 Ilaria Parenti1†Elsa Leitão1†Alma Kuechler1Laurent Villard2,3Cyril Goizet4,5,6Cécile Courdier4,5,6Allan Bayat7,8,9Alessandra Rossi8,10Sophie Julia11Ange-Line Bruel12,13Frédéric Tran Mau-Them12,13Sophie Nambot13Daphné Lehalle12,13Marjolaine Willems14,15James Lespinasse16Jamal Ghoumid17,18Roseline Caumes17,18Thomas Smol17,19Salima El Chehadeh20Elise Schaefer20Marie-Thérèse Abi-Warde21Boris Keren22Alexandra Afenjar23Anne-Claude Tabet24Jonathan Levy24Anna Maruani25Ángel Aledo-Serrano26Waltraud Garming27Clara Milleret-Pignot28Anna Chassevent29Marije Koopmans30Nienke E. Verbeek30Richard Person31Rebecca Belles32Gary Bellus32Bonnie A. Salbert32Frank J. Kaiser1,33Laure Mazzola34,35Philippe Convers34,35Laurine Perrin36Amélie Piton37,38,39,40Gert Wiegand41,42Andrea Accogli43,44Francesco Brancati45,46Fabio Benfenati47,48Nicolas Chatron49,50David Lewis-Smith51,52Rhys H. Thomas51,52Federico Zara53,54Pasquale Striano53,54Gaetan Lesca49,50Christel Depienne1*
Ilaria Parenti1†Elsa Leitão1†Alma Kuechler1Laurent Villard2,3Cyril Goizet4,5,6Cécile Courdier4,5,6Allan Bayat7,8,9Alessandra Rossi8,10Sophie Julia11Ange-Line Bruel12,13Frédéric Tran Mau-Them12,13Sophie Nambot13Daphné Lehalle12,13Marjolaine Willems14,15James Lespinasse16Jamal Ghoumid17,18Roseline Caumes17,18Thomas Smol17,19Salima El Chehadeh20Elise Schaefer20Marie-Thérèse Abi-Warde21Boris Keren22Alexandra Afenjar23Anne-Claude Tabet24Jonathan Levy24Anna Maruani25Ángel Aledo-Serrano26Waltraud Garming27Clara Milleret-Pignot28Anna Chassevent29Marije Koopmans30Nienke E. Verbeek30Richard Person31Rebecca Belles32Gary Bellus32Bonnie A. Salbert32Frank J. Kaiser1,33Laure Mazzola34,35Philippe Convers34,35Laurine Perrin36Amélie Piton37,38,39,40Gert Wiegand41,42Andrea Accogli43,44Francesco Brancati45,46Fabio Benfenati47,48Nicolas Chatron49,50David Lewis-Smith51,52Rhys H. Thomas51,52Federico Zara53,54Pasquale Striano53,54Gaetan Lesca49,50Christel Depienne1*- 1Institute of Human Genetics, University Hospital Essen, University Duisburg-Essen, Essen, Germany
- 2INSERM, MMG, Faculté de Médecine, Aix-Marseille University, Marseille, France
- 3Département de Génétique Médicale, APHM, Hôpital d'Enfants de La Timone, Marseille, France
- 4Service de Génétique Médicale, Bordeaux, France
- 5Centre de Référence Maladies Rares Neurogénétique, Service de Génétique Médicale, Bordeaux, France
- 6NRGEN Team, INCIA, CNRS UMR 5287, University of Bordeaux, Bordeaux, France
- 7Institute for Regional Health Services, University of Southern Denmark, Odense, Denmark
- 8Department of Epilepsy Genetics and Personalized Medicine, Danish Epilepsy Center, Dianalund, Denmark
- 9Department of Drug Design and Pharmacology, University of Copenhagen, Copenhagen, Denmark
- 10Pediatric Clinic, IRCCS Policlinico San Matteo Foundation, University of Pavia, Pavia, Italy
- 11Service de Génétique Médicale, Pôle de Biologie, CHU de Toulouse - Hôpital Purpan, Toulouse, France
- 12Unité Fonctionnelle Innovation en Diagnostic Génomique des Maladies Rares, FHU-TRANSLAD, CHU Dijon Bourgogne, Dijon, France
- 13UMR1231 GAD, Inserm - Université Bourgogne-Franche Comté, Dijon, France
- 14Department of Medical Genetics, Rare diseases and Personalized Medicine, CHU Montpellier, University of Montpellier, Montpellier, France
- 15Inserm U1298, INM, CHU Montpellier, University of Montpellier, Montpellier, France
- 16Service de Cytogenetique, Centre Hospitalier de Chambéry, Chambéry, France
- 17Univ. Lille, ULR7364 RADEME, Lille, France
- 18CHU Lille, Clinique de Génétique, Guy Fontaine, Lille, France
- 19CHU Lille, Institut de Génétique Médicale, Lille, France
- 20Service de Génétique Médicale, Institut de Génétique Médicale d'Alsace (IGMA), Hôpitaux Universitaires de Strasbourg, Hôpital de Hautepierre, Strasbourg, France
- 21Département de NeuroPédiatrie, Hôpitaux Universitaires de Strasbourg, Strasbourg, France
- 22APHP, Département de Génétique, UF de Génomique du Développement, Département de Génétique, Groupe Hospitalier Pitié-Salpêtrière, Sorbonne Université, Paris, France
- 23Département de Génétique, Centre de Référence déficiences Intellectuelles de Causes Rares, APHP, Hôpital Armand Trousseau, Sorbonne Université, Paris, France
- 24APHP, Département de Génétique, Hôpital Robert-Debré, Paris, France
- 25Department of Child and Adolescent Psychiatry, Robert Debré Hospital, APHP, Paris, France
- 26Epilepsy and Neurogenetics Program, Neurology Department, Ruber Internacional Hospital, Madrid, Spain
- 27Sozialpädiatrisches Zentrum, Kinder-und Jugendklinik Gelsenkirchen, Gelsenkirchen, Germany
- 28Service de Pédiatrie, CH de Mâcon, Mâcon, France
- 29Department of Neurogenetics, Kennedy Krieger Institute, Baltimore, MD, United States
- 30Department of Genetics, Utrecht University Medical Center, Utrecht, Netherlands
- 31GeneDx, Gaithersburg, MD, United States
- 32Medical Genetics, Geisinger Medical Center, Danville, PA, United States
- 33Essener Zentrum für Seltene Erkrankungen (EZSE), Universitätsklinikum Essen, Essen, Germany
- 34Department of Neurology, University Hospital, Lyon Neuroscience Research Center (CRNL), INSERM U1028, CNRS UMR5292, Lyon, France
- 35Department of Neurology, University Hospital, Saint-Etienne, France
- 36Department of Paediatric Physical Medicine and Rehabilitation, CHU Saint-Étienne, Hôpital Bellevue, Rhône-Alpes Reference Centre for Neuromuscular Diseases, Saint-Étienne, France
- 37Institut de Génétique et de Biologie Moléculaire et Cellulaire, Illkirch, France
- 38Centre National de la Recherche Scientifique, UMR7104, Illkirch, France
- 39Institut National de la Santé et de la Recherche Médicale, U964, Illkirch, France
- 40Université de Strasbourg, Illkirch, France
- 41Division of Pediatric Neurology, Department of Pediatrics, Asklepios Klinik Nord-Heidberg, Hamburg, Germany
- 42Department of Pediatric and Adolescent Medicine II (Neuropediatrics, Social Pediatrics), University Medical Centre Schleswig-Holstein, Kiel, Germany
- 43Department of Specialized Medicine, Division of Medical Genetics, McGill University Health Centre, Montreal, Qc, Canada
- 44Department of Human Genetics, Faculty of Medicine, McGill University, Montreal, Qc, Canada
- 45Department of Life, Human Genetics, Health and Environmental Sciences, University of L’Aquila, L’Aquila, Italy
- 46IRCCS San Raffaele Roma, Rome, Italy
- 47Center for Synaptic Neuroscience and Technology, Istituto Italiano di Tecnologia, Geneva, Italy
- 48IRCCS Ospedale Policlinico San Martino, Geneva, Italy
- 49Service de Genetique, Hospices Civils de Lyon, Bron, France
- 50Institute NeuroMyoGène, Laboratoire Physiopathologie et Génétique du Neurone et du Muscle, CNRS UMR 5261 -INSERM U1315, Université de Lyon - Université Claude Bernard Lyon 1, Lyon, France
- 51Translational and Clinical Research Institute, Newcastle University, Newcastle Upon Tyne, United Kingdom
- 52Department of Clinical Neurosciences, Newcastle Upon Tyne Hospitals NHS Foundation Trust, Newcastle Upon Tyne, United Kingdom
- 53IRCCS G. Gaslini, Genova, Italy
- 54Department of Neurology, Rehabilitation, Ophtalmology, Genetics, Maternal and Child Health, University of Genova, Genova, Italy
Synapsin-I (SYN1) is a presynaptic phosphoprotein crucial for synaptogenesis and synaptic plasticity. Pathogenic SYN1 variants are associated with variable X-linked neurodevelopmental disorders mainly affecting males. In this study, we expand on the clinical and molecular spectrum of the SYN1-related neurodevelopmental disorders by describing 31 novel individuals harboring 22 different SYN1 variants. We analyzed newly identified as well as previously reported individuals in order to define the frequency of key features associated with these disorders. Specifically, behavioral disturbances such as autism spectrum disorder or attention deficit hyperactivity disorder are observed in 91% of the individuals, epilepsy in 82%, intellectual disability in 77%, and developmental delay in 70%. Seizure types mainly include tonic-clonic or focal seizures with impaired awareness. The presence of reflex seizures is one of the most representative clinical manifestations related to SYN1. In more than half of the cases, seizures are triggered by contact with water, but other triggers are also frequently reported, including rubbing with a towel, fever, toothbrushing, fingernail clipping, falling asleep, and watching others showering or bathing. We additionally describe hyperpnea, emotion, lighting, using a stroboscope, digestive troubles, and defecation as possible triggers in individuals with SYN1 variants. The molecular spectrum of SYN1 variants is broad and encompasses truncating variants (frameshift, nonsense, splicing and start-loss variants) as well as non-truncating variants (missense substitutions and in-frame duplications). Genotype-phenotype correlation revealed that epileptic phenotypes are enriched in individuals with truncating variants. Furthermore, we could show for the first time that individuals with early seizures onset tend to present with severe-to-profound intellectual disability, hence highlighting the existence of an association between early seizure onset and more severe impairment of cognitive functions. Altogether, we present a detailed clinical description of the largest series of individuals with SYN1 variants reported so far and provide the first genotype-phenotype correlations for this gene. A timely molecular diagnosis and genetic counseling are cardinal for appropriate patient management and treatment.
Introduction
Synapsins are a family of presynaptic phosphoproteins mainly expressed in the central and peripheral nervous system (Ueda and Greengard 1977; De Camilli, Cameron, and Greengard 1983). They play a fundamental role during the early stages of neuronal development, where they regulate neurite outgrowth, synaptic development, function, and plasticity through the regulation of the number of synaptic vesicles (SVs) available for neurotransmitter release (Cesca et al. 2010).
Synapsins exist in both invertebrates and vertebrates. Mammals possess three synapsin genes named SYN1, SYN2 and SYN3. SYN1 is located on chromosome X, SYN2 and SYN3 are on autosomes. SYN1 and SYN2 each produce two different protein isoforms through alternative splicing (SYN1a, SYN1b, SYN2a, SYN2b). The different isoforms share a common N-terminal region and diverge at the C-terminus (Hosaka and Sudhof 1998; Sudhof et al. 1989). The N-terminus of synapsins is composed of domains A-C. Domain A comprises residues that are target of phosphorylation and therefore important for the regulation of the activity of the protein. Domain B is relatively poorly conserved and is considered a linker between domains A and C. The last N-terminal domain, namely domain C, is composed of 300 amino acids and is essential for the activity of the protein. The domains of the C-terminal region (D-J) are instead highly variable and differ between both the primary transcripts and the splice variants (Longhena et al. 2021; Cesca et al. 2010). Synapsin protein isoforms display partially overlapping functions but are also characterized by distinctive localization and expression patterns, as well as different post-translational modifications (Guarnieri et al. 2017; Longhena et al. 2021). The finely orchestrated balance between the isoforms is essential for proper brain functions. Importantly, the vast majority of neurons within the central nervous system express at least one synapsin isoform (De Camilli, Cameron, and Greengard 1983).
Alteration of synapsin function results in neurodevelopmental phenotypes in both humans and mice. In mice, the disruption of a single synapsin gene has only a mild impact on learning and behavior, while double or triple gene knockouts display additive effects which lead to more severe phenotypes (Etholm and Heggelund 2009; Cambiaghi et al. 2013). Late-onset seizures triggered by sensitive stimuli occur in Syn1−/−, Syn2−/−, as well as in Syn1/Syn2 and Syn1/Syn2/Syn3 double and triple knockout mice, but not in Syn3−/− mice (Corradi et al. 2008; Etholm et al. 2012; Rosahl et al. 1995; Gitler et al. 2004; Feng et al. 2002). In humans, variants in SYN1 have been independently associated with epilepsy, learning disabilities and behavioral disorders (OMIM #300491) (Garcia et al. 2004; Fassio et al. 2011) or non-syndromic intellectual disability (OMIM #300115) (Guarnieri et al. 2017). In mice, alterations of Syn2, and to a lesser extent of Syn1, are associated with autistic-like features and rare seizures (Etholm et al. 2012; Greco et al. 2013; Michetti et al. 2017). Autistic behavioral features seem to predominate in mice and human individuals with syn2/SYN2 variants, whereas focal-onset seizures seem more characteristic of syn1/SYN1 (Etholm et al. 2012). The evidence for an association between rare variants in SYN2 and epilepsy remains weak as the missense variants carried by the single described case has subsequently been reported in gnomAD (Corradi et al. 2014). Early suggestions of an association between epilepsy and a common intron variant in SYN2 (Cavalleri et al. 2007) have become less compelling in larger genome-wide association studies (International League Against Epilepsy Consortium on Complex, 2018). Finally, disruption of Syn3 also alters social behaviors in mice (Michetti et al. 2017; Greco et al. 2013) but a clear association between SYN3 and human disease has not yet been established, although de novo variants of unknown significance in this gene have been reported in two patients with developmental disorders (Eldomery et al. 2017; Turner et al. 2019).
Here, we focus on the X-linked gene SYN1. Pathogenic variants in this gene have been recurrently reported in individual cases and families (Garcia et al. 2004; Fassio et al. 2011; Guarnieri et al. 2017; Xiong et al. 2021; Zhou et al. 2021; Butler et al. 2017; Lindy et al. 2018; Nguyen et al. 2015; Rossi et al. 2017; Peron et al. 2018; Fernandez-Marmiesse et al. 2019; Darvish et al. 2020; Ibarluzea et al. 2020; Mojarad et al. 2021; van der Ven et al. 2021; Stranneheim et al. 2021; Yang et al. 2020; Cabana et al. 2018). Epilepsy, learning disabilities, speech delay, intellectual disability (ID), and autism spectrum disorder (ASD) are the most frequent clinical manifestations associated with SYN1 variants (Longhena et al. 2021; John et al. 2021; Accogli et al. 2021), but an extensive comparison of the clinical features and genotype-phenotype correlations are missing. Recent studies have highlighted a specific association of reflex epilepsy with SYN1 variants (Nguyen et al. 2015; Peron et al. 2018; Accogli et al. 2021), but the percentage of patients exhibiting this phenotype and the full clinical spectrum associated with SYN1 remains undetermined. Reflex epilepsy can be defined as the reproducible provocation of seizures in response to a specific motor, sensory, or cognitive stimulus, with or without the occurrence of spontaneous seizures. In patients with SYN1 variants, seizures are frequently triggered by bathing or showering. Importantly, SYN1-related disorders are characterized by extensive clinical heterogeneity and intrafamilial variability.
Herein, we report 31 unpublished individuals from 22 families harboring variants in SYN1 and review variants and clinical features published in the literature in order to deep-phenotype SYN1-related disorders and explore potential genotype-phenotype correlations.
Materials and methods
SYN1 study cohort
Our cohort was collected through an International collaboration (EuroEPINOMICS RES consortium) and GeneMatcher (Sobreira et al. 2015). Gene panels and exome sequencing were performed at the respective institutions. All procedures were performed following the ethical standards of the institutional and/or national research committee, and in conformity with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. After anonymization of patients’ data, each referring physician provided detailed developmental, neurological, and behavioral history of the patients. Informed consent was obtained from all individuals included in this study or their legal guardians. Clinical features of the previously published cases were retrieved from the corresponding publications.
SYN1 variants
Variants were mapped on the SYN1 NM_006950.3 RefSeq transcript using HGVS recommendations (den Dunnen et al. 2016) and classified according to ACMG Guidelines (Richards et al. 2015). All variants have been submitted to the ClinVar Database and have been assigned the following accession numbers: SCV002558865 - SCV002558886.
Known SYN1 ENST00000295987.7 (corresponding to NM_006950.3) variants were retrieved from gnomAD v2.1.1 (Karczewski et al. 2020), restricting to loss-of-function, missense and synonymous single nucleotide variants or indels. For each gnomAD variant, we calculated the number of females and males carrying the variant. The combined annotation-dependent depletion (CADD) score (Rentzsch et al. 2019) (https://cadd.gs.washington.edu/score) was calculated for all variants using GRCh37-v1.6 genomic coordinates.
Putative effects of the variants on splicing were calculated with the SpliceAI neural network (Jaganathan et al. 2019). The probability that a variant affects splicing is expressed as score cutoffs, namely >0.5 (recommended) and >0.8 (high precision).
Pathogenic and likely pathogenic SYN1 variants were retrieved from HGMD Professional (Qiagen, Hilden, Germany). Three variants reported in the HGMD database were excluded from the total count: p.(Ala51Gly) and p.(Thr567Ala) were previously deemed benign due to minor allele frequency (MAF) thresholds and computational evidence (Abouelhoda et al. 2016; Thauvin-Robinet et al. 2019), whereas p.(Gly240Arg) shows an incongruent pattern of inheritance (Fernandez-Marmiesse et al. 2019).
In silico SYN1 protein analysis
Amino acid positions of each protein region of SYN1 were retrieved from Uniprot (The UniProt Consortium et al., 2021). Evolutionary conservation of the SYN1 amino acids affected by missense substitutions was retrieved from Alamut Visual Plus™ (SOPHiA GENETICS, Lausanne, Switzerland). Alignment of amino acids across synapsins was carried out with Clustal Omega (Goujon et al. 2010; Sievers et al. 2011).
The putative effects of the missense substitutions on protein stability were predicted with DynaMut2 (Rodrigues, Pires, and Ascher 2021) and mCSM (Pires, Ascher, and Blundell 2014). The AlphaFold SYN1 protein structure (AF-P17600-F1-model_v3) was used for the analysis. Residues with a confidence score <70 based on AlphaFold predictions were excluded from the analysis.
Statistics
Fisher’s tests were performed to determine associations 1) between truncating variants and the manifestation of each main clinical feature, 2) between sex and the manifestation of the clinical features of interest, and 3) between sex and the existence of putatively damaging variants using gnomAD data. Odds ratios were log2 transformed and indicate enrichment or depletion of genes, for positive or negative values, respectively. p-values were adjusted for multiple comparisons using Bonferroni correction.
Mann-Whitney tests followed by Bonferroni correction for multiple testing were used to compare the age of epilepsy onset between individuals with different levels of ID.
Results
Classification of SYN1 variants
Twenty-two different genetic alterations of SYN1 were identified in a total of 31 individuals. Detailed information about each variant is available in Supplementary Table S1. The identified variants comprise nine frameshift variants, three nonsense variants, one start-loss, one splice site variant, seven missense substitutions, and one in-frame duplication. The newly identified variants almost double the total number of reported SYN1 alterations, raising it to 46 (13 frameshift, eight nonsense, 18 missense, four altering splice sites, one start-loss and two in-frame duplications). All variants reported so far are indicated on the corresponding transcript and protein domain in Figure 1. As shown in this Figure, all variants alter amino acids shared by both SYN1 isoforms (SYN1a and SYN1b), while the variable C-terminal domains E and F are spared. No apparent clustering of variants of unknown significance (VUS) or likely pathogenic missense substitutions can be observed.
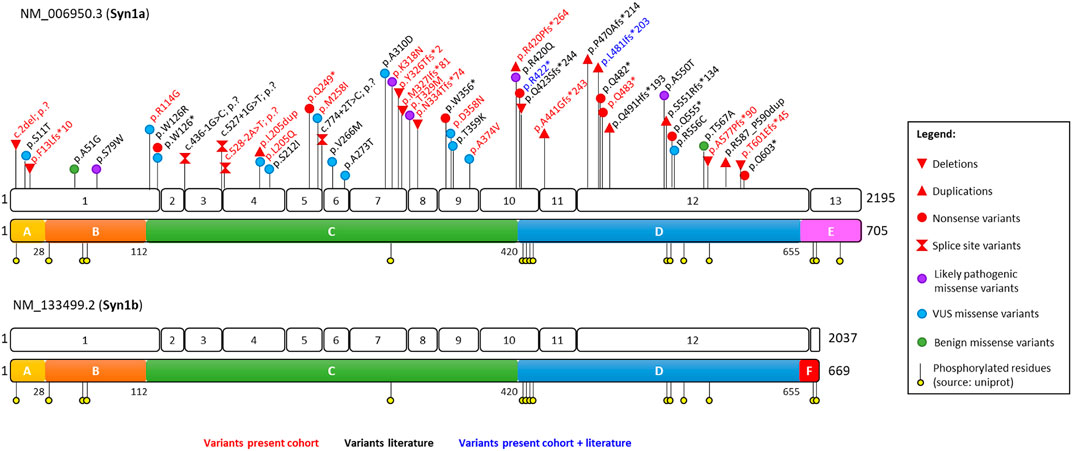
FIGURE 1. Schematic representation of the SYN1 gene and SYN1 protein with positions of the identified variants relative to exon and domains distribution. Variants identified in the present cohort are depicted in red, those reported in the literature in black, and those reported both in the literature and in our cohort in blue. The conserved A (N-terminal highly phosphorylated domain), B (linker), and C (functional domain) domains are depicted in yellow, orange, and green, respectively; the variable D, E, and F domains in blue, pink, and red. Classification of the variants was performed based on ACMG criteria. Missense substitutions are subdivided in likely pathogenic, VUS, and benign based on the presence of functional data and their frequency in the healthy population.
CADD scores were calculated for all variants and range from 20.3 to 40. As expected, truncating variants are associated with higher CADD scores, whereas the two in-frame duplications show the lowest scores. Variants were additionally classified based on ACMG guidelines. Of the 46 variants, 26 were classified as pathogenic, five as likely pathogenic and 15 as variants of unknown significance (VUS) (Supplementary Table S1). Missense substitutions and in-frame duplications fall within the category of VUS, unless functional studies (PS3) or a de novo origin with confirmed maternity and paternity (PS2) are reported.
Likely pathogenic and VUS missense substitutions were evaluated based on the conservation of the affected amino acids across species and synapsin isoforms (Supplementary Figures S1, 2). Except for p.(Arg114Gly), all missense substitutions affect evolutionarily conserved amino acids (Supplementary Figure S1). Arginine 114 also differs among SYN1, SYN2, and SYN3. Eleven missense variants (p.(Ser11Thr), p.(Ser79Trp), p.(Trp126Arg), p.(Met258Ile), p.(Val266Met), p.(Ala273Thr), p.(Lys318Asn), p.(Thr329Met), p.(Asp358Asn), p.(Thr359Lys), and p.(Ala374Val)) alter residues that are conserved in all synapsins. The following substitutions affect amino acids that are conserved in two out of three synapsins: p.(Leu205Gln), p.(Ser212Ile), p.(Arg420Gln), and p.(Arg556Cys). The two remaining missense variants, p.(Ala310Asp) and p.(Ala550Thr), affect residues that are specific for SYN1a and SYN1b (Supplementary Figure S2). According to the SpliceAI neural network, none of the missense substitutions is predicted to affect splice site junctions (Supplementary Table S1). We subsequently employed DynaMut2 and mCSM to predict the putative effects of missense substitutions on protein stability. The predictions of these two bioinformatic tools are in agreement with each other for all the analyzed variants and suggest that four variants (p.(Trp126Arg), p.(Lys318Asn), p.(Leu205Gln), and p.(Arg114Gly)) might lead to destabilization of the SYN1 protein (Supplementary Table S1).
All putatively disease-causing SYN1 variants are either absent from gnomAD or present at a very low allele frequency (<0.0001%). All SYN1 variants available in gnomAD were subsequently retrieved to compare a large population of males and females and the distribution of variants across different protein regions. Putatively damaging SYN1 variants (non-synonymous variants with CADD score ≥22) affect 78 residues, while non-deleterious variants (synonymous or non-synonymous variants with CADD score <22) alter codons corresponding to 140 residues (Figure 2). Notably, males are depleted in putatively damaging SYN1 variants compared to females (p = 7.3 × 10–4, OR = 0.67, Fisher’s test). Regions A and B are almost devoid of putatively damaging gnomAD variants while in the other protein regions, 17 residues are altered by putatively damaging variants in multiple female individuals whereas only 10 are altered in multiple males (Figure 2).
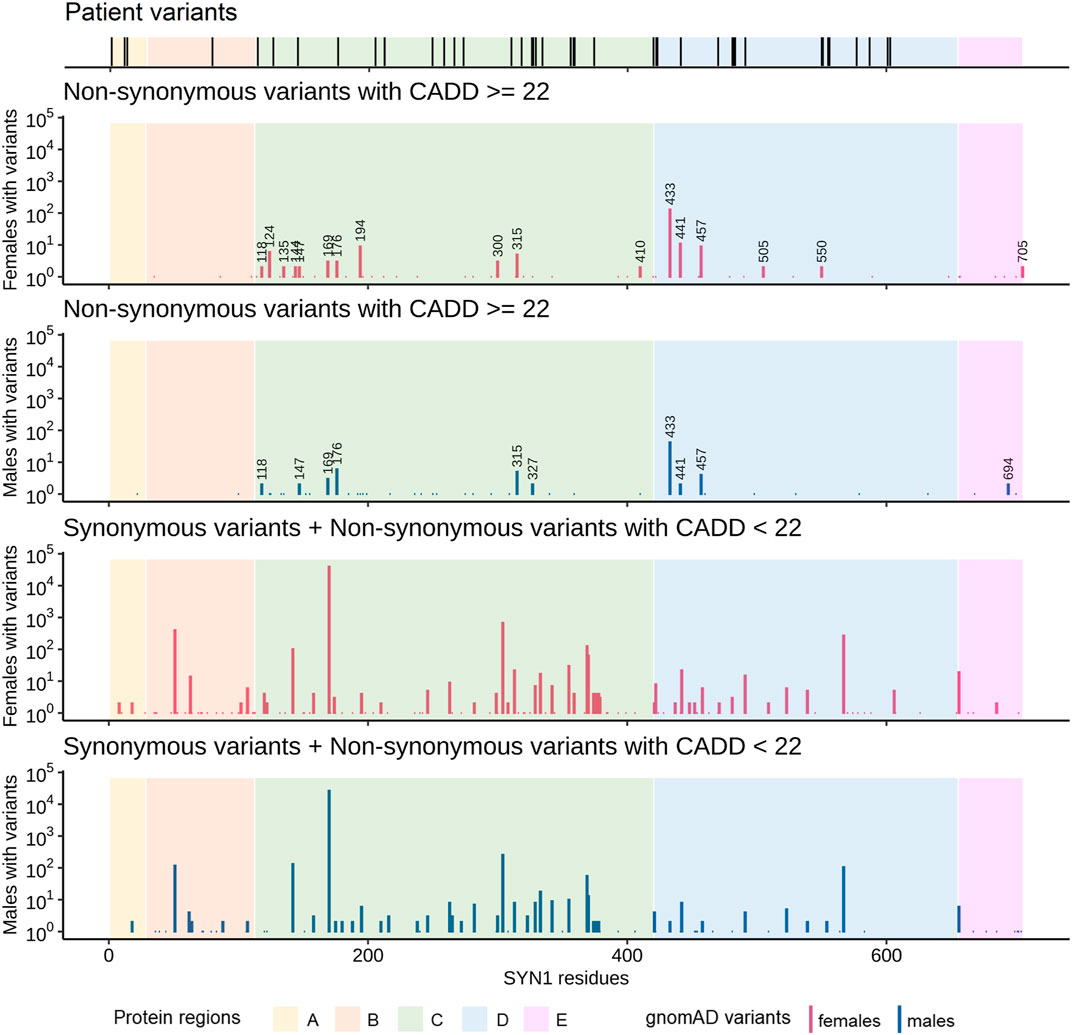
FIGURE 2. Comparison of the distribution of the SYN1 variants identified in our cohort and/or in the literature with the variants reported in gnomAD. The number of individuals with gnomAD variants was plotted for each combined codon. gnomAD variants were stratified by both sex and damage potential (based on variant type and CADD score). Specifically, gnomAD variants were subdivided in the following two groups: one comprising synonymous variants and non-synonymous variants with CADD scores lower than 22 and one comprising non-synonymous variants with CADD scores equal or higher than 22 (putatively damaging gnomAD variants). Numbers above bars represent the positions of residues affected by putatively damaging non-synonymous variants in at least two males or two females. Males are depleted in putatively damaging SYN1 variants compared to females (p = 7.3 × 10–4, OR = 0.67, Fisher’s test). Regions A and B are almost devoid of putatively damaging variants in gnomAD.
SYN1 variants predominantly affect males and result in clinical heterogeneity
This cohort comprises 29 males and two females with SYN1 variants (Supplementary Table S2). Taking previously published patients into account, the number of individuals with SYN1-related neurodevelopmental disorders for whom clinical information is available is 83 (71 males and 12 females; Table 1) (Garcia et al. 2004; Fassio et al. 2011; Nguyen et al. 2015; Guarnieri et al. 2017; Peron et al. 2018; Fernandez-Marmiesse et al. 2019; Darvish et al. 2020; Ibarluzea et al. 2020; Accogli et al. 2021; Mojarad et al. 2021; van der Ven et al. 2021; Xiong et al. 2021; Yang et al. 2020; Zhou et al. 2021).
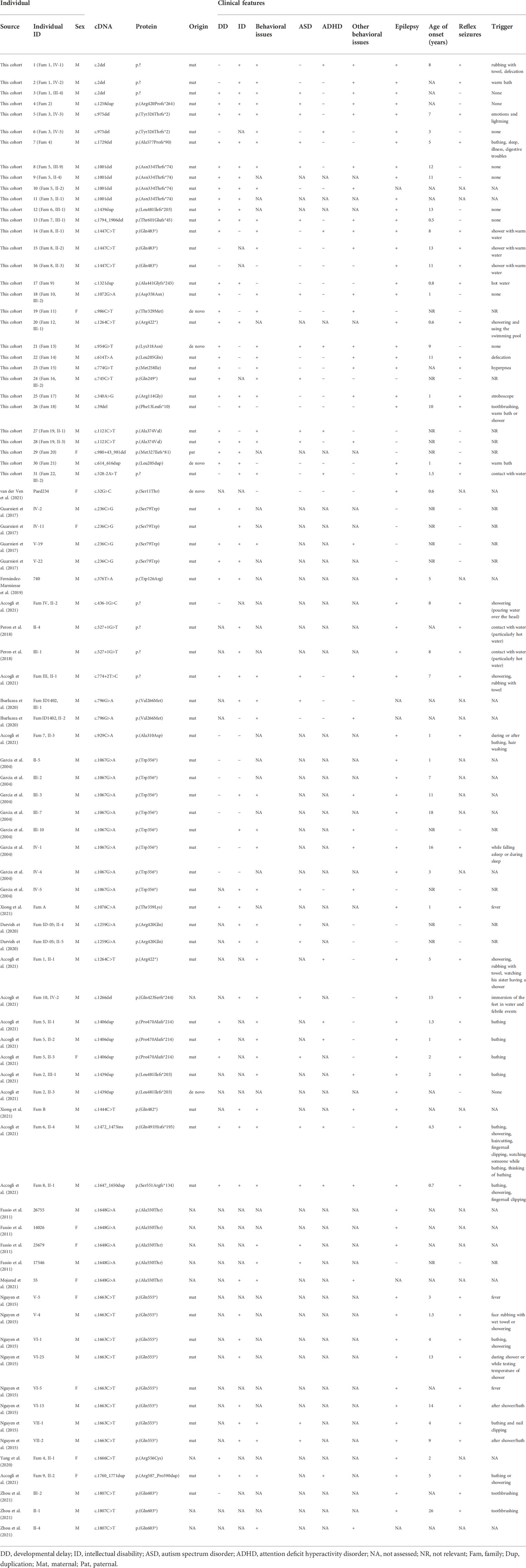
TABLE 1. Main clinical features of individuals with SYN1 variants.
Most SYN1 variants were identified in families with multiple affected family members. Four SYN1 variants arose de novo in the index patient (Table 1). One additional de novo variant in an unaffected mother was previously reported (Accogli et al. 2021). Of the four de novo variants, two were identified in female individuals (Individual 19 in our cohort and Paed234 in van der Ven et al. 2021) and two in males, both part of our cohort. One male subject was hemizygous for the variant (Individual 21), while the other male individual displayed somatic mosaicism in blood (Individual 30) (Supplementary Table S2). Remarkably, somatic mosaicism in blood was identified also in the unaffected father of the female Individual 29.
Overall, this study brings the total number of families with SYN1 variants to 50 (22 from our cohort and 28 from the literature). Pedigrees of the families from our cohort are depicted in Figure 3. In familial cases, males show a more homogeneous clinical presentation at the more severe end of the SYN1-phenotypic spectrum. Conversely, females can display a broad clinical spectrum, ranging from unaffected to equally affected as males. Nevertheless, male individuals within the same family can also show different phenotypes or severity of the displayed clinical features, leading to the hypothesis that additional factors might act as phenotypic modifiers.
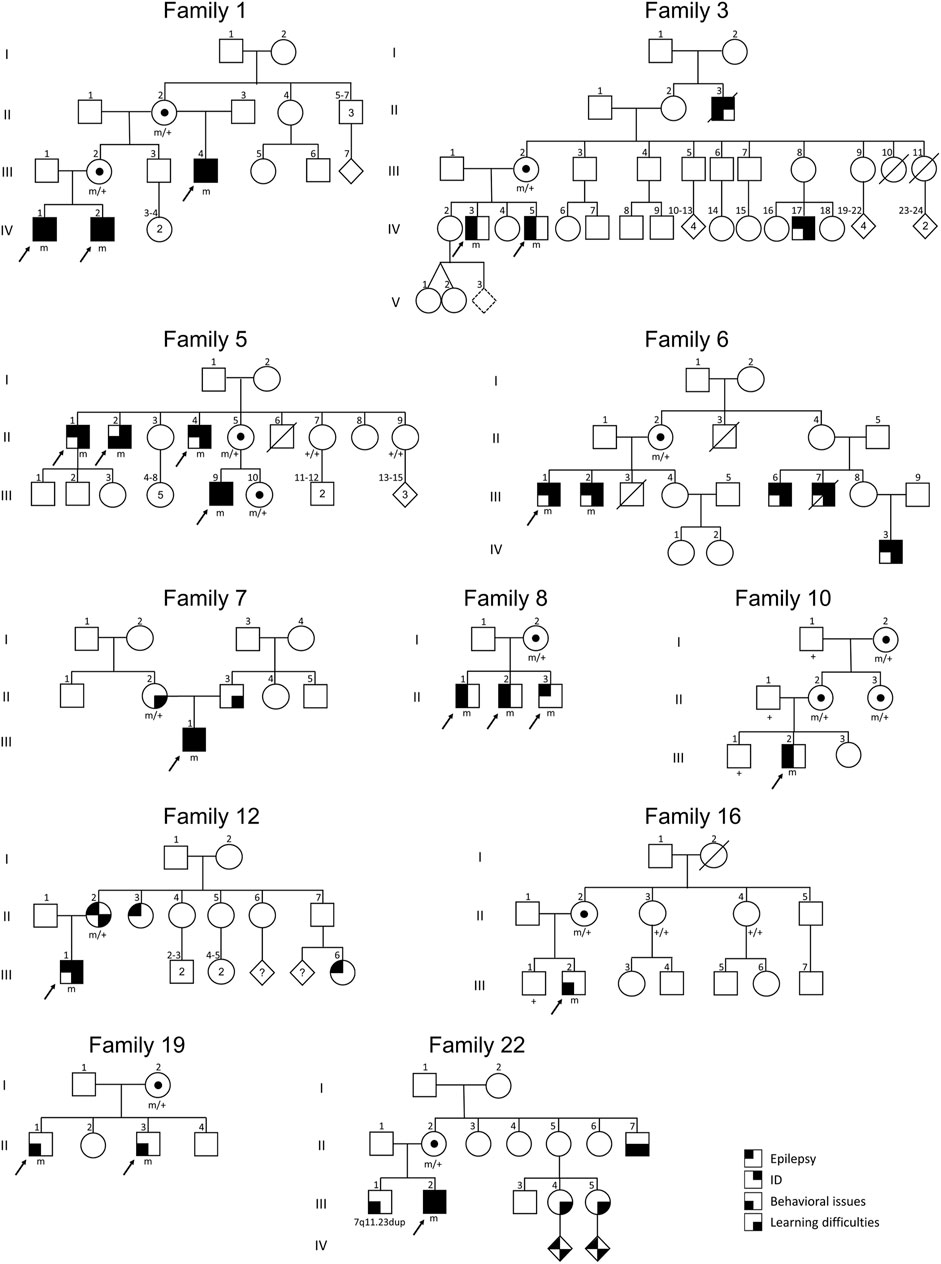
FIGURE 3. Pedigrees of the SYN1 familial cases of our cohort. Black arrows indicate the index cases initially referred to the genetic center. Individuals within the same family tree are variably affected, pointing to the existence of incomplete penetrance and clinical heterogeneity.
When assessed, X-inactivation analysis of the healthy female carriers within families did not reveal any skewing (Supplementary Table S2). Notably, Individual 29, who inherited the SYN1 variant from her mosaic father, displays complete skewing of the X-inactivation.
Altogether, our data suggest that SYN1 variants predominantly affect male individuals, whereas females exhibit a wider clinical heterogeneity and incomplete penetrance, possibly related to the status of X-inactivation in the brain. Extensive intrafamilial variability is also reported.
SYN1-related disorders are characterized by developmental delay, intellectual disability, behavioral disturbances, and epilepsy
A detailed clinical description of each individual in our cohort was provided by the referring physician and can be found in Supplementary Table S2. A summary of the main clinical features of all individuals (from our cohort and the literature) is provided in Table 1.
In our cohort, pregnancy and infancy were typically uneventful. Birth parameters were similar to those of the general population. Developmental delay (DD) was reported in 77% (24/31) of the individuals (Figure 4A), typically affecting speech acquisition more than gross motor development. Walking independently was achieved at a median age of 17.5 months (range 12–36 months), whereas the median age of pronouncing the first words was 24 months (ranging from 12 months to 12 years of age). At the latest clinical examination, ten individuals (age range: 3.5–50 years) were either non-verbal or could only pronounce a few words or short sentences. ID was present in 74% (20/27) of the subjects for whom assessment was possible. The degree of ID could be assessed for 16 of the 20 individuals and was mild in ten, moderate in three, and severe in three. Six individuals showed signs of regression; in five of them, regression started after the onset of epilepsy. Epilepsy was one of the cardinal features, reported in 83% (25/30) of the individuals. Triggers of seizures were reported in 60% (15/25) of those with epilepsy. Behavioral disturbances were present in 85% (22/26) of the individuals. More specifically, ASD was diagnosed in 42% (11/26) and attention deficit hyperactivity disorder (ADHD) in 23% (6/26) of the total number of individuals of our cohort. Additional behavioral issues were observed in 65% (17/26) of the cases and comprise poor eye contact, aggressivity, impulsivity and sleep disorders. At the latest clinical examination, weight and height were mostly in the normal range for a given age and sex. Five individuals exhibited macrocephaly (>2 SD). When performed, brain magnetic resonance imaging (MRI) and neurologic examinations were normal. Specifically, MRI was normal in 70% (14/20) of the individuals of our cohort and the clinical neurological examination in 88% (22/25). Facial dysmorphic features were rarely reported and were rather unspecific.
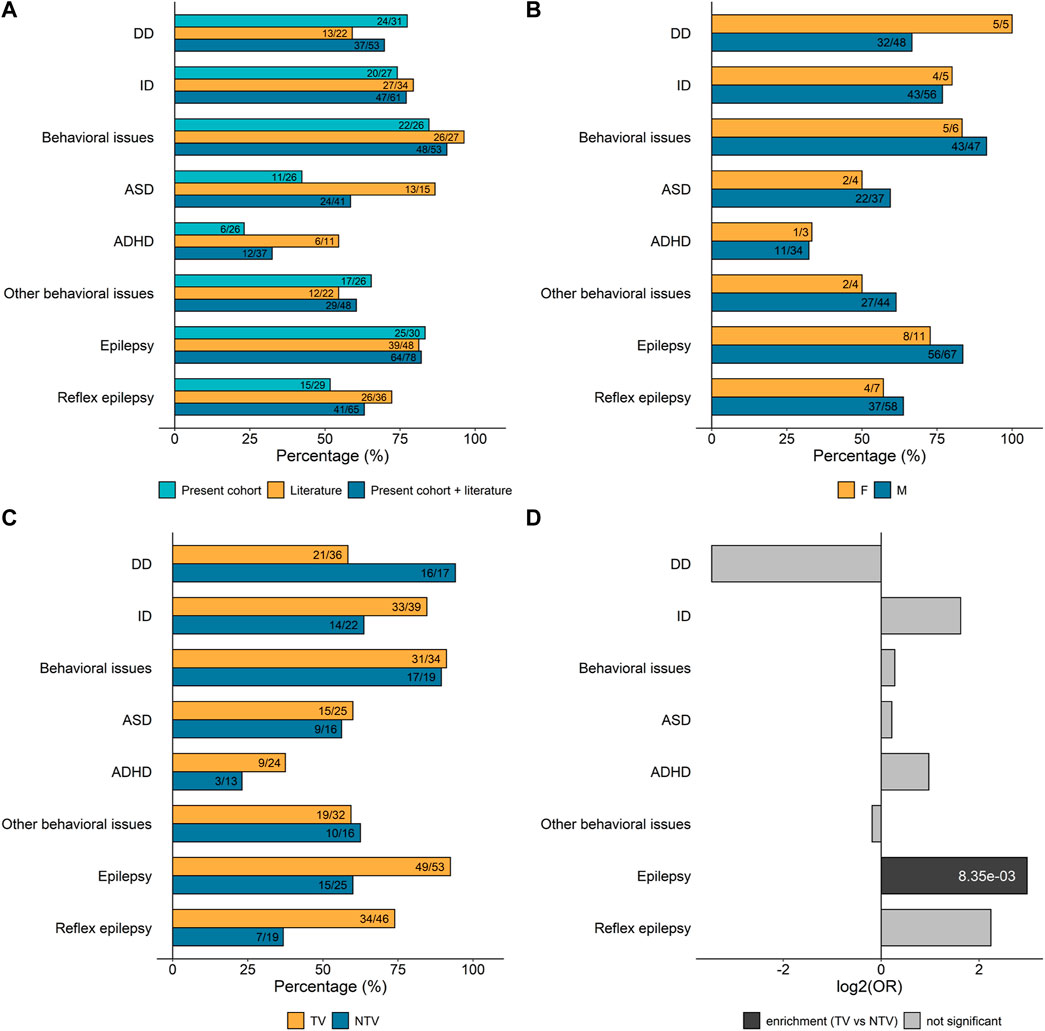
FIGURE 4. Frequency of the main clinical features associated with SYN1 variants. Numbers inside the bars are ratios between the number of patients presenting with the specific clinical sign and the number of patients for which information on that specific feature was available. (A) Percentage of SYN1 patients in our cohort and/or in the literature manifesting the clinical features of interest. (B) Percentage of male and female SYN1 patients manifesting specific clinical features. Data from patients in our cohort and the literature were combined. (C) Percentage of patients with either truncating variants (TV) or non-truncating variants (NTV) manifesting specific clinical features. Data from patients in our cohort and the literature were combined. TV: nonsense, frameshift and splicing variants; NTV: missense substitutions and in-frame duplications. (D) Per clinical feature enrichment/depletion of TV versus NTV. Only significant p-values are shown inside bars (Fisher’s test followed by Bonferroni correction for multiple testing; Supplementary Table S4). DD, developmental delay; ID, intellectual disability; ASD, autism spectrum disorder; ADHD, attention deficit hyperactivity disorder.
The frequency of the main clinical features (DD, ID, behavioral disturbances, and epilepsy) was subsequently calculated on the total number of individuals with SYN1 variants for whom clinical information was available: altogether, DD was present in 70% of the total subjects (37/53), ID in 77% (47/61), ASD in 59% (24/41), ADHD in 32% (12/37), epilepsy in 82% (64/78), and reflex epilepsy in 63% (41/65) (Figure 4A). Importantly, individuals with SYN1 variants can present with a variable combination and severity of these clinical signs.
Next, we investigated whether the clinical features of interest are enriched in association with a specific variant type or with the sex of the individual. Males represent the vast majority of affected individuals, with 71 males and 12 females with SYN1 putatively pathogenic variants reported so far. Although hemizygous males tend to show a more homogeneous and severe clinical presentation, no specific clinical feature was found to be enriched in males compared to females (Figure 4B, Supplementary Table S3). We then compared the frequency of ID, DD, behavioral disturbances, and epilepsy in individuals with truncating variants (TV; frameshift, nonsense, start-loss, and splicing variants) and non-truncating variants (NTV; including both missense substitutions and in-frame duplications) (Figures 4C,D). Subjects with TV presented more frequently with seizures than subjects with NTV (p = 8.35 × 10–3, Fisher Test, Supplementary Table S4). Reflex seizures were also more frequent in individuals with TV, and conversely, DD was more often observed in individuals with NTV, although these two differences were not statistically significant after adjusting for multiple testing (Figures 4C,D; Supplementary Table S4).
Lastly, we examined whether the presence of the main clinical features associated with SYN1 genetic alterations correlates with the location of the variant at the protein level. As shown in Figure 5, no correlation was identified between the position of the variant or its predicted functional consequences and any specific clinical feature.

FIGURE 5. Combined clinical features observed in patients with specific SYN1 variants. T: truncating variants (nonsense, frameshift and splicing variants); N: non-truncating variants (missense substitutions and in-frame duplications). Black squares indicate that a given clinical feature was described in association with the corresponding variant. The protein region has been subdivided in the four protein domains affected by the variants, namely domains A-D. No association can be detected between the position of the variant and the presence of a given clinical feature.
Of note, six individuals were each reported to carry a single VUS in additional genes (Supplementary Table S2). Of these, three genes (TMTC1, SUPT5H, and MYCBP2) had not been associated to any disease until now. The remaining three genes KMT2C, RELN, and BRCA2, had been previously associated with Kleefstra syndrome (OMIM #617768), Familial Temporal Lobe Epilepsy (OMIM #616436), and with different types of cancer, respectively. We cannot rule out that the presence of these subsidiary variants may have an effect on the phenotypic outcome and might therefore contribute to the SYN1-associated phenotypic variability.
SYN1 variants are frequently associated with reflex epilepsy mainly triggered by contact with water
Epilepsy represents one of the paramount features of SYN1-related disorders. Epileptic seizures were reported in 82% (64/78) of the total number of individuals with putatively disease-causing SYN1 variants. The onset of seizures ranges from 6 months to 26 years of life, with a median age of onset of 7 years old. In 29% of cases with seizures, seizure-onset occurred before the second year of life and in 55% within the first 5 years of age. Ninety percent of individuals manifest their first seizures before the age of thirteen (Supplementary Figure S3). Seizure frequency is also variable, with some individuals having seizures weekly or monthly and others being seizures-free for several months. In few individuals, seizures occurred only once or represented rare events. Seizures are typically tonic-clonic (focal-to-bilateral tonic-clonic or of unknown onset) or of focal-onset with impaired awareness (Fisher et al. 2017).
Electroencephalograms of individuals with epilepsy frequently revealed focal or diffuse slow spike waves or multifocal epileptiform discharges, mainly of frontal origin. Pharmacological treatment could successfully improve epilepsy in several patients. Pharmoresistance was reported in 11 individuals of our cohort.
Neurodevelopmental comorbidities (DD, ID, and ASD) did not correlate with the occurrence of epilepsy (Supplementary Table S5). However, the age at seizure onset showed some degree of correlation with the severity of ID: in individuals with severe-to-profound ID, seizures manifested at a median age of 1 year of life (range: 5 months to 5 years), much earlier than individuals with borderline-to-mild ID (median age 9.5 years) (p = 0.025, Mann-Whitney Test and Bonferroni correction for multiple testing) (Supplementary Table S6, Figure 6A).
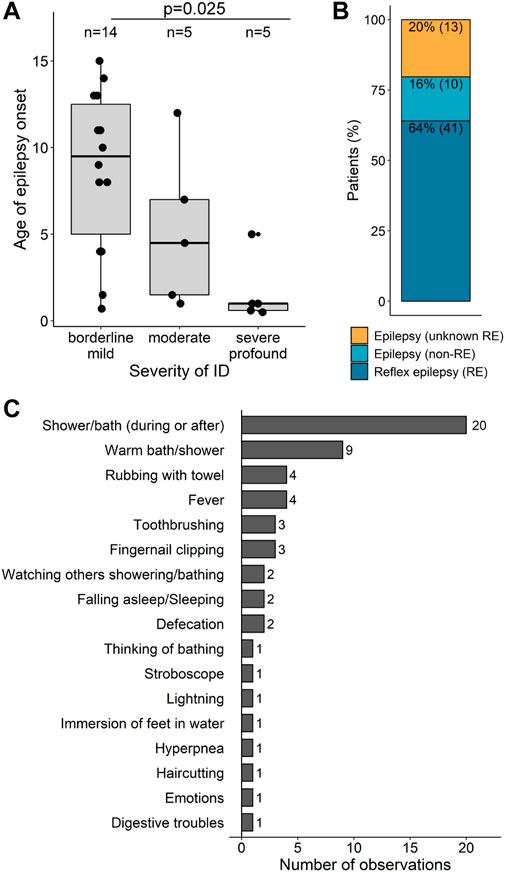
FIGURE 6. Epilepsy types and associations (A) Age of epilepsy onset according to the severity of ID. Patients were grouped into three categories of ID severity: borderline/mild (n = 14), moderate (n = 5) and severe/profound (n = 5). Each point represents one patient for which information was obtained. Box plot elements are defined as follows: center line: median; box limits: upper and lower quartiles; whiskers: 1.5× interquartile range; smaller point outside whiskers: outlier (B) Categorization of epilepsy types. Numbers inside the bars are shown as percentages and ratios for epilepsy patients. RE, reflex epilepsy. (C) Triggers reported in association with SYN1 variants sorted from the most to the less frequent.
More than half of the individuals with epilepsy (41/64, 64%) had reflex seizures. Unprovoked seizures were reported in 16% of the individuals (10/64). The presence of reflex seizures could not be confirmed or excluded in the remaining 20% (13/64) (Figure 6B). The most frequent triggers reported in association with SYN1 variants were showering, bathing, or contact with water (Figure 6C). In approximately one-third of these individuals, reflex seizures only occurred in the presence of warm water, whereas for the others the temperature of the water was irrelevant. Additional triggers include fever (n = 4), rubbing with towel (n = 4), toothbrushing (n = 3), fingernail clipping (n = 3), defecation (n = 2), falling asleep (n = 2), or even watching someone taking a shower (n = 2). Emotion, lightning, stroboscope, haircutting, thinking of bathing, immersion of feet in the water, digestive troubles, and hyperpnea were each reported as triggers in one individual each.
Discussion
The proper formation, function, and plasticity of synapses are highly dependent on the synapsin protein family. Three related synapsin genes that can generate at least five different protein isoforms are present in mammals (Longhena et al. 2021; Sudhof et al. 1989). Synapsins regulate the distribution and availability of neurotransmitter vesicles as well as neurotransmitter release upon binding to actin, which serves as a molecular scaffold system to allow a higher concentration of synapsins in proximity of presynaptic vesicles clusters (Garcia et al. 2004; Cesca et al. 2010; Longhena et al. 2021; Sankaranarayanan, Atluri, and Ryan 2003). By this, synapsins regulate SVs trafficking between the reserve pool and the readily releasable pool (Cesca et al., 2010; Guarnieri et al., 2017). Synapsin I is also implicated in the regulation of the final steps of docking and priming of SVs to the presynaptic plasma membrane (Fassio et al. 2006). Therefore, synapsins represent one of the key players in neurotransmission.
Synapsins are enriched in the brain and poorly or non-expressed in other tissues (De Camilli et al. 1979; De Camilli, Cameron, and Greengard 1983). However, within the brain, the three synapsin genes display only partially overlapping patterns of expression (Porton, Kao, and Greengard 1999; De Camilli, Cameron, and Greengard 1983). In light of this incomplete redundancy, accurate expression and balance of synapsin isoforms are instrumental for proper cellular and brain functions.
Correspondingly, alterations of synaptic homeostasis consequent to variants in SYN1 have been shown to lead to disease phenotypes mainly characterized by epilepsy, ID, DD, and ASD (Garcia et al. 2004; Accogli et al. 2021; Longhena et al. 2021). Due to the increasing accessibility of next-generation sequencing technologies, we were able to assemble a large cohort of individuals with SYN1 variants through international collaborations and Genematcher, thereby expanding the clinical and molecular spectrum of SYN1-related disorders. DD, ID, behavioral issues, and epilepsy (particularly reflex epilepsy) were corroborated as the main features of these disorders. ASD and reflex epilepsy were found to be more frequent in previously published cohorts than in our cohort. This could be accounted for by the gene-based inclusion criteria of our cohort in comparison to the phenotype-focus of previous publications. The advantage resulting from such a genotype-first approach is the more comprehensive recognition of the phenotypic spectrum associated with variants in a given gene.
Reflex epilepsy is emblematic of SYN1-related disorders (Nguyen et al. 2015; Peron et al. 2018; Accogli et al. 2021; Zhou et al. 2021). Contact with water, such as during bathing or showering, is the most frequent trigger in individuals with SYN1 variants. The temperature of the water may be relevant only for some subjects (Nguyen et al. 2015; Peron et al. 2018). As previously suggested, water temperature might play a confounding role, whereby the contact with water represents the real somatosensory stimulus that triggers the seizures (Accogli et al. 2021). Additionally, the precise contribution of water contact and temperature is complicated by other reported triggers including activities that are likely to occur around bath time. Herein, we expand on the type and number of possible triggers to include visual triggers (lighting, stroboscope lights), hyperpnea, emotions, as well as more unusual triggers such as digestive troubles or defecation. Particularly, seizures triggered by defecation were reported in two out of 15 individuals with reflex epilepsy of our cohort (13%) and warrant therefore special attention.
In our combined cohort, we did not detect enrichment of DD, ID, or ASD in individuals with epilepsy, suggesting that cognitive impairment processes are not a consequence of epileptic seizures and likely arise from parallel pathophysiological processes resulting from the SYN1 alteration. This is in line with previous evidence showing that social impairment becomes apparent before the onset of seizures in mouse models (Greco et al. 2013). Nevertheless, an association between the age at seizure onset and the severity of ID was noted, with individuals with severe-to-profound ID having seizures at an earlier median age than subjects with less severe cognitive impairment. Additionally, loss of acquired milestones also occurred in five individuals after the onset of seizures, while only one subject showed signs of regression before seizure onset. Altogether, these data suggest that, although the distinct clinical features observed in SYN1 individuals might develop independently from each other, seizure onset and frequency could still influence the course or the level of severity of other clinical manifestations.
We note that case ascertainment in many studies describing the clinical features of individuals with SYN1 variants has been weighted towards individuals with epilepsy. Identification of more individuals without epilepsy and comparison of the detailed longitudinal developmental trajectories of individuals carrying pathogenic SYN1 variants with and without epilepsy will help to clarify the role of seizures per se on neurodevelopment. The finding that epilepsy developed in 83% of SYN1 variant carriers published to date suggests that early molecular diagnosis of a SYN1 variant (for example, in children born within families known to carry a variant) should alert clinicians to the need for careful surveillance for possible seizures. Given that after a first seizure, a 10-year risk of subsequent seizures of 60% is sufficient to diagnose epilepsy and to tip the risk-benefit ratio in favor of commencing treatment (Fisher et al. 2014), the administration of anti-seizure medication after a first seizure should be considered. However, the possible ascertainment bias towards cases with epilepsy in the SYN1 literature means that we should not yet conclude that anti-seizure medication should be commenced prior to the presentation of seizures as it is likely that a significant proportion of carriers would not benefit from it.
SYN1 variants predominantly affect male individuals. The exact male:female ratio is however difficult to determine due to the paucity of detailed clinical information on female carriers. We have analyzed the reported phenotypes of 71 male and 12 female individuals. A higher frequency of DD and ID was observed in the limited female cohort, but this was not statistically significant. The ascertainment of the clinical features of a greater number of females with SYN1 variants will help to clarify whether these variants present differently in male and female individuals. Overall, female carriers have been reported to be either unaffected or to display a variable number and severity of clinical signs, hence pointing to the existence of clinical heterogeneity and incomplete penetrance. When assessed, X-inactivation analyses performed on blood DNA of female carriers did not show any tendency towards preferential skewing, although this might not reflect the status in the brain, i.e. the primarily affected organ of the SYN1-related disorders. In our cohort, Individual 29 presented with skewed X-inactivation in blood. Whether different degrees of X-inactivation in brain tissues might account for the distinct phenotypical manifestations of the female individuals is an intriguing possibility. Notably, SYN1 is not reported to escape X-inactivation in females (Balaton, Cotton, and Brown 2015).
Mosaicism had not been reported previously in association with SYN1. In our cohort, we identified two SYN1 variants in a mosaic state in blood DNA in two male individuals. Strikingly, the mosaic variant resulted in a classic phenotype characterized by the presence of epilepsy in Individual 30, whereas no clinical features were reported for the mosaic father of Individual 29. A different percentage of the mutant allele in brain tissues might explain the different phenotypes, similarly to what has been postulated regarding X-inactivation in female carriers, and as previously described in other epilepsy disorders like SCN1A-related Dravet syndrome (Depienne et al. 2010).
Multiple types of alterations were identified in our cohort and the literature, including frameshift duplications or deletions, nonsense variants, splicing variants, in-frame duplications, and missense substitutions (Garcia et al, 2004; Fassio et al., 2011; Butler et al., 2017; Nguyen et al., 2015; Guarnieri et al., 2017; Rossi et al., 2017; Lindy et al., 2018; Peron et al., 2018; Fernández-Marmiesse et al., 2019; Darvish et al., 2020; Ibarluzea et al., 2020; Accogli et al., 2021; Mojarad et al., 2021; Stranneheim et al., 2021; Van der Ven et al., 2021; Xiong et al., 2021; Yang et al., 2020; Zhou et al., 2021). We also report for the first time a deletion of a single nucleotide within the first codon of the SYN1 protein (Individuals 1–3). The loss of the start codon consequent to this deletion likely leads to alternative translation initiation and subsequent generation of an N-terminally truncated protein. SYN1 N-terminus is highly conserved and depleted from putatively pathogenic alterations within the gnomAD database. Additionally, several residues of this region are pivotal for the phosphorylation-dependent regulation of SYN1 activity (Cesca et al. 2010; Longhena et al. 2021). Hence, the start-loss variant identified in our cohort might impair the ability of SYN1 to interact with its binding partners, which is likely to affect the protein’s ability to carry out its normal function in SV dynamics.
Epilepsy was more frequently observed in individuals with TV (frameshift, nonsense, and splicing variants). Accordingly, functional studies have shown that SYN1 TV lead to higher network excitability and firing/bursting activity (Lignani et al. 2013). Decreased stability or expression of the mutated protein as well as alterations of its subcellular distribution or the inter- or intramolecular interactions were found to result in altered trafficking or release of the SVs, which could ultimately affect neural network excitability and induce the consequent epileptic phenotypes (Lignani et al. 2013; Fassio et al. 2011; Giannandrea et al. 2013).
Some missense substitutions were similarly shown to lead to reduced protein expression and altered SVs mobility or clustering (Fassio et al. 2011; Guarnieri et al. 2017; Darvish et al. 2020). Missense variants mainly fall within the category of VUS. Analysis of the conservation of the affected residues across species or synapsin isoforms together with bioinformatic predictions support a putative pathogenic role for all but one reported missense substitution. Namely, the p.(Arg114Gly) variant identified in Individual 25 of our cohort affects a residue that is poorly evolutionarily conserved, nor conserved across different synapsin proteins. For this reason, the functional/physiological relevance of this variant in the context of the individual’s phenotype remains uncertain. However, the prediction of the impact of this variant on protein stability suggests that the p.(Arg114Gly) substitution might be destabilizing. Notably, no additional disease-relevant variant was identified for this subject by targeted sequencing of an ID panel comprising 451 genes.
In conclusion, our study expands on the molecular spectrum of SYN1 variants and improves the clinical characterization of SYN1-related neurodevelopmental disorders. The newly reported data and unified analysis of the clinical and molecular features of all published carriers will refine genetic counselling and the personalization of medical care for individuals with SYN1-related neurodevelopmental disorders. Future studies will be required to understand the mechanisms by which specific pathogenic SYN1 variants result in a range of epileptic and neurodevelopmental phenotypes, and to clarify the mechanisms underlying the reflex seizures. These should be complemented by studies to assess the clinical and developmental trajectories of individuals with SYN1 variants and to examine how these are affected by co-morbid seizures and medication exposure with the ultimate aim of providing precision medicine.
Data availability statement
The datasets presented in this study can be found in online repositories. The link to the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, SUB11906611 (SCV002558865 - SCV002558886).
Ethics statement
The studies involving human participants were reviewed and approved by the Local ethics committees at the participating centers. Data were shared anonymously and do not contain identifiable information. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
Supervision: CD, GL; Conceptualization: CD, GL, EL, IP; Writing—original draft: CD, GL, EL, IP; Investigations, formal analysis, writing—review and editing: all authors.
Funding
This research was funded in whole, or in part, by the Wellcome Trust [203914/Z/16/Z] supporting DL-S. For the purpose of Open Access, the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission. CD is supported by Universitätsklinikum Essen and received grants from the Deutsche Forschungsgemeinschaft (DFG). AB is funded by a BRIDGE—Translational Excellence Programme grant funded by the Novo Nordisk Foundation, grant agreement number: NNF20SA0064340. FB received funding from the Italian Ministry of Health.
Acknowledgments
We thank the patients and their families for participating to this study. We acknowledge support from the Open Access Publication Fund of the University of Duisburg-Essen. One patient included in this study was diagnosed thanks to Defidiag (NCT04154891), a study supported by The French Ministry of Health and the Agence Nationale de la Recherche (ANR-10-IAHU-01).
Conflict of interest
Author RP was employed by GeneDx. RT reports Honoraria from Arvelle/Angelini, Bial, Eisai, GW Pharma/Jazz, Sanofi, UCB Pharma and Zogenix, and unrestricted funding support from Arvelle/Angelini and UNEEG. PS has served on a scientific advisory board for the Italian Agency of the Drug (AIFA); has received honoraria from GW pharma, Kolfarma s.r.l., Proveca Pharma Ltd, and Eisai Inc.; and has received research support from the Italian Ministry of Health (Ricerca Corrente 2022) and Fondazione San Paolo.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.1019715/full#supplementary-material
References
Abouelhoda, M., Faquih, T., El-Kalioby, M., and Alkuraya, F. S. (2016). Revisiting the morbid genome of Mendelian disorders. Genome Biol. 17, 235. doi:10.1186/s13059-016-1102-1
Accogli, A., Wiegand, G., Scala, M., Cerminara, C., Iacomino, M., Riva, A., et al. (2021). Clinical and Genetic Features in Patients With Reflex Bathing Epilepsy. Neurology 97, e577–e586. doi:10.1212/WNL.0000000000012298
Balaton, B. P., Cotton, A. M., and Brown, C. J. (2015). 'Derivation of consensus inactivation status for X-linked genes from genome-wide studies. Biol. Sex Differ. 6, 35. doi:10.1186/s13293-015-0053-7
Butler, K. M., da Silva, C., Alexander, J. J., Hegde, M., and Escayg, A. (2017). Diagnostic Yield From 339 Epilepsy Patients Screened on a Clinical Gene Panel. Pediatr. Neurol. 77, 61–66. doi:10.1016/j.pediatrneurol.2017.09.003
Cabana, J. F., Gilbert, G., Letourneau-Guillon, L., Safi, D., Rouleau, I., Cossette, P., and Nguyen, D. K. (2018). 'Effects of SYN1Q555X mutation on cortical gray matter microstructure. Hum. Brain Mapp. 39, 3428–48. doi:10.1002/hbm.24186
Cambiaghi, M., Cursi, M., Monzani, E., Benfenati, F., Comi, G., Minicucci, F., Valtorta, F., and Leocani, L. (2013). 'Temporal evolution of neurophysiological and behavioral features of synapsin I/II/III triple knock-out mice. Epilepsy Res. 103, 153–60. doi:10.1016/j.eplepsyres.2012.07.012
Cavalleri, G. L., Weale, M. E., Shianna, K. V., Singh, R., Lynch, J. M., Grinton, B., et al. (2007). 'Multicentre search for genetic susceptibility loci in sporadic epilepsy syndrome and seizure types: a case-control study. Lancet. Neurol. 6, 970–80. doi:10.1016/S1474-4422(07)70247-8
Cesca, F., Baldelli, P., Valtorta, F., and Benfenati, F. (2010). 'The synapsins: key actors of synapse function and plasticity. Prog. Neurobiol. 91, 313–48. doi:10.1016/j.pneurobio.2010.04.006
Corradi, A., Fadda, M., Piton, A., Patry, L., Marte, A., Rossi, P., et al. (2014). 'SYN2 is an autism predisposing gene: loss-of-function mutations alter synaptic vesicle cycling and axon outgrowth. Hum. Mol. Genet. 23, 90–103. doi:10.1093/hmg/ddt401
Corradi, A., Zanardi, A., Giacomini, C., Onofri, F., Valtorta, F., Zoli, M., and Benfenati, F. (2008). Synapsin-I- and synapsin-II-null mice display an increased age-dependent cognitive impairment. J. Cell Sci. 121, 3042–51. doi:10.1242/jcs.035063
Darvish, H., Azcona, L. J., Tafakhori, A., Mesias, R., Ahmadifard, A., Sanchez, E., et al. (2020). 'Phenotypic and genotypic characterization of families with complex intellectual disability identified pathogenic genetic variations in known and novel disease genes. Sci. Rep. 10, 968. doi:10.1038/s41598-020-57929-4
De Camilli, P., Cameron, R., and Greengard, P. (1983). 'Synapsin I (protein I), a nerve terminal-specific phosphoprotein. I. Its general distribution in synapses of the central and peripheral nervous system demonstrated by immunofluorescence in frozen and plastic sections. J. Cell Biol. 96, 1337–54. doi:10.1083/jcb.96.5.1337
De Camilli, P., Ueda, T., Bloom, F. E., Battenberg, E., and Greengard, P. (1979). 'Widespread distribution of protein I in the central and peripheral nervous systems'. Proc Natl Acad Sci U S A 76, 5977–81. doi:10.1073/pnas.76.11.5977
den Dunnen, J. T, Dalgleish, D. R, MaglottMcGowan-Jordan, R. K. Hart, M. S. Greenblatt, J.A. F., and SmithAntonarakis, T.S. E. (2016). .'HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat 37, 564–9. doi:10.1002/humu.22981Roux
Depienne, C., Trouillard, O., Gourfinkel-An, I., Saint-Martin, C., Bouteiller, D., Graber, D., et al. (2010). 'Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. J. Med. Genet. 47, 404–10. doi:10.1136/jmg.2009.074328
Eldomery, M. K., Coban-Akdemir, Z., Harel, T., Rosenfeld, J. A., Gambin, T., Stray-Pedersen, A., et al. (2017). 'Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 9, 26. doi:10.1186/s13073-017-0412-6
Etholm, L., Bahonjic, E., Walaas, S. I., Kao, H. T., and Heggelund, P. (2012). 'Neuroethologically delineated differences in the seizure behavior of synapsin 1 and synapsin 2 knock-out mice. Epilepsy Res. 99, 252–9. doi:10.1016/j.eplepsyres.2011.12.004
Etholm, L., and Heggelund, P. (2009). 'Seizure elements and seizure element transitions during tonic-clonic seizure activity in the synapsin I/II double knockout mouse: a neuroethological description. Epilepsy Behav. 14, 582–90. doi:10.1016/j.yebeh.2009.02.021
Fassio, A., Merlo, D., Mapelli, J., Menegon, A., Corradi, A., Mete, M., et al. (2006). The synapsin domain E accelerates the exoendocytotic cycle of synaptic vesicles in cerebellar Purkinje cells. J. Cell Sci. 119, 4257–68. doi:10.1242/jcs.03194
Fassio, A., Patry, L., Congia, S., Onofri, F., Piton, A., Gauthier, J., et al. (2011). 'SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum. Mol. Genet. 20, 2297–307. doi:10.1093/hmg/ddr122
Feng, J., Chi, P., Blanpied, T. A., Xu, Y., Magarinos, A. M., Ferreira, A., et al. (2002). Regulation of neurotransmitter release by synapsin III. J. Neurosci. 22, 4372–80. 20026433
Fernandez-Marmiesse, A., Roca, I., Diaz-Flores, F., Cantarin, V., Perez-Poyato, M. S., Fontalba, A., et al. (2019). 'Rare Variants in 48 Genes Account for 42% of Cases of Epilepsy With or Without Neurodevelopmental Delay in 246 Pediatric Patients. Front. Neurosci. 13, 1135. doi:10.3389/fnins.2019.01135
Fisher, R. S., Acevedo, C., Arzimanoglou, A., Bogacz, A., Cross, J. H., Elger, C. E., et al. (2014). 'ILAE official report: a practical clinical definition of epilepsy. Epilepsia 55, 475–82. doi:10.1111/epi.12550
Fisher, R. S., Cross, J. H., French, J. A., Higurashi, N., Hirsch, E., Jansen, F. E., et al. (2017). 'Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 58, 522–30. doi:10.1111/epi.13670
Garcia, C. C., Blair, H. J., Seager, M., Coulthard, A., Tennant, S., Buddles, M., et al. (2004). 'Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J. Med. Genet. 41, 183–6. doi:10.1136/jmg.2003.013680
Giannandrea, M., Guarnieri, F. C., Gehring, N. H., Monzani, E., Benfenati, F., Kulozik, A. E., and Valtorta, F. (2013). Nonsense-mediated mRNA decay and loss-of-function of the protein underlie the X-linked epilepsy associated with the W356× mutation in synapsin I. PLoS One 8, e67724. doi:10.1371/journal.pone.0067724
Gitler, D., Takagishi, Y., Feng, J., Ren, Y., Rodriguiz, R. M., Wetsel, W. C., et al. (2004). 'Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J. Neurosci. 24, 11368–80. doi:10.1523/JNEUROSCI.3795-04.2004
Goujon, M., McWilliam, H., Li, W., Valentin, F., Squizzato, S., Paern, J., and Lopez, R. (2010). 'A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–9. doi:10.1093/nar/gkq313
Greco, B., Manago, F., Tucci, V., Kao, H. T., Valtorta, F., and Benfenati, F. (2013). 'Autism-related behavioral abnormalities in synapsin knockout mice. Behav. Brain Res. 251, 65–74. doi:10.1016/j.bbr.2012.12.015
Guarnieri, F. C., Pozzi, D., Raimondi, A., Fesce, R., Valente, M. M., Delvecchio, V. S., et al. (2017). 'A novel SYN1 missense mutation in non-syndromic X-linked intellectual disability affects synaptic vesicle life cycle, clustering and mobility. Hum. Mol. Genet. 26, 4699–714. doi:10.1093/hmg/ddx352
Hosaka, M., and Sudhof, T. C. (1998). 'Synapsins I and II are ATP-binding proteins with differential Ca2+ regulation. J. Biol. Chem. 273, 1425–9. doi:10.1074/jbc.273.3.1425
Ibarluzea, N., Hoz, A. B., Villate, O., Llano, I., Ocio, I., Marti, I., et al. (2020). Targeted Next-Generation Sequencing in Patients with Suggestive X-Linked Intellectual Disability. Genes (Basel), 11.
International League Against Epilepsy Consortium on Complex, Epilepsies (2018). 'Genome-wide mega-analysis identifies 16 loci and highlights diverse biological mechanisms in the common epilepsies. Nat. Commun. 9, 5269. doi:10.1038/s41467-018-07524-z
Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J. F., Darbandi, S. F., Knowles, D., Li, Y. I., et al. (2019). Predicting Splicing from Primary Sequence with Deep Learning. Cell 176, 535–548. doi:10.1016/j.cell.2018.12.015
John, A., Ng-Cordell, E., Hanna, N., Brkic, D., and Baker, K. (2021). 'The neurodevelopmental spectrum of synaptic vesicle cycling disorders. J. Neurochem. 157, 208–28. doi:10.1111/jnc.15135
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alfoldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141, 456 humans. Nature 581, 434–43. doi:10.1038/s41586-020-2308-7
Lignani, G., Raimondi, A., Ferrea, E., Rocchi, A., Paonessa, F., Cesca, F., et al. (2013). 'Epileptogenic Q555X SYN1 mutant triggers imbalances in release dynamics and short-term plasticity. Hum. Mol. Genet. 22, 2186–99. doi:10.1093/hmg/ddt071
Lindy, A. S., Stosser, M. B., Butler, E., Downtain-Pickersgill, C., Shanmugham, A., Retterer, K., et al. (2018). 'Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia 59, 1062–71. doi:10.1111/epi.14074
Longhena, F., Faustini, G., Brembati, V., Pizzi, M., Benfenati, F., and Bellucci, A. (2021). 'An updated reappraisal of synapsins: structure, function and role in neurological and psychiatric disorders. Neurosci. Biobehav. Rev. 130, 33–60. doi:10.1016/j.neubiorev.2021.08.011
Michetti, C., Caruso, A., Pagani, M., Sabbioni, M., Medrihan, L., David, G., et al. (2017). The Knockout of Synapsin II in Mice Impairs Social Behavior and Functional Connectivity Generating an ASD-like Phenotype. Cereb. Cortex 27, 5014–23. doi:10.1093/cercor/bhx207
Mojarad, B. A., Yin, Y., Manshaei, R., Backstrom, I., Costain, G., Heung, T., et al. (2021). Genome sequencing broadens the range of contributing variants with clinical implications in schizophrenia. Transl. Psychiatry 11, 84. doi:10.1038/s41398-021-01211-2
Nguyen, D. K., Rouleau, I., Senechal, G., Ansaldo, A. I., Gravel, M., Benfenati, F., and Cossette, P. (2015). 'X-linked focal epilepsy with reflex bathing seizures: Characterization of a distinct epileptic syndrome. Epilepsia 56, 1098–108. doi:10.1111/epi.13042
Peron, A., Baratang, N. V., Canevini, M. P., Campeau, P. M., and Vignoli, A. (2018). 'Hot water epilepsy and SYN1 variants. Epilepsia 59, 2162–63. doi:10.1111/epi.14572
Pires, D. E., Ascher, D. B., and Blundell, T. L. (2014). 'mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30, 335–42. doi:10.1093/bioinformatics/btt691
Porton, B., Kao, H. T., and Greengard, P. (1999). 'Characterization of transcripts from the synapsin III gene locus. J. Neurochem. 73, 2266–71. doi:10.1046/j.1471-4159.1999.0732266.x
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). 'CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886-D894–D94. doi:10.1093/nar/gky1016
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). 'Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–24. doi:10.1038/gim.2015.30
Rodrigues, C. H. M., Pires, D. E. V., and Ascher, D. B. (2021). 'DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 30, 60–69. doi:10.1002/pro.3942
Rosahl, T. W., Spillane, D., Missler, M., Herz, J., Selig, D. K., Wolff, J. R., et al. (1995). 'Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375, 488–93. doi:10.1038/375488a0
Rossi, M., El-Khechen, D., Black, M. H., Farwell Hagman, K. D., Tang, S., and Powis, Z. (2017). 'Outcomes of Diagnostic Exome Sequencing in Patients With Diagnosed or Suspected Autism Spectrum Disorders. Pediatr. Neurol. 70, 34–43. doi:10.1016/j.pediatrneurol.2017.01.033
Sankaranarayanan, S., Atluri, P. P., and Ryan, T. A. (2003). 'Actin has a molecular scaffolding, not propulsive, role in presynaptic function. Nat. Neurosci. 6, 127–35. doi:10.1038/nn1002
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). 'Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. doi:10.1038/msb.2011.75
Sobreira, N., Schiettecatte, F., Valle, D., and Hamosh, A. (2015). 'GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 36, 928–30. doi:10.1002/humu.22844
Stranneheim, H., Lagerstedt-Robinson, K., Magnusson, M., Kvarnung, M., Nilsson, D., Lesko, N., et al. (2021). 'Integration of whole genome sequencing into a healthcare setting: high diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. 13, 40. doi:10.1186/s13073-021-00855-5
Sudhof, T. C., Czernik, A. J., Kao, H. T., Takei, K., Johnston, P. A., Horiuchi, A., et al. (1989). 'Synapsins: mosaics of shared and individual domains in a family of synaptic vesicle phosphoproteins. Science 245, 1474–80. doi:10.1126/science.2506642
Thauvin-Robinet, C., Thevenon, J., Nambot, S., Delanne, J., Kuentz, P., Bruel, A., et al. (2019). 'Secondary actionable findings identified by exome sequencing: expected impact on the organisation of care from the study of 700 consecutive tests. Eur. J. Hum. Genet. 27, 1197–214. doi:10.1038/s41431-019-0384-7
The UniProt Consortium (2021). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49, D480–D89.
Turner, T. N., Wilfert, A. B., Bakken, T. E., Bernier, R. A., Pepper, M. R., Zhang, Z., et al. (2019). 'Sex-Based Analysis of De Novo Variants in Neurodevelopmental Disorders. Am. J. Hum. Genet. 105, 1274–85. doi:10.1016/j.ajhg.2019.11.003
Ueda, T., and Greengard, P. (1977). 'Adenosine 3':5'-monophosphate-regulated phosphoprotein system of neuronal membranes. I. Solubilization, purification, and some properties of an endogenous phosphoprotein. J. Biol. Chem. 252, 5155–63. doi:10.1016/s0021-9258(17)40170-0
van der Ven, A. T., Johannsen, J., Kortum, F., Wagner, M., Tsiakas, K., Bierhals, T., et al. (2021). 'Prevalence and clinical prediction of mitochondrial disorders in a large neuropediatric cohort. Clin. Genet. 100, 766–70. doi:10.1111/cge.14061
Xiong, J., Duan, H., Chen, S., Kessi, M., He, F., Deng, X., et al. (2021). 'Familial SYN1 variants related neurodevelopmental disorders in Asian pediatric patients. BMC Med. Genomics 14, 182. doi:10.1186/s12920-021-01028-4
Yang, J. O., Choi, M. H., Yoon, J. Y., Lee, J. J., Nam, S. O., Jun, S. Y., et al. (2020). Corrigendum: Characteristics of Genetic Variations Associated With Lennox-Gastaut Syndrome in Korean Families. Front. Genet. 11, 669107. doi:10.3389/fgene.2021.669107
Keywords: SYN1, synapsins, reflex epilepsy, genotype-phenotype correlation, neurodevelopmental disorders, autism spectrum disorders
Citation: Parenti I, Leitão E, Kuechler A, Villard L, Goizet C, Courdier C, Bayat A, Rossi A, Julia S, Bruel A-L, Tran Mau-Them F, Nambot S, Lehalle D, Willems M, Lespinasse J, Ghoumid J, Caumes R, Smol T, El Chehadeh S, Schaefer E, Abi-Warde M-T, Keren B, Afenjar A, Tabet A-C, Levy J, Maruani A, Aledo-Serrano Á, Garming W, Milleret-Pignot C, Chassevent A, Koopmans M, Verbeek NE, Person R, Belles R, Bellus G, Salbert BA, Kaiser FJ, Mazzola L, Convers P, Perrin L, Piton A, Wiegand G, Accogli A, Brancati F, Benfenati F, Chatron N, Lewis-Smith D, Thomas RH, Zara F, Striano P, Lesca G and Depienne C (2022) The different clinical facets of SYN1-related neurodevelopmental disorders. Front. Cell Dev. Biol. 10:1019715. doi: 10.3389/fcell.2022.1019715
Received: 15 August 2022; Accepted: 20 October 2022;
Published: 08 December 2022.
Edited by:
Walter Erwin Kaufmann, Emory University, United StatesReviewed by:
Hans Van Bokhoven, Radboud University Medical Centre, NetherlandsGeorge Augustine, Nanyang Technological University, Singapore
Daniel Gitler, Ben-Gurion University of the Negev, Israel
Copyright © 2022 Parenti, Leitão, Kuechler, Villard, Goizet, Courdier, Bayat, Rossi, Julia, Bruel, Tran Mau-Them, Nambot, Lehalle, Willems, Lespinasse, Ghoumid, Caumes, Smol, El Chehadeh, Schaefer, Abi-Warde, Keren, Afenjar, Tabet, Levy, Maruani, Aledo-Serrano, Garming, Milleret-Pignot, Chassevent, Koopmans, Verbeek, Person, Belles, Bellus, Salbert, Kaiser, Mazzola, Convers, Perrin, Piton, Wiegand, Accogli, Brancati, Benfenati, Chatron, Lewis-Smith, Thomas, Zara, Striano, Lesca and Depienne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christel Depienne, christel.depienne@uni-due.de
†These authors have contributed equally to this work and share first authorship