 Ying Shi
Ying Shi Xinming Chen
Xinming Chen Chunling Huang
Chunling Huang Carol Pollock
Carol Pollock- 1Department of Nephrology, School of Medicine, Stanford University, Palo Alto, CA, United States
- 2Kolling Institute of Medical Research, Sydney Medical School, The University of Sydney, Sydney, NSW, Australia
Chronic kidney disease (CKD) is the end result of a plethora of renal insults, including repeated episodes of acute or toxic kidney injury, glomerular, or diabetic kidney disease. It affects a large number of the population worldwide, resulting in significant personal morbidity and mortality and economic cost to the community. Hence it is appropriate to focus on treatment strategies that interrupt the development of kidney fibrosis, the end result of all forms of CKD, in addition to upstream factors that may be specific to certain diseases. However, the current clinical approach to prevent or manage renal fibrosis remains unsatisfactory. The rising importance of receptor-interacting serine/threonine-protein kinase (RIPK) 3 in the inflammatory response and TGF-β1 signaling is increasingly recognized. We discuss here the biological functions of RIPK3 and its role in the development of renal fibrosis.
Introduction
Chronic kidney disease (CKD) is defined as a loss of glomerular filtration and or proteinuria, persisting for at least 3 months or structural abnormalities in the kidney. In the majority of cases, CKD eventually leads to end-stage kidney disease (ESKD) requiring renal replacement therapy or death will ensue. CKD affects a large proportion of the population and considerably more than is widely appreciated by the general public. In 2016–2017, 1.8 million hospitalizations in Australia were associated with CKD, which accounts for 16% of all hospital admissions in Australia. Of those hospitalizations, 80% were for regular dialysis (AIHW, 2019). Having CKD increases the length of stay, cost, and complications of non-CKD related hospital admissions. In the United States, the overall prevalence of CKD in the general adult population was 14.8% in 2011–2014 (United States Renal Data System, 2018). Regardless of the cause of the initial renal injury, progressive renal fibrosis is common to all forms of CKD, characterized pathologically by extracellular matrix (ECM) accumulation, myofibroblast activation, and inflammatory cell infiltration (Lee and Kalluri, 2010; Carew et al., 2012).
To date, inhibition of the renin-angiotensin-aldosterone system (RAAS) is the crucial strategy utilized to slow deterioration of renal functional decline. However, this influences intrarenal and extrarenal hemodynamics, and only secondarily reduces the development of renal fibrosis. It is primarily beneficial in patients with proteinuric renal disease, and at best, it delays the time to ESKD, leading to renal replacement therapy or death, by a factor of months. More recently, sodium-glucose linked transport inhibitors have been shown to reduce the development of end-stage kidney disease in patients with diabetic kidney disease (Ingelfinger and Rosen, 2019), and a recent trial in both diabetic and non-diabetic CKD was prematurely terminated in light of positive results in favor of the SGLT2 inhibitor (ClinicalTrials.gov, 2020). However, a treatment gap remains, and novel therapies directed toward reducing the ultimate fibrotic response in the kidney are urgently needed to arrest the progression of CKD and improve the outcome of patients.
Transforming Growth Factor BETA-1 (TGF-β1)
TGF-β is the prototype of a family of secreted polypeptide growth factors. Three isoforms of TGF-β have been identified in mammals, including TGF-β1, TGF-β2 and TGF-β3 (Yu et al., 2003). All TGF-βs are synthesized as homodimeric proproteins together with the latency-associated peptide (LAP), which binds to the TGF-β homodimer to promote the formation of the latent TGF-β binding protein (LTBP) (Robertson et al., 2015). The synthesized complex, consisting of TGF-β dimer, LAP dimer, and LTBP, remains inactive and stored in the ECM (Hinz, 2015). LTBP serves as a localizer to interact with the ECM (Annes et al., 2003). LAP inhibits TGF-β activity by preventing TGF-β binding to its receptors (Annes et al., 2003; Hinz, 2015). This mechanism controls free and, therefore, active TGFβ tissue levels. To cleave the TGF-β complex into the active component and release active TGF-β, one or more of a wide range of proteases, including plasmin, matrix metalloproteinase (MMP) 2, and MMP9, thrombospondin, integrins, and the cationic independent mannose 6 phosphate receptor, are needed (Annes et al., 2003).
It is well accepted that overexpression of active TGF-β1 induces a fibrotic response in multiple organs, including the kidney (Sanderson et al., 1995). TGF-β1 is a well-characterized key mediator in the pathogenesis of tubulointerstitial fibrosis, due to its direct and indirect effect on various cells types (Roberts, 1998; Wang et al., 2005; Bottinger, 2007). The direct action of TGF-β1 includes the transition of cells to a fibroblastic phenotype and synthesis of profibrotic proteins, such as collagens and fibronectin (Border et al., 1990; Haberstroh et al., 1993; Wilson et al., 1993). TGF-β1 also facilitates an indirect fibrotic response, via accelerating apoptosis of resident healthy cells and promoting resident and infiltrating cells to increase ECM deposition (Lebrin et al., 2005; Das et al., 2014; Mack and Yanagita, 2015). Inhibiting TGF-β1 in animal models of kidney disease attenuates fibroblast activation and ECM accumulation (Moon et al., 2006; Russo et al., 2007; Murphy et al., 2012; McGaraughty et al., 2017).
The central signal transduction in response to TGF-β1 is mediated by two specific receptors, TGF-β type II receptor (TGFβRII) and the TGF-β type I receptor (TGFβRI). TGF-β1 firstly binds with TGFβRII in an active form (homodimers) and recruits the low-affinity receptor (TGFβRI) by the ligand-bound high-affinity receptor (TGFβRII) (Groppe et al., 2008). The activation of TGFβRI initiates receptor signaling (Xu et al., 2012a) and phosphorylates the substrates, the Smad proteins. Specifically, TGF-β1 signaling stimulates receptor-regulated Smad (R-Smad) phosphorylation. This is followed by translocation of R-Smads and the common mediator Smad (Co-Smad) complexes in the nucleus to regulate gene transcription (Moustakas et al., 2001). By contrast, inhibitory Smads (I-Smads) antagonize the activity of the R-Smads by preventing phosphorylation of R-Smads (Hill, 1999).
TGF-β1-SMAD Pathway
Smads separate into different classes with regards to their functions: two TGF-β R-Smads (Smad2 and Smad3), three bone morphogenetic protein (BMP) R-Smads (Smad1, Smad5, and Smad8), one Co-Smad (Smad4) and two I-Smad (Smad6 and Smad7) (Hill, 1999; Heldin and Moustakas, 2012).
R-SMADS
Smad2 and Smad3 are extensively studied in the TGF-β1 facilitated fibrotic response using various animal models and in human kidney disease, including diabetic (Isono et al., 2002; Fujimoto et al., 2003; Li et al., 2004; Chung et al., 2010; Chen et al., 2011) and obstructive nephropathy (Terada et al., 2002; Lan et al., 2003; Sato et al., 2003; Huang et al., 2008a; Chung et al., 2009), remnant kidney disease (Hou et al., 2005; Yang et al., 2010), hypertensive nephropathy (Wang et al., 2006), drug-associated nephropathy, and immunologically mediated glomerulonephritis (Ka et al., 2007; Huang et al., 2008b).
TGF-β1/Smad3 signaling mediates transcription of multiple downstream genes, such as the collagen chains ColIa1, ColIa2, ColIIIa1, ColaVa2, ColVIa1, and ColVIa3, and tissue inhibitor of metalloproteinases (TIMP)-1 (Verrecchia et al., 2001). The deletion of Smad3 in mice suppresses fibrosis in rodent models of kidney disease (Fujimoto et al., 2003; Sato et al., 2003; Zhou et al., 2010).
Relative to Smad3, the function of Smad2 in renal fibrosis is not fully elucidated. Because of the unavailability of Smad2 knock out (KO) mice, conditional kidney tubular epithelial cells Smad2 KO mice were generated by crossing the Smad2 floxed mouse with the kidney-specific promoter (Cadherin 16)-driven Cre transgenic mouse (Shao et al., 2002). Unexpectedly, deletion of Smad2 in tubular cells significantly enhances fibrosis, with an associated elevated Smad3 signaling in the UUO mouse model (Meng et al., 2010). Similarly, Smad2-/- fibroblasts have an increased fibrotic response (Meng et al., 2010). Additional evidence has shown that Smad3, but not Smad2, mediates fibrotic process (Wang et al., 2006; Yang et al., 2009, 2010; Chung et al., 2010; Zhou et al., 2010). Hence Smad2 and Smad3 may have distinct roles in mediating the fibrosis upon exposure to TGF-β1.
Among the R-Smads, BMP R-Smads (1, 5, 8) mediate the development of kidney and renal cell cancer (Oxburgh and Robertson, 2002; Blank et al., 2008; Markic et al., 2010). The BMP-7-Smad1/5/8 pathway has been shown to accelerate ECM deposition in the kidneys of unilateral ureteral obstruction (UUO) rats (Cao et al., 2015). The activin receptor-like kinase (ALK)-1/Smad1/5 pathway may influence ECM protein expression in several cell types, such as rat myoblasts, hepatocytes, and human chondrocytes (Munoz-Felix et al., 2013). However, the role of BMP R-Smads in fibrotic disorders remains largely unknown.
CO-SMAD (SMAD4)
Smad4 promotes TGF-β1 signaling by dimerizing with R-Smads and facilitating nuclear translocation (Massague and Wotton, 2000; Gomez-Puerto et al., 2019). Deleting Smad4 from renal tubular cells alleviates renal fibrosis in a mouse model of UUO by suppressing Smad3 function (Meng et al., 2012). In mesangial cells, the loss of Smad4 inhibits TGF-β1 induced ECM accumulation (Tsuchida et al., 2003).
I-SMADS
Smad 6 and Smad7 are inhibitory mediators in the TGF-β1 signaling pathway. They provide a negative feedback loop through multiple mechanisms, including competing with R-Smads in activating the receptors by associating directly with TGFβRI (Hanyu et al., 2001; Nakayama et al., 2001), indirectly affecting the activity of TGFβRI by cooperation with BMPs (Murakami et al., 2003; Yan et al., 2009), interference in the formation of R-Smad/Co-Smad complex (Hata et al., 1998; Yan et al., 2016) and abolishing transcription in the nucleus (Lin et al., 2003; Zhang et al., 2007).
The deletion of Smad7 accelerates fibrogenesis in a number of mouse models, including UUO (Chung et al., 2009), diabetic (Chen et al., 2011), and hypertensive nephropathy (Liu et al., 2013). However, the importance of Smad6 in renal fibrogenesis is unclear.
Non-SMAD Pathways
TGF-β1 also independently and directly activates other pathways, such as Ras/Raf/extracellular-signal-regulated kinase (ERK)/ mitogen-activated protein kinase (MAPK) pathways, c-Jun N-terminal kinase (JNK), p38 MAPK signaling and Rho-like GTPase signaling pathways (Loeffler and Wolf, 2014).
TGF-β1 increases phosphorylation of tyrosine residues on TGFRs (I and II) and recruits ERK through Ras, Raf, and their downstream MAPK cascades. Specifically, ERK regulates target gene transcription through its downstream transcription factors in conjunction with Smads to control epithelial-mesenchymal transition (EMT) (Lee et al., 2007). ERK also regulates the activity of R-Smads, including Smad1, Smad2, and Smad3 (Kretzschmar et al., 1997, 1999; Funaba et al., 2002; Matsuura et al., 2005). Moreover, ERK is involved in the autoinduction of TGF-β1 via distinct transcriptional and translational mechanisms in tubular epithelial cells (Zhang et al., 2006). These studies suggest a dominant role of ERK in the non-Smad mediated transduction of TGF-β1.
The Rho-like GTPases, including RhoA, Rac, and Cdc42, play crucial roles in controlling dynamic cytoskeletal organization, cell motility, and gene expression through a variety of effectors (Jaffe and Hall, 2005). In addition to MAPK pathways, RhoA is a vital regulator, which can be activated by TGF-β1 via either Smad-dependent or independent pathways to promote stress fiber formation during EMT (Bhowmick et al., 2001a; Edlund et al., 2002).
JNK and p38 MAPK pathways are the best characterized non-Smad pathways involved in renal fibrosis. TGF-β1 can rapidly activate JNK and p38 MAPK via MAPK kinase (MKK) 4 and MKK 3/6, respectively (Frey and Mulder, 1997; Engel et al., 1999; Hanafusa et al., 1999; Hocevar et al., 1999; Sano et al., 1999; Bhowmick et al., 2001b; Yu et al., 2002). The activated JNK/p38 conjuncts with Smad2/3 to regulate apoptosis and EMT by controlling the activities of downstream transcription factors (Shibuya et al., 1998; Liao et al., 2001; Bakin et al., 2002; Yu et al., 2002; Edlund et al., 2003; Yamashita et al., 2008; Zhang, 2009). The phosphorylated JNK also regulates Smad 3 activity directly (Zhang, 2009; Grynberg et al., 2017).
Receptor-Interacting Serine/Threonine-Protein Kinase (RIPK) 3
The RIP kinase family contains seven members, each of which possesses a homologous kinase domain. To date, the functions of RIPK4–7 are poorly understood (Zhang et al., 2010). RIPK2 is a mediator of mucosal immunity. Extensive studies have clarified the importance and physiological roles of RIPK1 and RIPK3 in inflammation and cell death (Christofferson et al., 2014; Newton, 2015).
The RIPK3 gene is located on chromosome 14 in both humans and mice (Kasof et al., 2000; Shlomovitz et al., 2017). This gene encodes a 518 amino acid (aa) protein with a molecular mass of 57 kDa in humans (Sun et al., 1999), whereas it encodes a 486 aa protein of 53 kDa in mice (Pazdernik et al., 1999). RIPK3 is a threonine/serine protein kinase that shares almost 30% identity and 60% with the other two RIPK members, RIPK1 and RIPK2 (Sun et al., 1999; Yu et al., 1999). Compared with RIPK2, RIPK3, and RIPK1 have a unique C-terminal RIP homotypic interaction motif (RHIM) (Sun et al., 1999), which enables homotypic protein interactions (Sun et al., 2002).
To date, several phosphorylation sites of RIPK3 have been identified. The human Ser227 site (Thr231/Ser232 for mouse RIPK3) and Ser199 site (Ser204 in mouse) are particularly crucial for the activation of its downstream substrate in the necroptosis pathway, mixed-lineage kinase domain-like (MLKL) (He et al., 2009; Sun et al., 2012; Chen et al., 2013; McQuade et al., 2013).
RIPK3 in Necroptosis
In response to physiological signals and pathological stimuli, cell death is crucial to maintaining homeostasis. To date, several types of cell death have been identified. Among them, necrosis is a type of cell death characterized by loss of intracellular contents and the triggering of subsequent inflammatory response. For many years, necrosis was considered to be accidental and, therefore, unregulated cell death (Proskuryakov et al., 2003; Festjens et al., 2006; Zong and Thompson, 2006; Vandenabeele et al., 2010). The recognition that necroptosis is programmed necrosis that has provided new insights into necrosis-initiated cell death. Necroptosis is mediated by the dimerization of RIPK1-RIPK3, which forms the necrosome associated with the downstream expression of MLKL (Li et al., 2012; Newton and Manning, 2016; Weinlich et al., 2017). RIPK1 and RIPK3 both possess RHIM domains, with bilateral interaction between RIPK1 and RIPK3 (Li et al., 2012; Mompean et al., 2018). Subsequently, the necrosome facilitates the aggregation of phosphorylated RIPK3 and phosphorylation of MLKL by RIPK3 (Li et al., 2012; Newton and Manning, 2016; Weinlich et al., 2017). The phosphorylated MLKL translocates to the cell membrane and thus promotes necroptosis (Li et al., 2012).
In contrast to the obligatory involvement of RIPK3, RIPK1 is not always required to cause necroptosis. The M45-mutant strain of murine cytomegalovirus (MCMV) infection causes RIPK3 activation in the absence of activation of RIPK1 (Upton et al., 2010, 2012). There is also evidence that the RHIM-containing protein (ICP) 6 protein of herpes simplex virus 1 recruits RIPK3 directly and independent of RIPK1 (Wang et al., 2014b). In addition, RIPK1 may have dual influences on cell death by both promoting necroptosis and protecting cells from death under certain conditions (Filliol et al., 2017).
RIPK3 in Kidney Fibrosis
To date, few studies have investigated the role of RIPK3 in kidney fibrosis, and most of them have not dissected the role of RIPK3 from necroptosis. The majority of these studies used acute injury models where there is known increased necroptosis.
The RIPK1 inhibitor necrostatin-1 reduces renal ischemia and reperfusion injury (Shen et al., 2019) and sepsis-associated acute kidney injury (Dong et al., 2018). Lacking RIPK3 protects kidney tubular injury in the sepsis-induced acute kidney injury (Sureshbabu et al., 2018). The deletion of either RIPK3 or MLKL prevents kidney damage in the oxalate crystal-induced AKI (Mulay et al., 2016) and kidney ischemia-reperfusion injury mouse models (Moerke et al., 2019). However, blockade of MLKL in folic acid-induced AKI (Martin-Sanchez et al., 2017) and 7-day unilateral ureteral obstruction (UUO) models (Imamura et al., 2018) failed to protect against kidney injury. Hence blockade of RIPK1, RIPK3, or MLKL may have differential benefits depending on the model under study.
Necrostatin-1 reduces interstitial fibrosis in a mouse model of UUO (Xiao et al., 2017) by inhibiting necroptosis, associated with lower protein and mRNA expression of RIPK, RIPK3, and MLKL and TGF-β1. In parallel, collagen accumulation and fibroblast activation (Xiao et al., 2017) were reduced. This study showed the integral relationship between necroptosis and TGF-β1 activation leading to renal fibrosis.
RIPK3 in Apoptosis and Inflammation
Under certain conditions, RIPK3 also serves as a pro-apoptosis adaptor, which recruits RIPK1 and Fas-associated protein with death domain (FADD) to activate caspase 8 and thus apoptosis. This effect relies on the involvement of caspase 8 when RIPK3 is inactive, or MLKL is absent (Mandal et al., 2014; Newton et al., 2014). RIPK3 deficient animals develop normally, whereas mice expressing catalytically inactive RIPK3 D161N die around embryonic day 11.5 from increased RIPK1- and caspase-8-dependent apoptotic injury (Newton et al., 2014). Similar effects are observed in a study using compounds that selectively inhibit RIPK3 interaction with caspase 8 independent of pro-necrotic kinase activity and MLKL (Mandal et al., 2014).
Recent studies also identify that RIPK3 is an essential mediator in NOD-, LRR- and pyrin domain-containing protein (NLRP) 3 inflammasome formation (Wang et al., 2014a; Lawlor et al., 2015; Chen et al., 2018; Guo et al., 2019). In LPS-treated mouse bone marrow-derived dendritic cells, activation of the NLRP3 inflammasome was initiated by necroptosis (Kang et al., 2014). In a podocyte cell line, the RIPK3 specific inhibitor GSK’872 blocked both the necroptosis pathway and the NLRP3 inflammasome activation (Guo et al., 2019). These indicate the RIPK3 mediated NLRP3 inflammasome can be dependent of the necroptosis. Specifically, RIPK3–MLKL triggers NLRP3 activation when the activation of caspase 8 is reduced (Lawlor et al., 2015). In this setting, RIPK3 activity is required. RIPK3 can also promote NLRP3 inflammasome independent of the MLKL and RIPK3 kinase activity when caspase 8 is active (Lawlor et al., 2015). Collectively, RIPK3 mediated NLRP3 activation can be in both a necroptosis-independent and -dependent manner, depending on the levels of caspase-8 activity.
RIPK3 and TGF-β1
Necrostatin-1 in a mouse UUO model attenuates IL-1β, TNF-α, and TGF-β1 levels (Xiao et al., 2017). In contrast, another study demonstrated that RIPK3 deficiency in the same UUO model prevents renal fibrosis without altering the mRNA expression of interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and TGF-β1 (Imamura et al., 2018). These conflicting results may indicate that IL-1β, TNF-α, and TGF-β1 are “co-existing” as the downstream cytokines in the RIPK3 signaling. Mature IL-1β, the critical effector of the NLRP3 inflammasome (Jo et al., 2016; Kelley et al., 2019), has been demonstrated to increase TGF-β1 transcription (Lee et al., 2006). As described above, RIPK3 can regulate NLRP3 inflammasome (Lawlor et al., 2015). We, therefore, hypothesize that the “on/off switch” of RIPK3 in regulating TGF-β could be NLRP3 inflammasome activation (Figure 1). The trigger to promote NLRP3 inflammasome activation in RIPK3 signaling remains to be elucidated.
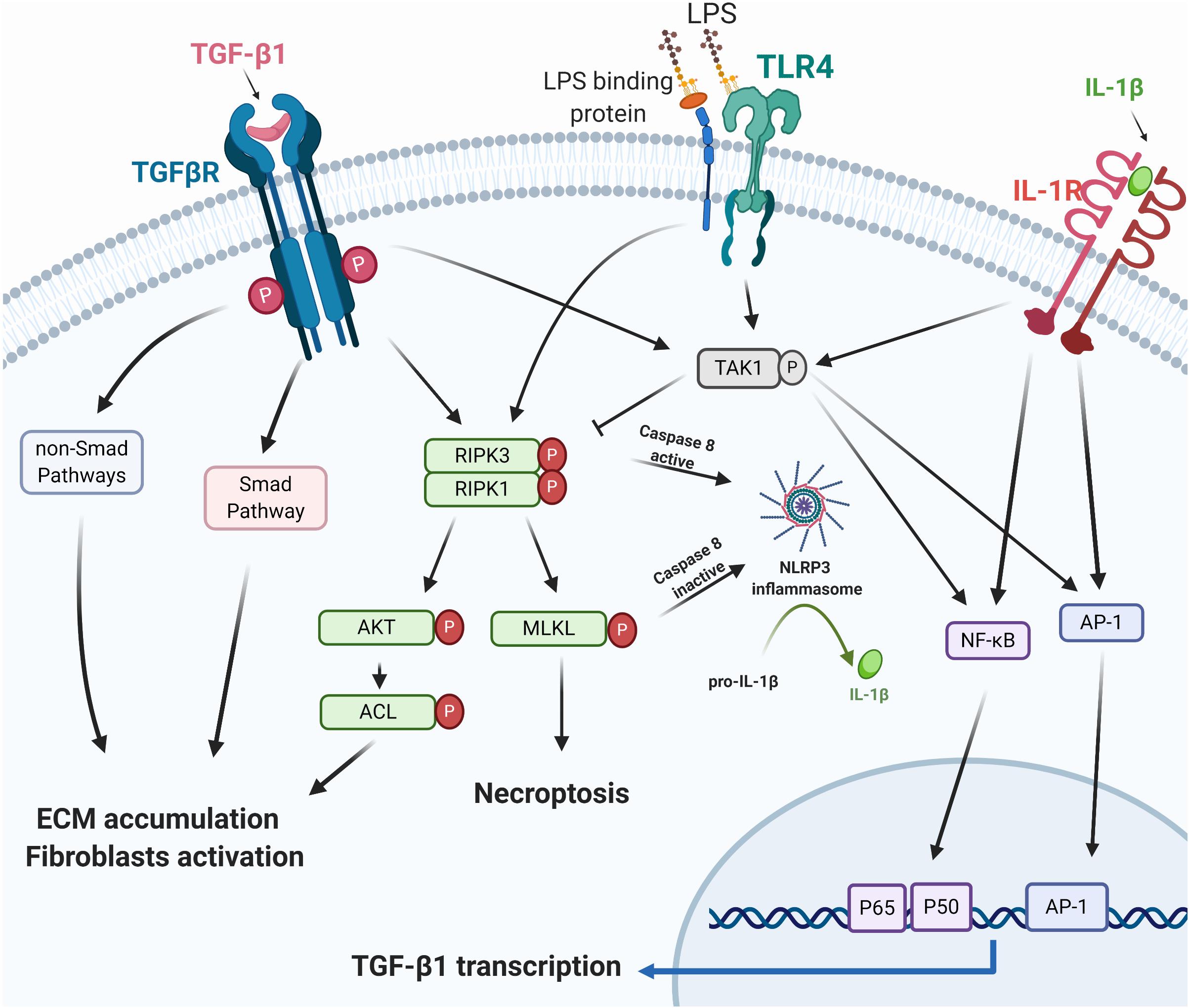
Figure 1. RIPK3 and TGF-β1. TGF-β1 exhibits its biological function via the canonical Smad/non-Smad pathways or TAK1/necrosome/AKT/ACL signaling to mediate ECM accumulation and fibroblast activation. Necroptosis or RIPK3 facilitates NLRP3 inflammasome assembly, triggers mature IL-1β secretion, and promotes the TGF-β1 transcription via the IL-1β regulated AP-1 and NFκB pathway (Lee et al., 2006). IL-1β, TGF-β, and TLR signaling pathways all activate TAK1 and its regulated inflammatory mediators (Kim and Choi, 2012; Fechtner et al., 2017). RIPK3, Receptor-interacting serine/threonine-protein kinase 3; TGF-β1, transforming growth factor beta-1; TAK1, TGF-β-activated kinase 1; AKT, protein kinase B; ACL, ATP citrate lyase; ECM, extracellular matrix; TLR4, toll-like receptor 4; LPS, lipopolysaccharides; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; IL-1β, interleukin-1β; AP-1, activator protein 1; NFκB, nuclear factor-kappa B. Created with BioRender.com.
In vitro studies using NIH 3T3 fibroblasts, RIPK3 targeted siRNA, the RIPK3 inhibitor GSK’872 or necrostatin-1 abolished TGF-β dependent ECM and α- smooth muscle actin (α-SMA) expression (Imamura et al., 2018), suggesting that the necrosome RIPK1/RIPK3 is a downstream regulator of TGF-β in stimulating ECM deposition and fibroblast activation. The necrosome/RIPK3- Protein Kinase B (AKT)-dependent ATP citrate lyase (ACL) pathway has previously been identified as downstream of TGF-β (Imamura et al., 2018).
TGF-β-activated kinase 1 (TAK1), interacts with TGF-β1 and contributes to the development and progression of organ fibrosis through TGF-β1/TAK1/MKK3/p38MAPK, TGF-β1/TAK1/MKK4/JNK, and TGF-β1/TAK1/NFκB pathways (Kim and Choi, 2012; Biesemann et al., 2015; Li et al., 2017; Wu et al., 2017; Zhou et al., 2018; Bao et al., 2019). Few studies of TAK1 on necroptosis have been reported, and these mostly report on RIPK1-dependent cell death. A recent study explored TAK1 regulated endothelial necroptosis in tumor progression and showed that TAK1 deficiency increases necroptosis and RIPK3 expression in endothelial cells in both in vitro and in vivo studies (Yang et al., 2019). Endothelial knockout of RIPK3 or MLKL abolishes the effects of TAK1-deficiency on the enhancement of necroptosis, suggesting an inhibitory role of TAK1 on necroptosis (Yang et al., 2019). TAK1 may, therefore, negatively regulate the necroptosis in the TGF-β1 signaling network (Figure 1).
Implications for Anti-Fibrotic Therapy
TGF-β1-specific, humanized, neutralizing monoclonal antibody added to RAAS inhibitors failed to slow the progression of diabetic nephropathy (Voelker et al., 2017). Therefore, targeting the full spectrum of downstream TGF- β1 signaling to prevent renal disease is unlikely to be fruitful, and the development of blockers of more targeted downstream pathways, such as the RIPK3/necroptotic pathway may be more beneficial.
To date, several small-molecule compounds (Li et al., 2014; Fauster et al., 2015; Martens et al., 2017, 2020; Park et al., 2018; Pan et al., 2019; Hart et al., 2020) have been developed to inhibit RIPK3 activity, providing an impressive toolbox for the investigation of the role of RIPK3 in diverse tissues. These inhibitors of RIPK3 can be divided into three types: ATP mimetic inhibitors targeting the active ATP-binding site in the kinases located between two catalytic domain lobes (type I), targeting the inactive states (type II), and unclassified inhibitors (Muller et al., 2015; Martens et al., 2020; Table 1).

Table 1. The RIPK3 inhibitors (Martens et al., 2020).
GSK’872 is the most widely used cell-permeable inhibitor of the RIPK3-selective kinase, with >1,000-fold selectivity over a vast majority of more than 300 other kinases (Kaiser et al., 2013). GSK’872 binds the kinase domain and inhibits kinase activity with high specificity, targeting a broad range of pro-necrotic stimuli (Mandal et al., 2014) and has been used to specifically inhibit RIPK3 (Lu et al., 2017; Chen et al., 2018; Imamura et al., 2018).
The serine/threonine kinase B-Raf V600E inhibitor dabrafenib is the only type I RIPK3 inhibitor approved for clinical use (Rheault et al., 2013; Li et al., 2014; Sugaya et al., 2019). Previous studies have reported that dabrafenib is a selective RIPK3 inhibitor in various models, including human hepatocytes (Li et al., 2014), mouse models of acetaminophen-caused liver injury (Li et al., 2014), and ischemic brain injury (Cruz et al., 2018). In addition, dabrafenib is a well-known inhibitor of B-Raf, which suppresses the downstream Ras/Raf/ERK/MAPK pathway (Spagnolo et al., 2014), which has been approved for clinical use for the treatment of non-small cell lung cancer expressing B-Raf V600E mutations and in melanoma (Odogwu et al., 2018). Inhibition of Raf kinase has found to attenuate renal fibrosis (Xu et al., 2012b; Chen et al., 2019).
Collectively, inhibition of RIPK3 is a promising anti-fibrotic strategy. RIPK3 facilitates necrosome and necroptosis. RIPK3 stimulates downstream activation of TGF-β1 cascades and regulates TGF-β1 transcription through NLRP3 inflammasome activation. Given inhibitors of RIPK3 are already approved for use in patients with non-small cell lung cancer and melanoma, an accelerated route to market in patients with CKD should be available if early phase clinical studies prove positive.
Author Contributions
YS wrote the manuscript. YS, CP, XC, and CH provided the critical discussion of the manuscript. YS and CP revised the manuscript.
Acknowledgments
Figure 1 was created with BioRender.com.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
AIHW (2018). Chronic Kidney Disease. Available online at: https://www.aihw.gov.au/reports/chronic-kidney-disease/chronic-kidney-disease-compendium/contents/how-many-australians-have-chronic-kidney-disease (accessed June 4, 2020).
Annes, J. P., Munger, J. S., and Rifkin, D. B. (2003). Making sense of latent TGFbeta activation. J. Cell Sci. 116(Pt 2), 217–224. doi: 10.1242/jcs.00229
Bakin, A. V., Rinehart, C., Tomlinson, A. K., and Arteaga, C. L. (2002). p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J. Cell Sci. 115(Pt 15), 3193–3206.
Bao, C., Yang, Z., Cai, Q., Li, Q., Li, H., and Shu, B. (2019). Incremental load training improves renal fibrosis by regulating the TGFbeta1/TAK1/MKK3/p38MAPK signaling pathway and inducing the activation of autophagy in aged mice. Int. J. Mol. Med. 44, 1677–1686. doi: 10.3892/ijmm.2019.4344
Bhowmick, N. A., Ghiassi, M., Bakin, A., Aakre, M., Lundquist, C. A., Engel, M. E., et al. (2001a). Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 12, 27–36. doi: 10.1091/mbc.12.1.27
Bhowmick, N. A., Zent, R., Ghiassi, M., McDonnell, M., and Moses, H. L. (2001b). Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38MAPK and epithelial plasticity. J. Biol. Chem. 276, 46707–46713. doi: 10.1074/jbc.M106176200
Biesemann, N., Mendler, L., Kostin, S., Wietelmann, A., Borchardt, T., and Braun, T. (2015). Myostatin induces interstitial fibrosis in the heart via TAK1 and p38. Cell Tissue Res. 361, 779–787. doi: 10.1007/s00441-015-2139-2
Blank, U., Seto, M. L., Adams, D. C., Wojchowski, D. M., Karolak, M. J., and Oxburgh, L. (2008). An in vivo reporter of BMP signaling in organogenesis reveals targets in the developing kidney. BMC Dev. Biol. 8:86. doi: 10.1186/1471-213X-8-86
Border, W. A., Okuda, S., Languino, L. R., and Ruoslahti, E. (1990). Transforming growth factor-beta regulates production of proteoglycans by mesangial cells. Kidney Int. 37, 689–695. doi: 10.1038/ki.1990.35
Bottinger, E. P. (2007). TGF-beta in renal injury and disease. Semin. Nephrol. 27, 309–320. doi: 10.1016/j.semnephrol.2007.02.009
Cao, J., Li, Y., Peng, Y., Zhang, Y., Li, H., Li, R., et al. (2015). Febuxostat prevents renal interstitial fibrosis by the activation of BMP-7 signaling and inhibition of USAG-1 expression in rats. Am. J. Nephrol. 42, 369–378. doi: 10.1159/000443023
Carew, R. M., Wang, B., and Kantharidis, P. (2012). The role of EMT in renal fibrosis. Cell Tissue Res. 347, 103–116. doi: 10.1007/s00441-011-1227-1
Chen, H. Y., Huang, X. R., Wang, W., Li, J. H., Heuchel, R. L., Chung, A. C., et al. (2011). The protective role of Smad7 in diabetic kidney disease: mechanism and therapeutic potential. Diabetes 60, 590–601. doi: 10.2337/db10-0403
Chen, J., Wang, S., Fu, R., Zhou, M., Zhang, T., Pan, W., et al. (2018). RIP3 dependent NLRP3 inflammasome activation is implicated in acute lung injury in mice. J. Transl. Med. 16:233. doi: 10.1186/s12967-018-1606-4
Chen, W., Zhou, Z., Li, L., Zhong, C. Q., Zheng, X., Wu, X., et al. (2013). Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J. Biol. Chem. 288, 16247–16261. doi: 10.1074/jbc.M112.435545
Chen, W. Y., Wu, S. Y., Lin, T. C., Lin, S. L., and Wu-Hsieh, B. A. (2019). Human dendritic cell-specific ICAM-3-grabbing non-integrin downstream signaling alleviates renal fibrosis via Raf-1 activation in systemic candidiasis. Cell Mol. Immunol. 16, 288–301. doi: 10.1038/s41423-018-0161-5
Christofferson, D. E., Li, Y., and Yuan, J. (2014). Control of life-or-death decisions by RIP1 kinase. Annu. Rev. Physiol. 76, 129–150. doi: 10.1146/annurev-physiol-021113-170259
Chung, A. C., Huang, X. R., Zhou, L., Heuchel, R., Lai, K. N., and Lan, H. Y. (2009). Disruption of the Smad7 gene promotes renal fibrosis and inflammation in unilateral ureteral obstruction (UUO) in mice. Nephrol. Dial. Transpl. 24, 1443–1454. doi: 10.1093/ndt/gfn699
Chung, A. C., Zhang, H., Kong, Y. Z., Tan, J. J., Huang, X. R., Kopp, J. B., et al. (2010). Advanced glycation end-products induce tubular CTGF via TGF-beta-independent Smad3 signaling. J. Am. Soc. Nephrol. 21, 249–260. doi: 10.1681/ASN.2009010018
ClinicalTrials.gov (2020). A Study to Evaluate the Effect of Dapagliflozin on Renal Outcomes and Cardiovascular Mortality in Patients With Chronic Kidney Disease (Dapa-CKD). Available online at: https://clinicaltrials.gov/ct2/show/NCT03036150 (accessed May 16, 2020).
Cruz, S. A., Qin, Z., Stewart, A. F. R., and Chen, H. H. (2018). Dabrafenib, an inhibitor of RIP3 kinase-dependent necroptosis, reduces ischemic brain injury. Neural Regen. Res. 13, 252–256. doi: 10.4103/1673-5374.226394
Das, R., Xu, S., Quan, X., Nguyen, T. T., Kong, I. D., Chung, C. H., et al. (2014). Upregulation of mitochondrial Nox4 mediates TGF-beta-induced apoptosis in cultured mouse podocytes. Am. J. Physiol. Renal Physiol. 306, F155–F167. doi: 10.1152/ajprenal.00438.2013
Dong, W., Li, Z., Chen, Y., Zhang, L., Ye, Z., Liang, H., et al. (2018). Necrostatin-1 attenuates sepsis-associated acute kidney injury by promoting autophagosome elimination in renal tubular epithelial cells. Mol. Med. Rep. 17, 3194–3199. doi: 10.3892/mmr.2017.8214
Edlund, S., Bu, S., Schuster, N., Aspenstrom, P., Heuchel, R., Heldin, N. E., et al. (2003). Transforming growth factor-beta1 (TGF-beta)-induced apoptosis of prostate cancer cells involves Smad7-dependent activation of p38 by TGF-beta-activated kinase 1 and mitogen-activated protein kinase kinase 3. Mol. Biol. Cell 14, 529–544. doi: 10.1091/mbc.02-03-0037
Edlund, S., Landstrom, M., Heldin, C. H., and Aspenstrom, P. (2002). Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell 13, 902–914. doi: 10.1091/mbc.01-08-0398
Engel, M. E., McDonnell, M. A., Law, B. K., and Moses, H. L. (1999). Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J. Biol. Chem. 274, 37413–37420. doi: 10.1074/jbc.274.52.37413
Fauster, A., Rebsamen, M., Huber, K. V., Bigenzahn, J. W., Stukalov, A., Lardeau, C. H., et al. (2015). A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis. 6:e001767. doi: 10.1038/cddis.2015.130
Fechtner, S., Fox, D. A., and Ahmed, S. (2017). Transforming growth factor beta activated kinase 1: a potential therapeutic target for rheumatic diseases. Rheumatology 56, 1060–1068. doi: 10.1093/rheumatology/kew301
Festjens, N., Vanden Berghe, T., and Vandenabeele, P. (2006). Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim. Biophys. Acta 1757, 1371–1387. doi: 10.1016/j.bbabio.2006.06.014
Filliol, A., Farooq, M., Piquet-Pellorce, C., Genet, V., Dimanche-Boitrel, M. T., Vandenabeele, P., et al. (2017). RIPK1 protects hepatocytes from death in Fas-induced hepatitis. Sci. Rep. 7:9205. doi: 10.1038/s41598-017-09789-8
Frey, R. S., and Mulder, K. M. (1997). Involvement of extracellular signal-regulated kinase 2 and stress-activated protein kinase/Jun N-terminal kinase activation by transforming growth factor beta in the negative growth control of breast cancer cells. Cancer Res. 57, 628–633.
Fujimoto, M., Maezawa, Y., Yokote, K., Joh, K., Kobayashi, K., Kawamura, H., et al. (2003). Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem. Biophys. Res. Commun. 305, 1002–1007. doi: 10.1016/s0006-291x(03)00885-4
Funaba, M., Zimmerman, C. M., and Mathews, L. S. (2002). Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J. Biol. Chem. 277, 41361–41368. doi: 10.1074/jbc.M204597200
Gomez-Puerto, M. C., Iyengar, P. V., Garcia de Vinuesa, A., Ten Dijke, P., and Sanchez-Duffhues, G. (2019). Bone morphogenetic protein receptor signal transduction in human disease. J. Pathol. 247, 9–20. doi: 10.1002/path.5170
Groppe, J., Hinck, C. S., Samavarchi-Tehrani, P., Zubieta, C., Schuermann, J. P., Taylor, A. B., et al. (2008). Cooperative assembly of TGF-beta superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol. Cell. 29, 157–168. doi: 10.1016/j.molcel.2007.11.039
Grynberg, K., Ma, F. Y., and Nikolic-Paterson, D. J. (2017). The JNK signaling pathway in renal fibrosis. Front. Physiol. 8:829. doi: 10.3389/fphys.2017.00829
Guo, C., Fu, R., Zhou, M., Wang, S., Huang, Y., Hu, H., et al. (2019). Pathogenesis of lupus nephritis: RIP3 dependent necroptosis and NLRP3 inflammasome activation. J. Autoimmun. 103:102286. doi: 10.1016/j.jaut.2019.05.014
Haberstroh, U., Zahner, G., Disser, M., Thaiss, F., Wolf, G., and Stahl, R. A. (1993). TGF-beta stimulates rat mesangial cell proliferation in culture: role of PDGF beta-receptor expression. Am. J. Physiol. 264(2 Pt 2), F199–F205. doi: 10.1152/ajprenal.1993.264.2.F199
Hanafusa, H., Ninomiya-Tsuji, J., Masuyama, N., Nishita, M., Fujisawa, J., Shibuya, H., et al. (1999). Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-beta-induced gene expression. J. Biol. Chem. 274, 27161–27167. doi: 10.1074/jbc.274.38.27161
Hanyu, A., Ishidou, Y., Ebisawa, T., Shimanuki, T., Imamura, T., and Miyazono, K. (2001). The N domain of Smad7 is essential for specific inhibition of transforming growth factor-beta signaling. J. Cell Biol. 155, 1017–1027. doi: 10.1083/jcb.200106023
Hart, A. C., Abell, L., Guo, J., Mertzman, M. E., Padmanabha, R., Macor, J. E., et al. (2020). Identification of RIPK3 Type II inhibitors using high-throughput mechanistic studies in hit triage. ACS Med. Chem. Lett. 11, 266–271. doi: 10.1021/acsmedchemlett.9b00065
Hata, A., Lagna, G., Massague, J., and Hemmati-Brivanlou, A. (1998). Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev. 12, 186–197. doi: 10.1101/gad.12.2.186
He, S., Wang, L., Miao, L., Wang, T., Du, F., Zhao, L., et al. (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111. doi: 10.1016/j.cell.2009.05.021
Heldin, C. H., and Moustakas, A. (2012). Role of smads in TGFbeta signaling. Cell Tissue Res. 347, 21–36. doi: 10.1007/s00441-011-1190-x
Hinz, B. (2015). The extracellular matrix and transforming growth factor-beta1: tale of a strained relationship. Matrix Biol. 47, 54–65. doi: 10.1016/j.matbio.2015.05.006
Hocevar, B. A., Brown, T. L., and Howe, P. H. (1999). TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 18, 1345–1356. doi: 10.1093/emboj/18.5.1345
Hou, C. C., Wang, W., Huang, X. R., Fu, P., Chen, T. H., Sheikh-Hamad, D., et al. (2005). Ultrasound-microbubble-mediated gene transfer of inducible Smad7 blocks transforming growth factor-beta signaling and fibrosis in rat remnant kidney. Am. J. Pathol. 166, 761–771. doi: 10.1016/s0002-9440(10)62297-3
Huang, X. R., Chung, A. C., Wang, X. J., Lai, K. N., and Lan, H. Y. (2008a). Mice overexpressing latent TGF-beta1 are protected against renal fibrosis in obstructive kidney disease. Am. J. Physiol. Renal Physiol. 295, F118–F127. doi: 10.1152/ajprenal.00021.2008
Huang, X. R., Chung, A. C., Zhou, L., Wang, X. J., and Lan, H. Y. (2008b). Latent TGF-beta1 protects against crescentic glomerulonephritis. J. Am. Soc. Nephrol. 19, 233–242. doi: 10.1681/ASN.2007040484
Imamura, M., Moon, J. S., Chung, K. P., Nakahira, K., Muthukumar, T., Shingarev, R., et al. (2018). RIPK3 promotes kidney fibrosis via AKT-dependent ATP citrate lyase. JCI Insight. 3:979. doi: 10.1172/jci.insight.94979
Ingelfinger, J. R., and Rosen, C. J. (2019). Clinical credence - SGLT2 inhibitors, diabetes, and chronic kidney disease. N. Engl. J. Med. 380, 2371–2373. doi: 10.1056/NEJMe1904740
Isono, M., Chen, S., Won, H., Carmen Iglesias-de la Cruz, M., and Ziyadeh, F. N. (2002). Smad pathway is activated in the diabetic mouse kidney and Smad3 mediates TGF-β-induced fibronectin in mesangial cells. Biochem. Biophys. Res. Commun. 296, 1356–1365. doi: 10.1016/S0006-291X(02)02084-3
Jaffe, A. B., and Hall, A. (2005). Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269. doi: 10.1146/annurev.cellbio.21.020604.150721
Jo, E. K., Kim, J. K., Shin, D. M., and Sasakawa, C. (2016). Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol. Immunol. 13, 148–159. doi: 10.1038/cmi.2015.95
Ka, S. M., Huang, X. R., Lan, H. Y., Tsai, P. Y., Yang, S. M., Shui, H. A., et al. (2007). Smad7 gene therapy ameliorates an autoimmune crescentic glomerulonephritis in mice. J. Am. Soc. Nephrol. 18, 1777–1788. doi: 10.1681/ASN.2006080901
Kaiser, W. J., Sridharan, H., Huang, C., Mandal, P., Upton, J. W., Gough, P. J., et al. (2013). Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 288, 31268–31279. doi: 10.1074/jbc.M113.462341
Kang, T. B., Yang, S. H., Toth, B., Kovalenko, A., and Wallach, D. (2014). Activation of the NLRP3 inflammasome by proteins that signal for necroptosis. Methods Enzymol. 545, 67–81. doi: 10.1016/B978-0-12-801430-1.00003-2
Kasof, G. M., Prosser, J. C., Liu, D., Lorenzi, M. V., and Gomes, B. C. (2000). The RIP-like kinase, RIP3, induces apoptosis and NF-kappaB nuclear translocation and localizes to mitochondria. FEBS Lett. 473, 285–291. doi: 10.1016/s0014-5793(00)01473-3
Kelley, N., Jeltema, D., Duan, Y., and He, Y. (2019). The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 20:328. doi: 10.3390/ijms20133328
Kim, S. I., and Choi, M. E. (2012). TGF-beta-activated kinase-1: new insights into the mechanism of TGF-beta signaling and kidney disease. Kidney Res. Clin. Pract. 31, 94–105. doi: 10.1016/j.krcp.2012.04.322
Kretzschmar, M., Doody, J., and Massague, J. (1997). Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature 389, 618–622. doi: 10.1038/39348
Kretzschmar, M., Doody, J., Timokhina, I., and Massague, J. (1999). A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev. 13, 804–816. doi: 10.1101/gad.13.7.804
Lan, H. Y., Mu, W., Tomita, N., Huang, X. R., Li, J. H., Zhu, H. J., et al. (2003). Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J. Am. Soc. Nephrol. 14, 1535–1548. doi: 10.1097/01.asn.0000067632.04658.b8
Lawlor, K. E., Khan, N., Mildenhall, A., Gerlic, M., Croker, B. A., D’Cruz, A. A., et al. (2015). RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 6:6282. doi: 10.1038/ncomms7282
Lebrin, F., Deckers, M., Bertolino, P., and Ten Dijke, P. (2005). TGF-beta receptor function in the endothelium. Cardiovasc. Res. 65, 599–608. doi: 10.1016/j.cardiores.2004.10.036
Lee, K. Y., Ito, K., Hayashi, R., Jazrawi, E. P., Barnes, P. J., and Adcock, I. M. (2006). NF-kappaB and activator protein 1 response elements and the role of histone modifications in IL-1beta-induced TGF-beta1 gene transcription. J. Immunol. 176, 603–615. doi: 10.4049/jimmunol.176.1.603
Lee, M. K., Pardoux, C., Hall, M. C., Lee, P. S., Warburton, D., Qing, J., et al. (2007). TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 26, 3957–3967. doi: 10.1038/sj.emboj.7601818
Lee, S. B., and Kalluri, R. (2010). Mechanistic connection between inflammation and fibrosis. Kidney Int. Suppl. 119, S22–S26. doi: 10.1038/ki.2010.418
Li, J., Liang, C., Zhang, Z. K., Pan, X., Peng, S., Lee, W. S., et al. (2017). TAK1 inhibition attenuates both inflammation and fibrosis in experimental pneumoconiosis. Cell Discov. 3:17023. doi: 10.1038/celldisc.2017.23
Li, J., McQuade, T., Siemer, A. B., Napetschnig, J., Moriwaki, K., Hsiao, Y. S., et al. (2012). The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350. doi: 10.1016/j.cell.2012.06.019
Li, J. H., Huang, X. R., Zhu, H. J., Oldfield, M., Cooper, M., Truong, L. D., et al. (2004). Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: implications for diabetic renal and vascular disease. FASEB J. 18, 176–178. doi: 10.1096/fj.02-1117fje
Li, J. X., Feng, J. M., Wang, Y., Li, X. H., Chen, X. X., Su, Y., et al. (2014). The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis. 5:e001278. doi: 10.1038/cddis.2014.241
Liao, J. H., Chen, J. S., Chai, M. Q., Zhao, S., and Song, J. G. (2001). The involvement of p38 MAPK in transforming growth factor beta1-induced apoptosis in murine hepatocytes. Cell Res. 11, 89–94. doi: 10.1038/sj.cr.7290072
Lin, X., Liang, Y. Y., Sun, B., Liang, M., Shi, Y., Brunicardi, F. C., et al. (2003). Smad6 recruits transcription corepressor CtBP to repress bone morphogenetic protein-induced transcription. Mol. Cell Biol. 23, 9081–9093. doi: 10.1128/mcb.23.24.9081-9093.2003
Liu, G. X., Li, Y. Q., Huang, X. R., Wei, L., Chen, H. Y., Shi, Y. J., et al. (2013). Disruption of Smad7 promotes ANG II-mediated renal inflammation and fibrosis via Sp1-TGF-beta/Smad3-NF.kappaB-dependent mechanisms in mice. PLoS One 8:e53573. doi: 10.1371/journal.pone.0053573
Loeffler, I., and Wolf, G. (2014). Transforming growth factor-β and the progression of renal disease. Nephrol. Dial. Transpl. 29(Suppl._1), i37–i45. doi: 10.1093/ndt/gft267
Lu, B., Gong, X., Wang, Z. Q., Ding, Y., Wang, C., Luo, T. F., et al. (2017). Shikonin induces glioma cell necroptosis in vitro by ROS overproduction and promoting RIP1/RIP3 necrosome formation. Acta Pharmacol. Sin. 38, 1543–1553. doi: 10.1038/aps.2017.112
Mack, M., and Yanagita, M. (2015). Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 87, 297–307. doi: 10.1038/ki.2014.287
Mandal, P., Berger, S. B., Pillay, S., Moriwaki, K., Huang, C., Guo, H., et al. (2014). RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 56, 481–495. doi: 10.1016/j.molcel.2014.10.021
Markic, D., Celic, T., Spanjol, J., Grskovic, A., Bobinac, D., and Fuckar, Z. (2010). Expression of bone morphogenetic protein-7, its receptors and Smad1/5/8 in normal human kidney and renal cell cancer. Coll. Antropol. 34(Suppl. 2), 149–153.
Martens, S., Hofmans, S., Declercq, W., Augustyns, K., and Vandenabeele, P. (2020). Inhibitors targeting RIPK1/RIPK3: old and new drugs. Trends Pharmacol. Sci. 41, 209–224. doi: 10.1016/j.tips.2020.01.002
Martens, S., Jeong, M., Tonnus, W., Feldmann, F., Hofmans, S., Goossens, V., et al. (2017). Sorafenib tosylate inhibits directly necrosome complex formation and protects in mouse models of inflammation and tissue injury. Cell Death Dis. 8:e02904. doi: 10.1038/cddis.2017.298
Martin-Sanchez, D., Ruiz-Andres, O., Poveda, J., Carrasco, S., Cannata-Ortiz, P., Sanchez-Nino, M. D., et al. (2017). Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced AKI. J. Am. Soc. Nephrol. 28, 218–229. doi: 10.1681/ASN.2015121376
Massague, J., and Wotton, D. (2000). Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 19, 1745–1754. doi: 10.1093/emboj/19.8.1745
Matsuura, I., Wang, G., He, D., and Liu, F. (2005). Identification and characterization of ERK MAP kinase phosphorylation sites in Smad3. Biochemistry 44, 12546–12553. doi: 10.1021/bi050560g
McGaraughty, S., Davis-Taber, R. A., Zhu, C. Z., Cole, T. B., Nikkel, A. L., Chhaya, M., et al. (2017). Targeting Anti-TGF-beta therapy to fibrotic kidneys with a dual specificity antibody approach. J. Am. Soc. Nephrol. 28, 3616–3626. doi: 10.1681/ASN.2017010013
McQuade, T., Cho, Y., and Chan, F. K. (2013). Positive and negative phosphorylation regulates RIP1- and RIP3-induced programmed necrosis. Biochem. J. 456, 409–415. doi: 10.1042/BJ20130860
Meng, X. M., Huang, X. R., Chung, A. C., Qin, W., Shao, X., Igarashi, P., et al. (2010). Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J. Am. Soc. Nephrol. 21, 1477–1487. doi: 10.1681/ASN.2009121244
Meng, X. M., Huang, X. R., Xiao, J., Chung, A. C., Qin, W., Chen, H. Y., et al. (2012). Disruption of Smad4 impairs TGF-beta/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int. 81, 266–279. doi: 10.1038/ki.2011.327
Moerke, C., Bleibaum, F., Kunzendorf, U., and Krautwald, S. (2019). Combined knockout of RIPK3 and MLKL reveals unexpected outcome in tissue injury and inflammation. Front. Cell Dev. Biol. 7:19. doi: 10.3389/fcell.2019.00019
Mompean, M., Li, W., Li, J., Laage, S., Siemer, A. B., Bozkurt, G., et al. (2018). The structure of the necrosome RIPK1-RIPK3 core, a human hetero-amyloid signaling complex. Cell 173, 1244–1253. doi: 10.1016/j.cell.2018.03.032
Moon, J. A., Kim, H. T., Cho, I. S., Sheen, Y. Y., and Kim, D. K. (2006). IN-1130, a novel transforming growth factor-beta type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. 70, 1234–1243. doi: 10.1038/sj.ki.5001775
Moustakas, A., Souchelnytskyi, S., and Heldin, C. H. (2001). Smad regulation in TGF-beta signal transduction. J. Cell Sci. 114(Pt 24), 4359–4369.
Mulay, S. R., Desai, J., Kumar, S. V., Eberhard, J. N., Thomasova, D., Romoli, S., et al. (2016). Cytotoxicity of crystals involves RIPK3-MLKL-mediated necroptosis. Nat. Commun. 7:10274. doi: 10.1038/ncomms10274
Muller, S., Chaikuad, A., Gray, N. S., and Knapp, S. (2015). The ins and outs of selective kinase inhibitor development. Nat. Chem. Biol. 11, 818–821. doi: 10.1038/nchembio.1938
Munoz-Felix, J. M., Gonzalez-Nunez, M., and Lopez-Novoa, J. M. (2013). ALK1-Smad1/5 signaling pathway in fibrosis development: friend or foe? Cytok. Growth Factor Rev. 24, 523–537. doi: 10.1016/j.cytogfr.2013.08.002
Murakami, G., Watabe, T., Takaoka, K., Miyazono, K., and Imamura, T. (2003). Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol. Biol. Cell 14, 2809–2817. doi: 10.1091/mbc.e02-07-0441
Murphy, S. R., Dahly-Vernon, A. J., Dunn, K. M., Chen, C. C., Ledbetter, S. R., Williams, J. M., et al. (2012). Renoprotective effects of anti-TGF-beta antibody and antihypertensive therapies in Dahl S rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 303, R57–R69. doi: 10.1152/ajpregu.00263.2011
Nakayama, T., Berg, L. K., and Christian, J. L. (2001). Dissection of inhibitory Smad proteins: both N- and C-terminal domains are necessary for full activities of Xenopus Smad6 and Smad7. Mech. Dev. 100, 251–262. doi: 10.1016/s0925-4773(00)00533-5
Newton, K. (2015). RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 25, 347–353. doi: 10.1016/j.tcb.2015.01.001
Newton, K., Dugger, D. L., Wickliffe, K. E., Kapoor, N., de Almagro, M. C., Vucic, D., et al. (2014). Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360. doi: 10.1126/science.1249361
Newton, K., and Manning, G. (2016). Necroptosis and inflammation. Annu. Rev. Biochem. 85, 743–763. doi: 10.1146/annurev-biochem-060815-014830
Odogwu, L., Mathieu, L., Blumenthal, G., Larkins, E., Goldberg, K. B., Griffin, N., et al. (2018). FDA approval summary: dabrafenib and trametinib for the treatment of metastatic non-small cell lung cancers harboring braf V600E mutations. Oncologist 23, 740–745. doi: 10.1634/theoncologist.2017-0642
Oxburgh, L., and Robertson, E. J. (2002). Dynamic regulation of smad expression during mesenchyme to epithelium transition in the metanephric kidney. Mech. Dev. 112, 207–211. doi: 10.1016/s0925-4773(01)00648-7
Pan, P., Cai, Z., Zhuang, C., Chen, X., and Chai, Y. (2019). Methodology of drug screening and target identification for new necroptosis inhibitors. J. Pharm. Anal. 9, 71–76. doi: 10.1016/j.jpha.2018.11.002
Park, H. H., Park, S. Y., Mah, S., Park, J. H., Hong, S. S., Hong, S., et al. (2018). HS-1371, a novel kinase inhibitor of RIP3-mediated necroptosis. Exp. Mol. Med. 50:125. doi: 10.1038/s12276-018-0152-8
Pazdernik, N. J., Donner, D. B., Goebl, M. G., and Harrington, M. A. (1999). Mouse receptor interacting protein 3 does not contain a caspase-recruiting or a death domain but induces apoptosis and activates NF-kappaB. Mol. Cell Biol. 19, 6500–6508. doi: 10.1128/mcb.19.10.6500
Proskuryakov, S. Y., Konoplyannikov, A. G., and Gabai, V. L. (2003). Necrosis: a specific form of programmed cell death? Exp. Cell Res. 283, 1–16. doi: 10.1016/s0014-4827(02)00027-7
Rheault, T. R., Stellwagen, J. C., Adjabeng, G. M., Hornberger, K. R., Petrov, K. G., Waterson, A. G., et al. (2013). Discovery of dabrafenib: a selective inhibitor of raf kinases with antitumor activity against B-Raf-driven tumors. ACS Med. Chem. Lett. 4, 358–362. doi: 10.1021/ml4000063
Robertson, I. B., Horiguchi, M., Zilberberg, L., Dabovic, B., Hadjiolova, K., and Rifkin, D. B. (2015). Latent TGF-beta-binding proteins. Matrix Biol. 47, 44–53. doi: 10.1016/j.matbio.2015.05.005
Russo, L. M., del Re, E., Brown, D., and Lin, H. Y. (2007). Evidence for a role of transforming growth factor (TGF)-beta1 in the induction of postglomerular albuminuria in diabetic nephropathy: amelioration by soluble TGF-beta type II receptor. Diabetes 56, 380–388. doi: 10.2337/db06-1018
Sanderson, N., Factor, V., Nagy, P., Kopp, J., Kondaiah, P., Wakefield, L., et al. (1995). Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc. Natl. Acad. Sci. U.S.A. 92, 2572–2576. doi: 10.1073/pnas.92.7.2572
Sano, Y., Harada, J., Tashiro, S., Gotoh-Mandeville, R., Maekawa, T., and Ishii, S. (1999). ATF-2 is a common nuclear target of Smad and TAK1 pathways in transforming growth factor-beta signaling. J. Biol. Chem. 274, 8949–8957. doi: 10.1074/jbc.274.13.8949
Sato, M., Muragaki, Y., Saika, S., Roberts, A. B., and Ooshima, A. (2003). Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Invest. 112, 1486–1494. doi: 10.1172/JCI19270
Shao, X., Somlo, S., and Igarashi, P. (2002). Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J. Am. Soc. Nephrol. 13, 1837–1846. doi: 10.1097/01.asn.0000016444.90348.50
Shen, B., Mei, M., Pu, Y., Zhang, H., Liu, H., Tang, M., et al. (2019). Necrostatin-1 attenuates renal ischemia and reperfusion injury via meditation of HIF-1alpha/mir-26a/TRPC6/PARP1 signaling. Mol. Ther. Nucleic Acids 17, 701–713. doi: 10.1016/j.omtn.2019.06.025
Shibuya, H., Iwata, H., Masuyama, N., Gotoh, Y., Yamaguchi, K., Irie, K., et al. (1998). Role of TAK1 and TAB1 in BMP signaling in early Xenopus development. EMBO J. 17, 1019–1028. doi: 10.1093/emboj/17.4.1019
Shlomovitz, I., Zargrian, S., and Gerlic, M. (2017). Mechanisms of RIPK3-induced inflammation. Immunol. Cell Biol. 95, 166–172. doi: 10.1038/icb.2016.124
Spagnolo, F., Ghiorzo, P., and Queirolo, P. (2014). Overcoming resistance to BRAF inhibition in BRAF-mutated metastatic melanoma. Oncotarget 5, 10206–10221. doi: 10.18632/oncotarget.2602
Sugaya, T., Kanno, H., Matsuda, M., Handa, K., Tateda, S., Murakami, T., et al. (2019). B-RAF(V600E) inhibitor dabrafenib attenuates RIPK3-mediated necroptosis and promotes functional recovery after spinal cord injury. Cells 8:82. doi: 10.3390/cells8121582
Sun, L., Wang, H., Wang, Z., He, S., Chen, S., Liao, D., et al. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. doi: 10.1016/j.cell.2011.11.031
Sun, X., Lee, J., Navas, T., Baldwin, D. T., Stewart, T. A., and Dixit, V. M. (1999). RIP3, a novel apoptosis-inducing kinase. J. Biol. Chem. 274, 16871–16875. doi: 10.1074/jbc.274.24.16871
Sun, X., Yin, J., Starovasnik, M. A., Fairbrother, W. J., and Dixit, V. M. (2002). Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J. Biol. Chem. 277, 9505–9511. doi: 10.1074/jbc.M109488200
Sureshbabu, A., Patino, E., Ma, K. C., Laursen, K., Finkelsztein, E. J., Akchurin, O., et al. (2018). RIPK3 promotes sepsis-induced acute kidney injury via mitochondrial dysfunction. JCI Insight. 3:411. doi: 10.1172/jci.insight.98411
Terada, Y., Hanada, S., Nakao, A., Kuwahara, M., Sasaki, S., and Marumo, F. (2002). Gene transfer of Smad7 using electroporation of adenovirus prevents renal fibrosis in post-obstructed kidney. Kidney Int. 61(Suppl.), S94–S98. doi: 10.1046/j.1523-1755.2002.0610s1094.x
Tsuchida, K., Zhu, Y., Siva, S., Dunn, S. R., and Sharma, K. (2003). Role of Smad4 on TGF-beta-induced extracellular matrix stimulation in mesangial cells. Kidney Int. 63, 2000–2009. doi: 10.1046/j.1523-1755.2003.00009.x
United States Renal Data System (2018). CKD in the General Population. Available online at: https://www.usrds.org/2017/view/v1_01.aspx (accessed September 29, 2018).
Upton, J. W., Kaiser, W. J., and Mocarski, E. S. (2010). Virus inhibition of RIP3-dependent necrosis. Cell Host Microb. 7, 302–313. doi: 10.1016/j.chom.2010.03.006
Upton, J. W., Kaiser, W. J., and Mocarski, E. S. (2012). DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microb. 11, 290–297. doi: 10.1016/j.chom.2012.01.016
Vandenabeele, P., Galluzzi, L., Vanden Berghe, T., and Kroemer, G. (2010). Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714. doi: 10.1038/nrm2970
Verrecchia, F., Chu, M. L., and Mauviel, A. (2001). Identification of novel TGF-beta/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J. Biol. Chem. 276, 17058–17062. doi: 10.1074/jbc.M100754200
Voelker, J., Berg, P. H., Sheetz, M., Duffin, K., Shen, T., Moser, B., et al. (2017). Anti-TGF-beta1 antibody therapy in patients with diabetic nephropathy. J. Am. Soc. Nephrol. 28, 953–962. doi: 10.1681/ASN.2015111230
Wang, W., Huang, X. R., Canlas, E., Oka, K., Truong, L. D., Deng, C., et al. (2006). Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ. Res. 98, 1032–1039. doi: 10.1161/01.RES.0000218782.52610.dc
Wang, W., Koka, V., and Lan, H. Y. (2005). Transforming growth factor-beta and Smad signalling in kidney diseases. Nephrology 10, 48–56. doi: 10.1111/j.1440-1797.2005.00334.x
Wang, X., Jiang, W., Yan, Y., Gong, T., Han, J., Tian, Z., et al. (2014a). RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat. Immunol. 15, 1126–1133. doi: 10.1038/ni.3015
Wang, X., Li, Y., Liu, S., Yu, X., Li, L., Shi, C., et al. (2014b). Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl. Acad. Sci. U.S.A. 111, 15438–15443. doi: 10.1073/pnas.1412767111
Weinlich, R., Oberst, A., Beere, H. M., and Green, D. R. (2017). Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 18, 127–136. doi: 10.1038/nrm.2016.149
Wilson, H. M., Reid, F. J., Brown, P. A., Power, D. A., Haites, N. E., and Booth, N. A. (1993). Effect of transforming growth factor-beta 1 on plasminogen activators and plasminogen activator inhibitor-1 in renal glomerular cells. Exp. Nephrol. 1, 343–350.
Wu, H., Zhou, J., Ou, W., Li, Y., Liu, M., and Yang, C. (2017). TAK1 as the mediator in the protective effect of propofol on renal interstitial fibrosis induced by ischemia/reperfusion injury. Eur. J. Pharmacol. 811, 134–140. doi: 10.1016/j.ejphar.2017.06.009
Xiao, X., Du, C., Yan, Z., Shi, Y., Duan, H., and Ren, Y. (2017). Inhibition of necroptosis attenuates kidney inflammation and interstitial fibrosis induced by unilateral ureteral obstruction. Am. J. Nephrol. 46, 131–138. doi: 10.1159/000478746
Xu, P., Liu, J., and Derynck, R. (2012a). Post-translational regulation of TGF-beta receptor and Smad signaling. FEBS Lett. 586, 1871–1884. doi: 10.1016/j.febslet.2012.05.010
Xu, T., Wang, N. S., Fu, L. L., Ye, C. Y., Yu, S. Q., and Mei, C. L. (2012b). Celecoxib inhibits growth of human autosomal dominant polycystic kidney cyst-lining epithelial cells through the VEGF/Raf/MAPK/ERK signaling pathway. Mol. Biol. Rep. 39, 7743–7753. doi: 10.1007/s11033-012-1611-2
Yamashita, M., Fatyol, K., Jin, C., Wang, X., Liu, Z., and Zhang, Y. E. (2008). TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 31, 918–924. doi: 10.1016/j.molcel.2008.09.002
Yan, X., Liao, H., Cheng, M., Shi, X., Lin, X., Feng, X. H., et al. (2016). Smad7 Protein interacts with receptor-regulated Smads (R-Smads) to inhibit transforming growth factor-beta (TGF-beta)/Smad signaling. J. Biol. Chem. 291, 382–392. doi: 10.1074/jbc.M115.694281
Yan, X., Lin, Z., Chen, F., Zhao, X., Chen, H., Ning, Y., et al. (2009). Human BAMBI cooperates with Smad7 to inhibit transforming growth factor-beta signaling. J. Biol. Chem. 284, 30097–30104. doi: 10.1074/jbc.M109.049304
Yang, F., Chung, A. C., Huang, X. R., and Lan, H. Y. (2009). Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension 54, 877–884. doi: 10.1161/HYPERTENSIONAHA.109.136531
Yang, F., Huang, X. R., Chung, A. C., Hou, C. C., Lai, K. N., and Lan, H. Y. (2010). Essential role for Smad3 in angiotensin II-induced tubular epithelial-mesenchymal transition. J. Pathol. 221, 390–401. doi: 10.1002/path.2721
Yang, L., Joseph, S., Sun, T., Hoffmann, J., Thevissen, S., Offermanns, S., et al. (2019). TAK1 regulates endothelial cell necroptosis and tumor metastasis. Cell Death Differ. 26, 1987–1997. doi: 10.1038/s41418-018-0271-8
Yu, L., Border, W. A., Huang, Y., and Noble, N. A. (2003). TGF-beta isoforms in renal fibrogenesis. Kidney Int. 64, 844–856. doi: 10.1046/j.1523-1755.2003.00162.x
Yu, L., Hebert, M. C., and Zhang, Y. E. (2002). TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 21, 3749–3759. doi: 10.1093/emboj/cdf366
Yu, P. W., Huang, B. C., Shen, M., Quast, J., Chan, E., Xu, X., et al. (1999). Identification of RIP3, a RIP-like kinase that activates apoptosis and NFkappaB. Curr. Biol. 9, 539–542.
Zhang, D., Lin, J., and Han, J. (2010). Receptor-interacting protein (RIP) kinase family. Cell Mol. Immunol. 7, 243–249. doi: 10.1038/cmi.2010.10
Zhang, M., Fraser, D., and Phillips, A. (2006). ERK, p38, and Smad signaling pathways differentially regulate transforming growth factor-beta1 autoinduction in proximal tubular epithelial cells. Am. J. Pathol. 169, 1282–1293. doi: 10.2353/ajpath.2006.050921
Zhang, S., Fei, T., Zhang, L., Zhang, R., Chen, F., Ning, Y., et al. (2007). Smad7 antagonizes transforming growth factor beta signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol. Cell Biol. 27, 4488–4499. doi: 10.1128/MCB.01636-06
Zhang, Y. E. (2009). Non-Smad pathways in TGF-beta signaling. Cell Res. 19, 128–139. doi: 10.1038/cr.2008.328
Zhou, J., Zhong, J., Huang, Z., Liao, M., Lin, S., Chen, J., et al. (2018). TAK1 mediates apoptosis via p38 involve in ischemia-induced renal fibrosis. Artif. Cells Nanomed. Biotechnol. 46, 1016–1025. doi: 10.1080/21691401.2018.1442841
Zhou, L., Fu, P., Huang, X. R., Liu, F., Chung, A. C., Lai, K. N., et al. (2010). Mechanism of chronic aristolochic acid nephropathy: role of Smad3. Am. J. Physiol. Renal Physiol. 298, F1006–F1017. doi: 10.1152/ajprenal.00675.2009
Keywords: RIPK3, Receptor interacting serine/threonine-protein kinase 3, renal fibrosis, TGF – β1, necroptosis, dabrafenib
Citation: Shi Y, Chen X, Huang C and Pollock C (2020) RIPK3: A New Player in Renal Fibrosis. Front. Cell Dev. Biol. 8:502. doi: 10.3389/fcell.2020.00502
Received: 13 December 2019; Accepted: 26 May 2020;
Published: 16 June 2020.
Edited by:
Diego Franco, University of Jaén, SpainReviewed by:
W. Wei-Lynn Wong, University of Zurich, SwitzerlandPaul J. Higgins, Albany Medical College, United States
Copyright © 2020 Shi, Chen, Huang and Pollock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Shi, eXNoaTYxMjVAc3RhbmZvcmQuZWR1; eXNoaTYxMjVAdW5pLnN5ZG5leS5lZHUuYXU=; Carol Pollock, Y2Fyb2wucG9sbG9ja0BzeWRuZXkuZWR1LmF1