 Li Hu1,2,3†Huiqin Li2†Meiting Zi2Wen Li4Jing Liu5Yang Yang6Daohong Zhou6Qing-Peng Kong2Yunxia Zhang1,3*Yonghan He2*
Li Hu1,2,3†Huiqin Li2†Meiting Zi2Wen Li4Jing Liu5Yang Yang6Daohong Zhou6Qing-Peng Kong2Yunxia Zhang1,3*Yonghan He2*- 1Department of Geriatrics, The Second Affiliated Hospital of Hainan Medical University, Haikou, China
- 2State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, China
- 3College of Basic Medicine and Life Sciences, Hainan Medical University, Haikou, China
- 4Department of Endocrinology, The Third People’s Hospital of Yunnan Province, Kunming, China
- 5Lab of Molecular Genetics of Aging and Tumor, Medical School, Kunming University of Science and Technology, Kunming, China
- 6Department of Pharmacodynamics, College of Pharmacy, University of Florida, Gainesville, FL, United States
Cellular senescence is a process that leads to a state of irreversible cell growth arrest induced by a variety of intrinsic and extrinsic stresses. Senescent cells (SnCs) accumulate with age and have been implicated in various age-related diseases in part via expressing the senescence-associated secretory phenotype. Elimination of SnCs has the potential to delay aging, treat age-related diseases and extend healthspan. However, once cells becoming senescent, they are more resistant to apoptotic stimuli. Senolytics can selectively eliminate SnCs by targeting the SnC anti-apoptotic pathways (SCAPs). They have been developed as a novel pharmacological strategy to treat various age-related diseases. However, the heterogeneity of the SnCs indicates that SnCs depend on different proteins or pathways for their survival. Thus, a better understanding of the underlying mechanisms for apoptotic resistance of SnCs will provide new molecular targets for the development of cell-specific or broad-spectrum therapeutics to clear SnCs. In this review, we discussed the latest research progresses and challenge in senolytic development, described the significance of regulation of senescence and apoptosis in aging, and systematically summarized the SCAPs involved in the apoptotic resistance in SnCs.
Introduction
Aging is a time-dependent functional decline that affects most living organisms. It is characterized by a progressive loss of physiological integrity, causing impaired function and increased vulnerability to death (López-Otín et al., 2013). Aging results from the time-dependent accumulation of a wide range of molecular and cellular damage over time. In humans, aging increases the risk of various age-related diseases. In 2013, López-Otín et al. identified and categorized the cellular and molecular hallmarks of aging, including genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion and altered intercellular communication (López-Otín et al., 2013). All of these hallmarks occur during aging and are interconnected. Among them, cellular senescence can link to almost all of other aging hallmarks, representing a good example to study healthy aging from basic research to therapeutics (Campisi et al., 2019; Di Micco et al., 2021).
Cellular senescence is a process that leads to a state of irreversible growth arrest in response to a variety of intrinsic and extrinsic stresses (Hernandez-Segura et al., 2017). Initially, the phenomenon was found when cultured cells were shown to undergo a limited number of cell divisions in vitro (Hayflick and Moorhead, 1961; Hayflick, 1965). Cellular senescence is different from cell quiescence which represents a transient and reversible cell cycle arrest. Cellular senescence can be a physiologically or pathologically relevant program depending on the specific situation (Burton and Krizhanovsky, 2014; Herranz and Gil, 2018). It normally functions as a vital tumor suppressive mechanism and also plays an important role in tissue damage repair (Demaria et al., 2014; Gorgoulis et al., 2019; Moiseeva et al., 2020). However, senescent cells (SnCs) have been implicated in various age-related diseases (Palmer et al., 2015; Ogrodnik et al., 2017; Aguayo-Mazzucato et al., 2019; Zhang et al., 2019). Accordingly, selective elimination of SnCs has been exploited as a novel strategy to treat the diseases. In addition, SnCs have also been implicated in infectious diseases. For example, virus infection can induce cellular senescence, which was found to be a pathogenic trigger of cytokine escalation and organ damage, and recently found to be associated with the COVID-19 severity in the elderly (Nehme et al., 2020; Gerdes et al., 2021; Lee et al., 2021; Mohiuddin and Kasahara, 2021). Clearance of virus-induced SnCs was considered as a novel treatment option against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Lee et al., 2021) and perhaps other viral infections (Camell et al., 2021).
One of the characteristics of SnCs is their ability to resist apoptosis. Until now, small molecules that can selectively kill SnCs, termed senolytics, were developed to target the proteins in the SnC anti-apoptotic pathways (SCAPs) (Childs et al., 2017; Kirkland and Tchkonia, 2017; Niedernhofer and Robbins, 2018). However, due to the high heterogeneity in gene expression and their diverse origins, SnCs may use different SCAPs to maintain their survival, making it difficult to use a single senolytic to kill all types of SnCs. Although significant progresses on the development of senolytics have been made, some proteins involved in the SCAPs have been overlooked, neither their potential of being senolytic targets have been investigated. Therefore, gaining more insights into the apoptosis-resistant mechanism of SnCs may greatly help to design or screen more effective senolytics that can be used to treat SnC-associated disorders. In this review, we systematically summarized the proteins or pathways involved in SnC apoptotic resistance documented in the publications since 1995 (Wang, 1995), when the first result of SnC resistance to apoptosis was reported.
SnCs Are Novel Therapeutic Targets for Aging and Age-Related Diseases
Cellular senescence is the irreversible growth arrest of individual mitotic cells. It was first described by Leonard Hayflick and Paul Moorhead (Hayflick and Moorhead, 1961). In their work they found that the cultured human fibroblasts lost the ability to proliferate, reaching permanent arrest after about 50 population doublings (Hayflick and Moorhead, 1961; Hayflick, 1965). This type of cellular senescence is referred to as replicative senescence, which is caused by telomere shortening after extensive cell proliferations. Another major type of cellular senescence is stress-induced premature cellular senescence, which is resulted from exposure to various genotoxic stressors, such as oxidative stress, and chemotherapy. In addition, the activation of various oncogenes can also induce senescence termed as oncogene-induced senescence, which is caused by DNA replicative stress (Di Micco et al., 2006). After being senescent, cells display a series of cellular and molecular changes, such as enlarged cell size, irregularly shaped cell body, increased lysosomal content, accumulation of mitochondria, enlarged nuclear size and increased DNA damage (Hernandez-Segura et al., 2018). The hallmarks of cellular senescence were well summarized elsewhere (Hernandez-Segura et al., 2018). Some of them, such as increased p16 and beta-galactosidase activity, are widely used for identifying SnCs in vitro and in vivo (Childs et al., 2017).
Cellular senescence is a key mechanism for the body to prevent the propagation of damaged cells (Herranz and Gil, 2018; Gorgoulis et al., 2019). In this regards, cellular senescence acts as a tumor suppressor mechanism (Sarkisian et al., 2007; Burton and Krizhanovsky, 2014), and it also shows beneficial effects in development (Muñoz-Espín et al., 2013) and tissue repair (Demaria et al., 2014). However, SnCs can accumulate when their production persists beyond the immune clearance capacity or the immune system is compromised. Under such circumstances, SnCs may play a causative role in aging and age-related diseases by inducing reactive oxygen species (ROS) and secreting a plethora of inflammatory mediators (e.g., cytokines and chemokines), growth factors, and extracellular proteases—termed as the senescence-associated secretory phenotype (SASP) (Demaria et al., 2017; Alimirah et al., 2020). Therefore, cellular senescence is both a physiologically and pathologically relevant program depending on the specific situation.
In 2011 and 2016, Baker et al. reported that elimination of SnCs by a transgenic approach delayed the onset of several age-related diseases and prolonged lifespan in progeroid and naturally aged mice, respectively (Baker et al., 2011, 2016). They provided solid evidence for that SnCs are causally implicated in generating age-related phenotypes and that clearance of SnCs can prevent or delay tissue dysfunction and extend healthspan. Later on, accumulating data demonstrate the involvement of SnCs in various age-related diseases and disorders (Childs et al., 2015, 2017; Kirkland and Tchkonia, 2017; Niedernhofer and Robbins, 2018), such as neurodegenerative diseases (Chinta et al., 2018; Zhang et al., 2019), diabetes (Aguayo-Mazzucato et al., 2019), atherosclerosis (Childs et al., 2016), osteoarthritis (Jeon et al., 2017), and tumors (Demaria et al., 2017; Takasugi et al., 2017). In sum, these findings suggest that SnCs are an emerging therapeutic target of aging and age-related diseases.
Targeting Senescent Cells With Senolytics and New Strategies
The evidence that elimination of SnCs by genetic strategy can extend the healthspan in mice prompts a gold rush in the development of pharmaceutical small molecules that can selectively kill SnCs to combat aging and age-related conditions. As these molecules are called ‘senolytics’, whereas those that can suppress SASP production, named senomorphics. Both senolytics and senomorphics have the potential to prevent or treat age-related diseases and to extend healthspan. However, more efforts were made on developing senolytics compared to senomorphics considering that senolytics have more favorable drug exposure, toxicity and durable effects (He et al., 2020c).
Until now, several classes of senolytics have been developed, including naturally occurring compounds and their derivatives [e.g., quercetin (Zhu et al., 2015), fisetin (Yousefzadeh et al., 2018), piperlongumine (Wang et al., 2016), EF24 (Li et al., 2019a)], cardiac glycosides [e.g., ouabain (Guerrero et al., 2019), digoxin (Triana-Martínez et al., 2019)], and targeted therapeutics [dasatinib (Zhu et al., 2015), ABT263 (Chang et al., 2016), HSP90 inhibitor (Fuhrmann-Stroissnigg et al., 2017), and FOXO4-p53 interfering peptide (Baar et al., 2017)] (Table 1). We have previously reviewed the progresses on naturally occurring and targeted senolytics (Li et al., 2019b; Ge et al., 2021). It is very encouraging that several senolytics have been approved to enter clinical trials and shown benefits as therapeutics for aging or age-related diseases (Childs et al., 2017; Kirkland and Tchkonia, 2017, 2020; Campisi et al., 2019; Thoppil and Riabowol, 2019; Di Micco et al., 2021). Even though, current senolytics still have some limitations in terms of potency, safety, specificity, broad-spectrum activity, pharmacodynamics and pharmacokinetics. For example, one of the most widely used senolytics, i.e. the combination of dasatinib and quercetin (D + Q), was well tolerated and could reduce SnC burden in patients (Thoppil and Riabowol, 2019; Kirkland and Tchkonia, 2020). However, quercetin is a polypharmacologic agent and its mechanisms of action have not been well defined. ABT263, a Bcl-2 and Bcl-xL dual inhibitor, has been extensively used as a potent and broad-spectrum senolytic agent (Chang et al., 2016). Its on-target toxicity of thrombocytopenia induced by Bcl-xL inhibition prevents its use in clinic even for tumor patients (Gandhi et al., 2011; Souers et al., 2013; Leverson et al., 2015; Ashkenazi et al., 2017). To overcome these challenges, some novel strategies, such as proteolysis-targeting chimera (PROTAC) technology (He et al., 2020b), chimeric antigen receptor (CAR) T cells (Amor et al., 2020), and β-galactosidase-modified prodrugs (Cai et al., 2020; Guerrero et al., 2020), have been developed to eliminate SnCs and shown promising therapeutic potential in the treatment of age-related diseases (Ge et al., 2021).

TABLE 1. Senolytic targets and agents.
One major characteristic of SnCs is that they are resistant to apoptosis. This is attributable to the upregulation of certain proteins in the SCAPs, which enables SnCs to accumulate during aging. Most of the abovementioned senolytics were developed to selectively kill SnCs by targeting the known SCAPs. However, the transcriptional heterogeneity (Hernandez-Segura et al., 2017) and the diverse tissue origins of SnCs, render SnCs depend on different SCAPs to maintain their survival, which makes it a challenge to develop certain senolytics to eliminate SnCs in various cell types. For example, we found that ABT263 is very potent to selectively kill senescent human lung diploid fibroblasts (HDF) (Chang et al., 2016; He et al., 2020b), but it does not work in senescent HDF isolated from human foreskins (Cho et al., 2020), suggesting that HDF from different tissue origins may have different apoptotic resistance mechanisms. Therefore, gaining deep insights into the apoptosis-resistant proteins or pathways of SnCs may help to develop cell-specific or broad-spectrum senolytics to clear SnCs. In the following sections, we will focus on reviewing cell apoptosis and its significance in aging and age-related diseases, as well as summarizing the emerging proteins or pathways involved in apoptotic resistance of SnCs.
The Significance of Apoptosis in Aging and Age-Related Diseases
Apoptosis is defined as a programmed cell death, which has received a lot of attention because of its importance during tissue development (Wanner et al., 2020), and maintenance of tissue homeostasis (Elmore, 2007; Günther et al., 2013; Kile, 2014; Singh et al., 2019). Apoptosis occurs when cells undergo exogenous stimuli or intrinsic signals and is required to maintain the integrity and homeostasis of tissues, and dysregulation of which is implicated in the development of various diseases (Singh et al., 2019). For example, increased cellular apoptosis is known to contribute to age-related diseases in the brain (Mattson et al., 2001). Unlike cellular necrosis, a way of cell death resulting from acute cellular injury, apoptosis does not induce an inflammation response to cause a bystander effect.
Aging has been associated with decreased apoptosis in some cell types (Figure 1A), such as adipose mesenchymal stem cells (Alt et al., 2012), and bone marrow mesenchymal stem cells (Wilson et al., 2010). In vivo, it has been reported that apoptotic potential was dramatically reduced in the liver of aged rats challenged with a DNA-damaging agent (Suh et al., 2002). Similarly, apoptosis induced by radiation was significantly reduced in peripheral blood lymphocytes isolated from aged mice (Polyak et al., 1997). In humans, the peripheral blood mononuclear cells (PBMCs) from old people and centenarians showed an increased resistance to apoptosis induced by 2-deoxy-D-ribose (dRib), which may contribute to immunosenescence (Monti et al., 2000). Consistently, there is a reduction in the markers of apoptosis in human serum during aging (Kavathia et al., 2009). In a few cases, the apoptosis is increased in some cell types during aging. For example, there is a significant loss of cells in the thymus and bone marrow, associated with an increase in the number of apoptotic lymphocytes during the aging process (Sainz et al., 2003). The discrepancy of apoptosis in different cell types during aging is likely due to the age-related disruptions in systemic and intracellular signaling combined with cell-autonomous effects (Tower, 2015). Because apoptosis is required for normal cell turnover and tissue homeostasis, dysregulation of apoptosis at old age is increasingly implicated in various age-related diseases, such as tumor (DePinho, 2000), and neurodegenerative diseases (Erekat, 2021). However, SnCs are highly resistant to apoptosis (Wang, 1995), likely enabling them accumulate during aging (Figure 1A), which acts as an important contributor to aging (Salminen et al., 2011), and age-related diseases (Kirkland and Tchkonia, 2017; Niedernhofer and Robbins, 2018; Thoppil and Riabowol, 2019; Di Micco et al., 2021; Ge et al., 2021). Thus, it is imperative to understand why SnCs are resistant to apoptosis. A better understanding of the underlying mechanisms for apoptotic resistance of SnCs is likely to provide novel molecular targets for the development of therapeutic senolytics to combat aging and age-related diseases (Figure 1B).
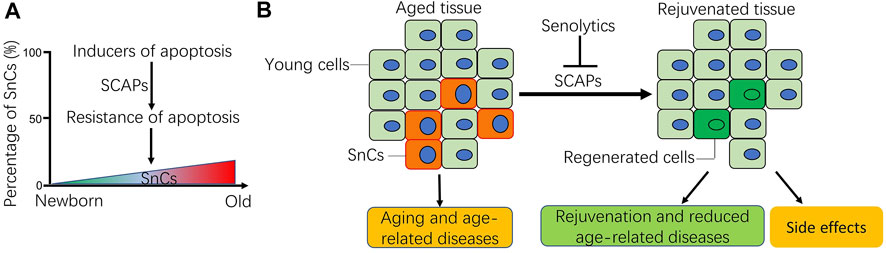
FIGURE 1. Schematic of apoptotic resistance of senescent cells (SnCs) in aging and age-related diseases. (A) During the aging process, aging-related disruptions in systemic and inter-cell signaling, together with cell-autonomous damage and mitochondrial malfunction result in either increased or decreased cell apoptosis depending on cellular context (Tower, 2015). As for SnCs, their percentage usually increases in tissues with age as they acquire resistance to apoptotic stimuli via the SnC anti-apoptotic pathways (SCAPs). (B) Accumulation of SnCs in tissues contributes to aging and the occurrence of age-related diseases. Senolytics can selectively clear the SnCs by targeting the SCAPs, which promote the regeneration of young cells, rejuvenate aged tissues and reduce age-related diseases. Sometimes, senolytics can also cause side effects, more caution should be exercised for the systemic use of senolytics for health benefits. Note: B was modified from our previous publication (Li et al., 2019a; Li et al., 2019b).
Apoptotic Resistance and the Underlying Mechanism in SnCs
Since the first observation of apoptotic resistance in SnC human fibroblasts was reported in 1995 (Wang, 1995), the phenomenon was later found in a variety of cell types when they became senescent, including senescent immune cells (Spaulding et al., 1999), keratinocytes (KCs) (Chaturvedi et al., 2004), human endometrium-derived mesenchymal stem cells (MESCs) (Burova et al., 2013) and hepatic stellate cells (HSC) (Kawada, 2006). There are various mechanisms that promote the survival of SnCs in vitro and in vivo, such as metabolic reprogramming, activation of unfolded protein response, and immune evasion (Soto-Gamez et al., 2019). In this review, we focus on the SCAPs, as most of the known senolytics were developed by targeting individual SCAPs (Figure 1B). Emerging SCAPs are reported along with the findings of SnC resistance to apoptosis in different cell types, providing new target candidates for developing either cell-specific or broad-spectrum senolytics. In this section, we review the findings of SnC apoptotic resistance reported since 1995 and tempt to broaden the knowledge of SCAPs for the development of senolytics. The known proteins or pathways responsible for apoptotic resistance in SnCs are listed in Table 2.
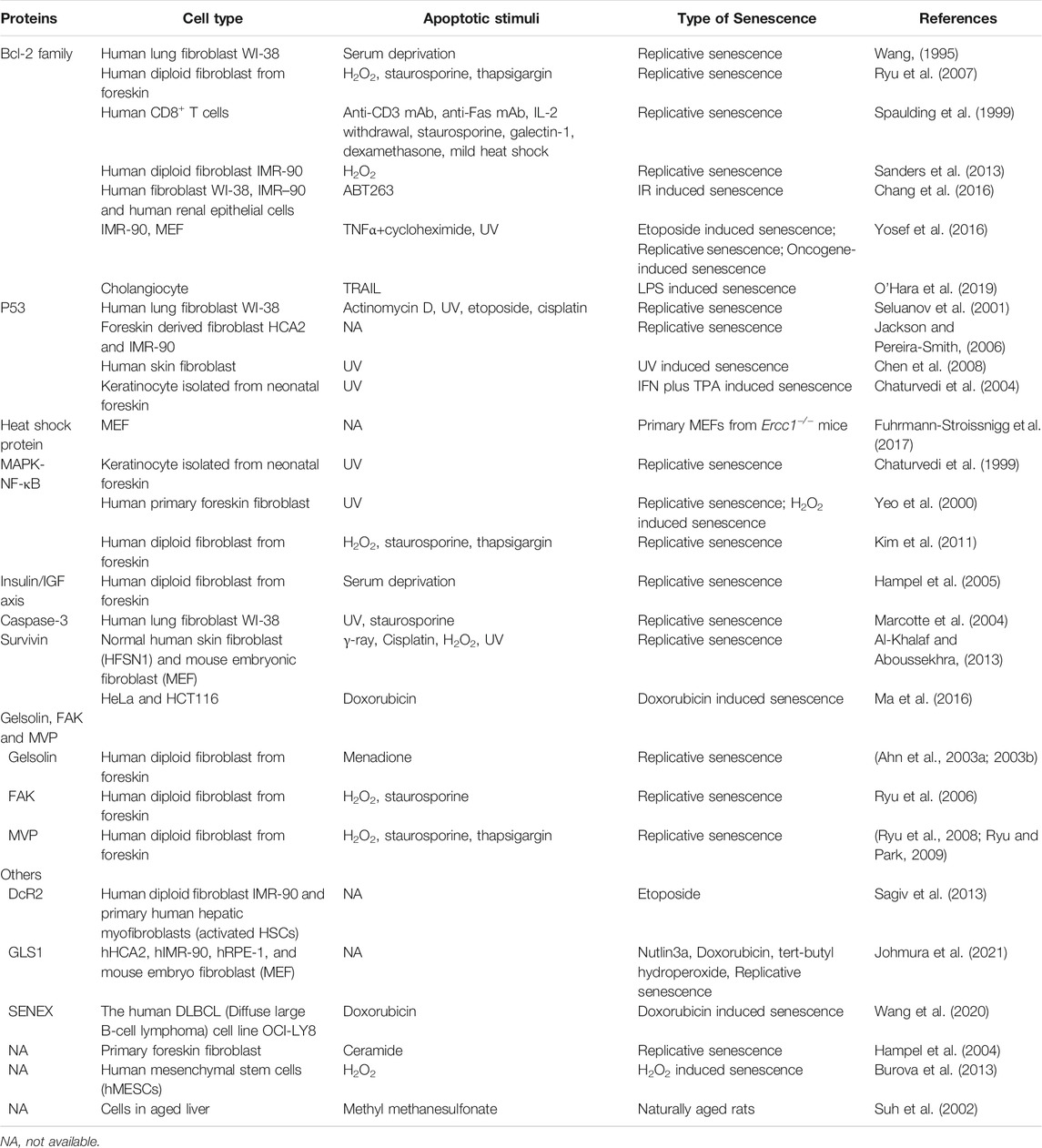
TABLE 2. Proteins implicated in the apoptotic resistance of senescent cells (SnCs).
The Bcl-2 Family Members
The Bcl-2 family proteins, consisting of antiapoptotic and proapoptotic proteins, play critical roles in the regulation of apoptosis (Youle and Strasser, 2008). The Bcl-2 antiapoptotic proteins are multi-BH-domain proteins including Bcl-2, Bcl-xL, Mcl-1, Bcl-w and Bfl1. They can inhibit apoptosis by binding to the multi-BH-domain and BH3-only proapoptotic proteins. Wang et al. first observed that aged human WI-38 cells were resistant to programmed cell death induced by serum deprivation. They found that the levels of Bcl-2 was undetectable in young WI-38 cells, but remained unchanged in senescent WI-38 cells after serum deprivation (Wang, 1995). This finding suggests that Bcl-2 might be primarily responsible for the resistance of cell death in senescent WI-38 cells. In HDF, several apoptotic stimuli such as H2O2, staurosporine and thapsigargin, can induce the downregulation of Bcl-2 and apoptosis in young cells but not in SnCs (Ryu et al., 2007). Spaulding et al. reported that replicative senescence in CD8+ T cells was associated with a significant increase in resistance to apoptosis, which was also associated with increased Bcl-2 expression (Spaulding et al., 1999). On the contrary, Bcl-2 was reduced in senescent HSC that were sensitive to TNF-α-induced cell death (Novo et al., 2006). Mechanistically, global and locus-specific histone modifications of chromatin regulate the gene expression of Bcl-2 and Bax in senescent fibroblasts, which in turn may mediate the resistance of apoptosis in SnCs (Sanders et al., 2013).
In contrast, Sasaki et al. observed the apoptotic resistance in several cell lines (human foreskin fibroblast strain HCA2, human fetal lung fibroblast strains TIG-1, TIG-3 and WI-38) when they became senescent (Sasaki et al., 2001). However, in these SnCs the Bcl-2 family members were not responsible for the resistance to apoptosis. Instead, they attributed the resistance to cell cycle arrest (Sasaki et al., 2001). Different from above findings, we and others demonstrated that Bcl-xL is primarily responsible for SnC resistance to apoptosis as inhibition of Bcl-xL with a Bcl-xL specific inhibitor (such as A-1331852 and A1155463) (Zhu et al., 2017) or a Bcl-2 and Bcl-xL dual inhibitor (such as ABT263 and ABT737) can effectively induce apoptosis in a variety of SnCs (Rochette and Brash, 2008; Chang et al., 2016; Yosef et al., 2016; Zhu et al., 2016; Baar et al., 2017; Zhu et al., 2017; He et al., 2020b), whereas inhibition of Bcl-2 alone with its specific inhibitor has no or weak effect on SnC survival (Chang et al., 2016; Yosef et al., 2016; He et al., 2020b). The induction of Bcl-xL in SnC and its role in mediating apoptotic resistance is likely regulated by the transcription factor ETS protooncogene 1 (ETS1) (O’Hara et al., 2019). Although ABT263 is one of the most potent and broad-spectrum senolytics identified to date (Zhu et al., 2015, 2017; Chang et al., 2016), Bcl-xL inhibition with ABT263 or other small molecular inhibitors induces platelet apoptosis and results in severe thrombocytopenia, which prevents their use in the clinic—even for tumor patients (Gandhi et al., 2011; Souers et al., 2013; Leverson et al., 2015; Ashkenazi et al., 2017). We have successfully used the PROTAC technology to overcome the on-target drug toxicity, and improved the senolytic activity of ABT263 (He et al., 2020b). As one of the most important Bcl-2 family members, Mcl-1 plays critical roles in suppressing apoptosis in tumor cells (Ramsey et al., 2018; Mukherjee et al., 2020; Seiller et al., 2020), which however may not affect the survival of SnCs, as inhibition of Mcl-1 with its specific inhibitor did not cause cell death in SnCs (Chang et al., 2016; He et al., 2020b). Of note is that Mcl-1 expression varies in SnCs induced by different stress-stimulus (Yosef et al., 2016), and it is a highly regulated protein with a short half life (Michels et al., 2005), implying that Mcl-1 may circumvent the inhibition of Bcl-2 or Bcl-xL and help SnCs to acquire apoptotic resistance. In sum, Bcl-2 family members are implicated in the apoptotic resistance of SnCs, but their expression levels and roles differ depending on cell types and senescence inducers.
The p53 Pathway
p53 is a well-known tumor suppressor that acts as a double-edged sword in the regulation of cellular senescence, aging, and tumor (Johmura and Nakanishi, 2016; Wu and Prives, 2018). The host defense function of p53 in protecting tissues against tumor growth via induction of apoptosis and cellular senescence has been widely studied in tumors. The p53 levels and activities increase when cells enter a presenescent state upon activation of the DNA damage response (DDR), triggering the initiation of cellular senescence (Hernandez-Segura et al., 2018). p53 can promote apoptosis through transcription-dependent and -independent mechanisms (Fridman and Lowe, 2003). The transcriptional pathway involves the induction of pro-apoptotic Bcl-2 family members (i.e., Bax, Bid, Noxa, and Puma), and repression of anti-apoptotic proteins (i.e., Bcl-2, Bcl-xL, and survivin). The non-transcriptional regulation of apoptosis by p53 includes its direct interaction with members of the Bcl-2 family anti-apoptotic proteins to control mitochondrial outer membrane permeabilization (MOMP) (Fridman and Lowe, 2003).
The role of p53 in regulating apoptotic resistance in SnCs has been reported in multiple cell types. Seluanov et al. studied the responses of cultured young and old WI-38 to a variety of genotoxic stresses and found that young cells were able to undergo p53-dependent and p53-independent apoptosis. In contrast, senescent fibroblasts were unable to undergo p53-dependent apoptosis. They concluded that stabilization of p53 in response to DNA damage is impaired in old fibroblasts, resulting in the resistance to apoptotic stimuli (Seluanov et al., 2001). Gansauge et al. also observed the same phenomenon in the same cell line (Gansauge et al., 1997). Similar findings were reported in senescent KCs (Chaturvedi et al., 2004), human skin fibroblasts (Chen et al., 2008) and HCA2 cells (Jackson and Pereira-Smith, 2006). The cellular levels of p53 usually increase in response to DNA damage stimuli. However, the increased levels of p53 subside when cells become fully senescent. This phenomenon was observed in senescent KCs (Kim et al., 2015), human fibroblast cell lines (Sisoula et al., 2011; Johmura and Nakanishi, 2016), human prostate epithelial and uroepithelial cells (Schwarze et al., 2001). The downregulation of p53 levels in SnCs may be attributable to the upregulation of C-terminus of Hsp70-interacting protein (CHIP) (Sisoula et al., 2011) and SCFFbxo22 (Johmura et al., 2016) E3 ligases, which promote p53 degradation through the ubiquitination-proteasome system. The reduction of p53 in SnCs may protect them from apoptosis and cause the accumulation of SnCs and higher prevalence of tumor during aging, which agrees with the finding that p53 activity was reduced along with accumulation of SnCs in aged tissues (Feng et al., 2007; Baker et al., 2016). Therefore, restoration of p53 activity has the potential to eliminate SnCs by inducing apoptosis. Baar et al. designed a peptide (termed as proxofim peptide) comprising part of the p53-interaction domain in FOXO4, which selectively induced p53 nuclear exclusion and cell-intrinsic apoptosis in SnCs (Baar et al., 2017; Zhang et al., 2020). However, there remain challenges using a peptide as a therapeutic. Inhibiting the interaction between MDM2 and p53 can also increase p53 stability and activity (Moll and Petrenko, 2003). UBX0101, an inhibitor of MDM2, has been shown to selectively kill SnCs in culture and treat post-traumatic osteoarthritis in vivo (Jeon et al., 2017). Nevertheless, UBX0101 was failed for the treatment of osteoarthritis in patients in a phase II clinical trial (Lane et al., 2021). We have previously reported that the ubiquitin-specific peptidase 7 (USP7) may be a novel target for senolysis as inhibition of USP7 can selectively induce apoptosis of SnCs by increasing the level of p53 (He et al., 2020a). However, USP7 is widely expressed in many tissues and has multiple substrates with different physiological functions (Zhou et al., 2018; Pozhidaeva and Bezsonova, 2019). It has yet to be determined whether USP7 can be safely used to clear SnCs without causing significant side effects.
Heat Shock Proteins
Heat shock proteins (HSPs) are evolutionarily conserved proteins whose expression is induced by different kinds of stresses (Jäättelä, 1999; Jego et al., 2013). They are involved in protein folding and maturation of various proteins and play an important role in regulation of cellular response to homeostatic challenges (Chatterjee and Burns, 2017). HSPs regulate protein assembly, secretion, transportation, translocation, and protein degradation. According to their molecular sizes, HSPs are grouped into six families, including HSP27, HSP40, HSP60, HSP70, HSP90 families, and the family of large HSPs (HSP110 and glucose-regulated protein 170, GRP170) (Chatterjee and Burns, 2017). When cells are subjected to stress, they can undergo either senescence or cell death and HSPs are involved in both two responses (Takayama et al., 2003; He et al., 2019; Srivastava et al., 2019; Kanugovi et al., 2020; Omer et al., 2020; Kanugovi Vijayavittal and Amere Subbarao, 2021). The role of HSPs in regulating cell apoptosis has been well reviewed elsewhere (Takayama et al., 2003; Lanneau et al., 2008). As the expression/activity of HSPs is significantly higher in tumors and responsive to different death stimuli (Jego et al., 2013), inhibition of HSPs has emerged as a novel therapeutic strategy for tumor therapy (Chatterjee and Burns, 2017).
Among the HSP families, HSP90 has been implicated as an anti-apoptotic and pro-survival factor in SnCs, and the inhibitor of HSP90 was identified as a new class of senolytic (Fuhrmann-Stroissnigg et al., 2017, 2018). Inhibition of HSP90 by its inhibitors, such as 17-DMAG, can sufficiently reduce the level of phosphorylated AKT and selectively induce apoptosis in SnCs (Fuhrmann-Stroissnigg et al., 2017, 2018). However, most of the known HSP90 inhibitors may have limited usage as senolytics because they can cause some dose-limiting toxicities and have poor pharmacokinetic profiles (Sanchez et al., 2020). These limitations may preclude their clinical use as an anti-aging agent as older people are more susceptible to adverse drug effects than younger individuals (He et al., 2020c).
The Autophagy-Lysosomal Pathway
Autophagy is an evolutionarily conserved process in eukaryotic cells, which is involved in scavenging and recycling senescent or damaged organelles and biological macromolecules by lysosomal degradation to maintain cellular homeostasis. Autophagy can be a physiologically or pathologically relevant program depending on the specific situation. In normal situations, autophagy can be induced by physiological signals such as starvation, to facilitate cell survival (Giampieri et al., 2019). Caloric restriction (CR) is one of the most important inducers of autophagy, leading to an increase of lifespan and delaying the onset of age-related diseases (Bergamini et al., 2007). Nevertheless, excessive autophagy is detrimental and results in “autophagic cell death” that describes a form of programmed cell death morphologically distinct from apoptosis. In autophagic programmed cell death, there is early degradation of organelles but preservation of cytoskeletal elements until late stages (Levine and Yuan, 2005). Different from necrosis, both apoptotic and autophagic cell death are characterized by the lack of a tissue inflammatory response. Autophagy have been linked to aging and various pathological conditions (Green et al., 2011; Giampieri et al., 2019), and thus it has become a major target for drug discovery and development (Kroemer, 2015; Leidal et al., 2018). However, as autophagy can act as both a cell survival and death mechanism, it is challenging to selectively turn on or turn off autophagic survival and death pathways in the treatment of autophagy-related diseases (Levine and Yuan, 2005). Beyond the abovementioned roles, autophagy has been shown to influence cellular immune responses (Ma et al., 2013). Particularly, autophagy can influence the antigenic profile of antigen-donor cells (ADCs) and their ability to release immunogenic signals (Michaud et al., 2011), as well as the survival, differentiation, and function of antigen-presenting cells (APCs) and T lymphocytes (Pua et al., 2007; Jia and He, 2011; Wildenberg et al., 2012; Fiegl et al., 2013). Autophagic responses in ADCs can enhance the release of “find-me” and “eat me” signals, which attract APC progenitors and facilitate antigen uptake, respectively (Ma et al., 2013). Theoretically, the autophagy-enhanced cellular immune responses may facilitate the immune system to recognize and eliminate SnCs, which has yet to be validated. The relationship between autophagy and cellular senescence is inconclusive (Gewirtz, 2013). Some studies suggest that autophagy is activated during senescence and inhibition of autophagy delays the senescence phenotype (Young et al., 2009; Gewirtz, 2013; Zhang et al., 2017), whereas increasing evidence reveals a negative correlation between them (Kang et al., 2011; Tai et al., 2017). For example, rapamycin, a mammalian target of rapamycin complex 1 (mTOR1) inhibitor and an autophagy activator, can efficiently suppress cellular senescence (Demidenko et al., 2009; Tai et al., 2017), and extend the life span of mice (Harrison et al., 2009). Fenofibrate (FN), a PPARα agonist used for dyslipidemia in humans, was reported to protect against cartilage degradation by reducing the number of senescent chondrocytes via inducing apoptosis in combination with increasing autophagic flux (Nogueira-Recalde et al., 2019). In addition, a BET family protein degrader was shown to provoke senolysis by targeting the attenuation of nonhomologous end joining (NHEJ) and autophagy in SnCs (Wakita et al., 2020). Lysosomes are degradative organelles essential for cell homeostasis that regulate various biological processes (Gómez-Sintes et al., 2016). Senescence-associated beta-galactosidase (SA-beta-gal) is the most common marker of lysosomal activity and one of the first tests used to determine senescence (Lee et al., 2006). Enhanced lysosomal activity can protect cells from oxidative stresses (Chakraborty et al., 2019; Nagakannan et al., 2020; Li et al., 2021), likely contributing to the resistance of apoptosis in SnCs. However, different forms of stress can induce lysosomal membrane permeabilization (LMP), leading to the translocation to the cytoplasm of intralysosomal components, such as cathepsins, inducing lysosomal-dependent cell death (LDCD) (Wang et al., 2018). Accumulating evidence indicate that aging significantly influences lysosomal activity by altering the physical and chemical properties of lysosomes (Gómez-Sintes et al., 2016). Most cells display an age-associated increase in lysosome number and size. The intracellular pH in SnCs was lowered by lysosomal membrane damage, but SnCs can resist the lowered pH by producing ammonia via glutaminolysis, which can neutralize the lower pH (Johmura et al., 2021). On the contrary, it was reported that lysosomes exhibited higher pH in aged animals than young mice (Liu et al., 2008). Lysosomal reacidification by inhibiting the ataxia telangiectasia mutated (ATM) induced functional recovery of the lysosome, which led to alleviated cellular senescence by accelerating the removal of dysfunctional mitochondria and recovering the mitochondrial function (Kang et al., 2017). Taken together, the autophagy-lysosomal pathway plays critical roles in maintaining cellular homeostasis, targeting the proteins in the pathway have become an attractive anti-aging strategy by either suppressing senescence or inducing apoptosis of SnCs.
Epigenetic Regulation
Alterations in the methylation of DNA or post-translational modification of histones, and other chromatin-associated proteins, can induce epigenetic changes that contribute to the aging process (López-Otín et al., 2013). We and others have previously reviewed the effects of DNA methylation on aging and longevity (Xiao et al., 2016; Sidler et al., 2017; Kane and Sinclair, 2019; Morris et al., 2019). Likewise, epigenetic changes have significant impact on the senescence phenotypes, notably the proliferative arrest and SASP (Nacarelli et al., 2017). For example, the INK4-ARF locus encodes proteins that drive cell growth arrest, and the polycomb group (PcG) proteins can epigenetically regulate the INK4-ARF locus, as well as catalyze histone modifications that promote changes in chromatin structure, leading to transcriptional repression (Simboeck et al., 2011). SIRT1, an NAD+-dependent protein deacetylase, suppresses the SASP through epigenetic gene regulation (Hayakawa et al., 2015; Hekmatimoghaddam et al., 2017). Besides the impacts on cellular senescence and SASP, epigenetic mechanisms are involved in the apoptotic resistance in SnCs. CpG nucleotide-rich regions in mammalian genomes are called CpG islands, which reside in very close proximity to gene promoters (Fatemi et al., 2005). In tumor cells, CpG islands at proapoptotic gene promoters are mostly hypermethylated due to methyltransferase (DNMT) overexpression (Roll et al., 2008). Hypermethylation blocks both intrinsic and extrinsic apoptosis by modulating the expression of major players of cell death cascade, as previously reviewed elsewhere (Elmallah and Micheau, 2019; Ozyerli-Goknar and Bagci-Onder, 2021). However, the expression of DNMT was found to be downregulated in SnCs although they harbour some features of the tumor epigenome (Cruickshanks et al., 2013). Thus, whether DNA methylation contributes to the apoptotic resistance in SnCs has yet to be investigated. In contrast, there is evidence showing that histone modification may affect the sensitivity of SnCs to cell death stimuli. Sanders et al. revealed that both global and locus-specific histone modifications of chromatin regulated altered Bcl-2:Bax gene expression in senescent fibroblasts, contributing to its apoptosis-resistant phenotype (Sanders et al., 2013). The heterochromatinization that surrounds and borders double-strand breaks (DSBs) enhances the pro-survival responses in SnCs, as evidenced by that HDAC inhibition triggered apoptosis (Di Micco et al., 2006; Paluvai et al., 2020). Panobinostat, an FDA-approved histone deacetylase inhibitor, was found to be a post-chemotherapy senolytic agent with the potential to kill persistent SnCs in non-small cell lung cancer (NSCLC) and head and neck squamous cell carcinoma (HNSCC) (Samaraweera et al., 2017). However, our preliminary data show that panobinostat was toxic to normal cells (data now shown). Therefore, whether the histone deacetylase inhibitors can be anti-aging senolytic targets has yet to be investigated.
The MAPK-NF-κB Axis
The nuclear factor κB (NF-κB) is a transcription factor complex consisting of homo- and heterodimers of five members of the Rel family including RelA (p65), RelB, c-Rel, NF-κB1 (p50/p105), and NF-κB2 (p52/p100). The NF-κB pathway transcriptionally controls a large set of target genes that play important roles in cell survival, inflammation, and immune responses (Hayden and Ghosh, 2008). Previous studies have correlated NF-κB signaling with cellular senescence. For example, indoxyl sulfate can induce cellular senescence via activation of the ROS-NF-κB-p53 pathway in proximal tubular cells (Shimizu et al., 2011). It has been well known that various DNA damage stimuli can activate NF-κB signaling, which stimulates the production of SASP in SnCs (Salminen et al., 2012), and regulates expression of genes that regulate apoptosis, cell cycle progression, and inflammation. Recently, we reported that SnCs not only expressed higher basal levels of various inflammatory cytokines and chemokines as a manifestation of the SASP, but also exhibited hyper-activation for the induction of a variety of inflammatory mediators in response to LPS, IL1β and TNFα stimulation (Budamagunta et al., 2021). This senescence-associated hyper-activation is mediated in part via the NF-κB pathway (Budamagunta et al., 2021). More importantly, NF-κB plays a crucial role in the induction of tumor resistance to apoptosis (Mortezaee et al., 2019). We previously reported that the curcumin analog EF24, a new senolytic which can downregulate anti-apoptotic family proteins, likely via the proteasome degradation of the Bcl-2 as the proteasome inhibitor MG132 partially prevented the degradation effect of EF24 (Li et al., 2019a). EF24 is also an anti-tumor agent that can kill melanoma cells effectively via downregulating the expression of Bcl-2 and IAP by inhibiting the NF-κB signaling (He et al., 2021). In an early study, senescent KCs were found to be resistant to apoptosis, which was associated with the anti-apoptotic role of NF-κB (Chaturvedi et al., 1999). NF-kB is upregulated in a variety of tissues with aging, and the inhibition of NF-kB has been shown to delay the onset of aging-related symptoms and pathologies such as diabetes, atherosclerosis, and tumor (Kanigur Sultuybek et al., 2019). Metformin is an anti-diabetic medication in type 2 diabetes (T2DM) (Dunn and Peters, 1995), and it has become a therapeutic candidate in the improvement of ageing and aging-related diseases (Hu et al., 2021). One of the major roles of metformin is to inhibit the NF-κB pathway, by which it reduce the susceptibility to age-related diseases (Kanigur Sultuybek et al., 2019). Many naturally-occurring compounds were reported to have anti-aging effects, either via triggering the apoptosis of SnCs (such as quercetin, fisetin, piperlongumine) (Li et al., 2019b), or inhibiting production of SASP via suppressing the NF-κB pathway (such as apigenin) (Lim et al., 2015; Perrott et al., 2017). In addition, some small chemical inhibitors of NF-κB, such as BAY 11-7082 (Pierce et al., 1997), also show good activity in reducing the expression of inflammatory cytokines (Lappas et al., 2005; Juliana et al., 2010; Zang et al., 2017), and protecting organs from oxidative injury (Kim et al., 2010; Kolati et al., 2015). However, more deep studies are needed to test whether NF-κB inhibitors could be safe anti-aging agents in the future.
The mitogen-activated protein kinase (MAPK) family can act as an upstream of the NF-κB pathway. MAP kinases are grouped into three families: stress-activated protein kinases (p38/SAPKs), Jun amino-terminal kinases (JNKs), and extracellular-signal-regulated kinases (ERKs). In cellular senescence, p38 induces the expression of the SASP largely by increasing NF-κB transcriptional activity (Freund et al., 2011; Budamagunta et al., 2021). JNKs play important roles in acquiring the resistance of senescent human fibroblasts to UV-induced DNA fragmentation (Yeo et al., 2000). In addition, ERK and p38 are also involved in the apoptotic resistance in SnCs (Kim et al., 2011). Kim et al. revealed that the nuclear translocation of activated ERK or p38 was implicated in the transduction of death signals, and that a decrease in nucleocytoplasmic trafficking of these proteins in SnCs caused the senescence-associated resistance to apoptosis (Kim et al., 2011). In conclusion, the MAPK-NF-κB axis plays important roles in mediating apoptotic resistance as well as modulating the production of various inflammatory cytokines in SnCs.
The Insulin/IGF Axis
The insulin/IGF signaling pathway plays a major role in determining the rate of aging in many species (Gems and Partridge, 2001; Cheng et al., 2005), which is an evolutionarily conserved mechanism from yeast to humans (Barbieri et al., 2003). In higher eukaryotes, signal transduction through the IGF pathway is modulated by IGF-binding protein (IGFBPs) family, among which IGFBP-3 regulates proliferation and survival in many mammalian cell types (Firth and Baxter, 2002). Moreover, another IGF protein IGFBP-7 can also induce cellular senescence and apoptosis through autocrine/paracrine pathways in melanocytes (Wajapeyee et al., 2008). However, why SnCs secret IGF proteins but can resist their pro-apoptotic effect is not known at the present. Hampel et al. found that IGFBP-3 accumulated in conditioned medium of senescent human fibroblasts, which may contribute to growth arrest of these cells. As IGFBP-3 can enhance apoptosis by activating intracellular regulators of apoptosis, whereas IGFBP-3 cannot be endocytosed by SnCs, which may contribute to the well-established apoptotic resistance of senescent human fibroblasts (Hampel et al., 2005). Further we also have previously reported that IGFBP-3 gene polymorphism rs11977526 with longevity in Chinese (He et al., 2014). These findings suggest that the insulin/IGF axis is an important determinant of aging, and acts as a mechanism by which SnCs acquire resistance to apoptosis.
Caspase-3
Caspases are a family of endoproteases that provide critical links in cell regulatory networks controlling inflammation and cell death. They are inactive until being cleaved at specific aspartate residues (McIlwain et al., 2013). Wang’s group reported the first observation of apoptotic resistance in senescent fibroblasts in 1995 (Wang, 1995). A few years later, they found that caspase-3 gene expression level was decreased in human senescent fibroblasts (WI-38) and could not be activated by UV or staurosporine treatment. They concluded that the resistance to apoptotic death seen in senescent fibroblasts was not simply due to increased Bcl-2 levels, but also attributable to a lack of caspase activity (Marcotte et al., 2004). However, in some cell types, cell death seems to be independent of caspase activity, such as in HDF isolated from skin, given that the treatment of HDF did not trigger the cleavage of caspase-3 or poly (ADP-ribose) polymerase (PARP) (Ryu et al., 2006).
Survivin
Survivin is an important anti-apoptotic protein. It is highly expressed in most tumors, which is also generally arised in cells of older individuals (Unruhe et al., 2016). Al-Khalaf et al. found that survivin and phospho-survivin were accumulated in aged normal human skin fibroblasts and mice organs, which may be attributable to HSP90-mediated stabilization of survivin. Inhibition of survivin by flavopiridol or shRNAs increased the apoptotic response of old fibroblasts to various genotoxic agents by restoring the pro-apoptotic Bax/Bcl-2 ratio and increasing the levels of cleaved caspase-3 and PARP (Al-Khalaf and Aboussekhra, 2013). Another study indicated that nuclear accumulation of Yes-associated protein (YAP) could promote the survival of senescent tumor cells by increasing the expression of survivin (Ma et al., 2016). Whether survivin can be a significant senolytic target has yet to determined.
Gelsolin, FAK and MVP
Park’s group showed that the protein and mRNA levels of gelsolin, a Ca2+-dependent actin regulatory protein, were increased in senescent HDF. Downregulation of gelsolin in senescent HDF resulted in increased sensitivity to menadione-induced apoptotic cell death (Ahn et al., 2003b; 2003a). The effect of gelsolin in mediating apoptotic resistance of SnCs was independent of Bcl-2 (Ahn et al., 2003a). Later, they reported that the focal adhesion kinase (FAK) might differently regulate apoptosis and focal adhesion formation in SnCs (Ryu et al., 2006). Furthermore, they identified a novel senolytic drug R406 that showed selective toxicity in HDF by inhibiting FAK and p38MAPK activity (Cho et al., 2020). They also found that the major vault protein (MVP) was markedly increased in senescent HDF as well as in aged organs, downregulation of MVP increased the sensitivity of senescent HDF to apoptosis by modulation of Bcl-2 expression (Ryu et al., 2008). MVP was a transcriptional target of p53 (An et al., 2009), being a potential therapeutic target for modulation of resistance to apoptosis (Ryu and Park, 2009). These new proteins involved in resistance to apoptosis expanded the scope of existed SCAPs, which have great potential to be senolytic targets.
The Extrinsic Apoptosis Pathway
The extrinsic apoptosis pathway is triggered by the binding of death ligands to their appropriate death receptors (DRs) on the cell surface (Sayers, 2011). The best-described death ligands belong to the TNF family of proteins, comprising TNF, FasL, and TRAIL, which are predominantly produced by immune cells such as T cells, NK cells, NKT cells, macrophages, and dendritic cells. The DRs include TNF receptor 1 (TNFR1), CD95 (Fas), and TNF-related apoptosis inducing ligand-receptor (Ashkenazi, 2008; Sayers, 2011; Nair et al., 2014). Upon ligand binding, DRs trimerizes and recruits specialized adaptor proteins via their death domain (DD), such as Fas-associated death domain (FADD), which transmits the death signal from the cell surface to the intracellular signaling pathways. In turn, DD can recruit pro-caspase-8 and form a death-inducing signaling complex (DISC) capable of activating inducer caspases‐8, which are then processed into its mature form. The mature caspases-8 subsequently activates downstream effector caspases such as caspase-3, and trigger apoptosis. DRs-mediated apoptosis can be inhibited by a protein called c-FLIP which can bind to DD and caspase-8, rendering them ineffective (French and Tschopp, 1999). In addition, decoy receptors (DcRs) have similar extracellular structure to DRs but lack an intracellular death domain, which can bind the ligand but cannot transmit death signals (Ashkenazi, 2002). Therefore, both c-FLIP and DcRs act as inhibitory mechanisms of extrinsic apoptosis. The expression of c-FLIP was reported to decline with age in normal thymus, and negatively associated with the levels of senescence markers in primary thymic epithelial cells, suggesting that cFLIP may not play a role in the apoptotic resistance of SnCs (Belharazem et al., 2017). Nevertheless, the expression of cFLIP was downregulated by senescence-secreted SASP via an activation of the Myc oncogene, which can sensitize pretransformed cells to TRAIL-induced apoptosis (Vjetrovic et al., 2014). These findings suggest that SnCs may not depend on cFLIP for survival, instead they may regulate cFLIP to prime pretransformed cells to undergo apoptosis. DcR2 (TNFRSF10D) have been shown to be upregulated in SnCs (Sagiv et al., 2013), which represents as a marker of cellular senescence (Chen et al., 2017). DcR2 may in part account for the resistance of SnCs to apoptosis, as suggested by that TRAIL induced twofold more killing in senescent IMR-90 cells with DcR2 silencing compared with the control (Sagiv et al., 2013). Similarly, DcR1 (TNFRSF10C) was also observed to be upregulated in SnCs and was viewed as a senescence marker (Shah et al., 2013; Wiley et al., 2017). As DcRs act as competitive inhibitors of death receptor signaling by death ligands (such as Fas or TRAIL), it is feasible to use the specific antibody of DcRs to enhance the killing effect of immune cells (such as NK cells) on SnCs. In addition, we can take advantage of the CAR-T strategy (Amor et al., 2020) to establish DcR-sepecific CAR T cells to kill SnCs as will be discussed later.
Surface Molecules and Immunosurveillance for Senolysis
Emerging molecules or proteins specifically present on the surface of SnCs have been discovered, which may play roles in the maintenance and survival of SnCs, targeting of which have been a novel senolytic strategy recently (Rossi and Abdelmohsen, 2021). The surface molecules have been shown to have a major role in the recognition and clearance of SnCs by the immune system. For example, SnCs express ligands for the NKG2D receptor of NK cells including MICA/B and ULBP2, which may drive the recognition and elimination of SnCs by NK-mediated cytotoxicity (Sagiv et al., 2016; Muñoz et al., 2019). SnCs express non-classical MHC molecule HLA-E, which interacts with the inhibitory receptor NKG2A of NK cells and highly differentiated CD8+ T cells to inhibit immune responses against SnCs. Accordingly, blocking the interaction between HLA-E and NKG2A boosts immune responses against SnCs (Pereira et al., 2019). Proteomics analysis identified dipeptidyl peptidase 4 (DPP4) as a novel protein marker on the surface of SnCs, targeting of which sensitized SnCs, but not dividing fibroblasts to cytotoxicity by natural killer cells (Kim et al., 2017). Amor et al. identified the urokinase-type plasminogen activator receptor (uPAR) as a cell-surface protein that is upregulated in SnCs in vitro and in vivo (Amor et al., 2020). They established uPAR-specific chimeric antigen receptor (CAR) T cells (CAR-T) to selectively clear SnCs, which extended the survival of mice with lung adenocarcinoma that were treated with MEK and CDK4/6 inhibitors to induce senescence, ameliorated liver fibrosis and improved liver function by clearing senescent hepatic stellate cells (Amor et al., 2020). In addition, Althubiti et al. have screened a series of the plasma membrane-associated proteins preferentially expressed on the surface of SnCs (Althubiti et al., 2014). The surface molecules and their implications in senolysis have been systematically summarized and reviewed (Rossi and Abdelmohsen, 2021). However, there are still some safety concerns on the use of immune system to eliminate SnCs. Although the surface molecules are highly expressed in SnCs, they can be expressed in proliferating cells, albeit at low levels, leading to side effects (Ravichandra et al., 2021). More researches are needed to verify the senolytic effect and evaluate the safety of immune clearance of SnCs.
Conclusions and Perspectives
Aging represents a progressive decline in the physiological properties of tissues/organs and the overall fitness of the organism. SnCs accumulate with age and play a casual role in aging as well age-related pathologies. One major characteristic of SnCs is their ability to resist apoptosis induced by different stimuli. For normal cells, increased resistance is an important safety mechanism against acute stress to maintain homeostasis. However, for SnCs, apoptotic resistance can lead to abnormal accumulation, which is detrimental to the organism. Senolytics and other novel strategies (PROTAC, β-galactosidase-modified prodrugs and CAR-T cells) were developed to selectively target SnCs and have shown great potential to delay aging and to treat age-related diseases. It is encouraging to know that several senolytics have been approved for clinical trial and have shown beneficial effects (Niedernhofer and Robbins, 2018; Thoppil and Riabowol, 2019; Kirkland and Tchkonia, 2020). The latest findings that senolytics can be used to prevent or treat virus infection-associated diseases, such as COVID-19 infection, expanding the applications of senolytics in clinic. Most of the current senolytics are discovered by targeting the SCAPs. However, the major challenge in developing senolytics is the high heterogenicity of SnCs, and their diverse origins. SnCs can resist apoptosis via different mechanisms depending on cell type and apoptotic stimuli. In this review, we systematically summarized the proteins or pathways involved in the apoptotic resistance of SnCs, which are not independent but rather interconnected. Inhibitors targeting some of the proteins or pathways have been developed as novel senolytics. However, whether the rest ones are senolytic targets has yet to be investigated. We believe that understanding of the underlying mechanisms for apoptotic resistance of SnCs can help to identify more targets that can be used to develop cell-specific or broad-spectrum therapeutic senolytics to clear SnCs. It should be noted that clearance of SnCs can cause undesirable side effects. For example, continuous or acute elimination of SnCs disrupted blood-tissue barriers with subsequent liver and perivascular tissue fibrosis and health deterioration (Grosse et al., 2020). More caution should be exercised for the systemic use of senolytics for health benefits (Figure 1B). We expect to witness more discoveries in novel and safe senolytics for the use as an intervention to treat SnC-related diseases in the near future.
Author Contributions
Conceptualization: YH and YZ; Writing-Original Draft Preparation: YH, LH, and HL; Writing-Review and Editing: YH, WL, JL, YY, DZ, and Q-PK; Literature collection: YH, HL, and LH.
Funding
This study was supported jointly and equally by grants from the National Key R&D Program of China (2018YFC2000400, 2018YFE0203700). Additional support was received from the Key Research Program (KFZD-SW-221), Key Research Program of Frontiers Science (QYZDB-SSW-SMC020), and Strategic Priority Research Program (XDPB17) of the Chinese Academy of Sciences, the National Natural Science Foundation of China (82171558, 31760310), the Basic Research Projects of Yunnan Province (202101AS070137, 202101BA070001-110), and the Key Research and Development Project of Hainan Province (ZDYF2021SHFZ227).
Conflict of Interest
DZ is a co-founder and a stockholder of Unity Biotechnology that develops senolytics as therapeutics for various age-related diseases. DZ and YH are inventors on several patent applications for the use of Bcl-xL and Bcl-2 inhibitors and PROTACs to clear SnCs.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank the members of the He laboratory for their thoughtful discussion and assistance and Zia Ur Rahman from Kong laboratory for editorial assistance.
References
Aguayo-Mazzucato, C., Andle, J., Lee, T. B., Midha, A., Talemal, L., Chipashvili, V., et al. (2019). Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cel Metab. 30, 129–142. doi:10.1016/j.cmet.2019.05.006
Ahn, J. S., Jang, I.-S., Kim, D.-I., Cho, K. A., Park, Y. H., Kim, K., et al. (2003a). Aging-associated Increase of Gelsolin for Apoptosis Resistance. Biochem. Biophysical Res. Commun. 312, 1335–1341. doi:10.1016/j.bbrc.2003.11.061
Ahn, J. S., Jang, I.-S., Rhim, J. H., Kim, K., Yeo, E.-J., and Park, S. C. (2003b). Gelsolin for Senescence-Associated Resistance to Apoptosis. Ann. N. Y Acad. Sci. 1010, 493–495. doi:10.1196/annals.1299.090
Al-Khalaf, H. H., and Aboussekhra, A. (2013). Survivin Expression Increases during Aging and Enhances the Resistance of Aged Human Fibroblasts to Genotoxic Stress. Age 35, 549–562. doi:10.1007/s11357-011-9378-2
Alimirah, F., Pulido, T., Valdovinos, A., Alptekin, S., Chang, E., Jones, E., et al. (2020). Cellular Senescence Promotes Skin Carcinogenesis through p38MAPK and p44/42MAPK Signaling. Cancer Res. 80, 3606–3619. doi:10.1158/0008-5472.CAN-20-0108
Alt, E. U., Senst, C., Murthy, S. N., Slakey, D. P., Dupin, C. L., Chaffin, A. E., et al. (2012). Aging Alters Tissue Resident Mesenchymal Stem Cell Properties. Stem Cel Res. 8, 215–225. doi:10.1016/j.scr.2011.11.002
Althubiti, M., Lezina, L., Carrera, S., Jukes-Jones, R., Giblett, S. M., Antonov, A., et al. (2014). Characterization of Novel Markers of Senescence and Their Prognostic Potential in Cancer. Cell Death Dis 5, e1528. doi:10.1038/cddis.2014.489
Amor, C., Feucht, J., Leibold, J., Ho, Y.-J., Zhu, C., Alonso-Curbelo, D., et al. (2020). Senolytic CAR T Cells Reverse Senescence-Associated Pathologies. Nature 583, 127–132. doi:10.1038/s41586-020-2403-9
An, H.-J., Ryu, S.-J., Kim, S.-Y., Choi, H.-R., Chung, J.-H., and Park, S.-C. (2009). Age Associated High Level of Major Vault Protein Is P53 Dependent. Cell Biochem. Funct. 27, 289–295. doi:10.1002/cbf.1571
Ashkenazi, A., Fairbrother, W. J., Leverson, J. D., and Souers, A. J. (2017). From Basic Apoptosis Discoveries to Advanced Selective BCL-2 Family Inhibitors. Nat. Rev. Drug Discov. 16, 273–284. doi:10.1038/nrd.2016.253
Ashkenazi, A. (2002). Targeting Death and Decoy Receptors of the Tumour-Necrosis Factor Superfamily. Nat. Rev. Cancer 2, 420–430. doi:10.1038/nrc821
Ashkenazi, A. (2008). Targeting the Extrinsic Apoptosis Pathway in Cancer. Cytokine Growth Factor. Rev. 19, 325–331. doi:10.1016/j.cytogfr.2008.04.001
Baar, M. P., Brandt, R. M. C., Putavet, D. A., Klein, J. D. D., Derks, K. W. J., Bourgeois, B. R. M., et al. (2017). Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 169, 132–147. doi:10.1016/j.cell.2017.02.031
Baker, D. J., Childs, B. G., Durik, M., Wijers, M. E., Sieben, C. J., Zhong, J., et al. (2016). Naturally Occurring p16Ink4a-Positive Cells Shorten Healthy Lifespan. Nature 530, 184–189. doi:10.1038/nature16932
Baker, D. J., Wijshake, T., Tchkonia, T., LeBrasseur, N. K., Childs, B. G., van de Sluis, B., et al. (2011). Clearance of p16Ink4a-Positive Senescent Cells Delays Ageing-Associated Disorders. Nature 479, 232–236. doi:10.1038/nature10600
Barbieri, M., Bonafè, M., Franceschi, C., and Paolisso, G. (2003). Insulin/IGF-I-signaling Pathway: an Evolutionarily Conserved Mechanism of Longevity from Yeast to Humans. Am. J. Physiology-Endocrinology Metab. 285, E1064–E1071. doi:10.1152/ajpendo.00296.2003
Barrera-Vázquez, O. S., Gómez-Verjan, J. C., and Magos-Guerrero, G. A. (2021). Chemoinformatic Screening for the Selection of Potential Senolytic Compounds from Natural Products. Biomolecules 11, 467. doi:10.3390/biom11030467
Belharazem, D., Grass, A., Paul, C., Vitacolonna, M., Schalke, B., Rieker, R. J., et al. (2017). Increased cFLIP Expression in Thymic Epithelial Tumors Blocks Autophagy via NF-Κb Signalling. Oncotarget 8, 89580–89594. doi:10.18632/oncotarget.15929
Bergamini, E., Cavallini, G., Donati, A., and Gori, Z. (2007). The Role of Autophagy in Aging: its Essential Part in the Anti-aging Mechanism of Caloric Restriction. Ann. New York Acad. Sci. 1114, 69–78. doi:10.1196/annals.1396.020
Budamagunta, V., Manohar-Sindhu, S., Yang, Y., He, Y., Traktuev, D. O., Foster, T. C., et al. (2021). Senescence-associated Hyper-Activation to Inflammatory Stimuli In Vitro. Aging 13, 19088–19107. doi:10.18632/aging.203396
Burova, E., Borodkina, A., Shatrova, A., and Nikolsky, N. (2013). Sublethal Oxidative Stress Induces the Premature Senescence of Human Mesenchymal Stem Cells Derived from Endometrium. Oxidative Med. Cell Longevity 2013, 1–12. doi:10.1155/2013/474931
Burton, D. G. A., and Krizhanovsky, V. (2014). Physiological and Pathological Consequences of Cellular Senescence. Cell. Mol. Life Sci. 71, 4373–4386. doi:10.1007/s00018-014-1691-3
Cai, Y., Zhou, H., Zhu, Y., Sun, Q., Ji, Y., Xue, A., et al. (2020). Elimination of Senescent Cells by β-galactosidase-targeted Prodrug Attenuates Inflammation and Restores Physical Function in Aged Mice. Cell Res 30, 574–589. doi:10.1038/s41422-020-0314-9
Camell, C. D., Yousefzadeh, M. J., Zhu, Y., Prata, L. G. P. L., Huggins, M. A., Pierson, M., et al. (2021). Senolytics Reduce Coronavirus-Related Mortality in Old Mice. Science 373, eabe4832. doi:10.1126/science.abe4832
Campisi, J., Kapahi, P., Lithgow, G. J., Melov, S., Newman, J. C., and Verdin, E. (2019). From Discoveries in Ageing Research to Therapeutics for Healthy Ageing. Nature 571, 183–192. doi:10.1038/s41586-019-1365-2
Chakraborty, D., Felzen, V., Hiebel, C., Stürner, E., Perumal, N., Manicam, C., et al. (2019). Enhanced Autophagic-Lysosomal Activity and Increased BAG3-Mediated Selective Macroautophagy as Adaptive Response of Neuronal Cells to Chronic Oxidative Stress. Redox Biol. 24, 101181. doi:10.1016/j.redox.2019.101181
Chang, J., Wang, Y., Shao, L., Laberge, R.-M., Demaria, M., Campisi, J., et al. (2016). Clearance of Senescent Cells by ABT263 Rejuvenates Aged Hematopoietic Stem Cells in Mice. Nat. Med. 22, 78–83. doi:10.1038/nm.4010
Chatterjee, S., and Burns, T. (2017). Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Ijms 18, 1978. doi:10.3390/ijms18091978
Chaturvedi, V., Qin, J.-Z., Denning, M. F., Choubey, D., Diaz, M. O., and Nickoloff, B. J. (1999). Apoptosis in Proliferating, Senescent, and Immortalized Keratinocytes. J. Biol. Chem. 274, 23358–23367. doi:10.1074/jbc.274.33.23358
Chaturvedi, V., Qin, J.-Z., Stennett, L., Choubey, D., and Nickoloff, B. J. (2004). Resistance to UV-Induced Apoptosis in Human Keratinocytes during Accelerated Senescence Is Associated with Functional Inactivation of P53. J. Cel. Physiol. 198, 100–109. doi:10.1002/jcp.10392
Chen, J., Chen, K.-H., Fu, B.-Q., Zhang, W., Dai, H., Lin, L.-R., et al. (2017). Isolation and Identification of Senescent Renal Tubular Epithelial Cells Using Immunomagnetic Beads Based on DcR2. Exp. Gerontol. 95, 116–127. doi:10.1016/j.exger.2017.04.008
Chen, W., Kang, J., Xia, J., Li, Y., Yang, B., Chen, B., et al. (2008). p53-related Apoptosis Resistance and Tumor Suppression Activity in UVB-Induced Premature Senescent Human Skin Fibroblasts. Int. J. Mol. Med. 21, 645–653. doi:10.3892/ijmm.21.5.645
Cheng, C.-L., Gao, T.-Q., Wang, Z., and Li, D.-D. (2005). Role of Insulin/insulin-like Growth Factor 1 Signaling Pathway in Longevity. Wjg 11, 1891–1895. doi:10.3748/wjg.v11.i13.1891
Childs, B. G., Baker, D. J., Wijshake, T., Conover, C. A., Campisi, J., and van Deursen, J. M. (2016). Senescent Intimal Foam Cells Are Deleterious at All Stages of Atherosclerosis. Science 354, 472–477. doi:10.1126/science.aaf6659
Childs, B. G., Durik, M., Baker, D. J., and van Deursen, J. M. (2015). Cellular Senescence in Aging and Age-Related Disease: from Mechanisms to Therapy. Nat. Med. 21, 1424–1435. doi:10.1038/nm.4000
Childs, B. G., Gluscevic, M., Baker, D. J., Laberge, R.-M., Marquess, D., Dananberg, J., et al. (2017). Senescent Cells: an Emerging Target for Diseases of Ageing. Nat. Rev. Drug Discov. 16, 718–735. doi:10.1038/nrd.2017.116
Chinta, S. J., Woods, G., Demaria, M., Rane, A., Zou, Y., McQuade, A., et al. (2018). Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson's Disease. Cel Rep. 22, 930–940. doi:10.1016/j.celrep.2017.12.092
Cho, H.-J., Yang, E. J., Park, J. T., Kim, J.-R., Kim, E.-C., Jung, K.-J., et al. (2020). Identification of SYK Inhibitor, R406 as a Novel Senolytic Agent. Aging 12, 8221–8240. doi:10.18632/aging.103135
Cruickshanks, H. A., McBryan, T., Nelson, D. M., Vanderkraats, N. D., Shah, P. P., van Tuyn, J., et al. (2013). Senescent Cells Harbour Features of the Cancer Epigenome. Nat. Cel Biol 15, 1495–1506. doi:10.1038/ncb2879
Demaria, M., O'Leary, M. N., Chang, J., Shao, L., Liu, S., Alimirah, F., et al. (2017). Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 7, 165–176. doi:10.1158/2159-8290.CD-16-0241
Demaria, M., Ohtani, N., Youssef, S. A., Rodier, F., Toussaint, W., Mitchell, J. R., et al. (2014). An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Developmental Cel 31, 722–733. doi:10.1016/j.devcel.2014.11.012
Demidenko, Z. N., Zubova, S. G., Bukreeva, E. I., Pospelov, V. A., Pospelova, T. V., and Blagosklonny, M. V. (2009). Rapamycin Decelerates Cellular Senescence. Cell Cycle 8, 1888–1895. doi:10.4161/cc.8.12.8606
Di Micco, R., Fumagalli, M., Cicalese, A., Piccinin, S., Gasparini, P., Luise, C., et al. (2006). Oncogene-induced Senescence Is a DNA Damage Response Triggered by DNA Hyper-Replication. Nature 444, 638–642. doi:10.1038/nature05327
Di Micco, R., Krizhanovsky, V., Baker, D., and d’Adda di Fagagna, F. (2021). Cellular Senescence in Ageing: from Mechanisms to Therapeutic Opportunities. Nat. Rev. Mol. Cel Biol 22, 75–95. doi:10.1038/s41580-020-00314-w
Dunn, C. J., and Peters, D. H. (1995). Metformin. Drugs 49, 721–749. doi:10.2165/00003495-199549050-00007
Elmallah, M. I. Y., and Micheau, O. (2019). Epigenetic Regulation of TRAIL Signaling: Implication for Cancer Therapy. Cancers 11, 850. doi:10.3390/cancers11060850
Elmore, S. (2007). Apoptosis: a Review of Programmed Cell Death. Toxicol. Pathol. 35, 495–516. doi:10.1080/01926230701320337
Erekat, N. S. (2021). Apoptosis and its Therapeutic Implications in Neurodegenerative Diseases. Clin. Anat. 35, 65–78. doi:10.1002/ca.23792
Fatemi, M., Pao, M. M., Jeong, S., Gal-Yam, E. N., Egger, G., Weisenberger, D. J., et al. (2005). Footprinting of Mammalian Promoters: Use of a CpG DNA Methyltransferase Revealing Nucleosome Positions at a Single Molecule Level. Nucleic Acids Res. 33, e176. doi:10.1093/nar/gni180
Feng, Z., Hu, W., Teresky, A. K., Hernando, E., Cordon-Cardo, C., and Levine, A. J. (2007). Declining P53 Function in the Aging Process: a Possible Mechanism for the Increased Tumor Incidence in Older Populations. Proc. Natl. Acad. Sci. 104, 16633–16638. doi:10.1073/pnas.0708043104
Fiegl, D., Kägebein, D., Liebler-Tenorio, E. M., Weisser, T., Sens, M., Gutjahr, M., et al. (2013). Amphisomal Route of MHC Class I Cross-Presentation in Bacteria-Infected Dendritic Cells. J.I. 190, 2791–2806. doi:10.4049/jimmunol.1202741
Firth, S. M., and Baxter, R. C. (2002). Cellular Actions of the Insulin-like Growth Factor Binding Proteins. Endocr. Rev. 23, 824–854. doi:10.1210/er.2001-0033
French, L. E., and Tschopp, J. (1999). Inhibition of Death Receptor Signaling by FLICE-Inhibitory Protein as a Mechanism for Immune Escape of Tumors. J. Exp. Med. 190, 891–894. doi:10.1084/jem.190.7.891
Freund, A., Patil, C. K., and Campisi, J. (2011). p38MAPK Is a Novel DNA Damage Response-independent Regulator of the Senescence-Associated Secretory Phenotype. EMBO J. 30, 1536–1548. doi:10.1038/emboj.2011.69
Fridman, J. S., and Lowe, S. W. (2003). Control of Apoptosis by P53. Oncogene 22, 9030–9040. doi:10.1038/sj.onc.1207116
Fuhrmann-Stroissnigg, H., Ling, Y. Y., Zhao, J., McGowan, S. J., Zhu, Y., Brooks, R. W., et al. (2017). Identification of HSP90 Inhibitors as a Novel Class of Senolytics. Nat. Commun. 8, 422. doi:10.1038/s41467-017-00314-z
Fuhrmann-Stroissnigg, H., Niedernhofer, L. J., and Robbins, P. D. (2018). Hsp90 Inhibitors as Senolytic Drugs to Extend Healthy Aging. Cell Cycle 17, 1048–1055. doi:10.1080/15384101.2018.1475828
Gandhi, L., Camidge, D. R., Ribeiro de Oliveira, M., Bonomi, P., Gandara, D., Khaira, D., et al. (2011). Phase I Study of Navitoclax (ABT-263), a Novel Bcl-2 Family Inhibitor, in Patients with Small-Cell Lung Cancer and Other Solid Tumors. Jco 29, 909–916. doi:10.1200/JCO.2010.31.6208
Gansauge, S., Gansauge, F., Gause, H., Poch, B., Schoenberg, M. H., and Beger, H. G. (1997). The Induction of Apoptosis in Proliferating Human Fibroblasts by Oxygen Radicals Is Associated with a P53- and p21WAF1CIP1 Induction. FEBS Lett. 404, 6–10. doi:10.1016/s0014-5793(97)00059-8
Ge, M., Hu, L., Ao, H., Zi, M., Kong, Q., and He, Y. (2021). Senolytic Targets and New Strategies for Clearing Senescent Cells. Mech. Ageing Development 195, 111468. doi:10.1016/j.mad.2021.111468
Gems, D., and Partridge, L. (2001). Insulin/IGF Signalling and Ageing: Seeing the Bigger Picture. Curr. Opin. Genet. Dev. 11, 287–292. doi:10.1016/s0959-437x(00)00192-1
Giampieri, F., Afrin, S., Forbes-Hernandez, T. Y., Gasparrini, M., Cianciosi, D., Reboredo-Rodriguez, P., et al. (2019). Autophagy in Human Health and Disease: Novel Therapeutic Opportunities. Antioxid. Redox Signaling 30, 577–634. doi:10.1089/ars.2017.7234
Gómez-Sintes, R., Ledesma, M. D., and Boya, P. (2016). Lysosomal Cell Death Mechanisms in Aging. Ageing Res. Rev. 32, 150–168. doi:10.1016/j.arr.2016.02.009
Gorgoulis, V., Adams, P. D., Alimonti, A., Bennett, D. C., Bischof, O., Bishop, C., et al. (2019). Cellular Senescence: Defining a Path Forward. Cell 179, 813–827. doi:10.1016/j.cell.2019.10.005
Green, D. R., Galluzzi, L., and Kroemer, G. (2011). Mitochondria and the Autophagy-Inflammation-Cell Death axis in Organismal Aging. Science 333, 1109–1112. doi:10.1126/science.1201940
Grosse, L., Wagner, N., Emelyanov, A., Molina, C., Lacas-Gervais, S., Wagner, K.-D., et al. (2020). Defined p16High Senescent Cell Types Are Indispensable for Mouse Healthspan. Cel Metab. 32, 87–99. doi:10.1016/j.cmet.2020.05.002
Guerrero, A., Guiho, R., Herranz, N., Uren, A., Withers, D. J., Martínez‐Barbera, J. P., et al. (2020). Galactose‐modified Duocarmycin Prodrugs as Senolytics. Aging Cell 19, e13133. doi:10.1111/acel.13133
Guerrero, A., Herranz, N., Sun, B., Wagner, V., Gallage, S., Guiho, R., et al. (2019). Cardiac Glycosides Are Broad-Spectrum Senolytics. Nat. Metab. 1, 1074–1088. doi:10.1038/s42255-019-0122-z
Günther, C., Neumann, H., Neurath, M. F., and Becker, C. (2013). Apoptosis, Necrosis and Necroptosis: Cell Death Regulation in the Intestinal Epithelium. Gut 62, 1062–1071. doi:10.1136/gutjnl-2011-301364
Hampel, B., Malisan, F., Niederegger, H., Testi, R., and Jansen-Dürr, P. (2004). Differential Regulation of Apoptotic Cell Death in Senescent Human Cells. Exp. Gerontol. 39, 1713–1721. doi:10.1016/j.exger.2004.05.010
Hampel, B., Wagner, M., Teis, D., Zwerschke, W., Huber, L. A., and Jansen-Durr, P. (2005). Apoptosis Resistance of Senescent Human Fibroblasts Is Correlated with the Absence of Nuclear IGFBP-3. Aging Cell 4, 325–330. doi:10.1111/j.1474-9726.2005.00180.x
Harrison, D. E., Strong, R., Sharp, Z. D., Nelson, J. F., Astle, C. M., Flurkey, K., et al. (2009). Rapamycin Fed Late in Life Extends Lifespan in Genetically Heterogeneous Mice. Nature 460, 392–395. doi:10.1038/nature08221
Hayakawa, T., Iwai, M., Aoki, S., Takimoto, K., Maruyama, M., Maruyama, W., et al. (2015). SIRT1 Suppresses the Senescence-Associated Secretory Phenotype through Epigenetic Gene Regulation. PLoS One 10, e0116480. doi:10.1371/journal.pone.0116480
Hayden, M. S., and Ghosh, S. (2008). Shared Principles in NF-Κb Signaling. Cell 132, 344–362. doi:10.1016/j.cell.2008.01.020
Hayflick, L., and Moorhead, P. S. (1961). The Serial Cultivation of Human Diploid Cell Strains. Exp. Cel Res. 25, 585–621. doi:10.1016/0014-4827(61)90192-6
Hayflick, L. (1965). The Limited In Vitro Lifetime of Human Diploid Cell Strains. Exp. Cel Res. 37, 614–636. doi:10.1016/0014-4827(65)90211-9
He, M. Y., Xu, S. B., Qu, Z. H., Guo, Y. M., Liu, X. C., Cong, X. X., et al. (2019). Hsp90β Interacts with MDM2 to Suppress P53‐dependent Senescence during Skeletal Muscle Regeneration. Aging Cell 18, e13003. doi:10.1111/acel.13003
He, Y.-H., Lu, X., Yang, L.-Q., Xu, L.-Y., and Kong, Q.-P. (2014). Association of the Insulin-like Growth Factor Binding Protein 3 (IGFBP-3) Polymorphism with Longevity in Chinese Nonagenarians and Centenarians. Aging 6, 944–951. doi:10.18632/aging.100703
He, Y., Li, W., Lv, D., Zhang, X., Zhang, X., Ortiz, Y. T., et al. (2020a). Inhibition of USP7 Activity Selectively Eliminates Senescent Cells in Part via Restoration of P53 Activity. Aging Cell 19, e13117. doi:10.1111/acel.13117
He, Y., Li, W., Zhang, J., Yang, Y., Qian, Y., and Zhou, D. (2021). The Curcumin Analog EF24 Is Highly Active against Chemotherapy- Resistant Melanoma Cells. Ccdt 21, 608–618. doi:10.2174/1568009621666210303092921
He, Y., Zhang, X., Chang, J., Kim, H.-N., Zhang, P., Wang, Y., et al. (2020b). Using Proteolysis-Targeting Chimera Technology to Reduce Navitoclax Platelet Toxicity and Improve its Senolytic Activity. Nat. Commun. 11, 1996. doi:10.1038/s41467-020-15838-0
He, Y., Zheng, G., and Zhou, D. (2020c). “Senolytic Drug Development,” in “Senolytic Drug Development,” in Senolytics In Disease, Ageing And Longevity. Editors D. Muñoz-Espin, and M. Demaria (Cham: Springer International Publishing), 3–20. doi:10.1007/978-3-030-44903-2_1
Hekmatimoghaddam, S., Dehghani Firoozabadi, A., Zare-Khormizi, M. R., and Pourrajab, F. (2017). Sirt1 and Parp1 as Epigenome Safeguards and microRNAs as SASP-Associated Signals, in Cellular Senescence and Aging. Ageing Res. Rev. 40, 120–141. doi:10.1016/j.arr.2017.10.001
Hernandez-Segura, A., de Jong, T. V., Melov, S., Guryev, V., Campisi, J., and Demaria, M. (2017). Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 27, 2652–2660. doi:10.1016/j.cub.2017.07.033
Hernandez-Segura, A., Nehme, J., and Demaria, M. (2018). Hallmarks of Cellular Senescence. Trends Cel Biol. 28, 436–453. doi:10.1016/j.tcb.2018.02.001
Herranz, N., and Gil, J. (2018). Mechanisms and Functions of Cellular Senescence. J. Clin. Invest. 128, 1238–1246. doi:10.1172/JCI95148
Hu, D., Xie, F., Xiao, Y., Lu, C., Zhong, J., Huang, D., et al. (2021). Metformin: A Potential Candidate for Targeting Aging Mechanisms. Aging Dis. 12, 480–493. doi:10.14336/AD.2020.0702
Jäättelä, M. (1999). Heat Shock Proteins as Cellular Lifeguards. Ann. Med. 31, 261–271. doi:10.3109/07853899908995889
Jackson, J. G., and Pereira-Smith, O. M. (2006). p53 Is Preferentially Recruited to the Promoters of Growth Arrest Genes P21 and GADD45 during Replicative Senescence of normal Human Fibroblasts. Cancer Res. 66, 8356–8360. doi:10.1158/0008-5472.CAN-06-1752
Jana, B., Kim, S., Chae, J. B., Chung, H., Kim, C., and Ryu, J. H. (2021). Mitochondrial Membrane Disrupting Molecules for Selective Killing of Senescent Cells. Chembiochem 22, 3391–3397. doi:10.1002/cbic.202100412
Jego, G., Hazoumé, A., Seigneuric, R., and Garrido, C. (2013). Targeting Heat Shock Proteins in Cancer. Cancer Lett. 332, 275–285. doi:10.1016/j.canlet.2010.10.014
Jeon, O. H., Kim, C., Laberge, R.-M., Demaria, M., Rathod, S., Vasserot, A. P., et al. (2017). Local Clearance of Senescent Cells Attenuates the Development of post-traumatic Osteoarthritis and Creates a Pro-regenerative Environment. Nat. Med. 23, 775–781. doi:10.1038/nm.4324
Jia, W., and He, Y.-W. (2011). Temporal Regulation of Intracellular Organelle Homeostasis in T Lymphocytes by Autophagy. J.I. 186, 5313–5322. doi:10.4049/jimmunol.1002404
Johmura, Y., and Nakanishi, M. (2016). Multiple Facets of P53 in Senescence Induction and Maintenance. Cancer Sci. 107, 1550–1555. doi:10.1111/cas.13060
Johmura, Y., Sun, J., Kitagawa, K., Nakanishi, K., Kuno, T., Naiki-Ito, A., et al. (2016). SCFFbxo22-KDM4A Targets Methylated P53 for Degradation and Regulates Senescence. Nat. Commun. 7, 10574. doi:10.1038/ncomms10574
Johmura, Y., Yamanaka, T., Omori, S., Wang, T.-W., Sugiura, Y., Matsumoto, M., et al. (2021). Senolysis by Glutaminolysis Inhibition Ameliorates Various Age-Associated Disorders. Science 371, 265–270. doi:10.1126/science.abb5916
Juliana, C., Fernandes-Alnemri, T., Wu, J., Datta, P., Solorzano, L., Yu, J.-W., et al. (2010). Anti-inflammatory Compounds Parthenolide and Bay 11-7082 Are Direct Inhibitors of the Inflammasome. J. Biol. Chem. 285, 9792–9802. doi:10.1074/jbc.M109.082305
Kane, A. E., and Sinclair, D. A. (2019). Epigenetic Changes during Aging and Their Reprogramming Potential. Crit. Rev. Biochem. Mol. Biol. 54, 61–83. doi:10.1080/10409238.2019.1570075
Kang, H. T., Lee, K. B., Kim, S. Y., Choi, H. R., and Park, S. C. (2011). Autophagy Impairment Induces Premature Senescence in Primary Human Fibroblasts. PLoS One 6, e23367. doi:10.1371/journal.pone.0023367
Kang, H. T., Park, J. T., Choi, K., Kim, Y., Choi, H. J. C., Jung, C. W., et al. (2017). Chemical Screening Identifies ATM as a Target for Alleviating Senescence. Nat. Chem. Biol. 13, 616–623. doi:10.1038/nchembio.2342
Kanigur Sultuybek, G., Soydas, T., and Yenmis, G. (2019). NF‐κB as the Mediator of Metformin's Effect on Ageing and Ageing‐related Diseases. Clin. Exp. Pharmacol. Physiol. 46, 413–422. doi:10.1111/1440-1681.13073
Kanugovi, A. V., Joseph, C., Siripini, S., Paithankar, K., and Amere, S. S. (2020). Compromising the Constitutive P16 INK4a Expression Sensitizes Human Neuroblastoma Cells to Hsp90 Inhibition and Promotes Premature Senescence. J. Cel Biochem 121, 2770–2781. doi:10.1002/jcb.29493
Kanugovi Vijayavittal, A., and Amere Subbarao, S. (2021). The Conformation-specific Hsp90 Inhibition Interferes with the Oncogenic RAF Kinase Adaptation and Triggers Premature Cellular Senescence, Hence, Acts as a Tumor Suppressor Mechanism. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1868, 118943. doi:10.1016/j.bbamcr.2020.118943
Kavathia, N., Jain, A., Walston, J., Beamer, B. A., and Fedarko, N. S. (2009). Serum Markers of Apoptosis Decrease with Age and Cancer Stage. Aging 1, 652–663. doi:10.18632/aging.100069
Kawada, N. (2006). Human Hepatic Stellate Cells Are Resistant to Apoptosis: Implications for Human Fibrogenic Liver Disease. Gut 55, 1073–1074. doi:10.1136/gut.2005.090449
Kile, B. T. (2014). The Role of Apoptosis in Megakaryocytes and Platelets. Br. J. Haematol. 165, 217–226. doi:10.1111/bjh.12757
Kim, K. M., Noh, J. H., Bodogai, M., Martindale, J. L., Yang, X., Indig, F. E., et al. (2017). Identification of Senescent Cell Surface Targetable Protein DPP4. Genes Dev. 31, 1529–1534. doi:10.1101/gad.302570.117
Kim, R. H., Kang, M. K., Kim, T., Yang, P., Bae, S., Williams, D. W., et al. (2015). Regulation of P53 during Senescence in normal Human Keratinocytes. Aging Cell 14, 838–846. doi:10.1111/acel.12364
Kim, S. Y., Ryu, S. J., Kang, H. T., Choi, H. R., and Park, S. C. (2011). Defective Nuclear Translocation of Stress-Activated Signaling in Senescent Diploid Human Fibroblasts: a Possible Explanation for Aging-Associated Apoptosis Resistance. Apoptosis 16, 795–807. doi:10.1007/s10495-011-0612-2
Kim, Y. S., Kim, J. S., Kwon, J. S., Jeong, M. H., Cho, J. G., Park, J. C., et al. (2010). BAY 11-7082, a Nuclear Factor-.KAPPA.B Inhibitor, Reduces Inflammation and Apoptosis in a Rat Cardiac Ischemia-Reperfusion Injury Model. Int. Heart J. 51, 348–353. doi:10.1536/ihj.51.348
Kirkland, J. L., and Tchkonia, T. (2017). Cellular Senescence: A Translational Perspective. EBioMedicine 21, 21–28. doi:10.1016/j.ebiom.2017.04.013
Kirkland, J. L., and Tchkonia, T. (2020). Senolytic Drugs: from Discovery to Translation. J. Intern. Med. 288, 518–536. doi:10.1111/joim.13141
Kolati, S. R., Kasala, E. R., Bodduluru, L. N., Mahareddy, J. R., Uppulapu, S. K., Gogoi, R., et al. (2015). BAY 11-7082 Ameliorates Diabetic Nephropathy by Attenuating Hyperglycemia-Mediated Oxidative Stress and Renal Inflammation via NF-Κb Pathway. Environ. Toxicol. Pharmacol. 39, 690–699. doi:10.1016/j.etap.2015.01.019
Kroemer, G. (2015). Autophagy: a Druggable Process that Is Deregulated in Aging and Human Disease. J. Clin. Invest. 125, 1–4. doi:10.1172/JCI78652
Lane, N., Hsu, B., Visich, J., Xie, B., Khan, A., and Dananberg, J. (2021). A Phase 2, Randomized, Double-Blind, Placebo-Controlled Study of Senolytic Molecule UBX0101 in the Treatment of Painful Knee Osteoarthritis. Osteoarthritis and Cartilage 29, S52–S53. doi:10.1016/j.joca.2021.02.077
Lanneau, D., Brunet, M., Frisan, E., Solary, E., Fontenay, M., and Garrido, C. (2008). Heat Shock Proteins: Essential Proteins for Apoptosis Regulation. J. Cell. Mol. Med. 12, 743–761. doi:10.1111/j.1582-4934.2008.00273.x
Lappas, M., Yee, K., Permezel, M., and Rice, G. E. (2005). Sulfasalazine and BAY 11-7082 Interfere with the Nuclear Factor-Κb and IκB Kinase Pathway to Regulate the Release of Proinflammatory Cytokines from Human Adipose Tissue and Skeletal Muscle In Vitro. Endocrinology 146, 1491–1497. doi:10.1210/en.2004-0809
Lee, B. Y., Han, J. A., Im, J. S., Morrone, A., Johung, K., Goodwin, E. C., et al. (2006). Senescence-associated β-galactosidase Is Lysosomal β-galactosidase. Aging Cell 5, 187–195. doi:10.1111/j.1474-9726.2006.00199.x
Lee, S., Yu, Y., Trimpert, J., Benthani, F., Mairhofer, M., Richter-Pechanska, P., et al. (2021). Virus-induced Senescence Is a Driver and Therapeutic Target in COVID-19. Nature 599, 283–289. doi:10.1038/s41586-021-03995-1
Leidal, A. M., Levine, B., and Debnath, J. (2018). Autophagy and the Cell Biology of Age-Related Disease. Nat. Cel Biol 20, 1338–1348. doi:10.1038/s41556-018-0235-8
Leverson, J. D., Phillips, D. C., Mitten, M. J., Boghaert, E. R., Diaz, D., Tahir, S. K., et al. (2015). Exploiting Selective BCL-2 Family Inhibitors to Dissect Cell Survival Dependencies and Define Improved Strategies for Cancer Therapy. Sci. Transl. Med. 7, 279ra40. doi:10.1126/scitranslmed.aaa4642
Levine, B., and Yuan, J. (2005). Autophagy in Cell Death: an Innocent Convict? J. Clin. Invest. 115, 2679–2688. doi:10.1172/JCI26390
Li, W., He, Y., Zhang, R., Zheng, G., and Zhou, D. (2019a). The Curcumin Analog EF24 Is a Novel Senolytic Agent. Aging 11, 771–782. doi:10.18632/aging.101787
Li, W., Qin, L., Feng, R., Hu, G., Sun, H., He, Y., et al. (2019b). Emerging Senolytic Agents Derived from Natural Products. Mech. Ageing Development 181, 1–6. doi:10.1016/j.mad.2019.05.001
Li, Z., Zhu, Y.-T., Xiang, M., Qiu, J.-L., Luo, S.-Q., and Lin, F. (2021). Enhanced Lysosomal Function Is Critical for Paclitaxel Resistance in Cancer Cells: Reversed by Artesunate. Acta Pharmacol. Sin 42, 624–632. doi:10.1038/s41401-020-0445-z
Lim, H., Park, H., and Kim, H. P. (2015). Effects of Flavonoids on Senescence-Associated Secretory Phenotype Formation from Bleomycin-Induced Senescence in BJ Fibroblasts. Biochem. Pharmacol. 96, 337–348. doi:10.1016/j.bcp.2015.06.013
Liu, J., Lu, W., Reigada, D., Nguyen, J., Laties, A. M., and Mitchell, C. H. (2008). Restoration of Lysosomal pH in RPE Cells from Cultured Human and ABCA4−/−Mice: Pharmacologic Approaches and Functional Recovery. Invest. Ophthalmol. Vis. Sci. 49, 772–780. doi:10.1167/iovs.07-0675
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The Hallmarks of Aging. Cell 153, 1194–1217. doi:10.1016/j.cell.2013.05.039
Ma, K., Xu, Q., Wang, S., Zhang, W., Liu, M., Liang, S., et al. (2016). Nuclear Accumulation of Yes-Associated Protein (YAP) Maintains the Survival of Doxorubicin-Induced Senescent Cells by Promoting Survivin Expression. Cancer Lett. 375, 84–91. doi:10.1016/j.canlet.2016.02.045
Ma, Y., Galluzzi, L., Zitvogel, L., and Kroemer, G. (2013). Autophagy and Cellular Immune Responses. Immunity 39, 211–227. doi:10.1016/j.immuni.2013.07.017
Marcotte, R., Lacelle, C., and Wang, E. (2004). Senescent Fibroblasts Resist Apoptosis by Downregulating Caspase-3. Mech. Ageing Development 125, 777–783. doi:10.1016/j.mad.2004.07.007
Mattson, M. P., Duan, W., Pedersen, W. A., and Culmsee, C. (2001). Neurodegenerative Disorders and Ischemic Brain Diseases. Apoptosis 6, 69–81. doi:10.1023/a:1009676112184
McIlwain, D. R., Berger, T., and Mak, T. W. (2013). Caspase Functions in Cell Death and Disease. Cold Spring Harbor Perspect. Biol. 5, a008656. doi:10.1101/cshperspect.a008656
Michaud, M., Martins, I., Sukkurwala, A. Q., Adjemian, S., Ma, Y., Pellegatti, P., et al. (2011). Autophagy-dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science 334, 1573–1577. doi:10.1126/science.1208347
Michels, J., Johnson, P. W. M., and Packham, G. (2005). Mcl-1. Int. J. Biochem. Cel Biol. 37, 267–271. doi:10.1016/j.biocel.2004.04.007
Mohiuddin, M., and Kasahara, K. (2021). The Emerging Role of Cellular Senescence in Complications of COVID-19. Cancer Treat. Res. Commun. 28, 100399. doi:10.1016/j.ctarc.2021.100399
Moiseeva, O., Guillon, J., and Ferbeyre, G. (2020). Senescence: A Program in the Road to Cell Elimination and Cancer. Semin. Cancer Biol. S1044-579X (20), 30277–7. doi:10.1016/j.semcancer.2020.12.017
Monti, D., Salvioli, S., Capri, M., Malorni, W., Straface, E., Cossarizza, A., et al. (2000). Decreased Susceptibility to Oxidative Stress-Induced Apoptosis of Peripheral Blood Mononuclear Cells from Healthy Elderly and Centenarians. Mech. Ageing Dev. 121, 239–250. doi:10.1016/s0047-6374(00)00220-7
Morris, B. J., Willcox, B. J., and Donlon, T. A. (2019). Genetic and Epigenetic Regulation of Human Aging and Longevity. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1865, 1718–1744. doi:10.1016/j.bbadis.2018.08.039
Mortezaee, K., Najafi, M., Farhood, B., Ahmadi, A., Shabeeb, D., and Musa, A. E. (2019). NF‐κB Targeting for Overcoming Tumor Resistance and normal Tissues Toxicity. J. Cel Physiol 234, 17187–17204. doi:10.1002/jcp.28504
Mukherjee, N., Amato, C. M., Skees, J., Todd, K. J., Lambert, K. A., Robinson, W. A., et al. (2020). Simultaneously Inhibiting BCL2 and MCL1 Is a Therapeutic Option for Patients with Advanced Melanoma. Cancers 12, 2182. doi:10.3390/cancers12082182
Muñoz, D. P., Yannone, S. M., Daemen, A., Sun, Y., Vakar-Lopez, F., Kawahara, M., et al. (2019). Targetable Mechanisms Driving Immunoevasion of Persistent Senescent Cells Link Chemotherapy-Resistant Cancer to Aging. JCI Insight 4, 124716. doi:10.1172/jci.insight.124716
Muñoz-Espín, D., Cañamero, M., Maraver, A., Gómez-López, G., Contreras, J., Murillo-Cuesta, S., et al. (2013). Programmed Cell Senescence during Mammalian Embryonic Development. Cell 155, 1104–1118. doi:10.1016/j.cell.2013.10.019
Nacarelli, T., Liu, P., and Zhang, R. (2017). Epigenetic Basis of Cellular Senescence and its Implications in Aging. Genes 8, 343. doi:10.3390/genes8120343
Nagakannan, P., Tabeshmehr, P., and Eftekharpour, E. (2020). Oxidative Damage of Lysosomes in Regulated Cell Death Systems: Pathophysiology and Pharmacologic Interventions. Free Radic. Biol. Med. 157, 94–127. doi:10.1016/j.freeradbiomed.2020.04.001
Nair, P., Lu, M., Petersen, S., and Ashkenazi, A. (2014). Apoptosis Initiation through the Cell-Extrinsic Pathway. Methods Enzymol. 544, 99–128. doi:10.1016/B978-0-12-417158-9.00005-4
Nehme, J., Borghesan, M., Mackedenski, S., Bird, T. G., and Demaria, M. (2020). Cellular Senescence as a Potential Mediator of COVID‐19 Severity in the Elderly. Aging Cell 19, e13237. doi:10.1111/acel.13237
Niedernhofer, L. J., and Robbins, P. D. (2018). Senotherapeutics for Healthy Ageing. Nat. Rev. Drug Discov. 17, 377. doi:10.1038/nrd.2018.44
Nogueira-Recalde, U., Lorenzo-Gómez, I., Blanco, F. J., Loza, M. I., Grassi, D., Shirinsky, V., et al. (2019). Fibrates as Drugs with Senolytic and Autophagic Activity for Osteoarthritis Therapy. EBioMedicine 45, 588–605. doi:10.1016/j.ebiom.2019.06.049
Novo, E., Marra, F., Zamara, E., Valfrè di Bonzo, L., Monitillo, L., Cannito, S., et al. (2006). Overexpression of Bcl-2 by Activated Human Hepatic Stellate Cells: Resistance to Apoptosis as a Mechanism of Progressive Hepatic Fibrogenesis in Humans. Gut 55, 1174–1182. doi:10.1136/gut.2005.082701
O'Hara, S. P., Splinter, P. L., Trussoni, C. E., Guicciardi, M. E., Splinter, N. P., Al Suraih, M. S., et al. (2019). The Transcription Factor ETS1 Promotes Apoptosis Resistance of Senescent Cholangiocytes by Epigenetically Up-Regulating the Apoptosis Suppressor BCL2L1. J. Biol. Chem. 294, 18698–18713. doi:10.1074/jbc.RA119.010176
Ogrodnik, M., Miwa, S., Tchkonia, T., Tiniakos, D., Wilson, C. L., Lahat, A., et al. (2017). Cellular Senescence Drives Age-dependent Hepatic Steatosis. Nat. Commun. 8, 15691. doi:10.1038/ncomms15691
Omer, A., Patel, D., Moran, J. L., Lian, X. J., Di Marco, S., and Gallouzi, I.-E. (2020). Autophagy and Heat-Shock Response Impair Stress Granule Assembly during Cellular Senescence. Mech. Ageing Development 192, 111382. doi:10.1016/j.mad.2020.111382
Ozsvari, B., Nuttall, J. R., Sotgia, F., and Lisanti, M. P. (2018). Azithromycin and Roxithromycin Define a New Family of “Senolytic” Drugs that Target Senescent Human Fibroblasts. Aging 10, 3294–3307. doi:10.18632/aging.101633
Ozyerli-Goknar, E., and Bagci-Onder, T. (2021). Epigenetic Deregulation of Apoptosis in Cancers. Cancers 13, 3210. doi:10.3390/cancers13133210
Palmer, A. K., Tchkonia, T., LeBrasseur, N. K., Chini, E. N., Xu, M., and Kirkland, J. L. (2015). Cellular Senescence in Type 2 Diabetes: A Therapeutic Opportunity. Diabetes 64, 2289–2298. doi:10.2337/db14-1820
Paluvai, H., Di Giorgio, E., and Brancolini, C. (2020). The Histone Code of Senescence. Cells 9, 466. doi:10.3390/cells9020466
Pereira, B. I., Devine, O. P., Vukmanovic-Stejic, M., Chambers, E. S., Subramanian, P., Patel, N., et al. (2019). Senescent Cells Evade Immune Clearance via HLA-E-Mediated NK and CD8+ T Cell Inhibition. Nat. Commun. 10, 2387. doi:10.1038/s41467-019-10335-5
Perrott, K. M., Wiley, C. D., Desprez, P.-Y., and Campisi, J. (2017). Apigenin Suppresses the Senescence-Associated Secretory Phenotype and Paracrine Effects on Breast Cancer Cells. Geroscience 39, 161–173. doi:10.1007/s11357-017-9970-1
Pierce, J. W., Schoenleber, R., Jesmok, G., Best, J., Moore, S. A., Collins, T., et al. (1997). Novel Inhibitors of Cytokine-Induced IκBα Phosphorylation and Endothelial Cell Adhesion Molecule Expression Show Anti-inflammatory Effects In Vivo. J. Biol. Chem. 272, 21096–21103. doi:10.1074/jbc.272.34.21096
Polyak, K., Wu, T.-T., Hamilton, S. R., Kinzler, K. W., and Vogelstein, B. (1997). Less Death in the Dying. Cell Death Differ 4, 242–246. doi:10.1038/sj.cdd.4400226
Pozhidaeva, A., and Bezsonova, I. (2019). USP7: Structure, Substrate Specificity, and Inhibition. DNA Repair 76, 30–39. doi:10.1016/j.dnarep.2019.02.005
Pua, H. H., Dzhagalov, I., Chuck, M., Mizushima, N., and He, Y.-W. (2007). A Critical Role for the Autophagy Gene Atg5 in T Cell Survival and Proliferation. J. Exp. Med. 204, 25–31. doi:10.1084/jem.20061303
Ramsey, H. E., Fischer, M. A., Lee, T., Gorska, A. E., Arrate, M. P., Fuller, L., et al. (2018). A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 8, 1566–1581. doi:10.1158/2159-8290.CD-18-0140
Ravichandra, A., Filliol, A., and Schwabe, R. F. (2021). Chimeric Antigen Receptor T Cells as Senolytic and Antifibrotic Therapy. Hepatology 73, 1227–1229. doi:10.1002/hep.31596
Rochette, P. J., and Brash, D. E. (2008). Progressive Apoptosis Resistance Prior to Senescence and Control by the Anti-apoptotic Protein BCL-xL. Mech. Ageing Development 129, 207–214. doi:10.1016/j.mad.2007.12.007
Roll, J. D., Rivenbark, A. G., Jones, W. D., and Coleman, W. B. (2008). DNMT3b Overexpression Contributes to a Hypermethylator Phenotype in Human Breast Cancer Cell Lines. Mol. Cancer 7, 15. doi:10.1186/1476-4598-7-15
Rossi, M., and Abdelmohsen, K. (2021). The Emergence of Senescent Surface Biomarkers as Senotherapeutic Targets. Cells 10, 1740. doi:10.3390/cells10071740
Ryu, S. J., An, H. J., Oh, Y. S., Choi, H. R., Ha, M. K., and Park, S. C. (2008). On the Role of Major Vault Protein in the Resistance of Senescent Human Diploid Fibroblasts to Apoptosis. Cel Death Differ 15, 1673–1680. doi:10.1038/cdd.2008.96
Ryu, S. J., Cho, K. A., Oh, Y. S., and Park, S. C. (2006). Role of Src-specific Phosphorylation Site on Focal Adhesion Kinase for Senescence-Associated Apoptosis Resistance. Apoptosis 11, 303–313. doi:10.1007/s10495-006-3978-9
Ryu, S. J., Oh, Y. S., and Park, S. C. (2007). Failure of Stress-Induced Downregulation of Bcl-2 Contributes to Apoptosis Resistance in Senescent Human Diploid Fibroblasts. Cel Death Differ 14, 1020–1028. doi:10.1038/sj.cdd.4402091
Ryu, S. J., and Park, S. C. (2009). Targeting Major Vault Protein in Senescence-Associated Apoptosis Resistance. Expert Opin. Ther. Targets 13, 479–484. doi:10.1517/14728220902832705
Sagiv, A., Biran, A., Yon, M., Simon, J., Lowe, S. W., and Krizhanovsky, V. (2013). Granule Exocytosis Mediates Immune Surveillance of Senescent Cells. Oncogene 32, 1971–1977. doi:10.1038/onc.2012.206
Sagiv, A., Burton, D. G. A., Moshayev, Z., Vadai, E., Wensveen, F., Ben-Dor, S., et al. (2016). NKG2D Ligands Mediate Immunosurveillance of Senescent Cells. Aging 8, 328–344. doi:10.18632/aging.100897
Sainz, R. M., Mayo, J. C., Reiter, R. J., Tan, D. X., and Rodriguez, C. (2003). Apoptosis in Primary Lymphoid Organs with Aging. Microsc. Res. Tech. 62, 524–539. doi:10.1002/jemt.10414
Salminen, A., Kauppinen, A., and Kaarniranta, K. (2012). Emerging Role of NF-Κb Signaling in the Induction of Senescence-Associated Secretory Phenotype (SASP). Cell Signal. 24, 835–845. doi:10.1016/j.cellsig.2011.12.006
Salminen, A., Ojala, J., and Kaarniranta, K. (2011). Apoptosis and Aging: Increased Resistance to Apoptosis Enhances the Aging Process. Cel. Mol. Life Sci. 68, 1021–1031. doi:10.1007/s00018-010-0597-y
Samaraweera, L., Adomako, A., Rodriguez-Gabin, A., and McDaid, H. M. (2017). A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci. Rep. 7, 1900. doi:10.1038/s41598-017-01964-1
Sanchez, J., Carter, T. R., Cohen, M. S., and Blagg, B. S. J. (2020). Old and New Approaches to Target the Hsp90 Chaperone. Ccdt 20, 253–270. doi:10.2174/1568009619666191202101330
Sanders, Y. Y., Liu, H., Zhang, X., Hecker, L., Bernard, K., Desai, L., et al. (2013). Histone Modifications in Senescence-Associated Resistance to Apoptosis by Oxidative Stress. Redox Biol. 1, 8–16. doi:10.1016/j.redox.2012.11.004
Sarkisian, C. J., Keister, B. A., Stairs, D. B., Boxer, R. B., Moody, S. E., and Chodosh, L. A. (2007). Dose-dependent Oncogene-Induced Senescence In Vivo and its Evasion during Mammary Tumorigenesis. Nat. Cel Biol 9, 493–505. doi:10.1038/ncb1567
Sasaki, M., Kumazaki, T., Takano, H., Nishiyama, M., and Mitsui, Y. (2001). Senescent Cells Are Resistant to Death Despite Low Bcl-2 Level. Mech. Ageing Development 122, 1695–1706. doi:10.1016/s0047-6374(01)00281-0
Sayers, T. J. (2011). Targeting the Extrinsic Apoptosis Signaling Pathway for Cancer Therapy. Cancer Immunol. Immunother. 60, 1173–1180. doi:10.1007/s00262-011-1008-4
Schwarze, S. R., Shi, Y., Fu, V. X., Watson, P. A., and Jarrard, D. F. (2001). Role of Cyclin-dependent Kinase Inhibitors in the Growth Arrest at Senescence in Human Prostate Epithelial and Uroepithelial Cells. Oncogene 20, 8184–8192. doi:10.1038/sj.onc.1205049
Schwarzenbach, C., Tatsch, L., Brandstetter Vilar, J., Rasenberger, B., Beltzig, L., Kaina, B., et al. (2021). Targeting C-IAP1, C-IAP2, and Bcl-2 Eliminates Senescent Glioblastoma Cells Following Temozolomide Treatment. Cancers 13, 3585. doi:10.3390/cancers13143585
Seiller, C., Maiga, S., Touzeau, C., Bellanger, C., Kervoëlen, C., Descamps, G., et al. (2020). Dual Targeting of BCL2 and MCL1 Rescues Myeloma Cells Resistant to BCL2 and MCL1 Inhibitors Associated with the Formation of BAX/BAK Hetero-Complexes. Cel Death Dis 11, 316. doi:10.1038/s41419-020-2505-1
Seluanov, A., Gorbunova, V., Falcovitz, A., Sigal, A., Milyavsky, M., Zurer, I., et al. (2001). Change of the Death Pathway in Senescent Human Fibroblasts in Response to DNA Damage Is Caused by an Inability to Stabilize P53. Mol. Cel Biol 21, 1552–1564. doi:10.1128/MCB.21.5.1552-1564.2001
Shah, P. P., Donahue, G., Otte, G. L., Capell, B. C., Nelson, D. M., Cao, K., et al. (2013). Lamin B1 Depletion in Senescent Cells Triggers Large-Scale Changes in Gene Expression and the Chromatin Landscape. Genes Dev. 27, 1787–1799. doi:10.1101/gad.223834.113
Shimizu, H., Bolati, D., Adijiang, A., Muteliefu, G., Enomoto, A., Nishijima, F., et al. (2011). NF-κB Plays an Important Role in Indoxyl Sulfate-Induced Cellular Senescence, Fibrotic Gene Expression, and Inhibition of Proliferation in Proximal Tubular Cells. Am. J. Physiology-Cell Physiol. 301, C1201–C1212. doi:10.1152/ajpcell.00471.2010
Sidler, C., Kovalchuk, O., and Kovalchuk, I. (2017). Epigenetic Regulation of Cellular Senescence and Aging. Front. Genet. 8, 138. doi:10.3389/fgene.2017.00138
Simboeck, E., Ribeiro, J. D., Teichmann, S., and Di Croce, L. (2011). Epigenetics and Senescence: Learning from the INK4-ARF Locus. Biochem. Pharmacol. 82, 1361–1370. doi:10.1016/j.bcp.2011.07.084
Singh, R., Letai, A., and Sarosiek, K. (2019). Regulation of Apoptosis in Health and Disease: the Balancing Act of BCL-2 Family Proteins. Nat. Rev. Mol. Cel Biol 20, 175–193. doi:10.1038/s41580-018-0089-8
Sisoula, C., Trachana, V., Patterson, C., and Gonos, E. S. (2011). CHIP-dependent P53 Regulation Occurs Specifically during Cellular Senescence. Free Radic. Biol. Med. 50, 157–165. doi:10.1016/j.freeradbiomed.2010.10.701
Soto-Gamez, A., Quax, W. J., and Demaria, M. (2019). Regulation of Survival Networks in Senescent Cells: From Mechanisms to Interventions. J. Mol. Biol. 431, 2629–2643. doi:10.1016/j.jmb.2019.05.036
Souers, A. J., Leverson, J. D., Boghaert, E. R., Ackler, S. L., Catron, N. D., Chen, J., et al. (2013). ABT-199, a Potent and Selective BCL-2 Inhibitor, Achieves Antitumor Activity while Sparing Platelets. Nat. Med. 19, 202–208. doi:10.1038/nm.3048
Spaulding, C., Guo, W., and Effros, R. B. (1999). Resistance to Apoptosis in Human CD8+ T Cells that Reach Replicative Senescence after Multiple Rounds of Antigen-specific Proliferation☆. Exp. Gerontol. 34, 633–644. doi:10.1016/s0531-5565(99)00033-9
Srivastava, S., Vishwanathan, V., Birje, A., Sinha, D., and D’Silva, P. (2019). Evolving Paradigms on the Interplay of Mitochondrial Hsp70 Chaperone System in Cell Survival and Senescence. Crit. Rev. Biochem. Mol. Biol. 54, 517–536. doi:10.1080/10409238.2020.1718062
Suh, Y., Lee, K.-A., Kim, W.-H., Han, B.-G., Vijg, J., and Park, S. C. (2002). Aging Alters the Apoptotic Response to Genotoxic Stress. Nat. Med. 8, 3–4. doi:10.1038/nm0102-3
Tai, H., Wang, Z., Gong, H., Han, X., Zhou, J., Wang, X., et al. (2017). Autophagy Impairment with Lysosomal and Mitochondrial Dysfunction Is an Important Characteristic of Oxidative Stress-Induced Senescence. Autophagy 13, 99–113. doi:10.1080/15548627.2016.1247143
Takasugi, M., Okada, R., Takahashi, A., Virya Chen, D., Watanabe, S., and Hara, E. (2017). Small Extracellular Vesicles Secreted from Senescent Cells Promote Cancer Cell Proliferation through EphA2. Nat. Commun. 8, 15729. doi:10.1038/ncomms15728
Takayama, S., Reed, J. C., and Homma, S. (2003). Heat-shock Proteins as Regulators of Apoptosis. Oncogene 22, 9041–9047. doi:10.1038/sj.onc.1207114
Thoppil, H., and Riabowol, K. (2019). Senolytics: A Translational Bridge between Cellular Senescence and Organismal Aging. Front. Cel Dev. Biol. 7, 367. doi:10.3389/fcell.2019.00367
Tower, J. (2015). Programmed Cell Death in Aging. Ageing Res. Rev. 23, 90–100. doi:10.1016/j.arr.2015.04.002
Triana-Martínez, F., Picallos-Rabina, P., Da Silva-Álvarez, S., Pietrocola, F., Llanos, S., Rodilla, V., et al. (2019). Identification and Characterization of Cardiac Glycosides as Senolytic Compounds. Nat. Commun. 10, 4731. doi:10.1038/s41467-019-12888-x
Unruhe, B., Schröder, E., Wünsch, D., and Knauer, S. K. (2016). An Old Flame Never Dies: Survivin in Cancer and Cellular Senescence. Gerontology 62, 173–181. doi:10.1159/000432398
Vjetrovic, J., Shankaranarayanan, P., Mendoza‐Parra, M. A., and Gronemeyer, H. (2014). Senescence‐secreted Factors Activate M Yc and Sensitize Pretransformed Cells to TRAIL‐induced Apoptosis. Aging Cell 13, 487–496. doi:10.1111/acel.12197
Wajapeyee, N., Serra, R. W., Zhu, X., Mahalingam, M., and Green, M. R. (2008). Oncogenic BRAF Induces Senescence and Apoptosis through Pathways Mediated by the Secreted Protein IGFBP7. Cell 132, 363–374. doi:10.1016/j.cell.2007.12.032
Wakita, M., Takahashi, A., Sano, O., Loo, T. M., Imai, Y., Narukawa, M., et al. (2020). A BET Family Protein Degrader Provokes Senolysis by Targeting NHEJ and Autophagy in Senescent Cells. Nat. Commun. 11, 1935. doi:10.1038/s41467-020-15719-6
Wang, E. (1995). Senescent Human Fibroblasts Resist Programmed Cell Death, and Failure to Suppress Bcl2 Is Involved. Cancer Res. 55, 2284–2292.
Wang, F., Gómez-Sintes, R., and Boya, P. (2018). Lysosomal Membrane Permeabilization and Cell Death. Traffic 19, 918–931. doi:10.1111/tra.12613
Wang, J., Tao, Q., Pan, Y., Wanyan, Z., Zhu, F., Xu, X., et al. (2020). Stress‐induced Premature Senescence Activated by the SENEX Gene Mediates Apoptosis Resistance of Diffuse Large B‐cell Lymphoma via Promoting Immunosuppressive Cells and Cytokines. Immun. Inflamm. Dis. 8, 672–683. doi:10.1002/iid3.356
Wang, Y., Chang, J., Liu, X., Zhang, X., Zhang, S., Zhang, X., et al. (2016). Discovery of Piperlongumine as a Potential Novel lead for the Development of Senolytic Agents. Aging 8, 2915–2926. doi:10.18632/aging.101100
Wanner, E., Thoppil, H., and Riabowol, K. (2020). Senescence and Apoptosis: Architects of Mammalian Development. Front. Cel Dev. Biol. 8, 620089. doi:10.3389/fcell.2020.620089
Wildenberg, M. E., Vos, A. C. W., Wolfkamp, S. C. S., Duijvestein, M., Verhaar, A. P., Te Velde, A. A., et al. (2012). Autophagy Attenuates the Adaptive Immune Response by Destabilizing the Immunologic Synapse. Gastroenterology 142, 1493–1503. doi:10.1053/j.gastro.2012.02.034
Wiley, C. D., Flynn, J. M., Morrissey, C., Lebofsky, R., Shuga, J., Dong, X., et al. (2017). Analysis of Individual Cells Identifies Cell-To-Cell Variability Following Induction of Cellular Senescence. Aging Cell 16, 1043–1050. doi:10.1111/acel.12632
Wilson, A., Shehadeh, L. A., Yu, H., and Webster, K. A. (2010). Age-related Molecular Genetic Changes of Murine Bone Marrow Mesenchymal Stem Cells. BMC Genomics 11, 229. doi:10.1186/1471-2164-11-229
Wissler Gerdes, E. O., Vanichkachorn, G., Verdoorn, B. P., Hanson, G. J., Joshi, A. Y., Murad, M. H., et al. (2021). Role of Senescence in the Chronic Health Consequences of COVID-19. Translational Res. S1931-5244 (21), 00259. doi:10.1016/j.trsl.2021.10.003
Wu, D., and Prives, C. (2018). Relevance of the P53-MDM2 axis to Aging. Cel Death Differ 25, 169–179. doi:10.1038/cdd.2017.187
Xiao, F.-H., Kong, Q.-P., Perry, B., and He, Y.-H. (2016). Progress on the Role of DNA Methylation in Aging and Longevity. Brief. Funct. Genomics 15, elw009–459. doi:10.1093/bfgp/elw009
Yang, D., Tian, X., Ye, Y., Liang, Y., Zhao, J., Wu, T., et al. (2021). Identification of GL-V9 as a Novel Senolytic Agent against Senescent Breast Cancer Cells. Life Sci. 272, 119196. doi:10.1016/j.lfs.2021.119196
Yeo, E. J., Hwang, Y. C., Kang, C. M., Choy, H. E., and Park, S. C. (2000). Reduction of UV-Induced Cell Death in the Human Senescent Fibroblasts. Mol. Cell 10, 415–422.
Yosef, R., Pilpel, N., Tokarsky-Amiel, R., Biran, A., Ovadya, Y., Cohen, S., et al. (2016). Directed Elimination of Senescent Cells by Inhibition of BCL-W and BCL-XL. Nat. Commun. 7, 11190. doi:10.1038/ncomms11190
Youle, R. J., and Strasser, A. (2008). The BCL-2 Protein Family: Opposing Activities that Mediate Cell Death. Nat. Rev. Mol. Cel Biol 9, 47–59. doi:10.1038/nrm2308
Young, A. R. J., Narita, M., Ferreira, M., Kirschner, K., Sadaie, M., Darot, J. F. J., et al. (2009). Autophagy Mediates the Mitotic Senescence Transition. Genes Dev. 23, 798–803. doi:10.1101/gad.519709
Yousefzadeh, M. J., Zhu, Y., McGowan, S. J., Angelini, L., Fuhrmann-Stroissnigg, H., Xu, M., et al. (2018). Fisetin Is a Senotherapeutic that Extends Health and Lifespan. EBioMedicine 36, 18–28. doi:10.1016/j.ebiom.2018.09.015
Zang, J., Sha, M., Zhang, C., Ye, J., Zhang, K., and Gao, J. (2017). Senescent Hepatocyte Secretion of Matrix Metalloproteinases Is Regulated by Nuclear Factor-Κb Signaling. Life Sci. 191, 205–210. doi:10.1016/j.lfs.2017.10.023
Zhang, C., Xie, Y., Chen, H., Lv, L., Yao, J., Zhang, M., et al. (2020). FOXO4-DRI Alleviates Age-Related Testosterone Secretion Insufficiency by Targeting Senescent Leydig Cells in Aged Mice. Aging 12, 1272–1284. doi:10.18632/aging.102682
Zhang, P., Kishimoto, Y., Grammatikakis, I., Gottimukkala, K., Cutler, R. G., Zhang, S., et al. (2019). Senolytic Therapy Alleviates Aβ-Associated Oligodendrocyte Progenitor Cell Senescence and Cognitive Deficits in an Alzheimer's Disease Model. Nat. Neurosci. 22, 719–728. doi:10.1038/s41593-019-0372-9
Zhang, X., Zhang, S., Liu, X., Wang, Y., Chang, J., Zhang, X., et al. (2018). Oxidation Resistance 1 Is a Novel Senolytic Target. Aging Cell 17, e12780. doi:10.1111/acel.12780
Zhang, Z., Yao, Z., Zhao, S., Shao, J., Chen, A., Zhang, F., et al. (2017). Interaction between Autophagy and Senescence Is Required for Dihydroartemisinin to Alleviate Liver Fibrosis. Cel Death Dis 8, e2886. doi:10.1038/cddis.2017.255
Zhou, J., Wang, J., Chen, C., Yuan, H., Wen, X., and Sun, H. (2018). USP7: Target Validation and Drug Discovery for Cancer Therapy. Mc 14, 3–18. doi:10.2174/1573406413666171020115539
Zhu, Y., Doornebal, E. J., Pirtskhalava, T., Giorgadze, N., Wentworth, M., Fuhrmann-Stroissnigg, H., et al. (2017). New Agents that Target Senescent Cells: the Flavone, Fisetin, and the BCL-XL Inhibitors, A1331852 and A1155463. Aging 9, 955–963. doi:10.18632/aging.101202
Zhu, Y., Tchkonia, T., Fuhrmann‐Stroissnigg, H., Dai, H. M., Ling, Y. Y., Stout, M. B., et al. (2016). Identification of a Novel Senolytic Agent, Navitoclax, Targeting the Bcl‐2 Family of Anti‐apoptotic Factors. Aging Cell 15, 428–435. doi:10.1111/acel.12445
Keywords: aging, apoptosis, resistance, senescent cell, senolytic
Citation: Hu L, Li H, Zi M, Li W, Liu J, Yang Y, Zhou D, Kong Q-P, Zhang Y and He Y (2022) Why Senescent Cells Are Resistant to Apoptosis: An Insight for Senolytic Development. Front. Cell Dev. Biol. 10:822816. doi: 10.3389/fcell.2022.822816
Received: 26 November 2021; Accepted: 26 January 2022;
Published: 16 February 2022.
Edited by:
Guang Yao, University of Arizona, United StatesReviewed by:
Joseph William Landry, Virginia Commonwealth University, United StatesOliver Bischof, Délégation Ile-de-France Ouest et Nord (CNRS), France
Copyright © 2022 Hu, Li, Zi, Li, Liu, Yang, Zhou, Kong, Zhang and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yonghan He, heyonghan@mail.kiz.ac.cn; Yunxia Zhang, zhangyunxia@hainmc.edu.cn
†These authors have contributed equally to this work