 Yixin Xie
Yixin Xie Yongnan Li2
Yongnan Li2 Hong Ding
Hong Ding- 1Department of Cardiology, Lanzhou University Second Hospital, Lanzhou, China
- 2Department of Cardiac Surgery, Lanzhou University Second Hospital, Lanzhou, China
Significance: Cardiovascular diseases are seen to be a primary cause of death, and their prevalence has significantly increased across the globe in the past few years. Several studies have shown that cell death is closely linked to the pathogenesis of cardiovascular diseases. Furthermore, many molecular and cellular mechanisms are involved in the pathogenesis of the cardiac cell death mechanism. One of the factors that played a vital role in the pathogenesis of cardiac cell death mechanisms included the early growth response-1 (Egr-1) factor.
Recent Advances: Studies have shown that abnormal Egr-1 expression is linked to different animal and human disorders like heart failure and myocardial infarction. The biosynthesis of Egr-1 regulates its activity. Egr-1 can be triggered by many factors such as serum, cytokines, hormones, growth factors, endotoxins, mechanical injury, hypoxia, and shear stress. It also displays a pro-apoptotic effect on cardiac cells, under varying stress conditions. EGR1 mediates a broad range of biological responses to oxidative stress and cell death by combining the acute changes occurring in the cellular environment with sustained changes in gene expression.
Future Directions: The primary regulatory role played by the Egr-1-targeting DNAzymes, microRNAs, and oligonucleotide decoy strategies in cardiovascular diseases were identified to provide a reference to identify novel therapeutic targets for cardiovascular diseases.
1. Introduction
Early growth response factor 1 (Egr-1) is an early gene, belonging to the EGR family that codes for a Cys2-His2 zinc finger protein (1). It is located on human chromosome 5q23-q31 (2). EGR1, also called NGFI-A (3), AT225, ZIF268 (4), TIS8, G0S30, KROX-24, and ZNF225, is an 80 kDa DNA-binding transcription factor with 543 residues that regulates transcription. EGR1 contains a three Cys2-His2 subtype zinc finger structure, an activation regulatory region, as well as a suppression regulatory region, which are located between 332 and 416 amino acids, close to the carboxyl terminus (5). It specifically identifies and binds to the target genes, and regulates their transcription. The Egr-1 promoter includes serum response elements, which preferentially bind to the GC-rich elements. They also regulate the interaction between different growth factors and this sequence to initiate the Egr-1 gene expression via different mechanisms that involved a co-activator and co-repressor (6). EGR1 binds to the DNA motifs [with a sequence of 5′-GCG(T/G) GGGCG-3′] with the help of the Cys2-His2-type zinc fingers. The C-terminal zinc finger binds to the 5′-GCG motif, while the majority of N-terminal zinc fingers bind to the 3′-GCG motif, and the middle zinc finger interacts with the middle TGG motif (Figure 1). Egr-1 gets activated (rapidly and transiently) in different human cell types in response to varying agonist and environmental factors (7). Egr-1 is triggered by a variety of stimuli, such as serum, cytokines, hormones, growth factors, endotoxins, mechanical damage, hypoxia, and shear stress (8–11). The products of the Egr-1-activated target genes play key roles in cell proliferation, differentiation, mitosis, and cell death pathways (12). Egr-1 expression is associated with many factors linked to cardiovascular pathologies such as atherosclerosis, cardiac hypertrophy, intimal thickening after acute arterial injury, and angiogenesis (13–15). Egr-1 also plays a role in doxorubicin-induced cardiomyopathy. Some rat model-based studies have shown that Egr-1 gene inhibition decreases the pathological effects of acute myocardial infarction (AMI) (16, 17). The Egr-1 phenotypes are seen to be cell type-specific, and Egr-1 overexpression stimulates cell apoptosis. Additionally, researchers have used catalytic and non-catalytic nucleic acid methods such as DNAzymes, microRNAs, and oligonucleotide decoys in animal models in conjunction with Egr-1-deficient mice to obtain novel insights into the regulatory role played by Egr-1 in cardiovascular diseases. Thus, Egr-1 can serve as a unique target for therapeutic intervention.
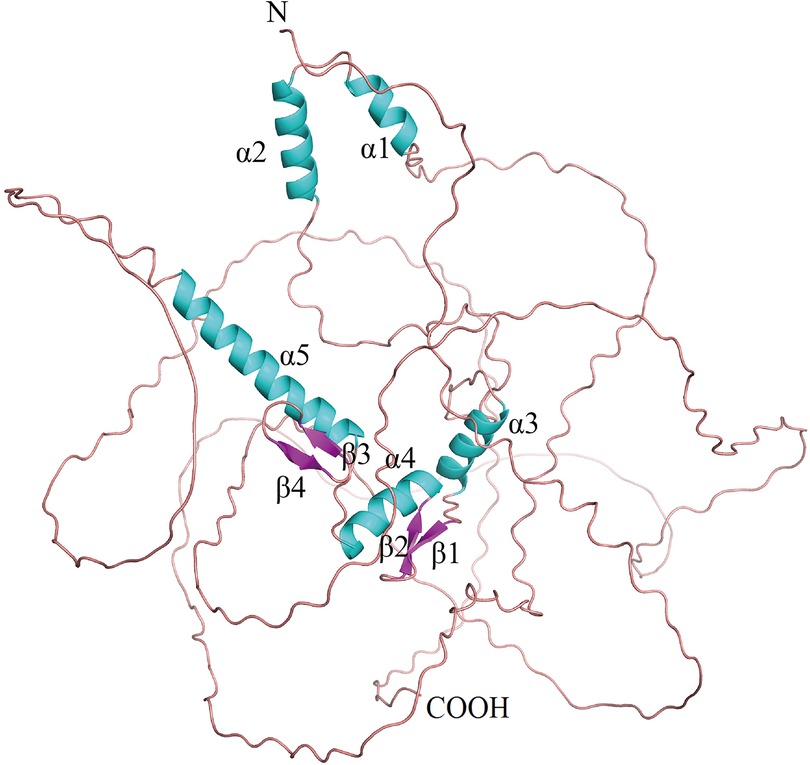
Figure 1. Structure diagram of Egr-1.
2. Egr-1 is involved in cardiac cell death
2.1. Egr-1 and apoptosis
Egr-1 regulates the expression of many genes in cardiac cells (18). Studies have shown that Egr-1 causes cardiac cell apoptosis through various mechanisms. In comparison to necrosis, apoptotic cells are immediately identified and cleared by adjoining phagocytes to prevent inflammation. Apoptosis affects the cardiovascular system and leads to the onset and progression of many cardiovascular disorders. It also helps in clearing the non-myocyte components and cardiomyocytes, which cause heart failure (19) (Table 1).
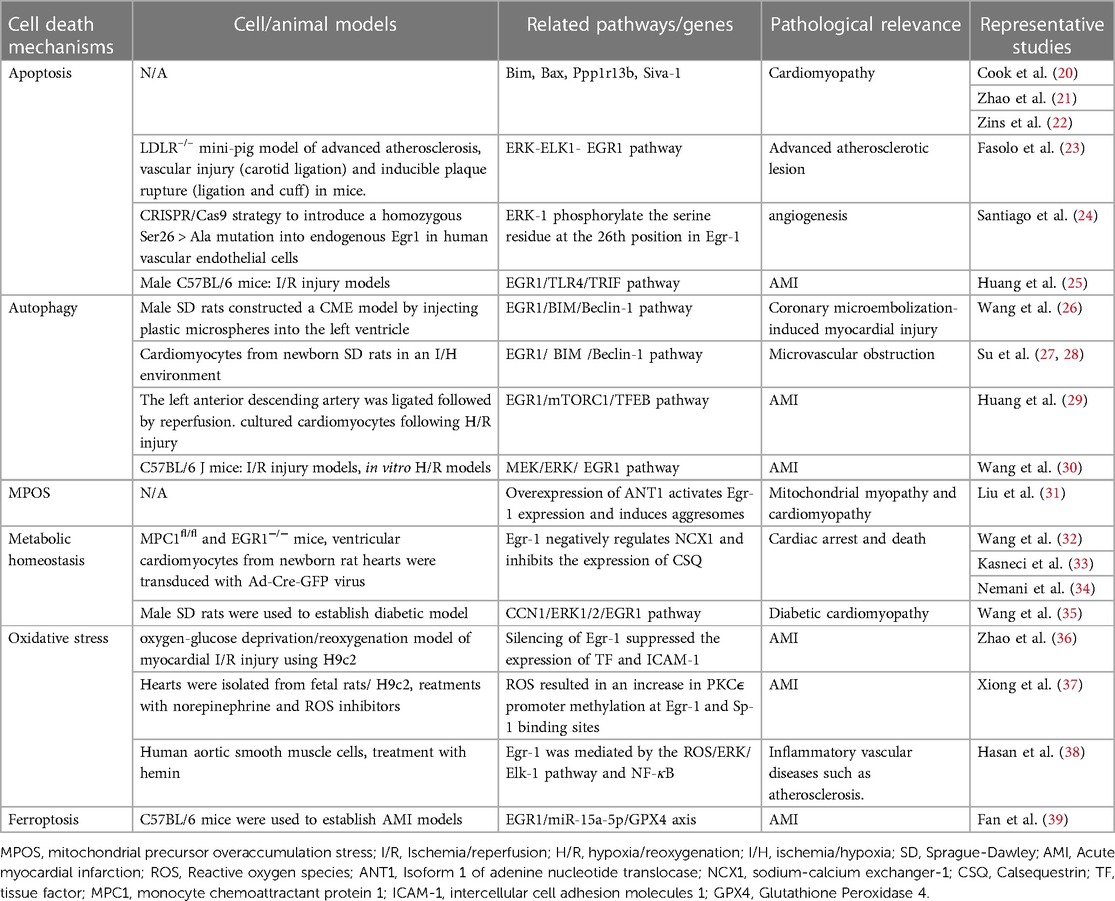
Table 1. Egr-1 is involved in cardiac cell death.
EGR1 can promote apoptosis by binding and stimulating the levels of different promoters of the apoptosis-based factors, like BIM (Bcl-2 Like 11), BAX (BCL2 Associated X, Apoptosis Regulator), ASPP (Apoptosis Stimulating P53 Protein), and SIVA1 (SIVA1 Apoptosis Induced Factor). The proteins encoded by Bax and Bim genes belong to the BCL2 protein family. The BCL2 family of proteins forms heterodimers or homodimers and participates in a variety of cellular activities as anti-apoptotic or pro-apoptotic regulators (40–42). On the other hand, Ppp1r13b (protein phosphatase 1 regulatory subunit 13b) encodes for ASPP that regulates cell death (43). Siva-1 encodes for an E3 ubiquitin ligase enzyme that regulates cell proliferation, cell cycle progression, and apoptosis (44).
Mitochondria are known to be key sites for the integration of pro-apoptotic and anti-apoptotic proteins in cardiac cells (45). Studies have shown that upregulated EGR1 can bind to the Bax and Bim promoters for increasing the BAX and BIM protein levels in cardiac cells that are stressed by injury (46–48). BAX, BIM, and other pro-apoptotic proteins bind to the mitochondrial membrane and interact with mitochondrial Voltage-dependent anion channels (VDAC) to increase the channel opening rate. This further increases membrane permeability, release of cytochrome (Cyt) into the cytoplasm, activation of the Caspase family and metabolic hydrolases, and finally promotes apoptosis (49). BAX forms heterodimers with the BCL2 protein, which inhibits apoptosis and autophagy and acts as an activator of apoptosis. The binding and ratio of the BAX and BCL2 proteins determine the death or survival of cells after apoptosis stimulation (20). BAX level is regulated by Tp53/p53 (tumor protein p53) and it is seen to participate in Tp53-mediated apoptosis (50, 51). EGR1 is involved in the upstream transcriptional regulation of Tp53 (52). It affects TP53 level via the Tp53 promoter, and then TP53 activates EGR1 to form a feedback loop (53, 54). On the other hand, Egr-1 activates the MAPK (Mitogen-Activated Pprotein Kinase)-ELK1 (ETS Transcription Factor ELK1)-EGR1 pathways to promote P21 (Cyclin-Dependent Kinase Inhibitor 1A) level without TP53. Studies have shown that Tp53 primarily targets P21, which regulates the cell cycle, promotes DNA repair, and induces apoptosis (55, 56).
Egr-1 specifically targets ASPP, as it plays a crucial role in the immediate up-regulation of ASPP. Additionally, it maintains the basic expression of the Ppp1r13b gene during non-stress conditions (21). Furthermore, Ppp1r13b stimulates EGR1 protein levels within a positive feedback loop. Initially, EGR1 is activated by multiple stimuli, and then, it binds to EBS, which is located in the Ppp1r13b promoter region, thereby transactivating the expression of Ppp1r13b in the nucleus. Elevated ASPP level were primarily localized in the cytoplasm, which inhibited the proteasome-mediated degradation of EGR1 and promoted a nuclear import of EGR1. Activated EGR1 can also promote apoptosis by transactivating the proapoptotic targets, such as Egr-1 itself. In an earlier study, the researchers identified a novel EGR1/ASPP inter-regulatory loop and determined the proapoptotic function of cytoplasmic ASPP by stabilizing EGR1 and inhibiting the autophagy promoter ATG5 (Autophagy protein 5) -ATG12/ATG16 (57, 58). The Tp53 controls TP53 by increasing its DNA binding and transactivation capabilities on proapoptotic gene promoters. Ppp1r13b interacts with the Tp53 gene and plays a crucial role in regulating apoptosis.
Studies have shown that during the in vitro apoptosis induction phase of cardiac fibroblasts, the continuous Egr-1 expression causes the downstream transcriptional regulation of the pro-apoptotic gene, i.e., Siva-1. Normal cardiac fibroblasts do not express the Siva-1 (22, 59). The Siva-1 gene codes for the SIVA1 protein that binds to the cytoplasmic tail of the members of the tumor necrosis factor (TNF) receptor superfamily (such as CD27 and a glucocorticoid-induced TNF receptor) that are activated by ligands (60). This protein also contains a domain that is similar to the death domain. It has been demonstrated that Siva-1 mediates proliferating cell nuclear antigen (PCNA) ubiquitination in response to ultraviolet-induced DNA damage and triggers CD27-mediated apoptosis. SIVA1 is a transcriptional target of the Tp53 gene, and it induces the oxidative stress-induced apoptosis process (61). Additionally, the Siva-1 gene encodes for an E3 ubiquitin ligase that suppresses the anti-apoptotic activity of BCL2, leads to caspase-dependent apoptosis, localizes in the mitochondria, and regulates cell cycle progression, cell proliferation, and apoptosis (62, 63).
Egr-1 controls the level of hundreds of proteins in cardiac cells and implements the above-mentioned processes to bind to different apoptosis-related factor promoters. One important functional switch is the phosphorylation of EGR1. Santiago et al. found that ERK1 (Mitogen-Activated Protein Kinase 3) could phosphorylate the serine residue at the 26th position (Ser26) in EGR1 in human vascular endothelial cells, which has a protective effect against apoptosis. If Ser26 is mutated, endothelial cells will undergo apoptosis (24). In one study, Fasolo et al. determined the effect of the long non-coding RNA myocardial invasion-associated transcript (MIAT) on advanced atherosclerotic lesions. They discovered that Egr-1 controlled the proliferation and death of smooth muscle cells (SMCs). They also noted that MIAT controlled the proliferation and apoptosis of the SMCs in the carotid artery via the ERK- ELK1-EGR1 pathway (23). Studies have found that during myocardial ischemia/reperfusion (I/R), the up-regulated Egr-1 gene activates TLR4 (Toll Like Receptor 4)/TRIF (TIR Domain Containing Adaptor Molecule 1) signaling pathway, increases neutrophil recruitment, intensifies cell apoptosis, and further aggravates cardiac function injury (25). Another study showed that Egr-1 could be inhibited by the JAK (Janus Kinase)/STAT (Signal Transducer And Activator Of Transcription) pathway, which decreased the myocardial I/R injury (64).
2.2. Egr-1 and autophagy
Egr-1 not only contributes to apoptosis but also autophagy. The regulation of cardiomyocyte homeostasis is enhanced by autophagy, which is crucial for cardiac physiology (65). Myocardial ischemia-related cell damage can be reduced by autophagy. It has been demonstrated that inhibiting autophagy would exacerbate heart hypertrophy in patients. In general, autophagy promotes the survival of cells by accelerating their metabolic cycle and allowing their adaptation to their surroundings. However, autophagy of the majority of organelles and cytoplasm in the phagocytes results in cell death. In patients with heart failure, excessive autophagy results in type II cell death, or autophagic cardiomyocyte death (66). Cardiomyocyte death following I/R damage is correlated to impaired autophagy flux. In an earlier study, the researchers noted an elevated EGR1 level in a coronary microembolism rat model, which led to the conclusion that Egr-1 helps in regulating autophagy and apoptosis (26). Cytoplasmic components, such as protein aggregates, damaged organelles, and lipid droplets, were encapsulated by the double-layer membrane vesicles to form autophagosomes, which could fuse with the lysosomes to form autophagic lysosomes, degrade the enclosed contents, and assist their recycling (67).
MicroRNAs (miRNAs) regulate and interact with autophagy and apoptosis. Earlier studies have shown that numerous cardiovascular disorders are related to the regulation of EGR1 by miRNAs (68). It was also concluded that miRNAs control EGR1 for mediating autophagy in cardiac cells. Furthermore, a few independent studies showed that BIM suppresses autophagy independent of its pro-apoptotic activity (69). The findings also showed that BIM performs a dual role in suppressing autophagy and promoting apoptosis. BIM directly interacts with the key autophagy regulator, BCL1, to inhibit autophagy (27, 28); which indicates that it could be involved in the pathogenesis of the disease. When cardiac cells are triggered by different environmental factors, the EGR1/BIM/Beclin-1 pathway is activated, thus inhibiting myocardial autophagy and inducing cell death. Furthermore, when an in vitro model of myocardial I/R injury and hypoxia/reoxygenation (H/R) was studied, the researchers noted that circRNAs influenced autophagy by regulating EGR1. Silencing circZNF512 attenuated its ability to bind to miR-181d-5p, and targeted 3′-UTR, thereby impairing EGR1 production, increasing cardiomyocyte autophagy, and inhibiting apoptosis, thus decreasing the myocardial tissue damage. The crosstalk between circZNF512, miR-181d-5p, and EGR1 activated the mTOR (Mechanistic Target Of Rapamycin Kinase)C1/TFEB (Transcription Factor EB) signaling pathway and elevated the mTORC1 level, while the TFEB level was decreased. Furthermore, it was seen that CircZNF512-mediated miR-181d-5p suppression restricted the cardiomyocyte autophagy and increased the myocardial I/R damage (29), with the help of the EGR11/mTORC1/TFEB-based mechanism. Additionally, ERK1/2 refers to a crucial upstream signal molecule that regulates Egr-1 expression and forms the basis for myocardial I/R. Inhibiting Egr-1 expression can lessen the MEK/ERK activation-induced myocardial I/R damage (30) (Figure 2).
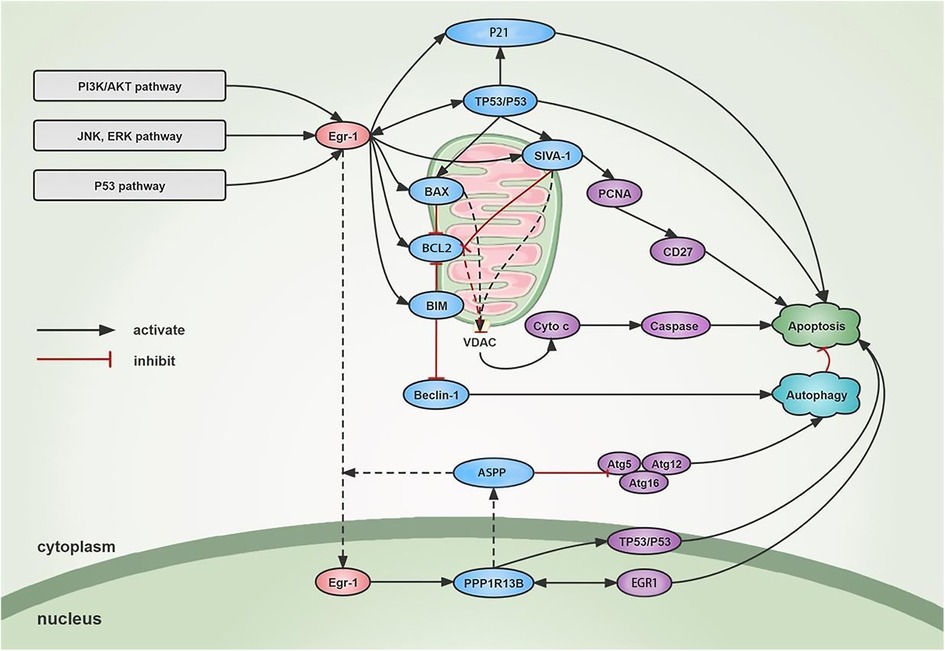
Figure 2. Mechanism of Egr-1 on apoptosis and autophagy. Egr-1 is activated through different signaling cascades. On one hand, Egr-1 stimulates the binding of pro-apoptotic proteins such as BIM, BAX, and SIVA1 to the mitochondrial membrane, increases the opening of VDAC, leads to increased membrane permeability, releases cytochrome, and activates Caspase family and metabolic hydrolases to promote apoptosis. SIVA1 can also mediate the ubiquitination of PCNA and induce CD27-mediated apoptosis. Egr-1 also transactivates the expression of Ppp1r13b in the nucleus. The increased ASPP is mainly localized in the cytoplasm and promotes the nuclear import of EGR1 to promote apoptosis. At the same time, ASPP binds to ATG5-ATG12 and inhibits the binding of ATG5-ATG12 to ATG16 and the formation of ATG16 complex, thus inhibiting cytoprotective autophagy and inducing apoptosis. On the other hand, Egr-1 can activate TP53 in a feedback loop to further activate EGR1 to induce apoptosis. In addition, Egr-1 can induce the P21 and promote cell apoptosis independently or dependently on TP53. Autophagy and apoptosis interact with one another, and Egr-1 can inhibit Beclin-1 through activating BIM and finally inhibit autophagy of cardiac cells.
2.3. Egr-1 and other mechanisms of cell death
According to earlier research, mitochondrial precursor overaccumulation stress (mPOS), which is defined as the dangerous accumulation of unimported mitochondrial proteins in the cytosol, kills cells by a mechanism independent of bioenergetics (70). Liu et al. observed that the overexpression of mitochondrial carrier proteins, notably isoform 1 of adenine nucleotide translocase (ANT1), could lead to the development of numerous cytoplasmic attackers that contained the unimported mitochondrial proteins (31). Dilated cardiomyopathy was linked to the expression of extremely unstable variants of ANT1-induced aggregation and the overexpression of ANT1. It was noted that the Egr-1 gene was maximally upregulated in response to ANT1 overexpression (71–73). Therefore, different mitochondrial stress factors could activate the nuclear transcription factor, EGR1, via ANT1 overexpression, which further increased the mPOS-related cell-killing mechanism, thereby causing cardiomyopathy (74).
In addition to mPOS, metabolic homeostasis (especially calcium homeostasis) can also lead to cardiomyocyte death. Calcium is transferred outside the cardiomyocytes via the sodium-calcium exchanger-1 (NCX1), where they bind to calsequestrin (CSQ) and are re-stored in the sarcoendoplasmic reticulum. In the past, researchers have reported that Egr-1 negatively regulates NCX1 level, which helps in controlling calcium homeostasis, both in vivo and in vitro. Subsequently, it was discovered that Egr-1 mostly inhibits CSQ level. Since CSQ is primarily responsible for the storage and release of calcium ions, CSQ inhibition can harm cardiomyocytes and impair cardiac function. The researchers also found that diabetic cardiomyopathy patients experienced issues related to calcium balance and mitochondrial malfunction, which caused the loss of cardiac cells. Inhibiting the production of EGR1 and controlling mitochondrial calcium homeostasis are two mechanisms by which the MOTS-c (Mitochondrial Open Reading Frame Of The 12S rRNA-c) can stop cardiomyocyte death. The above-mentioned calcium homeostasis is not the only factor that influences cardiomyocyte death; mitochondrial metabolic homeostasis is also important. In cardiomyocytes, Ca2+ ions enter the mitochondria via the mitochondrial Ca2+ (MCU), which stimulates the tricarboxylic acid (TCA) cycle to increase ATP production (32, 33, 35, 75–78). An Egr-1-mediated gene that codes for MICU1 (Mitochondrial Calcium Uptake 1), the gatekeeper of the mitochondrial calcium uniporter, is transcriptionally upregulated by the mitochondria in response to metabolic balance. After analyzing the MCU-mediated mitochondrial matrix Ca (mCa) uptake during metabolic stress, it was noted that Egr-1 regulates the Micu1 promoter, induces MICU1 level during mitochondrial stress, inhibits basal mCa accumulation, and lowers mitochondrial bioenergetics, thereby, preventing mCa overload and ensuing cardiac cell death (34).
Cardiomyocyte death and oxidative stress are also closely related. Oxidative stress negatively affects the development of ischemic heart disease, which leads to irreversible damage and even death of myocardial cells (79). Egr-1 is a redox-sensitive gene and is involved in the pathophysiology of cardiovascular diseases (80–83). Oxidative stress occurs when cellular reductases cannot protect the cells from increased Reactive oxygen species (ROS), which induce EGR1 protein level. This further imbalanced the redox state, which increased DNA and protein damage. Furthermore, higher concentrations of the oxidants can activate different signaling pathways, which, in turn, target the promoters of “redox-sensitive” genes. Notably, heart failure is characterized by the activation of the sympathetic nervous system, which increases oxidative stress in the cardiovascular system (84, 85). Protein kinase Cε (PKCε) plays an important role in cardioprotection, where the PKCε gene inhibition increases susceptibility to cardiac I/R injury (86). Egr-1 exhibits a regulatory effect on PKCε (37). In their study, Xiong et al. found that norepinephrine-induced ROS increased the methylation of the Egr-1 and Sp-1 binding sites in the PKCε promoter, which led to PKCε repression, finally, leading to cardiac function impairment (87). Also, it has been noted that free heme is one of the main causes of ROS in the cardiovascular system. Free heme is released from hemoglobin due to bleeding or hemolysis, leading to oxidative stress and cell death (88, 89). Hasan et al. observed that Hemin (oxidized heme) upregulates the vascular smooth muscle cells (VSMCs) in a redox-sensitive manner via the ROS/ERK and NF-κB (Nuclear Factor Kappa B) pathways, which results in thrombotic or atherosclerotic lesions (38). Also, it has been discovered that Egr-1 inhibition can shield cardiac cells from oxygen-glucose deprivation/reperfusion (OGD/R)-induced injury (36). Studies also showed that dimethyl fumarate protects the cardiac cells against myocardial I/R injury by blocking NOX-4 (NADPH oxidase 4)-mediated ROS production. A few researchers stated that Egr-1 interacts with two additional mechanical stress-related MAPKs, including JNKs. These JNKs, sometimes called stress-activated protein kinases or P38 subtypes, are all activated due to mechanical, oxidative, or environmental stress (90).
In 2012, the researchers identified ferroptosis as a novel cell death mechanism, which was characterized by excessive intracellular lipid peroxide concentrations (91). GPX4 (Glutathione Peroxidase 4) is considered a major factor that inhibits ferroptosis, as it protects cell integrity by eliminating cellular lipid peroxidation and maintaining the balance of the intracellular redox state. Several recent studies have demonstrated that ferroptosis plays a vital role in the development of cardiovascular diseases, such as myocardial infarction, cardiomyopathy, myocardial I/R injury, and heart failure (92). A few findings revealed that GPX4 level was significantly decreased during the early and middle myocardial infarction stages. The researchers also noted that silencing GPX4 could induce ferroptosis of myocardial cells (93). Ferroptosis is characterized by intracellular redox imbalance. A few earlier reports indicated that Egr-1 could be involved in oxidative stress injury, and it was hypothesized that Egr-1 may be involved in the development of ferroptosis. Studies were conducted using an in vitro ferroptosis model, and the results indicated that patients with AMI displayed an increased Egr-1 expression (39). Egr-1 inhibition decreased the miR-15a-5p level and elevated the GPX4 level. It further increases SOD (Superoxide Dismutase) activity, lowers ROS level, and decreases MDA (malondialdehyde) levels and cardiac cell death rates. These findings imply that miR-15a-5p expression is regulated by Egr-1 downregulation, which inhibits ferroptosis.
3. Egr-1 plays a vital role in the pathogenesis of cardiovascular disease
Egr-1 plays a crucial role in cardiovascular biology, and its expression is related to several aspects of cardiovascular pathology. Egr-1 can promote or reduce the synthesis of many pro-inflammatory and anti-inflammatory protein mediators that bind to the complimentary motifs on the DNA of the gene of interest, and are involved in the cell death mechanisms. These mediators primarily regulate angiogenesis, which helps in the healing and regeneration of injured tissues under physiological conditions. It was reported that these mediators actively promote tissue destruction under pathological conditions. According to earlier findings, the Egr-1 was involved in the pathogenesis of atherosclerosis, from the development of foam cells to the onset of acute cardiovascular and cerebrovascular ischemia events. Egr-1 is also believed to be a possible aggregator of other heterogeneous atherosclerotic risk factors, including hyperlipidemia, aberrant hemorheology (observed in hypertension), and other infectious factors. Egr-1 is overexpressed during AMI, which reduces cardiomyocyte energy loss and mass cardiomyocyte death. Targeting Egr-1 can decrease the degenerative effects of AMI in rats. Egr-1 also mediates doxorubicin-induced cardiomyopathy. Therefore, it is important to understand the Egr-1 regulation mechanism, as it could help in the future treatment of cardiovascular diseases.
3.1. MiRNAs regulation of Egr-1 mRNA
MiRNAs are small non-coding RNAs that are involved in gene regulation. Recent studies have revealed that many miRNAs regulate vascular homeostasis and play unique regulatory roles in cardiovascular diseases (94–96). The analysis of dysregulated gene expression in miR-208a mutant mice revealed higher EGR1 and FOS (Fos Proto-Oncogene) levels in the heart. This finding suggests that miR-208 regulates the Egr-1 gene response to cardiac stress. MiR-499 was significantly enriched in the human heart ventricles during microRNA screening (97). Also, substantial changes were noted in miR-499 levels in heart samples collected from aortic stenosis patients with heart failure and pressure overload (98). Furthermore, miR-499 transgenic mice experienced or were susceptible to cardiac dysfunction. Egr-1 is a critical component of the transcriptional response hierarchy to cardiac stress (99), which may explain why miR-499 levels significantly affect the Egr-1 gene response. Recent research has demonstrated that the Egr-1-mediated miR-99 could be regarded as a critical factor in AKT1 (AKT Serine/Threonine Kinase 1) level, which, in turn, controls important cell death pathways involved in the transformation of normal hypertrophy into pathological hypertrophy (100). EGR1 is a key transcriptional activator in the pathological hypertrophy process that might promote PTEN (Phosphatase And Tensin Homolog), which is a negative regulator of AKT (101, 102). EGR1 regulates AKT via two mechanisms: post-transcriptionally via the miR-99 family and post-translationally via PTEN. Egr-1 knockdown converts pathological hypertrophy to physiological hypertrophy by activating the AKT pathway. The miR-99 family regulates Akt/mTOR/IGF1 (Insulin Like Growth Factor 1) via Egr-1-mediated expression, which governs both pathological and healthy hypertrophy (103). AKT1 silencing reduces EGR1 phosphorylation, which decreases miR-99 transcription, resulting in lower apoptosis and cytotoxicity levels. Plasma cholesterol influences a variety of cardiovascular diseases, including atherosclerosis and coronary artery disease (104). Since miR-27a regulates the cholesterol production pathway, it could be hypothesized that a putative EGR1 binding site existed near the miR-27a promoter region. Computational and experimental studies have demonstrated that Egr-1 regulates miR-27a at basal and high cholesterol levels (105). Overexpression of miR-15a-5p promotes ferroptosis in cardiomyocytes, which, in turn, exacerbates hypoxic injury. In the case of AMI, suppressing Egr-1 can limit miR-15a-5p levels, boost GPX4 protein level, and decrease ferroptosis and myocardial damage (39). Recent evidence suggests that miR-146a inhibits Egr-1 transcription and expression, in vivo and in vitro, and attenuates AMI-induced myocardial injury through the TLR4/NF-κB pathway (106).
3.2. DNAzymes target Egr-1 mRNA in cardiovascular diseases
DNAzymes are a new generation of catalytic oligodeoxynucleotides that can be used as an effective gene-silencing strategy. They can be used for overcoming the disadvantages of oligonucleotides and ribozymes and present an effective in vivo gene targeting technique (107). These agents break phosphodiester linkages between specific purines and pyrimidines by precisely base-pairing and de-esterifying the DNAzymes with the target mRNA. Studies have shown that DNAzymes that target Egr-1 are biologically effective in treating AMI and other cardiovascular conditions (108). The proliferation of Endothelial cells (ECs) and VSMCs, in addition to the neointimal formation, is inhibited by Egr-1-targeting DNAzymes (109). Egr-1 also plays a significant role in the injury responses displayed by other cells related to the cardiovascular system (110). ED5 was the first DNAzyme that was successfully tested using an animal model (111). It was noted that ED5 inhibited the EGR1 level and regulated the cardiovascular diseases caused by intimal thickening after permanent carotid artery ligation in rats. The data indicated that ED5-mediated Egr-1 downregulates EGR1 after myocardial I/R and attenuates the level of the myocardial intercellular cell adhesion molecules (ICAM) -1 and neutrophil adhesion during myocardial injury and oxidative stress in cardiomyocytes. ED5 also mediates Egr-1-related inhibition of cyclin D1, monocyte chemoattractant protein (MCP) -1, tissue factor (TF), cyclin-dependent kinase 4 (CDK4), macrophage inflammatory protein (MIP) -2, and Plasminogen activator inhibitor (PAI)-1 (112). Inhibition of these downstream targets can decrease the cell death rate caused by myocardial ischemia (11). Thus, intracoronary inhibition of Egr-1 by targeted DNAzymes may reduce the size of the infarct by modulating the downstream effector molecules (113, 114). Human pulmonary arterial hypertension (PAH) is associated with elevated Platelet-derived growth factor (PDGF)-BB and Transforming growth factor (TGF)-b1 levels (115). These factors also promote the proliferation of VSMCs and ECs. In vivo studies showed that intravenous DNAzymes suppressed pulmonary vasculature remodeling, such as the development of occlusive neointimal lesions, and decreased PAH progression by downregulating EGR1 and lowering PDGF-BB and TGF-b1 level. Thus, Egr-1-targeting DNAzymes are involved in the initiation and progression of pulmonary vascular remodeling in flow-related pulmonary hypertension and could serve as a potential target for PAH therapy in the future (116).
3.3. Oligonucleotide decoys target the EGR1 protein
Synthetic double-stranded decoy oligonucleotides (ODNs) have also been used to target EGR1 at the protein level. In this technique, ODNs containing the DNA-binding component of the transcription factor bind to the DNA and inhibit the function of the transcription factor. ODNs have been utilized to prevent the expression of some genes. ODNs target particular regions of selected mRNAs and impede translation through base-pair hybridization, thus preventing the synthesis of a particular protein (117, 118). Studies have investigated the increasing effect of the sense and antisense oligodeoxynucleotides on the agonist-induced Egr-1 mRNA and proteins. ODNs block the induction of VSMCs by PDGF-BB and angiotensin II. In one study, the carotid arteries in rabbits were damaged with a balloon, and the results showed that there was a decrease in vascular inflammation and neointimal hyperplasia. They also showed that elevated expression of the EGR1-dependent genes was suppressed by transfection of decoy ODNs targeting EGR1 (119). In another study, EGR1 decoy ODNs were designed and synthesized. They were then transfected into balloon-damaged carotid arteries and primary cultures of rat VSMCs, and their ability to bind to Egr-1 was assessed. The competitive binding of Egr-1 decoy ODNs to EGR1 lowered EGR1 level, which was mediated via cell proliferation-related genes such as cyclin D1, CDK4, and PCNA. This further inhibited neointimal hyperplasia and lowered the formation of VSMCs in rats with balloon-damaged arteries (120). Additional research has demonstrated that decoy strategies are quite efficient in the treatment of AMI and cardiac rejection (121, 122). Studies that were conducted using the pig and rat models indicated that NF-κB decoy ODNs effectively suppressed neointimal development following arterial balloon injury. In an earlier study, the researchers could successfully transfect the NF-κB decoy ODNs at the site of coronary stent placement. Their findings showed that these NF-κB decoy ODNs exhibited a safe and beneficial effect on preventing restenosis after percutaneous coronary treatment since they prevented the apoptosis of ECs under hypoxia (121). To summarize, the use of EGR1 decoy ODNs as a novel therapy tool is critical for the prevention and treatment of coronary heart disease and restenosis after angioplasty (123, 124) (Figure 3).
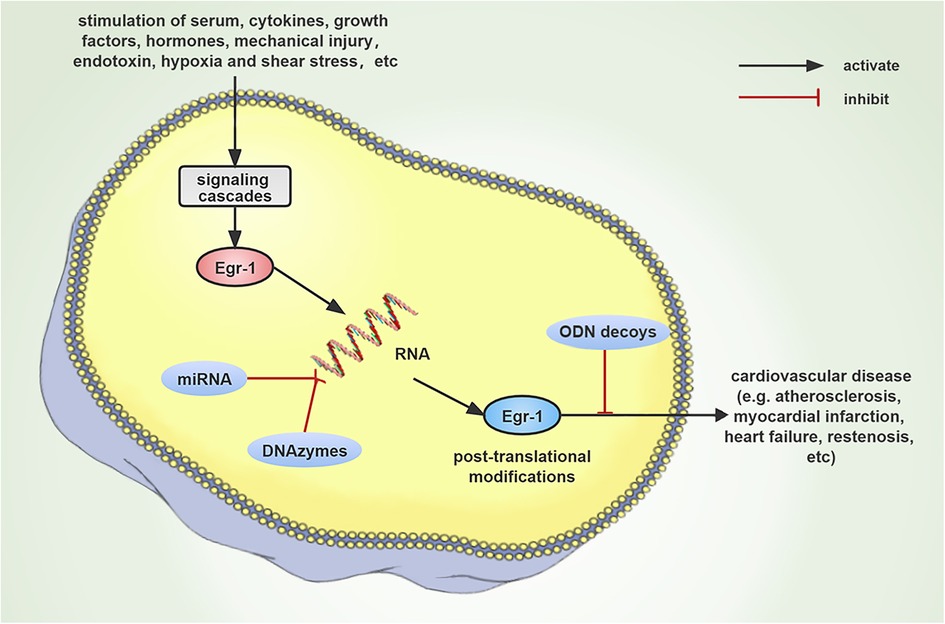
Figure 3. Schematic diagram that describes the progression of cardiovascular disease occurring due to the induction of Egr-1 expression by numerous stimuli. Since Egr-1 plays a major regulatory function in cardiovascular disease, it could be anticipated that Egr-1 targeting DNAzymes, miRNAs, and ODNs decoy strategies could emerge as new therapeutic targets.
4. Concluding remarks
In conclusion, studies conducted using in vitro tests, transgenic animal models, and human diseases revealed that cardiovascular diseases were caused by a variety of cellular and molecular pathways that affected the cell death processes in cardiac cells. Egr-1 is an essential component of the cardiac cell death signaling pathways related to apoptosis, autophagy, mPOS, and ferroptosis. Egr-1 is also involved in preserving heart homeostasis and assists in the development of cardiovascular disorders. The development of new signaling pathways and mediators in cardiovascular disease, as well as their role in tissue damage, has aided in the development of strategies for their specific targeting. Catalytic and non-catalytic nucleic acid approaches, such as DNAzymes, miRNAs, and ODNs decoys, have been used to investigate the primary regulatory role of Egr-1 in cardiovascular diseases, using animal models and Egr-1-deficient mice. In conclusion, Egr-1 can be regarded as a potential therapeutic target for cardiovascular diseases.
Author contributions
YL, JC, HD, XZ and YX developed the concept and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (82060080), Gansu science and Technology Department Project (20YF3WA016, 21JR11RA116), Project of Lanzhou Science and Technology Bureau (2019-RC36) and Cuiying Scientific and Technological Innovation Program of Lanzhou University Second Hospital (CY2022-MS-A06 and CY2022-QN-A17). This study was also supported by the Gansu heart rehabilitation engineering research center (CRQI-C00535) and Medical Postgraduate Training Innovation Development Project (lzuyxcx-2022-128).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Khachigian LM, Collins T. Early growth response factor 1: a pleiotropic mediator of inducible gene expression. J Mol Med (Berl). (1998) 76(9):613–6. doi: 10.1007/s001090050258
2. Sukhatme VP, Cao XM, Chang LC, Tsai-Morris CH, Stamenkovich D, Ferreira PC, et al. A zinc finger-encoding gene coregulated with C-fos during growth and differentiation, and after cellular depolarization. Cell. (1988) 53(1):37–43. doi: 10.1016/0092-8674(88)90485-0
3. Milbrandt J. A nerve growth factor-induced gene encodes a possible transcriptional regulatory factor. Science (New York, NY). (1987) 238(4828):797–9. doi: 10.1126/science.3672127
4. Christy BA, Lau LF, Nathans D. A gene activated in mouse 3t3 cells by serum growth factors encodes a protein with “zinc finger” sequences. Proc Natl Acad Sci U S A. (1988) 85(21):7857–61. doi: 10.1073/pnas.85.21.7857
5. Lemaire P, Revelant O, Bravo R, Charnay P. Two mouse genes encoding potential transcription factors with identica L DNA-binding domains are activated by growth factors in cultured cell S. Proc Natl Acad Sci U S A. (1988) 85(13):4691–5. doi: 10.1073/pnas.85.13.4691
6. Cao X, Mahendran R, Guy GR, Tan YH. Detection and characterization of cellular Egr-1 binding to its recogn ition site. J Biol Chem. (1993) 268(23):16949–57. doi: 10.1016/S0021-9258(19)85286-9
7. Arner E, Daub CO, Vitting-Seerup K, Andersson R, Lilje B, Drabløs F, et al. Transcribed enhancers lead waves of coordinated transcription in trans itioning mammalian cells. Science (New York, NY). (2015) 347(6225):1010–4. doi: 10.1126/science.1259418
8. Decker EL, Nehmann N, Kampen E, Eibel H, Zipfel PF, Skerka C. Early growth response proteins (Egr) and nuclear factors of activated T cells (Nfat) form heterodimers and regulate proinflammatory cytokine gene expression. Nucleic Acids Res. (2003) 31(3):911–21. doi: 10.1093/nar/gkg186
9. Guha M, O'Connell MA, Pawlinski R, Hollis A, McGovern P, Yan SF, et al. Lipopolysaccharide activation of the Mek-Erk1/2 pathway in human monoc ytic cells mediates tissue factor and tumor necrosis factor alpha expr ession by inducing Elk-1 phosphorylation and Egr-1 expression. Blood. (2001) 98(5):1429–39. doi: 10.1182/blood.v98.5.1429
10. Hjoberg J, Le L, Imrich A, Subramaniam V, Mathew SI, Vallone J, et al. Induction of early growth-response factor 1 by platelet-derived growth factor in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. (2004) 286(4):L817–25. doi: 10.1152/ajplung.00190.2003
11. Yan SF, Fujita T, Lu J, Okada K, Shan Zou Y, Mackman N, et al. Egr-1, a master switch coordinating upregulation of divergent gene fam ilies underlying ischemic stress. Nat Med. (2000) 6(12):1355–61. doi: 10.1038/82168
12. Krones-Herzig A, Mittal S, Yule K, Liang H, English C, Urcis R, et al. Early growth response 1 acts as a tumor suppressor in vivo and in vitr O via regulation of P53. Cancer Res. (2005) 65(12):5133–43. doi: 10.1158/0008-5472.CAN-04-3742
13. Du B, Fu C, Kent KC, Bush H, Schulick AH, Kreiger K, et al. Elevated Egr-1 in human atherosclerotic cells transcriptionally repres ses the transforming growth factor-Beta type ii receptor. J Biol Chem. (2000) 275(50):39039–47. doi: 10.1074/jbc.M005159200
14. Khachigian LM. Early growth response-1 in cardiovascular pathobiology. Circ Res. (2006) 98(2):186–91. doi: 10.1161/01.RES.0000200177.53882.c3
15. Wu M, Melichian DS, de la Garza M, Gruner K, Bhattacharyya S, Barr L, et al. Essential roles for early growth response transcription factor Egr-1 in tissue fibrosis and wound healing. Am J Pathol. (2009) 175(3):1041–55. doi: 10.2353/ajpath.2009.090241
16. Bhattacharyya S, Ishida W, Wu M, Wilkes M, Mori Y, Hinchcliff M, et al. A non-smad mechanism of fibroblast activation by transforming growth factor-Beta Via C-Abl and Egr-1: selective modulation by imatinib mesylate. Oncogene. (2009) 28(10):1285–97. doi: 10.1038/onc.2008.479
17. Xu J, Du WW, Wu N, Li F, Li X, Xie Y, et al. The circular Rna circnlgnmediates doxorubicin-inducedcardiac remodelin G and fibrosis. Mol Ther Nucleic Acids. (2022) 28:175–89. doi: 10.1016/j.omtn.2022.03.007
18. Fu M, Zhu X, Zhang J, Liang J, Lin Y, Zhao L, et al. Egr-1 target genes in human endothelial cells identified by microarray analysis. Gene. (2003) 315:33–41. doi: 10.1016/s0378-1119(03)00730-3
19. Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. (2019) 99(4):1765–817. doi: 10.1152/physrev.00022.2018
20. Cook SA, Sugden PH, Clerk A. Regulation of Bcl-2 family proteins during development and in response to oxidative stress in cardiac myocytes: association with changes in mitochondrial membrane potential. Circ Res. (1999) 85(10):940–9. doi: 10.1161/01.res.85.10.940
21. Zhao K, Yu M, Zhu Y, Liu D, Wu Q, Hu Y. Egr-1/Aspp1 inter-regulatory loop promotes apoptosis by inhibiting Cyt O-protective autophagy. Cell Death Dis. (2017) 8(6):e2869. doi: 10.1038/cddis.2017.268
22. Zins K, Pomyje J, Hofer E, Abraham D, Lucas T, Aharinejad S. Egr-1 upregulates siva-1 expression and induces cardiac fibroblast apo ptosis. Int J Mol Sci. (2014) 15(1):1538–53. doi: 10.3390/ijms15011538
23. Fasolo F, Jin H, Winski G, Chernogubova E, Pauli J, Winter H, et al. Long noncoding Rna miat controls advanced atherosclerotic lesion formation and plaque destabilization. Circulation. (2021) 144(19):1567–83. doi: 10.1161/circulationaha.120.052023
24. Santiago FS, Li Y, Khachigian LM. Serine 26 in early growth response-1 is critical for endothelial proliferation, migration, and network formation. J Am Heart Assoc. (2021) 10(18):e020521. doi: 10.1161/jaha.120.020521
25. Huang C, Qu Y, Feng F, Zhang H, Shu L, Zhu X, et al. Cardioprotective effect of circ_Smg6 knockdown against myocardial ischemia/reperfusion injury correlates with Mir-138-5p-mediated Egr1/Tlr4/Trif inactivation. Oxid Med Cell Longev. (2022) 2022:1927260. doi: 10.1155/2022/1927260
26. Wang XT, Wu XD, Lu YX, Sun YH, Zhu HH, Liang JB, et al. Egr-1 is involved in coronary microembolization-induced myocardial inj ury via bim/beclin-1 pathway-mediated autophagy inhibition and apoptos is activation. Aging. (2018) 10(11):3136–47. doi: 10.18632/aging.101616
27. Su B, Wang X, Sun Y, Long M, Zheng J, Wu W, et al. Mir-30e-3p promotes cardiomyocyte autophagy and inhibits apoptosis via regulating Egr-1 during ischemia/hypoxia. Biomed Res Int. (2020) 2020:7231243. doi: 10.1155/2020/7231243
28. Su B, Wang XT, Sun YH, Long MY, Zheng J, Wu WH, et al. Ischemia/hypoxia inhibits cardiomyocyte autophagy and promotes apoptos is via the Egr-1/Bim/Beclin-1 pathway. J Geriatr Cardiol. (2020) 17(5):284–93. doi: 10.11909/j.issn.1671-5411.2020.05.004
29. Huang C, Shu L, Zhang H, Zhu X, Huang G, Xu J. Circ_Znf512-mediated Mir-181d-5p inhibition limits cardiomyocyte autophagy and promotes myocardial ischemia/reperfusion injury through an Egr1/Mtorc1/Tfeb-based mechanism. J Med Chem. (2022) 65(3):1808–21. doi: 10.1021/acs.jmedchem.1c00745
30. Wang A, Zhang H, Liang Z, Xu K, Qiu W, Tian Y, et al. U0126 attenuates ischemia/reperfusion-induced apoptosis and autophagy in myocardium through Mek/Erk/Egr-1 pathway. Eur J Biochem. (2016) 788:280–5. doi: 10.1016/j.ejphar.2016.06.038
31. Liu J, Jin X, Liu KJ, Liu W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci. (2012) 32(9):3044–57. doi: 10.1523/jneurosci.6409-11.2012
32. Wang C, Dostanic S, Servant N, Chalifour LE. Egr-1 negatively regulates expression of the sodium-calcium exchanger-1 in cardiomyocytes in vitro and in vivo. Cardiovasc Res. (2005) 65(1):187–94. doi: 10.1016/j.cardiores.2004.09.026
33. Kasneci A, Kemeny-Suss NM, Komarova SV, Chalifour LE. Egr-1 negatively regulates calsequestrin expression and calcium dynamics in ventricular cells. Cardiovasc Res. (2009) 81(4):695–702. doi: 10.1093/cvr/cvn357
34. Nemani N, Dong Z, Daw CC, Madaris TR, Ramachandran K, Enslow BT, et al. Mitochondrial pyruvate and fatty acid flux modulate Micu1-dependent co ntrol of Mcu activity. Sci Signaling. (2020) 13(628):eaaz6206. doi: 10.1126/scisignal.aaz6206
35. Wang M, Wang G, Pang X, Ma J, Yuan J, Pan Y, et al. Mots-C repairs myocardial damage by inhibiting the Ccn1/Erk1/2/Egr1 pathway in diabetic rats. Front Nutr. (2022) 9:1060684. doi: 10.3389/fnut.2022.1060684
36. Zhao J, Cheng Z, Quan X, Xie Z, Zhang L, Ding Z. Dimethyl fumarate protects cardiomyocytes against oxygen-glucose deprivation/reperfusion (Ogd/R)-induced inflammatory response and damages via inhibition of Egr-1. Int Immunopharmacol. (2020) 86:106733. doi: 10.1016/j.intimp.2020.106733
37. Chen M, Xiong F, Zhang L. Promoter methylation of Egr-1 site contributes to fetal hypoxia-mediated Pkcε gene repression in the developing heart. Am J Physiol Regul Integr Comp Physiol. (2013) 304(9):R683–9. doi: 10.1152/ajpregu.00461.2012
38. Hasan RN, Schafer AI. Hemin upregulates Egr-1 expression in vascular smooth muscle cells via reactive oxygen species Erk-1/2-Elk-1 and Nf-Kappab. Circ Res. (2008) 102(1):42–50. doi: 10.1161/circresaha.107.155143
39. Fan K, Huang W, Qi H, Song C, He C, Liu Y, et al. The Egr-1/Mir-15a-5p/Gpx4 axis regulates ferroptosis in acute myocardi al infarction. Eur J Biochem. (2021) 909:174403. doi: 10.1016/j.ejphar.2021.174403
40. Cosentino K, Hertlein V, Jenner A, Dellmann T, Gojkovic M, Peña-Blanco A, et al. The interplay between Bax and Bak tunes apoptotic pore growth to contr ol mitochondrial-DNA-mediated inflammation. Mol Cell. (2022) 82(5):933–49.e9. doi: 10.1016/j.molcel.2022.01.008
41. Zhang H, Kim JK, Edwards CA, Xu Z, Taichman R, Wang C-Y. Clusterin inhibits apoptosis by interacting with activated bax. Nat Cell Biol. (2005) 7(9):909–15. doi: 10.1038/ncb1291
42. Zhu R, Li L, Nguyen B, Seo J, Wu M, Seale T, et al. Flt3 tyrosine kinase inhibitors synergize with Bcl-2 inhibition to eli minate Flt3/Itd acute leukemia cells through bim activation. Signal Transduction Targeted Ther. (2021) 6(1):186. doi: 10.1038/s41392-021-00578-4
43. Samuels-Lev Y, O'Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, et al. Aspp proteins specifically stimulate the apoptotic function of P53. Mol Cell. (2001) 8(4):781–94. doi: 10.1016/s1097-2765(01)00367-7
44. Park IK, Blum W, Baker SD, Caligiuri MA. E3 ubiquitin ligase Cbl-B activates the P53 pathway by targeting Siva1, a negative regulator of Arf, in Flt3 inhibitor-resistant acute myelo id leukemia. Leukemia. (2017) 31(2):502–5. doi: 10.1038/leu.2016.293
45. Green DR, Reed JC. Mitochondria and apoptosis. Science (New York, NY). (1998) 281(5381):1309–12. doi: 10.1126/science.281.5381.1309
46. Biala AK, Kirshenbaum LA. The interplay between cell death signaling pathways in the heart. Trends Cardiovasc Med. (2014) 24(8):325–31. doi: 10.1016/j.tcm.2014.08.002
47. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the Bcl-2 protein family: implications for phy siology and therapy. Nat Rev Mol Cell Biol. (2014) 15(1):49–63. doi: 10.1038/nrm3722
48. Flores-Romero H, Hohorst L, John M, Albert M-C, King LE, Beckmann L, et al. Bcl-2-Family protein tbid can act as a bax-like effector of apoptosis. EMBO J. (2022) 41(2):e108690. doi: 10.15252/embj.2021108690
49. Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. Vdac, a multi-functional mitochondrial protein regulating cell life an D death. Mol Aspects Med. (2010) 31(3):227–85. doi: 10.1016/j.mam.2010.03.002
50. Leri A, Liu Y, Malhotra A, Li Q, Stiegler P, Claudio PP, et al. Pacing-Induced heart failure in dogs enhances the expression of P53 and P53-dependent genes in ventricular myocytes. Circulation. (1998) 97(2):194–203. doi: 10.1161/01.cir.97.2.194
51. Asiri YA. Probucol attenuates cyclophosphamide-induced oxidative apoptosis, P53 and bax signal expression in rat cardiac tissues. Oxid Med Cell Longev. (2010) 3(5):308–16. doi: 10.4161/oxim.3.5.13107
52. Nair P, Muthukkumar S, Sells SF, Han SS, Sukhatme VP, Rangnekar VM. Early growth response-1-dependent apoptosis is mediated by P53. J Biol Chem. (1997) 272(32):20131–8. doi: 10.1074/jbc.272.32.20131
53. Miah S, Zadeh SN, Yuan XM, Li W. Expression of Egr1 and P53 in human carotid plaques and apoptosis induced by 7-oxysterol or P53. Exp Toxicol Pathol. (2013) 65(5):677–82. doi: 10.1016/j.etp.2012.08.002
54. Wang Y, Liu C, Wang J, Zhang Y, Chen L. Iodine-131 induces apoptosis in human cardiac muscle cells through the P53/Bax/Caspase-3 and Pidd/Caspase-2/ T-Bid/Cytochrome c/Caspase-3 signaling pathway. Oncol Rep. (2017) 38(3):1579–86. doi: 10.3892/or.2017.5813
55. Kojic S, Nestorovic A, Rakicevic L, Protic O, Jasnic-Savovic J, Faulkner G, et al. Cardiac transcription factor Nkx2.5 interacts with P53 and modulates its activity. Arch Biochem Biophys. (2015) 569:45–53. doi: 10.1016/j.abb.2015.02.001
56. Liao XD, Wang XH, Jin HJ, Chen LY, Chen Q. Mechanical stretch induces mitochondria-dependent apoptosis in neonatal rat cardiomyocytes and G2/M accumulation in cardiac fibroblasts. Cell Res. (2004) 14(1):16–26. doi: 10.1038/sj.cr.7290198
57. He J, Yu JJ, Xu Q, Wang L, Zheng JZ, Liu LZ, et al. Downregulation of Atg14 by Egr1-Mir152 sensitizes ovarian cancer cells to cisplatin-induced apoptosis by inhibiting cyto-protective autophag Y. Autophagy. (2015) 11(2):373–84. doi: 10.1080/15548627.2015.1009781
58. Yang Y, Zhang Y, Yang J, Zhang M, Tian T, Jiang Y, et al. Interdependent nuclear co-trafficking of Aspp1 and P53 aggravates cardiac ischemia/reperfusion injury. Circ Res. (2023) 132(2):208–22. doi: 10.1161/circresaha.122.321153
59. Chu F, Borthakur A, Sun X, Barkinge J, Gudi R, Hawkins S, et al. The siva-1 putative amphipathic helical region (Sah) is sufficient to bind to Bcl-Xl and sensitize cells to Uv radiation induced apoptosis. Apoptosis. (2004) 9(1):83–95. doi: 10.1023/B:APPT.0000012125.01799.4c
60. Prasad KV, Ao Z, Yoon Y, Wu MX, Rizk M, Jacquot S, et al. Cd27, a member of the tumor necrosis factor receptor family, induces a poptosis and binds to siva, a proapoptotic protein. Proc Natl Acad Sci U S A. (1997) 94(12):6346–51. doi: 10.1073/pnas.94.12.6346
61. Fortin A, MacLaurin JG, Arbour N, Cregan SP, Kushwaha N, Callaghan SM, et al. The proapoptotic gene siva is a direct transcriptional target for the tumor suppressors P53 and E2f1. J Biol Chem. (2004) 279(27):28706–14. doi: 10.1074/jbc.M400376200
62. Teixeira FR, Manfiolli AO, Soares CS, Baqui MMA, Koide T, Gomes MD. The F-Box protein Fbxo25 promotes the proteasome-dependent degradation of Elk-1 protein. J Biol Chem. (2013) 288(39):28152–62. doi: 10.1074/jbc.M113.504308
63. Yin L, Zhang J, Sun Y. Early growth response-1 is a new substrate of the Gsk3β-Fbxw7 axis. Neoplasia (New York, NY). (2022) 34:100839. doi: 10.1016/j.neo.2022.100839
64. Mudaliar H, Rayner B, Billah M, Kapoor N, Lay W, Dona A, et al. Remote ischemic preconditioning attenuates Egr-1 expression following myocardial ischemia reperfusion injury through activation of the Jak-Stat pathway. Int J Cardiol. (2017) 228:729–41. doi: 10.1016/j.ijcard.2016.11.198
65. Jimenez RE, Kubli DA, Gustafsson ÅB. Autophagy and mitophagy in the myocardium: therapeutic potential and C oncerns. Br J Pharmacol. (2014) 171(8):1907–16. doi: 10.1111/bph.12477
66. Dong Y, Chen H, Gao J, Liu Y, Li J, Wang J. Molecular machinery and interplay of apoptosis and autophagy in Corona Ry heart disease. J Mol Cell Cardiol. (2019) 136:27–41. doi: 10.1016/j.yjmcc.2019.09.001
67. Salazar G, Cullen A, Huang J, Zhao Y, Serino A, Hilenski L, et al. Sqstm1/P62 and Ppargc1a/Pgc-1alpha at the interface of autophagy and V ascular senescence. Autophagy. (2020) 16(6):1092–110. doi: 10.1080/15548627.2019.1659612
68. Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular rnas. Nat Rev Genet. (2019) 20(11):675–91. doi: 10.1038/s41576-019-0158-7
69. Mo B, Wu X, Wang X, Xie J, Ye Z, Li L. Mir-30e-5p mitigates hypoxia-induced apoptosis in human stem cell-derived cardiomyocytes by suppressing Bim. Int J Biol Sci. (2019) 15(5):1042–51. doi: 10.7150/ijbs.31099
70. Wang X, Chen XJ. A cytosolic network suppressing mitochondria-mediated proteostatic Str Ess and cell death. Nature. (2015) 524(7566):481–4. doi: 10.1038/nature14859
71. Dörner A, Schultheiss HP. The myocardial expression of the adenine nucleotide translocator isofo Rms is specifically altered in dilated cardiomyopathy. Herz. (2000) 25(3):176–80. doi: 10.1007/s000590050004
72. Dörner A, Schulze K, Rauch U, Schultheiss HP. Adenine nucleotide translocator in dilated cardiomyopathy: pathophysio logical alterations in expression and function. Mol Cell Biochem. (1997) 174(1-2):261–9. doi: 10.1023/A:1006825028170
73. Sylvén C, Lin L, Jansson E, Sotonyi P, Fu LX, Waagstein F, et al. Ventricular adenine nucleotide translocator mrna is upregulated in Dil Ated cardiomyopathy. Cardiovasc Res. (1993) 27(7):1295–9. doi: 10.1093/cvr/27.7.1295
74. Coyne LP, Chen XJ. Mpos is a novel mitochondrial trigger of cell death—implications for neurodegeneration. FEBS Lett. (2018) 592(5):759–75. doi: 10.1002/1873-3468.12894
76. Pagliarini DJ, Rutter J. Hallmarks of a new era in mitochondrial biochemistry. Gene Dev. (2013) 27(24):2615–27. doi: 10.1101/gad.229724.113
77. Sugiura A, McLelland G-L, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. (2014) 33(19):2142–56. doi: 10.15252/embj.201488104
78. Williams GSB, Boyman L, Chikando AC, Khairallah RJ, Lederer WJ. Mitochondrial calcium uptake. Proc Natl Acad Sci U S A. (2013) 110(26):10479–86. doi: 10.1073/pnas.1300410110
79. Forsberg L, de Faire U, Morgenstern R. Oxidative stress, human genetic variation, and disease. Arch Biochem Biophys. (2001) 389(1):84–93. doi: 10.1006/abbi.2001.2295
80. Huang RP, Adamson ED. Characterization of the DNA-binding properties of the early growth res ponse-1 (Egr-1) transcription factor: evidence for modulation by a red ox mechanism. DNA Cell Biol. (1993) 12(3):265–73. doi: 10.1089/dna.1993.12.265
81. Nose K, Ohba M. Functional activation of the Egr-1 (early growth response-1) gene by H ydrogen peroxide. Biochem J. (1996) 316(Pt 2):381–3. doi: 10.1042/bj3160381
82. Nose K, Shibanuma M, Kikuchi K, Kageyama H, Sakiyama S, Kuroki T. Transcriptional activation of early-response genes by hydrogen peroxid E in a mouse osteoblastic cell line. Eur J Biochem. (1991) 201(1):99–106. doi: 10.1111/j.1432-1033.1991.tb16261.x
83. Paron I, D'Elia A, D'Ambrosio C, Scaloni A, D'Aurizio F, Prescott A, et al. A proteomic approach to identify early molecular targets of oxidative stress in human epithelial Lens cells. Biochem J. (2004) 378(Pt 3):929–37. doi: 10.1042/BJ20031190
84. Liang C, Rounds NK, Dong E, Stevens SY, Shite J, Qin F. Alterations by norepinephrine of cardiac sympathetic nerve terminal function and myocardial Beta-adrenergic receptor sensitivity in the ferret: normalization by antioxidant vitamins. Circulation. (2000) 102(1):96–103. doi: 10.1161/01.cir.102.1.96
85. Münzel T, Gori T, Bruno RM, Taddei S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur Heart J. (2010) 31(22):2741–8. doi: 10.1093/eurheartj/ehq396
86. Lawrence J, Chen M, Xiong F, Xiao D, Zhang H, Buchholz JN, et al. Foetal nicotine exposure causes pkcε gene repression by promoter methylation in rat hearts. Cardiovasc Res. (2011) 89(1):89–97. doi: 10.1093/cvr/cvq270
87. Xiong F, Xiao D, Zhang L. Norepinephrine causes epigenetic repression of pkcε gene in rodent hearts by activating Nox1-dependent reactive oxygen Species production. Faseb J. (2012) 26(7):2753–63. doi: 10.1096/fj.11-199422
88. Jeney V, Balla J, Yachie A, Varga Z, Vercellotti GM, Eaton JW, et al. Pro-Oxidant and cytotoxic effects of circulating heme. Blood. (2002) 100(3):879–87. doi: 10.1182/blood.v100.3.879
89. Jin N, Hatton ND, Harrington MA, Xia X, Larsen SH, Rhoades RA. H(2)O(2)-induced Egr-1, Fra-1, and C-Jun gene expression is mediated by tyrosine kinase in aortic smooth muscle cells. Free Radic Biol Med. (2000) 29(8):736–46. doi: 10.1016/s0891-5849(00)00376-2
90. Hebert MA, Serova LI, Sabban EL. Single and repeated immobilization stress differentially trigger induction and phosphorylation of several transcription factors and mitogen-activated protein kinases in the rat locus Coeruleus. J Neurochem. (2005) 95(2):484–98. doi: 10.1111/j.1471-4159.2005.03386.x
91. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. (2021) 22(4):266–82. doi: 10.1038/s41580-020-00324-8
92. Wu X, Li Y, Zhang S, Zhou X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics. (2021) 11(7):3052–9. doi: 10.7150/thno.54113
93. Park TJ, Park JH, Lee GS, Lee JY, Shin JH, Kim MW, et al. Quantitative proteomic analyses reveal that Gpx4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. (2019) 10(11):835. doi: 10.1038/s41419-019-2061-8
94. Callis TE, Pandya K, Seok HY, Tang R-H, Tatsuguchi M, Huang Z-P, et al. Microrna-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest. (2009) 119(9):2772–86. doi: 10.1172/JCI36154
95. van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a Mi crorna. Science (New York, NY). (2007) 316(5824):575–9. doi: 10.1126/science.1139089
96. Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microrna that target S Hand2 during cardiogenesis. Nature. (2005) 436(7048):214–20. doi: 10.1038/nature03817
97. Kura B, Szeiffova Bacova B, Kalocayova B, Sykora M, Slezak J. Oxidative stress-responsive micrornas in heart injury. Int J Mol Sci. (2020) 21(1):358. doi: 10.3390/ijms21010358
98. Wang W, Li T, Gao L, Li Y, Sun Y, Yao H-C. Plasma mir-208b and mir-499: potential biomarkers for severity of coro nary artery disease. Dis Markers. (2019) 2019:9842427. doi: 10.1155/2019/9842427
99. Saadane N, Alpert L, Chalifour LE. Altered molecular response to adrenoreceptor-induced cardiac hypertrop hy in egr-1-deficient mice. Am J Physiol Heart Circ Physiol. (2000) 278(3):H796–805. doi: 10.1152/ajpheart.2000.278.3.H796
100. Ramasamy S, Velmurugan G, Rekha B, Anusha S, Shanmugha Rajan K, Shanmugarajan S, et al. Egr-1 mediated cardiac mir-99 family expression diverges physiological hypertrophy from pathological hypertrophy. Exp Cell Res. (2018) 365(1):46–56. doi: 10.1016/j.yexcr.2018.02.016
101. Gupta MP, Gupta M, Zak R, Sukhatme VP. Egr-1, a Serum-inducible zinc finger protein, regulates transcription of the rat cardiac alpha-myosin heavy chain gene. J Biol Chem. (1991) 266(20):12813–6. doi: 10.1016/S0021-9258(18)98762-4
102. Yu J, Zhang SS, Saito K, Williams S, Arimura Y, Ma Y, et al. Pten regulation by akt-Egr1-arf-pten axis. EMBO J. (2009) 28(1):21–33. doi: 10.1038/emboj.2008.238
103. Soci UPR, Cavalcante BRR, Improta-Caria AC, Roever L. The epigenetic role of mirnas in endocrine crosstalk between the cardi ovascular system and adipose tissue: a bidirectional view. Front Cell Dev Biol. (2022) 10:910884. doi: 10.3389/fcell.2022.910884
104. Sathiyakumar V, Kapoor K, Jones SR, Banach M, Martin SS, Toth PP. Novel therapeutic targets for managing dyslipidemia. Trends Pharmacol Sci. (2018) 39(8):733–47. doi: 10.1016/j.tips.2018.06.001
105. Khan AA, Agarwal H, Reddy SS, Arige V, Natarajan B, Gupta V, et al. Microrna 27a is a key modulator of cholesterol biosynthesis. Mol Cell Biol. (2020) 40(9):e00470-19. doi: 10.1128/mcb.00470-19
106. Pan J, Alimujiang M, Chen Q, Shi H, Luo X. Exosomes derived from mir-146a-modified adipose-derived stem cells attenuate acute myocardial infarction-induced myocardial damage via downregulation of early growth response factor 1. J Cell Biochem. (2019) 120(3):4433–43. doi: 10.1002/jcb.27731
107. McConnell EM, Cozma I, Mou Q, Brennan JD, Lu Y, Li Y. Biosensing with dnazymes. Chem Soc Rev. (2021) 50(16):8954–94. doi: 10.1039/d1cs00240f
108. Lowe HC, Fahmy RG, Kavurma MM, Baker A, Chesterman CN, Khachigian LM. Catalytic oligodeoxynucleotides define a key regulatory role for early growth response factor-1 in the porcine model of coronary in-stent Re stenosis. Circ Res. (2001) 89(8):670–7. doi: 10.1161/hh2001.097867
109. Lowe HC, Chesterman CN, Khachigian LM. Catalytic antisense DNA molecules targeting egr-1 inhibit neointima Fo rmation following permanent ligation of rat common carotid arteries. Thromb Haemostasis. (2002) 87(1):134–40. doi: 10.1055/s-0037-1612956
110. Brand T, Sharma HS, Fleischmann KE, Duncker DJ, McFalls EO, Verdouw PD, et al. Proto-Oncogene expression in porcine myocardium subjected to ischemia and reperfusion. Circ Res. (1992) 71(6):1351–60. doi: 10.1161/01.res.71.6.1351
111. Santoro SW, Joyce GF. A general purpose rna-cleaving DNA enzyme. Proc Natl Acad Sci U S A. (1997) 94(9):4262–6. doi: 10.1073/pnas.94.9.4262
112. Xiang G, Schuster MD, Seki T, Witkowski P, Eshghi S, Itescu S. Downregulated expression of plasminogen activator inhibitor-1 augments myocardial neovascularization and reduces cardiomyocyte apoptosis aft er acute myocardial infarction. J Am Coll Cardiol. (2005) 46(3):536–41. doi: 10.1016/j.jacc.2005.04.047
113. Bhindi R, Fahmy RG, McMahon AC, Khachigian LM, Lowe HC. Intracoronary delivery of dnazymes targeting human Egr-1 reduces infar ct size following myocardial ischaemia reperfusion. J Pathol. (2012) 227(2):157–64. doi: 10.1002/path.2991
114. Bhindi R, Khachigian LM, Lowe HC. Dnazymes targeting the transcription factor egr-1 reduce myocardial in farct size following ischemia-reperfusion in rats. J Thromb Haemost. (2006) 4(7):1479–83. doi: 10.1111/j.1538-7836.2006.02022.x
115. Evans CE, Cober ND, Dai Z, Stewart DJ, Zhao Y-Y. Endothelial cells in the pathogenesis of pulmonary arterial hypertensi on. Eur Respir J. (2021) 58(3):2003957. doi: 10.1183/13993003.03957-2020
116. van der Feen DE, Dickinson MG, Bartelds B, Borgdorff MAJ, Sietsma H, Lévy M, et al. Egr-1 identifies neointimal remodeling and relates to progression in H Uman pulmonary arterial hypertension. J Heart Lung Transpl. (2016) 35(4):481–90. doi: 10.1016/j.healun.2015.12.004
117. Klabenkova K, Fokina A, Stetsenko D. Chemistry of peptide-oligonucleotide conjugates: a review. Molecules (Basel, Switzerland). (2021) 26(17):5420. doi: 10.3390/molecules26175420
118. Stadlbauer THW, Wagner AH, Hölschermann H, Fiedel S, Fingerhuth H, Tillmanns H, et al. Ap-1 and Stat-1 decoy oligodeoxynucleotides attenuate transplant vascu lopathy in rat cardiac allografts. Cardiovasc Res. (2008) 79(4):698–705. doi: 10.1093/cvr/cvn135
119. Ohtani K, Egashira K, Usui M, Ishibashi M, Hiasa KI, Zhao Q, et al. Inhibition of neointimal hyperplasia after balloon injury by cis-eleme Nt “decoy” of early growth response gene-1 in hypercholesterolemic rab bits. Gene Ther. (2004) 11(2):126–32. doi: 10.1038/sj.gt.3302153
120. Han W, Liu GN. Egr-1 decoy odns inhibit vascular smooth muscle cell proliferation and neointimal hyperplasia of balloon-injured arteries in rat. Life Sci. (2010) 86(7-8):234–43. doi: 10.1016/j.lfs.2009.12.005
121. Yamasaki K, Asai T, Shimizu M, Aoki M, Hashiya N, Sakonjo H, et al. Inhibition of nfkappab activation using cis-element “decoy” of nfkappa B binding site reduces neointimal formation in porcine balloon-injured coronary artery model. Gene Ther. (2003) 10(4):356–64. doi: 10.1038/sj.gt.3301875
122. Yoshimura S, Morishita R, Hayashi K, Yamamoto K, Nakagami H, Kaneda Y, et al. Inhibition of intimal hyperplasia after balloon injury in rat carotid artery model using cis-element “decoy” of nuclear factor-kappab bindin G site as a novel molecular strategy. Gene Ther. (2001) 8(21):1635–42. doi: 10.1038/sj.gt.3301566
123. Mahjoubin-Tehran M, Teng Y, Jalili A, Aghaee-Bakhtiari SH, Markin AM, Sahebkar A. Decoy technology as a promising therapeutic tool for atherosclerosis. Int J Mol Sci. (2021) 22(9):4420. doi: 10.3390/ijms22094420
Keywords: early growth response-1, cell death, apoptosis, autophagy, cardiovascular diseases, transcription factors
Citation: Xie Y, Li Y, Chen J, Ding H and Zhang X (2023) Early growth response-1: Key mediators of cell death and novel targets for cardiovascular disease therapy. Front. Cardiovasc. Med. 10:1162662. doi: 10.3389/fcvm.2023.1162662
Received: 10 February 2023; Accepted: 13 March 2023;
Published: 28 March 2023.
Edited by:
Rajesh Katare, University of Otago, New ZealandReviewed by:
Fujie Zhao, University of Alabama at Birmingham, United StatesSundar Krishnasamy, SASTRA University, India
© 2023 Xie, Li, Chen, Ding and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaowei Zhang emhhbmd4dzcyN0AxNjMuY29t
Specialty Section: This article was submitted to Cardiovascular Biologics and Regenerative Medicine, a section of the journal Frontiers in Cardiovascular Medicine