acquired resistance of HR+/HER2- metastatic breast cancer to clinically approved CDK4/6 inhibitors are responsible for disease progression in the majority of patients. Identifying reliable prognostic biomarkers that enable novel treatment combinations is the key to treatment success (Shah et al., 2018). Multiple studies have demonstrated that one of the most common mechanisms of cancer cell resistance to CDK4/6 inhibitors is loss of function of Rb (mutation, hyperphosphorylation or deletion) (Dean et al., 2012; Malorni et al., 2016; Vijayaraghavan et al., 2017). However, although studies have detected Rb inactivation by hyperphosphorylation in breast cancer cells resistant to endocrine therapy and in patients’ tumors receiving adjuvant endocrine treatment (Thangavel et al., 2011; Malorni et al., 2016), large clinical trials couldn’t indicate a statistically relevant link between Rb and resistance to CDK4/6 inhibitors (Turner et al., 2019; Finn et al., 2020). An alternative mechanism of resistance to CDK4/6 inhibitors is CDK6 overexpression. CDK6 amplification mediated resistance to abemaciclib in breast cancer cells (Yang et al., 2017), while elevated CDK6 protein levels were associated with acquired resistance to endocrine treatment (Correction, 2016).
Instead of monotherapy, the development of novel therapeutic combinations based on the mechanisms of resistance has been an emerging area. Several preclinical studies and clinical trials proposed the synergistic effect of different agents either with only the CDK4/6 inhibitors or in combination with endocrine therapy. The main two combinatorial strategies involve growth factors that either activate upstream of the cyclin D-CDK4/6-Rb pathway or pathway members, such as mTOR, PI3K, AKT, RAF and MEK, and enhance the cytostatic effect of CDK4/6 inhibitors or the apoptosis of cancer cells (Cheng et al., 1998; Vora et al., 2014; Goel et al., 2016; Herrera-Abreu et al., 2016; Jansen et al., 2017; Formisano et al., 2019; Alves et al., 2021; Lelliott et al., 2021; Zhao et al., 2021). Combination of CDK4/6 inhibitors and immunotherapy is another strategy to induce anti-cancer immune responses by using anti-PD-1/PD-L1 inhibitors (Deng et al., 2018; Schaer et al., 2018; Yuan et al., 2021). A synergistic effect of abemaciclib and the PD-L1 checkpoint blocker led to complete tumor rejection compared to tumor growth delay by the abemaciclib monotherapy and enhanced adaptive and innate immune activation (Schaer et al., 2018).
Induction of autophagy by CDK4/6 inhibitors is a common resistance mechanism in cancer treatment (Chittaranjan et al., 2014; Wang et al., 2016). Therefore, the combinational effect of hydroxychloroquine (HCQ), a late-stage autophagy inhibitor, with palbociclib and abemaciclib is being examined in breast cancer cells, mice, and phase I/II or phase II clinical trials (NCT03774472, NCT04523857, and NCT04841148) (Vijayaraghavan et al., 2017). Co-treatment of palbociclib with HCQ induced irreversible growth inhibition and elevated levels of senescence in breast cancer cells and decreased tumor volume in both the treatment and recovery phase after the treatment was stopped (Vijayaraghavan et al., 2017).
The interplay between CDK4/6 and mitogen-activated protein kinase (MAPK) inhibitors was explored by several studies and clinical trials. Most of the solid tumors with resistance to CDK4/6 inhibitors, are characterized by alterations in MAPK key genes, such as RAS, RAF, MEK and ERK, which represent promising therapeutic targets (Stern, 2018). In addition, MAPKs are major regulators of cyclin D1, and abnormal MAPK expression levels lead to cancer progression and resistance to therapies (Plotnikov et al., 2011). This dependence on MAPK activation suggests that combinatorial treatment with MAPK and CDK4/6 inhibitors is a potentially effective cancer treatment strategy. Indeed, preclinical studies revealed that xenograft models with BRAF, KRAS or NRAS mutations were more sensitive to this combinatorial treatment and induced sustained tumor regression (Yadav et al., 2014; Yoshida et al., 2016; Pek et al., 2017; Chen et al., 2018; Kim et al., 2018; Martin et al., 2018; Teh et al., 2018). In addition, numerous clinical trials are performed with this combinatorial treatment in various cancer types by using ribociclib, palbociclib, abemaciclib or dalpiciclib as CDK4/6 inhibitors and different MAPK inhibitors (Table 1).
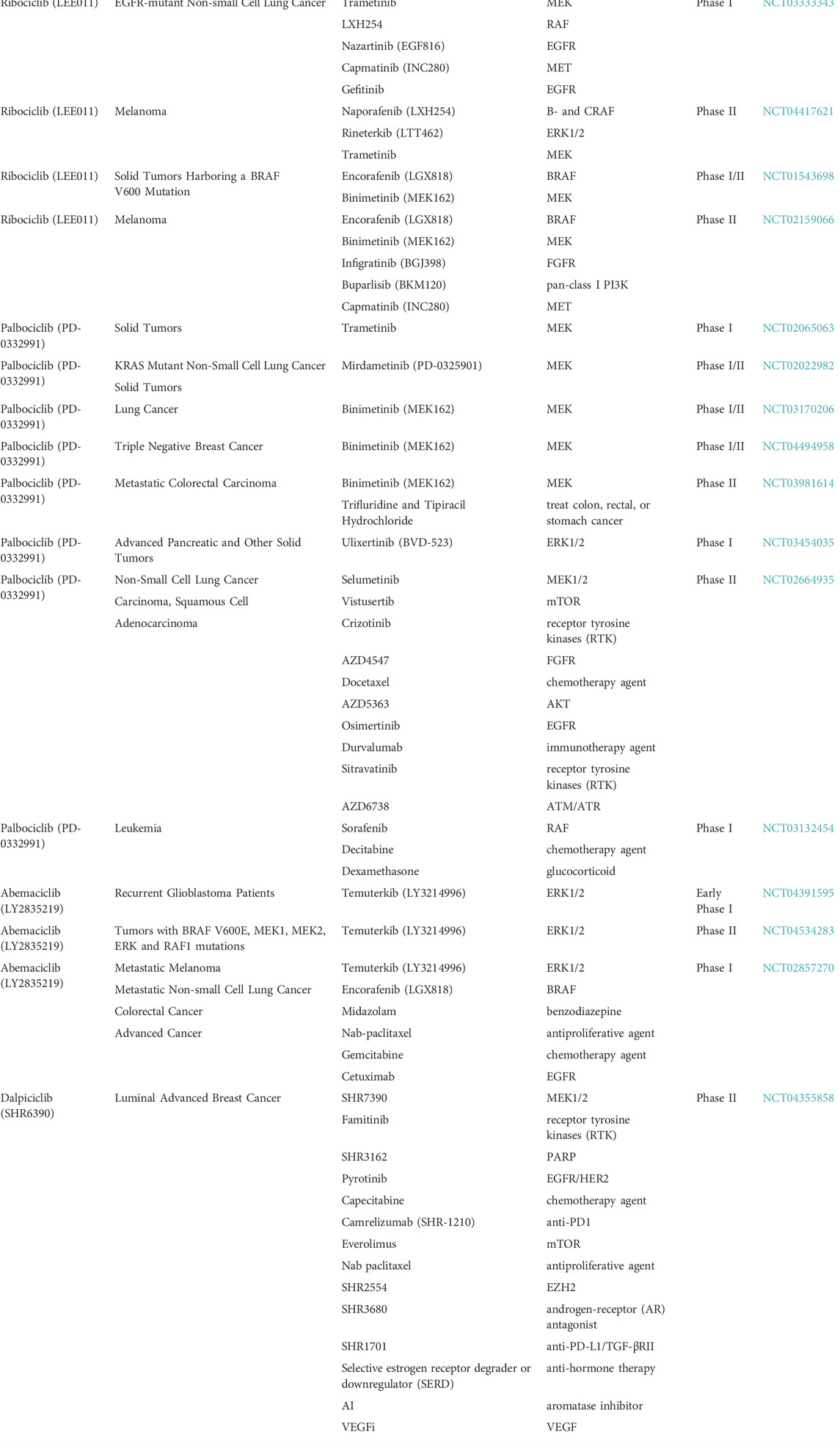
TABLE 1. Clinical trials with CDK4/6 inhibitors in combination with MAPK inhibitors.
Even though the majority of patients respond to the standard first-line drug therapy with CDK4/6 inhibitors combined with endocrine therapy, there is an increasing need to validate novel biomarkers to escape intrinsic or acquired multiple resistance mechanisms. Currently developed combinatorial therapies have made remarkable progress in the efficacy of CDK4/6 inhibitors for HR+/HER2- metastatic breast cancer therapy. Further comprehensive research into potential resistance mechanisms may lead to novel combinatorial treatment strategies and consequently the use of CDK4/6 inhibitors for additional diseases.
The world of kinases is complicated by the fact that many of them perform kinase-independent functions in addition to their kinase-dependent ones. To stay with the example of RAF kinases: Several kinase-independent functions have been described for CRAF, such as its interaction with the Rho effector Rok-α that affects keratinocyte and fibroblast migration (Ehrenreiter et al., 2005), or its binding to ASK1 that inhibits ASK1’s pro-apoptotic function (Chen et al., 2001). Moreover, the suppression of ERBB3-AKT-driven lung cancer spreading requires both kinase-dependent and independent functions of ARAF. Whereas ARAF suppresses activation of AKT kinase in a kinase-dependent manner, it inhibits the expression of ERBB3 in a kinase-independent manner (Mooz et al., 2022). Here, ARAF-dependent regulation of ERBB3 expression was mediated by the transcription factor KLF5. Not only were we surprised by the different regulatory mechanisms of ARAF with respect to the ERBB3-AKT signaling axis, but also by the observation that ARAF, previously classified more as an oncogene, acts as a tumor suppressor in a subset of lung cancers. We could show that ARAF has a suppressive effect on lung colonization of cells injected into the tail vein of mice and we observed that low expression of ARAF correlates with poor survival of lung cancer patients, highlighting the clinical relevance. Besides ARAF, CRAF has also been shown to have tumor suppressive properties in hepatocellular carcinoma (Jeric et al., 2016). Therefore, to develop rational therapies, it is important to also consider the tissue-specific role of “oncogenic” kinases in a kinase-dependent and kinase-independent manner. Better understanding of how the switch between oncogenic and tumor-suppressive kinase function works will in turn enable a variety of treatment options.
The study of pseudokinases has revealed the ways in which kinase-independent functions can affect biological processes. Approximately 10% of the members forming the human kinase group are explicitly classified as pseudokinases because they lack the necessary catalytic groups (Manning et al., 2002), while others contain multiple somatic mutations (Forbes et al., 2010; Hu et al., 2021) of known or unknown consequences directly or indirectly disrupting the activity of functional motifs. Manning and co-workers (Scheeff et al., 2009) have characterized the integrity and activity of functional motifs in pseudokinases. Particularly, they have predicted the integrity of glycine-rich loops (“intact,” “degraded” or “plausible degradation”) or their variations in atypical kinases. They have also characterized the VAIK, HRD and DFG motifs and predicted whether they are inactive. Pseudokinases were also subclassified into four groups based on their nucleotide-binding properties (Murphy et al., 2014): 1) devoid of detectable nucleotide or cation binding, 2) cation-independent nucleotide binding, 3) cation binding, and 4) nucleotide binding enhanced by cations.
Although pseudokinases lack kinase activity, a growing number of them have been shown to play key roles in regulating various cellular processes (Figure 2), including MAPK signaling, actin polymerization, and the development of malignancies (Fukuda et al., 2009; Scheeff et al., 2009; Ghatak et al., 2013; Toms et al., 2013; Murphy et al., 2015; Stefely et al., 2015; Martin et al., 2022). Three principal mechanisms of kinase-independent actions have emerged, which may also occur in combination in some pseudokinases: 1) Allosteric regulation of kinase activity through heterodimerization of pseudokinase and kinase. 2) Action as scaffolding proteins that recruit signaling proteins to regulate biological endpoints such as cell migration and invasion. 3) Action as molecular switches. Notably, each of these functions can theoretically also occur in kinases in addition to their kinase activity (Jacobsen and Murphy, 2017).
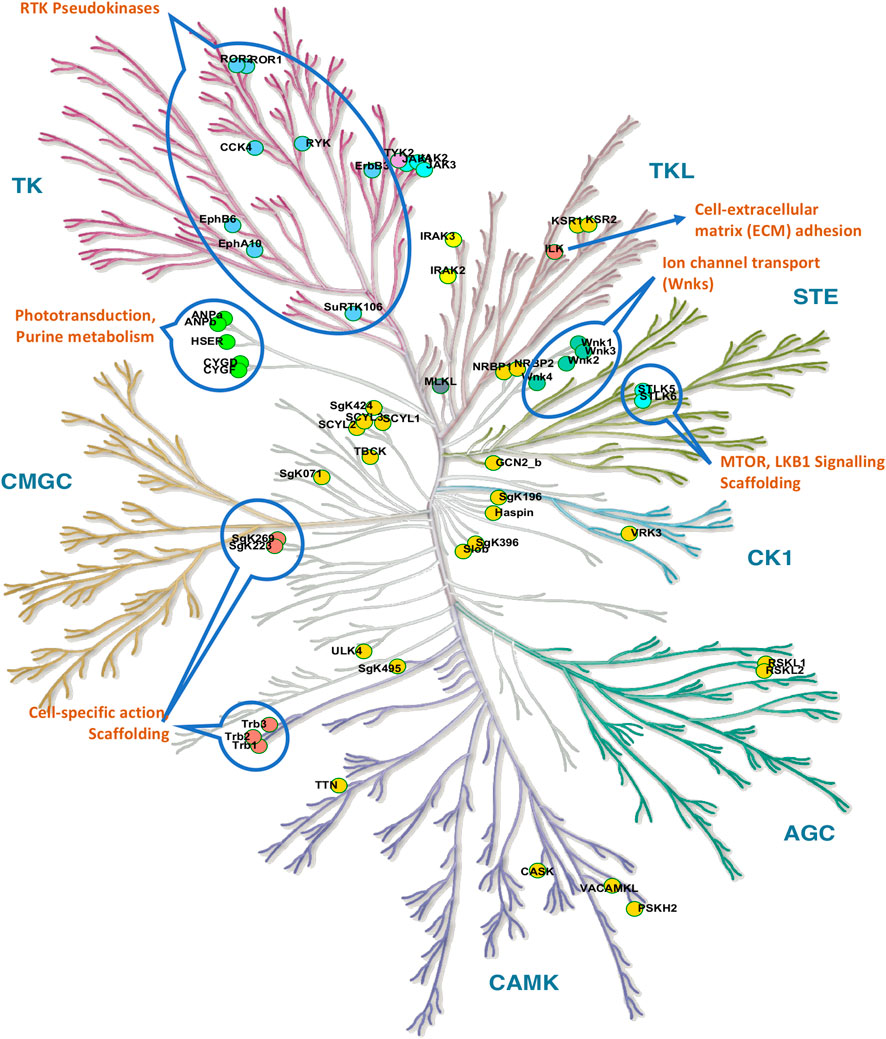
FIGURE 2. Visualization of pseudokinases. The kinome tree of pseudokinases shows a range of functions including scaffolding (The Tribbles TRIB1-3), cell-specific actions, visual signal transduction (phototransduction), purine metabolism, Ephrin receptors, ion channel transport (Wnk1-4), and others. The pseudokinases ADCK3 (atypical) and PAN3 (other) are not shown. The kinome tree illustration is reproduced from Kinmap portal by Cell Signaling Technology, Inc.
The transition from an inactive, monomeric configuration of the kinase domain to an active, dimeric configuration is essential for kinase activation and achieved through an allosteric regulatory mechanism (Engelman et al., 2007; Hu et al., 2013; Lavoie et al., 2014; Dhawan et al., 2016). Pseudokinases are often the result of gene duplication and exhibit substantial homology with their family members, with whom they frequently cooperate (Adrain and Freeman, 2012; Murphy et al., 2017; Ribeiro et al., 2019). Similarly, the ability to allosterically regulate kinases has been preserved in many pseudokinases. For example, the Kinase Suppressors of Ras 1 and 2 (KSR1/2) are well-studied pseudokinases of the RAF family and required for maximal activation of the Raf/MEK/ERK kinase cascade (Rajakulendran et al., 2009; Brennan et al., 2011; Lavoie et al., 2018). They do this by acting both as a scaffold bringing together the components of the cascade (see section “scaffolding function”) and by allosterically activating their RAF kinase family members. Dimerization and subsequent autophosphorylation of RAF kinases is a prerequisite for their kinase activity and controlled by GTP-bound RAS engaged through their RAS-binding domains (RBD). Besides homodimerization, RAF family members may also be activated by dimerizing with KSR1/2. KSR1/2 lack RBDs and thus the heterodimerization relies on a different regulatory principle that rather involves the RAF substrate MEK. Indeed, binding of MEK to the kinase domain of KSR1/2 induces conformational changes in KSR1/2 that drive in turn “side-to-side” heterodimerization with BRAF enabling the allosteric transactivation of BRAF by KSR1/2 (Lavoie et al., 2018). It is assumed that BRAF-KSR dimerization serves to establish the active conformation of BRAF as a prerequisite for subsequent cis-auto- phosphorylation of the BRAF activation loop, which is required for full activation of the dimer (Hu et al., 2013). Of note, the MEK molecule that induces BRAF-KSR dimerization is not the one targeted by the activated BRAF but rather a free “substrate” MEK molecule is engaged for phosphorylation (Lavoie et al., 2018). The FDA-approved MEK inhibitor trametinib, which disrupts RAF-MEK binding, was recently shown to stabilize KSR-MEK binding by directly engaging KSR at the MEK interface (Khan Z. M. et al., 2020). Thus, KSR is a critical factor to be considered in the evaluation and development of clinical MEK inhibitors. KSR1 is itself an advantageous target for the development of new therapeutics. KSR1 can be functionally inhibited with small molecules (e.g., APS-2-79) that stabilize KSR1 in an inactive conformation that blocks heterodimerization with BRAF (Lozano et al., 2003; Dhawan et al., 2016). However, efficacy and potential of this approach for the treatment of Ras-driven cancers remain to be demonstrated.
Another well-studied example of a pseudokinase that allosterically regulates its kinase-active family members is ErbB3/HER3, a well-documented ligand-dependent dimer- ization partner of epidermal growth factor receptor (EGFR), ErbB2/HER2, and c-Met (Engelman et al., 2007). By forming an asymmetric kinase dimer, HER3 stabilizes its dimer partner in the active conformation (Zhang et al., 2006; Jura et al., 2009). ErbB3/HER3 overexpression is associated with lung, colon, gastric, and other cancers (Amin et al., 2010) and has been also found to be responsible for resistance to HER2, IGF1R, and EGFR inhibitors in the treatment of several types of cancers (Erjala et al., 2006; Sergina et al., 2007; Desbois-Mouthon et al., 2009; Miller et al., 2009).
A dynamic switch-like conformational change is inherent to the activation of eukaryotic kinases leading to phosphorylation of their substrates. It is typically controlled by phosphorylation of the activation loop phosphate (Zhang et al., 2009) but can also occur in response to the binding of partners. Mixed lineage kinase domain-like (MLKL) is a paradigm example of a pseudokinase that acts as a molecular switch but when turned on results in multimerization and necroptotic cell death rather than phosphorylation-dependent signaling (Zhang et al., 2009; Murphy et al., 2013; Petrie et al., 2018). Necroptosis is a lytic form of programmed cell death characterized by the rupture of cells, often triggering inflammatory responses. It is associated with numerous human diseases, including cancer and inflammatory diseases, but has no function in development, making it an attractive therapeutic target. Key molecules of necroptosis are the kinases RIPK1 and RIPK3 that form the necroptotic complex (also called necrosome) (Cho et al., 2009; Li et al., 2012), and their phosphorylation target MLKL, which is considered the executioner of necroptosis (Zhang et al., 2009; Wu et al., 2013; Su et al., 2014). MLKL comprises an N-terminal four-helix bundle (4HB) domain connected to a C-terminal pseudokinase domain (PsKD), which lacks two of the three conserved catalytic residues (Zhang et al., 2009; Su et al., 2014). In the inactive state MKLK is monomeric and the 4HB domain, which is required for lipid engagement and membrane permeabilization, engages in inhibitory intramolecular interactions with the PsKD. Upon stimulation, RIPK3-mediated phosphorylation of the PsKD (Thr356 and Ser357) leads to the switch-like conformational change that frees the 4HB domain from intramolecular inhibition and results in the formation of pro-necroptotic MLKL tetramers that translocate to the cell membrane to cause cell expansion, rupture of the membrane and cell death (Zhang et al., 2009; Cai et al., 2014; Chen et al., 2014; Su et al., 2014; Wang et al., 2014; Petrie et al., 2018). While in some diseases (ischemic injury, inflammation, etc.) the goal is to inhibit necroptosis, in cancer therapy the goal is to activate it. In contrast to apoptosis, necroptosis triggers inflammation or causes an innate immune response through the release of damage-associated molecular patterns (DAMPs) and might thus trigger antitumor immunity in cancer therapy to defend against tumor progression (Su et al., 2016; Gong et al., 2019). Though lacking any catalytic activity, MLKL has retained its ability to bind nucleotides and it has been found that ATP-binding to MLKL as well as MLKL PsKD mutants, including some identified in cancers (Forbes et al., 2017), stabilized the monomeric OFF state. In contrast, mutations mimicking RIP3-mediated phosphorylation within the PsKD promoted the ON state (Petrie et al., 2018), however, in human MLKL they were not sufficient to induce necroptosis in a cell model. This knowledge has been the basis for the development of small molecules (Rübbelke et al., 2020; Rübbelke et al., 2021).
Another example for a switch-like function is provided by the Janus kinase (JAK) family of non-receptor tyrosine kinases that transduce extracellular cytokine signals through the JAK-STAT pathway and controls biological processes such as immunity, cell division, and cell death particularly in hematopoiesis. Besides playing critical roles in host defense and autoimmunity, JAKs are associated with cancers especially hematologic malignancies (James et al., 2005; Levine et al., 2005; Erdogan et al., 2022). JAKs contain both an active kinase domain (Jak homology 1, JH1) and a pseudokinase domain (Jak homology 2, JH2) in tandem. Structural studies and molecular dynamics simulations on JAK2 revealed that JH2 inhibits the kinase activity of JH1 by intramolecular interactions that stabilize the kinase domain in an inactive state (Saharinen et al., 2000; Saharinen and Silvennoinen, 2002; Shan et al., 2014). The switch to the active state occurs upon cytokine binding leading to a receptor rearrangement that facilitates JAK2 trans-phosphorylation of activation-loop tyrosines 1007–1008 in JH1. The importance of this switch is illustrated by the fact that mutations in JH2 are the most common activating somatic mutations underlying hematologic malignancies (Haan et al., 2010). Structural studies revealed that the most frequent of these mutations (V657F in Jak1 and V617F in Jak2) promotes the rearrangement of the pseudokinase domain allowing JH2 to adopt the active conformation (Toms et al., 2013). In fact, only recently Glassman et al. resolved the structure of full-length active JAK1 bound to intracellular domain regions of a cytokine receptor and investigated the structural impact of the oncogenic mutation V657F, showing that this mutation is positioned at the central pseudokinase interdimer interface (Glassman et al., 2022).
Pseudokinases are often multidomain proteins able to recruit multiple components of a signaling pathway to efficiently elicit a specific cellular response. For example, besides its function as allosteric activator of RAF kinases, KSR also acts as a scaffold providing docking sites for MEK, ERK, 14-3-3 proteins, caveolin-1, IMP, and phosphatases that collectively regulate flux through the RAF-MEK-ERK cascade. Notably, consistent with the function as scaffold excess levels of KSR disrupt signaling as members of the cascade are “diluted” rather than concentrated on a single KSR molecule.
Another intriguing example of a pseudokinase scaffold is the Tribbles family (TRIB1, TRIB2 and TRIB3) (Eyers et al., 2017), which is implicated in a wide variety of cancers as well as drug resistance. The structural studies on the family member TRIB1 revealed that the C-terminal tail binds to a pocket formed by helix αC in the N-lobe of the pseudokinase domain in a way that resembles an autoinhibitory conformation of conventional kinases precluding “substrate” binding by the highly conserved DQXVP motif (Murphy et al., 2015; Jamieson et al., 2018). One of the best characterized proteins that binds to this motif is the E3 Ub-ligase COP1, but it is also reported that the tribble family act as cell-type specific scaffolds for MEK/MAPK, AKT and NFκB signaling using other motifs (Wei et al., 2012; Yu et al., 2019). Unlike for COP1, the underlying mechanisms here are less well understood but may also involve autoinhibitory interactions between C-terminal tail and the pseudokinase domain.
The oncogenic PEAK family (PEAK1/SgK269, PEAK2/SgK223/Pragmin, and PEAK3) also functions as scaffolds for different cellular processes, particularly morphology, and migration (O'Rourke and Daly, 2018; Roche et al., 2019). Their molecular and structural features enable PEAKs to operate in a spatially and temporally regulated way: Besides being multidomain proteins offering a variety of docking sites and regulatory phosphorylation sites, PEAK proteins dimerize in a unique way via their split helical dimerisation (SHED) domains (Patel et al., 2017; Ha and Boggon, 2018) making the pseudokinase domain accessible from the outside to allow additional homo- and heterotypic interactions that are key to their function(s). The molecular mechanisms by which PEAKs regulate cytoskeletal and adhesion signaling are complex and context-dependent rendering therapeutic targeting challenging.
Finally, the integrin-linked kinase (ILK) is an evolutionarily conserved, intracellular pseudokinase scaffold with widespread expression and a central component of cell-extracellular matrix (ECM) adhesions where it facilitates bidirectional signaling between the ECM and intracellular sites (Hannigan et al., 2005; Fukuda et al., 2009; Qin and Wu, 2012). To this end, ILK forms a heterotrimer (the PINCH-ILK-parvin complex) through protein-protein interactions via multiple sites including the pseudoactive site. ILK activity is stimulated by adhesion to the ECM as well as growth factors in a PI3K-dependent manner. ILK overexpression induces epithelial-mesenchymal transition (EMT) by inhibiting E-cadherin expression and generates a tumorigenic phenotype through activation of nuclear β-catenin. It also promotes cell survival by stimulating the phosphorylation of AKT on Ser473. ILK activation promotes VEGF expression in tumor cells and has a crucial role in endothelial activation and angiogenesis. Therefore, ILK is one of the most versatile pseudokinases that can drive cell proliferation, anchorage and growth-factor independence, angiogenesis, cell death evasion (which may trigger MLKL-driven necroptosis), cell or tissue invasion and metastasis.
Inhibition of ILK has been a key strategy for many cancer treatments (Martin et al., 2022). Due to the complexity of protein-protein interactions and a plethora of affected pathways, these programs exhibit various levels of success in their development—these include chronic lymphocytic leukemia (CLL), breast cancer, and various cancer cell lines in vitro. Knockout of ILK sensitizes breast cancer to SRC inhibitors such as bosutinib (Beetham et al., 2022). When used independently, SRC inhibitor programs have not been successful but a combination therapy regime involving simultaneous inhibition of ILK and SRC would be more promising in future.
Pseudokinases were long considered undruggable. However, as indicated above, solid evidence suggests that pseudokinases are very similar to canonical kinases in their “active/ON” conformation, which offers approaches for inhibitor development. The most promising pseudokinases to target were those that retained their ability to bind nucleotides, as the ATP-binding site is considered the most “druggable” pocket in protein kinases. Pseudokinases functioning by allosteric activation of their dimer partners are currently the predominant target for interventions using small-molecules that act as ATP-competitive inhibitors binding to their pseudoactive site (Dhawan et al., 2016; Kim, 2020; Lu et al., 2020; Mace and Murphy, 2021). Yet, also the scaffolding and switch-like functions of pseudokinases could be addressed in this way, examples being TRIB2 (Bailey et al., 2015; Machado et al., 2020) and MLKL (Hildebrand et al., 2014), respectively.
The major pharmacological challenge of targeting the pseudoactive sites with small molecules is basically the same as for active kinases and, that is, selectivity and effectiveness (Ma et al., 2016). Moreover, many pseudokinases did not retain their nucleotide-binding ability, such as the PEAK family, or even lack a defined binding pocket that could be targeted by ATP-competitive molecules. Therefore, other binding surfaces outside the kinase-domain are searched for (Rübbelke et al., 2021), and novel therapeutic strategies, such as ProTACs or hydrophobic tagging (HyT) that induce degradation of specific proteins are also being pursued (Kung and Jura, 2019).
The analysis of pseudokinases for biomarker validation and drug development is greatly aided by the use of databases that allow the review of the pathophysiological effects of somatic mutations. For example, the ProKinO ontology resource (Gosal et al., 2011; McSkimming et al., 2015) provides a downloadable list of annotated pseudokinase domains and sequences. Somatic mutations of pseudokinases in cancer are available from the KinaseMD and COSMIC databases (Forbes et al., 2010; Forbes et al., 2017; Hu et al., 2021). Despite the tremendous recent developments in multiomics analysis, there is still a dark kinome that deserves further attention which shall open further avenues of therapeutic interventions. Further characterization of the biology of these understudied kinases and pseudokinases are much needed to cater the unmet medical needs in cancers. It is equally important to elucidate the role of these druggable family members in the immune system, as targeting these kinases should not dampen the immune responses for effective antitumor therapy.
KRI, KR, and PV served as lead authors and PV also prepared the figures. PK and KB contributed to the writing of the sections of the manuscript.
KR’s laboratory is supported by grants from CRC 1292, TP05, EKFS (SUNMAPK), and Deutsche Krebs Hilfe (DKH 70113042) and Forschungszentrum fuer Immuntherapie of the University Medical Center Mainz to KR. KR was supported through a Heisenberg professorship of the DFG (RA1739/4-1) and a GFK fellowship.
We thank Daniela Hoeller for valuable inputs and critical reading of this manuscript. We thank members of the KR lab for fruitful discussion on this topic.
KR is the founder and MD of KHR Biotec GmbH. PV is the founder and CEO of Svastia Genetics, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adrain, C., and Freeman, M. (2012). New lives for old: Evolution of pseudoenzyme function illustrated by iRhoms. Nat. Rev. Mol. Cell Biol. 13 (8), 489–498. doi:10.1038/nrm3392
Alves, C. L., Ehmsen, S., Terp, M. G., Portman, N., Tuttolomondo, M., Gammelgaard, O. L., et al. (2021). Co-targeting CDK4/6 and AKT with endocrine therapy prevents progression in CDK4/6 inhibitor and endocrine therapy-resistant breast cancer. Nat. Commun. 12 (1), 5112. doi:10.1038/s41467-021-25422-9
Amin, D. N., Campbell, M. R., and Moasser, M. M. (2010). The role of HER3, the unpretentious member of the HER family, in cancer biology and cancer therapeutics. Seminars Cell & Dev. Biol. 21 (9), 944–950. doi:10.1016/j.semcdb.2010.08.007
Anders, L., Ke, N., Hydbring, P., Choi, Y. J., Widlund, H. R., Chick, J. M., et al. (2011). A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 20 (5), 620–634. doi:10.1016/j.ccr.2011.10.001
Angst, D., Gessier, F., Janser, P., Vulpetti, A., Wälchli, R., Beerli, C., et al. (2020). Discovery of LOU064 (remibrutinib), a potent and highly selective covalent inhibitor of bruton's tyrosine kinase. J. Med. Chem. 63 (10), 5102–5118. doi:10.1021/acs.jmedchem.9b01916
Baar, M. P., Brandt, R. M. C., Putavet, D. A., Klein, J. D. D., Derks, K. W. J., Bourgeois, B. R. M., et al. (2017). Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169 (1), 132–147. e16. doi:10.1016/j.cell.2017.02.031
Bailey, F. P., Byrne, D. P., Oruganty, K., Eyers, C. E., Novotny, C. J., Shokat, K. M., et al. (2015). The Tribbles 2 (TRB2) pseudokinase binds to ATP and autophosphorylates in a metal-independent manner. Biochem. J. 467 (1), 47–62. doi:10.1042/bj20141441
Barvian, M., Boschelli, D. H., Cossrow, J., Dobrusin, E., Fattaey, A., Fritsch, A., et al. (2000). Pyrido[2,3-d]pyrimidin-7-one inhibitors of cyclin-dependent kinases. J. Med. Chem. 43 (24), 4606–4616. doi:10.1021/jm000271k
Beaver, J. A., Amiri-Kordestani, L., Charlab, R., Chen, W., Palmby, T., Tilley, A., et al. (2015). FDA approval: Palbociclib for the treatment of postmenopausal patients with estrogen receptor-positive, HER2-negative metastatic breast cancer. Clin. Cancer Res. 21 (21), 4760–4766. doi:10.1158/1078-0432.ccr-15-1185
Beetham, H., Griffith, B. G. C., Murina, O., Loftus, A. E. P., Parry, D. A., Temps, C., et al. (2022). Loss of integrin-linked kinase sensitizes breast cancer to SRC inhibitors. Cancer Res. 82 (4), 632–647. doi:10.1158/0008-5472.can-21-0373
Braig, M., Lee, S., Loddenkemper, C., Rudolph, C., Peters, A. H. F. M., Schlegelberger, B., et al. (2005). Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436 (7051), 660–665. doi:10.1038/nature03841
Brennan, D. F., Dar, A. C., Hertz, N. T., Chao, W. C. H., Burlingame, A. L., Shokat, K. M., et al. (2011). A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature 472 (7343), 366–369. doi:10.1038/nature09860
Burnett, G., and Kennedy, E. P. (1954). The enzymatic phosphorylation of proteins. J. Biol. Chem. 211 (2), 969–980. doi:10.1016/s0021-9258(18)71184-8
Cai, Z., Jitkaew, S., Zhao, J., Chiang, H.-C., Choksi, S., Liu, J., et al. (2014). Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 16 (1), 55–65. doi:10.1038/ncb2883
Cargnello, M., and Roux, P. P. (2011). Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75 (1), 50–83. doi:10.1128/mmbr.00031-10
Chang, E., Weinstock, C., Zhang, L., Fiero, M. H., Zhao, M., Zahalka, E., et al. (2022). FDA approval summary: Tivozanib for relapsed or refractory renal cell carcinoma. Clin. Cancer Res. 28 (3), 441–445. doi:10.1158/1078-0432.ccr-21-2334
Chellappan, S. P., Hiebert, S., Mudryj, M., Horowitz, J. M., and Nevins, J. R. (1991). The E2F transcription factor is a cellular target for the RB protein. Cell 65 (6), 1053–1061. doi:10.1016/0092-8674(91)90557-f
Chen, J., Fujii, K., Zhang, L., Roberts, T., and Fu, H. (2001). Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proc. Natl. Acad. Sci. U.S.A. 98 (14), 7783–7788. doi:10.1073/pnas.141224398
Chen, S.-H., Gong, X., Zhang, Y., Van Horn, R. D., Yin, T., Huber, L., et al. (2018). RAF inhibitor LY3009120 sensitizes RAS or BRAF mutant cancer to CDK4/6 inhibition by abemaciclib via superior inhibition of phospho-RB and suppression of cyclin D1. Oncogene 37 (6), 821–832. doi:10.1038/onc.2017.384
Chen, X., Li, W., Ren, J., Huang, D., He, W.-t., Song, Y., et al. (2014). Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 24 (1), 105–121. doi:10.1038/cr.2013.171
Cheng, M., Sexl, V., Sherr, C. J., and Roussel, M. F. (1998). Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1). Proc. Natl. Acad. Sci. U.S.A. 95 (3), 1091–1096. doi:10.1073/pnas.95.3.1091
Chien, Y., Scuoppo, C., Wang, X., Fang, X., Balgley, B., Bolden, J. E., et al. (2011). Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 25 (20), 2125–2136. doi:10.1101/gad.17276711
Chittaranjan, S., Bortnik, S., Dragowska, W. H., Xu, J., Abeysundara, N., Leung, A., et al. (2014). Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin. Cancer Res. 20 (12), 3159–3173. doi:10.1158/1078-0432.ccr-13-2060
Cho, Y., Challa, S., Moquin, D., Genga, R., Ray, T. D., Guildford, M., et al. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137 (6), 1112–1123. doi:10.1016/j.cell.2009.05.037
Cohen, P., Cross, D., and Jänne, P. A. (2021). Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 20 (7), 551–569. doi:10.1038/s41573-021-00195-4
Cohen, P. (2002). Protein kinases - The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 1 (4), 309–315. doi:10.1038/nrd773
Collett, M. S., and Erikson, R. L. (1978). Protein kinase activity associated with the avian sarcoma virus src gene product. Proc. Natl. Acad. Sci. U.S.A. 75 (4), 2021–2024. doi:10.1073/pnas.75.4.2021
Correction (2016). Correction: Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 76 (19), 5907. doi:10.1158/0008-5472.CAN-16-1853
Cowan-Jacob, S. W. (2006). Structural biology of protein tyrosine kinases. Cell. Mol. Life Sci. 63 (22), 2608–2625. doi:10.1007/s00018-006-6202-8
Dean, J. L., McClendon, A. K., Hickey, T. E., Butler, L. M., Tilley, W. D., Witkiewicz, A. K., et al. (2012). Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 11 (14), 2756–2761. doi:10.4161/cc.21195
Dean, J. L., Thangavel, C., McClendon, A. K., Reed, C. A., and Knudsen, E. S. (2010). Therapeutic CDK4/6 inhibition in breast cancer: Key mechanisms of response and failure. Oncogene 29 (28), 4018–4032. doi:10.1038/onc.2010.154
Degirmenci, U., Wang, M., and Hu, J. (2020). Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells 9 (1), 198. doi:10.3390/cells9010198
Deng, J., Wang, E. S., Jenkins, R. W., Li, S., Dries, R., Yates, K., et al. (2018). CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 8 (2), 216–233. doi:10.1158/2159-8290.cd-17-0915
Desbois-Mouthon, C., Baron, A., Blivet-Van Eggelpoël, M.-J., Fartoux, L., Venot, C., Bladt, F., et al. (2009). Insulin-like growth factor-1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/AKT signaling pathway: Rational basis for cotargeting insulin-like growth factor-1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin. Cancer Res. 15 (17), 5445–5456. doi:10.1158/1078-0432.ccr-08-2980
Dhawan, N. S., Scopton, A. P., and Dar, A. C. (2016). Small molecule stabilization of the KSR inactive state antagonizes oncogenic Ras signalling. Nature 537 (7618), 112–116. doi:10.1038/nature19327
Di Paolo, J. A., Huang, T., Balazs, M., Barbosa, J., Barck, K. H., Bravo, B. J., et al. (2011). Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat. Chem. Biol. 7 (1), 41–50. doi:10.1038/nchembio.481
Durrant, D. E., and Morrison, D. K. (2018). Targeting the Raf kinases in human cancer: The Raf dimer dilemma. Br. J. Cancer 118 (1), 3–8. doi:10.1038/bjc.2017.399
Ehrenreiter, K., Piazzolla, D., Velamoor, V., Sobczak, I., Small, J. V., Takeda, J., et al. (2005). Raf-1 regulates Rho signaling and cell migration. J. Cell Biol. 168 (6), 955–964. doi:10.1083/jcb.200409162
Engelman, J. A., Zejnullahu, K., Mitsudomi, T., Song, Y., Hyland, C., Park, J. O., et al. (2007). MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316 (5827), 1039–1043. doi:10.1126/science.1141478
Erdogan, F., Radu, T. B., Orlova, A., Qadree, A. K., Araujo, E. D., Israelian, J., et al. (2022). JAK-STAT core cancer pathway: An integrative cancer interactome analysis. J. Cell. Mol. Medi 26 (7), 2049–2062. doi:10.1111/jcmm.17228
Erjala, K., Sundvall, M., Junttila, T. T., Zhang, N., Savisalo, M., Mali, P., et al. (2006). Signaling via ErbB2 and ErbB3 associates with resistance and epidermal growth factor receptor (EGFR) amplification with sensitivity to EGFR inhibitor gefitinib in head and neck squamous cell carcinoma cells. Clin. Cancer Res. 12 (13), 4103–4111. doi:10.1158/1078-0432.ccr-05-2404
Eyers, P. A., Keeshan, K., and Kannan, N. (2017). Tribbles in the 21st century: The evolving roles of tribbles pseudokinases in biology and disease. Trends Cell Biol. 27 (4), 284–298. doi:10.1016/j.tcb.2016.11.002
Fabbro, D., Cowan-Jacob, S. W., and Moebitz, H. (2015). Ten things you should know about protein kinases: IUPHAR review 14. Br. J. Pharmacol. 172 (11), 2675–2700. doi:10.1111/bph.13096
Finn, R. S., Liu, Y., Zhu, Z., Martin, M., Rugo, H. S., Diéras, V., et al. (2020). Biomarker analyses of response to cyclin-dependent kinase 4/6 inhibition and endocrine therapy in women with treatment-naïve metastatic breast cancer. Clin. Cancer Res. 26 (1), 110–121. doi:10.1158/1078-0432.ccr-19-0751
Forbes, S. A., Tang, G., Bindal, N., Bamford, S., Dawson, E., Cole, C., et al. (2010). COSMIC (the catalogue of somatic mutations in cancer): A resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 38, D652–D657. doi:10.1093/nar/gkp995
Forbes, S. A., Beare, D., Boutselakis, H., Bamford, S., Bindal, N., Tate, J., et al. (2017). Cosmic: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 45 (D1), D777–D783. doi:10.1093/nar/gkw1121
Formisano, L., Lu, Y., Servetto, A., Hanker, A. B., Jansen, V. M., Bauer, J. A., et al. (2019). Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun. 10 (1), 1373. doi:10.1038/s41467-019-09068-2
Fry, D. W., Harvey, P. J., Keller, P. R., Elliott, W. L., Meade, M., Trachet, E., et al. (2004). Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 3 (11), 1427–1438. doi:10.1158/1535-7163.1427.3.11
Fukuda, K., Gupta, S., Chen, K., Wu, C., and Qin, J. (2009). The pseudoactive site of ILK is essential for its binding to α-parvin and localization to focal adhesions. Mol. Cell 36 (5), 819–830. doi:10.1016/j.molcel.2009.11.028
Gabizon, R., and London, N. (2020). A fast and clean BTK inhibitor. J. Med. Chem. 63 (10), 5100–5101. doi:10.1021/acs.jmedchem.0c00597
Ghatak, S., Morgner, J., and Wickström, S. A. (2013). Ilk: A pseudokinase with a unique function in the integrin-actin linkage. Biochem. Soc. Trans. 41 (4), 995–1001. doi:10.1042/bst20130062
Glassman, C. R., Tsutsumi, N., Saxton, R. A., Lupardus, P. J., Jude, K. M., and Garcia, K. C. (2022). Structure of a Janus kinase cytokine receptor complex reveals the basis for dimeric activation. Science 376 (6589), 163–169. doi:10.1126/science.abn8933
Goel, S., Wang, Q., Watt, A. C., Tolaney, S. M., Dillon, D. A., Li, W., et al. (2016). Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell 29 (3), 255–269. doi:10.1016/j.ccell.2016.02.006
Gong, X., Litchfield, L. M., Webster, Y., Chio, L.-C., Wong, S. S., Stewart, T. R., et al. (2017). Genomic aberrations that activate D-type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor abemaciclib. Cancer Cell 32 (6), 761–776. doi:10.1016/j.ccell.2017.11.006
Gong, Y., Fan, Z., Luo, G., Yang, C., Huang, Q., Fan, K., et al. (2019). The role of necroptosis in cancer biology and therapy. Mol. Cancer 18 (1), 100. doi:10.1186/s12943-019-1029-8
Gosal, G., Kochut, K. J., and Kannan, N. (2011). ProKinO: An ontology for integrative analysis of protein kinases in cancer. PLoS One 6 (12), e28782. doi:10.1371/journal.pone.0028782
Guinney, J., Dienstmann, R., Wang, X., de Reyniès, A., Schlicker, A., Soneson, C., et al. (2015). The consensus molecular subtypes of colorectal cancer. Nat. Med. 21 (11), 1350–1356. doi:10.1038/nm.3967
Guo, Y., Liu, Y., Hu, N., Yu, D., Zhou, C., Shi, G., et al. (2019). Discovery of zanubrutinib (BGB-3111), a novel, potent, and selective covalent inhibitor of bruton's tyrosine kinase. J. Med. Chem. 62 (17), 7923–7940. doi:10.1021/acs.jmedchem.9b00687
Ha, B. H., and Boggon, T. J. (2018). The crystal structure of pseudokinase PEAK1 (Sugen kinase 269) reveals an unusual catalytic cleft and a novel mode of kinase fold dimerization. J. Biol. Chem. 293 (5), 1642–1650. doi:10.1074/jbc.ra117.000751
Haan, C., Behrmann, I., and Haan, S. (2010). Perspectives for the use of structural information and chemical genetics to develop inhibitors of Janus kinases. J. Cell Mol. Med. 14 (3), 504–527. doi:10.1111/j.1582-4934.2010.01018.x
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell 144 (5), 646–674. doi:10.1016/j.cell.2011.02.013
Hannigan, G., Troussard, A. A., and Dedhar, S. (2005). Integrin-linked kinase: A cancer therapeutic target unique among its ILK. Nat. Rev. Cancer 5 (1), 51–63. doi:10.1038/nrc1524
Herrera-Abreu, M. T., Palafox, M., Asghar, U., Rivas, M. A., Cutts, R. J., Garcia-Murillas, I., et al. (2016). Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 76 (8), 2301–2313. doi:10.1158/0008-5472.can-15-0728
Hildebrand, J. M., Tanzer, M. C., Lucet, I. S., Young, S. N., Spall, S. K., Sharma, P., et al. (2014). Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. U.S.A. 111 (42), 15072–15077. doi:10.1073/pnas.1408987111
Hu, J., Stites, E. C., Yu, H., Germino, E. A., Meharena, H. S., Stork, P. J. S., et al. (2013). Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 154 (5), 1036–1046. doi:10.1016/j.cell.2013.07.046
Hu, R., Xu, H., Jia, P., and Zhao, Z. (2021). KinaseMD: Kinase mutations and drug response database. Nucleic Acids Res. 49 (D1), D552–D561. doi:10.1093/nar/gkaa945
Jacobsen, A. V., and Murphy, J. M. (2017). The secret life of kinases: Insights into non-catalytic signalling functions from pseudokinases. Biochem. Soc. Trans. 45 (3), 665–681. doi:10.1042/bst20160331
James, C., Ugo, V., Le Couédic, J.-P., Staerk, J., Delhommeau, F., Lacout, C., et al. (2005). A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434 (7037), 1144–1148. doi:10.1038/nature03546
Jamieson, S. A., Ruan, Z., Burgess, A. E., Curry, J. R., McMillan, H. D., Brewster, J. L., et al. (2018). Substrate binding allosterically relieves autoinhibition of the pseudokinase TRIB1. Sci. Signal 11 (549), eaau0597. doi:10.1126/scisignal.aau0597
Jansen, V. M., Bhola, N. E., Bauer, J. A., Formisano, L., Lee, K.-M., Hutchinson, K. E., et al. (2017). Kinome-wide RNA interference screen reveals a role for PDK1 in acquired resistance to CDK4/6 inhibition in ER-positive breast cancer. Cancer Res. 77 (9), 2488–2499. doi:10.1158/0008-5472.can-16-2653
Jeric, I., Maurer, G., Cavallo, A. L., Raguz, J., Desideri, E., Tarkowski, B., et al. (2016). A cell-autonomous tumour suppressor role of RAF1 in hepatocarcinogenesis. Nat. Commun. 7, 13781. doi:10.1038/ncomms13781
Jura, N., Shan, Y., Cao, X., Shaw, D. E., and Kuriyan, J. (2009). Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc. Natl. Acad. Sci. U.S.A. 106 (51), 21608–21613. doi:10.1073/pnas.0912101106
Khan, S., He, Y., Zhang, X., Yuan, Y., Pu, S., Kong, Q., et al. (2020). PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene 39 (26), 4909–4924. doi:10.1038/s41388-020-1336-y
Khan, Z. M., Real, A. M., Marsiglia, W. M., Chow, A., Duffy, M. E., Yerabolu, J. R., et al. (2020). Structural basis for the action of the drug trametinib at KSR-bound MEK. Nature 588 (7838), 509–514. doi:10.1038/s41586-020-2760-4
Kim, E. S. (2017). Abemaciclib: First global approval. Drugs 77 (18), 2063–2070. doi:10.1007/s40265-017-0840-z
Kim, H.-O. (2020). Development of JAK inhibitors for the treatment of immune-mediated diseases: Kinase-targeted inhibitors and pseudokinase-targeted inhibitors. Arch. Pharm. Res. 43 (11), 1173–1186. doi:10.1007/s12272-020-01282-7
Kim, S., Tiedt, R., Loo, A., Horn, T., Delach, S., Kovats, S., et al. (2018). The potent and selective cyclin-dependent kinases 4 and 6 inhibitor ribociclib (LEE011) is a versatile combination partner in preclinical cancer models. Oncotarget 9 (81), 35226–35240. doi:10.18632/oncotarget.26215
Knudsen, E. S., Pruitt, S. C., Hershberger, P. A., Witkiewicz, A. K., and Goodrich, D. W. (2019). Cell cycle and beyond: Exploiting new RB1 controlled mechanisms for cancer therapy. Trends Cancer 5 (5), 308–324. doi:10.1016/j.trecan.2019.03.005
Kornev, A. P., Haste, N. M., Taylor, S. S., and Ten Eyck, L. F. (2006). Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. U.S.A. 103 (47), 17783–17788. doi:10.1073/pnas.0607656103
Kung, J. E., and Jura, N. (2019). Prospects for pharmacological targeting of pseudokinases. Nat. Rev. Drug Discov. 18 (7), 501–526. doi:10.1038/s41573-019-0018-3
Lavoie, H., Li, J. J., Thevakumaran, N., Therrien, M., and Sicheri, F. (2014). Dimerization-induced allostery in protein kinase regulation. Trends Biochem. Sci. 39 (10), 475–486. doi:10.1016/j.tibs.2014.08.004
Lavoie, H., Sahmi, M., Maisonneuve, P., Marullo, S. A., Thevakumaran, N., Jin, T., et al. (2018). MEK drives BRAF activation through allosteric control of KSR proteins. Nature 554 (7693), 549–553. doi:10.1038/nature25478
Lelliott, E. J., Mangiola, S., Ramsbottom, K. M., Zethoven, M., Lim, L., Lau, P. K. H., et al. (2021). Combined BRAF, MEK, and CDK4/6 inhibition depletes intratumoral immune-potentiating myeloid populations in melanoma. Cancer Immunol. Res. 9 (2), 136–146. doi:10.1158/2326-6066.cir-20-0401
Levine, R. L., Wadleigh, M., Cools, J., Ebert, B. L., Wernig, G., Huntly, B. J. P., et al. (2005). Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7 (4), 387–397. doi:10.1016/j.ccr.2005.03.023
Li, J., McQuade, T., Siemer, A. B., Napetschnig, J., Moriwaki, K., Hsiao, Y.-S., et al. (2012). The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150 (2), 339–350. doi:10.1016/j.cell.2012.06.019
Logan, J. E., Mostofizadeh, N., Desai, A. J., von Euw, E., Conklin, D., Konkankit, V., et al. (2013). PD-0332991, a potent and selective inhibitor of cyclin-dependent kinase 4/6, demonstrates inhibition of proliferation in renal cell carcinoma at nanomolar concentrations and molecular markers predict for sensitivity. Anticancer Res. 33 (8), 2997–3004.
Lozano, J., Xing, R., Cai, Z., Jensen, H. L., Trempus, C., Mark, W., et al. (2003). Deficiency of kinase suppressor of Ras1 prevents oncogenic ras signaling in mice. Cancer Res. 63 (14), 4232–4238.
Lu, X., Smaill, J. B., and Ding, K. (2020). New promise and opportunities for allosteric kinase inhibitors. Angew. Chem. Int. Ed. 59 (33), 13764–13776. doi:10.1002/anie.201914525
Luo, R. X., Postigo, A. A., and Dean, D. C. (1998). Rb interacts with histone deacetylase to repress transcription. Cell 92 (4), 463–473. doi:10.1016/s0092-8674(00)80940-x
Ma, B., Marcotte, D., Paramasivam, M., Michelsen, K., Wang, T., Bertolotti-Ciarlet, A., et al. (2016). ATP-competitive MLKL binders have No functional impact on necroptosis. PLoS One 11 (11), e0165983. doi:10.1371/journal.pone.0165983
Mace, P. D., and Murphy, J. M. (2021). There's more to death than life: Noncatalytic functions in kinase and pseudokinase signaling. J. Biol. Chem. 296, 100705. doi:10.1016/j.jbc.2021.100705
Machado, S., Silva, A., De Sousa-Coelho, A. L., Duarte, I., Grenho, I., Santos, B., et al. (2020). Harmine and piperlongumine revert TRIB2-mediated drug resistance. Cancers (Basel) 12 (12), 3689. doi:10.3390/cancers12123689
Malorni, L., Piazza, S., Ciani, Y., Guarducci, C., Bonechi, M., Biagioni, C., et al. (2016). A gene expression signature of retinoblastoma loss-of-function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget 7 (42), 68012–68022. doi:10.18632/oncotarget.12010
Malumbres, M., and Barbacid, M. (2001). To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 1 (3), 222–231. doi:10.1038/35106065
Manning, G., Whyte, D. B., Martinez, R., Hunter, T., and Sudarsanam, S. (2002). The protein kinase complement of the human genome. Science 298 (5600), 1912–1934. doi:10.1126/science.1075762
Mariaule, G., and Belmont, P. (2014). Cyclin-dependent kinase inhibitors as marketed anticancer drugs: Where are we now? A short survey. Molecules 19 (9), 14366–14382. doi:10.3390/molecules190914366
Markham, A. (2021). Mobocertinib: First approval. Drugs 81 (17), 2069–2074. doi:10.1007/s40265-021-01632-9
Martin, C. A., Cullinane, C., Kirby, L., Abuhammad, S., Lelliott, E. J., Waldeck, K., et al. (2018). Palbociclib synergizes with BRAF and MEK inhibitors in treatment naïve melanoma but not after the development of BRAF inhibitor resistance. Int. J. Cancer 142 (10), 2139–2152. doi:10.1002/ijc.31220
Martin, I. M., Nava, M. M., Wickström, S. A., and Gräter, F. (2022). ATP allosterically stabilizes integrin-linked kinase for efficient force generation. Proc. Natl. Acad. Sci. U. S. A. 119 (11), e2106098119. doi:10.1073/pnas.2106098119
Martin, J., Anamika, K., and Srinivasan, N. (2010). Classification of protein kinases on the basis of both kinase and non-kinase regions. PLoS One 5 (9), e12460. doi:10.1371/journal.pone.0012460
Marzec, M., Kasprzycka, M., Lai, R., Gladden, A. B., Wlodarski, P., Tomczak, E., et al. (2006). Mantle cell lymphoma cells express predominantly cyclin D1a isoform and are highly sensitive to selective inhibition of CDK4 kinase activity. Blood 108 (5), 1744–1750. doi:10.1182/blood-2006-04-016634
Mathieu, L. N., Larkins, E., Akinboro, O., Roy, P., Amatya, A. K., Fiero, M. H., et al. (2022). FDA approval summary: Capmatinib and tepotinib for the treatment of metastatic NSCLC harboring MET exon 14 skipping mutations or alterations. Clin. Cancer Res. 28 (2), 249–254. doi:10.1158/1078-0432.ccr-21-1566
McSkimming, D. I., Dastgheib, S., Talevich, E., Narayanan, A., Katiyar, S., Taylor, S. S., et al. (2015). ProKinO: A unified resource for mining the cancer kinome. Hum. Mutat. 36 (2), 175–186. doi:10.1002/humu.22726
Melnikova, I., and Golden, J. (2004). Targeting protein kinases. Nat. Rev. Drug Discov. 3 (12), 993–994. doi:10.1038/nrd1600
Metz, K. S., Deoudes, E. M., Berginski, M. E., Jimenez-Ruiz, I., Aksoy, B. A., Hammerbacher, J., et al. (2018). Coral: Clear and customizable visualization of human kinome data. Cell Syst. 7 (3), 347–350. doi:10.1016/j.cels.2018.07.001
Miller, T. W., Pérez-Torres, M., Narasanna, A., Guix, M., Stål, O., Pérez-Tenorio, G., et al. (2009). Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 69 (10), 4192–4201. doi:10.1158/0008-5472.can-09-0042
Mooz, J., Riegel, K., Ps, H., Sadanandam, A., Marini, F., Klein, M., et al. (2022). ARAF suppresses ERBB3 expression and metastasis in a subset of lung cancers. Sci. Adv. 8 (11), eabk1538. doi:10.1126/sciadv.abk1538
Morgan, D. O. (1997). Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 13, 261–291. doi:10.1146/annurev.cellbio.13.1.261
Murali, B., Ren, Q., Luo, X., Faget, D. V., Wang, C., Johnson, R. M., et al. (2018). Inhibition of the stromal p38MAPK/MK2 pathway limits breast cancer metastases and chemotherapy-induced bone loss. Cancer Res. 78 (19), 5618–5630. doi:10.1158/0008-5472.can-18-0234
Murphy, J. M., Czabotar, P. E., Hildebrand, J. M., Lucet, I. S., Zhang, J.-G., Alvarez-Diaz, S., et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39 (3), 443–453. doi:10.1016/j.immuni.2013.06.018
Murphy, J. M., Farhan, H., and Eyers, P. A. (2017). Bio-zombie: The rise of pseudoenzymes in biology. Biochem. Soc. Trans. 45 (2), 537–544. doi:10.1042/bst20160400
Murphy, J. M., Nakatani, Y., Jamieson, S. A., Dai, W., Lucet, I. S., and Mace, P. D. (2015). Molecular mechanism of CCAAT-enhancer binding protein recruitment by the TRIB1 pseudokinase. Structure 23 (11), 2111–2121. doi:10.1016/j.str.2015.08.017
Murphy, J. M., Zhang, Q., Young, S. N., Reese, M. L., Bailey, F. P., Eyers, P. A., et al. (2014). A robust methodology to subclassify pseudokinases based on their nucleotide-binding properties. Biochem. J. 457 (2), 323–334. doi:10.1042/bj20131174
Nagar, B., Bornmann, W. G., Pellicena, P., Schindler, T., Veach, D. R., Miller, W. T., et al. (2002). Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 62 (15), 4236–4243.
O'Rourke, R. L., and Daly, R. J. (2018). The pseudokinases SgK269 and SgK223: A novel oncogenic alliance in human cancer. Cell Adhesion Migr. 12 (6), 524–528. doi:10.1080/19336918.2017.1394570
Otto, T., and Sicinski, P. (2017). Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 17 (2), 93–115. doi:10.1038/nrc.2016.138
Patel, O., Griffin, M. D. W., Panjikar, S., Dai, W., Ma, X., Chan, H., et al. (2017). Structure of SgK223 pseudokinase reveals novel mechanisms of homotypic and heterotypic association. Nat. Commun. 8 (1), 1157. doi:10.1038/s41467-017-01279-9
Pek, M., Yatim, S. M. J. M., Chen, Y., Li, J., Gong, M., Jiang, X., et al. (2017). Oncogenic KRAS-associated gene signature defines co-targeting of CDK4/6 and MEK as a viable therapeutic strategy in colorectal cancer. Oncogene 36 (35), 4975–4986. doi:10.1038/onc.2017.120
Petrie, E. J., Sandow, J. J., Jacobsen, A. V., Smith, B. J., Griffin, M. D. W., Lucet, I. S., et al. (2018). Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat. Commun. 9 (1), 2422. doi:10.1038/s41467-018-04714-7
Plotnikov, A., Zehorai, E., Procaccia, S., and Seger, R. (2011). The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochimica Biophysica Acta (BBA) - Mol. Cell Res. 1813 (9), 1619–1633. doi:10.1016/j.bbamcr.2010.12.012
Powell, K., and Prasad, V. (2021). Concerning FDA approval of trilaciclib (Cosela) in extensive-stage small-cell lung cancer. Transl. Oncol. 14 (11), 101206. doi:10.1016/j.tranon.2021.101206
Qin, J., and Wu, C. (2012). Ilk: A pseudokinase in the center stage of cell-matrix adhesion and signaling. Curr. Opin. Cell Biol. 24 (5), 607–613. doi:10.1016/j.ceb.2012.06.003
Rajakulendran, T., Sahmi, M., Lefrançois, M., Sicheri, F., and Therrien, M. (2009). A dimerization-dependent mechanism drives RAF catalytic activation. Nature 461 (7263), 542–545. doi:10.1038/nature08314
Ribeiro, A. J. M., Das, S., Dawson, N., Zaru, R., Orchard, S., Thornton, J. M., et al. (2019). Emerging concepts in pseudoenzyme classification, evolution, and signaling. Sci. Signal 12 (594), eaat9797. doi:10.1126/scisignal.aat9797
Riegel, K., Schloder, J., Sobczak, M., Jonuleit, H., Thiede, B., Schild, H., et al. (2019). RAF kinases are stabilized and required for dendritic cell differentiation and function. Cell Death Differ. 27 (4), 1300–1315. doi:10.1038/s41418-019-0416-4
Roche, S., Lecointre, C., Simon, V., and Labesse, G. (2019). SHEDding light on the role of Pragmin pseudo-kinases in cancer. Am. J. Cancer Res. 9 (2), 449–454.
Rodier, F., Coppé, J.-P., Patil, C. K., Hoeijmakers, W. A. M., Muñoz, D. P., Raza, S. R., et al. (2009). Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11 (8), 973–979. doi:10.1038/ncb1909
Roskoski, R. (2015). A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 100, 1–23. doi:10.1016/j.phrs.2015.07.010
Roskoski, R. (2016). Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 103, 26–48. doi:10.1016/j.phrs.2015.10.021
Roskoski, R. (2022). Properties of FDA-approved small molecule protein kinase inhibitors: A 2022 update. Pharmacol. Res. 175, 106037. doi:10.1016/j.phrs.2021.106037
Rübbelke, M., Fiegen, D., Bauer, M., Binder, F., Hamilton, J., King, J., et al. (2020). Locking mixed-lineage kinase domain-like protein in its auto-inhibited state prevents necroptosis. Proc. Natl. Acad. Sci. U.S.A. 117 (52), 33272–33281. doi:10.1073/pnas.2017406117
Rübbelke, M., Hamilton, J., Binder, F., Bauer, M., King, J., Nar, H., et al. (2021). Discovery and structure-based optimization of fragments binding the mixed lineage kinase domain-like protein executioner domain. J. Med. Chem. 64 (21), 15629–15638. doi:10.1021/acs.jmedchem.1c00686
Saharinen, P., and Silvennoinen, O. (2002). The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J. Biol. Chem. 277 (49), 47954–47963. doi:10.1074/jbc.m205156200
Saharinen, P., Takaluoma, K., and Silvennoinen, O. (2000). Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol. Cell Biol. 20 (10), 3387–3395. doi:10.1128/mcb.20.10.3387-3395.2000
Schaer, D. A., Beckmann, R. P., Dempsey, J. A., Huber, L., Forest, A., Amaladas, N., et al. (2018). The CDK4/6 inhibitor abemaciclib induces a T cell inflamed tumor microenvironment and enhances the efficacy of PD-L1 checkpoint blockade. Cell Rep. 22 (11), 2978–2994. doi:10.1016/j.celrep.2018.02.053
Scheeff, E. D., Eswaran, J., Bunkoczi, G., Knapp, S., and Manning, G. (2009). Structure of the pseudokinase VRK3 reveals a degraded catalytic site, a highly conserved kinase fold, and a putative regulatory binding site. Structure 17 (1), 128–138. doi:10.1016/j.str.2008.10.018
Sergina, N. V., Rausch, M., Wang, D., Blair, J., Hann, B., Shokat, K. M., et al. (2007). Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 445 (7126), 437–441. doi:10.1038/nature05474
Shah, M., Nunes, M. R., and Stearns, V. (2018). CDK4/6 inhibitors: Game changers in the management of hormone receptor-positive advanced breast cancer? Oncol. Willist. Park) 32 (5), 216–222.
Shan, Y., Gnanasambandan, K., Ungureanu, D., Kim, E. T., Hammarén, H., Yamashita, K., et al. (2014). Molecular basis for pseudokinase-dependent autoinhibition of JAK2 tyrosine kinase. Nat. Struct. Mol. Biol. 21 (7), 579–584. doi:10.1038/nsmb.2849
Sherr, C. J., and Roberts, J. M. (2004). Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 18 (22), 2699–2711. doi:10.1101/gad.1256504
Stefely, J. A., Reidenbach, A. G., Ulbrich, A., Oruganty, K., Floyd, B. J., Jochem, A., et al. (2015). Mitochondrial ADCK3 employs an atypical protein kinase-like fold to enable coenzyme Q biosynthesis. Mol. Cell 57 (1), 83–94. doi:10.1016/j.molcel.2014.11.002
Stern, D. F. (2018). Keeping tumors out of the MAPK fitness zone. Cancer Discov. 8 (1), 20–23. doi:10.1158/2159-8290.cd-17-1243
Su, L., Quade, B., Wang, H., Sun, L., Wang, X., and Rizo, J. (2014). A plug release mechanism for membrane permeation by MLKL. Structure 22 (10), 1489–1500. doi:10.1016/j.str.2014.07.014
Su, Z., Yang, Z., Xie, L., DeWitt, J. P., and Chen, Y. (2016). Cancer therapy in the necroptosis era. Cell Death Differ. 23 (5), 748–756. doi:10.1038/cdd.2016.8
Suntsova, M., Gaifullin, N., Allina, D., Reshetun, A., Li, X., Mendeleeva, L., et al. (2019). Atlas of RNA sequencing profiles for normal human tissues. Sci. Data 6 (1), 36. doi:10.1038/s41597-019-0043-4
Syed, Y. Y. (2017). Ribociclib: First global approval. Drugs 77 (7), 799–807. doi:10.1007/s40265-017-0742-0
Tate, S. C., Cai, S., Ajamie, R. T., Burke, T., Beckmann, R. P., Chan, E. M., et al. (2014). Semi-mechanistic pharmacokinetic/pharmacodynamic modeling of the antitumor activity of LY2835219, a new cyclin-dependent kinase 4/6 inhibitor, in mice bearing human tumor xenografts. Clin. Cancer Res. 20 (14), 3763–3774. doi:10.1158/1078-0432.ccr-13-2846
Teh, J. L. F., Cheng, P. F., Purwin, T. J., Nikbakht, N., Patel, P., Chervoneva, I., et al. (2018). In vivo E2F reporting reveals efficacious schedules of MEK1/2-CDK4/6 targeting and mTOR-S6 resistance mechanisms. Cancer Discov. 8 (5), 568–581. doi:10.1158/2159-8290.cd-17-0699
Thangavel, C., Dean, J. L., Ertel, A., Knudsen, K. E., Aldaz, C. M., Witkiewicz, A. K., et al. (2011). Therapeutically activating RB: Reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr. Relat. Cancer 18 (3), 333–345. doi:10.1530/erc-10-0262
Toms, A. V., Deshpande, A., McNally, R., Jeong, Y., Rogers, J. M., Kim, C. U., et al. (2013). Structure of a pseudokinase-domain switch that controls oncogenic activation of Jak kinases. Nat. Struct. Mol. Biol. 20 (10), 1221–1223. doi:10.1038/nsmb.2673
Tsai, J., Lee, J. T., Wang, W., Zhang, J., Cho, H., Mamo, S., et al. (2008). Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. U.S.A. 105 (8), 3041–3046. doi:10.1073/pnas.0711741105
Turner, N. C., Liu, Y., Zhu, Z., Loi, S., Colleoni, M., Loibl, S., et al. (2019). Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor-positive metastatic breast cancer. J. Clin. Oncol. 37 (14), 1169–1178. doi:10.1200/jco.18.00925
Vijayaraghavan, S., Karakas, C., Doostan, I., Chen, X., Bui, T., Yi, M., et al. (2017). CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat. Commun. 8, 15916. doi:10.1038/ncomms15916
Vora, S. R., Juric, D., Kim, N., Mino-Kenudson, M., Huynh, T., Costa, C., et al. (2014). CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 26 (1), 136–149. doi:10.1016/j.ccr.2014.05.020
Walker, A. J., Wedam, S., Amiri-Kordestani, L., Bloomquist, E., Tang, S., Sridhara, R., et al. (2016). FDA approval of palbociclib in combination with fulvestrant for the treatment of hormone receptor-positive, HER2-negative metastatic breast cancer. Clin. Cancer Res. 22 (20), 4968–4972. doi:10.1158/1078-0432.ccr-16-0493
Wang, B., Varela-Eirin, M., Brandenburg, S. M., Hernandez-Segura, A., van Vliet, T., Jongbloed, E. M., et al. (2022). Pharmacological CDK4/6 inhibition reveals a p53-dependent senescent state with restricted toxicity. EMBO J. 41 (6), e108946. doi:10.15252/embj.2021108946
Wang, H., Sun, L., Su, L., Rizo, J., Liu, L., Wang, L.-F., et al. (2014). Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54 (1), 133–146. doi:10.1016/j.molcel.2014.03.003
Wang, R., Zhang, Q., Peng, X., Zhou, C., Zhong, Y., Chen, X., et al. (2016). Stellettin B induces G1 arrest, apoptosis and autophagy in human non-small cell lung cancer A549 cells via blocking PI3K/Akt/mTOR pathway. Sci. Rep. 6, 27071. doi:10.1038/srep27071
Wei, S.-C., Rosenberg, I. M., Cao, Z., Huett, A. S., Xavier, R. J., and Podolsky, D. K. (2012). Tribbles 2 (Trib2) is a novel regulator of toll-like receptor 5 signaling. Inflamm. Bowel Dis. 18 (5), 877–888. doi:10.1002/ibd.22883
Weinberg, R. A. (1995). The retinoblastoma protein and cell cycle control. Cell 81 (3), 323–330. doi:10.1016/0092-8674(95)90385-2
Wu, J., Huang, Z., Ren, J., Zhang, Z., He, P., Li, Y., et al. (2013). Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 23 (8), 994–1006. doi:10.1038/cr.2013.91
Wu, P., Nielsen, T. E., and Clausen, M. H. (2016). Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discov. Today 21 (1), 5–10. doi:10.1016/j.drudis.2015.07.008
Yadav, V., Burke, T. F., Huber, L., Van Horn, R. D., Zhang, Y., Buchanan, S. G., et al. (2014). The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol. Cancer Ther. 13 (10), 2253–2263. doi:10.1158/1535-7163.mct-14-0257
Yang, C., Li, Z., Bhatt, T., Dickler, M., Giri, D., Scaltriti, M., et al. (2017). Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 36 (16), 2255–2264. doi:10.1038/onc.2016.379
Yao, Z., Gao, Y., Su, W., Yaeger, R., Tao, J., Na, N., et al. (2019). RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat. Med. 25 (2), 284–291. doi:10.1038/s41591-018-0274-5
Yao, Z., Murali, B., Ren, Q., Luo, X., Faget, D. V., Cole, T., et al. (2020). Therapy-induced senescence drives bone loss. Cancer Res. 80 (5), 1171–1182. doi:10.1158/0008-5472.can-19-2348
Yoshida, A., Lee, E. K., and Diehl, J. A. (2016). Induction of therapeutic senescence in vemurafenib-resistant melanoma by extended inhibition of CDK4/6. Cancer Res. 76 (10), 2990–3002. doi:10.1158/0008-5472.can-15-2931
Yu, J.-m., Sun, W., Wang, Z.-h., Liang, X., Hua, F., Li, K., et al. (2019). TRIB3 supports breast cancer stemness by suppressing FOXO1 degradation and enhancing SOX2 transcription. Nat. Commun. 10 (1), 5720. doi:10.1038/s41467-019-13700-6
Yuan, Y., Lee, J. S., Yost, S. E., Frankel, P. H., Ruel, C., Egelston, C. A., et al. (2021). Phase I/II trial of palbociclib, pembrolizumab and letrozole in patients with hormone receptor-positive metastatic breast cancer. Eur. J. Cancer 154, 11–20. doi:10.1016/j.ejca.2021.05.035
Zhang, D.-W., Shao, J., Lin, J., Zhang, N., Lu, B.-J., Lin, S.-C., et al. (2009). RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325 (5938), 332–336. doi:10.1126/science.1172308
Zhang, X., Gureasko, J., Shen, K., Cole, P. A., and Kuriyan, J. (2006). An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125 (6), 1137–1149. doi:10.1016/j.cell.2006.05.013
Keywords: kinome, RAF, MAPK, CDK4/6, inhibitors, cancer, molecular switch, allosteric regulation
Citation: Riegel K, Vijayarangakannan P, Kechagioglou P, Bogucka K and Rajalingam K (2022) Recent advances in targeting protein kinases and pseudokinases in cancer biology. Front. Cell Dev. Biol. 10:942500. doi: 10.3389/fcell.2022.942500
Received: 12 May 2022; Accepted: 27 June 2022;
Published: 22 July 2022.
Edited by:
Jiancheng Hu, National Cancer Centre Singapore, SingaporeReviewed by:
Junyu Xiao, Peking University, ChinaCopyright © 2022 Riegel, Vijayarangakannan, Kechagioglou, Bogucka and Rajalingam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Krishnaraj Rajalingam, a3Jpc2huYUB1bmktbWFpbnouZGU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.