
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 29 June 2023
Sec. Coronary Artery Disease
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1194402
 Guo-Yang Liu1,2,3,†
Guo-Yang Liu1,2,3,† Wan-Li Xie1,2,3,†Yan-Ting Wang1,2,3Lu Chen1,2,3
Wan-Li Xie1,2,3,†Yan-Ting Wang1,2,3Lu Chen1,2,3 Zhen-Zhen Xu1,2,3
Zhen-Zhen Xu1,2,3 Yong Lv1,2,3
Yong Lv1,2,3 Qing-Ping Wu1,2,3*
Qing-Ping Wu1,2,3*
Calpain is a conserved cysteine protease readily expressed in several mammalian tissues, which is usually activated by Ca2+ and with maximum activity at neutral pH. The activity of calpain is tightly regulated because its aberrant activation will nonspecifically cleave various proteins in cells. Abnormally elevation of Ca2+ promotes the abnormal activation of calpain during myocardial ischemia-reperfusion, resulting in myocardial injury and cardiac dysfunction. In this paper, we mainly reviewed the effects of calpain in various programmed cell death (such as apoptosis, mitochondrial-mediated necrosis, autophagy-dependent cell death, and parthanatos) in myocardial ischemia-reperfusion. In addition, we also discussed the abnormal activation of calpain during myocardial ischemia-reperfusion, the effect of calpain on myocardial repair, and the possible future research directions of calpain.
Heart disease is the leading cause of death, and coronary heart disease (CHD) accounts for a large proportion of deaths (1). Mammalian terminally differentiated cardiomyocytes have limited self-replacement capacity and cannot be replenished after injury, resulting in cardiac dysfunction and heart failure (2). Coronary artery recanalization and coronary artery bypass grafting provide the most effective treatment for patients with cardiac artery occlusion (3), reduced heart damage during ischemia. However, the ischemic heart will have a series of pathophysiological reactions after blood supply reconstruction, which will worsen myocardial damage, cause cardiomyocyte death, and is known as reperfusion injury. Cardiac reperfusion injury has not been effectively solved. How to reduce reperfusion injury and reduce the occurrence of heart failure caused by reperfusion injury is an urgent clinical problem to be solved. Calcium overload during myocardial ischemia-reperfusion (I/R) is one of the crucial mechanisms of myocardial reperfusion injury (4). Calcium overload can activate calcium-dependent protein hydrolases, such as calpain, a cysteine protease, which can nonspecifically cleave various proteins, leading to cell damage and death. Cell death is the central link of ischemia-reperfusion injury. Cell death can be subdivided into programmed cell death (PCD) and non-programmed cell death. Unlike non-programmed cell death, such as classical necrosis, PCD depends on specific molecular mechanisms (5). This provides the foundation for interventions to reduce injury. Accumulating evidence indicates that calpain regulates PCD and is critically associated with myocardial I/R injury (6). Therefore, we summarize the role of calpain in various PCD in myocardial I/R injury and stress that the pathways of PCD are complex and interact under the action of calpain. In addition, we also described the process of abnormal activation of calpain caused by the increase of intracellular Ca2+ during I/R, the effect of calpain on myocardial repair, analyzed the possible reasons for different results of inhibiting calpain in the basic experiment, and proposed the possible future research directions of calpain.
Ca2+ acts as the second messenger and participates in multiple biological processes in cells. Ca2+ plays a decisive role in the systolic function of the heart. Generally, the intracellular Ca2+ content is much lower than that in extracellular (7). In normal physiological conditions, intracellular Ca2+ content is usually lower than 0.05 μmol/L (8). Ca2+ in cardiomyocytes mainly comes from extracellular and sarcoplasmic reticulum (SR) (9). When the myocardial cell membrane is depolarization, the L-type Ca2+ channels (LTCC) on the cell membrane are open, issues in the flow of extracellular Ca2+ into the cells. When intracellular Ca2+ concentration is higher than 10 μmol/L, ryanodine receptors (RyR) in the SR are activated, Ca2+ in SR flow to the cytoplasm, which is called Ca2+-induced Ca2+ release (10). Ca2+-induced Ca2+ release is the core of the calcium signal transduction mechanism (11). In general, the increase of Ca2+ will lead to myocardial contraction, and then Ca2+ in the myocardial diastolic phase is transported back to the SR through SR Ca2+ ATPase (SERCA) or transferer to extracellular through Na+/Ca2+ exchanger (NCX) in the plasma membrane. However, during ischemia, ATP deficiency inhibited Na+/K+-ATPase and reduced Na+ excretion, anaerobic glycolysis led to cytoplasmic acidification, and the increased H+ was discharged through Na+/H+ exchanger (NHX) to promote extracellular Na+ influx. Together with Na+/HCO3− cotransporter and persistent Na+ channels, all mechanisms increase intracellular Na+ content (12). Abnormally elevated intracellular Na+ changed the membrane potential leading to NCX reverse exchange, which increased intracellular Ca2+ concentration (13). Moreover, the reperfusion blood flow took away the extracellular H+, which led to increased gradient difference of H+ between intracellular and extracellular. The combined effects were strengthened, and the intracellular Ca2+ concentration would further increase (6). In addition, studies have shown that Ca2+-sensitive RyR is disrupted during myocardial I/R (14). The leakage of Ca2+ in the SR is also the cause of calcium overload.
Calpain is an intracellular non-lysosomal cysteine protease, one of the few proteases that the second messenger Ca2+ can directly activate. Due to the proteolysis ability of calpain being finite, calpain mainly plays a role in regulating rather than removing substrates in cells (15). Up to now, 15 types of calpain isoforms have been identified in the human body (The distribution of calpain in the human body is shown in Table 1) (15–17), among which calpain-1 (μ-calpain) and calpain-2 (m-calpain) are the most common, they are activated by micromolar and millimolar concentrations of Ca2+ in vitro experiments, respectively (18). Usually, intracellular Ca2+ concentration can only activate calpain-1 in physiological conditions. However, in pathological conditions, such as ischemia-reperfusion, intracellular Ca2+ concentration is higher than that in normal conditions. Abnormally elevated Ca2+ are combined with other mechanisms, such as phospholipids or phosphorylation, to activate calpain-2 (19). Calpain is usually in the form of an inactive enzyme that binds to calpastatin, an endogenous calpain inhibitor, to form an isomerase dimer, which can be located in the cytoplasm, endoplasmic reticulum, or mitochondria (20). Calpastatin inhibits calpain activation by binding to calpain through its calpain inhibitor domain (21). When the intracellular Ca2+ concentration was increased, calpain was separated from calpastatin and allowed to shift to the membrane (22). Calpain transfers to the cell membrane, binding to phospholipids to reduce the required Ca2+ concentration for activation or directly to the vicinity of Ca2+ channels. The activation of calpain is also affected by oxidative stress (23, 24). The reactive oxygen species produced by oxidative stress can directly inhibit ATPase, reduce the outflow of Ca2+ and destroy the calcium channels on the SR, resulting in the Ca2+ in the SR flowing into the cytoplasm, thereby activating calpain. After reaching the threshold of Ca2+ concentration for activation, the inactive enzyme was self-melted into the active form (18), stayed on the membrane, or was released into the cytoplasm to hydrolyze substrate proteins. Intracellular acidification significantly inhibits the activation of calpain (25). Therefore, calpain usually has the highest activity at the reperfusion stage after the pH value returns to normal. Calpastatin is another major factor affecting the activation of calpain in addition to the concentration of Ca2+ (26). Calpastatin plays a role in preventing calpain's excessive activation with the increased Ca2+ concentration. In contrast, calpastatin will be degraded by activated calpain or proteasome during myocardial ischemia-reperfusion (25, 27), issue in increased activation of calpain. Due to the activation of calpain being associated with Ca2+, which is an uneven distribution in cells, the subcellular localization of calpain may be related to its activation. Recent studies have found an interaction between the integrated membrane protein tiny tim50 (Ttm50) and calpain. This interaction promotes the directional movement of calpain to the Golgi body or endoplasmic reticulum, which is responsible for calcium storage. Moreover, Ttm50 increases the sensitivity of calpain to Ca2+ and enhances the activation of calpain (28). The possible mechanism of Ttm50 promoting calpain activation is: calpain is guided to move to a relatively high calcium environment in cells, and the conformation of calpain is changed to increase its affinity to Ca2+ so that calpain can be effectively activated in physiological conditions (19). Of course, there are other mechanisms to reduce the dependence of calpain activation on Ca2+ concentration, such as calpain phosphorylation and phospholipid-mediated calpain autolysis. Several phosphatidylinositols have been found to increase the autolysis of calpain on the cell membrane (29). ERK directly or indirectly activates calpain through phosphorylation of calpain or reduced Ca2+ concentration required for activation (30, 31). Up to now, the activation and regulation mechanism of calpain has not been fully understood, which needs further exploration (The activation mechanism of calpain during I/R is detailed in Figure 1).
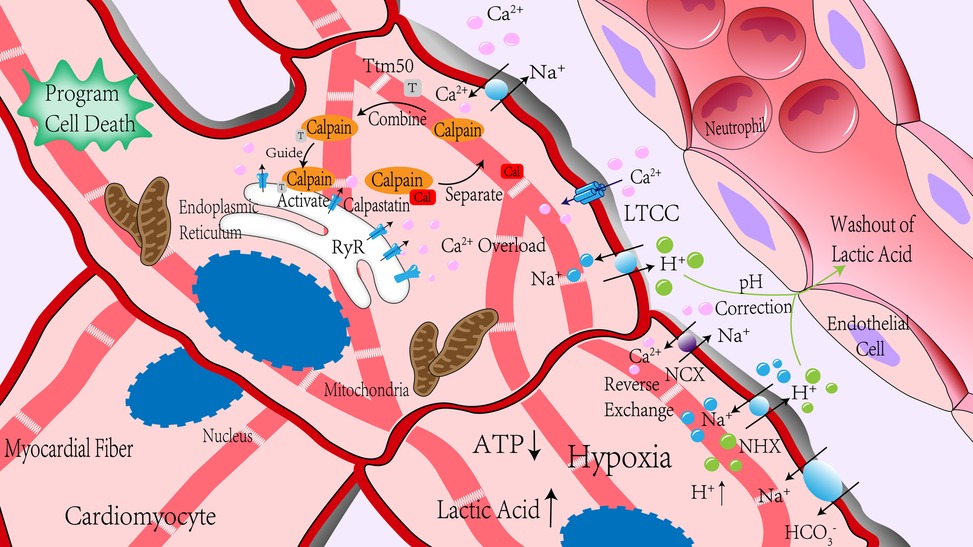
Figure 1. Calpain was significantly activated during ischemia-reperfusion. In the process of myocardial ischemia, due to anaerobic metabolism, ATP production is insufficient, lactic acid production is increased, pH value is decreased, H+ content is increased, Na+/H+ exchange is increased, and ATP deficiency leads to the insufficient activity of channel protein that mediates Na+ outflow, so intracellular Na+ content is increased. Changes in membrane potential caused by changes in Na+ content led to reverse exchange of Na+/Ca2+ exchanger, and ischemia-reperfusion led to RyR receptor injury, which promotes Ca2+ outflow in the sarcoplasmic reticulum. Together, these effects increase intracellular Ca2+ concentration rapidly. Reperfusion blood flow took away extracellular H+, and the concentration difference between intracellular and extracellular H+ further increased, enhancing the above effects. After reperfusion, the intracellular pH value gradually increased. Under the action of Ca2+, calpain was separated from its endogenous inhibitor, calpastatin. With the help of Ttm50, calpain migrated to the endoplasmic reticulum and was further activated in the high concentration of Ca2+ around the endoplasmic reticulum. NCX, Na+/Ca2+ exchanger; NHX, Na+/H+ exchanger; RyR, ryanodine receptors; LTCC, L-type Ca2+ channels.
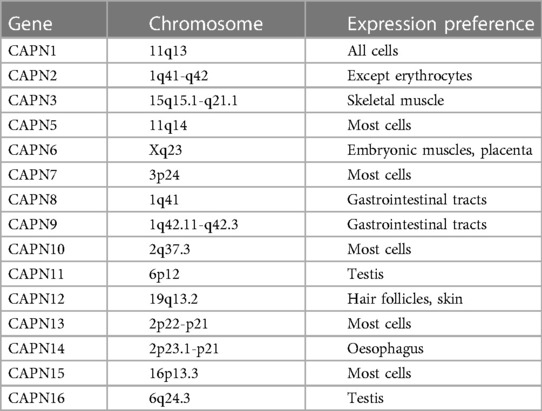
Table 1. Distribution of calpain in the human body.
Calpain is a regulated protein that can perform limited protein hydrolysis to regulate the function of substrate (15). The Calpain system participates in maintaining the cell cycle, cell movement, and cell signal transduction and regulates cell differentiation, migration, and various programmed cell death processes. Calpain plays a crucial role in activation or degradation of proteins by signal-dependent cleavage of substrate proteins (32, 33). Calpain will be activated significantly during myocardial ischemia reperfusion (34). Calpain has inconsistent binding sites and cleavage sites on a wide range of substrates, which has a wide range of effects on cell biology. Therefore, basic and clinical researchers often include calpain in their research. Excessive activation or insufficient activation of calpain may lead to abnormal accumulation or excessive degradation of intracellular proteins, resulting in various cell damage and pathological conditions (19). It has been proved that myocardial I/R injury is related to the dysfunction of calpain (34–36). During I/R, a large amount of calpain is usually activated during the reperfusion period of normal intracellular pH value, at which the activity of calpain is the highest (25). The expression and activity of calpain in cardiomyocytes treated with hypoxia-reoxygenation and I/R were increased (34), and calpain activation on the cell membrane was positively correlated with myocardial injury (36). Inhibiting calpain activity by drug Inhibitors or genetic knockout reduces myocardial I/R injury (37, 38). The review of Hiroyuki Sorimachi details the research on the effect of intervening calpain on ischemia-reperfusion injury (15).
Cell death is a critical event in myocardial I/R injury. Since specific molecular pathways mediate PCD, there is the possibility of inhibiting these signaling pathways to reduce myocardial damage and dysfunction caused by I/R. It has been confirmed that apoptosis, mitochondrial-mediated necrosis, necroptosis, pyroptosis, parthanatos, and autophagy-dependent cell death play significant roles in myocardial I/R injury (5). The following contents discuss the function of calpain in various PCD (The effects of calpain on key molecules in various programmed cellular pathways during I/R are detailed in Table 2).
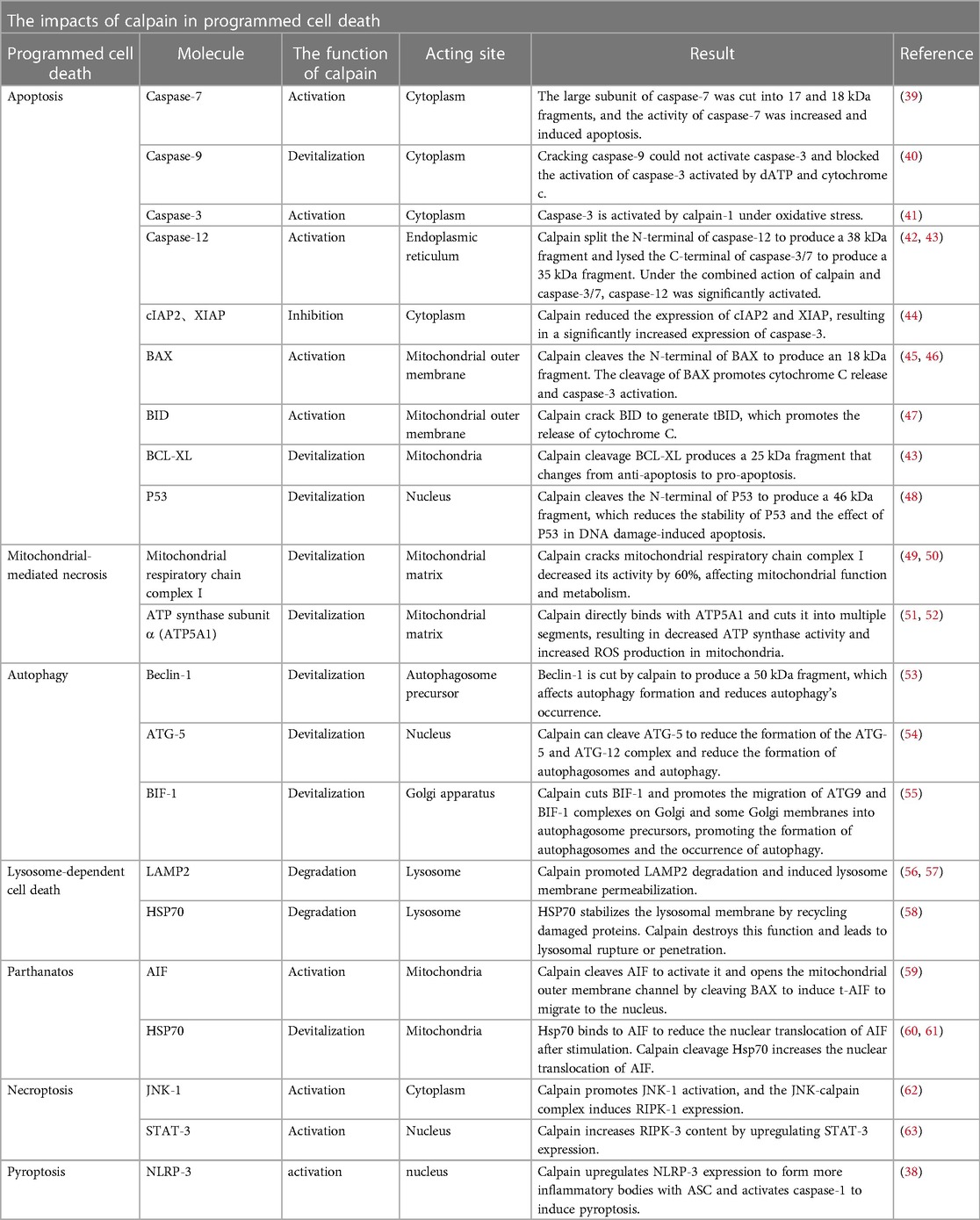
Table 2. The effects of calpain in programmed cell death.
Apoptosis is a highly controlled process of cell death, independently carried out by entirely healthy or sublethally injured cells in response to physiological or pathological stimulation (64). Several experiments have shown that inhibition of apoptosis during I/R can reduce myocardial infarction size in mice (65–67). Apoptosis signal can be transmitted through the cell surface death receptor pathway (external pathway) and mitochondrial pathway (internal pathway). Caspase plays a vital role in apoptosis (68). The caspase family mainly has three subclasses: promoter (caspase-8, 9, 10), effector (caspase-3, 6, 7), and inflammatory factors (caspase-1, 4, 5). Calpain can directly act on caspase family proteins to influence apoptosis—for example, calpain cleavages caspase-3 and caspase-7 to activate apoptosis (39–41). Calpain also can cleavage caspase-12 to trigger apoptosis during endoplasmic reticulum stress (42). Caspase-3 also cleavages endogenous calpastatin after activation, enhancing calpain activity (69). Calpain can cleavage inhibitor apoptosis protein (IAP), such as cellular IAP2 (cIAP2), and X chromosome-linked IAP (XIAP), to increase the expression of caspase-3 and promote the occurrence of apoptosis (44). IAP acts as a caspase inhibitor in cells (70) and binds to caspase-3,7 and 9 through its baculovirus IAP repeat (BIR) domain to block caspase activation and negatively regulate intrinsic and extrinsic apoptosis pathways (71).
Calpain can affect apoptosis not only by directly interfering with caspases but also by cutting essential BCL-2 family proteins in the mitochondrial apoptotic pathway (45, 72, 73). BCL-2 family proteins are divided into three subtypes: pro-survival BCL-2 family members, such as BCL-2 and BCL-XL; the multi-BH-domain pro-apoptotic members, such as BAX, BAK, and BOK; the pro-apoptotic “BH3-only” proteins, such as BIM, BID, and BAD (74). The three subtypes interact to regulate the permeability of the mitochondrial outer membrane (75). The main process of mitochondrial apoptotic is BIM, BID activated by intracellular death signal, activated BIM, BID stimulates BAX, BAK and inactivates pro-survival BCL-2 protein. BAX or BAK can form a channel on the outer membrane of mitochondria. Cytochrome C, apoptotic peptidase activating factor 1 (APAF1), and deoxyribonucleotide complexes enter the cytoplasm through the channel and assemble into apoptotic body complexes (76), which activate caspase-9, and its downstream caspase-3 induces apoptosis (77). Under the stimulation of ischemia-reperfusion injury, calpain-1 cuts BID into t-BID to activate it (47). T-BID can induce apoptosis by affecting mitochondrial outer membrane permeability in the following three ways: (i) Mediated BAX and BAK oligomerization; (ii) Self-forming homodimer; (iii) Induced mitochondrial permeability transition pore (mPTP) opening and remolded mitochondrial inner membrane (78). Decreasing calpain-1 expression by specific siRNA can significantly decrease isoproterenol-induced expression of Bax, t-Bid and cytochrome C release in cardiomyocytes (79). In addition, calpain can also cleave the N-terminal of BAX into an 18 kDa fragment to activate BAX and promote cytochrome C release from mitochondria to induce apoptosis (45).
However, some researchers have shown that calpain-2 can be used as a negative regulator of caspase activation and apoptosis. Activation of calpain-2 not only failed to cleave and activate caspase-3 but also inhibited it, which was mainly related to the inactivation of caspase-9 by calpain cleaving, blocking the activation of caspase-3 by dATP and cytochrome C (40). In addition, calpain can hydrolyze P53 protein to inhibit apoptosis induced by DNA damage (48). The content of P53 protein increased rapidly during myocardial I/R. Inhibition of P53 significantly attenuated the I/R injury (80). In summary, there may be two effects of calpain on apoptosis induced by myocardial I/R, and different calpain subtypes may have different effects on caspase (The mechanism of calpain affecting apoptosis is shown in Figure 2).
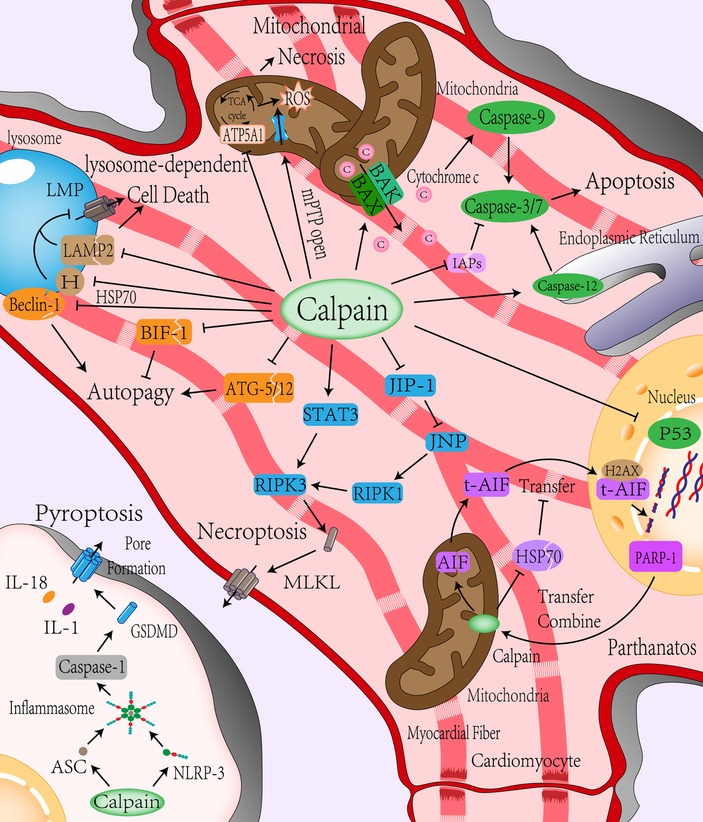
Figure 2. Role of calpain in programmed cell death. (i) Apoptosis: Calpain can directly lyse and activate caspase-3,7, and 12 to mediate cell apoptosis and reduce IAPs’ inhibition of caspase activity by cracking IAPs. Calpain can directly activate BAX/BAK or indirectly activate BAX/BAK by activating BID to induce the release of proapoptotic substances such as cytochrome C in the formation channel of the mitochondrial outer membrane to mediate the occurrence of apoptosis and reduce the response of cells to DNA damage by inhibiting P53. (ii) Mitochondrial-mediated cell necrosis: Calpain can directly promote mitochondrial inner membrane mPTP channel opening, cleavage of ATP synthase, result in energy disorders, ATP synthesis decreased, increased ROS generation, aggravate intracellular energy conflicts, and promote mitochondrial-mediated cell necrosis. (iii) Autophagy: Calpain inhibits autophagy by cleavage of Beclin-1 and autophagy-related protein ATG-5/12, which is involved in autophagosome formation, or inhibits apoptosis by cleavage of apoptosis-related protein BIF-1 to promote autophagy reversely. (iv) Lysosome-dependent cell death: Calpain can affect the stability of the lysosomal membrane by cleavage of LAMP2 and HSP70, causing lysosomes to permeate or rupture and mediating lysosome-dependent cell death. (v) Parthanatos: DNA damage during ischemia-reperfusion leads to PARP-1 activation, which is transferred to the mitochondrial membrane to activate AIF by Calpain cleavage. AIF is activated and then transferred from mitochondria to the nucleus through the cytoplasm. DNA is decomposed into large segments by calpain with the help of histone H2AX and cyclophilin. Calpain also cleaves HSP70 to reduce the stability of AIF and helps AIF transfer to the nucleus. (vi) Necroptosis: Calpain can increase the expression of RIPK1 by cleaving JIP-1, inhibiting JNP, and promoting the expression of RIPK3 by increasing the expression of STAT3, aggregation of MLKL leading to necroptosis. (vii) Pyroptosis: Calpain can up-regulate the expression of NLRP-3 to form more inflammatory bodies with ASC, then activate caspase-1 to mediate pyroptosis. mPTP, mitochondrial permeability transition pore; ATP5A1, ATP synthase subunit α; IAPs, inhibitor apoptosis protein; ATG, autophagy-related proteins; BIF-1, Bax-interacting factor 1; LMP, lysosomal membrane permeabilization; HSP70, Heat-shock protein70; LAMP2, lysosomal associated membrane protein 2; PARP-1, poly ADP-ribose polymerase 1; AIF, apoptosis-inducing factor mitochondria-associated 1, also known as AIFM1; H2AX, histone; RIPK1/3, receptor-interacting protein kinases 1/3; MLKL, mixed line kinase domain like; JNK1, c-Jun-N-terminal kinase 1; JIP-1, JNK-interacting protein-1; STAT3, signal transducer and activator of transcription 3; GSDMD, gasdermin D; NLRP-3, nucleotide-binding oligomerization domain-like receptor with a pyrin domain 3; ASC, apoptosis-associated speck-like protein.
Mitochondrial-mediated necrosis is caused by intracellular microenvironment disorders, such as severe oxidative stress and intracellular Ca2+ overload, usually presented as necrosis (81). The critical event of mitochondrial-mediated necrosis is the opening of mitochondrial permeability transition pore (mPTP) that is located on the mitochondrial inner membrane. mPTP opening is mainly mediated by Ca2+ concentration and mitochondrial membrane potential. An increase in Ca2+ concentration and depolarization of mitochondrial membrane potential will promote mPTP opening. The mPTP opening leads to increased permeability of the mitochondrial inner membrane that results in a dissipation of the proton gradient across the inner membrane. Water entered the mitochondrial matrix along the permeability gradient, resulting in mitochondrial swelling. Since the area of the mitochondrial inner membrane is much larger than that of the outer membrane, the expansion of the inner membrane is often accompanied by the loss of the integrity of the outer membrane. BAK and BAX mediate the formation of large enough pores in the outer membrane to allow the extrusion of the inner membrane. Moreover, proton leakage leads to the disappearance of mitochondrial membrane potential and the inhibition of ATP synthase leads to oxidative phosphorylation uncoupling, which lead to mitochondrial dysfunction. Dysfunctional mitochondria cannot produce enough energy to close mPTP, eventually leading to necrosis (82). The specific mechanism of necrosis caused by the opening of mPTP is not precise, which may be due to the increase of glycolysis rate cannot offset the depletion of cell energy and leads to the acidification of the cytoplasm. ATP deficiency damages the enzymes that pump Na+, H+, and Ca2+ out of the cell and the enzymes that pump Ca2+ into the endoplasmic reticulum. The sharp loss of Na+ and Ca2+ gradient inside and outside the cell causes cell membrane collapse and cell death with necrosis characteristics (82). Compared with apoptosis, the process of intracellular energy consumption (such as DNA repair and protein translation) during necrosis does not terminate. On the contrary, energy consumption has been intensified, and the intracellular energy contradiction has been aggravated. Therefore, a sharp decrease in ATP is also a sign of necrosis (5). Calpain exists not only in the cytoplasm but also in the mitochondria. Studies have shown that calpain activity in cardiac mitochondria increases during I/R (83). Ca2+ and ROS are considered to be two key activators for mPTP opening (84). Calpain induces Ca2+-induced mPTP opening through cleavage of complex I subunits (ND6 and NDUFC2) on the electron transport chain (85), The destruction of complex 1 leads to sensitization of mPTP (86). In addition, the increased calpain activity in mitochondrial can impair mitochondrial respiratory chain complex I (49, 50) and ATP synthase, such as ATP synthase subunit α (ATP5A1), these will cause mitochondrial dysfunction and increase the production of reactive oxygen species (ROS) (51, 52). Recent experiments have proven that overexpression of calpain inhibitors can maintain the activities of ATP5A1 and ATP synthase, prevent mitochondrial ROS production and reduce necrosis after hypoxia and reoxygenation (34). In summary, calpain not only promotes the opening of mPTP but also acts on ATP synthase in the process of mitochondrial-mediated necrosis, affecting mitochondrial energy production, aggravating intracellular energy contradiction, and promoting cell necrosis (The mechanism of calpain involving mitochondrial-mediated necrosis is shown in Figure 2).
Autophagy-dependent cell death is a PCD that depends on autophagy and its components (81). Autophagy, a function of conservative evolution, transports endogenous or exogenous substances in the cytoplasm to lysosomes for degradation. It plays a crucial role in quality control, stress alleviation, and recovery of cell homeostasis. It is usually used as a means of survival (87). However, in some pathophysiological environments, autophagy also can cause cell death, known as autophagy-dependent cell death (88). It is unclear whether autophagy-dependent cell death is caused by the high autophagy rate (excessive catabolism of intracellular components) or the change in the nature of the autophagy process itself. Therefore, this paper mainly introduces the role of calpain in autophagy-related molecules. Autophagy is mainly divided into macroautophagy, microautophagy, and chaperone-mediated autophagy. Autophagy usually refers to macroautophagy (88). Autophagy occurs mainly through two steps: autophagosome formation and transfer of autophagic substrates to lysosome (89). Beclin-1, part of the class III phosphatidylinositol-3-kinase (PI3K) complex, is responsible for phosphatidylinositol (PI) phosphorylation to form autophagosome precursors, which is indispensable in the formation of autophagosomes (90). Autophagy during reperfusion is closely related to Beclin-1. Autophagy is attenuated in Beclin-1 knockout mice during myocardial reperfusion (91). Calpain also plays a particular role in autophagy (92). Calpain can lyse Beclin-1 to reduce autophagy activity (53). Calpain also cleaves autophagy-related proteins (ATGs) to influence autophagy (54), ATGs are closely related to autophagosome maturation and autophagy (93), and ATGs play different roles in different stages of autophagy. For example, ATG-5 is involved in forming autophagosomes that encapsulate proteins or organelles. ATG-5 is the cutting substrate of calpain. After being cut, ATG-5 not only inhibits the formation of autophagosomes but also translocates to mitochondria and binds with BCL-XL, resulting in the release of cytochrome C (94). In addition, calpain affects autophagy by cleaving apoptosis-related molecules such as BAX-interacting factor 1 (BIF-1) (55). Although most studies have shown that calpain affects autophagy, most experiments only focus on the effect of calpain on the autophagy mechanism. Autophagy and autophagy-dependent cell death and the relationship between calpain and autophagy-dependent cell death need further research (The mechanism of calpain affecting autophagy-dependent cell death is shown in Figure 2).
Lysosome-dependent cell death is a PCD caused by the permeability of the lysosome membrane; proteolytic enzymes are released. The upstream mechanism of lysosomal membrane permeabilization (LMP) has not been fully understood. Previous studies have indicated that LMP mainly occurs after mitochondrial outer membrane permeabilization, which constitutes an intrinsic apoptosis-associated phenomenon. However, under certain experimental conditions, lysosomal membrane permeabilization can precede mitochondrial permeability, thereby mediating lysosomal-dependent cell death. The mechanism may be related to the recruitment of BAX to the lysosome membrane for pore formation (81). The stability of the lysosome membrane is mainly associated with the cholesterol content of the lysosome and member 1A of heat shock protein family A (HSPA1A; best known as HSP70) (95). Calpain affects lysosomal membrane permeability through various mechanisms (96). Calpain can cleave lysosomal associated membrane protein 2 (LAMP2) on the lysosomal membrane. High glycosylation of LAMP2 can prevent the lysosomal membrane from being dissolved by lysosomal enzyme (97). When LAMP2 is cleaved by calpain, lysosomal membrane permeabilization and proteolytic enzyme release lead to lysosome-dependent cell death (56, 57). Heat shock protein 70 (HSP70) plays a crucial role in maintaining lysosome stability (98). It can increase the activity of acid sphingomyelinase (ASM) by binding to the lysosomal anionic phospholipid bis monoacylglycerol phosphate (BMP) and promote the conversion of sphingomyelin from lysosome membrane to ceramide. Ceramide is one of the sphingolipids, these lipids serve structural roles in biomembranes (99). On the lysosomal membrane, ceramide enhances membrane acyl chain order and increases lateral packing of lipids in vitro (100). The content of ceramide affects the spatial conformation of lysosomal membrane, increases the fusion ability of lysosomal membrane with intracellular vesicles and cell membrane, and thus affects the composition and volume of lysosomal membrane, which is helpful to maintain the stability of lysosome (101). Interestingly, HSP70 is a calpain substrate in vivo and vitro (98, 102). HSP70 is more likely to be cleaved by calpain after carbonylation. Hsp70 is the regulator of calpain-mediated lysosomal permeabilization or rupture after I/R injury (58). Although there is no direct evidence to prove that lysosome-dependent cell death mediates the cell death of cardiomyocytes during ischemia-reperfusion injury, it may be hidden in autophagy-dependent cell death or apoptosis due to its close relationship with autophagy. Therefore, it is still described here (The mechanism of calpain affecting lysosome-dependent cell death is shown in Figure 2).
Parthanatos is a PCD mediated by poly ADP-ribose polymerase 1 (PARP-1) and is mainly activated by oxidative stress-induced DNA damage and chromatin decomposition (103). PARP-1 is a chromatin-related nuclear protein that plays a vital role in the repair of DNA single-strand or double-strand breaks. PARP-1 promotes DNA repair by identifying DNA ends and uses nicotinamide adenine dinucleotide (NAD+) and ATP to form poly (ADP-ribose)-sylation to change chromatin structure. However, DNA base modification affected by oxidative stress, hypoxia, and inflammatory stimuli can lead to excessive activation of PARP-1 (104). Over-activated PARP-1 consumes much NAD+ and ATP, leading to mitochondrial transmembrane potential dissipation. Over-consumption of intracellular energy leads to energy collapse and amplification of necrosis (105). Some scholars believe that parthanatos is a step before cell death. It is unclear whether specific inhibition of parthanatos can benefit myocardial I/R injury. Apoptosis-inducing factor mitochondria-associated 1 (AIFM1, also known as AIF) is necessary for parthanatos to perform. Over-activated PARP1 binds to AIFM1, leading to the transfer of AIFM1 from mitochondria to the nucleus and causing lethal chromatin dissolution (106). Calpain is directly involved in the occurrence of parthanatos in cells. PARP-1 is activated after DNA damage and is transferred to the mitochondrial membrane after activation. Calpain-1 in the mitochondrial intermembrane space is used to shear and activate AIF (107). The activated AIF is transferred to the nucleus, decomposed DNA into large fragments with histone H2AX and acyclic proteins (59). The release of t-AIF from mitochondria also requires calpain cleavage of BID to activate BAX formation channels (108). In addition, HSP70 can increase the stability of AIF and reduce nuclear transfer (60). Calpain can promote the transport of AIF from mitochondria to cytoplasm or nucleus by degrading HSP70 (61). Therefore, calpain may be synergistic in the induction of parthanatos during ischemia-reperfusion injury (109) (The mechanism of calpain affecting parthanatos is shown in Figure 2).
In the past, necrosis was believed to be an accidental or passive type of cell death. However, with the deepening of research, it was found that cell death factors such as tumor necrosis factor-α (TNF-α) could cause a kind of “programmed” necrosis, namely necroptosis (110). After the binding and activating of cell death factors with corresponding receptors, receptor-interacting protein kinase 1 (RIPK1) and receptor-interacting protein kinase 3 (RIPK3) were recruited into the cells to form a “necrosome.” Subsequently, the mixed line kinase domain like (MLKL) was activated (5) and shifted to the cell membrane to create a channel leading to membrane permeabilization (111). Studies have shown that calpain regulates necroptosis (62). Calpain can degrade c-Jun-N-terminal kinase 1 (JNK1) specific inhibitor, JNK-interacting protein-1 (JIP-1), induce JNK activation, and increase the expression of RIPK1 (62). Calpain also can increase the expression of RIPK3 by affecting the phosphorylation of signal transducer and activator of transcription 3 (STAT3), promote the phosphorylation of MLKL, and induce cardiomyocyte necroptosis (63). Inhibition of calpain can reduce the number of activated AIF and the occurrence of necroptosis (112) (The mechanism of calpain affecting necroptosis is shown in Figure 2).
Pyroptosis is a form of PCD closely related to innate immune response, characterized by cell membrane permeabilization and extracellular release of inflammatory factors. The central part of Pyroptosis is the activation of gasdermin D (GSDMD). GSDMD has an inhibitory -COOH terminal and death-promoting -NH2 terminal, which interacts to maintain the inactivation of GSDMD (113). Cells form inflammatory bodies after identifying various stressors (such as DAMPs and PAMPs), which recruit and activate inflammatory caspases (108). Inflammatory caspases can cut and activate GSDMD. After GSDMD activation, pores are formed on the cell membrane, releasing cellular contents, such as inflammatory factors, and ultimately Pyroptosis. Research has shown that inhibition of GSDMD activation during myocardial ischemia-reperfusion can reduce the occurrence of Pyroptosis and myocardial injury (114). The occurrence of Pyroptosis requires the involvement of inflammatory caspases such as caspase-1 and inflammatory bodies (115, 116). In myocardial I/R, calpain can up-regulate the expression of NLRP-3, making it form more inflammatory bodies with ASC to activate caspase-1 and induce Pyroptosis, causing myocardial injury (38) (The mechanism of calpain affecting Pyroptosis is shown in Figure 2).
During reperfusion, intracellular Ca2+ overload and increased pH increased calpain activity, leading to PCD by hydrolyzing various proteins. This is a feature of acute reperfusion injury (6). Calpain is at the regulatory point of positive feedback in most death processes. The increase of Ca2+ caused by I/R activates calpain, resulting in cell damage. The damage promotes further activation of calpain, which in turn aggravates the injury and eventually leads to PCD. Therefore, inhibiting calpain may reduce the heart damage caused by I/R, which has been verified in many experiments using drugs or gene inhibitors to reduce the activity of calpain (37, 38).
Calpain not only plays a significant role in acute myocardial I/R injury but also plays a vital role in myocardial remodeling after injury. Although the mechanism of over-expression of calpain remains to be determined in the late reperfusion period, it is reported that calpain remains high in the late reperfusion period (117, 118). High calpain expression in late reperfusion is involved in adverse remodeling after myocardial infarction. Inhibition of calpain within 24 h after transient coronary occlusion produces cardioprotective effects independent of the acute injury, reduces myocardial hypertrophy and fibrosis induced by I/R and attenuates adverse cardiac remodeling after infarction (119). Overexpression of calpastatin inhibits angiotensin-induced myocardial and perivascular inflammation and fibrosis (120). Removal of calpain-1 and calpain-2 by specific knockout of calpain small subunit 1 (Capn4) in cardiomyocytes reduces ventricular dilatation, dysfunction, and mortality after 30 days of infarction (121). Similar results were obtained in transgenic mice overexpressing endogenous calpastatin (117). On the contrary, deleting calpastatin increases calpain-induced cardiac remodeling, systolic dysfunction, and mortality after infarction (122). It can be seen that calpain harms myocardial remodeling. In general, inhibition of calpain can reduce myocardial damage caused by ischemia-reperfusion.
However, there are also reports that calpain inhibition can adversely affect cardiomyocytes (26, 123). In a recent study, overexpression of calpastatin resulted in impaired scar healing, cardiac rupture, and increased mortality (124). In addition, cardiac-specific knockout of the calpain subunit Capn4 affects isoproterenol and hemodynamic pressure-induced cardiac remodeling in mice, attenuating ventricular function (123).
Because calpain the mechanism of action has not been identified and the basic experimental results of calpain intervention are unstable, the treatment of calpain intervention has not been widely used in the clinic. There may be the following reasons for the different intervention results in calpain in basic experiments: (i) At present. There is still a lack of specific inhibitors of calpain. Most studies have adopted a wide range of non-selective calpain inhibition methods, ignoring the different subtypes of calpain that may have different effects. Javier Inserte et al. also put forward similar views (6); (ii) Calpain regulates many essential proteins of the heart, and the impact of calpain on these proteins is often ignored in the experiment. Among them, the most famous is spectrin. Spectrin is a critical membrane cytoskeletal maintenance protein, which plays a crucial role in assembling actin cytoskeleton, cross-linked actin filaments in various cell types, and scaffolds for assembling large protein complexes involving structural integrity, mechanical sensation, and cell signal transduction (125). The calpain-dependent protein hydrolysis of spectrin is closely related to cell death (15); (iii) Some death mechanisms have not been fully clarified. On the one hand, the existing death mechanisms are not sufficiently detailed—for example, the relationship between autophagy and autophagy-dependent cell death. Calpain can inhibit the occurrence of autophagy by degrading autophagy-related proteins, but in some cases, the damage after inhibition of calpain is more robust than before. The possible reason is that calpain leads to increased autophagy or changes in the nature of autophagy, resulting in autophagy-dependent cell death or lysosome-dependent cell death; On the one hand, new ways of death are still being discovered, such as copper death, cell death caused by acylation of TCA cycle proteins (126), some key proteins in TCA cycle have also been shown to be substrates of calpain in some studies (49, 50), calpain may also play a role in the occurrence of copper death; (iv) Disorder of substrate cleavage by calpain. Calpain cleavage does not target a specific molecule, which is a complete result after affecting all molecules. For example, calpain eventually leads to apoptosis, which may directly affect apoptosis-related proteins to mediate apoptosis or inhibit autophagy-related proteins from promoting apoptosis. The results may be similar. Cells eventually undergo apoptosis. However, the intermediate pathways may be different. We cannot quantify whether this apoptosis is mediated by calpain activating apoptosis-related proteins or inhibiting autophagy-related proteins; both work together or are mediated by other pathways. Therefore, if the ample cleavage of calpain is ignored, the isolated environment only considers the influence on one or more of the pathways, which may lead to a significant deviation in the experimental results.
Studies have reported that the calpain cleavage site is structurally dependent, not sequence-dependent. Calpain cleavage is uncertain. Why calpain eventually mediates apoptosis rather than autophagy or pyroptosis in some cases? Is there a pathway affecting the cleavage trend of calpain? However, there is no report on the related mechanism. If it can be found that something affects the tendency of calpain cleavage, this may have a significant impact on the future treatment of calpain. In addition, since we cannot ignore the extensive cleavage caused by calpain, can we analyze the action environment of calpain? With the development of proteomics, spatial transcriptomics, and protein structure prediction, we may know all proteins that can be cut by calpain at a particular time. Suppose apoptosis-related proteins account for the vast majority of the proteins that can be cut by calpain in cells at a specific time. Will we inhibit apoptosis by inhibiting calpain at this time? Therefore, we guess that the action time window of calpain may also significantly impact the experimental results. There is still a large blank in the study of calpain. With the progress of science and technology and the further understanding of various death mechanisms, the regulatory role of calpain will be further analyzed. The mode of action, targeted therapy, and treatment time of calpain may be essential research directions in the future.
G-YL and W-LX contributed to this manuscript's conceptual design, writing, and editing. Q-PW revised the manuscript and commented on previous versions. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant No. 81873952), the National Key Research and Development Program of China (Grant No. 2018YFC2001903), and the National Natural Science Foundation of China (Grant No. 81901948).
The authors thank Q-PW and W-LX for their teaching.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1194402/full#supplementary-material.
1. Lee YH, Fang J, Schieb L, Park S, Casper M, Gillespie C. Prevalence and trends of coronary heart disease in the United States, 2011 to 2018. JAMA Cardiol. (2022) 7:459–62. doi: 10.1001/jamacardio.2021.5613
2. Zhang J, Liang R, Wang K, Zhang W, Zhang M, Jin L, et al. Novel CaMKII-delta inhibitor hesperadin exerts dual functions to ameliorate cardiac ischemia/reperfusion injury and inhibit tumor growth. Circulation. (2022) 145:1154–68. doi: 10.1161/CIRCULATIONAHA.121.055920
3. Bhatt DL, Lopes RD, Harrington RA. Diagnosis and treatment of acute coronary syndromes: a review. J Am Med Assoc. (2022) 327:662–75. doi: 10.1001/jama.2022.0358
4. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. (2007) 357:1121–35. doi: 10.1056/NEJMra071667
5. Del Re DP, Amgalan D, Linkermann A, Liu QH, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. (2019) 99:1763–817. doi: 10.1152/physrev.00022.2018
6. Inserte J, Hernando V, Garcia-Dorado D. Contribution of calpains to myocardial ischaemia/reperfusion injury. Cardiovasc Res. (2012) 96:23–31. doi: 10.1093/cvr/cvs232
8. Matsumura Y, Saeki E, Otsu K, Morita T, Takeda H, Kuzuya T, et al. Intracellular calcium level required for calpain activation in a single myocardial cell. J Mol Cell Cardiol. (2001) 33:1133–42. doi: 10.1006/jmcc.2001.1373
9. Eisner DA, Caldwell JL, Trafford AW, Hutchings DC. The control of diastolic calcium in the heart: basic mechanisms and functional implications. Circ Res. (2020) 126:395–412. doi: 10.1161/CIRCRESAHA.119.315891
10. Dewenter M, von der Lieth A, Katus HA, Backs J. Calcium signaling and transcriptional regulation in cardiomyocytes. Circ Res. (2017) 121:1000–20. doi: 10.1161/CIRCRESAHA.117.310355
11. Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. (2008) 70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455
12. Garcia-Dorado D, Ruiz-Meana M, Inserte J, Rodriguez-Sinovas A, Piper HM. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc Res. (2012) 94:168–80. doi: 10.1093/cvr/cvs116
13. Heusch G, Gersh BJ. The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: a continual challenge. Eur Heart J. (2017) 38:774–84. doi: 10.1093/eurheartj/ehw224
14. Fauconnier J, Roberge S, Saint N, Lacampagne A. Type 2 ryanodine receptor: a novel therapeutic target in myocardial ischemia/reperfusion. Pharmacol Ther. (2013) 138:323–32. doi: 10.1016/j.pharmthera.2013.01.015
15. Ono Y, Saido TC, Sorimachi H. Calpain research for drug discovery: challenges and potential. Nat Rev Drug Discov. (2016) 15:854–76. doi: 10.1038/nrd.2016.212
16. Ono Y, Sorimachi H. Calpains: an elaborate proteolytic system. Biochim Biophys Acta. (2012) 1824:224–36. doi: 10.1016/j.bbapap.2011.08.005
17. Mahaman YAR, Huang F, Kessete Afewerky H, Maibouge TMS, Ghose B, Wang X. Involvement of calpain in the neuropathogenesis of Alzheimer’s disease. Med Res Rev. (2019) 39:608–30. doi: 10.1002/med.21534
18. Cong J, Goll DE, Peterson AM, Kapprell HP. The role of autolysis in activity of the Ca2+-dependent proteinases (mu-calpain and m-calpain). J Biol Chem. (1989) 264:10096–103. doi: 10.1016/S0021-9258(18)81771-9
19. Metwally E, Zhao G, Zhang YQ. The calcium-dependent protease calpain in neuronal remodeling and neurodegeneration. Trends Neurosci. (2021) 44:741–52. doi: 10.1016/j.tins.2021.07.003
20. Franco SJ, Huttenlocher A. Regulating cell migration: calpains make the cut. J Cell Sci. (2005) 118:3829–38. doi: 10.1242/jcs.02562
21. Hanna RA, Campbell RL, Davies PL. Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature. (2008) 456:409–12. doi: 10.1038/nature07451
22. Randriamboavonjy V, Kyselova A, Fleming I. Redox regulation of calpains: consequences on vascular function. Antioxid Redox Signal. (2019) 30:1011–26. doi: 10.1089/ars.2018.7607
23. Dargelos E, Brule C, Stuelsatz P, Mouly V, Veschambre P, Cottin P, et al. Up-regulation of calcium-dependent proteolysis in human myoblasts under acute oxidative stress. Exp Cell Res. (2010) 316:115–25. doi: 10.1016/j.yexcr.2009.07.025
24. Pal S, Sarkar A, Pal PB, Sil PC. Protective effect of arjunolic acid against atorvastatin induced hepatic and renal pathophysiology via MAPK, mitochondria and ER dependent pathways. Biochimie. (2015) 112:20–34. doi: 10.1016/j.biochi.2015.02.016
25. Hernando V, Inserte J, Sartorio CL, Parra VM, Poncelas-Nozal M, Garcia-Dorado D. Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion. J Mol Cell Cardiol. (2010) 49:271–9. doi: 10.1016/j.yjmcc.2010.02.024
26. Galvez AS, Diwan A, Odley AM, Hahn HS, Osinska H, Melendez JG, et al. Cardiomyocyte degeneration with calpain deficiency reveals a critical role in protein homeostasis. Circ Res. (2007) 100:1071–8. doi: 10.1161/01.RES.0000261938.28365.11
27. Pedrozo Z, Sanchez G, Torrealba N, Valenzuela R, Fernandez C, Hidalgo C, et al. Calpains and proteasomes mediate degradation of ryanodine receptors in a model of cardiac ischemic reperfusion. Biochim Biophys Acta. (2010) 1802:356–62. doi: 10.1016/j.bbadis.2009.12.005
28. Metwally E, Zhao G, Wang Q, Zhang YQ. Ttm50 facilitates calpain activation by anchoring it to calcium stores and increasing its sensitivity to calcium. Cell Res. (2021) 31:433–49. doi: 10.1038/s41422-020-0388-4
29. Saido TC, Shibata M, Takenawa T, Murofushi H, Suzuki K. Positive regulation of mu-calpain action by polyphosphoinositides. J Biol Chem. (1992) 267:24585–90. doi: 10.1016/S0021-9258(18)35804-6
30. Glading A, Chang P, Lauffenburger DA, Wells A. Epidermal growth factor receptor activation of calpain is required for fibroblast motility and occurs via an ERK/MAP kinase signaling pathway. J Biol Chem. (2000) 275:2390–8. doi: 10.1074/jbc.275.4.2390
31. Leloup L, Daury L, Mazeres G, Cottin P, Brustis JJ. Involvement of the ERK/MAP kinase signalling pathway in milli-calpain activation and myogenic cell migration. Int J Biochem Cell Biol. (2007) 39:1177–89. doi: 10.1016/j.biocel.2007.03.003
32. Nian H, Ma B. Calpain-calpastatin system and cancer progression. Biol Rev Camb Philos Soc. (2021) 96:961–75. doi: 10.1111/brv.12686
33. Zatz M, Starling A. Calpains and disease. N Engl J Med. (2005) 352:2413–23. doi: 10.1056/NEJMra043361
34. Zheng D, Cao T, Zhang LL, Fan GC, Qiu J, Peng TQ. Targeted inhibition of calpain in mitochondria alleviates oxidative stress-induced myocardial injury. Acta Pharmacol Sin. (2021) 42:909–20. doi: 10.1038/s41401-020-00526-y
35. Zheng D, Wang G, Li S, Fan GC, Peng T. Calpain-1 induces endoplasmic reticulum stress in promoting cardiomyocyte apoptosis following hypoxia/reoxygenation. Biochim Biophys Acta. (2015) 1852:882–92. doi: 10.1016/j.bbadis.2015.01.019
36. Lu HT, Feng RQ, Tang JK, Zhou JJ, Gao F, Ren J. CaMKII/calpain interaction mediates ischemia/reperfusion injury in isolated rat hearts. Cell Death Dis. (2020) 11:388. doi: 10.1038/s41419-020-2605-y
37. Chen Q, Thompson J, Hu Y, Dean J, Lesnefsky EJ. Inhibition of the ubiquitous calpains protects complex I activity and enables improved mitophagy in the heart following ischemia-reperfusion. Am J Physiol Cell Physiol. (2019) 317:C910–21. doi: 10.1152/ajpcell.00190.2019
38. Yue RC, Lu SZ, Luo Y, Wang T, Liang H, Zeng J, et al. Calpain silencing alleviates myocardial ischemia-reperfusion injury through the NLRP3/ASC/caspase-1 axis in mice. Life Sci. (2019) 233:116631. doi: 10.1016/j.lfs.2019.116631
39. Gafni J, Cong X, Chen SF, Gibson BW, Ellerby LM. Calpain-1 cleaves and activates caspase-7. J Biol Chem. (2009) 284:25441–9. doi: 10.1074/jbc.M109.038174
40. Chua BT, Guo K, Li P. Direct cleavage by the calcium-activated protease calpain can lead to inactivation of caspases. J Biol Chem. (2000) 275:5131–5. doi: 10.1074/jbc.275.7.5131
41. Yamada KH, Kozlowski DA, Seidl SE, Lance S, Wieschhaus AJ, Sundivakkam P, et al. Targeted gene inactivation of calpain-1 suppresses cortical degeneration due to traumatic brain injury and neuronal apoptosis induced by oxidative stress. J Biol Chem. (2012) 287:13182–93. doi: 10.1074/jbc.M111.302612
42. Martinez JA, Zhang Z, Svetlov SI, Hayes RL, Wang KK, Larner SF. Calpain and caspase processing of caspase-12 contribute to the ER stress-induced cell death pathway in differentiated PC12 cells. Apoptosis. (2010) 15:1480–93. doi: 10.1007/s10495-010-0526-4
43. Nakagawa T, Yuan JY. Cross-talk between two cysteine protease families: activation of caspase-12 by calpain in apoptosis. J Cell Biol. (2000) 150:887–94. doi: 10.1083/jcb.150.4.887
44. Zhu J, Jin M, Wang J, Zhang H, Wu Y, Li D, et al. TNFalpha induces ca(2+) influx to accelerate extrinsic apoptosis in hepatocellular carcinoma cells. J Exp Clin Cancer Res. (2018) 37:43. doi: 10.1186/s13046-018-0714-6
45. Gao G, Dou QP. N-terminal cleavage of bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes bcl-2-independent cytochrome c release and apoptotic cell death. J Cell Biochem. (2000) 80:53–72. doi: 10.1002/1097-4644(20010101)80:1%3C53::AID-JCB60%3E3.0.CO;2-E
46. Yeo JK, Cha SD, Cho CH, Kim SP, Cho JW, Baek WK, et al. Se-methylselenocysteine induces apoptosis through caspase activation and bax cleavage mediated by calpain in SKOV-3 ovarian cancer cells. Cancer Lett. (2002) 182:83–92. doi: 10.1016/s0304-3835(02)00075-7
47. Chen M, He HP, Zhan SX, Krajewski S, Reed JC, Gottlieb RA. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J Biol Chem. (2001) 276:30724–8. doi: 10.1074/jbc.M103701200
48. Kubbutat MH, Vousden KH. Proteolytic cleavage of human p53 by calpain: a potential regulator of protein stability. Mol Cell Biol. (1997) 17:460–8. doi: 10.1128/MCB.17.1.460
49. Thompson J, Hu Y, Lesnefsky EJ, Chen Q. Activation of mitochondrial calpain and increased cardiac injury: beyond AIF release. Am J Physiol Heart Circ Physiol. (2016) 310:H376–384. doi: 10.1152/ajpheart.00748.2015
50. Shintani-Ishida K, Yoshida K. Mitochondrial m-calpain opens the mitochondrial permeability transition pore in ischemia-reperfusion. Int J Cardiol. (2015) 197:26–32. doi: 10.1016/j.ijcard.2015.06.010
51. Ni R, Zheng D, Wang Q, Yu Y, Chen R, Sun T, et al. Deletion ofcapn4 protects the heart against endotoxemic injury by preventing ATP synthase disruption and inhibiting mitochondrial superoxide generation. Circ Heart Fail. (2015) 8:988–96. doi: 10.1161/CIRCHEARTFAILURE.115.002383
52. Ni R, Zheng D, Xiong S, Hill DJ, Sun T, Gardiner RB, et al. Mitochondrial calpain-1 disrupts ATP synthase and induces superoxide generation in type 1 diabetic hearts: a novel mechanism contributing to diabetic cardiomyopathy. Diabetes. (2016) 65:255–68. doi: 10.2337/db15-0963
53. Russo R, Berliocchi L, Adornetto A, Varano GP, Cavaliere F, Nucci C, et al. Calpain-mediated cleavage of beclin-1 and autophagy deregulation following retinal ischemic injury in vivo. Cell Death Dis. (2011) 2:e144. doi: 10.1038/cddis.2011.29
54. Xia HG, Zhang L, Chen G, Zhang T, Liu J, Jin M, et al. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy. (2010) 6:61–6. doi: 10.4161/auto.6.1.10326
55. Marcassa E, Raimondi M, Anwar T, Eskelinen EL, Myers MP, Triolo G, et al. Calpain mobilizes Atg9/bif-1 vesicles from Golgi stacks upon autophagy induction by thapsigargin. Biol Open. (2017) 6:551–62. doi: 10.1242/bio.022806
56. Villalpando Rodriguez GE, Torriglia A. Calpain 1 induce lysosomal permeabilization by cleavage of lysosomal associated membrane protein 2. Biochim Biophys Acta. (2013) 1833:2244–53. doi: 10.1016/j.bbamcr.2013.05.019
57. Arnandis T, Ferrer-Vicens I, Garcia-Trevijano ER, Miralles VJ, Garcia C, Torres L, et al. Calpains mediate epithelial-cell death during mammary gland involution: mitochondria and lysosomal destabilization. Cell Death Differ. (2012) 19:1536–48. doi: 10.1038/cdd.2012.46
58. Sahara S, Yamashima T. Calpain-mediated Hsp70.1 cleavage in hippocampal CA1 neuronal death. Biochem Biophys Res Commun. (2010) 393:806–11. doi: 10.1016/j.bbrc.2010.02.087
59. Baritaud M, Boujrad H, Lorenzo HK, Krantic S, Susin SA. Histone H2AX: the missing link in AIF-mediated caspase-independent programmed necrosis. Cell Cycle. (2010) 9:3166–73. doi: 10.4161/cc.9.16.12887
60. Matsumori Y, Hong SM, Aoyama K, Fan Y, Kayama T, Sheldon RA, et al. Hsp70 overexpression sequesters AIF and reduces neonatal hypoxic/ischemic brain injury. J Cereb Blood Flow Metab. (2005) 25:899–910. doi: 10.1038/sj.jcbfm.9600080
61. Yamashima T, Oikawa S. The role of lysosomal rupture in neuronal death. Prog Neurobiol. (2009) 89:343–58. doi: 10.1016/j.pneurobio.2009.09.003
62. Bollino D, Balan I, Aurelian L. Valproic acid induces neuronal cell death through a novel calpain-dependent necroptosis pathway. J Neurochem. (2015) 133:174–86. doi: 10.1111/jnc.13029
63. Kim H, Zamel R, Bai XH, Lu C, Keshavjee S, Keshavjee S, et al. Ischemia-reperfusion induces death receptor-independent necroptosis via calpain-STAT3 activation in a lung transplant setting. Am J Physiol Lung Cell Mol Physiol. (2018) 315:L595–608. doi: 10.1152/ajplung.00069.2018
64. Scarabelli TM, Gottlieb RA. Functional and clinical repercussions of myocyte apoptosis in the multifaceted damage by ischemia/reperfusion injury: old and new concepts after 10 years of contributions. Cell Death Differ. (2004) 11(Suppl 2):S144–152. doi: 10.1038/sj.cdd.4401544
65. Lee P, Sata M, Lefer DJ, Factor SM, Walsh K, Kitsis RN. Fas pathway is a critical mediator of cardiac myocyte death and MI during ischemia-reperfusion in vivo. Am J Physiol Heart Circ Physiol. (2003) 284:H456–63. doi: 10.1152/ajpheart.00777.2002
66. Chen Z, Chua CC, Ho YS, Hamdy RC, Chua BH. Overexpression of bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol Heart Circ Physiol. (2001) 280:H2313–2320. doi: 10.1152/ajpheart.2001.280.5.H2313
67. Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard M, et al. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. (2001) 104:330–5. doi: 10.1161/01.cir.104.3.330
68. Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. (2004) 5:897–907. doi: 10.1038/nrm1496
69. Wang KK, Posmantur R, Nadimpalli R, Nath R, Mohan P, Nixon RA, et al. Caspase-mediated fragmentation of calpain inhibitor protein calpastatin during apoptosis. Arch Biochem Biophys. (1998) 356:187–96. doi: 10.1006/abbi.1998.0748
70. Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, et al. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. (1998) 17:2215–23. doi: 10.1093/emboj/17.8.2215
71. Fulda S. Molecular pathways: targeting inhibitor of apoptosis proteins in cancer–from molecular mechanism to therapeutic application. Clin Cancer Res. (2014) 20:289–95. doi: 10.1158/1078-0432.CCR-13-0227
72. Gil-Parrado S, Fernandez-Montalvan A, Assfalg-Machleidt I, Popp O, Bestvater F, Holloschi A, et al. Ionomycin-activated calpain triggers apoptosis. A probable role for bcl-2 family members. J Biol Chem. (2002) 277:27217–26. doi: 10.1074/jbc.M202945200
73. Lee JK, Kang S, Wang XD, Rosales JL, Gao X, Byun HG, et al. HAP1 loss confers L-asparaginase resistance in ALL by downregulating the calpain-1-bid-caspase-3/12 pathway. Blood. (2019) 133:2222–32. doi: 10.1182/blood-2018-12-890236
74. Delbridge ARD, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. (2016) 16:99–109. doi: 10.1038/nrc.2015.17
75. Kvansakul M, Hinds MG. The bcl-2 family: structures, interactions and targets for drug discovery. Apoptosis. (2015) 20:136–50. doi: 10.1007/s10495-014-1051-7
76. Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell. (2002) 9:423–32. doi: 10.1016/s1097-2765(02)00442-2
77. Bleicken S, Landeta O, Landajuela A, Basanez G, Garcia-Saez AJ. Proapoptotic bax and bak proteins form stable protein-permeable pores of tunable size. J Biol Chem. (2013) 288:33241–52. doi: 10.1074/jbc.M113.512087
78. Smith MA, Schnellmann RG. Calpains, mitochondria, and apoptosis. Cardiovasc Res. (2012) 96:32–7. doi: 10.1093/cvr/cvs163
79. Li W, Yang J, Lyu Q, Wu G, Lin S, Yang Q, et al. Taurine attenuates isoproterenol-induced H9c2 cardiomyocytes hypertrophy by improving antioxidative ability and inhibiting calpain-1-mediated apoptosis. Mol Cell Biochem. (2020) 469:119–32. doi: 10.1007/s11010-020-03733-7
80. Chen Q, Thompson J, Hu Y, Lesnefsky EJ. Cardiomyocyte specific deletion of p53 decreases cell injury during ischemia-reperfusion: role of mitochondria. Free Radic Biol Med. (2020) 158:162–70. doi: 10.1016/j.freeradbiomed.2020.06.006
81. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature committee on cell death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
82. Bonora M, Giorgi C, Pinton P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat Rev Mol Cell Biol. (2022) 23:266–85. doi: 10.1038/s41580-021-00433-y
83. Chen Q, Paillard M, Gomez L, Ross T, Hu Y, Xu A, et al. Activation of mitochondrial mu-calpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion. Biochem Biophys Res Commun. (2011) 415:533–8. doi: 10.1016/j.bbrc.2011.10.037
84. Seidlmayer LK, Juettner VV, Kettlewell S, Pavlov EV, Blatter LA, Dedkova EN. Distinct mPTP activation mechanisms in ischaemia-reperfusion: contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc Res. (2015) 106:237–48. doi: 10.1093/cvr/cvv097
85. Arrington DD, Van Vleet TR, Schnellmann RG. Calpain 10: a mitochondrial calpain and its role in calcium-induced mitochondrial dysfunction. Am J Physiol Cell Physiol. (2006) 291:C1159–1171. doi: 10.1152/ajpcell.00207.2006
86. Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC Jr, Suthammarak W, Gong G, et al. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. (2013) 18:239–50. doi: 10.1016/j.cmet.2013.07.002
87. Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med. (2020) 383:1564–76. doi: 10.1056/NEJMra2022774
88. Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ. (2019) 26:605–16. doi: 10.1038/s41418-018-0252-y
89. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. (2018) 19:349–64. doi: 10.1038/s41580-018-0003-4
90. Fernandez AF, Sebti S, Wei Y, Zou Z, Shi M, McMillan KL, et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature. (2018) 558:136–40. doi: 10.1038/s41586-018-0162-7
91. Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and beclin 1 in mediating autophagy. Circ Res. (2007) 100:914–22. doi: 10.1161/01.RES.0000261924.76669.36
92. Demarchi F, Bertoli C, Copetti T, Tanida I, Brancolini C, Eskelinen EL, et al. Calpain is required for macroautophagy in mammalian cells. J Cell Biol. (2006) 175:595–605. doi: 10.1083/jcb.200601024
93. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. (2019) 176:11–42. doi: 10.1016/j.cell.2018.09.048
94. Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. (2006) 8:1124–32. doi: 10.1038/ncb1482
95. Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nat Rev Cancer. (2005) 5:886–97. doi: 10.1038/nrc1738
96. Gerónimo-Olvera C, Montiel T, Rincon-Heredia R, Castro-Obregón S, Massieu L. Autophagy fails to prevent glucose deprivation/glucose reintroduction-induced neuronal death due to calpain-mediated lysosomal dysfunction in cortical neurons. Cell Death Dis. (2017) 8:e2911. doi: 10.1038/cddis.2017.299
97. Sudhakar JN, Lu HH, Chiang HY, Suen CS, Hwang MJ, Wu SY, et al. Lumenal galectin-9-Lamp2 interaction regulates lysosome and autophagy to prevent pathogenesis in the intestine and pancreas. Nat Commun. (2020) 11:4286. doi: 10.1038/s41467-020-18102-7
98. Yamashima T. Can ‘calpain-cathepsin hypothesis’ explain Alzheimer neuronal death? Ageing Res Rev. (2016) 32:169–79. doi: 10.1016/j.arr.2016.05.008
99. Morad SA, Cabot MC. Ceramide-orchestrated signalling in cancer cells. Nat Rev Cancer. (2013) 13:51–65. doi: 10.1038/nrc3398
100. Holopainen JM, Subramanian M, Kinnunen PK. Sphingomyelinase induces lipid microdomain formation in a fluid phosphatidylcholine/sphingomyelin membrane. Biochemistry. (1998) 37:17562–70. doi: 10.1021/bi980915e
101. Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I, et al. Hsp70 stabilizes lysosomes and reverts niemann-pick disease-associated lysosomal pathology. Nature. (2010) 463:549–53. doi: 10.1038/nature08710
102. Nakajima E, David LL, Bystrom C, Shearer TR, Azuma M. Calpain-specific proteolysis in primate retina: contribution of calpains in cell death. Invest Ophthalmol Vis Sci. (2006) 47:5469–75. doi: 10.1167/iovs.06-0567
103. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. (2019) 29:347–64. doi: 10.1038/s41422-019-0164-5
104. Curtin NJ, Szabo C. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol Aspects Med. (2013) 34:1217–56. doi: 10.1016/j.mam.2013.01.006
105. Fatokun AA, Dawson VL, Dawson TM. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol. (2014) 171:2000–16. doi: 10.1111/bph.12416
106. Wang YF, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA, et al. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci Signal. (2011) 4:ra20. doi: 10.1126/scisignal.2000902
107. Ozaki T, Yamashita T, Ishiguro S. Mitochondrial m-calpain plays a role in the release of truncated apoptosis-inducing factor from the mitochondria. Biochim Biophys Acta. (2009) 1793:1848–59. doi: 10.1016/j.bbamcr.2009.10.002
108. Cabon L, Galan-Malo P, Bouharrour A, Delavallee L, Brunelle-Navas MN, Lorenzo HK, et al. BID regulates AIF-mediated caspase-independent necroptosis by promoting BAX activation. Cell Death Differ. (2012) 19:245–56. doi: 10.1038/cdd.2011.91
109. Chiu SC, Huang SY, Tsai YC, Chen SP, Pang CY, Lien CF, et al. Poly (ADP-ribose) polymerase plays an important role in intermittent hypoxia-induced cell death in rat cerebellar granule cells. J Biomed Sci. (2012) 19:29. doi: 10.1186/1423-0127-19-29
110. Zhang T, Zhang Y, Cui MY, Jin L, Wang YM, Lv FX, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. (2016) 22:175–82. doi: 10.1038/nm.4017
111. Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. (2014) 24:105–21. doi: 10.1038/cr.2013.171
112. Chen S, Yan J, Deng HX, Long LL, Hu YJ, Wang M, et al. Inhibition of calpain on oxygen glucose deprivation-induced RGC-5 necroptosis. J Huazhong Univ Sci Technolog Med Sci. (2016) 36:639–45. doi: 10.1007/s11596-016-1639-y
113. Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science. (2020) 369:1633–7. doi: 10.1126/science.abb9818
114. Shi H, Gao Y, Dong Z, Yang J, Gao R, Li X, et al. GSDMD-mediated cardiomyocyte pyroptosis promotes myocardial I/R injury. Circ Res. (2021) 129:383–96. doi: 10.1161/CIRCRESAHA.120.318629
115. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. (2005) 73:1907–16. doi: 10.1128/IAI.73.4.1907-1916.2005
116. He W-T, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. (2015) 25:1285–98. doi: 10.1038/cr.2015.139
117. Ye T, Wang Q, Zhang Y, Song X, Yang D, Li D, et al. Over-expression of calpastatin inhibits calpain activation and attenuates post-infarction myocardial remodeling. PLoS One. (2015) 10:e0120178. doi: 10.1371/journal.pone.0120178
118. Takahashi M, Tanonaka K, Yoshida H, Koshimizu M, Daicho T, Oikawa R, et al. Possible involvement of calpain activation in pathogenesis of chronic heart failure after acute myocardial infarction. J Cardiovasc Pharmacol. (2006) 47:413–21. doi: 10.1097/01.fjc.0000210074.56614.3b
119. Poncelas M, Inserte J, Aluja D, Hernando V, Vilardosa U, Garcia-Dorado D. Delayed, oral pharmacological inhibition of calpains attenuates adverse post-infarction remodelling. Cardiovasc Res. (2017) 113:950–61. doi: 10.1093/cvr/cvx073
120. Kim DH, Beckett JD, Nagpal V, Seman-Senderos MA, Gould RA, Creamer TJ, et al. Calpain 9 as a therapeutic target in TGFbeta-induced mesenchymal transition and fibrosis. Sci Transl Med. (2019) 11:eaau2814. doi: 10.1126/scitranslmed.aau2814
121. Ma J, Wei M, Wang Q, Li J, Wang H, Liu W, et al. Deficiency of Capn4 gene inhibits nuclear factor-kappaB (NF-kappaB) protein signaling/inflammation and reduces remodeling after myocardial infarction. J Biol Chem. (2012) 287:27480–9. doi: 10.1074/jbc.M112.358929
122. Kudo-Sakamoto Y, Akazawa H, Ito K, Takano J, Yano M, Yabumoto C, et al. Calpain-dependent cleavage of N-cadherin is involved in the progression of post-myocardial infarction remodeling. J Biol Chem. (2014) 289:19408–19. doi: 10.1074/jbc.M114.567206
123. Taneike M, Mizote I, Morita T, Watanabe T, Hikoso S, Yamaguchi O, et al. Calpain protects the heart from hemodynamic stress. J Biol Chem. (2011) 286:32170–7. doi: 10.1074/jbc.M111.248088
124. Wan F, Letavernier E, Le Saux CJ, Houssaini A, Abid S, Czibik G, et al. Calpastatin overexpression impairs postinfarct scar healing in mice by compromising reparative immune cell recruitment and activation. Am J Physiol Heart Circ Physiol. (2015) 309:H1883–1893. doi: 10.1152/ajpheart.00594.2015
125. Ribeiro ED, Pinotsis N, Ghisleni A, Salmazo A, Konarev PV, Kostan J, et al. The structure and regulation of human muscle alpha-actinin. Cell. (2014) 159:1447–60. doi: 10.1016/j.cell.2014.10.056
Keywords: programmed cell death, myocardial ischemia-reperfusion injury, calpain, apoptosis, parthanatos
Citation: Liu G-Y, Xie W-L, Wang Y-T, Chen L, Xu Z-Z, Lv Y and Wu Q-P (2023) Calpain: the regulatory point of myocardial ischemia-reperfusion injury. Front. Cardiovasc. Med. 10:1194402. doi: 10.3389/fcvm.2023.1194402
Received: 27 March 2023; Accepted: 13 June 2023;
Published: 29 June 2023.
Edited by:
Marco Mennuni, Azienda Ospedaliero Universitaria Maggiore della Carità, ItalyReviewed by:
Ki Ho Park, University of Virginia, United States© 2023 Liu, Xie, Wang, Chen, Xu, Lv and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing-Ping Wu d3FwMTk2OEAxMjYuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.