
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 23 February 2024
Sec. Heart Failure and Transplantation
Volume 11 - 2024 | https://doi.org/10.3389/fcvm.2024.1251780
This article is part of the Research Topic Current Proceedings in Magnetocardiology View all 8 articles
 Nicolas Musigk1
Nicolas Musigk1 Phillip Suwalski1
Phillip Suwalski1 Ainoosh Golpour1
Ainoosh Golpour1 DeLisa Fairweather2,3,4
DeLisa Fairweather2,3,4 Karin Klingel5
Karin Klingel5 Pilar Martin6
Pilar Martin6 Andrea Frustaci7
Andrea Frustaci7 Leslie T. Cooper2
Leslie T. Cooper2 Thomas F. Lüscher8,9
Thomas F. Lüscher8,9 Ulf Landmesser1Bettina Heidecker1*
Ulf Landmesser1Bettina Heidecker1*
Infiltration of the myocardium with various cell types, cytokines and chemokines plays a crucial role in the pathogenesis of cardiomyopathies including inflammatory cardiomyopathies and myocarditis. A more comprehensive understanding of the precise immune mechanisms involved in acute and chronic myocarditis is essential to develop novel therapeutic approaches. This review offers a comprehensive overview of the current knowledge of the immune landscape in cardiomyopathies based on etiology. It identifies gaps in our knowledge about cardiac inflammation and emphasizes the need for new translational approaches to improve our understanding thus enabling development of novel early detection methods and more effective treatments.
Graphical Abstract
Cardiomyopathy may result from various etiologies associated with a reduction in left ventricular ejection fraction (LVEF) (1). Myocarditis is characterized by inflammation of the myocardium and can progress to chronic inflammatory cardiomyopathy or dilated cardiomyopathy (DCM) in susceptible individuals (2, 3). Immune cell infiltration (as illustrated in Figures 1–3 showcasing representative EMB findings), cytokines and chemokines play a central role in this process (2, 3). Resident mononuclear immune cells in the pericardium have been reported to amplify or regulate the heart-specific adaptive immune responses (4).
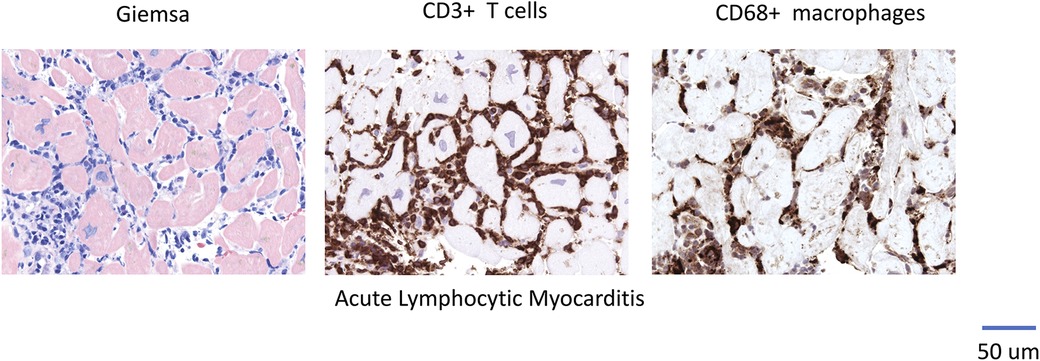
Figure 1. Representative EMB Findings in Acute Lymphocytic Myocarditis.
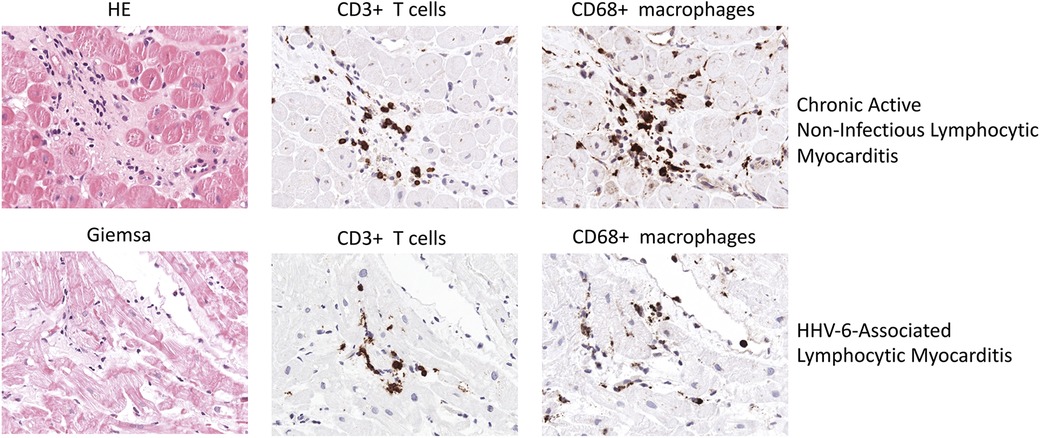
Figure 2. Representative EMB findings in chronic active Non-infectious lymphocytic myocarditis and HHV-6-assoziated lymphocytic myocarditis.
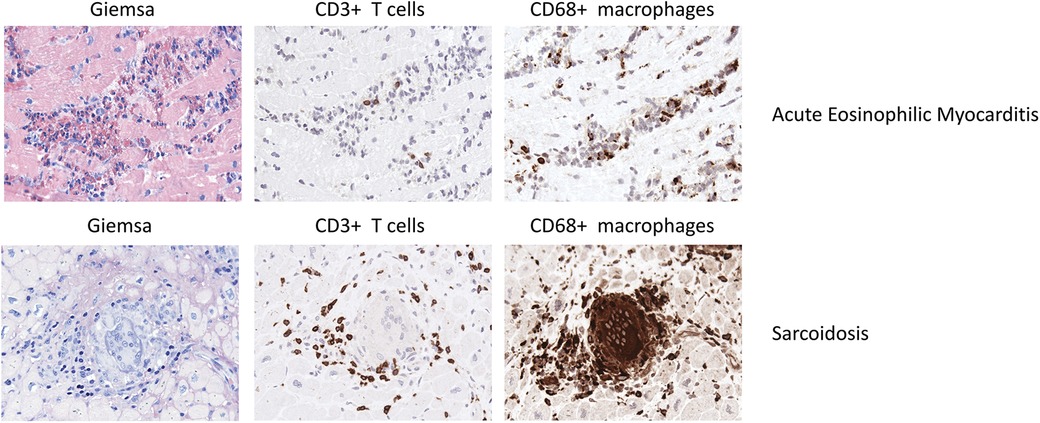
Figure 3. Representative EMB findings in acute eosinophilic myocarditis and sarcoidosis.
The precise prevalence of myocarditis remains uncertain. Myocarditis has been documented to occur in approximately 10–106 cases per 100,000 people globally (5). Patients suffering from cardiomyopathies may present with a spectrum of symptoms ranging from chest pain, dyspnea, palpitations or syncope to cardiogenic shock, in particular in the context of fulminant myocarditis (3, 6, 7).
Inflammatory cardiomyopathy may be triggered by many factors including infections, drugs, autoimmune conditions, and toxins and maintained through dysregulation of the immune system, which plays a critical role in the development and progression of the disease (8–11).
Viral infections are considered to be the most common cause of myocarditis, while numerous other etiologies of myocarditis and inflammatory cardiomyopathy have been reported (5, 12). Inflammatory infiltrates have been increasingly observed in endomyocardial biopsies (EMB) of various forms of non-ischemic cardiomyopathy including peripartum cardiomyopathy, takotsubo cardiomyopathy and cardiomyopathies associated with genetic variants of ion channels or structural proteins (8, 13, 14). Importantly, the release of proinflammatory and profibrotic cytokines, as well as other mediators by immune cells during acute myocarditis is critical for the progression from acute myocarditis to chronic inflammatory cardiomyopathy or DCM (15–21).
Recently there has been growing interest in the scientific community in immune cell-based and immunosuppressive therapies to restore immune homeostasis and to positively influence the clinical course at an early stage.
Gullestad et al. found that patients with congestive heart failure or idiopathic DCM that received intravenous immunoglobulin (IVIG) therapy produced elevated blood levels of the anti-inflammatory and anti-fibrotic mediators interleukin (IL)-10, IL-1 receptor antagonist and soluble tumor necrosis factor (TNF) receptor, leading to improved LVEF (22). It has also been shown in several studies that immunosuppressive therapy with steroids, azathioprine and/or cyclosporin leads to improved LVEF, less hospitalizations, less need for heart transplantation, or death in patients with chronic inflammatory cardiomyopathy (23–26). Targeting CD20+ B-lymphocytes with the CD20 antibody rituximab improved hemodynamics in a series of patients with inflammatory DCM or cardiac sarcoidosis (27, 28).
Animal models of myocarditis and cardiomyopathies have provided insight into the role of inflammation in such entities. The primary models used were autoimmune and coxsackievirus B3 (CVB3) murine models (29, 30). They revealed that the primary immune infiltrate in the heart during acute myocarditis in viral and autoimmune models, as well as in patient biopsies, are macrophages, followed by smaller percentages of T and B cells and other cell populations like natural killer, dendritic and mast cells (19, 20, 31, 32). Neutrophils are important in the pathogenesis of myocarditis, but they appear in the heart early after infection or damage and so are not often observed in EMB. Innate immune factors that are critical in driving acute myocardial inflammation and progression to chronic inflammatory cardiomyopathy such as complement, Toll-like receptor (TLR)2 and TLR4, inflammasome components like NLR-family pyrin domain-containing protein 3 (NLRP3) and cytokines including IL-1β, IL-18, IL-6 and transforming growth factor (TGF)β1 are released from mast cells and macrophages (6, 19, 20). Mast cells are often overlooked in cardiomyopathy, but they are critical for remodeling to occur as they release most of the enzymes needed to activate the cytokines and other factors involved in remodeling and fibrosis (15, 33, 34). Mast cell degranulation is also typically associated with pericarditis, which occurs frequently during myocarditis in animal models. Remodeling and fibrosis along the pericardium are important drivers of progression from myocarditis to DCM (21, 35).
However, T cells also play a critical role in the pathogenesis of inflammatory cardiomyopathy and are frequently found in EMB (36). Importantly, the chronic phase of cardiomyopathy is an endpoint with significantly fewer immune cells, whilst more T cells are present (37).
In a mouse model of CVB3 myocarditis, CD8+ depletion had no significant effect on disease progression (38). Furthermore, CVB3-induced cardiac injury and prevention of chronic myocarditis was found to be unrelated to perforin-mediated cytotoxicity in mice (39). T cells and T cell derived cytokines including IL-4 and IL-17 seem to play a major role in promoting chronic inflammatory cardiomyopathy (38, 40–44). Although interferons (IFNs) such as IFNγ increase acute myocarditis, they might also be of protective value, as they decrease viral replication and protect against chronic inflammatory cardiomyopathy by inhibiting profibrotic factors including IL-4 and IL-17 (21, 41, 44–47). CD4+ CD25+ FoxP3+ regulatory T cells (Treg) might have protective function, as a low number of Treg cells has been associated with worse DCM in autoimmune myocarditis (44, 48). Additionally, in viral and autoimmune models of myocarditis, females, as they have higher levels of Treg cells and other regulatory factors such as T cell Ig mucin (Tim)-3 and IL-4, exhibit notably lower rates of myocarditis compared to males (32, 49). Administering Treg cells prophylactically in a CVB3-induced myocarditis mouse model conferred protection against CVB3-induced myocarditis by exerting anti-inflammatory and antifibrotic effects (50).
There is mounting evidence that a detailed understanding of specific cell-cell interactions in myocarditis and inflammatory cardiomyopathy as well as involved signaling molecules will be paramount in the development of targeted novel therapies.
This narrative review aims to provide a comprehensive overview of the current knowledge on immune cells infiltrating the myocardium in myocarditis and non-ischemic cardiomyopathies that may be used as a resource for scientists investigating targeted cell-based therapies for cardiomyopathies.
Myocardial infiltration is observed following infection by different coxsackieviruses via the coxsackievirus-adenovirus receptor (CAR) (51). CVB3 virus RNA is recognized by TLR3, TLR4 and melanoma differentiation-associated protein (MDA)-5 (52). Damage-associated molecular patterns (DAMPs) initiate the immune response from antigen presenting cells including mast cells, macrophages and dendritic cells. In that context, histamine, complement, and IL-1β activate infiltration of a diverse range of immune cells to the heart (6), including monocytes (6), natural killer cells (53), neutrophils (54), lymphocytes (55), and macrophages (55, 56). Macrophages are the dominant immune infiltrate in males with lower levels of CD4+ and CD8+ T cells as well as γδ T cells (56), B cells (57) and mast cells (20, 32). Females have higher levels of T and B cells as well as Treg compared to males (58). B cells might play a role in progression to chronic disease, especially in females and autoimmune myocarditis (57), while Treg cells might protect from progression to chronic myocarditis (53). Furthermore, Ly6Chigh monocytes are detected in EMBs (59). Ly6Chigh monocytes play a crucial role in early inflammation during acute myocarditis and are recognized for their ability to produce abundant levels of proinflammatory cytokines such as IL-1β (60–63). Additionally, they possess phagocytic capabilities (60–63).
Also in enterovirus myocarditis, the effect of IFNβ therapy has been explored (64). As stated earlier, IFNs including IFNβ and IFNγ reduce CVB3 replication and remodeling, which prevents progression to chronic cardiomyopathy (46, 47). Elevated IFN responses are a key reason that C57BL/6 mice do not progress from myocarditis to DCM in animal models of myocarditis (46, 47). Other studies explored pocapavir, pleconaril and IVIG in neonates with enterovirus myocarditis (65–67). Consensus statements, however, do not recommend antiviral therapies in enterovirus myocarditis as there is not sufficient evidence (3).
Soluble anti-CAR antibody reduces the incidence of acute and chronic CVB3-induced myocarditis in mice by preventing viral infection (68, 69). In another study, progression to chronic cardiomyopathy was prevented by anti-mouse IL-1β therapy (70). A primary pathway increasing acute inflammation and promoting progression to DCM in white background male mice is the TLR4-induced IL-1β response (20, 70–72). IL-1β increases IL-6 that is needed for IL-17/Th17 responses that promote remodeling and chronic cardiomyopathy (73). Male mice had increased IL-17-related responses compared to females and showed more fibrosis leading to chronic cardiomyopathy in CVB3-induced myocarditis (74). However, IL-17 does not increase acute myocarditis but is important for the progression to chronic cardiomyopathy which also predominantly occurs in males (20, 75, 76). Additionally, in acute murine CVB3 myocarditis high expression levels of osteopontin were found to be associated with the development of extensive fibrosis that can be reduced by treatment with a vitamin D analog (77). A recent study demonstrated that administering eplerenone can regulate the acute immune response and protect against myocardial remodeling in CVB3-induced myocarditis (78). Similarly, we had previously shown in an animal model of myocardial infarction that inflammatory genes activated during ischemia were downregulated through treatment with eplerenone (79). This response to treatment was much greater in females (79).
Parvovirus B19 (B19V) infects endothelial cells (80) which may lead to apoptosis of cardiomyocytes (80, 81). Only high copy numbers of B19V DNA in the myocardium were found to be associated with acute lymphocytic myocarditis (82). It is likely that the toxic, non-structural viral protein NS1 triggers the release of proinflammatory cytokines (83), although the pathogenetic role of B19V in myocarditis is still controversial (84). Immune cell infiltration in patients with Parvovirus B19V myocarditis is dominated by macrophages and lymphocytes (85). CD4+ T cells play a role in acute Parvovirus B19V-related myocarditis (86). In individuals, striking CD8+ T cell responses were observed, which were sustained or even increased over many months after the resolution of acute disease (87).
Although no guidelines currently recommend the use of IVIG for patients with severe Parvovirus B19V viremia and associated complications such as transient aplastic crisis or chronic pure red cell aplasia, some studies suggest that IVIG may be beneficial in such cases (88). Other authors showed no significant improvement in cardiac systolic function in patients undergoing IVIG, thus rendering the potential benefits of IVIG uncertain (89). No therapy is recommended when B19V copy number is low and cardiac inflammation is absent in EMB (3). When EMB is positive for inflammation although B19V copy number is low, immunosuppressive therapy may be considered (90).
New therapeutic strategies are under investigation, including the synthetic nucleotide analogues cidofovir and brincidofovir as well as flavonoid molecules and hydroxyurea (88).
HCMV is usually acquired during childhood and is known to infect several cell types, such as endothelial cells, epithelial cells, and immune cells (91, 92). Usually, HCMV infection remains asymptomatic until the occurrence of immunosuppression, thereby posing a significant concern as a complication in individuals such as organ transplant recipients or Human Immunodeficiency Virus (HIV) patients (92).
Male BALB/c mice infected with murine CMV (MCMV) develop acute and chronic inflammatory myocarditis similar to CVB3 and autoimmune models, where macrophages predominate with fewer T-, B-, and other cells during the acute phase of the disease (37, 93, 94).
A reduction of viral burden might be achieved with the anti-herpesvirus drugs ganciclovir or acyclovir, although the efficacy in HCMV-induced myocarditis has not been studied directly (95, 96). Viral infection was reduced in MCMV-induced myocarditis if ganciclovir and cidofovir were administered during the innate immune response but not if it was given during acute myocarditis (97). A controlled trial suggested that a CMV hyperimmunoglobulin treatment may be effective in HCMV myocarditis (98). Antiviral therapy is not generally recommended for patients with virus-induced myocarditis and should be reserved for individual cases (3). Consultation with an infectious disease specialist is recommended before initiating antiviral therapy (3).
EBV invades cardiac tissue by initially infecting resting human B lymphocytes (96) and eventually infiltrating both the myocardium and pericardium (99). EMB from patients with EBV myocarditis typically reveals lymphocytic infiltration, consisting of predominantly CD8+ T cells but also CD4+ T cells (99). In this case, high numbers of EBV-encoded RNA copies were demonstrated in CD8+ T lymphocytes (99).
HHV6-induced myocarditis has been associated with a myocardial infiltration of CD4+ and CD8+ T cells (100). Figure 2 illustrates EMB findings representative for HHV6-associated lymphocytic myocarditis. In a case report, Rohayem and colleagues reported a patient suffering from lethal acute B19V and HHV6 coinfection who at autopsy was found to have diffuse infiltration of the myocardium with mononuclear cells and neutrophils as well as edema, degeneration and loss of myocardial cells (101). However, low HHV6 copy numbers are a common finding and are likely to have no clinical impact (102).
Chronic HCV infection may lead to cardiomyocyte hypertrophy and therefore hypertrophic cardiomyopathy due to expression of the HCV-core protein and overactivation of transcription factor AP-1 (103). The HCV-core protein plays a significant role in the viral nucleocapsids and has an impact on the transcription of cellular protooncogenes in hepatocytes (103, 104). HCV is also recognized to contribute to the development of insulin resistance and the generation of reactive oxygen species (105). The mechanisms that cause damage in relation to HCV-associated cardiomyopathy are not well understood. Hypotheses aiming to explain myocardial damage include direct mitochondrial disruption and oxidative stress to cardiomyocytes and a chronic systemic inflammatory state, which might lead to myocardial inflammation (103, 106). Furthermore, HCV core protein may lead to the activation of profibrotic pathways eventually leading to cardiac fibrosis (103, 106). In HCV-associated myocarditis, monocytes and CD68+ macrophages are the dominating cells infiltrating the heart (107).
HLA-DPB1*0901 and HLA-DRB1*1201 are associated with progression to chronic cardiomyopathy and persistence of HCV as well as the development of DCM (106). Mononuclear cells (in particular monocytes and CD68+ macrophages) are the primary target cells for treating extrahepatic manifestations in HCV infections (107).
Even though the viral genome typically persists in the myocardium after HCV infection, immunosuppression may be beneficial for HCV myocarditis, as it was shown to improve cardiac function (108).
In some cases of HIV-associated cardiomyopathy, diffuse myocardial damage with variable degrees of hypertrophy and degenerative changes leading to hydropic changes within the cardiomyocytes has been noted (109, 110). The HIV gp120 protein mediates myocardial injury and dysfunction through a nitric oxide dependent mechanism (111). Interstitial and endocardial fiber damage is increased, leading to fibrosis (109, 110, 112). T cells dominate the inflammatory infiltrate, with a majority of CD8+ T cells (109).
Cytokine-mediated cardiotoxicity and autoimmune response against components of the heart are likely involved in myocarditis following influenza virus infection (113). Monocytes, dendritic cells, and macrophages dominate the myocardial immune infiltration. The latter two secrete cytokines such as IFNs and TNF-α, contributing to increased acute inflammation (114, 115).
MERS-CoV-associated cardiomyopathy may be associated with myocyte hypertrophy, moderate coronary atherosclerosis and patchy myocardial fibrosis (116). There is limited data regarding myocardial immune infiltration. Lymphocytic infiltration (117, 118) predominantly with dendritic cells (119), macrophages (120), and T cells (121) has been described in MERS-CoV-induced myocarditis.
Edema and atrophy of myocardial fibers is commonly seen in myocarditis associated with SARS-CoV infection (122). Immune infiltration primarily consists of lymphocytes. Also, monocytes and plasma cells infiltrating the endothelium have been described (122). As the ACE2 receptor is the entry point for SARS-CoV into cells, the protein might represent a therapeutic target (123).
The role of SARS-CoV-2 on myocarditis was recently reviewed (124). Briefly, autopsy and histopathological findings suggest extensive lymphocytic infiltration in the myo- and pericardium after lethal SARS-CoV-2 infection (125, 126). An inflammatory state with predominantly CD68+ macrophages (124, 127) as well as enhanced monocyte recruitment (128) has been reported for cardiomyopathy associated with SARS-CoV-2 infection. Furthermore, a cytokine storm, dominated by IL-1β has been shown to play a major role in the pathophysiology of severe corona virus disease 2019 (COVID)-19 (129–131). IL-6 has been associated with cardiac dysfunction, as evidenced by reduced left ventricular function obtained with speckle tracking echocardiography in patients hospitalized due to COVID-19 (124, 132).
ACE2 receptor (124, 133) and transmembrane protease serine subtype 2 (TMPRSS2) (96, 124) interaction enables SARS-CoV-2 to enter its target cells (133). The interaction with the ACE2 receptor has been suggested as a potential direct cytotoxic effect of SARS-CoV-2 (133). While the possibility of direct damage to the heart by SARS-CoV-2 has been discussed (124), the prevailing absence of the virus within cardiomyocytes of COVID-19-associated myocarditis patients (134, 135) further reinforces the concept of cytokine-induced damage to the heart.
Several antiviral therapies are currently being investigated, such as protease inhibitors (e.g., lopinavir-ritonavir, darunavir), RNA polymerase inhibitors (remdesivir) and anti-cytokine agents (e.g., IL-6 receptor antagonists) (96). In patients with SARS-CoV-2, the IL-1β antagonist canakinumab improved clinical recovery and reduced cardiac injury at 28 days post infection (136, 137).
Borrelia species-induced myocarditis are associated with focal necrosis, hypertrophy and vacuolization of myocytes leading to fibrosis (138). Immune infiltrates are usually lymphocytic in nature (138), mostly composed of macrophages and lymphocytes (139), predominantly T cells (138). Mononuclear leucocytes were also seen in heart tissue (138).
Staphylococcus typically builds micro abscesses within the myocardium (140). Methicillin-resistant Staphylococcus aureus (MRSA)-induced myocarditis is characterized by focal myocyte necrosis (141).
The pathomechanisms of myocarditis following streptococcal upper airway infections are not well understood. Streptococcal toxins and cross-reactivity of IgG with streptococcus antigens and cardiac myosin have been postulated as possible causative mechanisms (142, 143). Mononuclear cells, especially lymphocytes (of which CD4+ T cells were predominating) are found in EMB (144). Neutrophil infiltration and micro abscesses containing bacteria have also been described (145).
Pneumococcus microlesions appear widely spread throughout the myocardium, but especially in the ventricles (146). Such microlesions manifest as areas within the myocardium with a reduced number of cardiomyocytes accompanied by an enlarged intercellular space filled with pneumococci (147). These lesions are further characterized by expansion of the intercellular space caused by extracellular vacuolation, the apparent loss of cardiomyocytes and the stark absence of infiltrating immune cells, as observed in both a BALB/c mouse model and human disease (147–149). Fibrosis is a common consequence of this response (148, 149).
Macrophages and neutrophils are subverted through biofilm production by pneumococcus (150).
Bacterial adhesin choline-binding protein A and its interaction with laminin receptor of vascular endothelial cells represents the cellular entry point for pneumococci (151). Also, cobinding of phosphorylcholine residues on the bacterial cell membrane to platelet-activating factor receptor is an additional activating mechanism (151). Neutralizing these interactions could be a possible pharmacological approach (151).
Meningococcus myocarditis is rare (152), but might occur during meningococcus sepsis (153, 154). There is no information regarding immune cell infiltration patterns in the heart available, but neutrophils and lymphocytes accumulate around meningeal vessels of patients suffering from meningococcal meningitis (155).
Myocarditis caused by gonococci usually occurs secondary to or during endocarditis (156, 157) leading to perivalvular abscesses (156, 158). Neutrophils are the dominant immune cells infiltrating the valves and spatially related myocardium during such an infection (158). Fibrosis is a common result of gonococcal myocarditis (158).
Following salmonella gastroenteritis, the heart and aorta may be a secondary target of inflammation (159). It remains uncertain whether this represents a sterile inflammation lacking bacterial myocardial infiltration, that occurs subsequent to the infection, or if it is a secondary manifestation directly linked to the salmonella infection (159). Non-typhoid salmonella might lead to multifocal biventricular inflammation in subepicardial and midmyocardial tissue (160). There is no data in the literature regarding the composition of the immune cell infiltrate.
Infiltration by Mycobacterium tuberculosis of the heart follows no specific pattern (161). Similar to tuberculosis of the lungs, myocardial involvement comes in 3 different types: tuberculomas, miliary tubercles or an uncommon diffuse infiltrative type (161). Commonly, the left ventricle is affected (162). Usually, a giant cell (163–165) and lymphocytic (164, 165) infiltrate accompanies the infection. Granulation (166) as well as scar tissue and fibrosis might result (163).
M. tuberculosis inactivated antigens are used as the adjuvant (mimic infection) in complete Freund's adjuvant that is required to induce disease in autoimmune models of myocarditis (29). The dominant immune response in natural infections and in experimental autoimmune myocarditis are macrophages and Th17-type responses (167–169). These findings suggest that M. tuberculosis can drive myocarditis and DCM in the context of an autoimmune response.
Myocarditis due to Mycoplasma pneumoniae infection has been described as myopericarditis (170). Fibrosis of the heart valves has been reported (171). Data are limited concerning EMB findings on cellular components of the immune infiltrate in the context of mycoplasma infection.
Brucella myocarditis is very rare (172) and tends to occur in the left ventricular myocardium, in particular subpericardial (173, 174). Following infection with Brucella, endocardial tissue might undergo calcification and become fibrotic (175). Data are limited regarding the type of immune infiltration.
Several other infections such as protozoa, fungi or parasites can cause myocarditis (3). As there is limited data currently available on specific inflammatory infiltrates in most of these entities, only the most common parasite causing myocarditis, Trypanosoma cruzi, is discussed.
Chagas disease is caused by Trypanosoma cruzi, which is a parasite that replicates within host cells, including cardiac myocytes (176). Cardiac involvement in Chagas disease typically leads to inflammation, fibrosis, diffuse ventricular wall motion abnormalities and arrhythmias especially when prolonged inflammation occurs (177, 178). The myocardium shows signs of necrosis, areas of myocellular hypertrophy and predominantly mononuclear cell infiltration (176). Macrophages, eosinophils, neutrophils and mast cells are found in myocardial tissue as well, but to a lesser extent (176). Proinflammatory cytokines such as TNFα and IFNγ promote inflammation which can further lead to autoimmunity (179). Furthermore, the persistence of the parasite is associated with high grade myocarditis (180), as tissue damage partly results directly from the pathogen itself (176).
Rheumatoid arthritis associated cardiomyopathy is characterized by fibrosis and citrullination within the myocardial intercellular space (181). The latter is a type of post-translational modification that converts arginine residues within proteins to citrulline residues (182). In rheumatoid arthritis, the immune system produces antibodies targeting those citrullinated proteins (182–185). Rheumatoid arthritis associated myocarditis is characterized by focal necrosis or granulomatous inflammation (186). There is no specific immune cell type dominating the immune infiltrate.
1.5% of patients with myocarditis are found to have necrotizing coronary vasculitis by EMB (187). In the case of eosinophilic granulomatosis with polyangiitis eosinophilis play the dominant role (188). As of today, data on the components of cellular infiltrates in other entities is very limited.
Myocarditis associated with connective tissue diseases such as systemic lupus erythematosus develops predominantly in the ventricular wall leading to edema and necrosis (189, 190). CD4+ T cells usually dominate the immune infiltrate (189).
Severe combined immunodeficient mice developed myocarditis dominated by CD4+ T cells, while depletion of CD4+ T cells suppresses the inflammation (189). In mice with myocarditis induced through the transfer of CD4+ T cells, Th1 and Th17 cells were found to infiltrate the myocardium (189).
Cardiac sarcoidosis primarily leads to subepicardial inflammation within the left ventricular septum. However, midmyocardial, subendocardial or transmural inflammation may also occur (191). Infiltration is predominantly granulomatous (192).
Th1 cell derived cytokines such as IL-2, IFNγ and IL-12 drive the granulomatous inflammation, while the infiltrate lacks Th2 cells (192–194). In a case report, Schoppet et al. described a high Th1 response as causative for multiple granulomas in EMB while a Th2 response has been associated with disease regression (195). Figure 3 illustrates EMB findings representative for cardiac sarcoidosis.
Cytokines derived from Th1 cells promote macrophage accumulation, with CD68+ CD163- M1 macrophages dominating (196–198). Macrophages play a major role in the formation of granulomas (192). In the early stages of the disease, macrophages tend to form multinucleated giant cells within granulomas of the foreign body type (192). In later stages, they form granulomas of the Langhans type (192). In the EMB, the presence of Schaumann bodies and asteroid bodies may be observed (192). Schaumann bodies are calcified protein structures that occur intracellularly, often within giant cells (192, 199). In contrast, asteroid bodies are comprised of non-collagenous filaments and myelinoid membranes and are also frequently found in multinucleated giant cells (192, 199). Both Schaumann and asteroid bodies are pathologic signs that may be found in sarcoidosis (192, 200).
M2 macrophage derived factors, such as TGF-β and chemokine CC motif ligand lead to fibrotic cardiac remodeling (196, 201, 202).
Anti-inflammatory treatment with glucocorticoids is recommended and other anti-inflammatory agents such as TNFα antibody can be considered referring to current consensus statements (203).
This topic has been recently reviewed (31, 124). Briefly, SARS-CoV-2 vaccine-related myocarditis is associated with myocardial edema and mostly subepicardial anterolateral and inferolateral involvement of the myocardium (204). A number of theories have been proposed for how vaccines, and mRNA vaccines in particular, could lead to myocarditis including innate immune activation of mast cells (124), myocyte necrosis (205, 206), cytokine-induced damage (207), and interstitial fibrosis (208). In EMB, SARS-CoV-2 vaccine-related myocarditis usually presents as lymphocytic myocarditis (209), although in some biopsies no inflammatory cells are found (204, 209). The inflammatory infiltrate primarily consists of macrophages and T cells (205, 206).
Various mechanisms underlying myocarditis following SARS-CoV-2 vaccination including cytokine related damage and spike glycoprotein antibodies cross-reacting with myocardial contractile proteins, as well as the influence of sex and gender through endocrine differences are discussed (207, 210). In young men, autoantibodies against the IL-1RA impairing the IL-1RA bioactivity in vitro were associated with low circulating levels of IL-1RA and were found in patients with biomarker evidence of cardiac damage and inflammation (211).
Autoimmune myocarditis may follow ischemic (212), surgical (213) or traumatic (214, 215) myocardial damage as a consequence of an immune response to released segregated antigens (i.e., cardiac myosin). Furthermore, autoimmune myocarditis has also been hypothesized to follow viral infections that release damaged heart tissue (37). The experimental autoimmune myocarditis (EAM) model closely follows the time-course, cellular infiltrate, sex differences and mechanisms of disease that have been identified with clinical myocarditis associated with viral infections (20, 216). Importantly, autoimmune models of myocarditis that use a mild viral infection (i.e., MCMV, CVB3) instead of complete Freund's adjuvant as the pathogen strongly mimic EAM and clinical viral myocarditis (20, 37, 124). Recognition of autoimmune myocarditis can be crucial for initiation of immunosuppressive therapy (217) with potential recovery of cardiac function.
The direct toxic effect of ethyl and acetaldehyde leads to ultrastructural alterations of the mitochondria and sarcoplasmatic reticulum of cardiac myocytes (218, 219). Protein metabolism is dysregulated at a molecular level leading to arrhythmias (218) and focal necrosis (218, 220). This causes a lymphocytic infiltrate, without a subtype of cells dominating (218, 221).
Chronic alcohol consumption directly influences immune cell concentration with a specific suppression of neutrophils (222–224). Alcohol use disorder (AUD) might also reduce lymphocytes (224–228) and their reactivity to mitogens (224, 229). Activation of T and B cells might be aggravated due to heightened antigen presentation (224, 230). After alcohol exposure T cells produce less IFN-γ (224, 231).
Oxidative stress that disrupts mitochondria and reduces ATP production is the main cause of drug-induced injury to the myocardium (219). In addition, reactive oxygen species interfere with mitochondrial DNA replication, which can further lead to myocardial damage (219, 232). In a postmortem examination, hearts of patients who developed cardiotoxicity as a result of cyclophosphamide medication featured hemorrhagic myocardial necrosis, interstitial edema, hemorrhage and fibrin deposition (233, 234). However, the precise mechanism of cyclophosphamide-induced injury to the myocardium is not yet clear (233, 234), but inflammatory cell infiltrates have been described (232). Treatment with doxorubicin also damages mitochondria and leads to a reduction of natural killer cell activity and stimulation of cytotoxic T cells (235). A decrease in macrophage differentiation has also been observed (235). EMB shows histiocytes dominating the infiltrate in anthracycline-induced cardiomyopathy (236, 237). Several other drugs are known to cause myocarditis rarely, such as antibiotics, diuretics and antidepressants (3).
The prevalence of ICI-induced myocarditis revolves around 1% in a multicenter registry and is generally considered underestimated (238). ICI-induced myocarditis is associated with a mononuclear, mainly lymphocytic infiltrate within the myocardium, myocyte degeneration (239, 240) and interstitial fibrosis (241). T cells are the dominating cellular phenotype (239, 240, 242), with similar levels of CD4+ and CD8+ T cells (239, 240). CD68+ macrophages are also involved, and antibody deposits might occur (239, 240).
In an A/J mouse model, Won et al. were able to show that treating naive mice with anti-programmed cell death protein (PD)-1 monoclonal antibody (PD-1 regulates/inhibits T cell responses) was able to induce myocarditis (243). Tumor cells or infectious agents were absent in this model (243). Troponin elevation, arrhythmias and lymphocytic infiltration of the myocardium was observed in this model (243). The investigators identified that T cells being reactive to cardiac myosin were elevated in the myocardium (243).
Lv et al. demonstrated that autoimmunity plays a role in ICI-induced myocarditis by demonstrating that CD4+ T cells specific for alpha myosin heavy chain were not effectively eliminated in the thymus of mice and humans after receiving immune checkpoint inhibitors (244). They identified high numbers of these autoreactive T cells in mice and in patients with myocarditis (244).
ICI therapy disrupts cytotoxic T-lymphocyte-associated protein (CTLA)-4 and PD-1 signaling on T cells, leading to a reduction in peripheral immune tolerance and an increased likelihood of T cell activation (242, 245, 246). Another possible mechanism for ICI-associated myocarditis is the proliferation of T cells that recognize an antigen shared by both the tumor and heart muscle (242, 245, 246).
In a further study, researchers observed that individuals with ICI myocarditis had an increased number of cytotoxic CD8+ T cells that expressed the CD45RA marker, which is found on naive CD8+ T cells (247). This contrasted with healthy individuals, who had lower levels of these cells (247). Sequencing of the T cell receptor indicated that the CD8+ CD45RA+ T cells were clonally augmented in patients with ICI myocarditis (247). An analysis of the gene expression patterns in Temra CD8+ T cells revealed that they have a cytotoxic and activated phenotype, as expected (247). Their interaction with innate immune cells was enhanced and anti-inflammatory regulating factors were missing, leading to more inflammation (247).
In HCM an accumulation of CD8+ T cells (248, 249), basophils, fibroblasts, and platelets has been described in the cardiac inflammatory infiltrate (248). Analysis of the immune infiltration in HCM patients vs. controls revealed a decrease in dendritic cells, macrophages, monocytes, and natural killer cells (249). A decrease in CD163 + LYVE1+ macrophages, which belong to the M2 macrophage subtype, may play a crucial role in the pathogenesis of the disease (248). Other authors have demonstrated a high abundance of neutrophils, as well as both naive and memory B cells, within the myocardium (250).
EMB of myocardial tissue in patients with peripartum cardiomyopathy revealed evidence of a mild cardiac infiltration of inflammatory cells; however, no discernible pattern of cells was described (14). Breastfeeding women were found to have increased levels of prolactin, which were correlated with heightened levels of CD8+ T cells in the circulation (251). The inhibition of prolactin release using the dopamine antagonist, bromocriptine, prevents the development of peripartum cardiomyopathy in mice, suggesting a potential role of prolactin in the pathogenesis of the disease (252, 253). McTiernan et al. reported a significant reduction in circulating natural killer cells, along with elevated levels of CD3+ CD4−CD8−CD38+ T cells in women with peripartum cardiomyopathy (254).
Recently, bromocriptine, a prolactin release inhibitor has been explored as a therapeutic option in peripartum cardiomyopathy. It has been shown that bromocriptine treatment improves LVEF in peripartum cardiomyopathy patients (255–259). The decrease of prolactin might reduce the prolactin-associated inflammation and might therefore explain the observed increase in LVEF.
The presence of patchy inflammatory infiltrates suggests a potential involvement of the immune system in the pathogenesis of arrhythmogenic cardiomyopathy (260, 261), with M1 and M2 macrophages in equal numbers dominating the lesion (262). Cardiac myocytes in Dsg2mut/mut hearts demonstrated positive immunoreactivity for IL-1β, TNFα, and MCP-1α (263). Additionally, infiltrating mononuclear inflammatory cells showed positive immunoreactivity for IL-1β and TNFα (263). Cardiac inflammation leads to fibrotic remodeling of the myocardium in a murine model of arrhythmogenic cardiomyopathy with a desmoglein 2 mutation (262). Clinically, affected individuals display replacement of the ventricular myocardium with fibrofatty tissue (260, 261, 264–266). The implementation of therapy aimed at inhibiting remodeling and fibrosis could potentially improve cardiovascular function and might limit disease progression but requires further investigation (267).
Fibrosis is a characteristic feature observed in EMB samples obtained from the myocardium of patients with Takotsubo Syndrome (TTS) (268). Clinical studies have documented the infiltration of monocytes and macrophages in myocardial tissue (268–270). According to single-cell RNA sequencing studies analyzing immune cells in myocardial tissue, TTS-like cardiomyopathy is associated with intricate activation of both innate and adaptive immune cells in the heart (268). Among these cells, macrophages were found to be predominate (268). When global macrophage depletion was induced through clodronate liposome administration or macrophage infiltration was blocked using a CCR2 antagonist or in CCR2-KO mice, cardiac function was improved in mice challenged with isoproterenol (268).
In diabetes, various pathophysiological factors contribute to the development of cardiomyopathy, such as systemic metabolic disturbances, inappropriate activation of the renin-angiotensin-aldosterone system, subcellular component abnormalities, oxidative stress, inflammation, and impaired immune modulation (271–273). During obesity or insulin resistance, there is a notable occurrence of macrophage polarization, particularly towards M1 macrophages, as well as activation of dendritic cells and T lymphocytes (271, 273). Recent studies have provided evidence suggesting that B cells have a significant impact on type 1 diabetes and its associated complications (274). High rates of B-cell depletion have been shown to delay the progression of disease-related pathology in non-obese type 1 diabetic mice and new-onset patients with the disease (274).
Administration of glucagon-like peptide-1 for a duration of 5 weeks resulted in improvement in LVEF and functional status in patients with chronic heart failure and comorbid diabetes, which suggests that insulin resistance is implicated in the pathogenesis of cardiomyopathy in diabetes mellitus (275). Those findings require further investigation and are therefore not implemented in current clinical practice.
Among metabolic entities, myocarditis causing cardiac dilation and dysfunction with heart failure is a possible manifestation of gout, particularly in patients with severe and untreated forms (276). Amorphous urate crystals can deposit inside the cardiomyocytes and induce a strong inflammatory response associated with macrophages and cell death (276). Activation of TLR4, NLRP3 and IL-1β in gout are known factors that drive the pathogenesis of myocarditis (277).
Fabry disease is characterized by glycosphingolipid (Gb) accumulation in cells, leading to a proinflammatory response that may impact disease progression and causes myocardial edema (278). In some cases, small coronary vessels, conduction tissue and subepicardial ganglia were infiltrated (278, 279). CD3+ T lymphocytes were identified as the primary component of the observed inflammatory infiltrates (278).
The chronic secretion of highly immunogenic Gb3 by affected Fabry cells may play a key role in the initiation of immune-mediated myocardial inflammation and subsequently cause interstitial damage (279).
The dystrophinopathies are a heterogenous group of X-linked neuromuscular disorders associated with cardiomyopathy and altered cardiac immune regulation (280). In Duchenne muscular dystrophy-associated DCM, pentraxin 3 has been shown to act as an inflammatory mediator, facilitating the functions of macrophages and dendritic cells and contributing to apoptosis and necrosis (281).
In this review, we provide a comprehensive summary of the current literature on the immune landscape of myocardial diseases specific to their causes. However, for many types of myocarditis and inflammatory cardiomyopathy little data is available based on histology from EMBs and there are no corresponding translational animal models to better understand the pathogenesis of disease.
We highlight in this review that many infections, in particular viral, and many different chemicals/drugs are able to induce myocarditis and inflammatory cardiomyopathy. Most of our understanding of the mechanisms that drive the pathogenesis of myocarditis and inflammatory cardiomyopathy or DCM was derived from animal models.
An EMB provides a snapshot of immune cells located within the myocardial tissue at the time of sample collection. Furthermore, EMB does help with the definite identification of etiologies such as giant cell myocarditis in which EMB findings help to initiate anti-inflammatory treatment (282, 283). In giant cell myocarditis, EMB is further utilized for prognosis estimation (282, 283). Although being helpful in the identification of patients who might benefit from anti-inflammatory treatment, an EMB does not provide information about pathophysiological processes within other areas of the heart and may not be obtained during the peak of myocardial inflammation in the contrary to animal models, where timing of sample collection can be very accurately determined. Cardiac magnetic resonance imaging (CMR) is the non-invasive gold-standard to diagnose myocarditis. It primarily identifies edema and fibrosis rather than directly detecting activity of inflammation. Animal models have revealed that edema and fibrosis occur after the peak of acute myocarditis as the disease progresses toward cardiomyopathy and so EMBs that are obtained at the time when CMR findings become visible are potentially capturing a later timepoint in the pathogenesis of disease. We need to keep this in mind as we consider pathogenic mechanisms. Additionally, investigators testing for immune cells in EMBs may not stain for all key immune cell types that animal models have identified to be important in the pathogenesis of disease such as mast cells, macrophages and neutrophils which may lead to T cells being reported more often in EMBs.
In viral myocarditis, mast cell, macrophage and T cell responses and cytokines released thereof dominate as drivers of disease. The idea of innate cytokine storm-related damage to the myocardium has been discussed as one pathogenetic mechanism in severe COVID-19 but has long been known to be a factor in other forms of myocarditis. The innate cell release of TNFα, IL-1β and IL-6 as well as complement activation are well documented in viral and autoimmune models of myocarditis, especially in males, where they increase acute and chronic myocarditis (124, 129–131, 284, 285). IL-6 increased by TLR4-released IL-1β drives T cells to a Th17 response that contributes along with IL-1β to remodeling and fibrosis that leads to chronic cardiomyopathy/DCM. In general, these mechanisms are also imaginable in bacterial or toxic myocarditis, where neutrophils augment the adaptive T cell response. Furthermore, myocarditis associated with autoimmune diseases such as connective tissue diseases, have a greater T and B cell/autoantibody response that is increased by estrogen in females who primarily develop these conditions. ICI-induced myocarditis is also dominated by an autoimmune T cell response that is created by drugs, that disinhibit PD-1 signaling. More research is needed to understand the immune infiltrate in cardiovascular diseases that are not traditionally considered as “inflammatory cardiomyopathies”, such as HCM or arrhythmogenic cardiomyopathy.
With the growing relevance of precision medicine and cellular therapies, it is imperative to gain a comprehensive understanding of the precise immune mechanisms involved in myocardial inflammation, in particular of the cytokines involved and the patterns they form, as this knowledge can inform the development of novel therapeutic approaches. Major gaps include the need to obtain better clinical data on the early and chronic immune response to various viruses, toxins and other causes of disease. Additionally, myocarditis and chronic cardiomyopathy occur predominantly in males and a better understanding of the role of sex and gender differences in disease is needed. As sex influences the levels of T and B cells as well as macrophages (20, 32, 56–58), sex is needed to be taken into consideration when developing targeted therapies. With the advent of new technologies that can identify gene profiles in distinct cell populations from EMB and animal models, a better understanding of similarities and differences in cardiomyopathy phenotypes should emerge that will inform novel therapy development. Thus, this review not only provides an overview of the immune landscape in myocardial diseases but also identifies research gaps in our understanding of cardiac inflammation. By highlighting these gaps, we aim to stimulate further translational investigation into the pathogenesis of myocardial inflammation and ultimately facilitate the development of more effective treatments.
A critical question in the field of cardiac therapeutics concerns the identification of patients who may benefit from anti-inflammatory therapy. To address this, new diagnostic methods are required to enable early detection or screening for inflammatory components in patients with heart failure, thereby allowing for timely initiation of anti-inflammatory treatment. Magnetocardiography (MCG) might emerge as a promising diagnostic tool for this purpose, offering the capability to measure the cardiac magnetic field (286–288). The interplay of electrons and ions and the directions of their respective movements shape the cardiac magnetic field and may deviate in disease (286–288). Even discrete alterations within the electromagnetic field are quantifiable, since small quantum interference device sensors (SQUIDs) were introduced into magnetocardiography (287, 288). It is noteworthy that obtaining MCG measurements require only one minute per patient (287, 288). Being a non-invasive and passive diagnostic tool, magnetocardiography entails no side effects (287, 288) which further renders it a potent tool with diverse possibilities for future research and clinical applications. Currently, the utility of MCG is being explored in screening for inflammatory cardiomyopathy and assessing early treatment response to anti-inflammatory treatment in respective patients (286–290).
The manuscript for submission to the journal was authored by NM, with PS and AG providing substantial research assistance. DF, KK, PM, AF, LC, TL, UL, and BH contributed to the research and the development of the manuscript. NM took the lead in writing the article, synthesizing the qualitative findings, and presenting them in a coherent manner. The other authors provided their expertise and insights to enhance the review, ensuring a comprehensive evaluation of the topic. Their contributions included critical discussions, reviewing and revising the manuscript for accuracy and clarity, and ensuring the overall quality of the review article. All authors contributed to the article and approved the submitted version.
This work was supported by National Institutes of Health (NIH) R01 HL164520, R21 AI145356, R21 AI152318, R21 AI154927 to DF; American Heart Association 20TPA35490415 to DF; NIH grants TL1 TR002380 to DF; the For Elyse Foundation to DF; and Mayo Clinic Center for Regenerative Medicine to DF.
PM is supported by MCIN-ISCIII-Fondo de Investigación Sanitaria (PI22/01759), (PMPTA22/00090-BIOCARDIOTOX) and Comunidad de Madrid (P2022/BMD-7209-INTEGRAMUNE-CM).
TL has no conflicts of interest related to this manuscript, but received educational and research grants from Abbott, Ablative Solutions, Amgen, AstraZeneca, Boehringer-Ingelheim, Daichi Sankyo, Novartis, Sanofi, Servier and Vifor and honoraria from Amgen, Dacadoo, Daichi-Sankyo, Glaxo, Smith & Kline, Firalis, Menarini Foundation, NeuroImmune, Novo Nordisk. BH is inventor on patents that use RNA for diagnosis of myocarditis. Patent protection is in process for MCG for diagnosis and measurement of therapy response in inflammatory cardiomyopathy and for cytokines as targets for therapy in patients with inflammatory cardiomyopathy and in heart failure.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Reviewer DP declared a past co-authorship with the authors BH, TL, and PS.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation. (1996) 93(5):841–2. doi: 10.1161/01.CIR.93.5.841
3. Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology working group on myocardial and pericardial diseases. Eur Heart J. (2013) 34(33):2636–48. 48a-48d. doi: 10.1093/eurheartj/eht210
4. Roberts LB, Lord GM, Howard JK. Heartbreakers or healers? Innate lymphoid cells in cardiovascular disease and obesity. Front Immunol. (2022) 13:903678. doi: 10.3389/fimmu.2022.903678
5. Golpour A, Patriki D, Hanson PJ, McManus B, Heidecker B. Epidemiological impact of myocarditis. J Clin Med. (2021) 10(4). doi: 10.3390/jcm10040603
6. Tschöpe C, Ammirati E, Bozkurt B, Caforio ALP, Cooper LT, Felix SB, et al. Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat Rev Cardiol. (2021) 18(3):169–93. doi: 10.1038/s41569-020-00435-x
7. Kociol RD, Cooper LT, Fang JC, Moslehi JJ, Pang PS, Sabe MA, et al. Recognition and initial management of fulminant myocarditis: a scientific statement from the American Heart Association. Circulation. (2020) 141(6):e69–92. doi: 10.1161/CIR.0000000000000745
8. Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. (2018) 18(12):733–44. doi: 10.1038/s41577-018-0065-8
9. Miranda AMA, Janbandhu V, Maatz H, Kanemaru K, Cranley J, Teichmann SA, et al. Single-cell transcriptomics for the assessment of cardiac disease. Nat Rev Cardiol. (2023) 20(5):289–308. doi: 10.1038/s41569-022-00805-7
10. Fine NM. Rare causes of autoimmune myocarditis: finding needles in a shifting haystack. JACC Case Rep. (2023) 9:101743. doi: 10.1016/j.jaccas.2023.101743
11. Bracamonte-Baran W, Čiháková D. Cardiac autoimmunity: myocarditis. Adv Exp Med Biol. (2017) 1003:187–221. doi: 10.1007/978-3-319-57613-8_10
12. Baboonian C, McKenna W. Eradication of viral myocarditis: is there hope?**editorials published in the Journal of the American College of Cardiologyreflect the views of the authors and do not necessarily represent the views of JACCor the American College of Cardiology. J Am Coll Cardiol. (2003) 42(3):473–6. doi: 10.1016/S0735-1097(03)00655-7
13. Siegismund CS, Escher F, Lassner D, Kühl U, Gross U, Fruhwald F, et al. Intramyocardial inflammation predicts adverse outcome in patients with cardiac AL amyloidosis. Eur J Heart Fail. (2018) 20(4):751–7. doi: 10.1002/ejhf.1039
14. Ofosu-Somuah A, Gattani R, Genovese L, Avstreih D, Shah P, Epps K, et al. Peripartum cardiomyopathy presenting with incessant ventricular arrhythmias. JACC Case Rep. (2022) 4(13):759–63. doi: 10.1016/j.jaccas.2022.03.006
15. Yang D, Liu HQ, Liu FY, Tang N, Guo Z, Ma SQ, et al. The roles of noncardiomyocytes in cardiac remodeling. Int J Biol Sci. (2020) 16(13):2414–29. doi: 10.7150/ijbs.47180
16. Hu YF, Chen YJ, Lin YJ, Chen SA. Inflammation and the pathogenesis of atrial fibrillation. Nat Rev Cardiol. (2015) 12(4):230–43. doi: 10.1038/nrcardio.2015.2
17. Zhang Y, Bauersachs J, Langer HF. Immune mechanisms in heart failure. Eur J Heart Fail. (2017) 19(11):1379–89. doi: 10.1002/ejhf.942
18. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. (2016) 119(1):91–112. doi: 10.1161/CIRCRESAHA.116.303577
19. Huber SA. Viral myocarditis and dilated cardiomyopathy: etiology and pathogenesis. Curr Pharm Des. (2016) 22(4):408–26. doi: 10.2174/1381612822666151222160500
20. Fairweather D, Beetler DJ, Musigk N, Heidecker B, Lyle MA, Cooper LT Jr, et al. Sex and gender differences in myocarditis and dilated cardiomyopathy: an update. Front Cardiovasc Med. (2023) 10:1129348. doi: 10.3389/fcvm.2023.1129348
21. Fairweather D, Frisancho-Kiss S, Yusung SA, Barrett MA, Davis SE, Gatewood SJ, et al. Interferon-gamma protects against chronic viral myocarditis by reducing mast cell degranulation, fibrosis, and the profibrotic cytokines transforming growth factor-beta 1, interleukin-1 beta, and interleukin-4 in the heart. Am J Pathol. (2004) 165(6):1883–94. doi: 10.1016/S0002-9440(10)63241-5
22. Gullestad L, Aass H, Fjeld JG, Wikeby L, Andreassen AK, Ihlen H, et al. Immunomodulating therapy with intravenous immunoglobulin in patients with chronic heart failure. Circulation. (2001) 103(2):220–5. doi: 10.1161/01.CIR.103.2.220
23. Chimenti C, Russo MA, Frustaci A. Immunosuppressive therapy in virus-negative inflammatory cardiomyopathy: 20-year follow-up of the TIMIC trial. Eur Heart J. (2022) 43(36):3463–73. doi: 10.1093/eurheartj/ehac348
24. Escher F, Kühl U, Lassner D, Poller W, Westermann D, Pieske B, et al. Long-term outcome of patients with virus-negative chronic myocarditis or inflammatory cardiomyopathy after immunosuppressive therapy. Clin Res Cardiol. (2016) 105(12):1011–20. doi: 10.1007/s00392-016-1011-z
25. Merken J, Hazebroek M, Van Paassen P, Verdonschot J, Van Empel V, Knackstedt C, et al. Immunosuppressive therapy improves both short- and long-term prognosis in patients with virus-negative nonfulminant inflammatory cardiomyopathy. Circ Heart Fail. (2018) 11(2):e004228. doi: 10.1161/CIRCHEARTFAILURE.117.004228
26. Wojnicz R, Nowalany-Kozielska E, Wojciechowska C, Glanowska G, Wilczewski P, Niklewski T, et al. Randomized, placebo-controlled study for immunosuppressive treatment of inflammatory dilated cardiomyopathy: two-year follow-up results. Circulation. (2001) 104(1):39–45. doi: 10.1161/01.CIR.104.1.39
27. Tschöpe C, Van Linthout S, Spillmann F, Posch MG, Reinke P, Volk HD, et al. Targeting CD20+ B-lymphocytes in inflammatory dilated cardiomyopathy with rituximab improves clinical course: a case series. Eur Heart J Case Rep. (2019) 3(3). doi: 10.1093/ehjcr/ytz131
28. Elwazir M, Krause ML, Bois JP, Christopoulos G, Kendi AT, Cooper JLT, et al. Rituximab for the treatment of refractory cardiac sarcoidosis: a single-center experience. J Card Fail. (2022) 28(2):247–58. doi: 10.1016/j.cardfail.2021.07.008
29. Myers JM, Fairweather D, Huber SA, Cunningham MW. Autoimmune myocarditis, valvulitis, and cardiomyopathy. Curr Protoc Immunol. (2013) Chapter 15:Unit 15.4.1–51. doi: 10.1002/0471142735.im1514s101
30. Błyszczuk P. Myocarditis in humans and in experimental animal models. Front Cardiovasc Med. (2019) 6:64. doi: 10.3389/fcvm.2019.00064
31. Heidecker B, Dagan N, Balicer R, Eriksson U, Rosano G, Coats A, et al. Myocarditis following COVID-19 vaccine: incidence, presentation, diagnosis, pathophysiology, therapy, and outcomes put into perspective. A clinical consensus document supported by the Heart Failure Association of the European Society of Cardiology (ESC) and the ESC working group on myocardial and pericardial diseases. Eur J Heart Fail. (2022) 24. doi: 10.1002/ejhf.2669
32. Frisancho-Kiss S, Davis SE, Nyland JF, Frisancho JA, Cihakova D, Barrett MA, et al. Cutting edge: cross-regulation by TLR4 and T cell ig mucin-3 determines sex differences in inflammatory heart disease. J Immunol. (2007) 178(11):6710–4. doi: 10.4049/jimmunol.178.11.6710
33. Case LK, Moussawi M, Roberts B, Noubade R, Huber SA, Teuscher C. Histamine H(1) receptor signaling regulates effector T cell responses and susceptibility to coxsackievirus B3-induced myocarditis. Cell Immunol. (2012) 272(2):269–74. doi: 10.1016/j.cellimm.2011.10.004
34. Kologrivova I, Shtatolkina M, Suslova T, Ryabov V. Cells of the immune system in cardiac remodeling: main players in resolution of inflammation and repair after myocardial infarction. Front Immunol. (2021) 12:664457. doi: 10.3389/fimmu.2021.664457
35. Bruno KA, Mathews JE, Yang AL, Frisancho JA, Scott AJ, Greyner HD, et al. BPA Alters estrogen receptor expression in the heart after viral infection activating cardiac mast cells and T cells leading to perimyocarditis and fibrosis. Front Endocrinol (Lausanne). (2019) 10:598. doi: 10.3389/fendo.2019.00598
36. Eriksson U, Ricci R, Hunziker L, Kurrer MO, Oudit GY, Watts TH, et al. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat Med. (2003) 9(12):1484–90. doi: 10.1038/nm960
37. Fairweather D, Kaya Z, Shellam GR, Lawson CM, Rose NR. From infection to autoimmunity. J Autoimmun. (2001) 16(3):175–86. doi: 10.1006/jaut.2000.0492
38. Opavsky MA, Penninger J, Aitken K, Wen WH, Dawood F, Mak T, et al. Susceptibility to myocarditis is dependent on the response of alphabeta T lymphocytes to coxsackieviral infection. Circ Res. (1999) 85(6):551–8. doi: 10.1161/01.RES.85.6.551
39. Klingel K, Schnorr JJ, Sauter M, Szalay G, Kandolf R. beta2-microglobulin-associated regulation of interferon-gamma and virus-specific immunoglobulin G confer resistance against the development of chronic coxsackievirus myocarditis. Am J Pathol. (2003) 162(5):1709–20. doi: 10.1016/S0002-9440(10)64305-2
40. Stephenson E, Savvatis K, Mohiddin SA, Marelli-Berg FM. T-cell immunity in myocardial inflammation: pathogenic role and therapeutic manipulation. Br J Pharmacol. (2017) 174(22):3914–25. doi: 10.1111/bph.13613
41. Afanasyeva M, Wang Y, Kaya Z, Park S, Zilliox MJ, Schofield BH, et al. Experimental autoimmune myocarditis in A/J mice is an interleukin-4-dependent disease with a Th2 phenotype. Am J Pathol. (2001) 159(1):193–203. doi: 10.1016/S0002-9440(10)61685-9
42. Afanasyeva M, Georgakopoulos D, Rose NR. Autoimmune myocarditis: cellular mediators of cardiac dysfunction. Autoimmun Rev. (2004) 3(7–8):476–86. doi: 10.1016/j.autrev.2004.08.009
43. Afanasyeva M, Georgakopoulos D, Belardi DF, Ramsundar AC, Barin JG, Kass DA, et al. Quantitative analysis of myocardial inflammation by flow cytometry in murine autoimmune myocarditis: correlation with cardiac function. Am J Pathol. (2004) 164(3):807–15. doi: 10.1016/S0002-9440(10)63169-0
44. Afanasyeva M, Georgakopoulos D, Belardi DF, Bedja D, Fairweather D, Wang Y, et al. Impaired up-regulation of CD25 on CD4+ T cells in IFN-gamma knockout mice is associated with progression of myocarditis to heart failure. Proc Natl Acad Sci U S A. (2005) 102(1):180–5. doi: 10.1073/pnas.0408241102
45. Fairweather D, Frisancho-Kiss S, Yusung SA, Barrett MA, Davis SE, Steele RA, et al. IL-12 protects against coxsackievirus B3-induced myocarditis by increasing IFN-gamma and macrophage and neutrophil populations in the heart. J Immunol. (2005) 174(1):261–9. doi: 10.4049/jimmunol.174.1.261
46. Abston ED, Coronado MJ, Bucek A, Bedja D, Shin J, Kim JB, et al. Th2 regulation of viral myocarditis in mice: different roles for TLR3 versus TRIF in progression to chronic disease. Clin Dev Immunol. (2012) 2012:129486. doi: 10.1155/2012/129486
47. Abston ED, Coronado MJ, Bucek A, Onyimba JA, Brandt JE, Frisancho JA, et al. TLR3 Deficiency induces chronic inflammatory cardiomyopathy in resistant mice following coxsackievirus B3 infection: role for IL-4. Am J Physiol Regul Integr Comp Physiol. (2013) 304(4):R267–77. doi: 10.1152/ajpregu.00516.2011
48. Chen P, Baldeviano GC, Ligons DL, Talor MV, Barin JG, Rose NR, et al. Susceptibility to autoimmune myocarditis is associated with intrinsic differences in CD4(+) T cells. Clin Exp Immunol. (2012) 169(2):79–88. doi: 10.1111/j.1365-2249.2012.04598.x
49. Frisancho-Kiss S, Coronado MJ, Frisancho JA, Lau VM, Rose NR, Klein SL, et al. Gonadectomy of male BALB/c mice increases Tim-3(+) alternatively activated M2 macrophages, Tim-3(+) T cells, Th2 cells and Treg in the heart during acute coxsackievirus-induced myocarditis. Brain Behav Immun. (2009) 23(5):649–57. doi: 10.1016/j.bbi.2008.12.002
50. Shi Y, Fukuoka M, Li G, Liu Y, Chen M, Konviser M, et al. Regulatory T cells protect mice against coxsackievirus-induced myocarditis through the transforming growth factor beta-coxsackie-adenovirus receptor pathway. Circulation. (2010) 121(24):2624–34. doi: 10.1161/CIRCULATIONAHA.109.893248
51. He Y, Chipman PR, Howitt J, Bator CM, Whitt MA, Baker TS, et al. Interaction of coxsackievirus B3 with the full length coxsackievirus-adenovirus receptor. Nat Struct Biol. (2001) 8(10):874–8. doi: 10.1038/nsb1001-874
52. Garmaroudi FS, Marchant D, Hendry R, Luo H, Yang D, Ye X, et al. Coxsackievirus B3 replication and pathogenesis. Future Microbiol. (2015) 10(4):629–53. doi: 10.2217/fmb.15.5
53. Klingel K, Fabritius C, Sauter M, Göldner K, Stauch D, Kandolf R, et al. The activating receptor NKG2D of natural killer cells promotes resistance against enterovirus-mediated inflammatory cardiomyopathy. J Pathol. (2014) 234(2):164–77. doi: 10.1002/path.4369
54. Xu D, Wang P, Yang J, Qian Q, Li M, Wei L, et al. Gr-1+ cells other than Ly6G+ neutrophils limit virus replication and promote myocardial inflammation and fibrosis following coxsackievirus B3 infection of mice. Front Cell Infect Microbiol. (2018) 8:157. doi: 10.3389/fcimb.2018.00157
55. Klingel K, Hohenadl C, Canu A, Albrecht M, Seemann M, Mall G, et al. Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proc Natl Acad Sci U S A. (1992) 89(1):314–8. doi: 10.1073/pnas.89.1.314
56. Lasrado N, Reddy J. An overview of the immune mechanisms of viral myocarditis. Rev Med Virol. (2020) 30(6):1–14. doi: 10.1002/rmv.2131
57. Kandolf R, Sauter M, Aepinus C, Schnorr JJ, Selinka HC, Klingel K. Mechanisms and consequences of enterovirus persistence in cardiac myocytes and cells of the immune system. Virus Res. (1999) 62(2):149–58. doi: 10.1016/S0168-1702(99)00041-6
58. Huber SA. Coxsackievirus B3-induced myocarditis: infection of females during the estrus phase of the ovarian cycle leads to activation of T regulatory cells. Virology. (2008) 378(2):292–8. doi: 10.1016/j.virol.2008.05.015
59. Miteva K, Pappritz K, El-Shafeey M, Dong F, Ringe J, Tschöpe C, et al. Mesenchymal stromal cells modulate monocytes trafficking in coxsackievirus B3-induced myocarditis. Stem Cells Transl Med. (2017) 6(4):1249–61. doi: 10.1002/sctm.16-0353
60. Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. (2007) 317(5838):666–70. doi: 10.1126/science.1142883
61. Geissmann F, Auffray C, Palframan R, Wirrig C, Ciocca A, Campisi L, et al. Blood monocytes: distinct subsets, how they relate to dendritic cells, and their possible roles in the regulation of T-cell responses. Immunol Cell Biol. (2008) 86(5):398–408. doi: 10.1038/icb.2008.19
62. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. (2007) 204(12):3037–47. doi: 10.1084/jem.20070885
63. Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. (2003) 19(1):59–70. doi: 10.1016/S1074-7613(03)00171-7
64. Kühl U, Lassner D, von Schlippenbach J, Poller W, Schultheiss HP. Interferon-Beta improves survival in enterovirus-associated cardiomyopathy. J Am Coll Cardiol. (2012) 60(14):1295–6. doi: 10.1016/j.jacc.2012.06.026
65. Yen MH, Huang YC, Chen MC, Liu CC, Chiu NC, Lien R, et al. Effect of intravenous immunoglobulin for neonates with severe enteroviral infections with emphasis on the timing of administration. J Clin Virol. (2015) 64:92–6. doi: 10.1016/j.jcv.2015.01.013
66. Abzug MJ, Michaels MG, Wald E, Jacobs RF, Romero JR, Sánchez PJ, et al. A randomized, double-blind, placebo-controlled trial of pleconaril for the treatment of neonates with enterovirus sepsis. J Pediatric Infect Dis Soc. (2016) 5(1):53–62. doi: 10.1093/jpids/piv015
67. Amdani SM, Kim HS, Orvedahl A, John AO, Said A, Simpson K. Successful treatment of fulminant neonatal enteroviral myocarditis in monochorionic diamniotic twins with cardiopulmonary support, intravenous immunoglobulin and pocapavir. BMJ Case Rep. (2018) 2018. doi: 10.1136/bcr-2017-224133
68. Pinkert S, Westermann D, Wang X, Klingel K, Dörner A, Savvatis K, et al. Prevention of cardiac dysfunction in acute coxsackievirus B3 cardiomyopathy by inducible expression of a soluble coxsackievirus-adenovirus receptor. Circulation. (2009) 120(23):2358–66. doi: 10.1161/CIRCULATIONAHA.108.845339
69. Pinkert S, Dieringer B, Klopfleisch R, Savvatis K, Van Linthout S, Pryshliak M, et al. Early treatment of coxsackievirus B3-infected animals with soluble coxsackievirus-adenovirus receptor inhibits development of chronic coxsackievirus B3 cardiomyopathy. Circ Heart Fail. (2019) 12(11):e005250. doi: 10.1161/CIRCHEARTFAILURE.119.005250
70. Kraft L, Erdenesukh T, Sauter M, Tschöpe C, Klingel K. Blocking the IL-1β signalling pathway prevents chronic viral myocarditis and cardiac remodeling. Basic Res Cardiol. (2019) 114(2):11. doi: 10.1007/s00395-019-0719-0
71. Fairweather D, Yusung S, Frisancho S, Barrett M, Gatewood S, Steele R, et al. IL-12 receptor beta 1 and toll-like receptor 4 increase IL-1 beta- and IL-18-associated myocarditis and coxsackievirus replication. J Immunol. (2003) 170(9):4731–7. doi: 10.4049/jimmunol.170.9.4731
72. Roberts BJ, Moussawi M, Huber SA. Sex differences in TLR2 and TLR4 expression and their effect on coxsackievirus-induced autoimmune myocarditis. Exp Mol Pathol. (2013) 94(1):58–64. doi: 10.1016/j.yexmp.2012.06.005
73. Myers JM, Cooper LT, Kem DC, Stavrakis S, Kosanke SD, Shevach EM, et al. Cardiac myosin-Th17 responses promote heart failure in human myocarditis. JCI Insight. (2016) 1(9). doi: 10.1172/jci.insight.85851
74. Li Z, Yue Y, Xiong S. Distinct Th17 inductions contribute to the gender bias in CVB3-induced myocarditis. Cardiovasc Pathol. (2013) 22(5):373–82. doi: 10.1016/j.carpath.2013.02.004
75. Cihakova D, Rose NR. Pathogenesis of myocarditis and dilated cardiomyopathy. Adv Immunol. (2008) 99:95–114. doi: 10.1016/S0065-2776(08)00604-4
76. Jain A, Norton N, Bruno KA, Cooper LT Jr, Atwal PS, Fairweather D. Sex differences, genetic and environmental influences on dilated cardiomyopathy. J Clin Med. (2021)10(11). doi: 10.3390/jcm10112289
77. Szalay G, Sauter M, Haberland M, Zuegel U, Steinmeyer A, Kandolf R, et al. Osteopontin: a fibrosis-related marker molecule in cardiac remodeling of enterovirus myocarditis in the susceptible host. Circ Res. (2009) 104(7):851–9. doi: 10.1161/CIRCRESAHA.109.193805
78. Tschöpe C, Van Linthout S, Jäger S, Arndt R, Trippel T, Müller I, et al. Modulation of the acute defence reaction by eplerenone prevents cardiac disease progression in viral myocarditis. ESC Heart Fail. (2020) 7(5):2838–52. doi: 10.1002/ehf2.12887
79. Kanashiro-Takeuchi RM, Heidecker B, Lamirault G, Dharamsi JW, Hare JM. Sex-specific impact of aldosterone receptor antagonism on ventricular remodeling and gene expression after myocardial infarction. Clin Transl Sci. (2009) 2(2):134–42. doi: 10.1111/j.1752-8062.2009.00094.x
80. Bültmann BD, Sotlar K, Klingel K. Parvovirus B19. N Engl J Med. (2004) 350(19):2006–7.; author reply −7. doi: 10.1056/NEJM200405063501920
81. Van Linthout S, Elsanhoury A, Klein O, Sosnowski M, Miteva K, Lassner D, et al. Telbivudine in chronic lymphocytic myocarditis and human parvovirus B19 transcriptional activity. ESC Heart Fail. (2018) 5(5):818–29. doi: 10.1002/ehf2.12341
82. Bock CT, Klingel K, Kandolf R. Human parvovirus B19-associated myocarditis. N Engl J Med. (2010) 362(13):1248–9. doi: 10.1056/NEJMc0911362
83. Duechting A, Tschöpe C, Kaiser H, Lamkemeyer T, Tanaka N, Aberle S, et al. Human parvovirus B19 NS1 protein modulates inflammatory signaling by activation of STAT3/PIAS3 in human endothelial cells. J Virol. (2008) 82(16):7942–52. doi: 10.1128/JVI.00891-08
84. Lotze U, Egerer R, Glück B, Zell R, Sigusch H, Erhardt C, et al. Low level myocardial parvovirus B19 persistence is a frequent finding in patients with heart disease but unrelated to ongoing myocardial injury. J Med Virol. (2010) 82(8):1449–57. doi: 10.1002/jmv.21821
85. Tavora F, Gonzalez-Cuyar LF, Dalal JS, O'Malley MT, Zhao R, Peng HQ, et al. Fatal parvoviral myocarditis: a case report and review of literature. Diagn Pathol. (2008) 3:21. doi: 10.1186/1746-1596-3-21
86. Verdonschot J, Hazebroek M, Merken J, Debing Y, Dennert R, Brunner-La Rocca HP, et al. Relevance of cardiac parvovirus B19 in myocarditis and dilated cardiomyopathy: review of the literature. Eur J Heart Fail. (2016) 18(12):1430–41. doi: 10.1002/ejhf.665
87. Norbeck O, Isa A, Pöhlmann C, Broliden K, Kasprowicz V, Bowness P, et al. Sustained CD8+ T-cell responses induced after acute parvovirus B19 infection in humans. J Virol. (2005) 79(18):12117–21. doi: 10.1128/JVI.79.18.12117-12121.2005
88. Manaresi E, Gallinella G. Advances in the development of antiviral strategies against parvovirus B19. Viruses. (2019) 11(7). doi: 10.3390/v11070659
89. Hazebroek MR, Henkens M, Raafs AG, Verdonschot JAJ, Merken JJ, Dennert RM, et al. Intravenous immunoglobulin therapy in adult patients with idiopathic chronic cardiomyopathy and cardiac parvovirus B19 persistence: a prospective, double-blind, randomized, placebo-controlled clinical trial. Eur J Heart Fail. (2021) 23(2):302–9. doi: 10.1002/ejhf.2082
90. Tschöpe C, Elsanhoury A, Schlieker S, Van Linthout S, Kühl U. Immunosuppression in inflammatory cardiomyopathy and parvovirus B19 persistence. Eur J Heart Fail. (2019) 21(11):1468–9. doi: 10.1002/ejhf.1560
91. Plachter B, Sinzger C, Jahn G. Cell types involved in replication and distribution of human cytomegalovirus. In: Maramorosch K, Murphy FA, Shatkin AJ, editors. Advances in Virus Research. 46. Amsterdam: Academic Press (1996). p. 195–261.
92. Dioverti MV, Razonable RR. Cytomegalovirus. Microbiol Spectr. (2016) 4(4). doi: 10.1128/microbiolspec.DMIH2-0022-2015
93. Ritter JT, Tang-Feldman YJ, Lochhead GR, Estrada M, Lochhead S, Yu C, et al. In vivo characterization of cytokine profiles and viral load during murine cytomegalovirus-induced acute myocarditis. Cardiovasc Pathol. (2010) 19(2):83–93. doi: 10.1016/j.carpath.2008.12.001
94. Lenzo JC, Fairweather D, Cull V, Shellam GR, James Lawson CM. Characterisation of murine cytomegalovirus myocarditis: cellular infiltration of the heart and virus persistence. J Mol Cell Cardiol. (2002) 34(6):629–40. doi: 10.1006/jmcc.2002.2003
95. Kühl U, Lassner D, Wallaschek N, Gross UM, Krueger GR, Seeberg B, et al. Chromosomally integrated human herpesvirus 6 in heart failure: prevalence and treatment. Eur J Heart Fail. (2015) 17(1):9–19. doi: 10.1002/ejhf.194
96. Schultheiss HP, Baumeier C, Aleshcheva G, Bock CT, Escher F. Viral myocarditis-from pathophysiology to treatment. J Clin Med. (2021) 10(22). doi: 10.3390/jcm10225240
97. Lenzo JC, Shellam GR, Lawson CM. Ganciclovir and cidofovir treatment of cytomegalovirus-induced myocarditis in mice. Antimicrob Agents Chemother. (2001) 45(5):1444–9. doi: 10.1128/AAC.45.5.1444-1449.2001
98. Maisch B, Pankuweit S, Funck R, Koelsch S, editors. Effective CMV hyperimmunoglobulin treatment in CMV myocarditis–a controlled treatment trial (abstract# 674). Proceedings of the European society of cardiology annual meeting: 28 Aug–1 Sept 2004; Munich; (2004).
99. Richter J, Quintanilla-Martinez L, Bienemann K, Zeus T, Germing U, Sander O, et al. An unusual presentation of a common infection. Infection. (2013) 41(2):565–9. doi: 10.1007/s15010-012-0321-y
100. Lusso P, Malnati M, De Maria A, Balotta C, DeRocco SE, Markham PD, et al. Productive infection of CD4 + and CD8 + mature human T cell populations and clones by human herpesvirus 6. Transcriptional down-regulation of CD3. J Immunol. (1991) 147(2):685–91. doi: 10.4049/jimmunol.147.2.685
101. Rohayem J, Dinger J, Fischer R, Klingel K, Kandolf R, Rethwilm A. Fatal myocarditis associated with acute parvovirus B19 and human herpesvirus 6 coinfection. J Clin Microbiol. (2001) 39(12):4585–7. doi: 10.1128/JCM.39.12.4585-4587.2001
102. Elsanhoury A, Kühl U, Stautner B, Klein O, Krannich A, Morris D, et al. The spontaneous course of human herpesvirus 6 DNA-associated myocarditis and the effect of immunosuppressive intervention. Viruses. (2022) 14(2). doi: 10.3390/v14020299
103. Omura T, Yoshiyama M, Hayashi T, Nishiguchi S, Kaito M, Horiike S, et al. Core protein of hepatitis C virus induces cardiomyopathy. Circ Res. (2005) 96(2):148–50. doi: 10.1161/01.RES.0000154263.70223.13
104. Ray RB, Lagging LM, Meyer K, Ray R. Hepatitis C virus core protein cooperates with ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. J Virol. (1996) 70(7):4438–43. doi: 10.1128/jvi.70.7.4438-4443.1996
105. Ampuero J, Romero-Gómez M. Assessing cardiovascular risk in hepatitis C: an unmet need. World J Hepatol. (2015) 7(19):2214–9. doi: 10.4254/wjh.v7.i19.2214
106. Sanchez MJ, Bergasa NV. Hepatitis C associated cardiomyopathy: potential pathogenic mechanisms and clinical implications. Med Sci Monit. (2008) 14(5):Ra55–63.18443562
107. Haykal M, Matsumori A, Saleh A, Fayez M, Negm H, Shalaby M, et al. Diagnosis and treatment of HCV heart diseases. Expert Rev Cardiovasc Ther. (2021) 19(6):493–9. doi: 10.1080/14779072.2021.1917383
108. Frustaci A, Calabrese F, Chimenti C, Pieroni M, Thiene G, Maseri A. Lone hepatitis C virus myocarditis responsive to immunosuppressive therapy. Chest. (2002) 122(4):1348–56. doi: 10.1378/chest.122.4.1348
109. Barbaro G, Di Lorenzo G, Grisorio B, Barbarini G. Cardiac involvement in the acquired immunodeficiency syndrome: a multicenter clinical-pathological study. Gruppo Italiano per lo Studio Cardiologico dei pazienti affetti da AIDS investigators. AIDS Res Hum Retroviruses. (1998) 14(12):1071–7. doi: 10.1089/aid.1998.14.1071
110. Klatt EC. Cardiovascular pathology in AIDS. Adv Cardiol. (2003) 40:23–48. doi: 10.1159/000073174
111. Kan H, Xie Z, Finkel MS. HIV Gp120 enhances NO production by cardiac myocytes through p38 MAP kinase-mediated NF-kappaB activation. Am J Physiol Heart Circ Physiol. (2000) 279(6):H3138–43. doi: 10.1152/ajpheart.2000.279.6.H3138
112. Choi H, Dey AK, Sharma G, Bhoite R, Burkholder G, Fedson S, et al. Etiology and pathophysiology of heart failure in people with HIV. Heart Fail Rev. (2021) 26(3):497–505. doi: 10.1007/s10741-020-10048-8
113. Van Linthout S, Klingel K, Tschöpe C. SARS-CoV-2-related myocarditis-like syndromes shakespeare’s question: what’s in a name? Eur J Heart Fail. (2020) 22(6):922–5. doi: 10.1002/ejhf.1899
114. Osterlund P, Pirhonen J, Ikonen N, Rönkkö E, Strengell M, Mäkelä SM, et al. Pandemic H1N1 2009 influenza A virus induces weak cytokine responses in human macrophages and dendritic cells and is highly sensitive to the antiviral actions of interferons. J Virol. (2010) 84(3):1414–22. doi: 10.1128/JVI.01619-09
115. Tatsumi K, Schmedes CM, Houston ER, Butler E, Mackman N, Antoniak S. Protease-activated receptor 4 protects mice from coxsackievirus B3 and H1N1 influenza A virus infection. Cell Immunol. (2019) 344:103949. doi: 10.1016/j.cellimm.2019.103949
116. Ng DL, Al Hosani F, Keating MK, Gerber SI, Jones TL, Metcalfe MG, et al. Clinicopathologic, immunohistochemical, and ultrastructural findings of a fatal case of Middle East respiratory syndrome coronavirus infection in the United Arab Emirates, April 2014. Am J Pathol. (2016) 186(3):652–8. doi: 10.1016/j.ajpath.2015.10.024
117. Cascella M, Rajnik M, Aleem A, Dulebohn SC, Di Napoli R. Features, Evaluation, and Treatment of Coronavirus (COVID-19). 2023 Aug 18. In: StatPearls [Internet]. Treasure Island, FL: StatPearls Publishing (2024). 32150360.
118. Alsaad KO, Hajeer AH, Al Balwi M, Al Moaiqel M, Al Oudah N, Al Ajlan A, et al. Histopathology of Middle East respiratory syndrome coronovirus (MERS-CoV) infection–clinicopathological and ultrastructural study. Histopathology. (2018) 72(3):516–24. doi: 10.1111/his.13379
119. Chu H, Zhou J, Wong BH, Li C, Cheng ZS, Lin X, et al. Productive replication of Middle East respiratory syndrome coronavirus in monocyte-derived dendritic cells modulates innate immune response. Virology. (2014) 454-455:197–205. doi: 10.1016/j.virol.2014.02.018
120. Zhou J, Chu H, Li C, Wong BH, Cheng ZS, Poon VK, et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J Infect Dis. (2014) 209(9):1331–42. doi: 10.1093/infdis/jit504
121. Chu H, Zhou J, Wong BH, Li C, Chan JF, Cheng ZS, et al. Middle East respiratory syndrome coronavirus efficiently infects human primary T lymphocytes and activates the extrinsic and intrinsic apoptosis pathways. J Infect Dis. (2016) 213(6):904–14. doi: 10.1093/infdis/jiv380
122. Ding Y, Wang H, Shen H, Li Z, Geng J, Han H, et al. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol. (2003) 200(3):282–9. doi: 10.1002/path.1440
123. Xiong TY, Redwood S, Prendergast B, Chen M. Coronaviruses and the cardiovascular system: acute and long-term implications. Eur Heart J. (2020) 41(19):1798–800. doi: 10.1093/eurheartj/ehaa231
124. Fairweather D, Beetler DJ, Di Florio DN, Musigk N, Heidecker B, Cooper LT Jr. COVID-19, myocarditis and pericarditis. Circ Res. (2023) 132(10):1302–19. doi: 10.1161/CIRCRESAHA.123.321878
125. Buja LM, Wolf DA, Zhao B, Akkanti B, McDonald M, Lelenwa L, et al. The emerging spectrum of cardiopulmonary pathology of the coronavirus disease 2019 (COVID-19): report of 3 autopsies from Houston, Texas, and review of autopsy findings from other United States cities. Cardiovasc Pathol. (2020) 48:107233. doi: 10.1016/j.carpath.2020.107233
126. Bradley BT, Maioli H, Johnston R, Chaudhry I, Fink SL, Xu H, et al. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington state: a case series. Lancet. (2020) 396(10247):320–32. doi: 10.1016/S0140-6736(20)31305-2
127. Basso C, Leone O, Rizzo S, De Gaspari M, van der Wal AC, Aubry MC, et al. Pathological features of COVID-19-associated myocardial injury: a multicentre cardiovascular pathology study. Eur Heart J. (2020) 41(39):3827–35. doi: 10.1093/eurheartj/ehaa664
128. Yang L, Nilsson-Payant BE, Han Y, Jaffré F, Zhu J, Wang P, et al. Cardiomyocytes recruit monocytes upon SARS-CoV-2 infection by secreting CCL2. Stem Cell Rep. (2021) 16(9):2274–88. doi: 10.1016/j.stemcr.2021.07.012
129. Shen Y, Chen M, Gu W, Wan J, Cheng Z, Shen K, et al. The molecular mechanism of cardiac injury in SARS-CoV-2 infection: focus on mitochondrial dysfunction. J Infect Public Health. (2023) 16(5):746–53. doi: 10.1016/j.jiph.2023.03.015
130. Arhontoulis DC, Kerr CM, Richards D, Tjen K, Hyams N, Jones JA, et al. Human cardiac organoids to model COVID-19 cytokine storm induced cardiac injuries. J Tissue Eng Regen Med. (2022) 16(9):799–811. doi: 10.1002/term.3327
131. Ryabkova VA, Churilov LP, Shoenfeld Y. Influenza infection, SARS, MERS and COVID-19: cytokine storm—the common denominator and the lessons to be learned. Clin Immunol. (2021) 223:108652. doi: 10.1016/j.clim.2020.108652
132. Minhas AS, Gilotra NA, Goerlich E, Metkus T, Garibaldi BT, Sharma G, et al. Myocardial work efficiency, a novel measure of myocardial dysfunction, is reduced in COVID-19 patients and associated with in-hospital mortality. Front Cardiovasc Med. (2021) 8:667721. doi: 10.3389/fcvm.2021.667721
133. Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res. (2020) 116(6):1097–100. doi: 10.1093/cvr/cvaa078
134. Ammirati E, Lupi L, Palazzini M, Hendren NS, Grodin JL, Cannistraci CV, et al. Prevalence, characteristics, and outcomes of COVID-19-associated acute myocarditis. Circulation. (2022) 145(15):1123–39. doi: 10.1161/CIRCULATIONAHA.121.056817
135. Escher F, Pietsch H, Aleshcheva G, Bock T, Baumeier C, Elsaesser A, et al. Detection of viral SARS-CoV-2 genomes and histopathological changes in endomyocardial biopsies. ESC Heart Fail. (2020) 7(5):2440–7. doi: 10.1002/ehf2.12805
136. Sheng CC, Sahoo D, Dugar S, Prada RA, Wang TKM, Abou Hassan OK, et al. Canakinumab to reduce deterioration of cardiac and respiratory function in SARS-CoV-2 associated myocardial injury with heightened inflammation (canakinumab in COVID-19 cardiac injury: the three C study). Clin Cardiol. (2020) 43(10):1055–63. doi: 10.1002/clc.23451
137. Cremer PC, Sheng CC, Sahoo D, Dugar S, Prada RA, Wang TKM, et al. Double-blind randomized proof-of-concept trial of canakinumab in patients with COVID-19 associated cardiac injury and heightened inflammation. Eur Heart J Open. (2021) 1(1):oeab002. doi: 10.1093/ehjopen/oeab002
138. Kubánek M, Šramko M, Berenová D, Hulínská D, Hrbáčková H, Malušková J, et al. Detection of Borrelia burgdorferi sensu lato in endomyocardial biopsy specimens in individuals with recent-onset dilated cardiomyopathy. Eur J Heart Fail. (2012) 14(6):588–96. doi: 10.1093/eurjhf/hfs027
139. Cadavid D, Bai Y, Hodzic E, Narayan K, Barthold SW, Pachner AR. Cardiac involvement in non-human primates infected with the lyme disease spirochete Borrelia burgdorferi. Lab Invest. (2004) 84(11):1439–50. doi: 10.1038/labinvest.3700177
140. Hofman P, Michiels JF, Rosenthal E, Tran AT, Taillan B, Ferrari E, et al. Acute Staphylococcus aureus myocarditis in AIDS. 2 cases. Arch Mal Coeur Vaiss. (1993) 86(12):1765–8.8024379
141. LeLeiko RM, Bower DJ, Larsen CP. MRSA-associated bacterial myocarditis causing ruptured ventricle and tamponade. Cardiology. (2008) 111(3):188–90. doi: 10.1159/000121602
142. O'Brien CE, Coulson JD, Sekar P, Resar JR, Nelson McMillan K. Non-rheumatic streptococcal myocarditis mimicking acute myocardial infarction in an adolescent male. Cardiol Young. (2018) 28(3):454–7. doi: 10.1017/S1047951117001524
143. Silva R, Puga L, Teixeira R, Lourenço C, Botelho A, Gonçalves L. Acute non-rheumatic myopericarditis: a rare complication of pharyngitis. Eur J Case Rep Intern Med. (2018) 5(12):000987. doi: 10.12890/2018_000987
144. Huber SA, Cunningham MW. Streptococcal M protein peptide with similarity to myosin induces CD4+ T cell-dependent myocarditis in MRL/++ mice and induces partial tolerance against coxsakieviral myocarditis. J Immunol. (1996) 156(9):3528–34. doi: 10.4049/jimmunol.156.9.3528
145. Hiraiwa H, Morimoto R, Ando R, Ito R, Araki T, Mizutani T, et al. Recurrent fulminant non-rheumatic streptococcal myocarditis proven by endomyocardial biopsy and autopsy. J Cardiol Cases. (2022) 26(1):62–5. doi: 10.1016/j.jccase.2022.02.004
146. Laboratory TJ. Pneumococcal bacteria can directly infect and damage the heart (2015). Available online at: https://www.jax.org/news-and-insights/2015/january/pneumococcal-bacteria-can-directly-infect-and-damage-the-heart (accessed December 01, 2022).
147. Brown AO, Mann B, Gao G, Hankins JS, Humann J, Giardina J, et al. Streptococcus pneumoniae translocates into the myocardium and forms unique microlesions that disrupt cardiac function. PLoS Pathog. (2014) 10(9):e1004383. doi: 10.1371/journal.ppat.1004383
148. Huppertz HI. Erkrankungen durch pneumokokken. Monatsschr Kinderheilkd. (2003) 151(4):384–90. doi: 10.1007/s00112-003-0674-5
149. de Egea V, Muñoz P, Valerio M, de Alarcón A, Lepe JA, Miró JM, et al. Characteristics and outcome of Streptococcus pneumoniae endocarditis in the XXI century: a systematic review of 111 cases (2000-2013). Medicine (Baltimore). (2015) 94(39):e1562. doi: 10.1097/MD.0000000000001562
150. Shenoy AT, Brissac T, Gilley RP, Kumar N, Wang Y, Gonzalez-Juarbe N, et al. Streptococcus pneumoniae in the heart subvert the host response through biofilm-mediated resident macrophage killing. PLoS Pathog. (2017) 13(8):e1006582. doi: 10.1371/journal.ppat.1006582
151. Brown AO, Millett ER, Quint JK, Orihuela CJ. Cardiotoxicity during invasive pneumococcal disease. Am J Respir Crit Care Med. (2015) 191(7):739–45. doi: 10.1164/rccm.201411-1951PP
152. Stange K, Damaschke HJ, Berwing K. Sekundär immunologisch bedingte myokarditis, perikarditis und pleuritis exsudativa infolge einer meningokokken-meningitis. Zeitschrift für Kardiologie. (2001) 90(3):197–202. doi: 10.1007/s003920170184
153. Aung M, Raith E, Williams E, Burrell AJ. Severe meningococcal serogroup W sepsis presenting as myocarditis: a case report and review of literature. J Intensive Care Soc. (2019) 20(2):182–6. doi: 10.1177/1751143718794127
154. Madelaine T, Cour M, Bohé J, Floccard B, Duperret S, Hernu R, et al. Invasive meningococcal disease-induced myocarditis in critically ill adult patients: initial presentation and long-term outcome. Intensive Care Med. (2017) 43(2):279–81. doi: 10.1007/s00134-016-4623-x
155. Corsini LM, Mishra V. 20—meningococcal Disease. In: Tyring SK, Lupi O, Hengge UR, editors. Tropical Dermatology 2nd ed. Amsterdam: Elsevier (2017). p. 236–42.
156. Ramos A, García-Pavía P, Orden B, Cobo M, Sánchez-Castilla M, Sánchez-Romero I, et al. Gonococcal endocarditis: a case report and review of the literature. Infection. (2014) 42(2):425–8. doi: 10.1007/s15010-013-0541-9
157. Nie S, Wu Y, Huang L, Pincus D, Tang YW, Lu X. Gonococcal endocarditis: a case report and literature review. Eur J Clin Microbiol Infect Dis. (2014) 33(1):23–7. doi: 10.1007/s10096-013-1921-x
158. de Campos FP, Kawabata VS, Bittencourt MS, Lovisolo SM, Felipe-Silva A, de Lemos AP. Gonococcal endocarditis: an ever-present threat. Autops Case Rep. (2016) 6(2):19–25. doi: 10.4322/acr.2016.037
159. Stelzer T, Heuss LT, Schorn R. Aortitis due to Salmonella. Dtsch Med Wochenschr. (2014) 139(16):835–8. doi: 10.1055/s-0034-1369931
160. Villablanca P, Mohananey D, Meier G, Yap JE, Chouksey S, Abegunde AT. Salmonella berta myocarditis: case report and systematic review of non-typhoid Salmonella myocarditis. World J Cardiol. (2015) 7(12):931–7. doi: 10.4330/wjc.v7.i12.931
161. Afzal A, Keohane M, Keeley E, Borzak S, Callender CW, Iannuzzi M. Myocarditis and pericarditis with tamponade associated with disseminated tuberculosis. Can J Cardiol. (2000) 16(4):519–21.10787468
162. Michira BN, Alkizim FO, Matheka DM. Patterns and clinical manifestations of tuberculous myocarditis: a systematic review of cases. Pan Afr Med J. (2015) 21:118. doi: 10.11604/pamj.2015.21.118.4282
163. Cowley A, Dobson L, Kurian J, Saunderson C. Acute myocarditis secondary to cardiac tuberculosis: a case report. Echo Res Pract. (2017) 4(3):K25–k9. doi: 10.1530/ERP-17-0024
164. Biedrzycki OJ, Baithun SI. TB-Related Sudden death (TBRSD) due to myocarditis complicating miliary TB: a case report and review of the literature. Am J Forensic Med Pathol. (2006) 27(4). doi: 10.1097/01.paf.0000233633.16185.32
165. Gautam M, Sogunuru G, Subramanyam G, Viswanath R. Tuberculous myocarditis presenting as a refractory ventricular tachycardia of biventricular origin. J Coll Med Sci-Nepal. (2011) 7(2):60–6. doi: 10.3126/jcmsn.v7i2.6686
166. Liu A, Nicol E, Hu Y, Coates A. Tuberculous endocarditis. Int J Cardiol. (2013) 167(3):640–5. doi: 10.1016/j.ijcard.2012.08.009
167. Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D, et al. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res. (2010) 106(10):1646–55. doi: 10.1161/CIRCRESAHA.109.213157
168. Barin JG, Baldeviano GC, Talor MV, Wu L, Ong S, Quader F, et al. Macrophages participate in IL-17-mediated inflammation. Eur J Immunol. (2012) 42(3):726–36. doi: 10.1002/eji.201141737
169. Hou X, Chen G, Bracamonte-Baran W, Choi HS, Diny NL, Sung J, et al. The cardiac microenvironment instructs divergent monocyte fates and functions in myocarditis. Cell Rep. (2019) 28(1):172–89.e7. doi: 10.1016/j.celrep.2019.06.007
170. Duthoit G, Ou P, Sidi D, Bonnet D. Mycoplasma pneumoniae myopericarditis in children. Arch Mal Coeur Vaiss. (2006) 99(5):511–3.16802744
171. Dawood H, Nasir S, Khair RM, Dawood M. Infective endocarditis secondary to Mycoplasma pneumoniae. Cureus. (2021) 13(8):e17461. doi: 10.7759/cureus.17461
172. Efe C, Can T, Ince M, Tunca H, Yildiz F, Sennaroglu E. A rare complication of Brucella infection: myocarditis and heart failure. Internal Medicine. (2009) 48(19):1773–4. doi: 10.2169/internalmedicine.48.2385
173. Pandit VR, Seshadri S, Valsalan R, Bahuleyan S, Vandana KE, Kori P. Acute brucellosis complicated by fatal myocarditis. Int J Infect Dis. (2010) 14(4):e358–60. doi: 10.1016/j.ijid.2009.05.013
174. Wendt S, Lippmann N, Fahnert J, Rodloff AC, Lübbert C. Brucella related myocarditis. Int J Infect Dis. (2018) 66:126–7. doi: 10.1016/j.ijid.2017.11.014
175. Li X, Wang T, Wang Y, Xie S, Tan W, Li P. Short- and long-term follow-up outcomes of patients with Brucella endocarditis: a systematic review of 207 Brucella endocarditis cases. Bioengineered. (2021) 12(1):5162–72. doi: 10.1080/21655979.2021.1962683
176. Bonney KM, Engman DM. Autoimmune pathogenesis of chagas heart disease: looking back, looking ahead. Am J Pathol. (2015) 185(6):1537–47. doi: 10.1016/j.ajpath.2014.12.023
177. Punukollu G, Gowda RM, Khan IA, Navarro VS, Vasavada BC. Clinical aspects of the Chagas’ heart disease. Int J Cardiol. (2007) 115(3):279–83. doi: 10.1016/j.ijcard.2006.03.004
178. Coelho LL, Pereira IR, de Souza Pereira MC, Mesquita L, Lannes-Vieira J, Adesse D, et al. Trypanosoma cruzi activates mouse cardiac fibroblasts in vitro leading to fibroblast-myofibroblast transition and increase in expression of extracellular matrix proteins. Parasit Vectors. (2018) 11(1):72. doi: 10.1186/s13071-018-2614-1
179. Cardillo F, de Pinho RT, Antas PR, Mengel J. Immunity and immune modulation in trypanosoma cruzi infection. Pathog Dis. (2015) 73(9):ftv082. doi: 10.1093/femspd/ftv082
180. Benvenuti LA, Roggério A, Freitas HFG, Mansur AJ, Fiorelli A, Higuchi ML. Chronic American trypanosomiasis: parasite persistence in endomyocardial biopsies is associated with high-grade myocarditis. Ann Trop Med Parasitol. (2008) 102(6):481–7. doi: 10.1179/136485908X311740
181. Giles JT, Fert-Bober J, Park JK, Bingham CO, Andrade F, Fox-Talbot K, et al. Myocardial citrullination in rheumatoid arthritis: a correlative histopathologic study. Arthritis Res Ther. (2012) 14(1):R39. doi: 10.1186/ar3752
182. Vossenaar ER, Després N, Lapointe E, van der Heijden A, Lora M, Senshu T, et al. Rheumatoid arthritis specific anti-sa antibodies target citrullinated vimentin. Arthritis Res Ther. (2004) 6(2):R142–50. doi: 10.1186/ar1149
183. Bang H, Egerer K, Gauliard A, Lüthke K, Rudolph PE, Fredenhagen G, et al. Mutation and citrullination modifies vimentin to a novel autoantigen for rheumatoid arthritis. Arthritis Rheum. (2007) 56(8):2503–11. doi: 10.1002/art.22817
184. Demoruelle MK, Deane K. Antibodies to citrullinated protein antigens (ACPAs): clinical and pathophysiologic significance. Curr Rheumatol Rep. (2011) 13(5):421–30. doi: 10.1007/s11926-011-0193-7
185. Spengler J, Lugonja B, Ytterberg AJ, Zubarev RA, Creese AJ, Pearson MJ, et al. Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol. (2015) 67(12):3135–45. doi: 10.1002/art.39313
186. Voskuyl AE. The heart and cardiovascular manifestations in rheumatoid arthritis. Rheumatology. (2006) 45(suppl_4):iv4–7. doi: 10.1093/rheumatology/kel313
187. Frustaci A, Alfarano M, Verardo R, Agrati C, Casetti R, Miraldi F, et al. Myocarditis-associated necrotizing coronary vasculitis: incidence, cause, and outcome. Eur Heart J. (2020) 42(16):1609–17. doi: 10.1093/eurheartj/ehaa973
188. Leone O, Pieroni M, Rapezzi C, Olivotto I. The spectrum of myocarditis: from pathology to the clinics. Virchows Arch. (2019) 475(3):279–301. doi: 10.1007/s00428-019-02615-8
189. Comarmond C, Cacoub P. Myocarditis in auto-immune or auto-inflammatory diseases. Autoimmun Rev. (2017) 16(8):811–6. doi: 10.1016/j.autrev.2017.05.021
190. Mavrogeni S, Sfikakis PP, Gialafos E, Bratis K, Karabela G, Stavropoulos E, et al. Cardiac tissue characterization and the diagnostic value of cardiovascular magnetic resonance in systemic connective tissue diseases. Arthritis Care Res (Hoboken). (2014) 66(1):104–12. doi: 10.1002/acr.22181
191. Okasha O, Kazmirczak F, Chen KA, Farzaneh-Far A, Shenoy C. Myocardial involvement in patients with histologically diagnosed cardiac sarcoidosis: a systematic review and meta-analysis of gross pathological images from autopsy or cardiac transplantation cases. J Am Heart Assoc. (2019) 8(10):e011253. doi: 10.1161/JAHA.118.011253
192. Lagana SM, Parwani AV, Nichols LC. Cardiac sarcoidosis: a pathology-focused review. Arch Pathol Lab Med. (2010) 134(7):1039–46. doi: 10.5858/2009-0274-RA.1
193. Moller DR, Forman JD, Liu MC, Noble PW, Greenlee BM, Vyas P, et al. Enhanced expression of IL-12 associated with Th1 cytokine profiles in active pulmonary sarcoidosis. J Immunol. (1996) 156(12):4952–60. doi: 10.4049/jimmunol.156.12.4952
194. Moller DR. Cells and cytokines involved in the pathogenesis of sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. (1999) 16(1):24–31.10207939
195. Schoppet M, Pankuweit S, Maisch B. Cardiac sarcoidosis: cytokine patterns in the course of the disease. Arch Pathol Lab Med. (2003) 127(9):1207–10. doi: 10.5858/2003-127-1207-CSCPIT
196. Kato S, Sakai Y, Okabe A, Kawashima Y, Kuwahara K, Shiogama K, et al. Histology of cardiac sarcoidosis with novel considerations arranged upon a pathologic basis. J Clin Med. (2022) 11(1):251. doi: 10.3390/jcm11010251
197. Honda Y, Nagai T, Ikeda Y, Sakakibara M, Asakawa N, Nagano N, et al. Myocardial immunocompetent cells and macrophage phenotypes as histopathological surrogates for diagnosis of cardiac sarcoidosis in Japanese. J Am Heart Assoc. (2016) 5(11). doi: 10.1161/JAHA.116.004019
198. Ten Berge B, Kleinjan A, Muskens F, Hammad H, Hoogsteden HC, Hendriks RW, et al. Evidence for local dendritic cell activation in pulmonary sarcoidosis. Respir Res. (2012) 13(1):33. doi: 10.1186/1465-9921-13-33
199. Ma Y, Gal A, Koss MN. The pathology of pulmonary sarcoidosis: update. Semin Diagn Pathol. (2007) 24(3):150–61. doi: 10.1053/j.semdp.2007.06.002
200. Sreeja C, Priyadarshini A, Premika Nachiammai N. Sarcoidosis—a review article. J Oral Maxillofac Pathol. 2022;26(2):242–53. doi: 10.4103/jomfp.jomfp_373_21
201. Müller-Quernheim J, Pfeifer S, Strausz J, Ferlinz R. Correlation of clinical and immunologic parameters of the inflammatory activity of pulmonary sarcoidosis. Am Rev Respir Dis. (1991) 144(6):1322–9. doi: 10.1164/ajrccm/144.6.1322
202. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. (2012) 18(7):1028–40. doi: 10.1038/nm.2807
203. Baughman RP, Valeyre D, Korsten P, Mathioudakis AG, Wuyts WA, Wells A, et al. ERS Clinical practice guidelines on treatment of sarcoidosis. Eur Respir J. (2021) 58(6):2004079. doi: 10.1183/13993003.04079-2020
204. Nassar M, Nso N, Gonzalez C, Lakhdar S, Alshamam M, Elshafey M, et al. COVID-19 vaccine-induced myocarditis: case report with literature review. Diabetes Metab Syndr. (2021) 15(5):102205. doi: 10.1016/j.dsx.2021.102205
205. Ehrlich P, Klingel K, Ohlmann-Knafo S, Hüttinger S, Sood N, Pickuth D, et al. Biopsy-proven lymphocytic myocarditis following first mRNA COVID-19 vaccination in a 40-year-old male: case report. Clin Res Cardiol. (2021) 110(11):1855–9. doi: 10.1007/s00392-021-01936-6
206. Nagasaka T, Koitabashi N, Ishibashi Y, Aihara K, Takama N, Ohyama Y, et al. Acute myocarditis associated with COVID-19 vaccination: a case report. J Cardiol Cases. (2022) 25(5):285–8. doi: 10.1016/j.jccase.2021.11.006
207. Muthukumar A, Narasimhan M, Li QZ, Mahimainathan L, Hitto I, Fuda F, et al. In-depth evaluation of a case of presumed myocarditis after the second dose of COVID-19 mRNA vaccine. Circulation. (2021) 144(6):487–98. doi: 10.1161/CIRCULATIONAHA.121.056038
208. Amemiya K, Kobayashi T, Kataoka Y, Iwai T, Nakagawa S, Morita Y, et al. Myocarditis after COVID-19 mRNA vaccination in three young adult males: significance of biopsy in vaccine-associated myocarditis. Pathol Int. (2022) 72(7):385–7. doi: 10.1111/pin.13234
209. McMurray JC, May JW, Cunningham MW, Jones OY. Multisystem inflammatory syndrome in children (MIS-C), a post-viral myocarditis and systemic vasculitis-A critical review of its pathogenesis and treatment. Front Pediatr. (2020) 8:626182. doi: 10.3389/fped.2020.626182
210. Heymans S, Cooper LT. Myocarditis after COVID-19 mRNA vaccination: clinical observations and potential mechanisms. Nat Rev Cardiol. (2022) 19(2):75–7. doi: 10.1038/s41569-021-00662-w
211. Thurner L, Kessel C, Fadle N, Regitz E, Seidel F, Kindermann I, et al. IL-1RA Antibodies in myocarditis after SARS-CoV-2 vaccination. N Engl J Med. (2022) 387(16):1524–7. doi: 10.1056/NEJMc2205667
212. Frustaci A, Chimenti C, Maseri A. Global biventricular dysfunction in patients with asymptomatic coronary artery disease may be caused by myocarditis. Circulation. (1999) 99(10):1295–9. doi: 10.1161/01.CIR.99.10.1295
213. Wan S, Yim AP, Wong CK, Arifi AA, Yip JH, Ng CS, et al. Expression of FHL2 and cytokine messenger RNAs in human myocardium after cardiopulmonary bypass. Int J Cardiol. (2002) 86(2-3):265–72. doi: 10.1016/S0167-5273(02)00331-5
214. Frustaci A, Placanica G, Galea N, Chimenti C. Auto-Reactive myocarditis and necrotizing coronary vasculitis after blunt chest trauma. Circ Cardiovasc Imaging. (2018) 11(9):e008078. doi: 10.1161/CIRCIMAGING.118.008078
215. Kalbitz M, Amann EM, Bosch B, Palmer A, Schultze A, Pressmar J, et al. Experimental blunt chest trauma-induced myocardial inflammation and alteration of gap-junction protein connexin 43. PLoS One. (2017) 12(11):e0187270. doi: 10.1371/journal.pone.0187270
216. Blanco-Domínguez R, Sánchez-Díaz R, de la Fuente H, Jiménez-Borreguero LJ, Matesanz-Marín A, Relaño M, et al. A novel circulating MicroRNA for the detection of acute myocarditis. N Engl J Med. (2021) 384(21):2014–27. doi: 10.1056/NEJMoa2003608
217. Frustaci A, Russo MA, Chimenti C. Randomized study on the efficacy of immunosuppressive therapy in patients with virus-negative inflammatory cardiomyopathy: the TIMIC study. Eur Heart J. (2009) 30(16):1995–2002. doi: 10.1093/eurheartj/ehp249
218. Wilke A, Kaiser A, Ferency I, Maisch B. Alcohol and myocarditis. Herz. (1996) 21(4):248–57.8805005
219. Varga ZV, Ferdinandy P, Liaudet L, Pacher P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am J Physiol Heart Circ Physiol. (2015) 309(9):H1453–67. doi: 10.1152/ajpheart.00554.2015
220. Maisch B. Alcoholic cardiomyopathy: the result of dosage and individual predisposition. Herz. (2016) 41(6):484–93. doi: 10.1007/s00059-016-4469-6
221. Schoppet M, Maisch B. Alcohol and the heart. Herz. (2001) 26(5):345–52. doi: 10.1007/PL00002037
222. Zhang P, Bagby GJ, Xie M, Stoltz DA, Summer WR, Nelson S. Acute ethanol intoxication inhibits neutrophil beta2-integrin expression in rats during endotoxemia. Alcohol Clin Exp Res. (1998) 22(1):135–41. doi: 10.1111/j.1530-0277.1998.tb03629.x
223. Bagby GJ, Zhang P, Stoltz DA, Nelson S. Suppression of the granulocyte colony-stimulating factor response to Escherichia coli challenge by alcohol intoxication. Alcohol Clin Exp Res. (1998) 22(8):1740–5. doi: 10.1111/j.1530-0277.1998.tb03974.x
224. Simet SM, Sisson JH. Alcohol’s effects on lung health and immunity. Alcohol Res. (2015) 37(2):199–208.26695745
225. Cook RT. Alcohol abuse, alcoholism, and damage to the immune system–a review. Alcohol Clin Exp Res. (1998) 22(9):1927–42.9884135
226. Szabo G. Consequences of alcohol consumption on host defence. Alcohol Alcohol. (1999) 34(6):830–41. doi: 10.1093/alcalc/34.6.830
227. Tønnesen H, Andersen JR, Pedersen AE, Kaiser AH. Lymphopenia in heavy drinkers–reversibility and relation to the duration of drinking episodes. Ann Med. (1990) 22(4):229–31. doi: 10.3109/07853899009148931
228. Mason CM, Dobard E, Zhang P, Nelson S. Alcohol exacerbates murine pulmonary tuberculosis. Infect Immun. (2004) 72(5):2556–63. doi: 10.1128/IAI.72.5.2556-2563.2004
229. Spinozzi F, Bertotto A, Rondoni F, Gerli R, Scalise F, Grignani F. T-lymphocyte activation pathways in alcoholic liver disease. Immunology. (1991) 73(2):140–6.1676985
230. Ness KJ, Fan J, Wilke WW, Coleman RA, Cook RT, Schlueter AJ. Chronic ethanol consumption decreases murine langerhans cell numbers and delays migration of langerhans cells as well as dermal dendritic cells. Alcohol Clin Exp Res. (2008) 32(4):657–68. doi: 10.1111/j.1530-0277.2007.00614.x
231. Chadha KC, Stadler I, Albini B, Nakeeb SM, Thacore HR. Effect of alcohol on spleen cells and their functions in C57BL/6 mice. Alcohol. (1991) 8(6):481–5. doi: 10.1016/S0741-8329(91)90187-2
232. Shi Y, Moon M, Dawood S, McManus B, Liu PP. Mechanisms and management of doxorubicin cardiotoxicity. Herz. (2011) 36(4):296–305. doi: 10.1007/s00059-011-3470-3
233. Morandi P, Ruffini PA, Benvenuto GM, La Vecchia L, Mezzena G, Raimondi R. Serum cardiac troponin I levels and ECG/Echo monitoring in breast cancer patients undergoing high-dose (7 g/m(2)) cyclophosphamide. Bone Marrow Transplant. (2001) 28(3):277–82. doi: 10.1038/sj.bmt.1703132
234. Higgins AY, O'Halloran TD, Chang JD. Chemotherapy-induced cardiomyopathy. Heart Fail Rev. (2015) 20(6):721–30. doi: 10.1007/s10741-015-9502-y
235. Renu K, VG A, PB T, Arunachalam S. Molecular mechanism of doxorubicin-induced cardiomyopathy—an update. Eur J Pharmacol. (2018) 818:241–53. doi: 10.1016/j.ejphar.2017.10.043
236. Takemura G, Fujiwara H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis. (2007) 49(5):330–52. doi: 10.1016/j.pcad.2006.10.002
237. Simmons A, Vacek JL, Meyers D. Anthracycline-induced cardiomyopathy. Postgrad Med. (2008) 120(4):67–72. doi: 10.3810/pgm.2008.11.1940
238. Mahmood SS, Fradley MG, Cohen JV, Nohria A, Reynolds KL, Heinzerling LM, et al. Myocarditis in patients treated with immune checkpoint inhibitors. J Am Coll Cardiol. (2018) 71(16):1755–64. doi: 10.1016/j.jacc.2018.02.037
239. Moslehi J, Lichtman AH, Sharpe AH, Galluzzi L, Kitsis RN. Immune checkpoint inhibitor-associated myocarditis: manifestations and mechanisms. J Clin Invest. (2021) 131(5). doi: 10.1172/JCI145186
240. Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. (2016) 375(18):1749–55. doi: 10.1056/NEJMoa1609214
241. Zhang L, Awadalla M, Mahmood SS, Nohria A, Hassan MZO, Thuny F, et al. Cardiovascular magnetic resonance in immune checkpoint inhibitor-associated myocarditis. Eur Heart J. (2020) 41(18):1733–43. doi: 10.1093/eurheartj/ehaa051
242. Hu JR, Florido R, Lipson EJ, Naidoo J, Ardehali R, Tocchetti CG, et al. Cardiovascular toxicities associated with immune checkpoint inhibitors. Cardiovasc Res. (2019) 115(5):854–68. doi: 10.1093/cvr/cvz026
243. Won T, Kalinoski HM, Wood MK, Hughes DM, Jaime CM, Delgado P, et al. Cardiac myosin-specific autoimmune T cells contribute to immune-checkpoint-inhibitor-associated myocarditis. Cell Rep. (2022) 41(6):111611. doi: 10.1016/j.celrep.2022.111611
244. Lv H, Havari E, Pinto S, Gottumukkala RV, Cornivelli L, Raddassi K, et al. Impaired thymic tolerance to α-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest. (2011) 121(4):1561–73. doi: 10.1172/JCI44583
245. Khunger A, Battel L, Wadhawan A, More A, Kapoor A, Agrawal N. New insights into mechanisms of immune checkpoint inhibitor-induced cardiovascular toxicity. Curr Oncol Rep. (2020) 22(7):65. doi: 10.1007/s11912-020-00925-8
246. Palaskas N, Lopez-Mattei J, Durand JB, Iliescu C, Deswal A. Immune checkpoint inhibitor myocarditis: pathophysiological characteristics, diagnosis, and treatment. J Am Heart Assoc. (2020) 9(2):e013757. doi: 10.1161/JAHA.119.013757
247. Zhu H, Galdos FX, Lee D, Waliany S, Huang YV, Ryan J, et al. Identification of pathogenic immune cell subsets associated with checkpoint inhibitor-induced myocarditis. Circulation. (2022) 146(4):316–35. doi: 10.1161/CIRCULATIONAHA.121.056730
248. Zhang XZ, Zhang S, Tang TT, Cheng X. Bioinformatics and immune infiltration analyses reveal the key pathway and immune cells in the pathogenesis of hypertrophic cardiomyopathy. Front Cardiovasc Med. (2021) 8:696321. doi: 10.3389/fcvm.2021.696321
249. Zhao W, Wu T, Zhan J, Dong Z. Identification of the immune Status of hypertrophic cardiomyopathy by integrated analysis of bulk- and single-cell RNA sequencing data. Comput Math Methods Med. (2022) 2022:7153491. doi: 10.1155/2022/7153491
250. Zheng X, Yang Y, Huang Fu C, Huang R. Identification and verification of promising diagnostic biomarkers in patients with hypertrophic cardiomyopathy associate with immune cell infiltration characteristics. Life Sci. (2021) 285:119956. doi: 10.1016/j.lfs.2021.119956
251. Koczo A, Marino A, Jeyabalan A, Elkayam U, Cooper LT, Fett J, et al. Breastfeeding, cellular immune activation, and myocardial recovery in peripartum cardiomyopathy. JACC Basic Transl Sci. (2019) 4(3):291–300. doi: 10.1016/j.jacbts.2019.01.010
252. Davis MB, Arany Z, McNamara DM, Goland S, Elkayam U. Peripartum cardiomyopathy: jACC state-of-the-art review. J Am Coll Cardiol. (2020) 75(2):207–21. doi: 10.1016/j.jacc.2019.11.014
253. Hilfiker-Kleiner D, Kaminski K, Podewski E, Bonda T, Schaefer A, Sliwa K, et al. A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell. (2007) 128(3):589–600. doi: 10.1016/j.cell.2006.12.036
254. McTiernan CF, Morel P, Cooper LT, Rajagopalan N, Thohan V, Zucker M, et al. Circulating T-cell subsets, monocytes, and natural killer cells in peripartum cardiomyopathy: results from the multicenter IPAC study. J Card Fail. (2018) 24(1):33–42. doi: 10.1016/j.cardfail.2017.10.012
255. Hilfiker-Kleiner D, Haghikia A, Berliner D, Vogel-Claussen J, Schwab J, Franke A, et al. Bromocriptine for the treatment of peripartum cardiomyopathy: a multicentre randomized study. Eur Heart J. (2017) 38(35):2671–9. doi: 10.1093/eurheartj/ehx355
256. Trongtorsak A, Kittipibul V, Mahabir S, Ibrahim M, Saint Croix GR, Hernandez GA, et al. Effects of bromocriptine in peripartum cardiomyopathy: a systematic review and meta-analysis. Heart Fail Rev. (2022) 27(2):533–43. doi: 10.1007/s10741-021-10185-8
257. Kumar A, Ravi R, Sivakumar RK, Chidambaram V, Majella MG, Sinha S, et al. Prolactin inhibition in peripartum cardiomyopathy: systematic review and meta-analysis. Curr Probl Cardiol. (2023) 48(2):101461. doi: 10.1016/j.cpcardiol.2022.101461
258. Azibani F, Pfeffer TJ, Ricke-Hoch M, Dowling W, Pietzsch S, Briton O, et al. Outcome in German and South African peripartum cardiomyopathy cohorts associates with medical therapy and fibrosis markers. ESC Heart Fail. (2020) 7(2):512–22. doi: 10.1002/ehf2.12553
259. Haghikia A, Podewski E, Libhaber E, Labidi S, Fischer D, Roentgen P, et al. Phenotyping and outcome on contemporary management in a German cohort of patients with peripartum cardiomyopathy. Basic Res Cardiol. (2013) 108(4):366. doi: 10.1007/s00395-013-0366-9
260. Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation. (1996) 94(5):983–91. doi: 10.1161/01.CIR.94.5.983
261. Campuzano O, Alcalde M, Iglesias A, Barahona-Dussault C, Sarquella-Brugada G, Benito B, et al. Arrhythmogenic right ventricular cardiomyopathy: severe structural alterations are associated with inflammation. J Clin Pathol. (2012) 65(12):1077–83. doi: 10.1136/jclinpath-2012-201022
262. Ng KE, Delaney PJ, Thenet D, Murtough S, Webb CM, Zaman N, et al. Early inflammation precedes cardiac fibrosis and heart failure in desmoglein 2 murine model of arrhythmogenic cardiomyopathy. Cell Tissue Res. (2021) 386(1):79–98. doi: 10.1007/s00441-021-03488-7
263. Chelko SP, Asimaki A, Lowenthal J, Bueno-Beti C, Bedja D, Scalco A, et al. Therapeutic modulation of the immune response in arrhythmogenic cardiomyopathy. Circulation. (2019) 140(18):1491–505. doi: 10.1161/CIRCULATIONAHA.119.040676
264. Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation. (1982) 65(2):384–98. doi: 10.1161/01.CIR.65.2.384
265. Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. (1988) 318(3):129–33. doi: 10.1056/NEJM198801213180301
266. Corrado D, Basso C, Judge DP. Arrhythmogenic cardiomyopathy. Circ Res. (2017) 121(7):784–802. doi: 10.1161/CIRCRESAHA.117.309345
267. van der Voorn SM, Te Riele A, Basso C, Calkins H, Remme CA, van Veen TAB. Arrhythmogenic cardiomyopathy: pathogenesis, pro-arrhythmic remodelling, and novel approaches for risk stratification and therapy. Cardiovasc Res. (2020) 116(9):1571–84. doi: 10.1093/cvr/cvaa084
268. Liao X, Chang E, Tang X, Watanabe I, Zhang R, Jeong HW, et al. Cardiac macrophages regulate isoproterenol-induced takotsubo-like cardiomyopathy. JCI Insight. (2022) 7(3). doi: 10.1172/jci.insight.156236
269. Scally C, Abbas H, Ahearn T, Srinivasan J, Mezincescu A, Rudd A, et al. Myocardial and systemic inflammation in acute stress-induced (takotsubo) cardiomyopathy. Circulation. (2019) 139(13):1581–92. doi: 10.1161/CIRCULATIONAHA.118.037975
270. Nef HM, Möllmann H, Kostin S, Troidl C, Voss S, Weber M, et al. Tako-Tsubo cardiomyopathy: intraindividual structural analysis in the acute phase and after functional recovery. Eur Heart J. (2007) 28(20):2456–64. doi: 10.1093/eurheartj/ehl570
271. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. (2016) 12(3):144–53. doi: 10.1038/nrendo.2015.216
272. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. (2018) 122(4):624–38. doi: 10.1161/CIRCRESAHA.117.311586
273. Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia. (2018) 61(1):21–8. doi: 10.1007/s00125-017-4390-4
274. Hinman RM, Cambier JC. Role of B lymphocytes in the pathogenesis of type 1 diabetes. Curr Diab Rep. (2014) 14(11):543. doi: 10.1007/s11892-014-0543-8
275. Sokos GG, Nikolaidis LA, Mankad S, Elahi D, Shannon RP. Glucagon-like peptide-1 infusion improves left ventricular ejection fraction and functional status in patients with chronic heart failure. J Card Fail. (2006) 12(9):694–9. doi: 10.1016/j.cardfail.2006.08.211
276. Frustaci A, Russo MA, Sansone L, Francone M, Verardo R, Grande C, et al. Heart failure from gouty myocarditis: a case report. Ann Intern Med. (2020) 172(5):363–5. doi: 10.7326/L19-0486
277. Mei Y, Dong B, Geng Z, Xu L. Excess uric acid induces gouty nephropathy through crystal formation: a review of recent insights. Front Endocrinol (Lausanne). (2022) 13:911968. doi: 10.3389/fendo.2022.911968
278. Frustaci A, Verardo R, Grande C, Galea N, Piselli P, Carbone I, et al. Immune-Mediated myocarditis in fabry disease cardiomyopathy. J Am Heart Assoc. (2018) 7(17):e009052. doi: 10.1161/JAHA.118.009052
279. Frustaci A, Scarpa M, da Riol RM, Agrati C, Finato N, Verardo R, et al. Fabry cardiomyopathy: gb3-induced auto-reactive panmyocarditis requiring heart transplantation. ESC Heart Fail. (2020) 7(3):1331–7. doi: 10.1002/ehf2.12723
280. Mavrogeni SI, Markousis-Mavrogenis G, Papavasiliou A, Papadopoulos G, Kolovou G. Cardiac involvement in duchenne muscular dystrophy and related dystrophinopathies. Methods Mol Biol. (2018) 1687:31–42. doi: 10.1007/978-1-4939-7374-3_3
281. Farini A, Villa C, Di Silvestre D, Bella P, Tripodi L, Rossi R, et al. PTX3 Predicts myocardial damage and fibrosis in duchenne muscular dystrophy. Front Physiol. (2020) 11:403. doi: 10.3389/fphys.2020.00403
282. Ekström K, Lehtonen J, Kandolin R, Räisänen-Sokolowski A, Salmenkivi K, Kupari M. Long-term outcome and its predictors in giant cell myocarditis. Eur J Heart Fail. (2016) 18(12):1452–8. doi: 10.1002/ejhf.606
283. Maleszewski JJ, Orellana VM, Hodge DO, Kuhl U, Schultheiss HP, Cooper LT. Long-term risk of recurrence, morbidity and mortality in giant cell myocarditis. Am J Cardiol. (2015) 115(12):1733–8. doi: 10.1016/j.amjcard.2015.03.023
284. Sultan RH, Abdallah M, Ali TM, Ahmed AE, Assal HH, Elesawy BH, et al. The associations between cytokine levels, kidney and heart function biomarkers, and expression levels of angiotensin-converting enzyme-2 and neuropilin-1 in COVID-19 patients. Vaccines (Basel). (2022) 10(7). doi: 10.3390/vaccines10071045
285. Zhang R, Chen X, Zuo W, Ji Z, Qu Y, Su Y, et al. Inflammatory activation and immune cell infiltration are main biological characteristics of SARS-CoV-2 infected myocardium. Bioengineered. (2022) 13(2):2486–97. doi: 10.1080/21655979.2021.2014621
286. Brala D, Thevathasan T, Grahl S, Barrow S, Violano M, Bergs H, et al. Application of magnetocardiography to screen for inflammatory cardiomyopathy and monitor treatment response. J Am Heart Assoc. (2023) 12(4):e027619. doi: 10.1161/JAHA.122.027619
287. Heidecker B. Quantum sensors: detect heart disease sooner. Nature. (2023) 619(7970):465. doi: 10.1038/d41586-023-02247-8
288. Heidecker B. Rediscovery of magnetocardiography for diagnostic screening and monitoring of treatment response in cardiology. Eur Heart J. (2023) 44(24):2140–2. doi: 10.1093/eurheartj/ehad213
289. Suwalski P, Golpour A, Musigk N, Wilke F, Landmesser U, Heidecker B. Case report: recurrence of inflammatory cardiomyopathy detected by magnetocardiography. Front Cardiovasc Med. (2023) 10:1225057. doi: 10.3389/fcvm.2023.1225057
Keywords: myocarditis, inflammatory cardiomyopathy, heart failure, cardiomyopathy, endomyocardial biopsy (EMB)
Citation: Musigk N, Suwalski P, Golpour A, Fairweather D, Klingel K, Martin P, Frustaci A, Cooper LT, Lüscher TF, Landmesser U and Heidecker B (2024) The inflammatory spectrum of cardiomyopathies. Front. Cardiovasc. Med. 11:1251780. doi: 10.3389/fcvm.2024.1251780
Received: 2 July 2023; Accepted: 29 January 2024;
Published: 23 February 2024.
Edited by:
Sang-Wook Kim, Chung-Ang University Hospital, Republic of KoreaReviewed by:
Sebastian Bannasch, Steinbeis Foundation, Germany© 2024 Musigk, Suwalski, Golpour, Fairweather, Klingel, Martin, Frustaci, Cooper, Lüscher, Landmesser and Heidecker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bettina Heidecker YmV0dGluYS5oZWlkZWNrZXJAZGh6Yy1jaGFyaXRlLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.