 Qian-Qian Liao
Qian-Qian Liao Qing-Qing Dong
Qing-Qing Dong Hui Zhang
Hui Zhang Hua-Pan Shu1
Hua-Pan Shu1- 1Department of Nephrology, Tongji Medical College, Union Hospital, Huazhong University of Science and Technology, Wuhan, China
- 2Department of Nephrology, The Second Affiliated Hospital, Chongqing Medical University, Chongqing, China
Serum- and glucocorticoid-induced kinase 3 (SGK3), which is ubiquitously expressed in mammals, is regulated by estrogens and androgens. SGK3 is activated by insulin and growth factors through signaling pathways involving phosphatidylinositol-3-kinase (PI3K), 3-phosphoinositide-dependent kinase-1 (PDK-1), and mammalian target of rapamycin complex 2 (mTORC2). Activated SGK3 can activate ion channels (TRPV5/6, SOC, Kv1.3, Kv1.5, Kv7.1, BKCa, Kir2.1, Kir2.2, ENaC, Nav1.5, ClC-2, and ClC Ka), carriers and receptors (Npt2a, Npt2b, NHE3, GluR1, GluR6, SN1, EAAT1, EAAT2, EAAT4, EAAT5, SGLT1, SLC1A5, SLC6A19, SLC6A8, and NaDC1), and Na+/K+-ATPase, promoting the transportation of calcium, phosphorus, sodium, glucose, and neutral amino acids in the kidney and intestine, the absorption of potassium and neutral amino acids in the renal tubules, the transportation of glutamate and glutamine in the nervous system, and the transportation of creatine. SGK3-sensitive transporters contribute to a variety of physiological and pathophysiological processes, such as maintaining calcium and phosphorus homeostasis, hydro-salinity balance and acid-base balance, cell proliferation, muscle action potential, cardiac and neural electrophysiological disturbances, bone density, intestinal nutrition absorption, immune function, and multiple substance metabolism. These processes are related to kidney stones, hypophosphorous rickets, multiple syndromes, arrhythmia, hypertension, heart failure, epilepsy, Alzheimer’s disease, amyotrophic lateral sclerosis, glaucoma, ataxia idiopathic deafness, and other diseases.
1 Introduction
Serum- and glucocorticoid-induced kinase (SGK), the AGC family of serine/threonine protein kinase, which is similar to the protein kinase B (PKB)/AKT family in exhibiting structure and sequence, is widely distributed in various tissues and organs of mammals, involving the regulation of substance transportation, hormone release, neuroexcitability, cell proliferation, and apoptosis (Lang et al., 2006a). The SGK family consists of three highly homologous mammalian isoforms, SGK1, SGK2, and SGK3, which are, respectively, encoded by three different genes (Lang et al., 2006a), but the catalytic domains of SGK3 and SGK2 have up to 80% amino acid sequence homology with SGK1 (Kobayashi and DeakM.MorriceN.Cohen, 1999). Although they are similar in function, there are differences. In this brief review, we focus on the distribution and regulation of SGK3, as well as the contribution of SGK3 to transporter-related diseases.
1.1 Structure and distribution of serum- and glucocorticoid-induced kinase-3
The primary structure of SGK3, known as cytokine-independent survival kinase (CISK), consists of a Phox-homology domain (PX) in the N terminus, a kinase domain (comprising Thr-320), and a protein kinase AGC C-terminal domain/hydrophobic motif (HM) (comprising Ser-486) (Xing et al., 2004). The PX domain is a phosphoinositide-binding domain, which can target the subcellular localization of SGK3 to membranes, especially early endosomes. SGK3 is distributed in almost all mammalian tissues and cell lines, including the heart, brain, lungs, skin, liver, pancreas, skeletal muscle, and kidney (Kobayashi and DeakM.MorriceN.Cohen, 1999; Yao et al., 2011). At the subcellular level, SGK3 is diffusely expressed throughout cells, covering the nucleus, cytoplasm, endosome, and plasma membrane (Lang et al., 2006a; He et al., 2011; Bago et al., 2014).
1.2 Regulatory mechanisms of serum- and glucocorticoid-induced kinase-3
To become functional, SGK3 is activated by phosphorylation through PI3K, PDK-1, and mTORC2 signaling pathways (Bago et al., 2014; Gasser et al., 2014; Chen et al., 2018). Insulin, growth factors (EGF, IGF1, FGF, and TPA), and oncogenic Ras can also activate SGK3 through the PI3K pathway (Malik et al., 2018). The PX domain of SGK3 binds phosphatidylinositol-3-phosphate (PI (3) P), which is activated by PI3K and targets SGK3 to the early endosome. Like other members of the AGC kinases, SGK3 is activated following the phosphorylation of its C-terminal/HM Ser-486 residue by mTORC2 and kinase domain T-loop Thr-320 residues by PDK-1 (Tessier and Woodgett, 2006; Bago et al., 2014). Unlike SGK1, although SGK3 belongs to serum- and glucocorticoid-induced kinase family, its expression is not regulated by serums and glucocorticoids (Kobayashi and DeakM.MorriceN.Cohen, 1999; Naray-Fejes-Toth et al., 2004). On the contrary, estrogen (Wang et al., 2011), androgen (Wang et al., 2014), insulin (Malik et al., 2018), and other hormones participate in the regulation of SGK3.
Numerous studies have demonstrated that SGK3 regulates downstream targets in cell survival, including B-cell lymphoma-extra-large (Bcl-xl), Bcl-2-associated death promoter (BAD), glycogen synthase kinase-3 (GSK3), forkhead family of transcription factors (FKHRs), and Flightless-1 (Fli-I) (Liu et al., 2000; Xu et al., 2009; Liu et al., 2012). Mainly, depending on GSK3-β, tuberous sclerosis factor 2 (TSC2), and proline-rich AKT substrate of 40 kDa (PRAS40), SGK3 leads to cell growth and proliferation (Bruhn et al., 2013). SGK3 also induces cell migration by downregulating the chemokine (C-X-C) receptor type-4 (CXCR4) through strong interaction and colocalization with ubiquitin ligase AIP4 (atrophin-1-interacting protein 4) (Slagsvold et al., 2006). In addition, the ubiquitin E3 ligase Nedd4-2 (neural precursor cell expressed developmentally down-regulated protein 4 subtype 2) is an important downstream target of SGK3 and has been shown to mediate the ubiquitination degradation process of various proteins (Yuan et al., 2018). In short, the activation of SGK3 plays an important role in cell proliferation, growth, survival, migration, and protein degradation (Hou et al., 2015).
1.3 Transporter-related diseases
Transmembrane transport of substances is widely involved in physiological and pathophysiological processes and is responsible for the transport of ions and small- and medium-size molecules. We can distinguish few types of transmembrane transports: active transport, passive transport, exocytosis, and endocytosis. The active transport is the energy-consuming process achieved by ATP hydrolysis including Ca2+-ATPase, Na+/K+-ATPase, and H+/K+-ATPase. The passive transport does not require energy including simple diffusion, facilitated diffusion, and osmosis, among which facilitated diffusion requires the participation of carriers, ion channels, and transporters. Ion channels, mainly contained voltage-gated channels and ligand-gated channels, are the primary mediators of muscle cell electrical signals and neuronal excitability including Na+, K+, Ca2+, and Cl− channels (Kulbacka et al., 2017). The transmembrane transport involves complex mechanisms and is decisive for cell biological functions. This process is determined by many factors, such as the internal and external environment of cells, transporters, carriers, and ion channels and the change in any link will cause the occurrence of diseases. For example, sodium and potassium ion channels are associated with many diseases, such as epilepsy, arrhythmias, and hypertension (Kulbacka et al., 2017). Aquaporins (AQPs) are related to congestive heart failure, hypertension, glaucoma, and other diseases (Julita et al., 2017).
1.4 Role of serum- and glucocorticoid-induced kinase-3 in transporter-related diseases
1.4.1 Serum- and glucocorticoid-induced kinase-3 and calcium–phosphorus metabolism
The strict control of calcium–phosphorus metabolism balance is the key to maintaining normal physiological functions. In skeletal tissues, calcium–phosphorus metabolism cooperates with osteoblasts and extracellular matrices to promote physiological mineralization. In soft tissues, it can prevent calcium-phosphate complexes from pathological or ectopic mineralization (Kirsch, 2006). Normally, calcium and phosphate that enter bodies through diet are mainly absorbed in the upper part of the small intestine and excreted through kidneys, intestines, and sweat glands. Thus, calcium–phosphorus metabolism is closely related to intestinal absorption, renal excretion, and bone remodeling in bodies (Peacock, 2010), depending on a variety of hormones, including parathyroid hormone (PTH) (Potts and Gardella, 2007), serum-ionized calcium and phosphate (Brown, 2007), and 1.25 (OH) 2D3 (Jurutka et al., 2001). In addition, the fibroblast growth factor-23 (FGF-23) and the FGF/Klotho-receptor complex reduce the reabsorption of phosphate by affecting sodium-dependent phosphate transport protein IIa (Npt2a) and inhibit the production of vitamin D3 by impairing 1-α hydroxylation of 25-hydroxyvitamin D, thus participating in calcium–phosphorus metabolism (Hruska et al., 2008; Liu et al., 2016) (Figure 1).
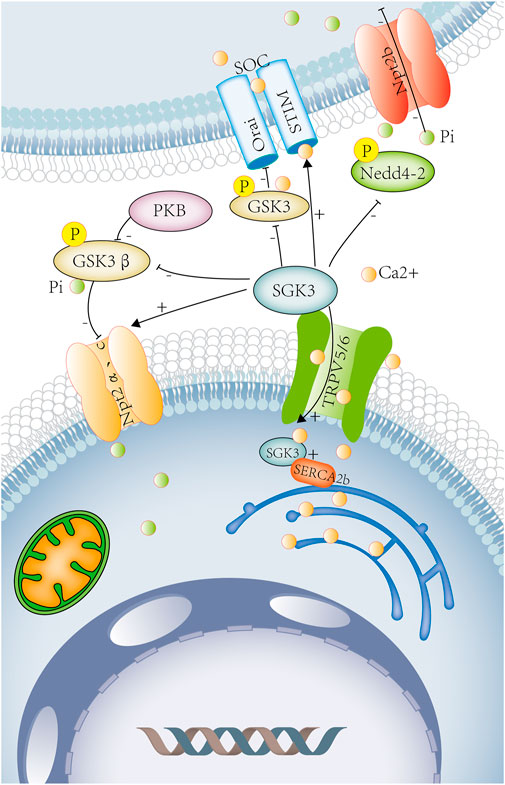
FIGURE 1. SGK3 and calcium–phosphorus metabolism. Calcium and phosphorus metabolism are closely related to intestinal absorption, renal excretion, and bone remodeling. SGK3 leads to calcium influx by increasing the number of calcium channels, TRPV5 and TRPV6, on the renal epithelial cell membrane and the brush-border membrane of intestinal epithelial cells. SGK3 affects the SOC channel by rising STIM2 and promotes the entry of Ca2+ into DC. It can also regulate the activity of SOC and bone mineral density by phosphorylating GSK3. SGK3 maintains ENR Ca2+ homeostasis by interacting with SERCA2b. SGK3 can directly regulate the activity of Npt2a. SGK3 can also phosphorylate and inhibit GSK3β activity through the PKB/SGK pathway, thereby up-regulating the activity of Npt2a. SGK3 can directly phosphorylate and inhibit the activity of Nedd4-2 and reduce the inhibition of the Npt2b activity by Nedd4-2. SGK3, serum/glucocorticoid-regulated kinase family member 3. TRPV5, transient receptor potential cation channel subfamily V member 5. TRPV6, transient receptor potential cation channel subfamily V member 6. SOC, store-operated channels. STIM2, stromal interaction molecule 2. ORAI, calcium release-activated calcium modulator 1. DC, dendritic cells. GSK3, glycogen synthase kinase-3. SERCA2b (ATP2a2) ATPase, Ca+ transporting, cardiac muscle, slow twitch 2. NPT2a, sodium-dependent phosphate transport protein. 2A.NPT2b, solute carrier family 34 member. 2a.GSK3β, glycogen synthase kinase 3 beta. PKB, rac-beta serine/threonine protein kinase. Orange circles represent calcium ions. Green circles represent phosphorus ions.
1.4.1.1 Renal calcium–phosphorus metabolism
The kidney is the main site of calcium and phosphate excretion. The serum calcium filtered from the glomerulus must be reabsorbed in more than 98% along nephrons, involving the proximal tubules, thick ascending limbs of Henle’s loops, distal tubules, and collecting ducts. On the apical membrane of the distal tubular epithelium, calcium passively enter into the epithelial cells by epithelial Ca2+ channels (TRPV5 or TRPV6) and binds to vitamin D3-sensitive calcium-binding proteins (calbindin-D28K). (Figure 1) On the basolateral membrane, calcium is actively extruded via the Na+/Ca2+ exchanger (NCX1) and Ca2+-ATPase (Hoenderop et al., 2000). (Figure 1) The epithelial Ca2+ channels, TRPV5 and TRPV6, play an important role in the renal and intestinal calcium absorption. It has been found in xenopus oocytes that SGK3 can significantly increase TRPV5 activity and the uptake of Ca2+, inducing calcium currents (ICa), an effect requiring the presence of PDZ domains of NHE regulating factors NHERF2 (Embark et al., 2004a) (Figure 1). In Xenopus oocytes, the expression of SGK3 together with TRPV6 resulted in significant further stimulation of calcium currents (ICa) but this process does not depend on PDZ domains of NHERF2 (Böhmer et al., 2007). (Figure 1) Therefore, SGK3 induces Ca2+ influx by increasing the number of calcium channels TRPV5/6 on renal epithelial cell membranes, which participates in the reabsorption of calcium in the kidney, and maintains calcium homeostasis.
The kidney plays a key role in the regulation of phosphate. About 70%–80% of the phosphate filtered by the glomerulus is reabsorbed in the proximal tubule of the kidney, which is mainly mediated by Npt2a and followed by Npt2c, Pit-2 (type III sodium-dependent phosphate transporter) (Lederer, 2014; Vervloet et al., 2017). (Figure 1) The study found that compared with SGK3 wild-type mice, SGK3 knockout mice had increased urinary phosphate excretion and decreased plasma FGF23, 1.25 (OH) 2D3. The expression of SGK3 together with Npt2a in Xenopus oocytes could significantly enhance phosphate-induced currents and the transport of renal tubular phosphate. Therefore, SGK3 is involved in the transport of renal tubular phosphate by regulating the activity of Npt2a (Bhandaru et al., 2011). SGK3 could also phosphorylate and inhibit the activity of GSK3 through the PKB/SGK pathway by subsequently increasing renal Npt2a activity and reducing renal calcium and phosphate loss (Foller et al., 2011). (Figure 1) FGF23 is secreted by osteoblasts to regulate homeostasis of phosphate, vitamin D, and bone mineralization, which can inhibit renal tubular type II sodium-dependent phosphate transporters (Npt2a and Npt2c), reducing renal phosphate reabsorption, and promoting phosphate excretion (Baum et al., 2005; Ide et al., 2018). (Figure 1) As previously described, plasma FGF23 was reduced in SGK3 knockout mice. SGK3 may participate in renal phosphate reabsorption through FGF23 (Figure 1).
1.4.1.2 Intestinal calcium–phosphorus metabolism
In the intestine, the absorption of calcium and phosphate is codependent, regulated by a variety of hormones, including 1.25 (OH) 2D3, glucocorticoid, growth hormone, and IGF-1. Among them, 1.25 (OH) 2D3 is a major factor affecting intestinal calcium and phosphate transportation, which increases intestinal calcium absorption by regulating the intestinal epithelial calcium channel TRPV6 and calcium-binding protein D9k (Christakos, 2012). In SGK3 knockout mice, plasma FGF23, 1.25 (OH) 2D3 was reduced (Bhandaru et al., 2011), thereby affecting intestinal calcium absorption. SGK3 can also directly regulate epithelial Ca2+ channels TRPV5 and TRPV6 on the brush-border membrane of intestinal epithelial cells and participate in intestinal calcium absorption (Embark et al., 2004a; Böhmer et al., 2007) (Figure 1).
The intestinal phosphate is absorbed through the following two pathways: passive transport (cell-tight junction) and active transport (mainly through sodium-dependent phosphate transporter Npt2b), and approximately 50% of the intestinal phosphate is absorbed and transported by the active pathway (Lederer, 2014; Hernando and Wagner, 2018). In Xenopus oocytes, co-expression of Nedd4-2 and Npt2b could attenuate Npt2b activity, while the effect reversed by the additional co-expression of SGK3 could directly phosphorylate and inhibit Nedd4-2 activity. Therefore, SGK3 can regulate the activity of Npt2b and participate in the absorption of intestinal phosphate by phosphorylating Nedd4-2 (Palmada et al., 2004a). (Figure 1) The plasma levels of 1.25(OH) 2D3 were reduced in SGK3 knockout mice (Bhandaru et al., 2011), and another study showed that 1.25 (OH) 2D3-induced increase in intestinal phosphate absorption is mediated by Npt2b (Xu et al., 2002). Intraperitoneal injection of 1.25 (OH) 2D3 and intragastric administration of 33P-labeled phosphate in rats can significantly increase plasma 33Pi levels, so 1.25 (OH) 2D3 could increase the intestinal transport of phosphate (Williams and DeLuca, 2007). Therefore, SGK3 may participate in intestinal phosphate absorption through 1.25 (OH) 2D3.
1.4.1.3 Bone calcium–phosphorus metabolism
Hereditary hypophosphatemic rickets (HR) is a group of renal phosphate wasting disorders due to genetic mutations. A novel mutation in the SGK3 gene was found in HR patients (Cebeci et al., 2020). SGK3 gene mutation makes the loss of the Thr-320 phosphorylation site and the disruption of the protein tertiary structure, resulting in increased urine phosphate, decreased serum phosphate, and bone deformity (Cebeci et al., 2020). In Bhandaru’s study, it was found that bone density of SGK3 knockout mice decreased, which may be related to the decrease in FGF23, 1.25 (OH) 2D3 in the plasma of mice after the SGK3 knockout (Bhandaru et al., 2011). Compared with GSK3 wt mice, the bone density of GSK3 ki mice is significantly reduced, and SGK3 could inhibit the activity of GSK3 through the PKB/SGK pathway to regulate bone density (Foller et al., 2011) (Figure 1).
1.4.1.4 Immune system calcium metabolism
Dendritic cells (DCs) are antigen-presenting cells that participate in both innate and adaptive immunities. Ca2+ signaling is involved in DC activation, maturation, migration, and the formation of immunological synapses with T cells (Shumilina et al., 2011). Ca2+ enters the DC partly accomplished by store-operated Ca2+ (SOC) channels, which consists of the plasma membrane Ca2+ channel Orai and the endoplasmic reticulum (ER) Ca2+ sensing subunit STIM. SGK3 could up-regulate the expression of STIM2, affecting the activity of SOC channels and DC migration, but the maturation and phagocytic capacity of DC is not affected by SGK3 (Schmid et al., 2012). Another study found that PKB/SGK-dependent GSK3 phosphorylation participated in the regulation of Ca2+ signaling in DCs, preventing DC maturation (Russo et al., 2013). Compared with DCs isolated from GSK3WT mice, DCs from GSK3KI mice had significantly reduced the levels of GSK3 phosphorylation and the expression of Orai1 and STIM2, the main components of the SOC channel, while the phosphorylation of GSK3 depends on the regulation of PBK/AKT and SGK. As shown earlier, SGK3 can regulate the release of Ca2+ and the activity of calcium channels (SOC) in DCs by phosphorylating GSK3 (Schmid et al., 2014) (Figure 1).
Mast cells play a decisive role in immunoglobulin E (IgE)-dependent allergic reactions, and the influx of extracellular calcium is the key to this process (Bradding et al., 2003). In vitro, mast cells were isolated from the bone marrow of sgk3−/− and sgk3+/+ mice. In sgk3−/− mast cells, after stimulation with IgE and related antigens, Ca2+ entry, Ca2+-activated K+ channel activity, and degranulation was significantly reduced. In vivo, in sgk3−/− mice, the serum histamine levels measured 30 min after the allergic reaction induced by intraperitoneal injection of anti-DNP IgE and DNP-HSA antigen was significantly lower than that in sgk3+/+ mice. The measured drop in the body temperature of sgk3−/− mice following antigen treatment was lower than that of sgk3+/+ mice, so the systemic allergic reaction of sgk3−/− mice was weakened. Therefore, SGK3 affects the immune function of mast cells by regulating antigen-stimulated Ca2+ entry and degranulation (Zemtsova et al., 2010) (Figure 1).
1.4.1.5 Endoplasmic reticulum calcium metabolism
The endoplasmic reticulum (EnR), an organelle, is the major site for protein folding and biosynthesis, and it is also a storage site for intracellular calcium. The Ca2+ pump sarcoplasmic/EnR calcium ATPase (SERCA) is a key enzyme that maintains high concentrations of Ca2+ in EnR and participates in the biosynthesis function of EnR. Studies have shown that SGK3 maintained EnR Ca2+ homeostasis through interaction with SERCA2b, thereby preventing it from excessive EnR stress (Wang et al., 2017) (Figure 1).
Taken together, SGK3 can regulate calcium–phosphorus metabolism through calcium channels TRPV5/6, SOC, and sodium-dependent phosphate transporter (Npt2a/2b) (Figure 1). Disorders of calcium-phosphorus metabolism can lead to various pathological changes, such as kidney stones, bone diseases, ectopic calcification, cardiovascular diseases, and abnormal immune functions (Taylor and Bushinsky, 2009).
In SGK3 and calcium–phosphorus metabolism, SGK3 induces calcium current, promotes calcium uptake, increases the absorption of calcium by renal epithelial cells and intestinal tract, and maintains calcium homeostasis by increasing the activity of TRPV5 or TRPV6. SGK3 can also affect the activity of SOC channels and DC migration by up-regulating STIM2 and is related to the immune function of mast cells, EnR Ca2+homeostasis. SGK3 can inactivate GSK3 and Nedd4-2 by phosphorylating them, thereby increasing the activity of Npt2a and Npt2b, enhancing the phosphate-induced current, promoting the renal tubular phosphate transport, intestinal phosphate absorption, and regulating bone mineral density (Table 1).
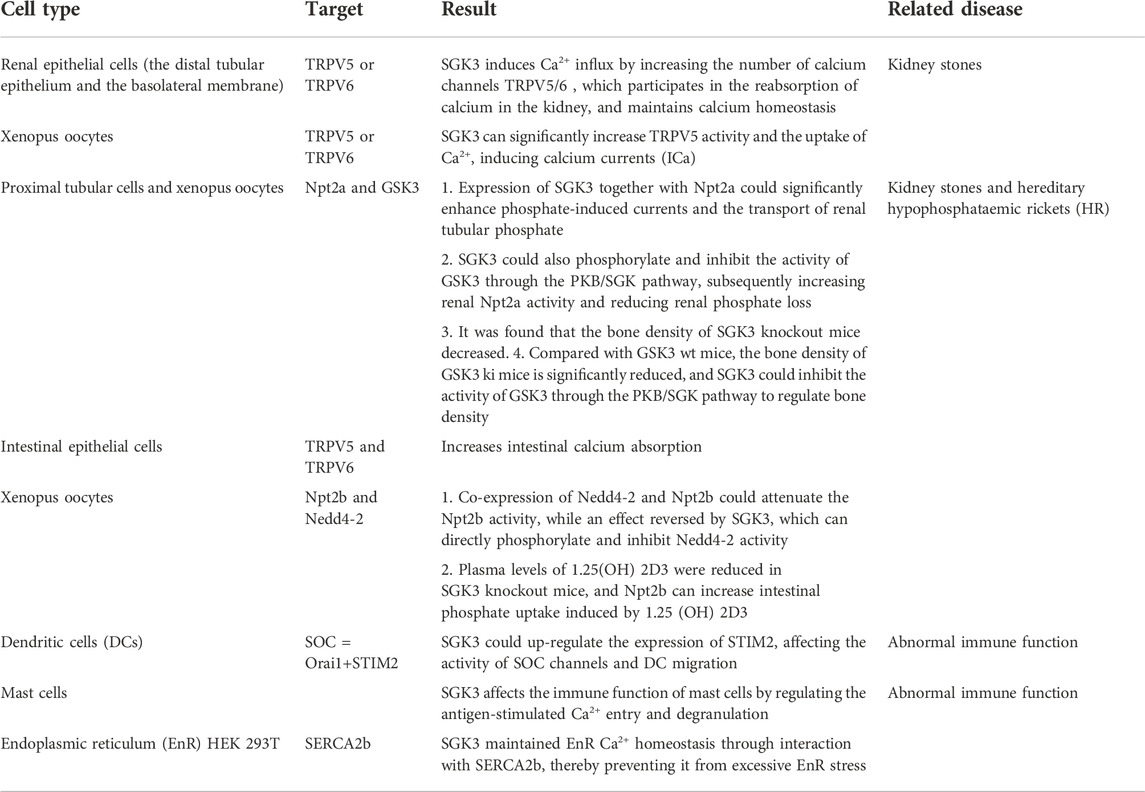
TABLE 1. SGK3 and calcium–phosphorus transport.
1.4.2 Serum- and glucocorticoid-induced kinase-3 and potassium transport
Potassium transport is closely related to potassium channels, potassium transporters, and Na+/K+-ATPase. The subtypes of potassium channels mainly include voltage-gated K+ channels (Kv), inwardly rectifying K+ channels (Kir), two-P K+ channels (K2p), and Ca2+-activated K+ channels (KCa). Potassium ions maintain cell membrane potential, participating in muscle-cell electrical signals and neuronal excitability and are associated with a variety of nervous and cardiovascular system diseases (Humphries and Dart, 2015).
1.4.2.1 Cardiovascular system potassium transport
Potassium ions are involved in the repolarization phase of myocardial action potential and are closely related to a variety of arrhythmias, which can also be used as targets for the treatment of certain arrhythmias (Humphries and Dart, 2015). In Xenopus oocytes, SGK3 could enhance Kir2.1-mediated currents and the channel protein abundance in the cell membrane (Munoz et al., 2014) (Figure 2). The loss-of-function mutation of potassium voltage-gated channel subfamily J member 2 (KCNJ2), the encoding gene of Kir2.1, could lead to Andersen–Tawil syndrome accompanied by periodic paralysis, cardiac arrhythmia, and dysmorphic features (Seebohm et al., 2012). SGK3 could also increase Kir2.2 inward currents, and the gain-of-function mutation of KCNJ could cause atrial fibrillation (Xia et al., 2005) (Figure 2). Consequently, SGK3 may participate in the occurrence of these diseases by regulating Kir2 inward currents and protein expressions.
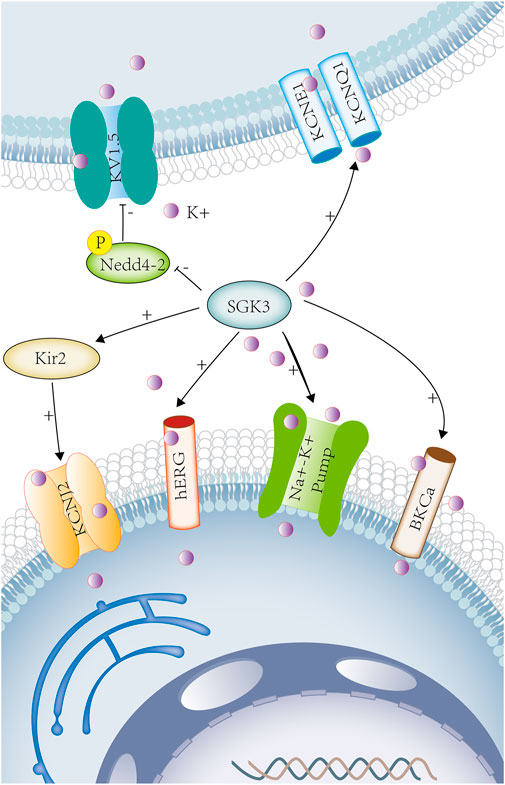
FIGURE 2. SGK3 and potassium transport. Potassium is involved in maintaining cellular electrical signals and cell excitability and is associated with a variety of neurological and cardiovascular diseases. SGK3 can enhance Kir2-mediated inward current. Kir2 gene encodes potassium voltage-gated channel subfamily J member 2 (KCNJ2). Sgk3 can up-regulate the activity and abundance of HERG. HERG (human ether-related gene-1, or kv11.1) belongs to the fast activation delay rectifier K+ current (IKr). Co-expression of SGK3 and KCNE1/KCNQ1 up-regulates KCNE1/KCNQ1-induced delayed potassium rectifier current (IKs). SGK3 can regulate KV15 by phosphorylating Nedd4-2. SGK3 can up-regulate Na+/K+-ATPase activity. The co-expression of BKCa and SGK3 can increase the activity of BKCa.SGK3 serum/glucocorticoid-regulated kinase family member 3. KIR2, inwardly rectifying potassium channel 2. KCNJ2, potassium inwardly rectifying channel subfamily J member 2. HERG, potassium voltage-gated channel subfamily H member 2. KCNE1, potassium voltage-gated channel subfamily E regulatory subunit 1. KCNQ1, potassium voltage-gated channel subfamily Q member 1 KV1.5(KCNA5), potassium voltage-gated channel subfamily A member 5. Na+-K+ ATPASE, ATPase Na+/K+ transporting subunit. BKCA, potassium large conductance calcium-activated channel. Purple circles represent potassium ion.
hERG (human ether-a-go-go-related gene 1, or Kv11.1) belongs to the rapidly activating delayed rectifier K+ current (IKr), which functions in conjunction with the auxiliary subunit KCNE2 (Eldstrom and Fedida, 2011). SGK3 could up-regulate the activity and abundance of hERG and change the duration of cardiac action potential. (Figure 2) The loss-of-function mutations of hERG could lead to long QT syndrome, fatal ventricular arrhythmia, or sudden death (Maier et al., 2006). KCNQ1 (a pore-forming tetramer) and KCNE1 (an auxiliary subunit) combine to form the delayed potassium rectifier current (IKS) (Larsson et al., 2018). KCNE1/KCNQ1 (Kv7.1) is widely expressed in cardiac myocytes, the stria vascularis of inner ears, renal tubules, and intestinal epithelial cells (Bleich and Warth, 2000). In Xenopus oocytes, the co-expression of SGK3 and KCNE1/KCNQ1 could up-regulate the KCNE1/KCNQ1-induced current, and the mutation of KCNE1/KCNQ1 led to Jervell and Lange–Nielsen syndrome, which is a disorder characterized by congenital profound sensorineural deafness, cardiac long QT syndrome, ventricular arrhythmias, and sudden cardiac death (Embark et al., 2003; Faridi et al., 2019). (Figure 2) SGK3 could also regulate the cardiac potassium channel Kv1.5 (KCNA5) by phosphorylating Nedd4-2 and participated in cardiac action potential (Ahmed et al., 2016a). (Figure 2)As previously described, potassium channels (hERG, KCNE1/KCNQ1, and Kv1.5) are involved in the repolarization phase of myocardial action potential.
Big Ca2+-activated K+ channel (BKCa) is a transmembrane protein, also known as BK, Slo1, KCa1.1, or MaxiK channel (Toro et al., 2014). It is widely expressed in vascular smooth muscle cells, cardiomyocytes, nerve cells, and skeletal muscle cells and regulates neuronal excitation, cell volume, vascular tone, and blood pressure (Ahmed et al., 2016b). The activation of BKCa channel results in increasing potassium outflow, vasodilation, and cardioprotection (Dong et al., 2016). In Xenopus oocytes, the co-expression of BKCa and SGK3 could increase the activity of BKCa and participate in the regulation of cell volume and neuroexcitability (Ahmed et al., 2016b). (Figure 2) The BKCa channel is associated with numerous cardiovascular diseases, such as hypertension, diabetes, metabolic syndrome, and heart failure, and is a potential therapeutic target for various cardiovascular diseases (Dong et al., 2016).
Na+/K+-ATPase is an important membrane protein that regulates the concentration of Na+ and K+ inside and outside the cell to maintain fluid and electrolyte balance in the body. In Xenopus oocytes, SGK3 could up-regulate Na+/K+-ATPase activity (Henke et al., 2002), which provides power for cardiac contraction and is related to obesity, diabetes, hypertension, and cardiac insufficiency (Yan et al., 2017). (Figure 2) In addition, SGK3 could also participate in the development of cardiac hypertrophy and fibrosis by regulating the phosphorylation of GSK3β (Wyatt et al., 2006).
As previously mentioned , SGK3 can regulate the potassium ion transport (Figure 2), participating in the repolarization of action potential, maintaining cell volume, and excitability, and is closely related to the occurrence and development of arrhythmia, sudden death, hypertension, obesity. and other diseases.
1.4.2.2 Nervous system potassium transport
Both SGK1 and SGK3 could up-regulate the voltage-gated K+ channel Kv1.3 by phosphorylating Nedd4-2 (Gamper et al., 2002). (Figure 2) Potassium channels regulate neuronal excitability and membrane potential, participating in learning and memory (Kourrich et al., 2001), and are closely related to early memory loss in Alzheimer’s disease (Alkon, 1999). In Xenopus oocytes, SGK3 could increase the activity of big Ca2+-activated K+ channel (BKCa) (Ahmed et al., 2016b). (Figure 2) The loss of BKCa channel function can also cause neuronal hyperexcitability, which may lead to seizures (N'Gouemo, 2014). SGK3 could also regulate the activity of Na+/K+-ATPase in neurons, the osmotic pressure and transmembrane potential inside and outside neurons, and maintaining integrity and excitability of neurons, and is related to depression (Henke et al., 2002; Gamaro et al., 2014) (Figure 2).
1.4.2.3 Renal potassium transport
In vitro, the co-expression of SGK3 and KCNE1/KCNQ1(Kv7.1) in Xenopus oocytes could up-regulate the activity of the potassium channel KCNE1/KCNQ1 (Embark et al., 2003). In vivo, KCNE1-deficient mice showed that the renal proximal tubular reabsorption function was mildly impaired because of decreased ability to maintain membrane potential in renal proximal tubules and increased urinary excretions including fluid, Na+, Cl−, and glucose (Vallon et al., 2001). Thus, SGK3 affects renal reabsorption by regulating potassium channels KCNE1/KCNQ1. (Figure 2)
In SGK3 and potassium transport, SGK3 can participate in up-regulating the activities of Kir2.1, Kir2.2, hERG, Kv7.1, BKCa, and Na+/K+- ATPase or indirectly enhance the potassium current induced by KCNA5 and Kv1.3 through the phosphorylation of Nedd4-2, participating in cardiac action potential, provide power for cardiac contraction, participate in renal proximal tubular membrane potential, maintain renal proximal tubular reabsorption function, and regulate neuronal excitability (Table 2).
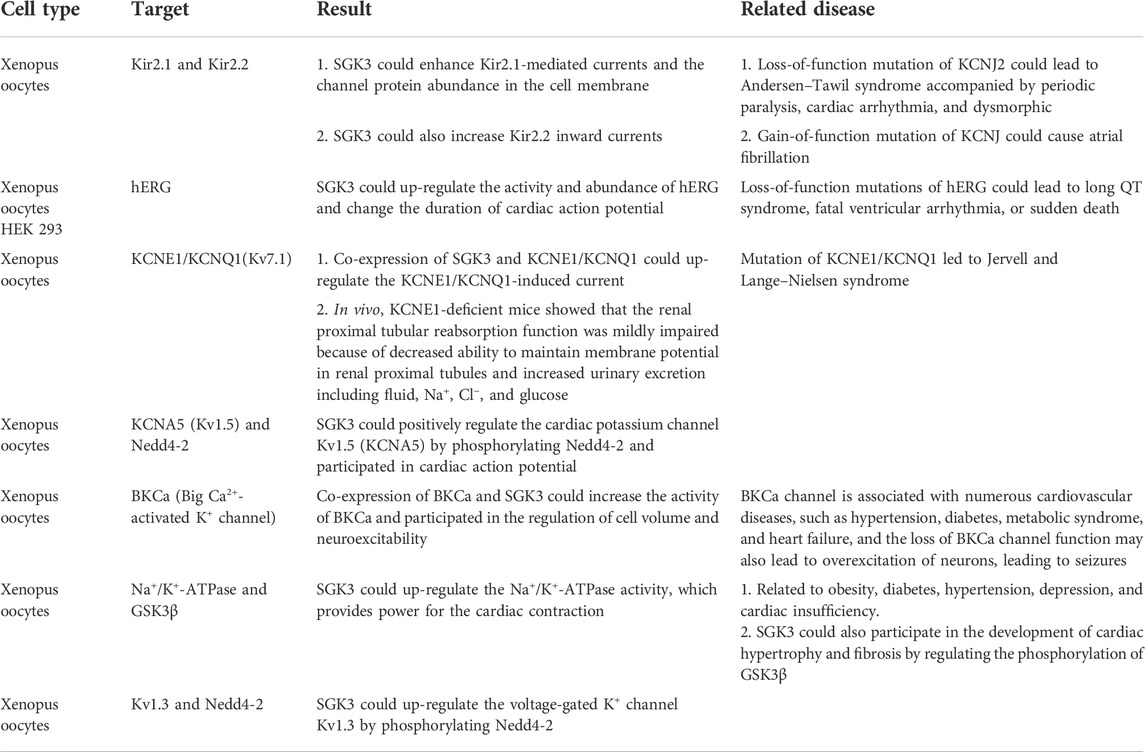
TABLE 2. SGK3 and potassium transport.
1.4.3 Serum- and glucocorticoid-induced kinase-3 and sodium transport
Sodium ions play an important role in the transmission of electrical signals in cells and participate in the depolarization of nerve and muscle action potentials. The abnormal structure or function of sodium channels usually leads to diseases such as failure of cardiovascular system and nervous system.
1.4.3.1 Renal and lung sodium transport
The epithelial sodium channel (ENaC) is mainly expressed in the kidney, lungs, salivary glands, and skin as well as maintaining water and salt homeostasis, which can affect the reabsorption of sodium in the kidney, regulating the epithelial surface liquid volume in the respiratory system (Hanukoglu and Hanukoglu, 2016). SGK3 could up-regulate the activity of ENaC (Figure 3) but it was not achieved by directly phosphorylating ENaC (Friedrich et al., 2003). SGK1 could indirectly regulate the activity and quantity of ENaC by phosphorylating Nedd4-2 (Debonneville et al., 2001). Therefore, SGK3 may, similar to SGK1, indirectly participate in the regulation of ENaC activity and the reabsorption of renal sodium and influences fluid balance and blood pressure (Hanukoglu and Hanukoglu, 2016).
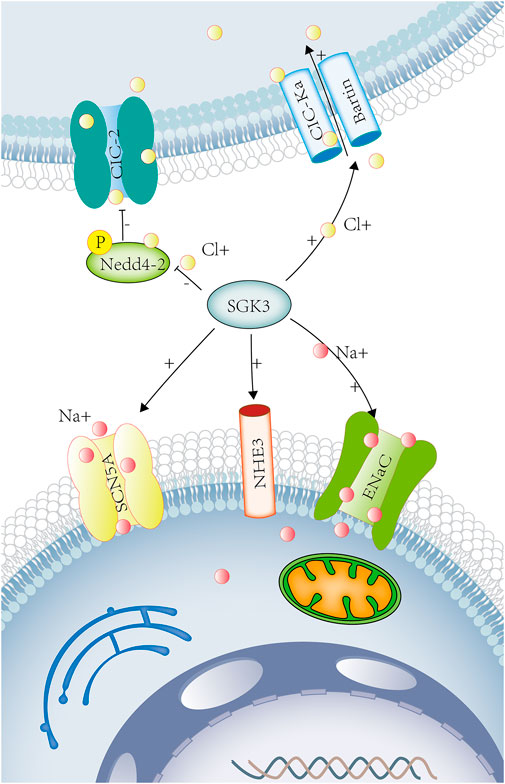
FIGURE 3. SGK3 and sodium chloride transport. Sodium ions participate in the nerve and muscle action potentials. Chloride ions participate in the regulation of neuronal excitability and cell volume. Sodium chloride maintains extracellular fluid osmotic pressure and participates in the regulation of acid-base balance in the body. SGK3 can raise the activity of ENaC. The Phox-homology (PX) domain of SGK3 can be colocated with NHE3 and participate in the acute activation of NHE3 by glucocorticoids. SGK3 can regulate myocardial sodium channel SCN5A (Nav1.5). SGK3 can raise CIC-Ka/Bartin-induced current. SGK3 indirectly regulates the activity and quantity of ClC-2 by phosphorylating Nedd4-2. SGK3, serum/glucocorticoid-regulated kinase family member 3. ENaC, sodium channel, nonvoltage-gated 1 alpha. NHE3, Na [+]/H [+] hydrogen exchanger 3. SCN5A, sodium voltage-gated channel alpha subunit 5. CIC-Ka/barttin chloride channel protein ClC-Ka/Bartin.CIC-2 chloride voltage-gated channel 2. Red circles represent sodium ions. Yellow circles represent chloride ions.
1.4.3.2 Intestinal sodium transport
Na+/H+ exchanger 3 (NHE3) is the main sodium transport protein in the intestine, which is located at the brush-border membrane of the intestinal epithelium and the endosomal compartment. The Phox-homology (PX) domain of SGK3 could be colocalized with NHE3 to recycling endosomes, participating in the acute activation of NHE3 by glucocorticoids. (Figure 3) Although SGK3 was not directly affected by glucocorticoids, the activation of SGK3 and NHE3 by glucocorticoids was dependent on the PI3K-PDK-1 pathway (He et al., 2011). Therefore, SGK3 takes part in Na+ reabsorption and H+ secretion of epithelial cells through NHE3 (Figure 3) and play a role in the sodium transport and acid-base balance.
1.4.3.3 Cardiac sodium transport
The voltage-gated sodium channel 1.5 (Nav1.5 and SCN5A) is abundantly expressed in skeletal muscles and myocardium. In Xenopus oocytes, the co-expression of SGK3 and SCN5A could enhance the activity of SCN5A. SGK3 could regulate the cardiac sodium channel Nav1.5 (Figure 3) and the depolarization of cardiac action potential, involving in various arrhythmia diseases (Boehmer et al., 2003a).
1.4.4 Serum- and glucocorticoid-induced kinase-3 and chloride transport
1.4.4.1 Renal and inner ear chloride transport
The voltage-gated chloride channels (ClC) include ClC-1-7, ClC-Ka, and ClC-Kb, in which ClC-Ka and ClC-Kb require the co-expression of barttin to become functional (Waldegger et al., 2002), only in the kidney and stria vascularis of the inner ear. In Xenopus oocytes, SGK3 up-regulated ClC-Ka/barttin-induced currents, while ubiquitin ligase Nedd4-2 down-regulated ClC-Ka/barttin activity, an effect reversed by SGK3 (Embark et al., 2004b) (Figure 3). The loss of function of ClC-Ka/barttin channels results in idiopathic deafness and Bartter syndrome, which is characterized by the renal dysfunction of concentration, accompanied by nephrogenic diabetes insipidus, hyponatremia, hypokalemia, metabolic alkalosis, and hypovolemia (Stolting et al., 2014).
1.4.4.2 Ubiquitous chloride transport
The chloride channel ClC-2 is widely distributed in the brain, heart, and gastrointestinal tract, involved in the regulation of neuronal excitability, chloride secretion, and cell volume. The amino acid sequence of ClC-2 contains a consensus site (Ser82) for phosphorylation by SGK3. In Xenopus oocytes, inhibiting the function of Ser82, SGK3 still increased the activity and number of ClC-2. SGK3 did not directly phosphorylate ClC-2 but indirectly regulated the activity and quantity of ClC-2 through the phosphorylation of Nedd4-2 (Palmada et al., 2004b) (Figure 3).
In SGK3 and sodium chloride transport, SGK3 is involved in up-regulating the activity of ENaC, NHE3, and Nav1.5, affecting the reabsorption of sodium by kidneys and intestines, regulating the depolarization of cardiac action potential, and maintaining hydro-salinity balance and acid-base balance. SGK3 indirectly regulates the activity and quantity of ClC Ka and ClC-2 through the phosphorylation of Nedd4-2, promotes the transport of chloride ions, and regulates the excitability of neurons (Table 3).
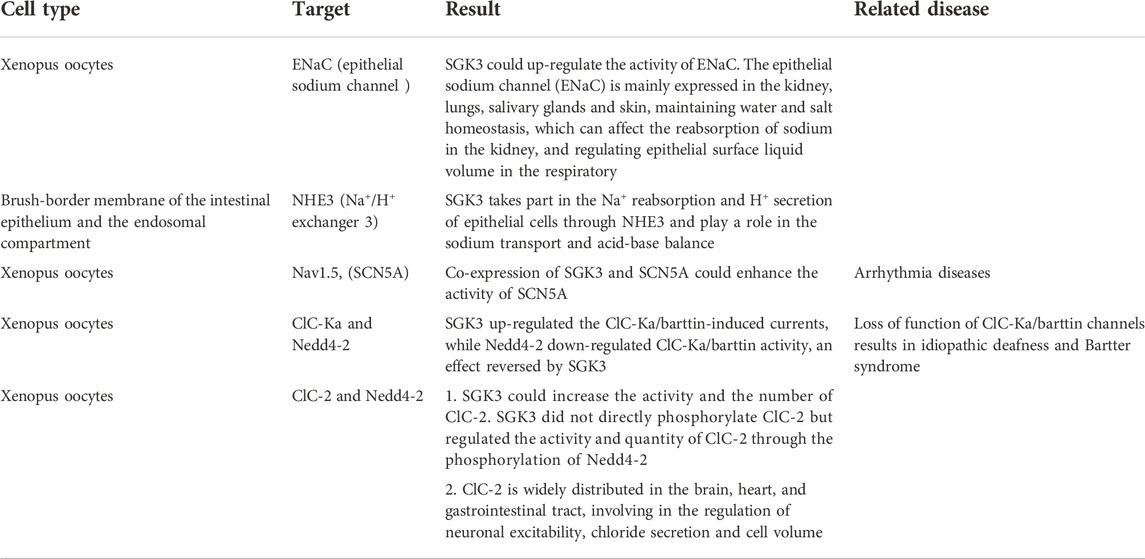
TABLE 3. SGK3 and sodium, chloride transport.
1.4.5 Serum- and glucocorticoid-induced kinase 3 and glucose and amino acid transport
1.4.5.1 Renal and intestinal glucose transport
The sodium-glucose cotransporter-1 (SGLT1) is involved in glucose (re)absorption in the kidney and intestines. In vitro, SGK3 could increase SGLT1 activity by phosphorylating Nedd4-2 and accelerate glucose absorption, which is related to diabetes and obesity (Dieter et al., 2004). (Figure 4) In vivo, compared with SGK3 wild-type mice, the SGLT1 function and intestinal glucose transport of SGK3 knockout mice were impaired (Sandu et al., 2005).
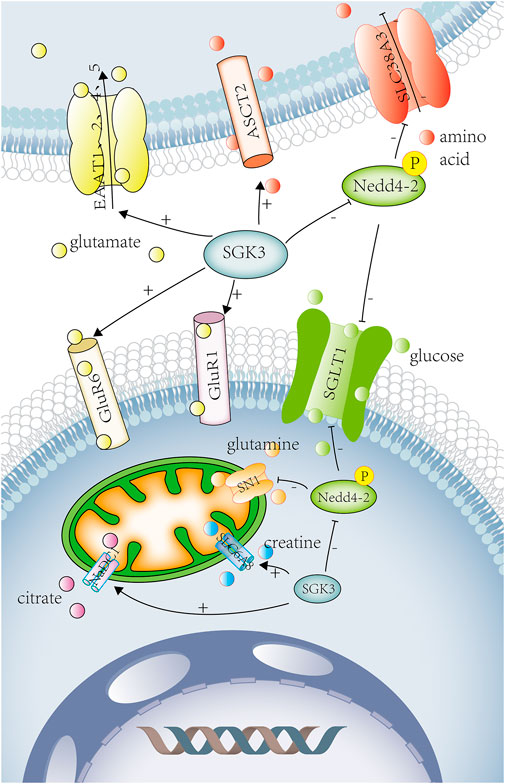
FIGURE 4. SGK3 and glucose ,amino acid ,other substance transport. SGK3 can increase SGLT1 activity and accelerate glucose absorption by phosphorylating Nedd4-2, which is related to diabetes and obesity. SGK3 can increase the expression and activity of ASCT2. SGK3 regulates the activity and expression of SLC6A19 by affecting the ubiquitin ligase Nedd4-2. SGK3 can increase the expression of glutamate receptor-1 (GluR1) in neuronal synapses and promote the glutamate transport. SGK3 can also raise the activity of GluR6. SGK3 can stimulate the expression of excitatory amino acid transporters (EAAT1, EAAT2, EAAT4, and EAAT5) and promote the uptake of glutamate. SGK3 can indirectly regulate SN1 (SLC38A3/SNAT3) activity through the phosphorylation and inhibition of Nedd4-2. SGK3 is an effective regulator of CreaT, which may regulate the uptake of creatine. SGK3 can enhance the activity of NaDC1, promote the reabsorption of citrate in renal tubules, and promote the occurrence of calcium urolithiasis. SGLT1, solute carrier family 5 member 1. Nedd4-2 E3, ubiquitin-protein ligase Nedd4. ASCT2, solute carrier family 1 member 5. SLC6A19, solute carrier family 6 member 19. GluR1, glutamate ionotropic receptor AMPA-type subunit 1. GluR6, glutamate ionotropic receptor kainate-type subunit 2. EAAT1, excitatory amino acid transporter-1. EAAT2, excitatory amino acid transporter-2. EAAT4, excitatory amino acid transporter-4. EAAT5, excitatory amino acid transporter-5. SN1, solute carrier family 38 member 3. SLC6A8(CreaT), solute carrier family 6 member 8. NaDC1, solute carrier family 13 member 2. Red circles represent amino acid. Yellow circles represent glutamate. Green circles represent glucose. Purple circle represents citrate. Orange circles represent glutamine. Blue circles represent creatine.
1.4.5.2 Renal and intestinal amino acid transport
SLC6A19 is a sodium-dependent neutral amino acid transporter, which covered the transport of neutral amino acids in the kidney and intestines. The amino acid sequence of SLC6A19 contains the consensus phosphorylation site 100Ser for SGK3, but SGK3 stimulated SLC6A19 activity and expression by influencing the ubiquitin ligase Nedd4-2 independently of 100Ser without modifying carrier substrate affinities (Bohmer et al., 2010). The Na+-dependent neutral amino acid transporter type-2 (ASCT2/SLC1A5) is widely expressed in lungs, kidneys, intestines, placenta, pancreas, and other tissues and plays an important role in the transport of neutral amino acid in the epithelial cells. SGK3 could up-regulate the expression and activity of ASCT2, thereby regulating the absorption of cellular nutrients (Palmada et al., 2005) (Figure 4).
1.4.5.3 Nervous system amino acid transport
SGK3 can increase the expression of glutamate receptor-1 (GluR1) in neuronal synapses and promote the transport of glutamate. In Xenopus oocytes, the co-expression of SGK3 and GluR1 significantly increased GluR1-current amplitudes and protein abundance (Strutz-Seebohm et al., 2006). SGK3 could also up-regulate the expression and activity of GluR1 in murine hippocampal neurons, and the hippocampal abundance of GluR1 was significantly lower in Sgk3−/− than in Sgk3+/+ mice (Strutz-Seebohm et al., 2005a). (Figure 4) GluR is the most important mediator of excitatory signaling in the central nervous system, and GluR1 is necessary for long-term potentiation (LTP) of hippocampal CA1 function, which participates in the retention of spatial memory (Lee et al., 2003). Therefore, SGK3 can up-regulate the GluR1 expression and activity to strengthen spatial memory. SGK3 could also up-regulate the activity of GluR6, maintaining neuronal excitability and memory consolidation (Strutz-Seebohm et al., 2005b). (Figure 4) Another study showed that SGK, as a downstream target of MAPK/ERK, could also participate in the formation of spatial memory (Lee et al., 2006). Compared with SGK3+/+, SGK3−/− mice led to subtle behavioral defects such as reduction of locomotor activity, shorter moving distance, slower speed, and slight impairment in recognition of spatial precision (Lang et al., 2006b).
SGK3 can stimulate the expression of excitatory amino acid transporters (EAAT1, EAAT2, EAAT4, and EAAT5) (Boehmer et al., 2003b; Bohmer et al., 2004; Boehmer et al., 2005; Boehmer et al., 2006) and contribute to glutamate uptake. (Figure 4) Glutamate is the main excitatory neurotransmitter in the central nervous system. When the functions of glutamate receptors and transporters are impaired, it has indeed been shown to promote the accumulation of extracellular glutamate, resulting in neuroexcitotoxicity and ultimately cell death. Increased glutamate concentration and decreased EAAT1 expression could be seen in the epileptogenic foci of intractable temporal lobe epilepsy (TLE) (Sarac et al., 2009). The loss of EAAT function and accumulation of glutamate is also closely related to Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), glaucoma, and diabetic retinopathy (Boehmer et al., 2005; Magi et al., 2019). These diseases may be related to the expression of the glutamate transporter regulated by SGK3.
Glutamine transporter SN1 (SLC38A3/SNAT3) is mainly expressed in astrocytes, hepatocytes, and kidneys, coupled with Na+ to participate in glutamine uptake and efflux. Glutamine is a precursor of purine, pyrimidines, glucose, and protein synthesis and participates in various metabolic processes, such as the glutamine/glutamate-γ-aminobutyric acid (GABA) cycle, hepatic and renal gluconeogenesis, and hepatic ammonia detoxification. Mice deficient in SN1 can cause ataxia and disorder in amino acid homeostasis and glucose metabolism (Chan et al., 2016). SGK3 can indirectly regulate SN1 activity through phosphorylation and inhibition of Nedd4-2 (Boehmer et al., 2003c) and participate in the occurrence of the aforementioned diseases. (Figure 4)
In short, SGK3 can tune GluR1, GluR6, EAAT1, EAAT2, EAAT4, EAAT5, and SN1 (Figure 4), participating in the transport of glutamate and glutamine in the nervous system, and play a role in the (patho) physiological process of various nervous system diseases (Table 4).
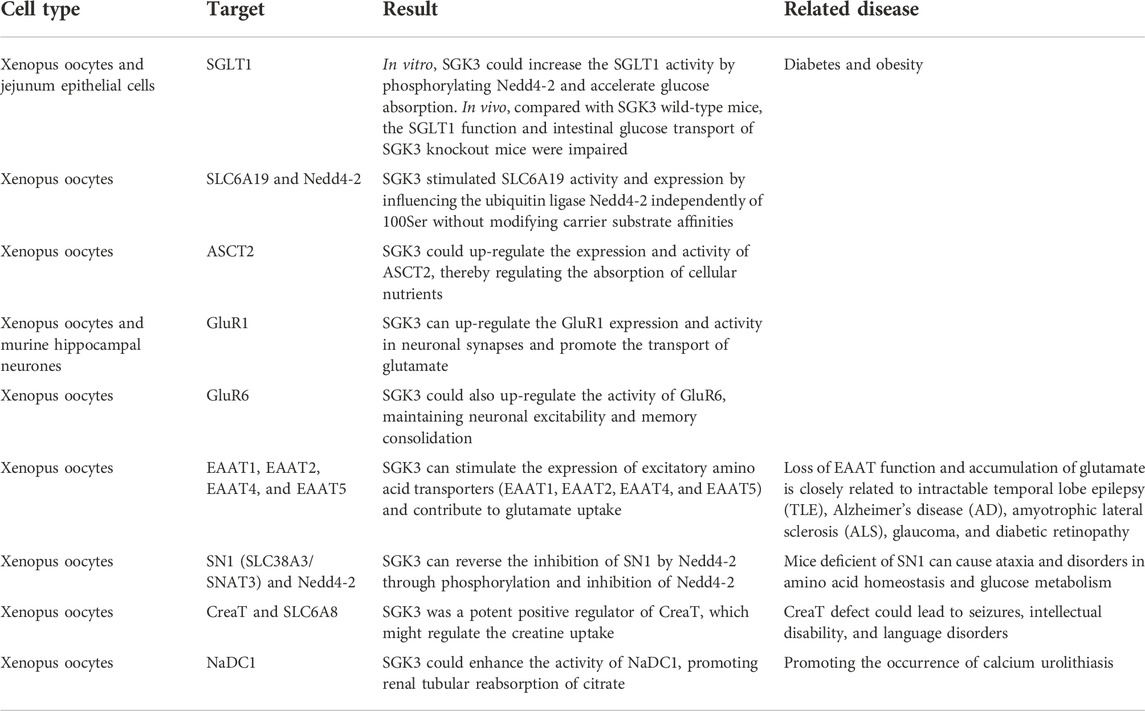
TABLE 4. SGK3 and glucose, amino acid, and other substance transport.
1.4.6 Serum- and glucocorticoid-induced kinase 3 and other substance transport
1.4.6.1 Creatine transport
Creatine transporter (CreaT, SLC6A8), a superfamily of Na+,Cl− coupled transporters, is widely expressed in the brain, kidney, heart, skeletal muscle, and intestinal tissues, involving in the transport of various neurotransmitters (such as dopamine, GABA, 5-hydroxytryptamine, and norepinephrine) and amino acids. SGK3 is a potent regulator of CreaT (Shojaiefard et al., 2005) (Figure 4), which might regulate the creatine uptake. CreaT defect could lead to seizures, intellectual disability, and language disorders (Battini et al., 2007).
1.4.6.2 Renal citrate transport
Citrate, known as citric acid, excreted into the urine can chelate with calcium, preventing calcium salt deposition, and thus inhibit the formation of urinary stones. The Na+ coupled dicarboxylate transporter 1 (NaDC1) participates in the renal tubular citrate transport. SGK3 could enhance the activity of NaDC1 (Figure 4), promoting the renal tubular reabsorption of citrate and the occurrence of calcium urolithiasis (Boehmer et al., 2004).
During the transportation of SGK3 with glucose, amino acids, and other substances, SGK3 enhances the activities of SGLT1 and SLC6A19 through the phosphorylation of Nedd4-1, promotes the absorption of glucose in the kidney and intestine, and regulates the transport of neutral amino acids in the kidney and intestine. SGK3 can up-regulate GluR1, GluR6, EAAT1, EAAT2, EAAT4, EAAT5, and SLC38A3, participate in the transport of glutamate and glutamine in the nervous system, and play a role in (pathological) physiological process of various nervous system diseases. SGK3 positively regulates CreaT and NaDC1 and promotes the uptake of creatine and the absorption of citric acid by renal tubules (Table 4).
2 Conclusion
In summary, this review highlights the function of SGK3 in calcium–phosphorus metabolism by regulating the (re)absorption of calcium and phosphate in the intestine and kidney and mineralization of the bone. We here describe that SGK3 involves in the generation of neuronal and muscle action potentials by influencing the activity of sodium and potassium channels and maintaining the cell electrical signal, excitability, volume, and fluid balance. The effects of SGK3 on the transport of glucose and amino acids can participate in the absorption of nutrients and is associated with a variety of metabolic diseases. Particular evidence points to its role in cell growth, proliferation, migration, and apoptosis, and SGK3 is closely related to the (patho) physiological processes of tumor growth, hair growth, nervous system diseases, and cardiovascular diseases. Additionally, SGK3 can be used as a prognostic biomarker and a novel therapeutic target in tumor growth.
It is suggested that the activated SGK3 can activate a large number of ion channels, carriers and receptors, and Na+/K+- ATPase, which involve a wide range of substance transport. It is also a very interesting topic to further explore how SGK3 regulates these molecular mechanisms. The following is a summary of mechanisms of SGK3 regulating activity.
S419DSGK3 can activate epithelial calcium channels TRPV5 and TRPV6. The key for SGK3 to activate TRPV5 is the presence of the PDZ domain containing scaffold protein NHERF2 that binds to the PDZ-binding motif on the carboxy-terminal tail of the channel. However, the deletion of the C-terminal PDZ-binding motif on TRPV6 will not affect the activation of TRPV6 by S419DSGK3. S419DSGK3 activates TRPV6 by enhancing its protein abundance at the cell surface (Böhmer et al., 2007).
SGK3 serine phosphorylation sites in GSK3 will inhibit GSK3 activity, thereby increasing renal Npt2a activity. SGK3 inhibits the ubiquitination degradation of Npt2b by Nedd4-2 through the phosphorylation inactivation of Nedd4-2, thereby increasing the activity of Npt2b (Palmada et al., 2004a; Bhandaru et al., 2011; Foller et al., 2011).
SGK3 may promote the transcription of STIM1 and STIM2 by regulating NF kB (Schmid et al., 2012).
In 293T cells, FLAG-SGK3 can pull down SERCA2b but SGK3 does not regulate the SERCA2b expression. ATPase activity is critical to SERCA2b. After GSK650394 inhibited SGK3 in EXE-R cells, the SERCA2b ATPase activity was significantly impaired. Therefore, SGK3 retains the function of SERCA2b (Wang et al., 2017).
PI (3.5) P2 can activate Kir2.2. The 45–78 amino acid residues at the C-terminal on Kir2.2 are very sensitive to SGK3. SGK3 phosphorylates S318 of PI (3.5) P2 to activate Kir2.2. SGK3 does not significantly affect the expression of Kir2.1 on the surface of Xenopus laevis oocytes, but the co-expression with Kir2.2 can increase Kir2.1 protein abundance at the cell surface, indicating that SGK3 can promote the transport of heterogeneous complexes composed of Kir2.1 and Kir2.2 channel proteins to the cell surface (Seebohm et al., 2012).
The PY motif on hERG is combined with the WW domain on Nedd4-2. SGK3 inactivates Nedd4-2 by phosphorylating Ser-444 (and Ser-338) residues in the Nedd4-2WW domain, thereby inhibiting the ubiquitination and internalization degradation of hERG by Nedd4-2, and finally increasing the Na channel abundance at the cell surface. According to the report, the GTP enzyme Rab11 could transport membrane proteins to the cell surface and interacted with hERG protein. SGK phosphorylates and activates PIKfyve, and PIKfyve interacts with phosphatidylinositol-3-phosphate to produce PI (3.5) P2. PIKfyve and its product PI (3.5) P2 can promote the Rab11-mediated insertion of the Kv7.1 channel protein into the plasma membrane. Similarly, SGK can also promote Rab11 to insert the recovered internalized hERG into the cell surface (Lamothe and Zhang., 2013).
In theory, SGK3 can change its activity by directly phosphorylating channel proteins or other signaling molecules. SGK3 can also inhibit the degradation of ubiquitinated target proteins by phosphorylating and inactivating Nedd4-2. However, it is unknown whether BKCa is targeted by Nedd4-2 (Ahmed et al., 2016b).
SGK3 can activate Na+/K+ ATPase, which may up-regulate K channels (Pao., 2012).
ENaC is composed of three subunits called α, β, and γ and the C-terminal of each subunit contains a proline-rich PPXY (PY) motif, wherein the C-terminal of α and γ subunits also contains the SGK consensus motif RXRXX (S/T). Ubiquitin-protein ligase Nedd4-2 recognizes the PY motif of ENaC and degrades ENaC through ubiquitinated ENaC. SGK3 inactivates Nedd4-2 by phosphorylating and inhibits Nedd4-2 to degrade ENaC. Because ENaC contains SGK3 phosphorylation consensus motifs, SGK3 is very likely to directly phosphorylate ENaC to affect its activity (Alexei Diakov and Korbmacher., 2004).
Unlike SGK1 and SGK2, the Phox-homologous domain at the end of SGK3 can locate SGK3 to the early endosome and this subcellular localization is important for the activation of NHE3 (Pao., 2012).
Nav1.5 contains S484 and S664 sites of the SGK3 phosphorylation consensus motif, which will change Nav1.5 gating properties. SGK3 can also inhibit the Nedd4-2 ubiquitination degradation of Nav1.5 through the phosphorylation inactivation of Nedd4-2 (Céline Marionneau et al., 2015).
SGK3 inhibits Nedd4-2 and then inhibits the ubiquitination degradation of CIC-Ka/barttin and CIC-2. In addition, SGK is still effective even if Nedd4-2 cannot interact with CIC-Ka/barttin. SGK may also affect CIC-Ka/barttin through other unknown mechanisms, for example, SGK3 directly phosphorylates the motif on CIC-Ka (Arg Val Arg Thr Thr-187Thr). CIC also contains SGK consensus sites (Ser82) (Embark et al., 2004b; Palmada et al., 2004b).
SGK3 phosphorylation inactivates Nedd4-2, which inhibits the ubiquitination clearance of SGLT1 in the cell membrane, thus increasing the abundance of the glucose transporter SGLT1 protein in the cell membrane (Dieter et al., 2004).
SGK3 can activate SLC6A19 by increasing the abundance of SLC6A19 proteins at the cell surface without changing the substrate affinity of SLC6A19 (Bohmer et al., 2010).
In Xenopus laevis oocytes, SGK3 may regulate the intermediate molecules of hASCT2 through phosphorylation or directly phosphorylating hASCT2 at non consensus sites, thus increasing the maximum transport rate without affecting the substrate affinity of hASCT2 to enhance the abundance of hASCT2 proteins at the cell surface and activate hASCT2 (Palmada et al., 2005).
SGK3 can increase GluR1 activity by increasing the abundance of GluR1 proteins at the cell surface. SGK3 can also partially up-regulate the GluR6 protein abundance in the cell membrane. However, GluR1 is more sensitive to SGK3, and GluR6 is more sensitive to SGK1 (Strutz-Seebohm et al., 2006).
SGK can directly phosphorylate the site on EAAT1 or inhibit Nedd4-2 to down regulate EAAT1 by phosphorylating and inactivating Nedd4-2. EAAT2 has no SGK consensus site. SGK3 improves the maximum transport rate and increases the abundance of EAAT2 proteins at the cell surface without affecting the affinity of EAAT2 for substrates (Boehmer et al., 2006).
SGK3 enhances glutamine-induced current by inhibiting Nedd4-2 and SN1 ubiquitination and degradation (Boehmer et al., 2003c).
SGK is unlikely to phosphorylate SLC6A8 directly. SGK3 can improve SLC6A8 activity by increasing the maximum transport rate of the carrier through a currently unknown protein (the protein itself targets the creatine transporter) (Shojaiefard et al., 2005).
SGK3 does not significantly change the concentration substrate affinity of the NaDC1 transporter but by an increase of its maximum transport rate to activate NaDC1 (Boehmer et al., 2004).
In the future, additional downstream target proteins as well as molecular and disease mechanisms will almost certainly be discovered in further research on SGK3, which will help in understanding the physiological and pathophysiological significance of SGK3 in detail and provide more pieces of evidence for this field.
Author contributions
QL, QD, and L-JY wrote the manuscript. All authors approved the final version of this manuscript.
Funding
This work was supported by the fund from the National Natural Science foundation of China (No. 81974102).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmed, M., Fezai, M., and Lang, F. (2016). SGK3 sensitivity of large-conductance Ca2+-activated K+ channel. Neurosignals. 24 (1), 113–124. doi:10.1159/000442618
Ahmed, M., Fezai, M., Uzcategui, N. L., Hosseinzadeh, Z., and Lang, F. (2016). SGK3 sensitivity of voltage gated K+ channel Kv1.5 (KCNA5). Cell. Physiol. biochem. 38 (1), 359–367. doi:10.1159/000438636
Alexei Diakov, C., and Korbmacher, C. (2004). A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the C terminus of the channel's α-subunit. J. Biol. Chem. 279 (37), 38134–38142. doi:10.1074/jbc.m403260200
Alkon, D. L. (1999). Ionic conductance determinants of synaptic memory nets and their implications for Alzheimer's disease. J. Neurosci. Res. 58 (1), 24–32. doi:10.1002/(sici)1097-4547(19991001)58:1<24::aid-jnr4>3.0.co;2-n
Bago, R., Malik, N., Munson, M. J., Prescott, A. R., Davies, P., Sommer, E., et al. (2014). Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem. J. 463 (3), 413–427. doi:10.1042/BJ20140889
Battini, R., Chilosi, A., Mei, D., Casarano, M., Alessandri, M. G., Leuzzi, V., et al. (2007). Mental retardation and verbal dyspraxia in a new patient with de novo creatine transporter (SLC6A8) mutation. Am. J. Med. Genet. A 143A (15), 1771–1774. doi:10.1002/ajmg.a.31827
Baum, M., Schiavi, S., Dwarakanath, V., and Quigley, R. (2005). Effect of fibroblast growth factor-23 on phosphate transport in proximal tubules. Kidney Int. 68 (3), 1148–1153. doi:10.1111/j.1523-1755.2005.00506.x
Bhandaru, M., Kempe, D. S., Rotte, A., Capuano, P., Pathare, G., Sopjani, M., et al. (2011). Decreased bone density and increased phosphaturia in gene-targeted mice lacking functional serum- and glucocorticoid-inducible kinase 3. Kidney Int. 80 (1), 61–67. doi:10.1038/ki.2011.67
Bleich, M., and Warth, R. (2000). The very small-conductance K+ channel KvLQT1 and epithelial function. Pflugers Arch. 440 (2), 202–206. doi:10.1007/s004240000257
Boehmer, C., Embark, H. M., Bauer, A., Palmada, M., Yun, C. H., Weinman, E. J., et al. (2004). Stimulation of renal Na+ dicarboxylate cotransporter 1 by Na+/H+ exchanger regulating factor 2, serum and glucocorticoid inducible kinase isoforms, and protein kinase B. Biochem. Biophys. Res. Commun. 313 (4), 998–1003. doi:10.1016/j.bbrc.2003.12.011
Boehmer, C., Henke, G., Schniepp, R., Palmada, M., Rothstein, J. D., Broer, S., et al. (2003). Regulation of the glutamate transporter EAAT1 by the ubiquitin ligase Nedd4-2 and the serum and glucocorticoid-inducible kinase isoforms SGK1/3 and protein kinase B. J. Neurochem. 86 (5), 1181–1188. doi:10.1046/j.1471-4159.2003.01937.x
Boehmer, C., Okur, F., Setiawan, I., Broer, S., and LangF., (2003). Properties and regulation of glutamine transporter SN1 by protein kinases SGK and PKB. Biochem. Biophys. Res. Commun. 306 (1), 156–162. doi:10.1016/s0006-291x(03)00921-5
Boehmer, C., Palmada, M., Rajamanickam, J., Schniepp, R., Amara, S., and Lang, F. (2006). Post-translational regulation of EAAT2 function by co-expressed ubiquitin ligase Nedd4-2 is impacted by SGK kinases. J. Neurochem. 97 (4), 911–921. doi:10.1111/j.1471-4159.2006.03629.x
Boehmer, C., Rajamanickam, J., Schniepp, R., Kohler, K., Wulff, P., Kuhl, D., et al. (2005). Regulation of the excitatory amino acid transporter EAAT5 by the serum and glucocorticoid dependent kinases SGK1 and SGK3. Biochem. Biophys. Res. Commun. 329 (2), 738–742. doi:10.1016/j.bbrc.2005.02.035
Boehmer, C., Wilhelm, V., Palmada, M., Wallisch, S., Henke, G., Brinkmeier, H., et al. (2003). Serum and glucocorticoid inducible kinases in the regulation of the cardiac sodium channel SCN5A. Cardiovasc. Res. 57 (4), 1079–1084. doi:10.1016/s0008-6363(02)00837-4
Böhmer, C., PalMadaM., , Kenngott, C., LindneR, R., KlausF., , Laufer, J., et al. (2007). Regulation of the epithelial calcium channel TRPV6 by the serum and glucocorticoid-inducible kinase isoforms SGK1 and SGK3. FEBS Lett. 581 (29), 5586–5590. doi:10.1016/j.febslet.2007.11.006
Bohmer, C., Philippin, M., Rajamanickam, J., Mack, A., Broer, S., Palmada, M., et al. (2004). Stimulation of the EAAT4 glutamate transporter by SGK protein kinase isoforms and PKB. Biochem. Biophys. Res. Commun. 324 (4), 1242–1248. doi:10.1016/j.bbrc.2004.09.193
Bohmer, C., Sopjani, M., Klaus, F., Lindner, R., Laufer, J., Jeyaraj, S., et al. (2010). The serum and glucocorticoid inducible kinases SGK1-3 stimulate the neutral amino acid transporter SLC6A19. Cell. Physiol. biochem. 25 (6), 723–732. doi:10.1159/000315092
Bradding, P., Okayama, Y., Kambe, N., and Saito, H. (2003). Ion channel gene expression in human lung, skin, and cord blood-derived mast cells. J. Leukoc. Biol. 73 (5), 614–620. doi:10.1189/jlb.1202602
Brown, E. M. (2007). The calcium-sensing receptor: Physiology, pathophysiology and CaR-based therapeutics. Subcell. Biochem. 45, 139–167. doi:10.1007/978-1-4020-6191-2_6
Bruhn, M. A., Pearson, R. B., Hannan, R. D., and Sheppard, K. E. (2013). AKT-independent PI3-K signaling in cancer - emerging role for SGK3. Cancer Manag. Res. 5, 281–292. doi:10.2147/CMAR.S35178
Cebeci, A. N., Zou, M., BinEssa, H. A., Alzahrani, A. S., Al-Rijjal, R. A., Al-Enezi, A. F., et al. (2020). Mutation of SGK3, a novel regulator of renal phosphate transport, causes autosomal dominant hypophosphatemic rickets. J. Clin. Endocrinol. Metab. 105 (6), dgz260. doi:10.1210/clinem/dgz260
Céline Marionneau, H., Abriel, H., et al. (2015). Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications. J. Mol. Cell. Cardiol. 82, 36–47. doi:10.1016/j.yjmcc.2015.02.013
Chan, K., Busque, S. M., Sailer, M., Stoeger, C., Broer, S., Daniel, H., et al. (2016). Loss of function mutation of the Slc38a3 glutamine transporter reveals its critical role for amino acid metabolism in the liver, brain, and kidney. Pflugers Arch. 468 (2), 213–227. doi:10.1007/s00424-015-1742-0
Chen, Y., Sun, Z., Qi, M., Wang, X., Zhang, W., Chen, C., et al. (2018). INPP4B restrains cell proliferation and metastasis via regulation of the PI3K/AKT/SGK pathway. J. Cell. Mol. Med. 22 (5), 2935–2943. doi:10.1111/jcmm.13595
Christakos, S. (2012). Recent advances in our understanding of 1, 25-dihydroxyvitamin D(3) regulation of intestinal calcium absorption. Arch. Biochem. Biophys. 523 (1), 73–76. doi:10.1016/j.abb.2011.12.020
Debonneville, C., Flores, S. Y., Kamynina, E., Plant, P. J., Tauxe, C., Thomas, M. A., et al. (2001). Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J. 20 (24), 7052–7059. doi:10.1093/emboj/20.24.7052
Dieter, M., Palmada, M., Rajamanickam, J., Aydin, A., Busjahn, A., Boehmer, C., et al. (2004). Regulation of glucose transporter SGLT1 by ubiquitin ligase nedd4-2 and kinases SGK1, SGK3, and PKB. Obes. Res. 12 (5), 862–870. doi:10.1038/oby.2004.104
Dong, D. L., Bai, Y. L., and Cai, B. Z. (2016). Calcium-activated potassium channels: Potential target for cardiovascular diseases. Adv. Protein Chem. Struct. Biol. 104, 233–261. doi:10.1016/bs.apcsb.2015.11.007
Eldstrom, J., and Fedida, D. (2011). The voltage-gated channel accessory protein KCNE2: Multiple ion channel partners, multiple ways to long QT syndrome. Expert Rev. Mol. Med. 13, e38. doi:10.1017/S1462399411002092
Embark, H. M., Bohmer, C., Palmada, M., Rajamanickam, J., Wyatt, A. W., Wallisch, S., et al. (2004). Regulation of CLC-Ka/barttin by the ubiquitin ligase Nedd4-2 and the serum- and glucocorticoid-dependent kinases. Kidney Int. 66 (5), 1918–1925. doi:10.1111/j.1523-1755.2004.00966.x
Embark, H. M., Bohmer, C., Vallon, V., Luft, F., and Lang, F. (2003). Regulation of KCNE1-dependent K(+) current by the serum and glucocorticoid-inducible kinase (SGK) isoforms. Pflugers Arch. 445 (5), 601–606. doi:10.1007/s00424-002-0982-y
Embark, H. M., Setiawan, I., Poppendieck, S., van de Graaf, S. F. J., Boehmer, C., Palmada, M., et al. (2004). Regulation of the epithelial Ca2+ channel TRPV5 by the NHE regulating factor NHERF2 and the serum and glucocorticoid inducible kinase isoforms SGK1 and SGK3 expressed in Xenopus oocytes. Cell. Physiol. biochem. 14 (4-6), 203–212. doi:10.1159/000080329
Faridi, R., Tona, R., Brofferio, A., Hoa, M., Olszewski, R., Schrauwen, I., et al. (2019). Mutational and phenotypic spectra of KCNE1 deficiency in Jervell and lange-nielsen syndrome and romano-ward syndrome. Hum. Mutat. 40 (2), 162–176. doi:10.1002/humu.23689
Foller, M., Kempe, D. S., Boini, K. M., Pathare, G., Siraskar, B., Capuano, P., et al. (2011). PKB/SGK-resistant GSK3 enhances phosphaturia and calciuria. J. Am. Soc. Nephrol. 22 (5), 873–880. doi:10.1681/ASN.2010070757
Friedrich, B., Feng, Y., Cohen, P., Risler, T., VandewAlle, A., Broer, S., et al. (2003). The serine/threonine kinases SGK2 and SGK3 are potent stimulators of the epithelial Na+ channel alpha, beta, gamma-ENaC. Pflugers Arch. 445 (6), 693–696. doi:10.1007/s00424-002-0993-8
Gamaro, G. D., Streck, E. L., Matte, C., Prediger, M. E., Wyse, A. T. S., and Dalmaz, C. (2014). Reduction of hippocampal Na+, K+-ATPase activity in rats subjected to an experimental model of depression. Neurochem. Res. 5, 1339–1344. doi:10.1023/a:1024988113978
Gamper, N., Fillon, S., Feng, Y., Friedrich, B., Lang, P. A., Henke, G., et al. (2002). K+ channel activation by all three isoforms of serum- and glucocorticoid-dependent protein kinase SGK. Pflugers Arch. 445 (1), 60–66. doi:10.1007/s00424-002-0873-2
Gasser, J. A., Inuzuka, H., Lau, A. W., Wei, W., Beroukhim, R., and Toker, A. (2014). SGK3 mediates INPP4B-dependent PI3K signaling in breast cancer. Mol. Cell 56 (4), 595–607. doi:10.1016/j.molcel.2014.09.023
Hanukoglu, I., and Hanukoglu, A. (2016). Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 579 (2), 95–132. doi:10.1016/j.gene.2015.12.061
He, P., Lee, S. J., Lin, S., Seidler, U., Lang, F., Fejes-Toth, G., et al. (2011). Serum- and glucocorticoid-induced kinase 3 in recycling endosomes mediates acute activation of Na+/H+ exchanger NHE3 by glucocorticoids. Mol. Biol. Cell 22 (20), 3812–3825. doi:10.1091/mbc.E11-04-0328
Henke, G., Setiawan, I., Bohmer, C., and Lang, F. (2002). Activation of Na+/K+-ATPase by the serum and glucocorticoid-dependent kinase isoforms. Kidney Blood Press. Res. 25 (6), 370–374. doi:10.1159/000068699
Hernando, N., and Wagner, C. A. (2018). Mechanisms and regulation of intestinal phosphate absorption. Compr. Physiol. 8 (3), 1065–1090. doi:10.1002/cphy.c170024
Hoenderop, J. G., Willems, P. H., and Bindels, R. J. (2000). Toward a comprehensive molecular model of active calcium reabsorption. Am. J. Physiol. Ren. Physiol. 278 (3), F352–F360. doi:10.1152/ajprenal.2000.278.3.F352
Hou, M., Lai, Y., He, W., Shen, H., and Ke, Z. (2015). SGK3 (CISK) may induce tumor angiogenesis (Hypothesis). Oncol. Lett. 10 (1), 23–26. doi:10.3892/ol.2015.3182
Hruska, K. A., Mathew, S., Lund, R., Qiu, P., and Pratt, R. (2008). Hyperphosphatemia of chronic kidney disease. Kidney Int. 74 (2), 148–157. doi:10.1038/ki.2008.130
Humphries, E. S., and Dart, C. (2015). Neuronal and cardiovascular potassium channels as therapeutic drug targets: Promise and pitfalls. J. Biomol. Screen. 20 (9), 1055–1073. doi:10.1177/1087057115601677
Ide, N., Ye, R., Courbebaisse, M., Olauson, H., Densmore, M. J., Larsson, T. E., et al. (2018). In vivo evidence for an interplay of FGF23/Klotho/PTH axis on the phosphate handling in renal proximal tubules. Am. J. Physiol. Ren. Physiol. 315 (5), F1261–F1270. doi:10.1152/ajprenal.00650.2017
Julita, K., Anna, C., Joanna, R., Joanna, W., Jolanta, S., and Marie-Pierre, R. (2017). Aquaporins in cardiovascular system. Adv. Exp. Med. Biol. 227, 39–58. doi:10.1007/978-3-319-56895-9_3
Jurutka, P. W., Whitfield, G. K., Hsieh, J. C., Thompson, P. D., Haussler, C. A., and Haussler, M. R. (2001). Molecular nature of the vitamin D receptor and its role in regulation of gene expression. Rev. Endocr. Metab. Disord. 2 (2), 203–216. doi:10.1023/a:1010062929140
Kirsch, T. (2006). Determinants of pathological mineralization. Curr. Opin. Rheumatol. 18 (2), 174–180. doi:10.1097/01.bor.0000209431.59226.46
Kobayashi, T., DeakM., , MorriceN., , and Cohen, P. (1999). Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Biochem. J. 344, 189–197. doi:10.1042/bj3440189
Kourrich, S., Mourre, C., and Soumireu-Mourat, B. (2001). Kaliotoxin, a Kv1.1 and Kv1.3 channel blocker, improves associative learning in rats. Behav. Brain Res. 120 (1), 35–46. doi:10.1016/s0166-4328(00)00356-9
Kulbacka, J., Choromanska, A., Rossowska, J., Wezgowiec, J., Saczko, J., and Rols, M. P. (2017). Cell membrane transport mechanisms: Ion channels and electrical properties of cell membranes. Adv. Anat. Embryol. Cell Biol. 227, 39–58. doi:10.1007/978-3-319-56895-9_3
Lang, F., Bohmer, C., Palmada, M., Seebohm, G., Strutz-Seebohm, N., and Vallon, V. (2006). (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol. Rev. 86 (4), 1151–1178. doi:10.1152/physrev.00050.2005
Lang, U. E., Wolfer, D. P., Grahammer, F., Strutz-Seebohm, N., Seebohm, G., Lipp, H. P., et al. (2006). Reduced locomotion in the serum and glucocorticoid inducible kinase 3 knock out mouse. Behav. Brain Res. 167 (1), 75–86. doi:10.1016/j.bbr.2005.08.017
Larsson, J. E., Larsson, H. P., and Liin, S. I. (2018). KCNE1 tunes the sensitivity of KV7.1 to polyunsaturated fatty acids by moving turret residues close to the binding site. Elife 7, e37257. doi:10.7554/eLife.37257
Lederer, E. (2014). Renal phosphate transporters. Curr. Opin. Nephrol. Hypertens. 2323 (55), 502502–502566. doi:10.1097/MNH.0000000000000053
Lee, C. T., Tyan, S. W., Ma, Y. L., Tsai, M. C., Yang, Y. C., and Lee, E. H. Y. (2006). Serum- and glucocorticoid-inducible kinase (SGK) is a target of the MAPK/ERK signaling pathway that mediates memory formation in rats. Eur. J. Neurosci. 23 (5), 1311–1320. doi:10.1111/j.1460-9568.2006.04650.x
Lee, H. K., Takamiya, K., Han, J. S., Man, H., Kim, C. H., Rumbaugh, G., et al. (2003). Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112 (5), 631–643. doi:10.1016/s0092-8674(03)00122-3
Liu, D., Yang, X., and Songyang, Z. (2000). Identification of CISK, a new member of the SGK kinase family that promotes IL-3-dependent survival. Curr. Biol. 10 (19), 1233–1236. doi:10.1016/s0960-9822(00)00733-8
Liu, E. S., Martins, J. S., Raimann, A., Chae, B. T., Brooks, D. J., Jorgetti, V., et al. (2016). 1, 25-dihydroxyvitamin D alone improves skeletal growth, microarchitecture, and strength in a murine model of XLH, despite enhanced FGF23 expression. J. Bone Min. Res. 31 (5), 929–939. doi:10.1002/jbmr.2783
Liu, M., Chen, L., Chan, T. H. M., Wang, J., Li, Y., Li, Y., et al. (2012). Serum and glucocorticoid kinase 3 at 8q13.1 promotes cell proliferation and survival in hepatocellular carcinoma. Hepatology 55 (6), 1754–1765. doi:10.1002/hep.25584
Magi, S., Piccirillo, S., and Amoroso, S. (2019). The dual face of glutamate: From a neurotoxin to a potential survival factor-metabolic implications in health and disease. Cell. Mol. Life Sci. 76 (8), 1473–1488. doi:10.1007/s00018-018-3002-x
Maier, G., Palmada, M., Rajamanickam, J., Shumilina, E., Bohmer, C., and Lang, F. (2006). Upregulation of HERG channels by the serum and glucocorticoid inducible kinase isoform SGK3. Cell. Physiol. biochem. 18 (4-5), 177–186. doi:10.1159/000097666
Malik, N., Macartney, T., Hornberger, A., Anderson, K. E., Tovell, H., Prescott, A. R., et al. (2018). Mechanism of activation of SGK3 by growth factors via the Class 1 and Class 3 PI3Ks. Biochem. J. 475 (1), 117–135. doi:10.1042/BCJ20170650
Munoz, C., Pakladok, T., Almilaji, A., Elvira, B., Decher, N., Shumilina, E., et al. (2014). Up-regulation of Kir2.1 (KCNJ2) by the serum & glucocorticoid inducible SGK3. Cell. Physiol. biochem. 33 (2), 491–500. doi:10.1159/000358629
N'Gouemo, P. (2014). BKCa channel dysfunction in neurological diseases. Front. Physiol. 5, 373. doi:10.3389/fphys.2014.00373
Naray-Fejes-Toth, A., Helms, M. N., Stokes, J. B., and Fejes-Toth, G. (2004). Regulation of sodium transport in mammalian collecting duct cells by aldosterone-induced kinase, SGK1: Structure/function studies. Mol. Cell. Endocrinol. 217 (1-2), 197–202. doi:10.1016/j.mce.2003.10.043
Palmada, M., Dieter, M., Boehmer, C., Waldegger, S., and Lang, F. (2004). Serum and glucocorticoid inducible kinases functionally regulate ClC-2 channels. Biochem. Biophys. Res. Commun. 321 (4), 1001–1006. doi:10.1016/j.bbrc.2004.07.064
Palmada, M., DieterM., , Speil, A., Bohmer, C., Mack, A. F., Wagner, H. J., et al. (2004). Regulation of intestinal phosphate cotransporter NaPi IIb by ubiquitin ligase Nedd4-2 and by serum- and glucocorticoid-dependent kinase 1. Am. J. Physiol. Gastrointest. Liver Physiol. 287 (1), G143–G150. doi:10.1152/ajpgi.00121.2003
Palmada, M., Speil, A., Jeyaraj, S., Bohmer, C., and Lang, F. (2005). The serine/threonine kinases SGK1, 3 and PKB stimulate the amino acid transporter ASCT2. Biochem. Biophys. Res. Commun. 331 (1), 272–277. doi:10.1016/j.bbrc.2005.03.159
Pao, A. (2012). SGK regulation of renal sodium transport. Curr. Opin. Nephrol. Hypertens. 21 (5), 534–540. doi:10.1097/MNH.0b013e32835571be
Peacock, M. (2010). Calcium metabolism in health and disease. Clin. J. Am. Soc. Nephrol. 5, S23–S30. doi:10.2215/CJN.05910809
Potts, J. T., and Gardella, T. J. (2007). Progress, paradox, and potential: Parathyroid hormone research over five decades. Ann. N. Y. Acad. Sci. 1117, 196–208. doi:10.1196/annals.1402.088
Russo, A., Schmid, E., Nurbaeva, M. K., Yang, W., Yan, J., Bhandaru, M., et al. (2013). PKB/SGK-dependent GSK3-phosphorylation in the regulation of LPS-induced Ca2+ increase in mouse dendritic cells. Biochem. Biophys. Res. Commun. 437 (3), 336–341. doi:10.1016/j.bbrc.2013.06.075
Sandu, C., Rexhepaj, R., Grahammer, F., McCormick, J. A., Henke, G., Palmada, M., et al. (2005). Decreased intestinal glucose transport in the sgk3-knockout mouse. Pflugers Arch. 451 (3), 437–444. doi:10.1007/s00424-005-1474-7
Sarac, S., Afzal, S., Broholm, H., Madsen, F. F., Ploug, T., and Laursen, H. (2009). Excitatory amino acid transporters EAAT-1 and EAAT-2 in temporal lobe and hippocampus in intractable temporal lobe epilepsy. APMIS 117 (4), 291–301. doi:10.1111/j.1600-0463.2009.02443.x
Schmid, E., Bhandaru, M., Nurbaeva, M. K., Yang, W., Szteyn, K., Russo, A., et al. (2012). SGK3 regulates Ca(2+) entry and migration of dendritic cells. Cell. Physiol. biochem. 30 (6), 1423–1435. doi:10.1159/000343330
Schmid, E., Yan, J., Nurbaeva, M. K., Russo, A., Yang, W., Faggio, C., et al. (2014). Decreased store operated Ca2+ entry in dendritic cells isolated from mice expressing PKB/SGK-resistant GSK3. PLoS One 9 (2), e88637. doi:10.1371/journal.pone.0088637
Seebohm, G., Strutz-Seebohm, N., Ursu, O. N., Preisig-Muller, R., Zuzarte, M., Hill, E. V., et al. (2012). Altered stress stimulation of inward rectifier potassium channels in Andersen-Tawil syndrome. FASEB J. 26 (2), 513–522. doi:10.1096/fj.11-189126
Lamothe, S. M., and Zhang, S. (2013). The serum- and glucocorticoid-inducible kinases SGK1 and SGK3 regulate hERG channel expression via ubiquitin ligase Nedd4-2 and GTPase Rab11. J. Biol. Chem. 288 (21), 15075–15084. doi:10.1074/jbc.M113.453670
Shojaiefard, M., Christie, D. L., and Lang, F. (2005). Stimulation of the creatine transporter SLC6A8 by the protein kinases SGK1 and SGK3. Biochem. Biophys. Res. Commun. 334 (3), 742–746. doi:10.1016/j.bbrc.2005.06.164
Shumilina, E., Huber, S. M., and Lang, F. (2011). Ca2+ signaling in the regulation of dendritic cell functions. Am. J. Physiol. Cell Physiol. 300 (6), C1205–C1214. doi:10.1152/ajpcell.00039.2011
Slagsvold, T., Marchese, A., Brech, A., and Stenmark, H. (2006). CISK attenuates degradation of the chemokine receptor CXCR4 via the ubiquitin ligase AIP4. EMBO J. 25 (16), 3738–3749. doi:10.1038/sj.emboj.7601267
Stolting, G., Fischer, M., and Fahlke, C. (2014). CLC channel function and dysfunction in health and disease. Front. Physiol. 5, 378. doi:10.3389/fphys.2014.00378
Strutz-Seebohm, N., Seebohm, G., Korniychuk, G., Baltaev, R., Ureche, O., Striegel, M., et al. (2006). Additive regulation of GluR1 by stargazin and serum- and glucocorticoid-inducible kinase isoform SGK3. Pflugers Arch. 452 (3), 276–282. doi:10.1007/s00424-005-0032-7
Strutz-Seebohm, N., Seebohm, G., Mack, A. F., Wagner, H. J., Just, L., Skutella, T., et al. (2005). Regulation of GluR1 abundance in murine hippocampal neurones by serum- and glucocorticoid-inducible kinase 3. J. Physiol. 565, 381–390. doi:10.1113/jphysiol.2004.079582
Strutz-Seebohm, N., Seebohm, G., Shumilina, E., Mack, A. F., Wagner, H. J., Lampert, A., et al. (2005). Glucocorticoid adrenal steroids and glucocorticoid-inducible kinase isoforms in the regulation of GluR6 expression. J. Physiol. 565, 391–401. doi:10.1113/jphysiol.2004.079624
Taylor, J. G., and Bushinsky, D. A. (2009). Calcium and phosphorus homeostasis. Blood Purif. 27 (4), 387–394. doi:10.1159/000209740
Tessier, M., and Woodgett, J. R. (2006). Role of the Phox homology domain and phosphorylation in activation of serum and glucocorticoid-regulated kinase-3. J. Biol. Chem. 281 (33), 23978–23989. doi:10.1074/jbc.M604333200
Toro, L., Li, M., Zhang, Z., Singh, H., Wu, Y., and Stefani, E. (2014). MaxiK channel and cell signalling. Pflugers Arch. 466 (5), 875–886. doi:10.1007/s00424-013-1359-0
Vallon, V., GrahammerF., , Richter, K., BleichM., , LangF., , Barhanin, J., et al. (2001). Role of KCNE1-dependent K+ fluxes in mouse proximal tubule. J. Am. Soc. Nephrol. 12 (10), 2003–2011. doi:10.1681/ASN.V12102003
Vervloet, M. G., Sezer, S., Massy, Z. A., Johansson, L., Cozzolino, M., Fouque, D., et al. (2017). The role of phosphate in kidney disease. Nat. Rev. Nephrol. 13 (1), 27–38. doi:10.1038/nrneph.2016.164
Waldegger, S., Jeck, N., Barth, P., Peters, M., Vitzthum, H., Wolf, K., et al. (2002). Barttin increases surface expression and changes current properties of ClC-K channels. Pflugers Arch. 444 (3), 411–418. doi:10.1007/s00424-002-0819-8
Wang, Y., Zhou, D., and Chen, S. (2014). SGK3 is an androgen-inducible kinase promoting prostate cancer cell proliferation through activation of p70 S6 kinase and up-regulation of cyclin D1. Mol. Endocrinol. 28 (6), 935–948. doi:10.1210/me.2013-1339
Wang, Y., Zhou, D., Phung, S., Masri, S., Smith, D., and Chen, S. (2011). SGK3 is an estrogen-inducible kinase promoting estrogen-mediated survival of breast cancer cells. Mol. Endocrinol. 25 (1), 72–82. doi:10.1210/me.2010-0294
Wang, Y., Zhou, D., Phung, S., Warden, C., Rashid, R., Chan, N., et al. (2017). SGK3 sustains ERα signaling and drives acquired aromatase inhibitor resistance through maintaining endoplasmic reticulum homeostasis. Proc. Natl. Acad. Sci. U. S. A. 114 (8), E1500–E1508. doi:10.1073/pnas.1612991114
Williams, K. B., and DeLuca, H. F. (2007). Characterization of intestinal phosphate absorption using a novel in vivo method. Am. J. Physiol. Endocrinol. Metab. 292 (6), E1917–E1921. doi:10.1152/ajpendo.00654.2006
Wyatt, A. W., Hussain, A., Amann, K., Klingel, K., Kandolf, R., Artunc, F., et al. (2006). DOCA-induced phosphorylation of glycogen synthase kinase 3beta. Cell. Physiol. biochem. 17 (3-4), 137–144. doi:10.1159/000092075
Xia, M., Jin, Q., Bendahhou, S., He, Y., Larroque, M. M., Chen, Y., et al. (2005). A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem. Biophys. Res. Commun. 332 (4), 1012–1019. doi:10.1016/j.bbrc.2005.05.054
Xing, Y., Liu, D., Zhang, R., Joachimiak, A., Songyang, Z., and Xu, W. (2004). Structural basis of membrane targeting by the Phox homology domain of cytokine-independent survival kinase (CISK-PX). J. Biol. Chem. 279 (29), 30662–30669. doi:10.1074/jbc.M404107200
Xu, H., Bai, L., Collins, J. F., and Ghishan, F. K. (2002). Age-dependent regulation of rat intestinal type IIb sodium-phosphate cotransporter by 1, 25-(OH)(2) vitamin D(3). Am. J. Physiol. Cell Physiol. 282 (3), C487–C493. doi:10.1152/ajpcell.00412.2001
Xu, J., Liao, L., Qin, J., Liu, D., and Songyang, Z. (2009). Identification of Flightless-I as a substrate of the cytokine-independent survival kinase CISK. J. Biol. Chem. 284 (21), 14377–14385. doi:10.1074/jbc.M807770200
Yan, X., Xun, M., Dou, X., Wu, L., Han, Y., and Zheng, J. (2017). Regulation of Na(+)-K(+)-ATPase effected high glucose-induced myocardial cell injury through c-Src dependent NADPH oxidase/ROS pathway. Exp. Cell Res. 357 (2), 243–251. doi:10.1016/j.yexcr.2017.05.023
Yao, L. J., McCormick, J. A., Wang, J., Yang, K. Y., Kidwai, A., Colussi, G. L., et al. (2011). Novel role for SGK3 in glucose homeostasis revealed in SGK3/Akt2 double-null mice. Mol. Endocrinol. 25 (12), 2106–2118. doi:10.1210/me.2010-0329
Yuan, Y. P., Zhao, H., Peng, L. Q., Li, Z. F., Liu, S., Yuan, C. Y., et al. (2018). The SGK3-triggered ubiquitin-proteasome degradation of podocalyxin (PC) and ezrin in podocytes was associated with the stability of the PC/ezrin complex. Cell Death Dis. 9 (11), 1114. doi:10.1038/s41419-018-1161-1
Keywords: serum- and glucocorticoid-induced kinase, SGK3, channel, substance metabolism, substance transport
Citation: Liao Q-Q, Dong Q-Q, Zhang H, Shu H-P, Tu Y-C and Yao L-J (2022) Contributions of SGK3 to transporter-related diseases. Front. Cell Dev. Biol. 10:1007924. doi: 10.3389/fcell.2022.1007924
Received: 31 July 2022; Accepted: 09 November 2022;
Published: 01 December 2022.
Edited by:
Xinhua Shu, Glasgow Caledonian University, United KingdomReviewed by:
Sharon Weiss, Tel Aviv University, IsraelChristopher Stroupe, Stroupe.net, United States
Copyright © 2022 Liao, Dong, Zhang, Shu, Tu and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li-Jun Yao, ZHJ5YW9saWp1bkAxNjMuY29t
†These authors have contributed equally to this work and share first authorship.