 Thomas Hilberath
Thomas Hilberath Anouska van Troost
Anouska van Troost Miguel Alcalde
Miguel Alcalde Frank Hollmann
Frank Hollmann
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Catal. , 23 May 2022
Sec. Biocatalysis
Volume 2 - 2022 | https://doi.org/10.3389/fctls.2022.882992
The use of water-miscible organic co-solvents in biocatalysis is a simple procedure for obtaining higher enzymatic activities toward hydrophobic substrates. However, effects on activity and stability have to be carefully evaluated, also with regard to the type and concentration of the respective co-solvent. In this contribution, we investigated and evaluated the effect of some common water-miscible co-solvents on the biocatalytic performance of the recombinant unspecific peroxygenase rAaeUPO from Agrocybe aegerita. rAaeUPO showed promising activities in the presence of high concentrations of the best co-solvent acetonitrile, which enabled to use higher substrate concentrations (≥100 mM). Employing high acetonitrile concentrations for UPO-mediated oxidation of ethylbenzene to (R)-1-phenylethanol was demonstrated under preparative scale conditions and led to product accumulation rates of 31 mM h−1.
Biocatalysis bears an enormous potential for the chemical industry to become more resource-saving and environmentally more benign (Ni et al., 2014; Sheldon and Woodley, 2018). However, to tap its full potential, a range of issues still have to be addressed. The hydrophobicity of most reagents of interest represents one of these challenges. As aqueous buffer systems still represent the reaction media of choice for biocatalysis, frequently, only low reagent concentrations can be realized. In fact, today, the majority of biocatalytic reactions reported by academic research groups deal with reagent concentrations below 100 mM (Holtmann and Hollmann, 2022). Typically, this corresponds to maximal product loadings of less than 15 g L−1 and wastewater amounts for more than 60 kg kg−1product. Obviously, this situation is far away from being economically and environmentally sustainable!
Among the various approaches to increase the reagent concentrations, water-miscible organic solvents are the most widespread strategies (van Schie et al., 2021). The presence of such co-solvents, however, can exceed unpredictable effects on the biocatalyst. For various enzymes, co-solvents have been reported to (negatively) influence their activity and stability (Klibanov, 2001; Serdakowski and Dordick, 2008; Doukyu and Ogino, 2010; Stepankova et al., 2013; Dutta Banik et al., 2016).
Fungal peroxygenases are heme-thiolate enzymes catalyzing a broad range of preparatively interesting conversions such as regio-, chemo- and enantioselective oxyfunctionalization reactions (Hobisch et al., 2020a). As extracellular enzymes, peroxygenases may be expected to be robust also under harsh reaction conditions such as the presence of high concentrations of organic solvents. Thus, the use of peroxygenases in two-liquid phase systems (Churakova et al., 2011; Tonin et al., 2021) or neat reaction media (Poraj-Kobielska et al., 2015; Fernández-Fueyo et al., 2016; Rauch et al., 2019; Hobisch et al., 2020b) has already been demonstrated, whereas the application in one-phase systems, in particular with high amounts of simple water-miscible solvents, still needs to be explored. In the current contribution we investigate the effect of a range of water-miscible organic co-solvents on fungal peroxygenases, using the recombinant evolved peroxygenase from Agrocybe aegerita (rAaeUPO) as a model enzyme.
The expression-engineered variant rAaeUPO (PaDa-I mutant) was typically used as concentrated supernatant or purified enzyme derived from a 2,500 L pilot-scale cultivation of recombinant Pichia pastoris X-33 expressing rAaeUPO (Molina-Espeja et al., 2014; Tonin et al., 2021). Purification was similarly performed according to previous studies (Molina-Espeja et al., 2014). rAaeUPO concentrations were determined in CO-difference spectra (see below) or via their absorption at 420 nm using the extinction coefficient (ε420 = 115 mM−1 cm−1) (Fernández-Fueyo et al., 2016).
The amount of rAaeUPO in concentrated supernatants was determined using CO-difference spectra. 950 µL of protein sample, diluted in 100 mM KPi-buffer if necessary, were filled into plastic cuvettes and placed in a photometer. After recording a blank (base subtraction), the sample was exposed with CO for a few seconds. Next, 50 µL of a 1 M sodium dithionite stock solution was added, and a difference spectrum between 400 and 500 nm was recorded. The measurements were continued until a constant absorption maximum was obtained. All measurements were performed in duplicates. The concentration of rAaeUPO was calculated based on the absorption values at 445 and 490 nm using the molar extinction coefficient of ε445 = 107 mM−1 cm−1 (Tieves et al., 2019a).
The peroxidase activity of rAaeUPO was determined in a continuous photometric assay against the substrate 2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid (ABTS) at 420 nm (ε420 = 36.0 mM−1 cm−1). AaeUPO (50 µL) in appropriate dilution (typically 50- or 100-fold) was mixed with 0.5 mM ABTS in 100 mM sodium citrate buffer, pH 4.4 in 2 mL plastic cuvettes. After short incubation at 25°C, measurements were started by the addition of 2 mM H2O2. The increase of absorption at 420 nm caused by ABTS formation was tracked for 90 s at 25°C in a Cary 60 UV–VIS spectrophotometer (Agilent). The initial slope (ΔA420/min) between 20–80 s was linear and used to calculate the volumetric activity [U/mL]. 1 U is defined as the amount of enzyme which is needed to convert 1 µmol substrate in 1 min under assay conditions. All measurements were performed in triplicates.
The activity of rAaeUPO against the primary alcohols methanol and ethanol was examined using the purpald assay (Dickinson and Jacobsen, 1970; Hopps, 2000). Reaction mixtures contained 1 µM rAaeUPO (concentrated supernatant or purified enzyme), 20 vol% co-solvent, and H2O2, which was added with a dosing rate of 4 mM h−1. The 200 mM H2O2-stock solution was freshly prepared from commercially available H2O2 (50 wt% solution, Merck). For analysis, 5–50 µL sample was diluted in 100 mM potassium phosphate buffer, pH 7.5 to a final volume of 200 µL in a 96-well plate. Afterward, 50 µL purpald (0.16 M in 2 M NaOH) was added, and samples were properly mixed. The samples were incubated at room temperature for at least 1 h, allowing the formation of a purple dye in the presence of aldehydes and oxygen.
rAaeUPO-mediated oxidation reactions were typically performed in 0.5 mL 100 mM potassium phosphate buffer, pH 6.0 in 2 mL glass vials. Reaction mixtures contained 1 µM rAaeUPO (concentrated supernatant), 10 mM ethylbenzene or cis-β-methyl styrene (in 10–97 vol% of acetonitrile), and H2O2, which was added with a dosing rate of 4 mM h−1. The 200 mM H2O2-stock solution was freshly prepared from commercially available H2O2 (50 wt% solution, Merck). The samples were incubated at 25°C and 600 min−1 for 0–3 h in an Eppendorf shaker.
In order to track the substrate oxidation with automated H2O2 addition, reactions in a 10 mL scale were prepared in glass vessels (Supplementary Figure S5). The glass devices were placed in an Eppendorf shaker equipped with a 50 mL falcon attachment, which was fixed in such a way that proper shaking and correct placement of the glass vials were enabled as it would the case for 50 mL falcons. Springe pumps containing a stock solution of 800 mM H2O2 (freshly prepared from a 50 wt% H2O2) were connected and automatically pumped 100 µL of H2O2 every hour (dosing rate: 8 mM h−1 H2O2).
Ethyl acetate (500 µL) containing 5 mM of the internal standard n-dodecane was used for extraction. After addition, samples were vortexed and centrifuged at 13.000 x g for 3 min at room temperature. The organic phase was transferred to a plastic vial with inlet and used for GC analysis.
The kinetic parameters Km and kcat for the rAaeUPO substrates ethylbenzene and 1-phenylethanol were determined in the presence of different concentrations of the co-solvent acetonitrile. Reaction mixtures (8 mL) in 10 mM KPi buffer (pH 7) contained 8.4 nM rAaeUPO (concentrated supernatant), 1.7 mM H2O2 (freshly diluted from a 50 wt% H2O2 stock solution), and the substrates ethylbenzene or 1-phenylethanol at concentrations ranging from 0.03–2 mM. Acetonitrile concentrations varied from 0–20 vol%. For reactions without acetonitrile, the maximum concentration of ethylbenzene was set at the solubility limit of 1.2 mM (150 mg/L, IPCS Inchem.). Samples were incubated at room temperature for 15 s, and then the reaction was stopped by adding 1 mL 50 vol% trifluoroacetic acid. Afterward, the samples were extracted with ethyl acetate containing 5 mM of the internal standard 1-octanol, dried over magnesium sulfate, and analyzed by GC. Km and vmax values were obtained by fitting the reaction rate (µM product per min) vs. the substrate concentration according to the equation of Michaelis–Menten by non-linear regression using Prism GraphPad, version 5 (Supplementary Figures S9–S11). The turnover number kcat was calculated by dividing vmax by the total UPO concentration.
The rAaeUPO-mediated hydroxylation of ethylbenzene on a 100 mL scale was performed in a SYSTAG jacketed lab reactor (250 mL operational volume) at 25°C and 300 rpm shaking speed. Reaction solution (100 mL) contained 100 mM KPi buffer (pH 6), 50 vol% acetonitrile, 10 µM rAaeUPO (concentrated supernatant), and 300 mM ethylbenzene. H2O2 was added continuously from a 4.5 M stock solution with a H2O2-dosing rate of 50 mM h−1 and was freshly prepared from a commercial 50 wt% H2O2 solution (Merck) prior to the experiment. At different time points, samples from the aqueous phase were withdrawn, extracted with ethyl acetate containing 5 mM n-dodecane (IS), and analyzed via chiral GC.
For GC analysis, a Shimadzu GC-14A/FID or Shimadzu GC-2010 plus/FID system was used. Achiral GC-analysis was performed on an Agilent CP-Sil 5 CB column (50 m × 0.53 mm × 1.0 μm) with N2 as carrier gas. Kinetic studies were analyzed on a CP-Wax 52 CB column (25 m × 0.25 mm × 1.2 µm) using N2 as carrier gas. Chiral GC-analysis for cis-β-methyl styrene and products was performed according to Rauch et al. on a Lipodex E column (Macherney Nagel) using helium as carrier gas (Rauch et al., 2019). Ethylbenzene and oxidized products thereof were analyzed on a CP-Chirasil-Dex CB (Agilent) (25 m × 0.32 mm × 0.25 μm) with helium as carrier gas (Tieves et al., 2019b). Substrate and product concentrations were calculated based on calibration curves. Because an authentic standard for (1R,2S)-cis-methyl styrene oxide was not available, the response factor between calibration curves of styrene and styrene epoxide was used to calculate the concentration of (1R,2S)-cis-methyl styrene oxide relative to cis-β-methyl styrene.
Temperature programs are described in key points as follows:
100°C hold 3 min and 20°C/min to 325°C hold 3 min; retention times: 3.18 min cis-β-methyl styrene and 4.5 min cis-methyl styrene oxide; potential minor side product peaks (not in negative control): 4.76, 6.08, and 6.42 min; retention times: 5.1 min ethylbenzene, 7.5 min 1-phenylethanol, and 7.7 min acetophenone; potential side product peaks (not in negative control): 5.45 min and IS n-dodecane 8.8 min.
135°C hold 4 min, 20°C/min to 195°C hold 5 min, and 30°C/min to 250°C hold 1 min; retention times: 3.3 min ethylbenzene, 9.4 min acetophenone, 11.5 min 1-phenylethanol, and IS 1-octanol 7.5 min.
100°C hold 15 min and 20°C/min to 220°C hold 1 min; retention times: 5.44 min cis-β-methyl styrene and 10.77 min (1R,2S)-cis-methyl styrene oxide.
120°C hold 2.6 min, 15°C/min to 135°C hold 3.3 min, and 25°C/min to 225°C hold 1.0 min; retention times: 2.47 min ethylbenzene, 6.38 min (R)-1-phenylethanol, 6.65 min (S)-1-phenylethanol, 4.30 min acetophenone, and IS n-dodecane 4.58 min.
To investigate the effect of water-soluble organic co-solvents on the activity of rAaeUPO, we evaluated a selection of commonly used solvents ranging from dimethyl sulfoxide (DMSO) (logP = −1.35) to isopropanol (logP = 0.05) at a substrate concentration of 10 mM. As model reactions, we chose the hydroxylation of ethylbenzene and the epoxidation of cis-β-methyl styrene (Figure 1) in the presence of 20 vol% of the respective solvents. It is worth mentioning that the enantioselectivity of both reactions was not significantly impaired by the co-solvent (Supplementary Figures S1, S2).
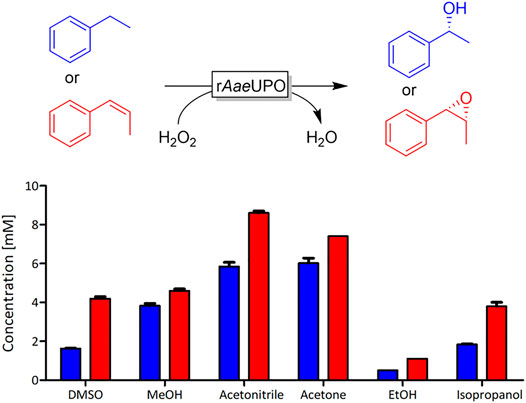
FIGURE 1. rAaeUPO-catalyzed oxyfunctionalization of ethylbenzene (blue) and cis-β-methyl styrene (red). Reaction conditions: [substrate] = 10 mM, [KPi, pH 6] = 100 mM, [co-solvent] = 20 vol%, [rAaeUPO concentrated supernatant] = 1 μM, and [H2O2-feed] = 4 mM/h; reaction time = 2.5 h for ethylbenzene and 3 h for cis-β-methyl styrene, total volume 0.5 mL, 25°C, 600 rpm, duplicate measurements.
Both model reactions were influenced by the presence of the solvents in a similar fashion. There was no obvious correlation between the polarities of the solvent (as expressed by their logP value). Specially for the alcohol-based solvents methanol (MeOH) and ethanol (EtOH), we suspected that possibly alcohol oxidation may compete with the model reaction investigated. Note that the molar surplus of alcohol solvent over the starting material was approximately 250-fold. In fact, using the purpald assay (Dickinson and Jacobsen, 1970; Hopps, 2000), we could qualitatively confirm the formation of formaldehyde and acetaldehyde from the corresponding alcohol oxidations catalyzed by rAaeUPO (Figure 2). We assume that also isopropanol at least to some extend can be converted by rAaeUPO but an analytical validation is yet pending. Also in case of DMSO, which is an inhibitor of peroxygenase activity, GC/MS analysis confirmed rAaeUPO-conversion of the solvent to the sulfone (Supplementary Figure S6). For acetone and acetonitrile, we did not observe any indication for co-solvent oxidation in our experiments and nearly full conversion (≥90%) of 10 mM substrate was achieved under the experimental conditions chosen.
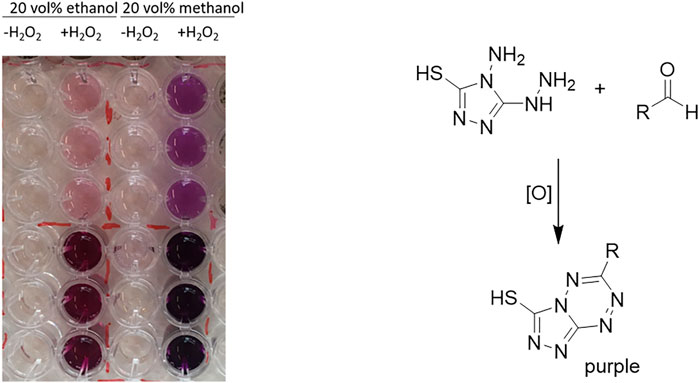
FIGURE 2. Purpald assay of rAaeUPO-catalyzed oxidations of methanol and ethanol. Reaction conditions: [MeOH or EtOH] = 20 vol%, [KPi, pH 6] = 100 mM, [purified rAaeUPO] = 1 μM, and [H2O2-feed] = 4 mM h−1; reaction time = 1.5 h. For analysis, the reaction mixtures were diluted 40 × fold (upper 3 rows) or 4 × fold (lower 3 rows) and then incubated for 1 h at room temperature with the purpald reagent. The negative control is left of the enzyme-containing sample.
We next investigated the influence of the acetonitrile concentration on the rAaeUPO-catalyzed hydroxylation reaction (Figure 3). Increasing the acetonitrile concentration up to approximately 50 vol% had a beneficial effect on product formation. Notably, even in the presence of very high acetonitrile concentrations of 90 vol% significant activity was detected. Possibly, the increasing product accumulation up to 50 vol% can be attributed to the increasing solubility of the reactants in the reaction medium thereby also minimizing the undesired evaporation. With increasing acetonitrile concentrations, also a larger amount of the alcohol product (1-phenylethanol) compared to the double oxidation product acetophenone was formed. We decided to investigate this effect by further determining the kinetic parameters Km and kcat for the substrates ethylbenzene and 1-phenylethanol at different acetonitrile concentrations (Table 1). It was found that increasing acetonitrile concentrations did not significantly affect the kinetic parameters for the substrate ethylbenzene. In contrary, higher Km and higher kcat values for the substrate 1-phenylethanol were observed when the acetonitrile amount was increased. Currently, we are lacking a full understanding of this behavior as the better solubility of the reactants as well as the solvent presence in the active site might contribute to these changes in kinetic parameters and the product concentrations, respectively. Thus, further investigations in this regard need to be performed. Nevertheless, the use of increased acetonitrile concentrations seems attractive in order to avoid over oxidation to the ketone and obtain an accumulation of the desired hydroxylation product.
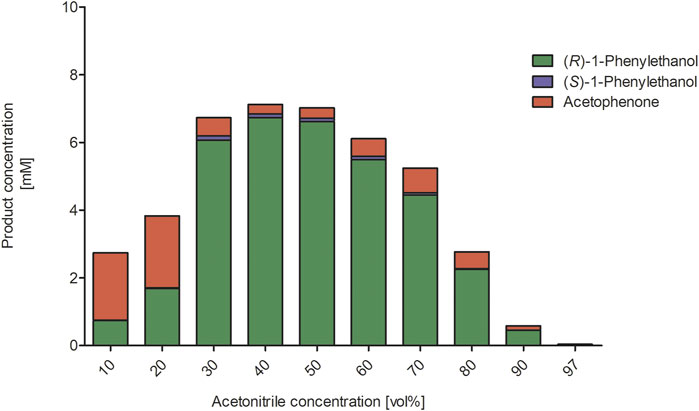
FIGURE 3. Influence of increasing acetonitrile concentrations on rAaeUPO-catalyzed hydroxylation of ethylbenzene. Reaction conditions: The reactions were performed in technical duplicates in potassium phosphate buffer (0.5 mL, 100 mM, pH 6.0) at 25°C and 600 min−1 for 2.5 h. Reaction mixtures contained substrate (10 mM in 10–97 vol% ACN), rAaeUPO (1 μM, concentrated supernatant) and up to 10 mM H2O2, which was added in 2 mM portions every hour. The concentration of the analytes derived from calibration curves. GC-analysis on the chiral column (CP-Chirasil-Dex CB).
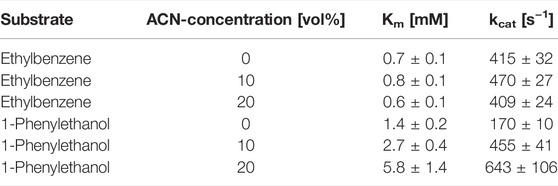
TABLE 1. Effect of increasing acetonitrile (ACN) concentrations on the kinetic parameters of rAaeUPO toward the substrates ethylbenzene or 1-phenylethanol. Reaction conditions: [ethylbenzene or 1-phenylethanol] = 0.3–2 mM, [KPi, pH 7] = 10 mM, [acetonitrile] = 0–20 vol%, [rAaeUPO concentrated supernatant] = 8.4 nM, [H2O2-concentration] = 1.7 mM; reaction time = 15 s, total volume 8 mL, room temperature, and triplicate measurements.
Increasing the co-solvent concentration beyond 50 vol% most likely has a negative impact on the structural integrity of the biocatalyst explaining the decreasing product accumulation (Figure 3). Similar observations were also made for the epoxidation of cis-β-methyl styrene, indicating that these effects are independent on the tested substrate (Supplementary Figure S7). To further investigate the influence of acetonitrile on the stability of rAaeUPO, we performed a range of pre-incubation experiments exposing the enzyme to varying concentrations of acetonitrile and determining at intervals the residual activity (Figure 4). While the enzyme, incubated in buffer without any acetonitrile, showed no significant decrease in catalytic activity over several days, a sharp drop was observed in the presence of 50 and 80 vol% acetonitrile, resulting in approximately 40 and 1% of residual enzyme activity after 1 h. In the presence of 50 vol% acetonitrile, rAaeUPO activity did not decrease further for several days. The dramatic decrease in catalytic activity was accompanied by a green precipitate evolving in the incubation mixture. We assume that the dramatic activity losses in the first few hours of incubation can be attributed to precipitation of the biocatalyst. Activity measurements of the recovered precipitate from 50 vol% acetonitrile resuspended in buffer indicated that the precipitated UPO was still active (Supplementary Figure S12). Thus, the loss of activity in 50 vol% acetonitrile is most likely not due to enzyme inactivation but rather to a decreased solubility of the enzyme in the acetonitrile/buffer mixture. Consequently, future studies might provide exciting possibilities to use acetonitrile for UPO precipitation to easily obtain active enzyme immobilisates and compare them to previous immobilization procedures (Rauch et al., 2019; Hobisch et al., 2020b).
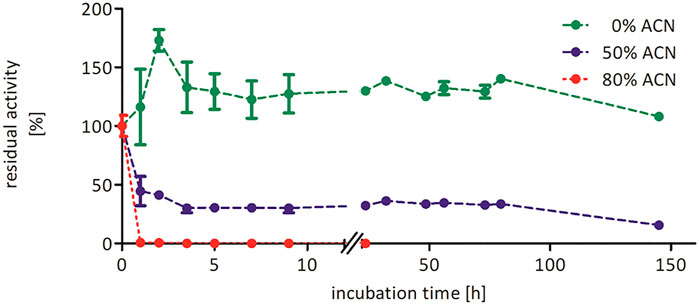
FIGURE 4. Stability of rAaeUPO (concentrated supernatant) in the presence of 0 vol% (green), 50 vol% (blue), or 80 vol% (red) acetonitrile. Enzyme (1 µM) was incubated in 100 mM KPi, pH 6.0, and indicated acetonitrile concentrations at 25°C and 600 rpm in glass vials with 10 mL starting volume. At indicated time points, a 200 µL aliquot was transferred to an Eppendorf tube, and enzyme activity was evaluated via ABTS assay. Data are normalized to the 0 h sample, which was taken immediately after mixing (t0 = 27.3 U/mL).
Complementary to the stability measurements, we followed the rAaeUPO-catalyzed oxidation of the model substrate ethylbenzene over time in the presence of high concentrations of acetonitrile (Figure 5). We observed a different effect of the tested acetonitrile concentration of 50 and 80 vol%. The product concentration increased linearly within 7 h in the presence of 50 vol% acetonitrile, whereas the reaction stopped earlier in the presence of 80 vol% acetonitrile. This resulted in a maximum target product concentration of 57 mM (R)-1-phenylethanol in 80 vol% acetonitrile. Looking at the stability measurements (Figure 4), the lower product formation in the presence of 80 vol% acetonitrile can be attributed to insufficient enzyme activity and stability in the presence of co-solvent. In this regard, it is still astonishing that over 50 mM (R)-1-phenylethanol are formed though the enzyme activity was less than 1% compared to its initial value according to our stability measurements. Using 50 vol% acetonitrile for the UPO-mediated oxidation of cis-β-methyl styrene resulted in similar trends, indicating that these conditions can also be applied for other UPO substrates (Supplementary Figure S8).
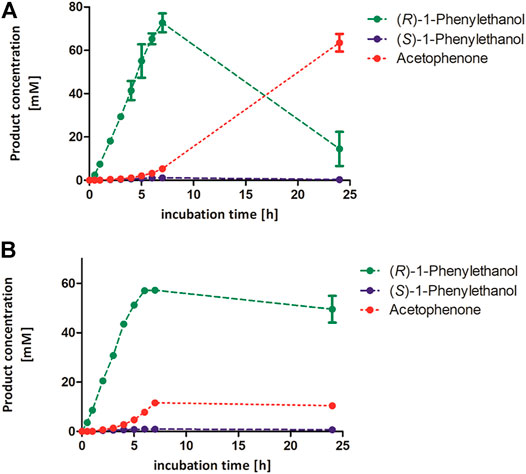
FIGURE 5. Product concentrations during conversion of 100 mM ethylbenzene by rAaeUPO applying 50 vol% (A) or 80 vol% acetonitrile (B). Reaction conditions: 100 mM potassium phosphate buffer, pH 6.0, 1 µM rAaeUPO (concentrated supernatant), and H2O2-dosing rate: 8 mM h−1. The concentration of both 1-phenylethanol enantiomers and acetophenone were calculated based on a calibration curve recorded earlier. Duplicates. GC-analysis was performed on the chiral column (CP-Chirasil-Dex CB).
Encouraged by these promising results, we decided to demonstrate the preparative-scale application using an ethylbenzene starting concentration of 300 mM (Figure 6). The reaction produced smoothly accumulating enantiomerically pure (R)-1-phenylethanol until nearly all starting material was consumed after 6 h. Only then, over oxidation to acetophenone occurred. Within 6 h, 188 mM of enantiomerically pure (R)-1-phenylethanol has been formed corresponding to a turnover number (TN = molProduct × molrAaeUPO−1) of 18.800 and a turnover frequency (TOF) of 0.9 s−1 (over 6 h). These turnover numbers were lower compared to previous results from our group, where for the same substrate and enzyme in a two-liquid phase system TNs of 340.000 were achieved (Tonin et al., 2021). However, it should be pointed out that the achieved concentration of 120 mM (R)-1-phenylethanol was obtained after 7 days of incubation resulting in a low product accumulation rate of 0.7 mM h−1. The initial (R)-1-phenylethanol accumulation rate in our experiments was more than 31 mM h−1 (3.8 gproduct h−1 L−1) and thus represent to the best of our knowledge one of the highest productivities reported for a biocatalytic oxyfunctionalization reaction to date.
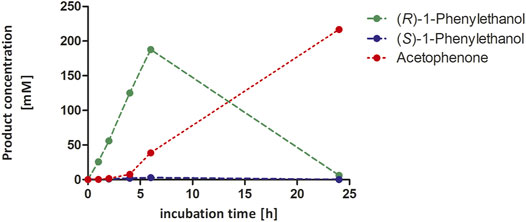
FIGURE 6. Hydroxylation of ethylbenzene to (R)-1-phenylethanol in presence of 50 vol% acetonitrile in 100 mL scale. Reaction conditions: 300 mM substrate concentration, 100 mM potassium phosphate buffer, pH 6.0, 10 µM rAaeUPO (concentrated supernatant), and H2O2-dosing rate: 50 mM h−1. The reaction was stirred at 300 rpm at 25°C. The concentration of all analytes derived from calibration curves. GC-analysis was performed on the chiral column (CP-Chirasil-Dex CB).
Overall 216 mM of acetophenone (double oxidation product) were obtained corresponding to 25.9 g L−1 and 73% yield on the starting material used. The product to the catalyst ratio was 70 gproduct g−1AaeUPO. As no starting material and only minor (R)-1-phenylethanol was detected after 24 h, we conclude that still evaporation of the reagents (especially the starting material) occurred. Nevertheless, these numbers bring peroxygenase-catalysis close to industrial requirements (Tufvesson et al., 2011).
From an environmental point of view, we believe that the application of acetonitrile represents an interesting alternative to pure aqueous media. A simple E-factor analysis (Sheldon, 2017; 2018) (Figure 7) revealed that in the experiment shown in Figure 6 approximately 35.2 gwaste g−1product have been generated. 98% of this waste has been caused by the solvents (19.3 gwater g−1product and 15.2 gacetonitrile g−1product). The non-reacted starting material and phosphate buffer contributed roughly 1 g g−1product each, whereas the contribution of the biocatalyst was almost negligible (<0.02 gAaeUPO g−1product). Hence, the overall E-factor compared to similar reactions could be reduced more than 7-fold (Xu et al., 2022). In this regard, the direction for further improvements of the reaction’s environmental footprint is also clear by further increasing the product concentration.
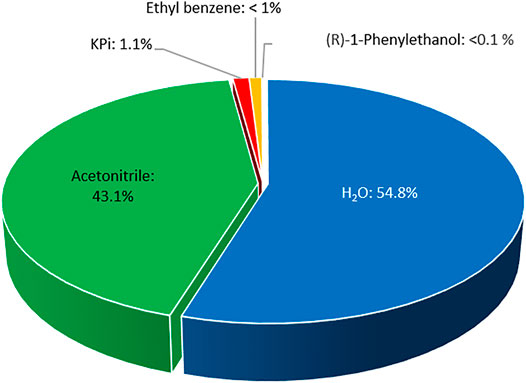
FIGURE 7. E-factor contributions.
Obviously, a simple E-factor analysis does not replace a more detailed environmental impact analysis taking the history of the reagents used and energy contributions into account (Tieves et al., 2019a). Nevertheless, this analysis already demonstrates the impact of increasing product concentrations.
Water-miscible organic solvents represent a simple measure to increase the substrate- and product concentrations in biocatalysis reactions and thereby decrease the solvent wastes generated. In this contribution, we demonstrated that the peroxygenase from Agrocybe aegerita exhibits superb activity and stability in the presence of acetonitrile, enabling significantly increased reagent payloads in AaeUPO-catalyzed oxyfunctionalization reactions. This feature makes the use of this peroxygenase already remarkable as other heme-containing enzymes such as the peroxygenase HspUPO or the frequently investigated cytochrome P450 BM3 get already inactivated in presence of low concentrations of water-miscible co-solvents of typically less than 20 vol% (Seng Wong et al., 2004; Rotilio et al., 2021). Further improvements may be even expected using evolved variants of rAaeUPO with even higher solvent tolerance (Martin-Diaz et al., 2021).
The productivities and product concentrations achieved in preliminary experiments using high co-solvent concentrations point toward economically feasible and environmentally benign preparative applications.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
TH planned, designed and carried out most experiments, analysed the data, and drafted the manuscript. AvT performed and evaluated kinetic experiments with different co-solvent concentrations. MA and FH acquired funding, gave advices in the research work, helped in drafting the manuscript, and revised the manuscript. All authors read and approved the final manuscript.
This work was performed as a part of the Bio Based Industries Joint Undertaking under the European Union’s Horizon 2020 Research and Innovation program (Grant Agreement No.: 886567, BIZENTE project).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Remco van Oosten for technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fctls.2022.882992/full#supplementary-material
Churakova, E., Kluge, M., Ullrich, R., Arends, I., Hofrichter, M., and Hollmann, F. (2011). Specific Photobiocatalytic Oxyfunctionalization Reactions. Angew. Chem. Int. Ed. 50 (45), 10716–10719. doi:10.1002/anie.201105308
Dickinson, R. G., and Jacobsen, N. W. (1970). A New Sensitive and Specific Test for the Detection of Aldehydes: Formation of 6-Mercapto-3-Substituted-S-Triazolo [4, 3-B]-S-Tetrazines. J. Chem. Soc. D (24), 1719–1720. doi:10.1039/c29700001719
Doukyu, N., and Ogino, H. (2010). Organic Solvent-Tolerant Enzymes. Biochem. Eng. J. 48 (3), 270–282. doi:10.1016/j.bej.2009.09.009
Dutta Banik, S., Nordblad, M., Woodley, J. M., and Peters, G. H. (2016). A Correlation between the Activity of Candida antarctica Lipase B and Differences in Binding Free Energies of Organic Solvent and Substrate. ACS Catal. 6 (10), 6350–6361. doi:10.1021/acscatal.6b02073
Fernández-Fueyo, E., Ni, Y., Gomez Baraibar, A., Alcalde, M., van Langen, L. M., and Hollmann, F. (2016). Towards Preparative Peroxygenase-Catalyzed Oxyfunctionalization Reactions in Organic Media. J. Mol. Catal. B Enzym. 134, 347–352. doi:10.1016/j.molcatb.2016.09.013
Hobisch, M., Holtmann, D., de Santos, P. G., Alcalde, M., Hollmann, F., and Kara, S. (2020a). Recent Developments in the Use of Peroxygenases–Exploring Their High Potential in Selective Oxyfunctionalisations. Biotechnol. Adv. 51, 107615. doi:10.1016/j.biotechadv.2020.107615
Hobisch, M., Schie, M. M. C. H., Kim, J., Røjkjær Andersen, K., Alcalde, M., Kourist, R., et al. (2020b). Solvent‐Free Photobiocatalytic Hydroxylation of Cyclohexane. ChemCatChem 12 (16), 4009–4013. doi:10.1002/cctc.202000512
Holtmann, D., and Hollmann, F. (2022). Is Water the Best Solvent for Biocatalysis? Mol. Catal. 517, 112035. doi:10.1016/j.mcat.2021.112035
Hopps, H. B. (2000). Purpald (R): a Reagent that Turns Aldehydes Purple. Aldrichim. Acta 33 (1), 28–30.
Klibanov, A. M. (2001). Improving Enzymes by Using Them in Organic Solvents. Nature 409 (6817), 241–246. doi:10.1038/35051719
Martin-Diaz, J., Molina-Espeja, P., Hofrichter, M., Hollmann, F., and Alcalde, M. (2021). Directed Evolution of Unspecific Peroxygenase in Organic Solvents. Biotechnol. Bioeng. 118 (8), 3002–3014. doi:10.1002/bit.27810
Molina-Espeja, P., Garcia-Ruiz, E., Gonzalez-Perez, D., Ullrich, R., Hofrichter, M., and Alcalde, M. (2014). Directed Evolution of Unspecific Peroxygenase from Agrocybe Aegerita. Appl. Environ. Microbiol. 80 (11), 3496–3507. doi:10.1128/aem.00490-14
Ni, Y., Holtmann, D., and Hollmann, F. (2014). How green Is Biocatalysis? to Calculate Is to Know. ChemCatChem 6 (4), 930–943. doi:10.1002/cctc.201300976
Poraj-Kobielska, M., Peter, S., Leonhardt, S., Ullrich, R., Scheibner, K., and Hofrichter, M. (2015). Immobilization of Unspecific Peroxygenases (EC 1.11.2.1) in PVA/PEG Gel and Hollow Fiber Modules. Biochem. Eng. J. 98, 144–150. doi:10.1016/j.bej.2015.02.037
Rauch, M. C. R., Tieves, F., Paul, C. E., Arends, I. W. C. E., Alcalde, M., and Hollmann, F. (2019). Peroxygenase‐Catalysed Epoxidation of Styrene Derivatives in Neat Reaction Media. ChemCatChem 11 (18), 4519–4523. doi:10.1002/cctc.201901142
Rotilio, L., Swoboda, A., Ebner, K., Rinnofner, C., Glieder, A., Kroutil, W., et al. (2021). Structural and Biochemical Studies Enlighten the Unspecific Peroxygenase from Hypoxylon Sp. EC38 as an Efficient Oxidative Biocatalyst. ACS Catal. 11, 11511–11525. doi:10.1021/acscatal.1c03065
Seng Wong, T., Arnold, F. H., and Schwaneberg, U. (2004). Laboratory Evolution of Cytochrome P450 BM-3 Monooxygenase for Organic Cosolvents. Biotechnol. Bioeng. 85 (3), 351–358. doi:10.1002/bit.10896
Serdakowski, A. L., and Dordick, J. S. (2008). Enzyme Activation for Organic Solvents Made Easy. Trends Biotechnol. 26 (1), 48–54. doi:10.1016/j.tibtech.2007.10.007
Sheldon, R. A., and Woodley, J. M. (2018). Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 118, 801–838. doi:10.1021/acs.chemrev.7b00203
Sheldon, R. A. (2017). The E Factor 25 Years on: the Rise of green Chemistry and Sustainability. Green. Chem. 19 (1), 18–43. doi:10.1039/c6gc02157c
Sheldon, R. A. (2018). Metrics of Green Chemistry and Sustainability: Past, Present, and Future. ACS Sustain. Chem. Eng. 6 (1), 32–48. doi:10.1021/acssuschemeng.7b03505
Stepankova, V., Bidmanova, S., Koudelakova, T., Prokop, Z., Chaloupkova, R., and Damborsky, J. (2013). Strategies for Stabilization of Enzymes in Organic Solvents. ACS Catal. 3 (12), 2823–2836. doi:10.1021/cs400684x
Tieves, F., Tonin, F., Fernández-Fueyo, E., Robbins, J. M., Bommarius, B., Bommarius, A. S., et al. (2019a). Energising the E-Factor: The E+-factor. Tetrahedron 75 (10), 1311–1314. doi:10.1016/j.tet.2019.01.065
Tieves, F., Willot, S. J. P., van Schie, M. M. C. H., Rauch, M. C. R., Younes, S. H. H., Zhang, W., et al. (2019b). Formate Oxidase (FOx) from Aspergillus oryzae: One Catalyst Enables Diverse H2O2-Dependent Biocatalytic Oxidation Reactions. Angew. Chem. Int. Ed. 58 (23), 7873–7877. doi:10.1002/anie.201902380
Tonin, F., Tieves, F., Willot, S., van Troost, A., van Oosten, R., Breestraat, S., et al. (2021). Pilot-Scale Production of Peroxygenase from Agrocybe Aegerita. Org. Process. Res. Dev. 25 (6), 1414–1418. doi:10.1021/acs.oprd.1c00116
Tufvesson, P., Lima-Ramos, J., Nordblad, M., and Woodley, J. M. (2011). Guidelines and Cost Analysis for Catalyst Production in Biocatalytic Processes. Org. Process. Res. Dev. 15 (1), 266–274. doi:10.1021/op1002165
van Schie, M. M. C. H., Spöring, J.-D., Bocola, M., Domínguez de María, P., and Rother, D. (2021). Applied Biocatalysis beyond Just Buffers - from Aqueous to Unconventional media. Options and Guidelines. Green. Chem. 23 (9), 3191–3206. doi:10.1039/d1gc00561h
Keywords: peroxygenase, oxyfunctionalization, co-solvent, hydroxylation, epoxidation, medium engineering
Citation: Hilberath T, van Troost A, Alcalde M and Hollmann F (2022) Assessing Peroxygenase-Mediated Oxidations in the Presence of High Concentrations of Water-Miscible Co-Solvents. Front. Catal. 2:882992. doi: 10.3389/fctls.2022.882992
Received: 24 February 2022; Accepted: 07 April 2022;
Published: 23 May 2022.
Edited by:
Francesca Paradisi, University of Bern, SwitzerlandReviewed by:
Vicente Gotor-Fernández, University of Oviedo, SpainCopyright © 2022 Hilberath, van Troost, Alcalde and Hollmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Hilberath, dC5oaWxiZXJhdGhAdHVkZWxmdC5ubA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.