 Caojie Liu
Caojie Liu Ning Kang
Ning Kang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol. , 20 September 2021
Sec. Cellular Biochemistry
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.710203
This article is part of the Research Topic Differentiation and Regulation of Bone Marrow Mesenchymal Stromal Cells View all 17 articles
Background: Regulation of gene expression is critical for stem cell differentiation, tissue development, and human health maintenance. Recently, epigenetic modifications of histone and chromatin remodeling have been verified as key controllers of gene expression and human diseases.
Objective: In this study, we review the role of chromodomain helicase DNA-binding (CHD) proteins in stem cell differentiation, cell fate decision, and several known human developmental disorders and cancers.
Conclusion: CHD proteins play a crucial role in stem cell differentiation and human diseases.
There are three classes of epigenetic modifier proteins, including chromatin writers, erasers, and readers. We center around a chromatin reader superfamily and review their functions and structures as well as known roles in human diseases. In eukaryotic organisms, ATP-dependent chromatin remodeling enzymes are commonly divided into three major superfamilies, including SWItch/sucrose non-fermentable (SWI/SNF), Imitation SWI (ISWI), and chromodomain helicase DNA-binding (CHD) proteins (Dann et al., 2017; Barisic et al., 2019).
CHD comprises nine proteins, which are classified as three subfamilies on account of domain homology. These nine CHDs all involve tandem chromatin organization modifier domains (chromodomain) as well as sucrose non-fermentable2 (SNF2)-like ATP-dependent helicase domains (Flanagan et al., 2005; Bajpai et al., 2010; He et al., 2016; Farnung et al., 2017). Recently, a study reviewed the structure of these four domains and the differences between CHDs (Mills, 2017; Goodman and Bonni, 2019). CHDs read and/or interpret histone modifications by specialized domains. When reading the chromatin state, CHDs disrupt the DNA–histone interaction via translocating the nucleosomes along the same or the other DNA strand (Clapier et al., 2017; Mashtalir et al., 2018).
CHDs share highly similar helicase-ATPase domains with the SWItch2/SNF2 superfamily (Noh et al., 2015; Mashtalir et al., 2018). These helicase-ATPase domains provide energy. At the same time, they promote disruption of histone-DNA contacts as mentioned above (Clapier et al., 2017; Mashtalir et al., 2018). Moreover, the three subfamilies described above are defined by specific domains (Figure 1). Briefly, subfamily I, including CHD1 and 2, share DNA-binding domains that demonstrate similar function with SWI3, ADA2. Subfamily II, including CHD3-5, contain the PHD zinc finger domain in addition to the three domains mentioned above, which promotes its binding to methylated histone residues and protein cofactors. Subfamily III, including CHD6-9, its specific SANT domain could promote non-specific DNA binding (Mills, 2017; Platt et al., 2017).
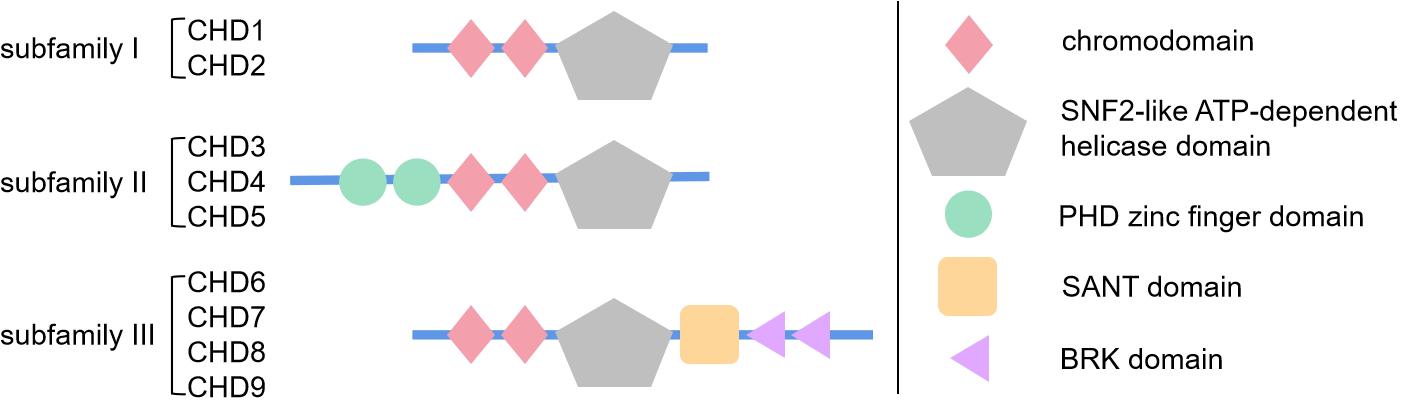
Figure 1. Schematic diagram showing the structure and specific domains of three subfamilies of chromodomain helicase DNA-binding (CHD) proteins. This figure briefly demonstrates the basic structure and domains of CHD protein family. As is reviewed in section “Introduction of CHD Superfamily” of this article, CHD proteins are mainly divided in three subfamilies by their specific domains, subfamily I with basic chromodomian and sucrose non-fermentable2 (SNF2)-like ATP-dependent helicase domain, subfamily II with additional PHD zinc finger domain, and subfamily III with additional SANT domain and BRK domain. The simple geometric patterns represent the protein domains, whose size or location are not strictly to scale, just for brief demonstration.
Chromodomains were first recognized in Drosophila heterochromatin protein 1. Heterochromatin protein 1 owns a chromodomain that binds to nucleosomes to facilitate a closed chromatin state as well as regulate homeotic genes (Flanagan et al., 2005; Farnung et al., 2017; Zhao et al., 2020). It is known nowadays that binding to methylated histone residues is the primary function of chromodomains. CHDs contain a special variant of the chromodomains with methy1-binding cages, which promote interactions with H3K4me (Flanagan et al., 2005; Dann et al., 2017; Barisic et al., 2019). For instance, CHD1 chromodomains interact with H3K4me, and CHD5 chromodomains bind to H3K27me3 (Dorighi and Tamkun, 2013; Link et al., 2018; Goodman and Bonni, 2019). Therefore, CHDs demonstrate special functions and preferences for active or repressive histone marks. CHD chromodomains are essential for proper gene expression and maintaining dynamic chromatin structures.
CHD superfamily proteins are essential to regulate gene expression. Thus, CHDs are crucial for the survival, maintenance, and proliferation of stem cells as well as regulating the cell fate of their daughter cells (Figure 2).
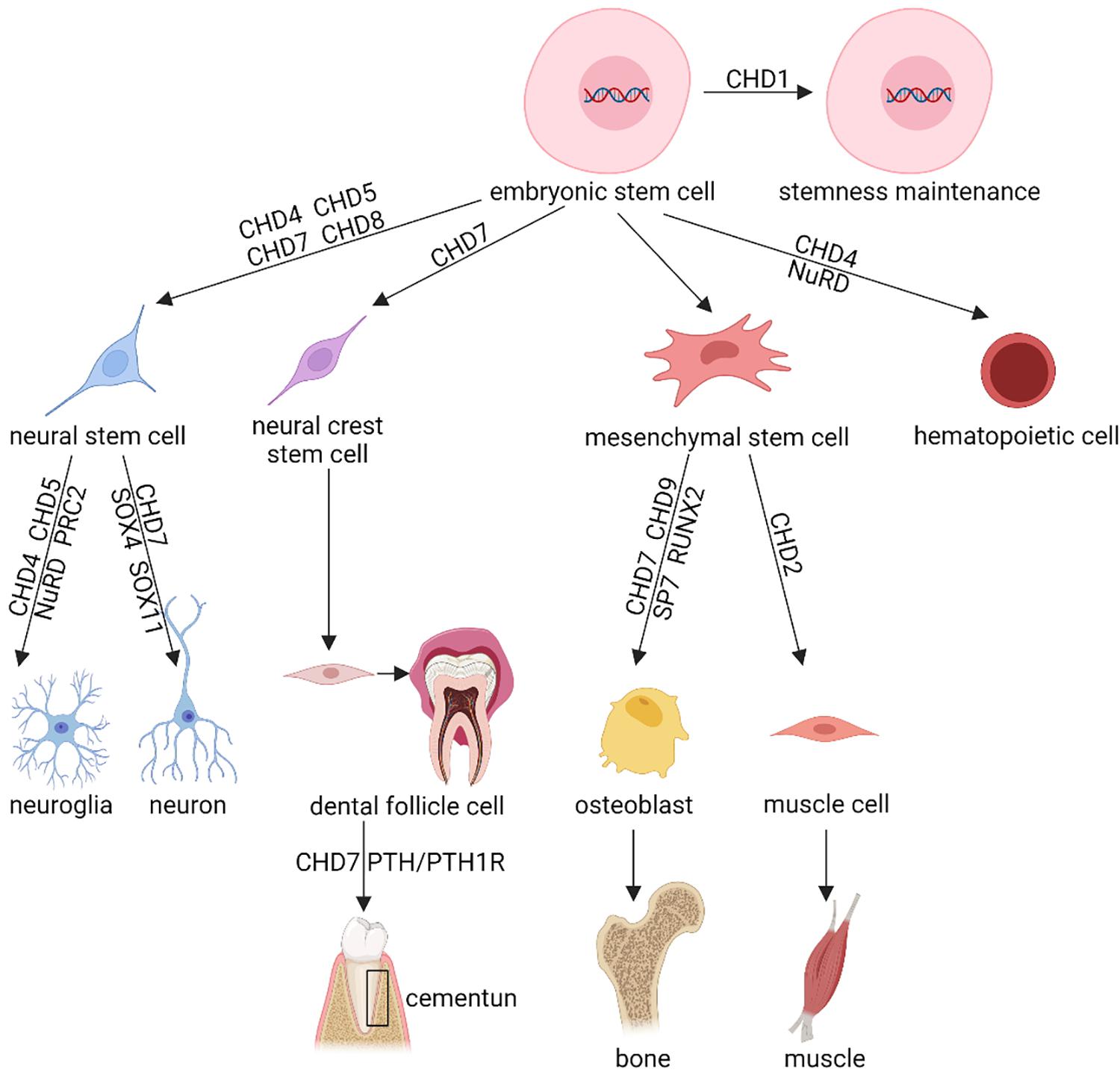
Figure 2. Schematic diagram showing the regulatory effects of CHD proteins in stem cell proliferation, differentiation, and functioning. This figure briefly demonstrates the known function of the CHD protein family in various stem cells, including the stemness maintenance, cellular differentiation, lineage commitment, cell fate decision, etc. Shown are representative stem cells, e.g., Embryonic Stem Cells (ESCs), Neural Stem Cells (NSCs), neural crest stem cells, Mesenchymal Stem Cells (MSCs), reviewed in section “CHD and Stem Cells” of this article. This diagram only exhibits the certified function of CHD protein family, the uncertain or controversial effects of CHDs have been omitted. NuRD, nucleosome remodeling and deacetylase; PRC2, Polycomb Repressive Complex 2.
ESCs demonstrate an open chromatin environment. Via activation or repression of different genetic pathways, ESCs differentiate toward mesenchymal, hematopoietic, neural, and other lineage cells. By maintaining open chromatin, CHD1 participates in the mediator complex, a regulator of ESCs (Gaspar-Maia et al., 2009; Bulut-Karslioglu et al., 2018). This complex is a multiprotein complex that pre-initiates gene transcription through binding to CHD1 and recruiting it to express genes (de Dieuleveult et al., 2016; Percharde et al., 2017). If lacking CHD1, chromatin would condense to form heterochromatin. In that case, due to increased ectodermal lineage gene expression and decreased endodermal lineage gene expression, pluripotent differentiation would be impaired (Lin et al., 2011; Koh et al., 2015; Suzuki et al., 2015). Furthermore, induction of CHD1 is necessary for efficient programming of induced pluripotent stem cells (de Dieuleveult et al., 2016; Percharde et al., 2017; Bulut-Karslioglu et al., 2018). Therefore, CHD1 is essential to maintain pluripotency in stem cells.
By regulating epigenetic and signaling pathways, CHD7 is also crucial for cell fate decisions. In mouse ESCs, the Chd7 gene is highly expressed and associated with gene expression active signals. In stem and progenitor cells, the euchromatic chromatin environment is poised between activation and repression (Yang et al., 2017). Different from active or inactive states, those promoters as well as enhancers typically express both active and inactive marks (Schnetz et al., 2009; Yang et al., 2017). Via binding to active as well as poised enhancers of ectodermal lineage genes, CHD7 plays an important role in histone modifying, transcription factor recruiting, and other chromatin remodeling (Platt et al., 2017; Goodman and Bonni, 2019). Meanwhile, it promotes open chromatin at enhancers of critical genes such as Sox2, Nanog, and Oct4 (Engelen et al., 2011; Puc and Rosenfeld, 2011; Fujita et al., 2016).
In brief, the CHD family is essential to regulating the function of ESCs.
NSCs produce both neuron and supporting cells and, thus, play a pivotal role in the nervous system as well as sensory organs. In the dentate gyrus of the hippocampus and subventricular zone of the forebrain, CHD4, CHD5, and CHD7 cooperate with signaling pathways and transcription factors, which are critical for the differentiation and function of NSC niches (Feng et al., 2017; Weiss et al., 2020; Parenti et al., 2021).
Binding to the polycomb repressive complex 2 (PRC2) during cortical neurogenesis, CHD4 was found to express in the murine subventricular zone neural progenitor (Mohd-Sarip et al., 2017; Pierson et al., 2019; Shieh et al., 2020). This complex represses the expression of glial fibrillary acidic protein gene (Gfap) as well as blocks glial differentiation (Dorighi and Tamkun, 2013; Sparmann et al., 2013). During neurogenesis, this complex promotes neuronal differentiation by inhibiting the Gfap locus (Sparmann et al., 2013). Moreover, CHD4, together with other nucleosome remodeling and deacetylase (NuRD) complex, can repress several genes that downregulate neuronal differentiation (Mohd-Sarip et al., 2017; Link et al., 2018; Pierson et al., 2019; Shieh et al., 2020).
CHD5 also binds to PRC2 and H3K27me3 in NSCs (Egan et al., 2013; Xie et al., 2015; Hayashi et al., 2016). In the subventricular and subgranular zones in the hippocampus, high expression of CHD5 is found in neuroblast cells as well as neural progenitors (Egan et al., 2013). CHD5 is pivotal for learning and memory. It is also found that depletion of Chd5 in the developing cortex leads to reduction of migratory neuroblasts (Egan et al., 2013).
CHD7 is also vital for fine function of NSCs. CHD7 is highly expressed in both the subgranular and subventricular zones in adult mice. In these areas, CHD7 colocalizes with markers of NSCs, neural progenitor cells, and neuroblasts (Feng et al., 2017; Goodman and Bonni, 2019). Studies in which Chd7 was conditionally deleted in the adult subventricular zone demonstrate that CHD7 deletion resulted in a reduction of mature dopaminergic NSCs (Feng et al., 2013). Such Chd7 deficiency also downregulates the expression of proneural genes, such as Sox4 and Sox11 (Feng et al., 2013; Brajadenta et al., 2019). Conditional knockout of Chd7 in the subgranular zone also reduces neurogenesis (Feng et al., 2013). Furthermore, in the otic placodes and olfactory, CHD7 promotes NSC progenitor proliferation (Jones et al., 2015; Ohta et al., 2016; Whittaker et al., 2017). CHD7 is critical for NSC function although the mechanisms by which CHD7 regulates NSC function remain to be determined.
As a kind of multipotent mesoderm-derived cell differentiating into myoblasts, adipocytes, osteoblasts, and chondrocytes, MSCs are shown to be regulated by CHD proteins (Mohd-Sarip et al., 2017). Several different CHD proteins regulate the differentiation of MSCs into four distinct lineages. CHD2 is critical for induction of myogenic cell fates (Harada et al., 2012; de Dieuleveult et al., 2016; Semba et al., 2017; Nieto-Estevez and Hsieh, 2018). CHD9 could bind to osteocalcin, which is one of the master transcriptional factors for bone development, and promote its expression (Shur et al., 2006a,b; de Dieuleveult et al., 2016). Recently, we found that CHD7 is essential for osteogenic differentiation of human MSCs. Depletion of CHD7 via siRNA impairs the osteogenesis potential of MSCs, and overexpression of CHD7 via lentivirus vector could promote the osteogenesis potential of MSCs. Mechanically, we found that CHD7 might bind to the enhancer of Sp7, and it also interacts with SMAD1, indicating that CHD7 is crucial to the osteogenesis potential of MSCs (Chen et al., 2016).
Besides the above-described biological processes, the CHD family also affect the development and functional maintenance of many organ systems, such as the hematopoietic and circulatory systems (Koh et al., 2015; Sperlazza et al., 2015; Zhen et al., 2017; Arends et al., 2019; Hsu et al., 2020; Tu et al., 2021). For example, CHD7 promotes the osteogenesis potential of dental follicle cells to form cementum by upregulating the PTH/PTH1R signaling pathway (Liu et al., 2020).
Due to the important regulatory role of the CHD family in the differentiation and function of NSCs, the deletion or mutation of CHD genes often leads to NDDs, featuring as intellectual disability (ID), autism spectrum disorders (ASDs), and epilepsy.
Several CHD, such as CHD2, CHD6, CHD7, and CHD8, were found non-sense, heterozygous, or other kinds of mutations in patients with ASD, ID, and epilepsy (Allen et al., 2013; Suls et al., 2013; Sugathan et al., 2014; Ellingford et al., 2021; Parenti et al., 2021). In patients with ASD and/or ID that associate with gastrointestinal disturbance and macrocephaly, 13 recurrent alleles of CHD8 were figured out (Bernier et al., 2014; Sugathan et al., 2014; Ellingford et al., 2021). Using RNA- and ChIP-sequencing, a recent study indicates that knockdown of CHD8 could not alter the neural ectodermal or morphology markers of neural progenitors, but could impair their gene expression (Sugathan et al., 2014; Goodman and Bonni, 2019). CHD8 was found to bind CHD7 as well as p53, regulate p53, and inhibit cell proapoptotic effects during development (Nishiyama et al., 2009; Hurley et al., 2021; Tu et al., 2021). Interestingly, CHD7 could also bind to and repress p53 (Van Nostrand et al., 2014; Corsten-Janssen and Scambler, 2017). Thus, some CHD proteins might share common target genes, interacting factors, and downstream mechanisms. CHD2 mutations were also observed in patients with epilepsy (Allen et al., 2013; Suls et al., 2013; Nieto-Estevez and Hsieh, 2018). All these data raise the possibility that the region of CHD mutation may be wider than the previous hypothesis. Due to the intricacy of CHD targets as well as associating partners, a main goal of future studies is making clear the mechanisms by which CHD mutation disturbs NSCs and neuronal development.
CHARGE is the acronym of an autosomal dominant genetic syndrome, which is characterized by six main symptoms, including ocular coloboma, congenital heart defects, choanal atresia, developmental retardation, genital anomalies, and ear anomalies (Pagon et al., 1981; Brajadenta et al., 2019). According to a Canadian study, this syndrome occurs in approximately 1 in 10,000 live births (Issekutz et al., 2005; Van Nostrand et al., 2014). CHD7 has been closely linked to this disorder because heterozygous mutations in this gene were found in more than 90% of these patients (Vissers et al., 2004; Ghaoui et al., 2015; Brajadenta et al., 2019). Therefore, CHD7 is also one of the most researched members among the CHD superfamily. The embryologic expression of CHD7 involves several sites, including eyes, olfactory bulb cells, inner ears, etc. (Vissers et al., 2004; Jamadagni et al., 2021). Additionally, high expression of CHD7 is also observed in undifferentiated neuroepithelium and neural crest mesenchyme as mentioned above. Thus, CHARGE is a monogenic disorder with variable expressivity (Jones et al., 2015; Yan et al., 2020). However, due to the multiplicity of CHD7 mutations as well as the variable expressivity of this syndrome, no critical genotype/phenotype correlations can be found (Bajpai et al., 2010; Butcher et al., 2017). In an in vitro study, intact recombinant CHD7 protein was purified and proved to be an ATP-dependent nucleosome remodeling factor (Bouazoune and Kingston, 2012; Yan et al., 2020). Interestingly, when CHARGE patients were administrated with recombinant CHD7 protein, their enzymatic activity, which is related to chromatin remodeling, reduced in a mutation-specific mode (Rother et al., 2020; Yan et al., 2020). This study supports the hypothesis that CHD7 haploinsufficiency is the main cause of CHARGE syndrome (Whittaker et al., 2017; Brajadenta et al., 2019). Although other functions of CHD7 should be discovered, these functions might help explain the clinical features in CHARGE patients.
CHD7 haploinsufficiency leads to dysfunction in sensory processes as well as impaired vision, hearing, balance, and olfaction. A mouse model has been established and analyzed to learn more about the role of CHD7 in CHARGE. Chd7 knockout mice embryos cannot survive over embryonic day 10.5, but heterozygous mice show several similar defects observed in CHARGE syndrome (Kim et al., 2008; Gage et al., 2015; Jones et al., 2015). Moreover, there is no CHARGE syndrome patient who is figured out to have CHD7 homozygous mutations (Vissers et al., 2004; Basson and van Ravenswaaij-Arts, 2015). These facts suggest that homozygous mutations in CHD7 might cause embryonic lethality, possibly due to the wide expression of the CHD7 gene in tissues affected in CHARGE syndrome (Yan et al., 2020). Using conditional Chd7flox allele mating with tissue-specific Cre transgenes, a recent study found that CHD7 is necessary for eye development in multiple embryonic tissues and also essential for lens development in the surface ectoderm (Gage et al., 2015; Goodman and Bonni, 2019). Chd7+/– mice showed hypoplasia and aplasia of posterior and lateral semicircular canals as well as innervation defects of the vestibular sensory epithelium (Yan et al., 2020; Jamadagni et al., 2021). In all, this evidence suggests that CHD7 might play a similar role in sensory tissues as in NSCs.
As two major phenotypes observed in CHARGE syndrome patients, hyposmia and anosmia mean decrease and loss of the smell sense, respectively (Brajadenta et al., 2019; Yan et al., 2020). Olfactory deficiency is usually accompanied by aplasia or hypoplasia of the olfactory bulbs (Ghaoui et al., 2015). Through behavioral assays and electrophysiological study, Chd7 heterozygous mice were found lacking in odor discrimination, olfactory bulb hypoplasia, and complete anosmia (Layman et al., 2009). As an essential gene in stem cell differentiation, high expression of CHD7 could be found in olfactory NSCs as well as progenitor cells (Whittaker et al., 2017; Jamadagni et al., 2021). Depletion of Chd7 results in a significant reduction of NSC proliferation in olfactory epithelials, thus leading to a decrease in olfactory receptor neurons and delayed recovery post damage (Feng et al., 2017; Whittaker et al., 2017). Moreover, efferent neurons in the olfactory epithelium as well as olfactory bulb neurogenesis from the subventricular NSC niches of such mutant mice are impaired, resulting in the reduction of tyrosine hydroxylase-positive interneurons in the olfactory bulb (Ohta et al., 2016; Feng et al., 2017). Taken together, olfactory processing counts on CHD7 function.
Through co-IP and chromatin immunoprecipitation studies, CHD7 was proved to interact with SOX2, which associated with several diseases, such as Feingold syndrome, Alagille syndrome, and Pallister–Hall syndrome (Engelen et al., 2011; Puc and Rosenfeld, 2011; Fujita et al., 2016). These syndromes share several similar phenotypes with CHARGE syndrome, including tracheoesophageal defects, genital abnormalities, semicircular canal hypoplasia, and pituitary and endocrine dysfunction (Puc and Rosenfeld, 2011; Stamou et al., 2020). It is reported that CHD7 binds to SOX2 because of the massive overlap in expression as well as function of these two proteins. Besides this, CHD7 and SOX2 share similar functions in the development of ectodermal lineages that are influenced in CHARGE syndrome (Schnetz et al., 2010; Doi et al., 2017). As one of the characters of CHARGE syndrome, craniofacial malformations are also commonly observed (Van Nostrand et al., 2014; He et al., 2016). It was found that Chd7 is necessary for proper craniofacial development via immunofluorescence and Cre lineage tracing (Sperry et al., 2014). Importantly, CHD7 also interacts with SMAD1 and could bind to the enhancer region of Sp7, which is a master transcription factor of osteogenic differentiation (Chen et al., 2016).
Besides this, CHD7 is also closely associated with Kallmann syndrome, which is a genetic heterogeneous congenital disease mainly characterized by idiopathic hypogonadotrophic hypogonadism (IHH) (Kim et al., 2008; Stamou et al., 2020). Such manifestation is largely caused by impaired gonadotropin-releasing hormone (GnRH) (Balasubramanian and Crowley, 2017; Stamou et al., 2020). The pathogenic mechanism of Kallmann syndrome is complicated and could be partially explained by several mutated genes, including the missence mutation of CHD7, leading to the alteration of the domain or function of CHD7 protein (Kim et al., 2008; Boehm et al., 2015). Some of the clinical symptoms of Kallmann syndrome, e.g., anosmia, IHH, heart defect, cleft lip, cleft palate, etc., also often appear in CHARGE syndrome, so these two syndromes are often compared in clinical work and need differential diagnosis (Ufartes et al., 2018; Stamou et al., 2020).
CHD proteins are also involved in cancers. Genomic and epigenomic changes are correlated and could predict the tumor phenotypes and progression.
CHD1 was proved to be frequently deleted in prostate cancer (Attard et al., 2016; Zhang et al., 2020). Researchers conducted and analyzed whole-genome, whole-transcriptome, and DNA methylation data from patients with primary prostate cancer and healthy controls. Deletions in CHD1 occurred in 18% of the tumors (Li et al., 2020). Further studies confirm that CHD1 plays a key role in myeloid-derived suppressor cell recruitment and find that CHD1/IL6 is a major regulator of the immunosuppressive tumor microenvironment in prostate cancer (Zhao et al., 2020).
CHD4 is closely associated with breast, endometrial, and colorectal cancer (Novillo et al., 2021). As a crucial ingredient in NuRD complex, the upstream regulating effects of CHD4 involves the recruitment of DNA methyl transferase and key transcriptional repressors (Hata et al., 2019; Wang et al., 2020). CHD4 could recruit inhibitory chromatin remodelers to the DNA damage repair sites and initiate and support the silencing of tumor suppressor gene. Such functions confirm the oncogenic effect of CHD4 (Chang et al., 2019; Novillo et al., 2021). In addition, CHD4, as a co-activator of hypoxia-inducible factor (HIF), is upregulated in human breast tumors and is related to the expression of HIF target genes (Shieh et al., 2020; Wang et al., 2020). Besides this, CHD4 was associated with poorer overall survival in breast cancer patients (Novillo et al., 2021).
According to the position, expression pattern, and function of CHD5 in neuroblastoma cells and xenograft cells, CHD5 was identified as a tumor suppressor gene (Kolla et al., 2014; Liu et al., 2018). CHD5 also functions as one of the tumor suppressor genes in other types of tumors, e.g., gliomas, breast, colon, lung, ovarian, and prostate cancers (Bagchi et al., 2007; Xie et al., 2015). Especially, low expression of CHD5 is strongly associated with poorer clinical and biological characteristics and prognosis (Kolla et al., 2014; Higashi et al., 2015).
To sum up, all of the associations among these diseases emphasize the variety of cell processes that require proper chromatin remodeling.
Here, we review the CHD superfamily as well as known functions in several kinds of stem cells. The CHD family is widely involved in and regulates many physiological and biochemical pathways in the organism, which is of great significance for normal growth, development, and functional maintenance of the body as well as the occurrence and development of several diseases. Future studies aiming at revealing the proper mechanisms by which CHDs mediate these effects will uncover more important cues about this important chromatin remodeler.
CL: drafting manuscript. PG: approval of article and funding secured. YG: literature summary, article revision, and funding secured. NK: concept, design, and funding secured. All authors contributed to the article and approved the submitted version.
This research was supported by the National Natural Science Foundation of China (NSFC Grant Nos. 81701009 and 82001017), the West China Hospital of Stomatology (Grant No. RCDWJS2020-23), and the China Postdoctoral Science Foundation Grant (Grant Nos. 2019TQ0218 and 2020M683328).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We appreciate the website BioRender for the technical support in schematic diagram drawing (https://app.biorender.com/).
Allen, A., Berkovic, S., Cossette, P., Delanty, N., Dlugos, D., Eichler, E., et al. (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. doi: 10.1038/nature12439
Arends, T., Dege, C., Bortnick, A., Danhorn, T., Knapp, J., Jia, H., et al. (2019). CHD4 is essential for transcriptional repression and lineage progression in B lymphopoiesis. Proc. Natl. Acad. Sci. U. S. A. 116, 10927–10936. doi: 10.1073/pnas.1821301116
Attard, G., Parker, C., Eeles, R., Schröder, F., Tomlins, S., Tannock, I., et al. (2016). Prostate cancer. Lancet 387, 70–82. doi: 10.1016/s0140-6736(14)61947-4
Bagchi, A., Papazoglu, C., Wu, Y., Capurso, D., Brodt, M., Francis, D., et al. (2007). CHD5 is a tumor suppressor at human 1p36. Cell 128, 459–475. doi: 10.1016/j.cell.2006.11.052
Bajpai, R., Chen, D., Rada-Iglesias, A., Zhang, J., Xiong, Y., Helms, J., et al. (2010). CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463, 958–962. doi: 10.1038/nature08733
Balasubramanian, R., and Crowley, W. (2017). Reproductive endocrine phenotypes relating to CHD7 mutations in humans. Am. J. Med. Genet. C Semin. Med. Genet. 175, 507–515. doi: 10.1002/ajmg.c.31585
Barisic, D., Stadler, M., Iurlaro, M., and Schübeler, D. (2019). Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature 569, 136–140. doi: 10.1038/s41586-019-1115-5
Basson, M., and van Ravenswaaij-Arts, C. (2015). Functional Insights into Chromatin Remodelling from Studies on CHARGE Syndrome. Trends Genet. 31, 600–611. doi: 10.1016/j.tig.2015.05.009
Bernier, R., Golzio, C., Xiong, B., Stessman, H., Coe, B., Penn, O., et al. (2014). Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276. doi: 10.1016/j.cell.2014.06.017
Boehm, U., Bouloux, P., Dattani, M., de Roux, N., Dodé, C., Dunkel, L., et al. (2015). Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism–pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 11, 547–564. doi: 10.1038/nrendo.2015.112
Bouazoune, K., and Kingston, R. (2012). Chromatin remodeling by the CHD7 protein is impaired by mutations that cause human developmental disorders. Proc. Natl. Acad. Sci. U. S. A. 109, 19238–19243. doi: 10.1073/pnas.1213825109
Brajadenta, G., Bilan, F., Gilbert-Dussardier, B., Kitzis, A., and Thoreau, V. (2019). A functional assay to study the pathogenicity of CHD7 protein variants encountered in CHARGE syndrome patients. Eur. J. Hum. Genet. 27, 1683–1691. doi: 10.1038/s41431-019-0465-7
Bulut-Karslioglu, A., Macrae, T., Oses-Prieto, J., Covarrubias, S., Percharde, M., Ku, G., et al. (2018). The Transcriptionally Permissive Chromatin State of Embryonic Stem Cells Is Acutely Tuned to Translational Output. Cell Stem Cell 22, 369–383.e8. doi: 10.1016/j.stem.2018.02.004
Butcher, D., Cytrynbaum, C., Turinsky, A., Siu, M., Inbar-Feigenberg, M., Mendoza-Londono, R., et al. (2017). CHARGE and Kabuki Syndromes: gene-Specific DNA Methylation Signatures Identify Epigenetic Mechanisms Linking These Clinically Overlapping Conditions. Am. J. Hum. Genet. 100, 773–788. doi: 10.1016/j.ajhg.2017.04.004
Chang, S., Yim, S., and Park, H. (2019). The cancer driver genes IDH1/2, JARID1C/KDM5C, and UTX/KDM6A: crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Exp. Mol. Med. 51, 1–17. doi: 10.1038/s12276-019-0230-6
Chen, Y., Wang, M., Chen, D., Wang, J., and Kang, N. (2016). Chromatin remodeling enzyme CHD7 is necessary for osteogenesis of human mesenchymal stem cells. Biochem. Biophy. Res. Commun. 478, 1588–1593. doi: 10.1016/j.bbrc.2016.08.161
Clapier, C., Iwasa, J., Cairns, B., and Peterson, C. (2017). Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 18, 407–422. doi: 10.1038/nrm.2017.26
Corsten-Janssen, N., and Scambler, P. (2017). Clinical and molecular effects of CHD7 in the heart. Am. J. Med. Genet. C Semin. Med. Genet. 175, 487–495. doi: 10.1002/ajmg.c.31590
Dann, G., Liszczak, G., Bagert, J., Müller, M., Nguyen, U., Wojcik, F., et al. (2017). ISWI chromatin remodellers sense nucleosome modifications to determine substrate preference. Nature 548, 607–611. doi: 10.1038/nature23671
de Dieuleveult, M., Yen, K., Hmitou, I., Depaux, A., Boussouar, F., and Dargham, D. Bou, et al. (2016). Genome-wide nucleosome specificity and function of chromatin remodellers in ES cells. Nature 530, 113–116. doi: 10.1038/nature16505
Doi, T., Ogata, T., Yamauchi, J., Sawada, Y., Tanaka, S., and Nagao, M. (2017). Chd7 Collaborates with Sox2 to Regulate Activation of Oligodendrocyte Precursor Cells after Spinal Cord Injury. J. Neurosci. 37, 10290–10309. doi: 10.1523/jneurosci.1109-17.2017
Dorighi, K., and Tamkun, J. (2013). The trithorax group proteins Kismet and ASH1 promote H3K36 dimethylation to counteract Polycomb group repression in Drosophila. Development 140, 4182–4192. doi: 10.1242/dev.095786
Egan, C., Nyman, U., Skotte, J., Streubel, G., Turner, S., O’Connell, D., et al. (2013). CHD5 is required for neurogenesis and has a dual role in facilitating gene expression and polycomb gene repression. Dev. Cell 26, 223–236. doi: 10.1016/j.devcel.2013.07.008
Ellingford, R., Panasiuk, M., de Meritens, E., Shaunak, R., Naybour, L., Browne, L., et al. (2021). Cell-type-specific synaptic imbalance and disrupted homeostatic plasticity in cortical circuits of ASD-associated Chd8 haploinsufficient mice. Mol. Psychiatry. doi: 10.1038/s41380-021-01070-9 [Online ahead of print].
Engelen, E., Akinci, U., Bryne, J., Hou, J., Gontan, C., Moen, M., et al. (2011). Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat. Genet. 43, 607–611. doi: 10.1038/ng.825
Farnung, L., Vos, S., Wigge, C., and Cramer, P. (2017). Nucleosome-Chd1 structure and implications for chromatin remodelling. Nature 550, 539–542. doi: 10.1038/nature24046
Feng, W., Kawauchi, D., Körkel-Qu, H., Deng, H., Serger, E., Sieber, L., et al. (2017). Chd7 is indispensable for mammalian brain development through activation of a neuronal differentiation programme. Nat. Commun. 8:14758. doi: 10.1038/ncomms14758
Feng, W., Khan, M., Bellvis, P., Zhu, Z., Bernhardt, O., Herold-Mende, C., et al. (2013). The chromatin remodeler CHD7 regulates adult neurogenesis via activation of SoxC transcription factors. Cell Stem Cell 13, 62–72. doi: 10.1016/j.stem.2013.05.002
Flanagan, J., Mi, L., Chruszcz, M., Cymborowski, M., Clines, K., Kim, Y., et al. (2005). Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature 438, 1181–1185. doi: 10.1038/nature04290
Fujita, K., Ogawa, R., and Ito, K. (2016). CHD7, Oct3/4, Sox2, and Nanog control FoxD3 expression during mouse neural crest-derived stem cell formation. FEBS J. 283, 3791–3806. doi: 10.1111/febs.13843
Gage, P., Hurd, E., and Martin, D. (2015). Mouse Models for the Dissection of CHD7 Functions in Eye Development and the Molecular Basis for Ocular Defects in CHARGE Syndrome. Invest. Ophthalmol. Vis. Sci. 56, 7923–7930. doi: 10.1167/iovs.15-18069
Gaspar-Maia, A., Alajem, A., Polesso, F., Sridharan, R., Mason, M., Heidersbach, A., et al. (2009). Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature 460, 863–868. doi: 10.1038/nature08212
Ghaoui, R., Cooper, S., Lek, M., Jones, K., Corbett, A., Reddel, S., et al. (2015). Use of Whole-Exome Sequencing for Diagnosis of Limb-Girdle Muscular Dystrophy: outcomes and Lessons Learned. JAMA Neurol. 72, 1424–1432. doi: 10.1001/jamaneurol.2015.2274
Goodman, J., and Bonni, A. (2019). Regulation of neuronal connectivity in the mammalian brain by chromatin remodeling. Curr. Opin. Neurobiol. 59, 59–68. doi: 10.1016/j.conb.2019.04.010
Harada, A., Okada, S., Konno, D., Odawara, J., Yoshimi, T., Yoshimura, S., et al. (2012). Chd2 interacts with H3.3 to determine myogenic cell fate. EMBO J. 31, 2994–3007. doi: 10.1038/emboj.2012.136
Hata, T., Rajabi, H., Takahashi, H., Yasumizu, Y., Li, W., Jin, C., et al. (2019). MUC1-C Activates the NuRD Complex to Drive Dedifferentiation of Triple-Negative Breast Cancer Cells. Cancer Res. 79, 5711–5722. doi: 10.1158/0008-5472.can-19-1034
Hayashi, M., Maehara, K., Harada, A., Semba, Y., Kudo, K., Takahashi, H., et al. (2016). Chd5 Regulates MuERV-L/MERVL Expression in Mouse Embryonic Stem Cells Via H3K27me3 Modification and Histone H3.1/H3.2. J. Cell. Biochem. 117, 780–792. doi: 10.1002/jcb.25368
He, D., Marie, C., Zhao, C., Kim, B., Wang, J., Deng, Y., et al. (2016). Chd7 cooperates with Sox10 and regulates the onset of CNS myelination and remyelination. Nat. Neurosci. 19, 678–689. doi: 10.1038/nn.4258
Higashi, M., Kolla, V., Iyer, R., Naraparaju, K., Zhuang, T., Kolla, S., et al. (2015). Retinoic acid-induced CHD5 upregulation and neuronal differentiation of neuroblastoma. Mol. Cancer 14:150. doi: 10.1186/s12943-015-0425-y
Hsu, J., Huang, H., Lee, C., Choudhuri, A., Wilson, N., Abraham, B., et al. (2020). CHD7 and Runx1 interaction provides a braking mechanism for hematopoietic differentiation. Proc. Natl. Acad. Sci. U. S. A. 117, 23626–23635. doi: 10.1073/pnas.2003228117
Hurley, S., Mohan, C., Suetterlin, P., Ellingford, R., Riegman, K., Ellegood, J., et al. (2021). Distinct, dosage-sensitive requirements for the autism-associated factor CHD8 during cortical development. Mol. Autism 12:16. doi: 10.1186/s13229-020-00409-3
Issekutz, K., Graham, J., Prasad, C., Smith, I., and Blake, K. (2005). An epidemiological analysis of CHARGE syndrome: preliminary results from a Canadian study. Am. J. Med. Genet. A 133A, 309–317. doi: 10.1002/ajmg.a.30560
Jamadagni, P., Breuer, M., Schmeisser, K., Cardinal, T., Kassa, B., Parker, J., et al. (2021). Chromatin remodeller CHD7 is required for GABAergic neuron development by promoting PAQR3 expression. EMBO Rep. 22:e50958. doi: 10.15252/embr.202050958
Jones, K., Sarić, N., Russell, J., Andoniadou, C., Scambler, P., and Basson, M. (2015). CHD7 maintains neural stem cell quiescence and prevents premature stem cell depletion in the adult hippocampus. Stem Cells 33, 196–210. doi: 10.1002/stem.1822
Kim, H., Kurth, I., Lan, F., Meliciani, I., Wenzel, W., Eom, S., et al. (2008). Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 83, 511–519. doi: 10.1016/j.ajhg.2008.09.005
Koh, F., Lizama, C., Wong, P., Hawkins, J., Zovein, A., and Ramalho-Santos, M. (2015). Emergence of hematopoietic stem and progenitor cells involves a Chd1-dependent increase in total nascent transcription. Proc. Natl. Acad. Sci. U. S. A. 112, E1734–E1743. doi: 10.1073/pnas.1424850112
Kolla, V., Zhuang, T., Higashi, M., Naraparaju, K., and Brodeur, G. (2014). Role of CHD5 in human cancers: 10 years later. Cancer Res. 74, 652–658. doi: 10.1158/0008-5472.can-13-3056
Layman, W., McEwen, D., Beyer, L., Lalani, S., Fernbach, S., Oh, E., et al. (2009). Defects in neural stem cell proliferation and olfaction in Chd7 deficient mice indicate a mechanism for hyposmia in human CHARGE syndrome. Hum. Mol. Genet. 18, 1909–1923. doi: 10.1093/hmg/ddp112
Li, J., Xu, C., Lee, H., Ren, S., Zi, X., Zhang, Z., et al. (2020). A genomic and epigenomic atlas of prostate cancer in Asian populations. Nature 580, 93–99. doi: 10.1038/s41586-020-2135-x
Lin, J., Lehmann, L., Bonora, G., Sridharan, R., Vashisht, A., Tran, N., et al. (2011). Mediator coordinates PIC assembly with recruitment of CHD1. Genes Dev. 25, 2198–2209. doi: 10.1101/gad.17554711
Link, S., Spitzer, R., Sana, M., Torrado, M., Völker-Albert, M., Keilhauer, E., et al. (2018). PWWP2A binds distinct chromatin moieties and interacts with an MTA1-specific core NuRD complex. Nat. Commun. 9:4300. doi: 10.1038/s41467-018-06665-5
Liu, C., Li, Q., Xiao, Q., Gong, P., and Kang, N. (2020). CHD7 Regulates Osteogenic Differentiation of Human Dental Follicle Cells via PTH1R Signaling. Stem Cells Int. 2020:8882857. doi: 10.1155/2020/8882857
Liu, Z., Su, D., Qi, X., and Ma, J. (2018). MiR-500a-5p promotes glioblastoma cell proliferation, migration and invasion by targeting chromodomain helicase DNA binding protein 5. Mol. Med. Rep. 18, 2689–2696. doi: 10.3892/mmr.2018.9259
Mashtalir, N., D’Avino, A., Michel, B., Luo, J., Pan, J., Otto, J., et al. (2018). Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 175, 1272–1288.e20. doi: 10.1016/j.cell.2018.09.032
Mills, A. (2017). The Chromodomain Helicase DNA-Binding Chromatin Remodelers: family Traits that Protect from and Promote Cancer. Cold Spring Harb. Perspect. Med. 7:a026450. doi: 10.1101/cshperspect.a026450
Mohd-Sarip, A., Teeuwssen, M., Bot, A., De Herdt, M., Willems, S., Baatenburg de Jong, R., et al. (2017). DOC1-Dependent Recruitment of NURD Reveals Antagonism with SWI/SNF during Epithelial-Mesenchymal Transition in Oral Cancer Cells. Cell Rep. 20, 61–75. doi: 10.1016/j.celrep.2017.06.020
Nieto-Estevez, V., and Hsieh, J. (2018). CHD2: one Gene, Many Roles. Neuron 100, 1014–1016. doi: 10.1016/j.neuron.2018.11.036
Nishiyama, M., Oshikawa, K., Tsukada, Y., Nakagawa, T., Iemura, S., Natsume, T., et al. (2009). CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat. Cell Biol. 11, 172–182. doi: 10.1038/ncb1831
Noh, K., Maze, I., Zhao, D., Xiang, B., Wenderski, W., Lewis, P., et al. (2015). ATRX tolerates activity-dependent histone H3 methyl/phos switching to maintain repetitive element silencing in neurons. Proc. Natl. Acad. Sci. U. S. A. 112, 6820–6827. doi: 10.1073/pnas.1411258112
Novillo, A., Fernández-Santander, A., Gaibar, M., Galán, M., Romero-Lorca, A., El Abdellaoui-Soussi, F., et al. (2021). Role of Chromodomain-Helicase-DNA-Binding Protein 4 (CHD4) in Breast Cancer. Front. Oncol. 11:633233. doi: 10.3389/fonc.2021.633233
Ohta, S., Yaguchi, T., Okuno, H., Chneiweiss, H., Kawakami, Y., and Okano, H. (2016). CHD7 promotes proliferation of neural stem cells mediated by MIF. Mol. Brain 9:96. doi: 10.1186/s13041-016-0275-6
Pagon, R., Graham, J., Zonana, J., and Yong, S. (1981). Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J. Pediatr. 99, 223–227. doi: 10.1016/s0022-3476(81)80454-4
Parenti, I., Lehalle, D., Nava, C., Torti, E., Leitão, E., Person, R., et al. (2021). Missense and truncating variants in CHD5 in a dominant neurodevelopmental disorder with intellectual disability, behavioral disturbances, and epilepsy. Hum. Genet. 140, 1109–1120. doi: 10.1007/s00439-021-02283-2
Percharde, M., Bulut-Karslioglu, A., and Ramalho-Santos, M. (2017). Hypertranscription in Development, Stem Cells, and Regeneration. Dev. Cell 40, 9–21. doi: 10.1016/j.devcel.2016.11.010
Pierson, T., Otero, M., Grand, K., Choi, A., Graham, J., Young, J., et al. (2019). The NuRD complex and macrocephaly associated neurodevelopmental disorders. Am. J. Med. Genet. C Semin. Med. Genet. 181, 548–556. doi: 10.1002/ajmg.c.31752
Platt, J., Kent, N., Kimmel, A., and Harwood, A. (2017). DictyosteliumRegulation of nucleosome positioning by a CHD Type III chromatin remodeler and its relationship to developmental gene expression in. Genome Res. 27, 591–600. doi: 10.1101/gr.216309.116
Puc, J., and Rosenfeld, M. (2011). SOX2 and CHD7 cooperatively regulate human disease genes. Nat. Genet. 43, 505–506. doi: 10.1038/ng.843
Rother, M., Pellegrino, S., Smith, R., Gatti, M., Meisenberg, C., Wiegant, W., et al. (2020). CHD7 and 53BP1 regulate distinct pathways for the re-ligation of DNA double-strand breaks. Nat. Commun. 11:5775. doi: 10.1038/s41467-020-19502-5
Schnetz, M., Bartels, C., Shastri, K., Balasubramanian, D., Zentner, G., Balaji, R., et al. (2009). Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res. 19, 590–601. doi: 10.1101/gr.086983.108
Schnetz, M., Handoko, L., Akhtar-Zaidi, B., Bartels, C., Pereira, C., Fisher, A., et al. (2010). CHD7 targets active gene enhancer elements to modulate ES cell-specific gene expression. PLoS Genet. 6:e1001023. doi: 10.1371/journal.pgen.1001023
Semba, Y., Harada, A., Maehara, K., Oki, S., Meno, C., Ueda, J., et al. (2017). Chd2 regulates chromatin for proper gene expression toward differentiation in mouse embryonic stem cells. Nucleic Acids Res. 45, 8758–8772. doi: 10.1093/nar/gkx475
Shieh, C., Jones, N., Vanle, B., Au, M., Huang, A., Silva, A., et al. (2020). GATAD2B-associated neurodevelopmental disorder (GAND): clinical and molecular insights into a NuRD-related disorder. Genet. Med. 22, 878–888. doi: 10.1038/s41436-019-0747-z
Shur, I., Socher, R., and Benayahu, D. (2006a). In vivo association of CReMM/CHD9 with promoters in osteogenic cells. J. Cell. Physiol. 207, 374–378. doi: 10.1002/jcp.20586
Shur, I., Solomon, R., and Benayahu, D. (2006b). Dynamic interactions of chromatin-related mesenchymal modulator, a chromodomain helicase-DNA-binding protein, with promoters in osteoprogenitors. Stem Cells 24, 1288–1293. doi: 10.1634/stemcells.2005-0300
Sparmann, A., Xie, Y., Verhoeven, E., Vermeulen, M., Lancini, C., Gargiulo, G., et al. (2013). The chromodomain helicase Chd4 is required for Polycomb-mediated inhibition of astroglial differentiation. EMBO J. 32, 1598–1612. doi: 10.1038/emboj.2013.93
Sperlazza, J., Rahmani, M., Beckta, J., Aust, M., Hawkins, E., Wang, S., et al. (2015). Depletion of the chromatin remodeler CHD4 sensitizes AML blasts to genotoxic agents and reduces tumor formation. Blood 126, 1462–1472. doi: 10.1182/blood-2015-03-631606
Sperry, E., Hurd, E., Durham, M., Reamer, E., Stein, A., and Martin, D. (2014). The chromatin remodeling protein CHD7, mutated in CHARGE syndrome, is necessary for proper craniofacial and tracheal development. Dev. Dyn. 243, 1055–1066. doi: 10.1002/dvdy.24156
Stamou, M., Ng, S., Brand, H., Wang, H., Plummer, L., Best, L., et al. (2020). A Balanced Translocation in Kallmann Syndrome Implicates a Long Noncoding RNA, RMST, as a GnRH Neuronal Regulator. J. Clin. Endocrinol. Metab. 105, e231–e244. doi: 10.1210/clinem/dgz011
Sugathan, A., Biagioli, M., Golzio, C., Erdin, S., Blumenthal, I., Manavalan, P., et al. (2014). CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. U. S. A. 111, E4468–E4477. doi: 10.1073/pnas.1405266111
Suls, A., Jaehn, J., Kecskés, A., Weber, Y., Weckhuysen, S., Craiu, D., et al. (2013). De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am. J. Hum. Genet. 93, 967–975. doi: 10.1016/j.ajhg.2013.09.017
Suzuki, S., Nozawa, Y., Tsukamoto, S., Kaneko, T., Manabe, I., Imai, H., et al. (2015). CHD1 acts via the Hmgpi pathway to regulate mouse early embryogenesis. Development 142, 2375–2384. doi: 10.1242/dev.120493
Tu, Z., Wang, C., Davis, A., Hu, M., Zhao, C., Xin, M., et al. (2021). Chromatin remodeler CHD8 governs hematopoietic stem/progenitor survival by regulating ATM-mediated P53 protein stability. Blood. doi: 10.1182/blood.2020009997 [Online ahead of print].
Ufartes, R., Schwenty-Lara, J., Freese, L., Neuhofer, C., Möller, J., Wehner, P., et al. (2018). Sema3a plays a role in the pathogenesis of CHARGE syndrome. Hum. Mol. Genet. 27, 1343–1352. doi: 10.1093/hmg/ddy045
Van Nostrand, J., Brady, C., Jung, H., Fuentes, D., Kozak, M., Johnson, T., et al. (2014). Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature 514, 228–232. doi: 10.1038/nature13585
Vissers, L., van Ravenswaaij, C., Admiraal, R., Hurst, J., de Vries, B., Janssen, I., et al. (2004). Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 36, 955–957. doi: 10.1038/ng1407
Wang, Y., Chen, Y., Bao, L., Zhang, B., Wang, J., Kumar, A., et al. (2020). CHD4 Promotes Breast Cancer Progression as a Coactivator of Hypoxia-Inducible Factors. Cancer Res. 80, 3880–3891. doi: 10.1158/0008-5472.can-20-1049
Weiss, K., Lazar, H., Kurolap, A., Martinez, A., Paperna, T., Cohen, L., et al. (2020). The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis. Genet. Med. 22, 389–397. doi: 10.1038/s41436-019-0612-0
Whittaker, D., Riegman, K., Kasah, S., Mohan, C., Yu, T., Pijuan-Sala, B., et al. (2017). The chromatin remodeling factor CHD7 controls cerebellar development by regulating reelin expression. J. Clin. Investig. 127, 874–887. doi: 10.1172/jci83408
Xie, C., Li, Z., Sun, H., Wang, F., Sun, Y., Zhao, W., et al. (2015). Mutual regulation between CHD5 and EZH2 in hepatocellular carcinoma. Oncotarget 6, 40940–40952. doi: 10.18632/oncotarget.5724
Yan, S., Thienthanasit, R., Chen, D., Engelen, E., Brühl, J., Crossman, D., et al. (2020). CHD7 regulates cardiovascular development through ATP-dependent and -independent activities. Proc. Natl. Acad. Sci. U. S. A. 117, 28847–28858. doi: 10.1073/pnas.2005222117
Yang, P., Oldfield, A., Kim, T., Yang, A., Yang, J., and Ho, J. (2017). Integrative analysis identifies co-dependent gene expression regulation of BRG1 and CHD7 at distal regulatory sites in embryonic stem cells. Bioinformatics 33, 1916–1920. doi: 10.1093/bioinformatics/btx092
Zhang, Z., Zhou, C., Li, X., Barnes, S., Deng, S., Hoover, E., et al. (2020). Loss of CHD1 Promotes Heterogeneous Mechanisms of Resistance to AR-Targeted Therapy via Chromatin Dysregulation. Cancer Cell 37, 584–598.e11. doi: 10.1016/j.ccell.2020.03.001
Zhao, D., Cai, L., Lu, X., Liang, X., Li, J., Chen, P., et al. (2020). Chromatin Regulator CHD1 Remodels the Immunosuppressive Tumor Microenvironment in PTEN-Deficient Prostate Cancer. Cancer Discov. 10, 1374–1387. doi: 10.1158/2159-8290.cd-19-1352
Keywords: epigenetic, chromodomain helicase DNA-binding protein, chromatin remodeling, histone modification, CHARGE syndrome
Citation: Liu C, Kang N, Guo Y and Gong P (2021) Advances in Chromodomain Helicase DNA-Binding (CHD) Proteins Regulating Stem Cell Differentiation and Human Diseases. Front. Cell Dev. Biol. 9:710203. doi: 10.3389/fcell.2021.710203
Received: 15 May 2021; Accepted: 29 July 2021;
Published: 20 September 2021.
Edited by:
Ce Dou, Army Medical University, ChinaReviewed by:
Fernando Moreira Simabuco, State University of Campinas, BrazilCopyright © 2021 Liu, Kang, Guo and Gong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuchen Guo, ZGVudC5ndW9AcXEuY29t; Ping Gong, ZGVudGlzdGdvbmdAaG90bWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.