
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 25 July 2022
Sec. Cell Death and Survival
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.918943
 Nannan Tan1†
Nannan Tan1† Tianhua Liu2†
Tianhua Liu2† Xiaoping Wang1†
Xiaoping Wang1† Mingyan Shao3Miao Zhang1
Mingyan Shao3Miao Zhang1 Weili Li3Guanjing Ling1
Weili Li3Guanjing Ling1 Jinchi Jiang1Qiyan Wang3Jing Li1
Jinchi Jiang1Qiyan Wang3Jing Li1 Chun Li4*Wei Wang5*
Chun Li4*Wei Wang5* Yong Wang1*
Yong Wang1*Mitophagy plays a vital role in the selective elimination of dysfunctional and unwanted mitochondria. As a receptor of mitophagy, FUN14 domain containing 1 (FUNDC1) is attracting considerably critical attention. FUNDC1 is involved in the mitochondria fission, the clearance of unfolded protein, iron metabolism in mitochondria, and the crosstalk between mitochondria and endoplasmic reticulum besides mitophagy. Studies have demonstrated that FUNDC1 is associated with the progression of ischemic disease, cancer, and metabolic disease. In this review, we systematically examine the recent advancements in FUNDC1 and the implications of this protein in health and disease.
Mitochondria regarded as the direct descendants of a bacterial endosymbiont are symbiotic with the host cell (Gray et al., 1999). To the best of our knowledge, approximately 99% of proteins used for maintaining mitochondrial function are regulated by nuclear genes, while only 13 proteins are coded by mitochondria (Song et al., 2021). Apart from providing energy for the host cell, mitochondria can sequester hazardous substances by creating an isolated space in cells (Song and Dorn, 2015). Furthermore, mitochondria are vulnerable when they participant in generating reactive oxygen species (ROS), iron metabolism, and lipid oxidation. Accumulation of damaged and superfluous mitochondria are detrimental to cells and organs (Liu et al., 2014; Zorov et al., 2014), suggesting that the process of clearance of mitochondria should not be overlooked.
Mitophagy that responsible for the selective elimination of dysfunctional and unwanted mitochondria was observed for the first time under electron microscopy by glucagon-stimulated stem cell activation (Deter and De Duve, 1967; Hirota et al., 2015). The past few years have witnessed an explosion in our understanding of mitophagy, which preserves mitochondrial function and cell homeostasis under diverse (patho-)physiological conditions such as hypoxia, starvation, and exposed cold stimulus (Hirota et al., 2015; Liu et al., 2021a).
The molecular mechanisms of mitophagy include PTEN induced putative kinase 1 (PINK1)-Parkin pathway and receptor-mediated pathway. PINK1-Parkin partnership mainly modulates the turnover of depolarized mitochondria (Springer and Macleod, 2016). When the mitochondrial membrane potential decreases, PINK1 firstly accumulates in the mitochondrial outer membrane, and then recruits Parkin, which is an E3 ubiquitin ligase, to the mitochondrial outer membrane, leading to the ubiquitination of various proteins in the mitochondrial outer membrane (Yoshii et al., 2011). Simultaneously, the autophagy receptor optineurin, which binds to light chain 3 (LC3), is recruited to ubiquitinated mitochondria and located to the outer membrane surface by phosphorylation of TANK-binding kinase 1. And then, the damaged mitochondria are engulfed into autophagy precursors, which are ultimately degraded by the conserved lysosomal pathway (Evans and Holzbaur, 2020).
Mitophagy receptors include BCL2 interacting protein three like (BNIP3L/NIX) and FUNDC1. FUNDC1mainly participates in the regulation of mitochondrial homeostasis under hypoxic (Liu et al., 2012a). Missed or mutated FUNDC1 may promote the progression of human disease. For example, the loss of FUNDC1 leads to mitochondrial fragmentation, and fails to repair infarcted hearts during the differentiation of cardiac progenitor cells (Lampert et al., 2019). Geng Guangfeng and his colleagues found that intervening FUNDC1 could simultaneously improve renal anemia and renal fibrosis (Geng et al., 2021). Moreover, Lei Liu and his colleagues gave a detailed description of the role of FUNDC1-mediated mitophagy in cardiovascular diseases (Liu et al., 2021b). Instances of disease ranging from ischemic disease to cancer, as well as metabolic related disease, could also be influenced by FUNDC1. Thus, this review aims to delineate the timely advancement of FUNDC1 and its role in human disease.
FUNDC1 is one of the paralogous subfamilies of FUN14 domain-containing protein family which is present in eukaryotes, archaea, and bacteria (Wu et al., 2017a). Human FUNDC1 is widely expressed in the body, especially in the heart (Zhang et al., 2017). It contains 155 amino acids and is mainly located in the outer membrane of mitochondria. Furthermore, FUNDC1 has three transmembrane regions, the N-terminal is exposed to the cytoplasm. The exposed part includes a LC3-interacting region (LIR, Y18-E-V-L21) which can bind to LC3 to regulate the occurrence of mitophagy (Liu et al., 2012a; Wu et al., 2017a). Eleven lysine residues sites such as K70 and K119, which are included in the transmembrane region of FUNDC1, can bind to optic atrophy 1 (OPA1) and MARCH5, respectively (Chen et al., 2017a; Wu et al., 2017a).
The mRNA level of FUNDC1 is downregulated under hypoxia condition (Wu et al., 2017a), but the specific transcriptional regulators of FUNDC1 are not deeply understood. As a nuclear transcription factor, nuclear transcription factor 1(NRF1) can participate in the activation of mitochondrial genes and transcription and translation of mtRNA (Carraway et al., 2010). After being activated by the peroxisome proliferator-activated receptor gamma coactivator 1alpha (PGC1α) and acetaldehyde dehydrogenase 2 (ALDH2), NRF1 can directly bind to 5’ promoter of FUNDC1, upregulating the level of FUNDC1, and further promoting mitophagy and mitochondrial biogenesis to maintain the normal function of mitochondria and cells (Li et al., 2020; Liu et al., 2021a). Moreover, FOXO3a that is a member of the fork head box class O (FOXO) family that are widely expressed transcription factors can regulate a variety of cellular physiological processes by targeting effector genes (Liu et al., 2018) and activate FUNDC1 to promote the occurrence of mitophagy in ISO-induced myocardial hypertrophy model (Liu et al., 2021c).
In normal condition, the LIR region of FUNDC1 is phosphorylated at Y18 and S13 sites by Src and CK2 kinase, respectively, to maintain inactivation. However, under long-term hypoxia condition, FUNDC1 is dephosphorylated at Y18 and S13 due to the inactivation of Src and CK2 kinase, leading to the occurrence of mitophagy (Liu et al., 2012a; Chen et al., 2014; Zhou et al., 2018a). MARCH5, an E3 ubiquitin ligase, is an important regulator of FUNDC1-mediated mitophagy. Interaction between MARCH5 and FUNDC1 means a crosstalk between ubiquitin- and mitophagy. Chen et al. (2017a), Chen et al. (2017b) suggested that MARCH5 homo-oligomers were disassembled under the early stage of hypoxia, and then MARCH5 bound to K119 site of FUNDC1, thereby making FUNDC1 ubiquitinated and degraded, which inhibiting mitophagy and avoiding improper clearness of undamaged mitochondria. As a negative factor for FUNDC1, micro-137 can directly target to FUNDC1 and then inhibit mitophagy (Li et al., 2014).
Phosphoglycerate mutase family member 5 (PGAM5) exists in the inner membrane of mitochondria and protects PINK1 from degradation. After being stimulated by carbonyl cyanide m-chlorophenyl hydrazine in models, PINK1 can migrate from the inner membrane to the outer membrane under the protection of PGAM5 and bind with Parkin to complete the subsequent mitophagy process. The loss of PGAM5 affects the occurrence of PINK1-mediated mitophagy (Lu et al., 2014). Similarly, PGAM5 is important to regulate FUNDC1-mediated mitophagy. Under normal condition, BCL2-like 1 (BCL2L1/Bcl-xL) binds to PGAM5 to inhibit the activation of PGAM5. While under hypoxia condition, BCL2L1 is hydrolyzed, and PGAM5 is released. Activated PGAM5 dephosphorylates FUNDC1 at S13 and promotes the interaction between FUNDC1 and LC3, thereby inducing mitophagy (Wu et al., 2014a; Kuang et al., 2016; Biswal et al., 2018). In addition, FUNDC1 is phosphorylated by unc-51 like autophagy activating kinase 1 (ULK1) at S17 site under hypoxic condition (Wu et al., 2014b). Wang Li and his colleagues found that mitophagy that was activated via ULK1-FUNDC1 pathway could prevent nerve cells from apoptosis (Wang et al., 2018). Specific details of factors are shown in Figure 1.
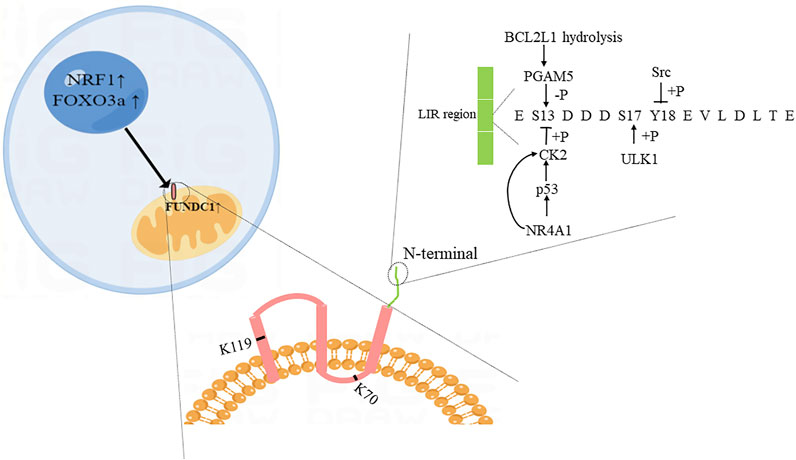
FIGURE 1. Diagram of possible structure and regulatory proteins of FUNDC1. The illustration was created by Figdraw (www.Figdraw.com).
Mitochondrial fission is strictly monitored in cells that can produce healthy mitochondria and induce mitophagy through the ROS produced by the division (Archer, 2013; Song and Dorn, 2015). It also participates in cytochrome C-mediated apoptosis pathologically (Cassidy-Stone et al., 2008). Dynamin-related protein 1 (DRP1, also known as DNM1L), which is recruited to the outer membrane of mitochondria, is essential to drive mitochondrial fission (Cerveny et al., 2007). In addition, as a dynamin-like guanosine triphosphatase, OPA1, which can be found as long-OPA1(L-OPA1) and short-OPA1(S-OPA1) forms, is located in the inner membrane of mitochondria and is vital for balancing mitochondrial fusion and fission (Wai et al., 2015). Seminal findings from Chen Ming and colleagues depicted a new molecular mechanism of mitochondria fission, which depended on the interaction between FUNDC1 and DNM1L, OPA1. Under normal condition, FUNDC1 interacted with OPA1 at the site K70. But OPA1 could be cleaved or degraded by yeast mitochondrial escape 1-like (YME1L) and OMA1 under mitochondrial stress condition. Then FUNDC1 and OPA1 separated, dephosphorylated FUNDC1 recruited DNM1L toward mitochondria, thus promoting mitochondrial fission (Chen et al., 2016).
Mitochondria-associated ER membranes (MAMs) are critical for the occurrence of mitochondrial fission. Under hypoxia condition, FUNDC1 is accumulated at MAMs by interacting with calnexin. As mitophagy proceeds, the association between FUNDC1 and calnexin becomes attenuated, and FUNDC1 recruits DRP1 toward mitochondria to complete mitochondrial fission (Wu et al., 2016a; Wu et al., 2016b). Mitochondrial fission and mitophagy are closely relevant. They seemed to be a continuous process, just as van der Bliek AM described in his paper (van der Bliek, 2016). Ubiquitin-specific peptidase 19 (USP19), identified as a deubiquitylase, is also an important regulator involved in MAMs-mediated mitochondrial fission (Volkmar et al., 2016). Under hypoxia condition, USP19 is recruited to ER, which is mainly responsible for removing the ubiquitin chain from FUNDC1. Then deubiquitinated FUNDC1 is stabilized at MAMs and recruits DRP1 toward the contact site to finish mitochondria fission (Chai et al., 2021). Under hypoxia condition, why does FUNDC1 interact with calnexin and then dissociate? Does this process recruit USP19 to MAMs or recruit DRP1 to mitochondria to interact with FUNDC1? Is USP19 able to interact with calnexin? How do MARCH5 and USP19 coordinate the ubiquitinated fate of FUNDC1? There are still no clear answers to these questions. Mitochondrial fission is vital for mitophagy. In order to get a better understanding of mitophagy, it is necessary to conduct in-depth research on the upstream (Chakrabarti and Higgs, 2021). Therefore, the molecular mechanism of FUNDC1-mediated mitochondrial fission and mitophagy is worthy of further study.
Iron is an essential component to maintain cellular metabolism and is crucial for the normal function of mitochondria because it participates in the electron transport chain (Huang et al., 2011; Dietz et al., 2021). Moreover, mitochondria as machineries where iron-sulfur clusters (ISCs) are assembled and exported are necessary for modulating iron metabolism in cells (Paul and Lill, 2015). Relative drugs are being exploited for treating human disease. For example, mitochondrially targeted deferoxamine (mitoDFO) impairs mitochondrial respiration and biogenesis of [Fe-S] clusters/heme in cancer cells, thereby suppressing the proliferation and migration of cancer cells (Sandoval-Acuña et al., 2021).
As a fundamental process in the cell, iron metabolism is closely related to mitophagy. Chiang et al. (2021) suggested that in the frataxin knockout mouse, mitophagy was enhanced when iron was accumulated in mitochondria, resulting in the dysfunction of mitochondria. But in pathogenic yeast Candida glabrata cells, mitophagy is also enhanced under iron-depletion condition (Nagi et al., 2016). The result of Schiavi Alfonso and his colleagues’ study was similar that iron depletion could induce mitophagy and then extended C. elegans lifespan (Schiavi et al., 2015). The studies mentioned above suggest that either iron deficiency or iron overload has an impact on mitophagy. Wu Hao and his colleagues found that if the machinery of ISCs was disrupted, iron regulatory proteins 1 (IRP1) would suppress the translation of Bcl-xL, causing PGAM5 separate from Bcl-xL, finally initiating FUNDC1-mediated mitophagy (Wu et al., 2020). Pei Zhaohui and his colleagues discovered that under short-term high fat intake, the deficiency of FUNDC1 induced metabolic and cardiac dysfunction through ferroptosis regulated by specificity protein 1 (SP1) -acyl-CoA synthetase long-chain family member 4 (ACLS4) axis (Pei et al., 2021). In short, the relationship between FUNDC1 and iron metabolism is close in cells.
Damaged or excess proteins can promote proteotoxic effects if they are not removed in time (Song, 2019). FUNDC1-mediated mitophagy is involved in the clearance of damaged proteins collaborating with ubiquitination. Under stress condition, FUNDC1 interacts with HSC70 belonging to the HSP70 family, which is a member of the heat shock protein (HSP)family (Hartl and Hayer-Hartl, 2002; Liu et al., 2012b), transporting misfolded proteins to mitochondria matrix, and the proteins are degraded in the presence of LonP1, which is a AAA protease. The formation of mitochondrion-associated protein aggregates (MAPAs) is triggered when the misfolded proteins are over-accumulation in the mitochondrial matrix. Then FUNDC1-FIS1-mediated mitophagy is activated to promote the clearance of unfolded proteins. If massive unfolded proteins are not timely disposed, cells will finally become senescent. Li et al. (2019a), Li et al. (2019b) pointed out that FUNDC1-LonP1 axis also played an important role in maintaining mitochondrial reprogramming and cellular plasticity in cancer cells (Cappellini et al., 2020). Wang Yue’s study confirmed that the coordination between FUNDC1-mediated mitophagy and unfolded proteins could attenuate inflammation-mediated myocardial injury in septic cardiomyopathy (Wang et al., 2021a).
The mitochondria generally maintain a relationship with other organelles in the cells. Such interactions could offer some forms of benefit. FUNDC1 plays a key role in the crosstalk between mitochondria and other organelles.
The most typical membrane contact site is the interaction between mitochondria and ER, known as mitochondria-associated ER membranes (MAMs) or mitochondria-ER contacts (MERCs), it maintains the homeostasis of Ca2+ level and lipid metabolism in two organelles (Rowland and Voeltz, 2012; Giacomello and Pellegrini, 2016). FUNDC1-mediated MAMs are involved in the transportation of calcium. Calcium is transported to mitochondria from ER through the inositol 1,4,5-triphosphate receptor (IP3R)-glucose regulated protein 75 (Grp75)-voltage-dependent anion channel (VDAC) complex, FUNDC1 which interacts with IP3R2 is responsible for facilitating MAMs stabilization. The cAMP-responsive element binding protein (CREB) is phosphorylated at S133 site due to mitochondrial retrograde calcium signal, which inducing CREB nuclear translocation, and then the transcription of fission one protein (FIS1) is activated. Subsequently, enhanced FIS1 promotes both mitochondrial fission and mitophagy (Wu et al., 2017b; Muñoz and Zorzano, 2017), as shown in Figure 2. Moreover, it has been suggested that FUNDC1 may maintain mitochondrial calcium homeostasis via the interaction between FUNDC1 and receptor subunit of human SCF (SKP1/cullin/F-box protein) ubiquitin ligase complex (FBXL2) (Ren et al., 2020).
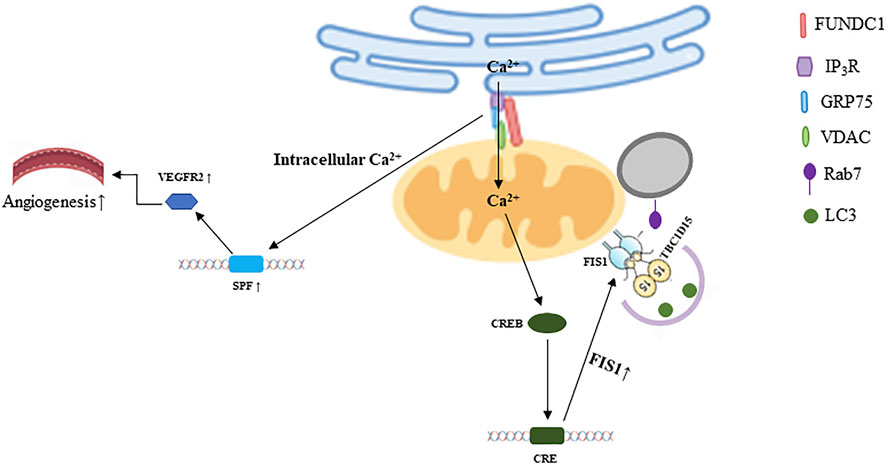
FIGURE 2. FUNDC1 mediated crosstalk among mitochondria and other organelles. The illustration was created by Figdraw (www.Figdraw.com).
FUNDC1-dependent MAMs are involved in the development of multiple diseases. For example, diabetes mellitus inhibits the activity of AMP-activated protein kinase (AMPK), which promotes the formation of FUNDC1-mediated MAMs, and then inducing mitochondrial dysfunction, leading to cardiomyopathy (Wu et al., 2019a). AMPK-FUNDC1-MAMs axis is also involved in the development of diabetic nephropathy (DN) (Wei et al., 2020). Excessive Ca2+ releasing from ER into mitochondria leads to cardiac damage and heart failure when FUNDC1 interacts with IP3R2 (63). Moreover, if the formation of FUNDC1-mediated MAMs is disrupted under angiogenic condition, the level of serum response factor (SRF) and vascular endothelial growth factor 2 (VEGFR2) will be downregulated due to the dyshomeostasis of Ca2+, which reducing the VEGF-induced angiogenesis (Wang et al., 2021b), as shown in Figure 2. Some studies documented that FUNDC1-mediated mitophagy could improve cerebral ischemia via inhibiting NLRP3 inflammasome. And the level of NLRP3 would be increased when FUNDC1 was knocked out, then inflammatory response was exacerbated, lung cells were further injured (Pan et al., 2021; Zheng et al., 2021). Furthermore, the activation of NLRP3 inflammasome compromises the recovery of HFD-induced vascular impairment (Elshaer et al., 2017; Mohamed et al., 2020). Considering that FUNDC1 can regulate NLRP3, and FUNDC1-mediated MAMs can promote angiogenesis, whether there is a link between FUNDC1-mediated MAMs and NLRP3 is worthy of mention. MAMs may be potential drug targets. For example, capsaicin improves DN via activating transient receptor potential cation channel subfamily V member 1 (TRPV1). The molecular mechanism is that AMPK is activated by transient Ca2+ level due to the activation of TRPV1, causing the reduction of FUNDC1, which downregulates the formation of MAMs (Wei et al., 2020).
Mitochondria and lysosome are important organelles to maintain cell homeostasis. Their dysfunction or interaction disorders are related to neurodegeneration and other human diseases (Audano et al., 2018; English and Hughes, 2019). The role of mitochondria-lysosome contacts in the function of mitochondria and lysosome has received increased attention in recent years. Apart from regulating mitochondrial fission, Wong et al. (2018) discovered that the morphology of lysosome would be impaired if the mitochondria-lysosome contact could not untether.
Rab7 is a small G protein belonging to the Rab family and is present on the lysosome, ER, and mitochondrial membranes (Jimenez-Orgaz et al., 2018). It is mainly involved in membrane trafficking (Progida et al., 2010). Furthermore, Rab7 is involved in regulating mitochondria-lysosome contact (Wong et al., 2018). Activated Rab7 maintains mitochondrial-lysosome contact, while inactivated Rab7 makes mitochondrial-lysosome contact dissociated. The activation of Rab7 is regulated by TBC1D15, which belongs to the TBC (Tre-2/Bub2/Cdc16) domain family and function as a GTPase-activating protein (GAP) for Rab GTPases (Chen et al., 2017c; Wong et al., 2018). TBC1D15 could be recruited to mitochondria by FIS1 (Onoue et al., 2013). And then, TBC1D15 makes Rab7-GTP translate to Rab7-GDP, which cannot bind to the lysosome, ultimately promoting the dissociation between mitochondria and lysosome (Wong et al., 2018). Autophagy is inhibited in a variety of diseases, but the formation of autophagosomes is normal or even enhanced. If there are not enough or normal functional lysosome to receive autophagosomes, autophagosomes will not be degraded and may lead to cytotoxicity (Cuervo, 2011). In view of that, Yu Wenjun and his colleagues proved that TBC1D15/FIS1/Rab7 pathway exerted protective effects on the function and morphology of infarct heart via untethering mitochondria-lysosome contact (Yu et al., 2020).
TBC1D15 could modulate autophagosome biogenesis via interacting with LC3/GABARAP family members (Yamano et al., 2014). And FIS1 regulates mitochondrial fission by recruiting lysosome toward mitochondria and is primarily involved in the clearance of damaged mitochondria (Kleele et al., 2021). Moreover, FUNDC1 can sense the inner mitochondrial stress via interacting with OPA1(38) and increase the level of Ca2+ in mitochondria, which is a sensor that promotes transcription of FIS1(64). Whether these proteins can coordinate fission and mitophagy and whether these processes could happen continuously, the mechanisms in integrating these processes are unclear. Thus, further in-depth studies are needed to explore whether the FUNDC1/FIS1/TBC1D15/Rab7 pathway participates in regulating mitochondria-lysosome contact, as shown in Figure 2.
Ischemic disease, which has high disability and mortality, especially heart and brain ischemia, occurs when blood vessels are blocked (Zhang et al., 2021). Although reperfusion is necessary to recover cell function, it can also provoke activation of deleterious processes to impair cell function (Kalogeris et al., 2016). Numerous studies demonstrated that mitophagy played a vital role in the physiopathology and treatment of I/R injury. The details are shown in Figure 3.
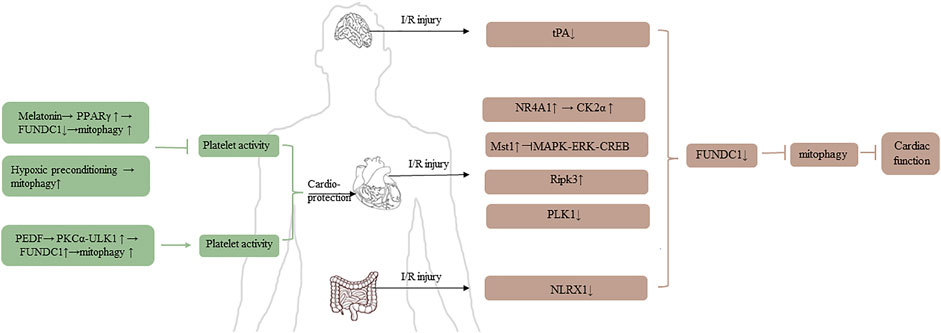
FIGURE 3. The role of FUNDC1 in I/R injury.
Platelet reactivity is related to mitochondrial mass and function, and it is crucial for I/R injury (Zhang et al., 2017; Davizon-Castillo et al., 2019). There are two studies to clarify the relationship between mitochondria and platelet. Zhou et al. (2017a) pointed out that the level of peroxisome proliferator-activated receptor γ (PPARγ) was downregulated in platelets under I/R injury condition, and then FUDNC1-mediated mitophagy was activated. Subsequently, the platelet was activated, which promoting the block of microcirculation, finally causing cardiomyocyte death, excessive inflammation response, and heart dysfunction. According to the report, melatonin can reverse heart injury via promoting the level of PPARγ, PPARγ-FUNDC1-mitophagy pathway could be an essential strategy for treating I/R injury. Coincidentally, Zhang et al. (2016), Zhang et al. (2017) used conditional knockout mice specifically lacking FUNDC1 in their platelets and found that hypoxic preconditioning had a cardioprotective effect because hypoxic induced mitophagy causing extensive mitochondrial degradation in platelets, and then compromising the activation of platelet, reducing the I/R injury. It seems that mitophagy could upregulate or downregulate the activity of platelet under different conditions. It is unclear whether the contradictory results are due to inconsistent intervention methods in the two experiments or whether they suggest other pathways other than FUNDC1- mediated mitophagy affecting platelet activity.
From the perspective of cardiac ischemia studies, it is generally believed that the regulation of mitophagy may be a potential therapy for cardioprotective effect. Zhou Hao and his colleagues’ study revolved around nuclear receptor subfamily four group A member 1 (NR4A1) and FUNDC1. Increased NR4A1 was observed in cardiac I/R model, upregulated NR4A1 induced FUNDC1 phosphorylation via activating CK2α, increased CK2α was harmful for mitochondria, and then mitophagy was suppressed, ultimately impairing cardiac function. Moreover, Zhou et al., (2018a), Zhou et al. (2018b) explored the relationship between NR4A1 and FUNDC1 in alcohol-related liver disease (ARLD). They revealed that NR4A1 was increased after alcohol treatment, upregulated NR4A1 could promote the disassociation of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and Ku80. Subsequently, disassociated DNA-PKcs bound to p53, activated p53 promoted the upregulation of CK2. Same results as the cardiac I/R model, FUNDC1-mediated mitophagy was suppressed, ultimately leading to the progression of ARLD (Zhou et al., 2019).
Mammalian STE20-like kinase 1 (Mst1) which is a negative regulator of cardiac function is upregulated in acute I/R injury model, inhibits MAPK-ERK-CREB pathway, consequently suppresses the level of FUNDC1, thus FUNDC1-mediated mitophagy is defective (Yu et al., 2019). Receptor-interacting protein kinase 3 (Ripk3) is another factor to damage cardiac function. Upregulated Ripk3 can interact with FUNDC1, inhibiting mitophagy, leading to caspase9-mediated apoptosis (Zhou et al., 2017b). The level of polo-like kinase 1 (PLK1) is downregulated in cardiac I/R injury model, but overexpression of PLK1 can relieve myocardial damage via p-AMPK/FUNDC1-mediated mitophagy (Mao et al., 2021). Pigment epithelial-derived factor is a protective factor for acute myocardial infarction. Under hypoxia condition, it can interact with PEDF-R, and then active PKC-α through increasing the level of palmitic acid and diacylglycerol. The activated PKC-α can promote FUNDC1-mediated mitophagy, which depends on ULK1 pathway, thus protecting cardiomyocytes (Li et al., 2018). In addition to myocardial ischemia, ULK1-FUNDC1-mediated mitophagy is also the mechanism of renoprotection using ischemia preconditioning in ischemic acute kidney injury (Wang et al., 2020).
mTORC1-ULK1-FUNDC1-mitophagy is an important axis to reduce ischemic injury. Electroacupuncture can not only attenuate myocardial ischemia-reperfusion injury but also cerebral ischemia-reperfusion injury via mitophagy inhibited by mTORC1-ULK1-FUNDC1 pathway (Mao et al., 2020; Xiao et al., 2020; Tian et al., 2021). By contrast, Cai Ying and his colleagues investigated that tissue-type plasminogen activator (tPA) was decreased in cerebral I/R model, causing FUNDC1 degraded and mitophagy inhibited, while exogenous tPA, which could induce FUNDC1-mediated mitophagy via phosphorylating AMPK, could reduce I/R injury (Cai et al., 2021).
Furthermore, FUNDC1-mediated mitophagy is also an underlying physiopathology mechanism in intestinal I/R injury. Li Shaoqin revealed that NOD-like receptor X1 (NLRX1) was decreased in intestinal I/R model, and the phosphorylation of FUNDC1 was enhanced. Thus, FUNDC1 could not interact with non-neuronal SNAP25-like protein homolog one and 2 (NIPSNAP one and 2), resulting in the suppression of mitophagy, ultimately contributing to intestinal morphological damage (Li et al., 2021).
High level of FUNDC1 promotes oxidative bioenergetics and supports tumor cell proliferation and growth (Cappellini et al., 2020). It might be an independent prognostic factor for overall survival and disease-free survival in patients with cervical cancer (Hou et al., 2017). Hydrogen peroxide is a regulator to increase the level of FUNDC1 in laryngeal cancer cells, mainly through activating ERK1/2 (Hui et al., 2019). Furthermore, FUNDC1-involved MAMs can promote calcium transport to cytoplasm, making nuclear factor of activated T cells 1 (NFATC1) dephosphorylated, and then Bmi1 polycomb ring finger oncogene (BMI1) is upregulated, thus promoting the progression of breast cancer (Wu et al., 2019b). FUNDC1 promotes the growth of hepatocellular carcinoma (HCC) tissues in the late stage of the disease (Li et al., 2019c), and the high level of FUNDC1 might also be a risk factor for HCC patients. However, Li Wenhui found that FUNDC1-mediated mitophagy partially inhibited the release of mtRNA and mtROS produced by damaged mitochondria, thereby preventing inflammasome hyperactivated, suppressing the initiation of HCC in early-stage (Li et al., 2019c). These inconsistent results might be interpreted using the dynamic function of autophagy. Micro-137 is a protective factor for breast cancer because it can target FUNDC1 to inhibit the occurrence of mitophagy (Hu et al., 2020).
Mitochondria are energy factories, participating in the occurrence of multiple metabolic-related events. In recent years, mitophagy, which is regarded as the key mechanism for mitochondrial quality control and function, has received increased attention in metabolic-related diseases.
Like the global knockout mice, Wu Hao’s study showed that severe mitochondrial abnormality, aggregated inflammation, obesity phenotypes, and insulin resistance phenotypes were observed when animals were fed high fat diet in the adipose tissue specific FUNDC1 knockout mice model. The mechanism is mainly related to the accumulated ROS in damaged mitochondria, because ROS can activate MAPK, which leads to white adipose tissue insulin resistance and systematic insulin resistance induced by elevated tumor necrosis factor (TNF), interleukin 6 (IL6) and downregulated adiponectin gene (ADIPOQ) (Wu et al., 2019c). However, in skeletal-muscle specific FUNDC1 knockout mice, the obesity and systemic insulin resistance induced by high-fat diet are improved. Fu et al. (2018) argued that FUNDC1 deficiency in skeletal muscle could evoke a retrograde response in muscle that promoted the thermogenic remodeling of adipose tissue through increasing the level of fibroblast growth factor 21(FGF21). The two seemingly contradictory results above suggest that FUNDC1 in mitochondria of diverse tissues may have different physiological roles, and FUNDC1 may also play other roles besides mediating mitophagy. It seems to be worth further investigating the phenotypes of mice in which FUNDC1 is knocked out simultaneously in skeletal muscle and adipose tissue.
FUNDC1 is a crucial molecule which contains multiple sites performing different functions. FUNDC1 is mainly involved in mitophagy. On this basis, it is also involved in promoting mitochondrial division, maintaining iron homeostasis in cells, removing unfolded proteins, and maintaining cell functions through interacting with endoplasmic reticulum or lysosome.(as shown in Figure 4). Further investigation of the mechanism of the crosstalk are still needed to help combat human diseases. FUNDC1 is involved in the physiological and pathological processes of I/R injury, cancers, and metabolic-related diseases. It seems clear that targeting FUNDC1 is a potential therapeutic option for numerous diseases related to mitochondria abnormality, although there are currently few small molecule drugs targeting FUNDC1.
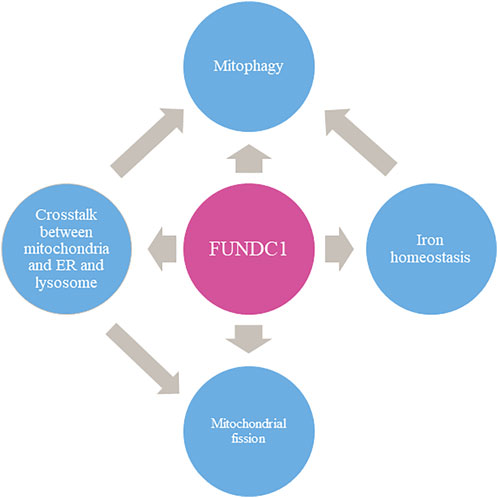
FIGURE 4. The multi-faced role of FUNDC1 in mitochondrial events.
NT, TL, and XW contributed to the conception and design of this article. MS, MZ, QW, and JL contributed to the language editing. WL, GL, and JJ contributed to draw the figures. YW, WW, and CL were responsible for the financial support, manuscript revision and final approval of the manuscript. All authors have read and agreed with the manuscript.
This study was funded by grants from National Natural Science Foundation of China (Nos. 82174364 and 81822049).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank the members of their laboratory and their collaborators for their research work.
Archer, S. L. (2013). Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 369 (23), 2236–2251. doi:10.1056/NEJMra1215233
Audano, M., Schneider, A., and Mitro, N. (2018). Mitochondria, lysosomes, and dysfunction: their meaning in neurodegeneration. J. Neurochem. 147 (3), 291–309. doi:10.1111/jnc.14471
Biswal, S., Barhwal, K. K., Das, D., Dhingra, R., Dhingra, N., Nag, T. C., et al. (2018). Salidroside mediated stabilization of Bcl -x(L) prevents mitophagy in CA3 hippocampal neurons during hypoxia. Neurobiol. Dis. 116, 39–52. doi:10.1016/j.nbd.2018.04.019
Cai, Y., Yang, E., Yao, X., Zhang, X., Wang, Q., Wang, Y., et al. (2021). FUNDC1-dependent mitophagy induced by tPA protects neurons against cerebral ischemia-reperfusion injury. Redox Biol. 38, 101792. doi:10.1016/j.redox.2020.101792
Cappellini, M. D., Musallam, K. M., and Taher, A. T. (2020). Iron deficiency anaemia revisited. J. Intern. Med. 287 (2), 153–170. doi:10.1111/joim.13004
Carraway, M. S., Suliman, H. B., Jones, W. S., Chen, C. W., Babiker, A., Piantadosi, C. A., et al. (2010). Erythropoietin activates mitochondrial biogenesis and couples red cell mass to mitochondrial mass in the heart. Circ. Res. 106 (11), 1722–1730. doi:10.1161/CIRCRESAHA.109.214353
Cassidy-Stone, A., Chipuk, J. E., Ingerman, E., Song, C., Yoo, C., Kuwana, T., et al. (2008). Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14 (2), 193–204. doi:10.1016/j.devcel.2007.11.019
Cerveny, K. L., Tamura, Y., Zhang, Z., Jensen, R. E., and Sesaki, H. (2007). Regulation of mitochondrial fusion and division. Trends Cell Biol. 17 (11), 563–569. doi:10.1016/j.tcb.2007.08.006
Chai, P., Cheng, Y., Hou, C., Yin, L., Zhang, D., Hu, Y., et al. (2021). USP19 promotes hypoxia-induced mitochondrial division via FUNDC1 at ER-mitochondria contact sites. J. Cell Biol. 220 (7), e202010006. doi:10.1083/jcb.202010006
Chakrabarti, R., and Higgs, H. N. (2021). Revolutionary view of two ways to split a mitochondrion. Nature 593 (7859), 346–347. doi:10.1038/d41586-021-01173-x
Chen, G., Han, Z., Feng, D., Chen, Y., Chen, L., Wu, H., et al. (2014). A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell 54 (3), 362–377. doi:10.1016/j.molcel.2014.02.034
Chen, M., Chen, Z., Wang, Y., Tan, Z., Zhu, C., Li, Y., et al. (2016). Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12 (4), 689–702. doi:10.1080/15548627.2016.1151580
Chen, Y. N., Gu, X., Zhou, X. E., Wang, W., Cheng, D., Ge, Y., et al. (2017). Crystal structure of TBC1D15 GTPase-activating protein (GAP) domain and its activity on Rab GTPases. Protein Sci. 26 (4), 834–846. a publication of the Protein Society. doi:10.1002/pro.3132
Chen, Z., Liu, L., Cheng, Q., Li, Y., Wu, H., Zhang, W., et al. (2017). Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. 18 (3), 495–509. doi:10.15252/embr.201643309
Chen, Z., Siraj, S., Liu, L., and Chen, Q. (2017). MARCH5-FUNDC1 axis fine-tunes hypoxia-induced mitophagy. Autophagy 13 (7), 1244–1245. doi:10.1080/15548627.2017.1310789
Chiang, S., Braidy, N., Maleki, S., Lal, S., Richardson, D. R., Huang, M. L., et al. (2021). Mechanisms of impaired mitochondrial homeostasis and NAD(+) metabolism in a model of mitochondrial heart disease exhibiting redox active iron accumulation. Redox Biol. 46, 102038. doi:10.1016/j.redox.2021.102038
Cuervo, A. M. (2011). Cell biology. Autophagy's top chef. Science 332 (6036), 1392–1393. doi:10.1126/science.1208607
Davizon-Castillo, P., McMahon, B., Aguila, S., Bark, D., Ashworth, K., Allawzi, A., et al. (2019). TNF-α-driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood 134 (9), 727–740. doi:10.1182/blood.2019000200
Deter, R. L., and De Duve, C. (1967). Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell Biol. 33 (2), 437–449. doi:10.1083/jcb.33.2.437
Dietz, J. V., Fox, J. L., and Khalimonchuk, O. (2021). Down the iron path: Mitochondrial iron homeostasis and beyond. Cells 10 (9), 2198. doi:10.3390/cells10092198
Elshaer, S. L., Mohamed, I. N., Coucha, M., Altantawi, S., Eldahshan, W., Bartasi, M. L., et al. (2017). Deletion of TXNIP mitigates high-fat diet-impaired angiogenesis and prevents inflammation in a mouse model of critical limb ischemia. Antioxidants (Basel, Switz. 6 (3), E47. doi:10.3390/antiox6030047
English, A. M., and Hughes, A. L. (2019). Knowing when to let go: Lysosomes regulate inter-mitochondrial tethering. Dev. Cell 50 (3), 259–260. doi:10.1016/j.devcel.2019.07.019
Evans, C. S., and Holzbaur, E. L. F. (2020). Lysosomal degradation of depolarized mitochondria is rate-limiting in OPTN-dependent neuronal mitophagy. Autophagy 16 (5), 962–964. doi:10.1080/15548627.2020.1734330
Fu, T., Xu, Z., Liu, L., Guo, Q., Wu, H., Liang, X., et al. (2018). Mitophagy directs muscle-adipose crosstalk to alleviate dietary obesity. Cell Rep. 23 (5), 1357–1372. doi:10.1016/j.celrep.2018.03.127
Geng, G., Liu, J., Xu, C., Pei, Y., Chen, L., Mu, C., et al. (2021). Receptor-mediated mitophagy regulates EPO production and protects against renal anemia. eLife. 10, e64480. doi:10.7554/eLife.64480
Giacomello, M., and Pellegrini, L. (2016). The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ. 23 (9), 1417–1427. doi:10.1038/cdd.2016.52
Gray, M. W., Burger, G., and Lang, B. F. (1999). Mitochondrial evolution. Science 283 (5407), 1476–1481. doi:10.1126/science.283.5407.1476
Hartl, F. U., and Hayer-Hartl, M. (2002). Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295 (5561), 1852–1858. doi:10.1126/science.1068408
Hirota, Y., Yamashita, S., Kurihara, Y., Jin, X., Aihara, M., Saigusa, T., et al. (2015). Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 11 (2), 332–343. doi:10.1080/15548627.2015.1023047
Hou, H., Er, P., Cheng, J., Chen, X., Ding, X., Wang, Y., et al. (2017). High expression of FUNDC1 predicts poor prognostic outcomes and is a promising target to improve chemoradiotherapy effects in patients with cervical cancer. Cancer Med. 6 (8), 1871–1881. doi:10.1002/cam4.1112
Hu, Q., Yuan, Y., Wu, Y., Huang, Y., Zhao, Z., Xiao, C., et al. (2020). MicroRNA-137 exerts protective effects on hypoxia-induced cell injury by inhibiting autophagy/mitophagy and maintaining mitochondrial function in breast cancer stem-like cells. Oncol. Rep. 44 (4), 1627–1637. doi:10.3892/or.2020.7714
Huang, M. L., Lane, D. J., and Richardson, D. R. (2011). Mitochondrial mayhem: the mitochondrion as a modulator of iron metabolism and its role in disease. Antioxid. Redox Signal. 15 (12), 3003–3019. doi:10.1089/ars.2011.3921
Hui, L., Wu, H., Wang, T. W., Yang, N., Guo, X., Jang, X. J., et al. (2019). Hydrogen peroxide-induced mitophagy contributes to laryngeal cancer cells survival via the upregulation of FUNDC1. Clin. Transl. Oncol. 21 (5), 596–606. doi:10.1007/s12094-018-1958-5
Jimenez-Orgaz, A., Kvainickas, A., Nägele, H., Denner, J., Eimer, S., Dengjel, J., et al. (2018). Control of RAB7 activity and localization through the retromer-TBC1D5 complex enables RAB7-dependent mitophagy. EMBO J. 37 (2), 235–254. doi:10.15252/embj.201797128
Kalogeris, T., Baines, C. P., Krenz, M., and Korthuis, R. J. (2016). Ischemia/reperfusion. Compr. Physiol. 7 (1), 113–170. doi:10.1002/cphy.c160006
Kleele, T., Rey, T., Winter, J., Zaganelli, S., Mahecic, D., Perreten Lambert, H., et al. (2021). Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 593 (7859), 435–439. doi:10.1038/s41586-021-03510-6
Kuang, Y., Ma, K., Zhou, C., Ding, P., Zhu, Y., Chen, Q., et al. (2016). Structural basis for the phosphorylation of FUNDC1 LIR as a molecular switch of mitophagy. Autophagy 12 (12), 2363–2373. doi:10.1080/15548627.2016.1238552
Lampert, M. A., Orogo, A. M., Najor, R. H., Hammerling, B. C., Leon, L. J., Wang, B. J., et al. (2019). BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 15 (7), 1182–1198. doi:10.1080/15548627.2019.1580095
Li, S., Zhou, Y., Gu, X., Zhang, X., and Jia, Z. (2021). NLRX1/FUNDC1/NIPSNAP1-2 axis regulates mitophagy and alleviates intestinal ischaemia/reperfusion injury. Cell Prolif. 54 (3), e12986. doi:10.1111/cpr.12986
Li, W., Li, Y., Siraj, S., Jin, H., Fan, Y., Yang, X., et al. (2019). FUN14 domain-containing 1-mediated mitophagy suppresses hepatocarcinogenesis by inhibition of inflammasome activation in mice. Hepatol. Baltim. Md) 69 (2), 604–621. doi:10.1002/hep.30191
Li, W., Yin, L., Sun, X., Wu, J., Dong, Z., Hu, K., et al. (2020). Alpha-lipoic acid protects against pressure overload-induced heart failure via ALDH2-dependent Nrf1-FUNDC1 signaling. Cell Death Dis. 11 (7), 599. doi:10.1038/s41419-020-02805-2
Li, W., Zhang, X., Zhuang, H., Chen, H. G., Chen, Y., Tian, W., et al. (2014). MicroRNA-137 is a novel hypoxia-responsive microRNA that inhibits mitophagy via regulation of two mitophagy receptors FUNDC1 and NIX. J. Biol. Chem. 289 (15), 10691–10701. doi:10.1074/jbc.M113.537050
Li, Y., Liu, L., Zhu, Y., and Chen, Q. (2019). Mitochondria organize the cellular proteostatic response and promote cellular senescence. Cell stress 3 (4), 110–114. doi:10.15698/cst2019.04.181
Li, Y., Liu, Z., Zhang, Y., Zhao, Q., Wang, X., Lu, P., et al. (2018). PEDF protects cardiomyocytes by promoting FUNDC1-mediated mitophagy via PEDF-R under hypoxic condition. Int. J. Mol. Med. 41 (6), 3394–3404. doi:10.3892/ijmm.2018.3536
Li, Y., Xue, Y., Xu, X., Wang, G., Liu, Y., Wu, H., et al. (2019). A mitochondrial FUNDC1/HSC70 interaction organizes the proteostatic stress response at the risk of cell morbidity. EMBO J. 38 (3), e98786. doi:10.15252/embj.201798786
Liu, B. Y., Li, L., Liu, G. L., Ding, W., Chang, W. G., Xu, T., et al. (2021). Baicalein attenuates cardiac hypertrophy in mice via suppressing oxidative stress and activating autophagy in cardiomyocytes. Acta Pharmacol. Sin. 42 (5), 701–714. doi:10.1038/s41401-020-0496-1
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14 (2), 177–185. doi:10.1038/ncb2422
Liu, L., Li, Y., and Chen, Q. (2021). The emerging role of FUNDC1-mediated mitophagy in cardiovascular diseases. Front. Physiol. 12, 807654. doi:10.3389/fphys.2021.807654
Liu, L., Li, Y., Wang, J., Zhang, D., Wu, H., Li, W., et al. (2021). Mitophagy receptor FUNDC1 is regulated by PGC-1α/NRF1 to fine tune mitochondrial homeostasis. EMBO Rep. 22 (3), e50629. doi:10.15252/embr.202050629
Liu, L., Sakakibara, K., Chen, Q., and Okamoto, K. (2014). Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 24 (7), 787–795. doi:10.1038/cr.2014.75
Liu, T., Daniels, C. K., and Cao, S. (2012). Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol. Ther. 136 (3), 354–374. doi:10.1016/j.pharmthera.2012.08.014
Liu, Y., Ao, X., Ding, W., Ponnusamy, M., Wu, W., Hao, X., et al. (2018). Critical role of FOXO3a in carcinogenesis. Mol. Cancer 17 (1), 104. doi:10.1186/s12943-018-0856-3
Lu, W., Karuppagounder, S. S., Springer, D. A., Allen, M. D., Zheng, L., Chao, B., et al. (2014). Genetic deficiency of the mitochondrial protein PGAM5 causes a Parkinson's-like movement disorder. Nat. Commun. 5, 4930. doi:10.1038/ncomms5930
Mao, C., Hu, C., Zhou, Y., Zou, R., Li, S., Cui, Y., et al. (2020). Electroacupuncture pretreatment against cerebral ischemia/reperfusion injury through mitophagy. Evid. Based. Complement. Altern. Med. 2020, 7486041. doi:10.1155/2020/7486041
Mao, S., Tian, S., Luo, X., Zhou, M., Cao, Z., Li, J., et al. (2021). Overexpression of PLK1 relieved the myocardial ischemia-reperfusion injury of rats through inducing the mitophagy and regulating the p-AMPK/FUNDC1 axis. Bioengineered 12 (1), 2676–2687. doi:10.1080/21655979.2021.1938500
Mohamed, I. N., Sheibani, N., and El-Remessy, A. B. (2020). Deletion of thioredoxin-interacting protein (TXNIP) abrogates high fat diet-induced retinal leukostasis, barrier dysfunction and microvascular degeneration in a mouse obesity model. Int. J. Mol. Sci. 21 (11), E3983. doi:10.3390/ijms21113983
Muñoz, J. P., and Zorzano, A. (2017). FUNDC1: A novel protein in cardiac health. Circulation 136 (23), 2267–2270. doi:10.1161/CIRCULATIONAHA.117.031417
Nagi, M., Tanabe, K., Nakayama, H., Ueno, K., Yamagoe, S., Umeyama, T., et al. (2016). Iron-depletion promotes mitophagy to maintain mitochondrial integrity in pathogenic yeast Candida glabrata. Autophagy 12 (8), 1259–1271. doi:10.1080/15548627.2016.1183080
Onoue, K., Jofuku, A., Ban-Ishihara, R., Ishihara, T., Maeda, M., Koshiba, T., et al. (2013). Fis1 acts as a mitochondrial recruitment factor for TBC1D15 that is involved in regulation of mitochondrial morphology. J. Cell Sci. 126 (Pt 1), 176–185. doi:10.1242/jcs.111211
Pan, P., Chen, J., Liu, X., Fan, J., Zhang, D., Zhao, W., et al. (2021). FUNDC1 regulates autophagy by inhibiting ROS-NLRP3 signaling to avoid apoptosis in the lung in a lipopolysaccharide-induced mouse model. Shock (Augusta, Ga) 56 (5), 773–781. doi:10.1097/SHK.0000000000001835
Paul, V. D., and Lill, R. (2015). Biogenesis of cytosolic and nuclear iron-sulfur proteins and their role in genome stability. Biochim. Biophys. Acta 1853 (6), 1528–1539. doi:10.1016/j.bbamcr.2014.12.018
Pei, Z., Liu, Y., Liu, S., Jin, W., Luo, Y., Sun, M., et al. (2021). FUNDC1 insufficiency sensitizes high fat diet intake-induced cardiac remodeling and contractile anomaly through ACSL4-mediated ferroptosis. Metabolism. 122, 154840. doi:10.1016/j.metabol.2021.154840
Progida, C., Cogli, L., Piro, F., De Luca, A., Bakke, O., Bucci, C., et al. (2010). Rab7b controls trafficking from endosomes to the TGN. J. Cell Sci. 123 (Pt 9), 1480–1491. doi:10.1242/jcs.051474
Ren, J., Sun, M., Zhou, H., Ajoolabady, A., Zhou, Y., Tao, J., et al. (2020). FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-dependent manner in obesity. Sci. Adv. 6 (38), eabc8561. doi:10.1126/sciadv.abc8561
Rowland, A. A., and Voeltz, G. K. (2012). Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat. Rev. Mol. Cell Biol. 13 (10), 607–625. doi:10.1038/nrm3440
Sandoval-Acuña, C., Torrealba, N., Tomkova, V., Jadhav, S. B., Blazkova, K., Merta, L., et al. (2021). Targeting mitochondrial iron metabolism suppresses tumor growth and metastasis by inducing mitochondrial dysfunction and mitophagy. Cancer Res. 81 (9), 2289–2303. doi:10.1158/0008-5472.CAN-20-1628
Schiavi, A., Maglioni, S., Palikaras, K., Shaik, A., Strappazzon, F., Brinkmann, V., et al. (2015). Iron-starvation-induced mitophagy mediates lifespan extension upon mitochondrial stress in C. elegans. Curr. Biol. 25 (14), 1810–1822. doi:10.1016/j.cub.2015.05.059
Song, J., Herrmann, J. M., and Becker, T. (2021). Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 22 (1), 54–70. doi:10.1038/s41580-020-00300-2
Song, M., and Dorn, G. W. (nd2015). Mitoconfusion: Noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab. 21 (2), 195–205. doi:10.1016/j.cmet.2014.12.019
Springer, M. Z., and Macleod, K. F. (2016). In brief: Mitophagy: mechanisms and role in human disease. J. Pathol. 240 (3), 253–255. doi:10.1002/path.4774
Tian, W., Zhu, M., Zhou, Y., Mao, C., Zou, R., Cui, Y., et al. (2021). Electroacupuncture pretreatment alleviates cerebral ischemia-reperfusion injury by regulating mitophagy via mTOR-ULK1/FUNDC1 Axis in rats. J. Stroke Cerebrovasc. Dis. 31 (1), 106202. doi:10.1016/j.jstrokecerebrovasdis.2021.106202
van der Bliek, A. M. (2016). Mitochondria just wanna have FUN(DC1). EMBO J. 35 (13), 1365–1367. doi:10.15252/embj.201694759
Volkmar, N., Fenech, E., and Christianson, J. C. (2016). New MAPS for misfolded proteins. Nat. Cell Biol. 18 (7), 724–726. doi:10.1038/ncb3381
Wai, T., García-Prieto, J., Baker, M. J., Merkwirth, C., Benit, P., Rustin, P., et al. (2015). Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350 (6265), aad0116. doi:10.1126/science.aad0116
Wang, C., Dai, X., Wu, S., Xu, W., Song, P., Huang, K., et al. (2021). FUNDC1-dependent mitochondria-associated endoplasmic reticulum membranes are involved in angiogenesis and neoangiogenesis. Nat. Commun. 12 (1), 2616. doi:10.1038/s41467-021-22771-3
Wang, J., Zhu, P., Li, R., Ren, J., and Zhou, H. (2020). Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 30, 101415. doi:10.1016/j.redox.2019.101415
Wang, L., Wang, P., Dong, H., Wang, S., Chu, H., Yan, W., et al. (2018). Ulk1/FUNDC1 prevents nerve cells from hypoxia-induced apoptosis by promoting cell autophagy. Neurochem. Res. 43 (8), 1539–1548. doi:10.1007/s11064-018-2568-x
Wang, Y., Jasper, H., Toan, S., Muid, D., Chang, X., Zhou, H., et al. (2021). Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol. 45, 102049. doi:10.1016/j.redox.2021.102049
Wei, X., Wei, X., Lu, Z., Li, L., Hu, Y., Sun, F., et al. (2020). Activation of TRPV1 channel antagonizes diabetic nephropathy through inhibiting endoplasmic reticulum-mitochondria contact in podocytes. Metabolism. 105, 154182. doi:10.1016/j.metabol.2020.154182
Wong, Y. C., Ysselstein, D., and Krainc, D. (2018). Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554 (7692), 382–386. doi:10.1038/nature25486
Wu, H., Wang, Y., Li, W., Chen, H., Du, L., Liu, D., et al. (2019). Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 15 (11), 1882–1898. doi:10.1080/15548627.2019.1596482
Wu, H., Wei, H., Zhang, D., Sehgal, S. A., Zhang, D., Wang, X., et al. (2020). Defective mitochondrial ISCs biogenesis switches on IRP1 to fine tune selective mitophagy. Redox Biol. 36, 101661. doi:10.1016/j.redox.2020.101661
Wu, H., Xue, D., Chen, G., Han, Z., Huang, L., Zhu, C., et al. (2014). The BCL2L1 and PGAM5 axis defines hypoxia-induced receptor-mediated mitophagy. Autophagy 10 (10), 1712–1725. doi:10.4161/auto.29568
Wu, L., Zhang, D., Zhou, L., Pei, Y., Zhuang, Y., Cui, W., et al. (2019). FUN14 domain-containing 1 promotes breast cancer proliferation and migration by activating calcium-NFATC1-BMI1 axis. EBioMedicine 41, 384–394. doi:10.1016/j.ebiom.2019.02.032
Wu, S., Lu, Q., Ding, Y., Wu, Y., Qiu, Y., Wang, P., et al. (2019). Hyperglycemia-Driven inhibition of AMP-activated protein kinase α2 induces diabetic cardiomyopathy by promoting mitochondria-associated endoplasmic reticulum membranes in vivo. Circulation 139 (16), 1913–1936. doi:10.1161/CIRCULATIONAHA.118.033552
Wu, S., Lu, Q., Wang, Q., Ding, Y., Ma, Z., Mao, X., et al. (2017). Binding of FUN14 domain containing 1 with inositol 1, 4, 5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation 136 (23), 2248–2266. doi:10.1161/CIRCULATIONAHA.117.030235
Wu, W., Li, W., Chen, H., Jiang, L., Zhu, R., Feng, D., et al. (2016). FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy 12 (9), 1675–1676. doi:10.1080/15548627.2016.1193656
Wu, W., Lin, C., Wu, K., Jiang, L., Wang, X., Li, W., et al. (2016). FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 35 (13), 1368–1384. doi:10.15252/embj.201593102
Wu, W., Tian, W., Hu, Z., Chen, G., Huang, L., Li, W., et al. (2014). ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 15 (5), 566–575. doi:10.1002/embr.201438501
Wu, X., Wu, F. H., Wu, Q., Zhang, S., Chen, S., Sima, M., et al. (2017). Phylogenetic and molecular evolutionary analysis of mitophagy receptors under hypoxic conditions. Front. Physiol. 8, 539. doi:10.3389/fphys.2017.00539
Xiao, Y., Chen, W., Zhong, Z., Ding, L., Bai, H., Chen, H., et al. (2020). Electroacupuncture preconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting mitophagy mediated by the mTORC1-ULK1-FUNDC1 pathway. Biomed. Pharmacother. = Biomedecine Pharmacother. 127, 110148. doi:10.1016/j.biopha.2020.110148
Yamano, K., Fogel, A. I., Wang, C., van der Bliek, A. M., and Youle, R. J. (2014). Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 3, e01612. doi:10.7554/eLife.01612
Yoshii, S. R., Kishi, C., Ishihara, N., and Mizushima, N. (2011). Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 286 (22), 19630–19640. doi:10.1074/jbc.M110.209338
Yu, W., Sun, S., Xu, H., Li, C., Ren, J., Zhang, Y., et al. (2020). TBC1D15/RAB7-regulated mitochondria-lysosome interaction confers cardioprotection against acute myocardial infarction-induced cardiac injury. Theranostics 10 (24), 11244–11263. doi:10.7150/thno.46883
Yu, W., Xu, M., Zhang, T., Zhang, Q., and Zou, C. (2019). Mst1 promotes cardiac ischemia-reperfusion injury by inhibiting the ERK-CREB pathway and repressing FUNDC1-mediated mitophagy. J. Physiol. Sci. 69 (1), 113–127. doi:10.1007/s12576-018-0627-3
Zhang, G., Wang, X., Rothermel, B. A., Lavandero, S., and Wang, Z. V. (2021). The integrated stress response in ischemic diseases. Cell Death Differ. 29, 750–757. doi:10.1038/s41418-021-00889-7
Zhang, W., Ren, H., Xu, C., Zhu, C., Wu, H., Liu, D., et al. (2016). Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. eLife 5, e21407. doi:10.7554/eLife.21407
Zhang, W., Siraj, S., Zhang, R., and Chen, Q. (2017). Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy 13 (6), 1080–1081. doi:10.1080/15548627.2017.1300224
Zheng, S., Jian, D., Gan, H., Wang, L., Zhao, J., Zhai, X., et al. (2021). FUNDC1 inhibits NLRP3-mediated inflammation after intracerebral hemorrhage by promoting mitophagy in mice. Neurosci. Lett. 756, 135967. doi:10.1016/j.neulet.2021.135967
Zhou, H., Li, D., Zhu, P., Hu, S., Hu, N., Ma, S., et al. (2017). Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARγ/FUNDC1/mitophagy pathways. J. Pineal Res. 63 (4), e12438. doi:10.1111/jpi.12438
Zhou, H., Wang, J., Zhu, P., Zhu, H., Toan, S., Hu, S., et al. (2018). NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2α. Basic Res. Cardiol. 113 (4), 23. doi:10.1007/s00395-018-0682-1
Zhou, H., Zhu, P., Guo, J., Hu, N., Wang, S., Li, D., et al. (2017). Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 13, 498–507. doi:10.1016/j.redox.2017.07.007
Zhou, H., Zhu, P., Wang, J., Toan, S., and Ren, J. (2019). DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal Transduct. Target. Ther. 4, 56. doi:10.1038/s41392-019-0094-1
Zhou, H., Zhu, P., Wang, J., Zhu, H., Ren, J., Chen, Y., et al. (2018). Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 25 (6), 1080–1093. doi:10.1038/s41418-018-0086-7
Keywords: FUNDC1, mitophagy, mitochondrial fission, endoplasmic reticulum, I/R injury
Citation: Tan N, Liu T, Wang X, Shao M, Zhang M, Li W, Ling G, Jiang J, Wang Q, Li J, Li C, Wang W and Wang Y (2022) The multi-faced role of FUNDC1 in mitochondrial events and human diseases. Front. Cell Dev. Biol. 10:918943. doi: 10.3389/fcell.2022.918943
Received: 14 April 2022; Accepted: 28 June 2022;
Published: 25 July 2022.
Edited by:
Patrice X. Petit, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Alexander Van Der Bliek, UCLA Health System, United StatesCopyright © 2022 Tan, Liu, Wang, Shao, Zhang, Li, Ling, Jiang, Wang, Li, Li, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chun Li, bGljaHVuMTk4NTAyMDRAMTYzLmNvbQ==; Wei Wang, d2FuZ3dlaTI2OTYwQDEyNi5jb20=; Yong Wang, d2FuZ3lvbmdAYnVjbS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.