 Esteban Gabory1Moses Njagi Mwaniki2
Esteban Gabory1Moses Njagi Mwaniki2 Nadia Pisanti2*
Nadia Pisanti2* Solon P. Pissis1,3Jakub Radoszewski4Michelle Sweering1Wiktor Zuba1
Solon P. Pissis1,3Jakub Radoszewski4Michelle Sweering1Wiktor Zuba1- 1Centrum Wiskunde & Informatica, Amsterdam, Netherlands
- 2Department of Computer Science, University of Pisa, Pisa, Italy
- 3Department of Computer Science, Vrije Universiteit, Amsterdam, Netherlands
- 4Institute of Informatics, University of Warsaw, Warsaw, Poland
Introduction: An elastic-degenerate (ED) string is a sequence of sets of strings. It can also be seen as a directed acyclic graph whose edges are labeled by strings. The notion of ED strings was introduced as a simple alternative to variation and sequence graphs for representing a pangenome, that is, a collection of genomic sequences to be analyzed jointly or to be used as a reference.
Methods: In this study, we define notions of matching statistics of two ED strings as similarity measures between pangenomes and, consequently infer a corresponding distance measure. We then show that both measures can be computed efficiently, in both theory and practice, by employing the intersection graph of two ED strings.
Results: We also implemented our methods as a software tool for pangenome comparison and evaluated their efficiency and effectiveness using both synthetic and real datasets.
Discussion: As for efficiency, we compare the runtime of the intersection graph method against the classic product automaton construction showing that the intersection graph is faster by up to one order of magnitude. For showing effectiveness, we used real SARS-CoV-2 datasets and our matching statistics similarity measure to reproduce a well-established clade classification of SARS-CoV-2, thus demonstrating that the classification obtained by our method is in accordance with the existing one.
1 Introduction
Many biomedical applications of bioinformatics face the twofold challenge of analyzing an ever-increasing number of genome sequences and the need to choose which genome should be used as the reference. Generalizing other definitions, in The computational Pangenomics Consortium (2018), a pangenome was defined as “any collection of genomic sequences to be analyzed jointly or to be used as a reference,” somewhat merging the two above-mentioned challenges into that of analyzing a pangenome. When projected within a single species, a pangenome represents a collection of sequences that are (part of whole) genomes originating from different individuals or strains of a single clade or population.
Currently, pangenomics constitutes an important paradigm shift within genomics to deal with the widespread availability of human sequencing data and the discovery of large-scale genomic variation in many eukaryotic species; see Paten et al. (2017); Liao et al. (2023). In contrast to a linear reference, a pangenome reference aims to compactly represent the variation within a population by encoding the commonalities and differences among the underlying sequences. This gives rise to different pangenome representations, usually edge- or node-labeled directed graphs (Baaijens et al., 2022; Carletti et al., 2019). The most widely-used pangenome representations are variation graphs (see Garrison et al., 2018a; Eizenga et al., 2021) and sequence graphs (see Rakocevic et al., 2019).
The computational challenges posed by pangenomes often result in a trade-off between the efficiency and accuracy of the methods and the information content of the chosen representation. A simpler acyclic alternative to the aforementioned representations is the notion of elastic-degenerate string (ED string); see Iliopoulos et al. (2021). When compared to more powerful representations, ED strings have the algorithmic advantage of supporting both theoretically (Aoyama et al., 2018; Bernardini et al., 2019; 2022)) and practically (Grossi et al., 2017; Pissis and Retha, 2018; Cisłak et al., 2018) fast on-line pattern matching, also for the approximate case (Bernardini et al., 2017; 2020). Moreover, they support fast dynamic-programming-based alignment, as shown in Mwaniki et al. (2023); Mwaniki and Pisanti (2022), and short-read mapping; see Büchler et al. (2023).
An ED string is a concatenation of
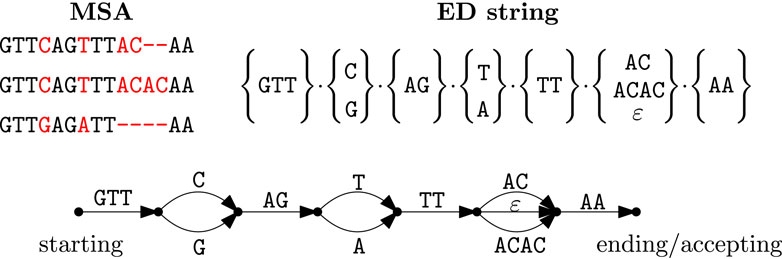
Figure 1. An example of an MSA (top left) and its corresponding (non-unique) ED string
Any pangenome representation aims to improve the downstream analyses of genomic data by removing biases inherent in the use of a linear single-genome representation (Baaijens et al., 2022). For example, pangenomes allow for representing haplotype-resolved data with genome phasing, as shown in Bonizzoni et al. (2016). Using linear genomes as a reference, determining at which chromosomal copy (i.e., paternal or maternal) the different alleles are located, may be erroneous or incomplete due to reference bias. Genotyping (the task of reconstructing the allele variants that characterize an individual) can be improved by using a pangenome representation as a reference, which removes the bias of using a single linear genome as a reference to map the reads coming from an individual’s sample. Pangenomes also allow for accurate read alignment as certain genome regions are important yet challenging to assemble using classic read alignment tools, because of the bias of using a single linear reference genome (Garrison et al., 2018b; Liao et al., 2023). This explains the high level of attention paid in recent years to the task of sequence-to-graph comparison; see, e.g., Büchler et al. (2023); Equi et al. (2023); Mwaniki et al. (2023); Li et al. (2020); Gibney et al. (2022); Jain et al. (2020); Rautiainen et al. (2019); Rautiainen and Marschal (2020). In phylogenetic analyses, the aim is to study the evolutionary relationships among different groups of organisms (e.g., species or population variants). This was traditionally performed using a sample sequence per organism that is somewhat representative of the group or population, and then inferring a tree or a graph based on pairwise distances or similarities among these samples. This task can be biased if the sample linear genome turns out not to be the most representative, whereas a pangenome can compactly represent the entire population.
Our Contributions. Here, we make an important step from the above-mentioned sequence-to-graph (i.e., sequence-to-pangenome) comparison towards graph-to-graph (pangenome-to-pangenome) comparison1. In particular, we assume that the two pangenomes to be compared are represented by means of two ED strings:
The output is the Matching Statistics array
Both distance measures can be trivially computed in
We also implemented the entire pipeline in C++ as a software tool for pangenome comparison, which is publicly available at https://github.com/urbanslug/junctions under GNU GPL v3.0.
We evaluated the efficiency and effectiveness of the developed methods using both synthetic and real datasets. For efficiency, we compared the runtime of the intersection graph against the classic product automaton construction. As expected, the intersection graph is faster by up to one order of magnitude. For effectiveness, we used real SARS-CoV-2 datasets and our Breakpoint Matching Statistics method to reproduce a well-established clades classification of SARS-CoV-2, thus demonstrating that the classification obtained by our method is in accordance with the existing classification.
In Section 2, we describe and analyze our methods. In Section 3, we present our results. We conclude this paper in Section 4.
2 Methods
In this section we recall from Gabory et al. (2023) the ED String Intersection problem for two ED strings (Section 2.1) and the notion of intersection graph (Section 2.2). We then extend the Matching Statistics problem on two standard strings (Gusfield, 1997) to two ED strings defining the ED Matching Statistics and Breakpoint Matching Statistics problems, and show how to solve them using an intersection graph (Section 2.3). We also formally define our similarity and distance measures for ED strings (Section 2.4).
Let us begin with some basic definitions and notations from Gabory et al. (2023). An alphabet
2.1 ED strings intersection
Let us start by formally defining the ED String Intersection problem.
ED String Intersection (EDSI)
Input: Two ED strings,
Output: YES if
This EDSI problem can be efficiently solved using the notion of compacted nondeterministic finite automaton (compacted NFA). A compacted NFA is a 5-tuple
We next define the path-automaton of an ED string.
Definition 1. (Path-automaton of an ED string). Let
In the following, we are interested only in the graph underlying the path-automaton, that is, the directed acyclic graph (DAG), where every node represents an explicit state and every labeled directed edge represents an extended transition of the path-automaton (inspect also Figure 1). Indeed, it should be noticed that the path-automata of the ED strings are DAGs (direct acyclic graphs) as they are always acyclic, but may contain
Checking whether two NFA have a nonempty intersection can be done in
Lemma 1. Gabory et al. (2023). Given two compacted NFA
Lemma 1 thus implies the following result.
Corollary 1. The compacted NFA representing the intersection of two path-automata with
Consequently, we can compute the compacted NFA of the intersection of two ED strings
2.2 The intersection graph
In this section we describe the notion of the intersection graph
Let us consider the two ED strings
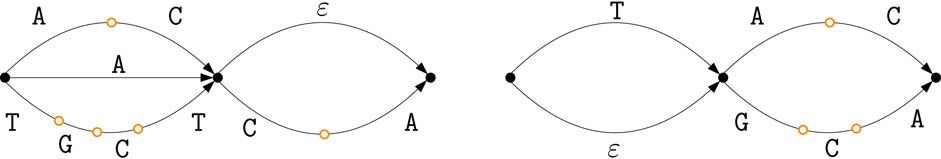
Figure 2. The two DAGs
The nodes of the intersection graph correspond to pairs
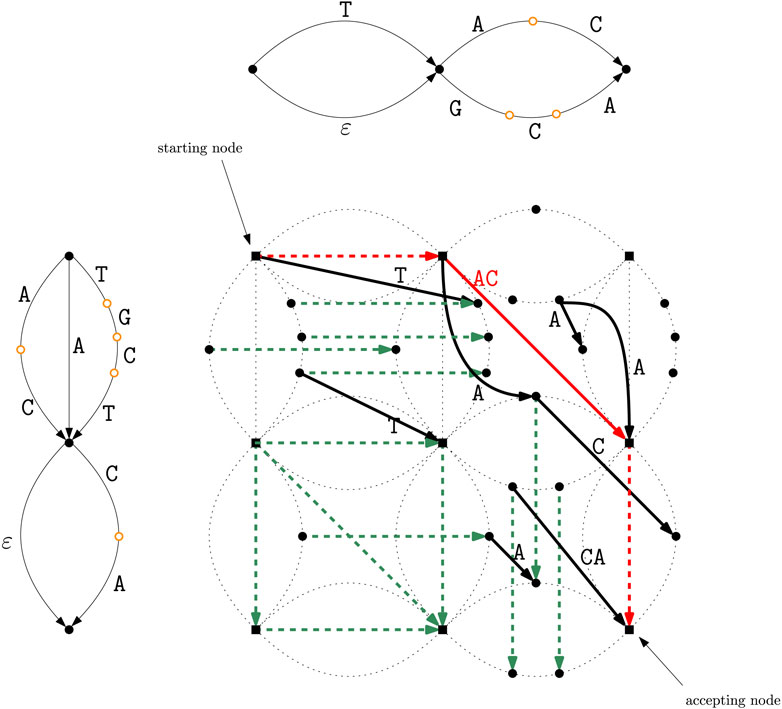
Figure 3. Intersection graph
2.3 Matching statistics for ED strings
When the ED strings
Let us first recall the classic Matching Statistics problem on standard strings.
Matching Statistics
Input: Two strings,
Output: For each
For example, the Matching Statistics of
2.3.1 ED matching statistics
The ED Matching Statistics between two ED strings
ED Matching Statistics
Input: Two ED strings,
Output: For each
Example 1. Let us consider again
We observe that, in the intersection graph
We consider a slightly augmented version of the intersection graph obtained from an uncompacted intersection automaton. We first construct the automaton as in Corollary 1, and then we additionally compute some extra nodes and transitions as follows: when we process a state corresponding to a pair
We observe that, even in this case, the overall number of the transition pair checks remains the same; therefore the total size of the constructed underlying graph
We then assign to each edge the weight
This ends the description of the proposed algorithm for the computation of ED Matching Statistics, which proves the following result.
Theorem 1. The ED MATCHING STATISTICS problem can be solved in
Figure 4 shows how the Matching Statistics of
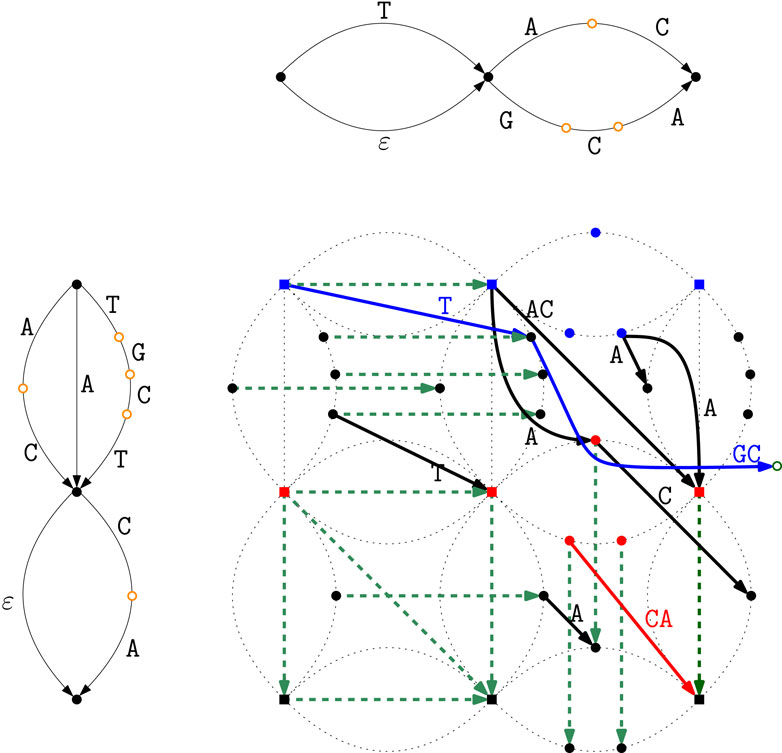
Figure 4. Matching Statistics of
2.3.2 Breakpoint Matching Statistics
The Breakpoint Matching Statistics between two ED strings
Formally, for any two ED strings,
A string
Let us now formalize the problem of computing the Breakpoint Matching Statistics that we solve in this section.
Breakpoint Matching Statistics
Input: Two ED strings,
Output: For each
The motivation for allowing a left-extension to an lb-match and not forcing
Example 2. Let
We now show that the intersection graph
Figure 5 shows the computation of the Breakpoint Matching Statistics in the intersection graph
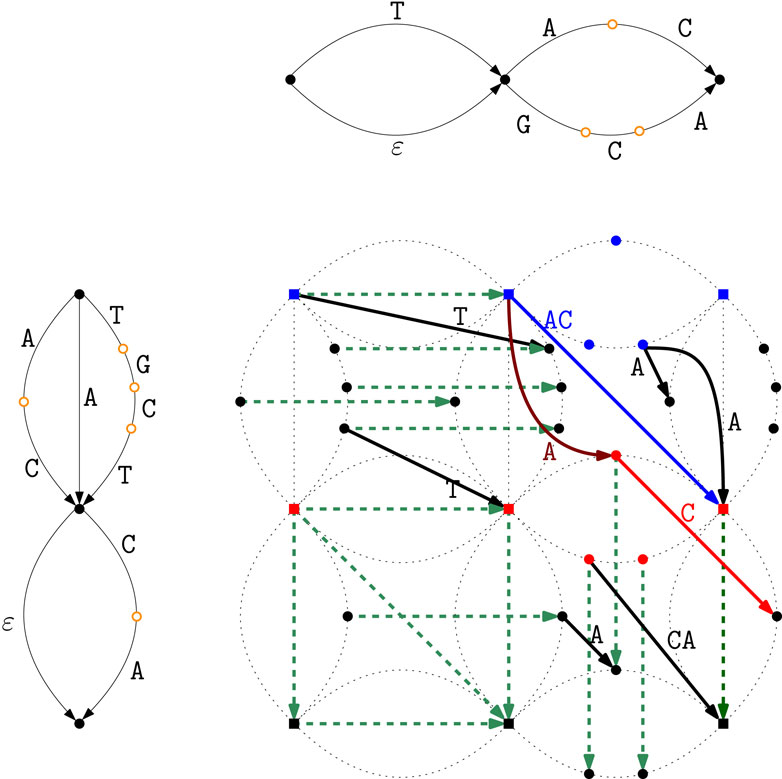
Figure 5. Breakpoint Matching Statistics computation in the intersection graph
Notice that the Breakpoint Matching Statistics require more restricted matches tī ED Matching Statistics. Indeed we have that for any two ED strings
Theorem 2. The Breakpoint Matching Statistics problem can be solved in
2.4 Our measures for comparing pangenomes
In this section we describe a similarity and a distance measure for pangenome comparison. These measures can be derived from either the
We consider both arrays
and
We now move further in order to design a notion of distance between pangenomes based on
and
thus guaranteeing that
and
Our
3 Experiments
We implemented the
3.1 Efficiency on simulated data
In this section, we compare the running time of our EDSI with that of the naïve method and with the parameters that define the characteristics of the input ED strings, with the purpose of validating the time efficiency of our algorithm and show how it actually improves in practice with respect to the baseline quadratic running time. In order to do that, we use synthetic data generated on the alphabet
The synthetic ED strings were generated using another tool of ours named SimED (https://github.com/urbanslug/simed). The tool starts by generating a random standard string of length
As aforementioned, the tool first generates a standard string uniformly at random, which we denote by
Starting from the same base sequence
We report a table for each set of parameters, and in each table, we show the data for different starting synthetic string lengths
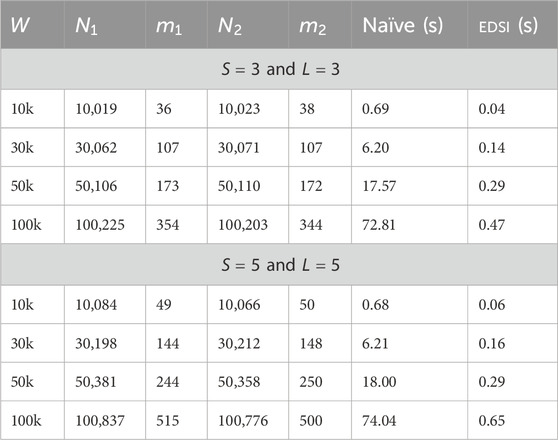
Table 1. Results with simulation parameters:
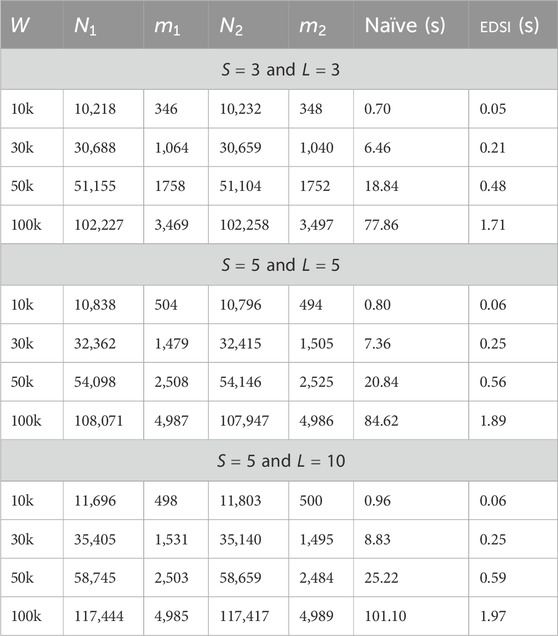
Table 2. Simulation parameters:
The tables show that the Naïve scales quadratically in the size of the ED strings while EDSI is much faster as
These experiments were conducted on a laptop with a 64 bit 11th Gen Intel(R) Core(TM) i7-11800H 8 core processor and 16 GB of RAM. Timings were recorded using std::chrono from the C++ standard library.
3.2 Efficiency on human genome data
In this section, we present some experiments for the running time of EDSI on real human genome data with variants. The goal is to show that our tool is fast even on real data, as the ratio between
The human leukocyte antigen (HLA) gene is contained in the major histocompatibility complex on the p arm (chromosomal region 6p21.33) of Chromosome VI which is known to be one of the most polymorphic regions of the human genome. The HLA gene is involved in immune system regulation (Crux and Elahi, 2017; Romero-Sánchez et al., 2021) and is found in the region between positions 31,353,872 and 31,367,067 (hence it is 13 kb long). ACTB is a highly conserved gene in humans that codes for several proteins involved in cell structure and integrity. For each genome fragment (HLA and ACTB data), and for each database (out of the three 1000G, TOPmed, and gnomAD), we selected from the .vcf file only the variants that are recorded in that specific database, and we updated the regions containing variation, as denoted in the.vcf file into sets containing both the original in the reference and the variants contained in.vcf, thus creating an ED string. We performed this in two ways: one for all variants of that database for that genome fragment, and one by selecting the SNPs variants only. We then used AEDSO (https://github.com/urbanslug/aedso) to build the ED strings. Data download links and commands used are available at https://github.com/urbanslug/junctions/blob/master/test_data/human.org.
In summary, we have two ED strings (one with all variants and one with SNPs only) per each database, and each genome fragment. We remark that all of these ED strings include the original non-mutated string in the language; hence for each pair the intersection is nonempty. This ensures detecting a running time of EDSI without a premature ending due to empty intersection: we ran EDSI for all pairs. For the HLA data, Table 3 shows the sizes (and types) of the ED strings and the running times of a few of these experiments. Table 4 shows results for the ACTB data. We observe that EDSI improves over Naïve by one order of magnitude whenever

Table 3. ED strings with databases and VCF and sizes, and time (in seconds) required by Naïve and by EDSI for HLA data.

Table 4. ED strings with databases and VCF and sizes, and time (in seconds) required by Naïve and by EDSI for ACTB data.
Finally, to conduct an experiment on these data with larger inputs, we picked a larger fragment of reference from Chromosome VI (spanning over the HLA region) of length 100Kb, and we repeated the same procedure as above. Table 5 presents the results of the experiment. We observe that even for these longer ED strings, EDSI is generally significantly faster than the Naïve method, especially on data such as that of the 1000G variants dataset–therein the ratio between
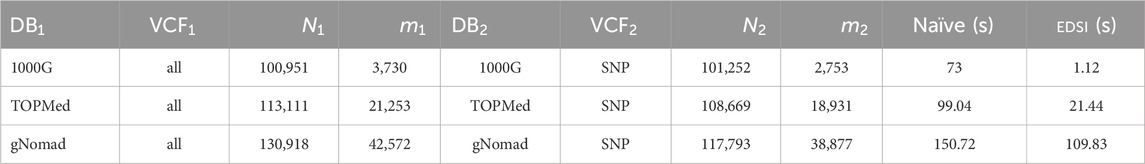
Table 5. ED strings with databases and VCF and sizes, and time (in seconds) required by Naïve and by EDSI for a fragment of data.
These experiments were conducted on a laptop with a 64 bit 11th Gen Intel(R) Core(TM) i7-11800H 8 core processor and 16 GB of RAM. Timings were recorded using std::chrono from the C++ standard library.
3.3 Similarity of SARS-CoV-2 clades
To demonstrate the effectiveness of the Breakpoint Matching Statistics and the similarity measure based on them, we computed the
We selected 35 SARS-CoV-2 clades as classified by NextStrain in https://nextstrain.org/ncov/open/global/alltime (2024) and, within each of them, we downloaded randomly selected individual genome samples (10 when available, and less otherwise), ruling out a few clades with too few samples: we were left with 31 clades. We provide the raw datasets at https://github.com/urbanslug/junctions/tree/master/test/_data/SARS/_CoV/_2.
For each clade, we constructed a multiple sequence alignment (MSA) using abPOA (Gao et al., 2020) and, from each such MSA, we generated the corresponding ED string using msa2eds7. Our tool msa2eds constructs ED strings from an MSA by simply collapsing 100% conserved columns (that is, columns with the same letter in all variants) into solid letters in the ED string, and into sets of distinct variants otherwise.
For all pairs
Figure 6 shows the graph generated using NetworkX’s spring_layout toolkit (Hagberg et al., 2008) when given all pairwise
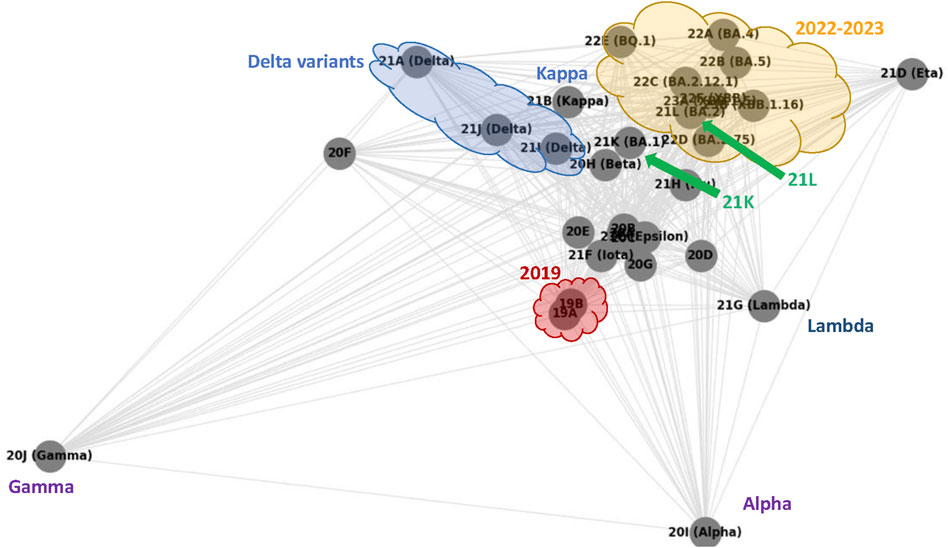
Figure 6. SARS-CoV-2 clades pairwise similarity graph generated according to average Breakpoint Matching Statistics. The annotation (all non grey nor black graphics and text) highlights similarities with Figure 7.
As our annotation shows, the graph in Figure 6 sketches a phylogenetic network of SARS-CoV-2 clades that essentially reproduces the NextStrain phylogeny shown in Figure 7. The former is a complete graph in which all edges are present, and their length is related to our similarity measure (the closer, the more similar); the latter is a typical unrooted phylogenetic tree structure in which clades similarities group subpopulations into subtrees (the closer the common ancestor is, the more similar).
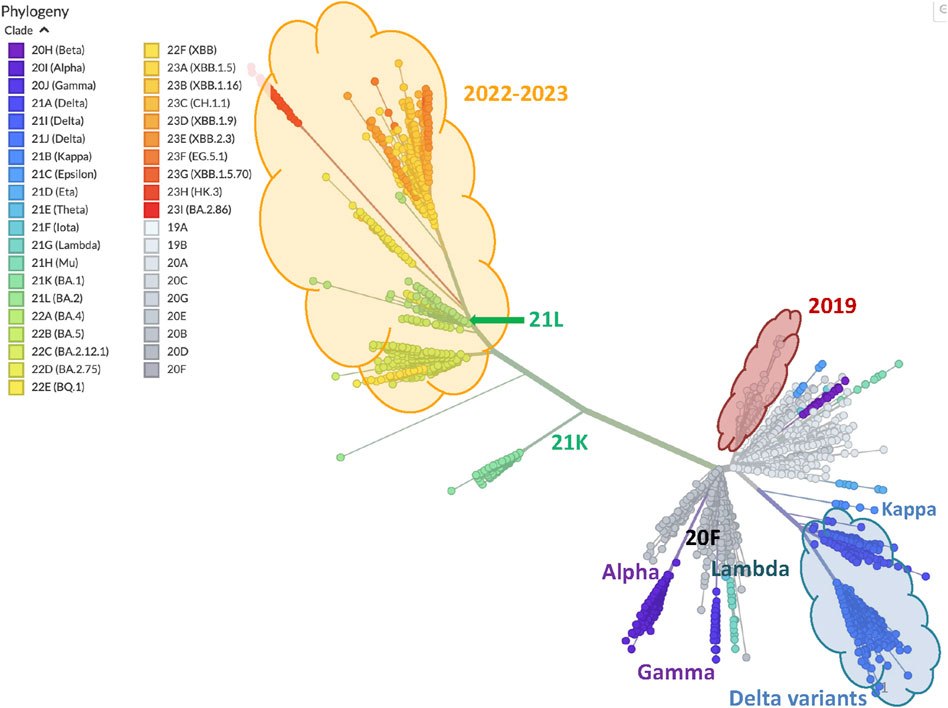
Figure 7. Phylogeny of 3357 SARS-CoV-2 genomes samples. The figure is generated and downloaded from Nextstrain https://nextstrain.org/ncov/open/global/all-time (2024), and some annotation is added here to highlight similarities with the graph of Figure 6.
Thus, we can conclude that our
4 Future work
We plan to investigate methods for local comparison of ED strings, that is, devising efficient methods to find fragments that are common to two or more ED strings (like the fragments we detect with Matching Statistics) but that are not necessarily identical in all of their occurrences (unlike those we detect with Matching Statistics). This could be achieved by means of a preliminary preprocessing filtering step such as those of Peterlongo et al. (2009) for edit distance and Peterlongo et al. (2005), (2008) for Hamming distance. This filtering step could possibly be paired with suitable notions of maximality in frequency (Federico and Pisanti, 2009) or in conservation (Grossi et al., 2009; 2011) for the common fragments in order to avoid redundancy in the output results.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
EG: Writing–original draft, Writing–review and editing. MM: Writing–original draft, Writing–review and editing. NP: Writing–original draft, Writing–review and editing. SP: Writing–original draft, Writing–review and editing. JR: Writing–original draft, Writing–review and editing. MS: Writing–original draft, Writing–review and editing. WZ: Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was partially supported by the PANGAIA, ALPACA and NETWORKS projects that have received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreements No. 872539, 956229 and 101034253, respectively. Nadia Pisanti was partially supported by MUR PRIN 2022 YRB97K PINC and by NextGeneration EU programme PNRR ECS00000017 Tuscany Health Ecosystem. Jakub Radoszewski was supported by the Polish National Science Center, grant no. 2022/46/E/ST6/00463.
Acknowledgments
We are also very grateful to NextStrain for maintaining and allowing access to their platform and the reports therein.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
1A preliminary step on degenerate strings (D strings), that is, a restricted version of ED strings, was made by Alzamel et al. (2018), (2020).
2A breakpoint in a genome is a location on a chromosome where DNA might get deleted, inverted, or swapped around Sankoff and Blanchette (1998).
3This can be done using the folklore product automaton construction (Lawson, 2004).
4We assume that
5For a nonempty
6https://nextstrain.org/ncov/open/global/all-time
7https://github.com/urbanslug/junctions
8A complete table with all pairwise
9Precise measurements are listed here: https://github.com/urbanslug/junctions/blob/master/test_data/covid.org.
References
Alzamel, M., Ayad, L. A. K., Bernardini, G., Grossi, R., Iliopoulos, C. S., Pisanti, N., et al. (2018). “Degenerate string comparison and applications,”. 18th international workshop on algorithms in bioinformatics, WABI 2018, August 20-22, 2018, Helsinki, Finland. Editors L. Parida, and E. Ukkonen (Schloss Dagstuhl: LIPIcs), 21, 1–21:14. 113 of LIPIcs. doi:10.4230/LIPIcs.WABI.2018.21
Alzamel, M., Ayad, L. A. K., Bernardini, G., Grossi, R., Iliopoulos, C. S., Pisanti, N., et al. (2020). Comparing degenerate strings. Fundam. Inf. 175, 41–58. doi:10.3233/FI-2020-1947
Aoyama, K., Nakashima, Y., I, T., Inenaga, S., Bannai, H., and Takeda, M. (2018). “Faster online elastic degenerate string matching,”. Annual symposium on combinatorial pattern matching, CPM 2018, july 2-4, 2018 - qingdao, China. Editors G. Navarro, D. Sankoff, and B. Zhu (Schloss Dagstuhl: LIPIcs), 9, 1–9:10. doi:10.4230/LIPIcs.CPM.2018.9
Apostolico, A., Guerra, C., Landau, G. M., and Pizzi, C. (2016). Sequence similarity measures based on bounded hamming distance. Theor. Comput. Sci. 638, 76–90. doi:10.1016/J.TCS.2016.01.023
Apostolico, A., Guerra, C., and Pizzi, C. (2014). “Alignment free sequence similarity with bounded hamming distance,” in Data compression conference, DCC 2014, snowbird, UT, USA, 26-28 march, 2014. Editors A. Bilgin, M. W. Marcellin, J. Serra-Sagristà, and J. A. Storer (IEEE), 183–192. doi:10.1109/DCC.2014.57
Baaijens, J. A., Bonizzoni, P., Boucher, C., Vedova, G. D., Pirola, Y., Rizzi, R., et al. (2022). Computational graph pangenomics: a tutorial on data structures and their applications. Nat. Comput. 21, 81–108. doi:10.1007/s11047-022-09882-6
Baudet, C., Lemaitre, C., Dias, Z., Gautier, C., Tannier, E., and Sagot, M. (2010). Cassis: detection of genomic rearrangement breakpoints. Bioinform 26, 1897–1898. doi:10.1093/bioinformatics/btq301
Bernardini, G., Gawrychowski, P., Pisanti, N., Pissis, S. P., and Rosone, G. (2019). “Even faster elastic-degenerate string matching via fast matrix multiplication,”. 46th international colloquium on automata, languages, and programming, ICALP 2019, july 9-12, 2019, patras, Greece. Editors C. Baier, I. Chatzigiannakis, P. Flocchini, and S. Leonardi (Schloss Dagstuhl: LIPIcs), 21, 1–21:15. doi:10.4230/LIPIcs.ICALP.2019.21
Bernardini, G., Gawrychowski, P., Pisanti, N., Pissis, S. P., and Rosone, G. (2022). Elastic-degenerate string matching via fast matrix multiplication. SIAM J. Comput. 51, 549–576. doi:10.1137/20M1368033
Bernardini, G., Pisanti, N., Pissis, S., and Rosone, G. (2017). “Pattern matching on elastic-degenerate text with errors,” in 24th international symposium on string processing and information retrieval (SPIRE), 74–90. doi:10.1007/978-3-319-67428-5_7
Bernardini, G., Pisanti, N., Pissis, S. P., and Rosone, G. (2020). Approximate pattern matching on elastic-degenerate text. Theor. Comput. Sci. 812, 109–122. doi:10.1016/j.tcs.2019.08.012
Bonizzoni, P., Dondi, R., Klau, G. W., Pirola, Y., Pisanti, N., and Zaccaria, S. (2016). On the minimum error correction problem for haplotype assembly in diploid and polyploid genomes. J. Comput. Biol. 23, 718–736. doi:10.1089/cmb.2015.0220
Büchler, T., Olbrich, J., and Ohlebusch, E. (2023). Efficient short read mapping to a pangenome that is represented by a graph of ED strings. Bioinformatics 39, btad320. doi:10.1093/bioinformatics/btad320
Carletti, V., Foggia, P., Garrison, E., Greco, L., Ritrovato, P., and Vento, M. (2019). “Graph-based representations for supporting genome data analysis and visualization: opportunities and challenges,” in Graph-based representations in pattern recognition - 12th IAPR-TC-15 international workshop, GbRPR 2019, tours, France, june 19-21, 2019, proceedings. Editors D. Conte, J. Ramel, and P. Foggia (Springer), 237–246. 11510 of Lecture Notes in Computer Science. doi:10.1007/978-3-030-20081-7_23
Cisłak, A., Grabowski, S., and Holub, J. (2018). SOPanG: online text searching over a pan-genome. Bioinformatics 34, 4290–4292. doi:10.1093/bioinformatics/bty506
Crux, N. B., and Elahi, S. (2017). Human leukocyte antigen (HLA) and immune regulation: how do classical and non-classical HLA alleles modulate immune response to human immunodeficiency virus and hepatitis C virus infections? Front. Immunol. 8, 832. doi:10.3389/fimmu.2017.00832
Eizenga, J. M., Novak, A. M., Kobayashi, E., Villani, F., Cisar, C., Heumos, S., et al. (2021). Efficient dynamic variation graphs. Bioinform 36, 5139–5144. doi:10.1093/bioinformatics/btaa640
Equi, M., Mäkinen, V., Tomescu, A. I., and Grossi, R. (2023). On the complexity of string matching for graphs. ACM Trans. Algorithms 19 (21), 1–25. doi:10.1145/3588334
Federico, M., and Pisanti, N. (2009). Suffix tree characterization of maximal motifs in biological sequences. Theor. Comput. Sci. 410, 4391–4401. doi:10.1016/J.TCS.2009.07.020
Gabory, E., Mwaniki, N. M., Pisanti, N., Pissis, S. P., Radoszewski, J., Sweering, M., et al. (2023). “Comparing elastic-degenerate strings: algorithms, lower bounds, and applications,”. 34th annual symposium on combinatorial pattern matching, CPM 2023, june 26-28, 2023, marne-la-vallée, France. Editors L. Bulteau, and Z. Lipták (Schloss Dagstuhl: LIPIcs), 11, 1–1120. doi:10.4230/LIPIcs.CPM.2023.11
Gao, Y., Liu, Y., Ma, Y., Liu, B., Wang, Y., and Xing, Y. (2020). abPOA: an SIMD-based C library for fast partial order alignment using adaptive band. Bioinformatics 37, 2209–2211. doi:10.1093/bioinformatics/btaa963
Garrison, E., Sirén, J., Novak, A. M., Hickey, G., Eizenga, J. M., Dawson, E. T., et al. (2018a). Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 36, 875–879. doi:10.1038/nbt.4227
Garrison, E., Sirén, J., Novak, A. M., Hickey, G., Eizenga, J. M., Dawson, E. T., et al. (2018b). Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 36, 875–879. doi:10.1038/nbt.4227
Gibney, D., Thankachan, S. V., and Aluru, S. (2022). On the hardness of sequence alignment on de bruijn graphs. J. Comput. Biol. 29, 1377–1396. doi:10.1089/cmb.2022.0411
Grossi, R., Iliopoulos, C. S., Liu, C., Pisanti, N., Pissis, S. P., Retha, A., et al. (2017). “On-line pattern matching on similar texts,”. 28th annual symposium on combinatorial pattern matching, CPM 2017, july 4-6, 2017, Warsaw, Poland. Editors J. Kärkkäinen, J. Radoszewski, and W. Rytter (Schloss Dagstuhl: LIPIcs), 9, 1–9:14. doi:10.4230/LIPIcs.CPM.2017.9
Grossi, R., Pietracaprina, A., Pisanti, N., Pucci, G., Upfal, E., and Vandin, F. (2009). “MADMX: a novel strategy for maximal dense motif extraction,” in Algorithms in bioinformatics, 9th international workshop, WABI 2009, Philadelphia, PA, USA, september 12-13, 2009. Proceedings. Editors S. Salzberg, and T. J. Warnow (Springer), 362–374. 5724 of Lecture Notes in Computer Science. doi:10.1007/978-3-642-04241-6_30
Grossi, R., Pietracaprina, A., Pisanti, N., Pucci, G., Upfal, E., and Vandin, F. (2011). MADMX: a strategy for maximal dense motif extraction. J. Comput. Biol. 18, 535–545. doi:10.1089/CMB.2010.0177
Gusfield, D. (1997). Algorithms on strings, trees, and sequences - computer science and computational biology. Cambridge University Press. doi:10.1017/cbo9780511574931
Hadfield, J., Megill, C., Bell, S. M., Huddleston, J., Potter, B., Callender, C., et al. (2018). Nextstrain: real-time tracking of pathogen evolution. Bioinform 34, 4121–4123. doi:10.1093/bioinformatics/bty407
Hagberg, A. A., Schult, D. A., and Swart, P. J. (2008). “Exploring network structure, dynamics, and function using networkx,” in Proceedings of the 7th Python in science conference. Editors G. Varoquaux, T. Vaught, and J. Millman (Pasadena, CA USA), 11–15.
Hopcroft, J. E., Motwani, R., and Ullman, J. D. (2003). Introduction to automata theory, languages, and computation - international edition. 2nd Edition. Addison-Wesley.
Iliopoulos, C. S., Kundu, R., and Pissis, S. P. (2021). Efficient pattern matching in elastic-degenerate strings. Inf. Comput. 279, 104616. doi:10.1016/j.ic.2020.104616
Jain, C., Zhang, H., Gao, Y., and Aluru, S. (2020). On the complexity of sequence-to-graph alignment. J. Comput. Biol. 27, 640–654. doi:10.1089/cmb.2019.0066
Leimeister, C., and Morgenstern, B. (2014). kmacs: the k-mismatch average common substring approach to alignment-free sequence comparison. Bioinform 30, 2000–2008. doi:10.1093/bioinformatics/btu331
Li, H., Feng, X., and Chu, C. (2020). The design and construction of reference pangenome graphs with minigraph. Genome Biol. 21, 265. doi:10.1186/s13059-020-02168-z
Liao, W.-W., Asri, M., Ebler, J., Doerr, D., Haukness, M., Hickey, G., et al. (2023). A draft human pangenome reference. Nature 617, 312–324. doi:10.1038/s41586-023-05896-x
Mwaniki, N. M., Garrison, E., and Pisanti, N. (2023). “Fast exact string to D-texts alignments,” in Proceedings of the 16th international joint conference on biomedical engineering systems and Technologies, BIOSTEC 2023, volume 3: BIOINFORMATICS, Lisbon, Portugal, february 16-18, 2023. Editors H. Ali, N. Deng, A. L. N. Fred, and H. Gamboa, 70–79. doi:10.5220/0011666900003414
Mwaniki, N. M., and Pisanti, N. (2022). “Optimal sequence alignment to ED-strings,” in Bioinformatics research and applications - 18th international symposium, ISBRA 2022, haifa, Israel, november 14-17, 2022, proceedings. Editors M. S. Bansal, Z. Cai, and S. Mangul (Springer), 204–216. 13760 of Lecture Notes in Computer Science. doi:10.1007/978-3-031-23198-8_19
Paten, B., Novak, A. M., Eizenga, J. M., and Garrison, E. (2017). Genome graphs and the evolution of genome inference. Genome Res. 27, 665–676. doi:10.1101/gr.214155.116
Peterlongo, P., Pisanti, N., Boyer, F., do Lago, A. P., and Sagot, M. (2008). Lossless filter for multiple repetitions with hamming distance. J. Discrete Algorithms 6, 497–509. doi:10.1016/J.JDA.2007.03.003
Peterlongo, P., Pisanti, N., Boyer, F., and Sagot, M. (2005). “Lossless filter for finding long multiple approximate repetitions using a new data structure, the bi-factor array,” in String processing and information retrieval, 12th international conference, SPIRE 2005, Buenos Aires, Argentina, november 2-4, 2005, proceedings. Editors M. P. Consens, and G. Navarro (Springer), 179–190. 3772 of Lecture Notes in Computer Science. doi:10.1007/11575832_20
Peterlongo, P., Sacomoto, G. A. T., do Lago, A. P., Pisanti, N., and Sagot, M. (2009). Lossless filter for multiple repeats with bounded edit distance. Algorithms Mol. Biol. 4, 3. doi:10.1186/1748-7188-4-3
Pissis, S. P., and Retha, A. (2018). “Dictionary matching in elastic-degenerate texts with applications in searching VCF files on-line,”. 17th international symposium on experimental algorithms, SEA 2018, june 27-29, 2018, L’aquila, Italy. Editor G. D’Angelo (Schloss Dagstuhl: LIPIcs), 16, 1–16:14. doi:10.4230/LIPIcs.SEA.2018.16
Pizzi, C. (2016). Missmax: alignment-free sequence comparison with mismatches through filtering and heuristics. Algorithms Mol. Biol. 11, 6. doi:10.1186/S13015-016-0072-X
Rakocevic, G., Semenyuk, V., Lee, W.-P., Spencer, J., Browning, J., Johnson, I. J., et al. (2019). Fast and accurate genomic analyses using genome graphs. Nat. Genet. 51, 354–362. doi:10.1038/s41588-018-0316-4
Rautiainen, M., Mäkinen, V., and Marschall, T. (2019). Bit-parallel sequence-to-graph alignment. Bioinform 35, 3599–3607. doi:10.1093/bioinformatics/btz162
Rautiainen, M., and Marschal, T. (2020). GraphAligner: rapid and versatile sequence-to-graph alignment. Genome Biol. 21, 253. doi:10.1186/s13059-020-02157-2
Romero-Sánchez, C., Hernández, N., Chila-Moreno, L., Jiménez, K., Padilla, D., Bello-Gualtero, J. M., et al. (2021). HLA-B allele, genotype, and haplotype frequencies in a group of healthy individuals in Colombia. J. Clin. Rheumatol. 27, S148–S152. doi:10.1097/rhu.0000000000001671
Sankoff, D., and Blanchette, M. (1998). Multiple genome rearrangement and breakpoint phylogeny. J. Comput. Biol. 5, 555–570. doi:10.1089/cmb.1998.5.555
Thankachan, S. V., Chockalingam, S. P., Liu, Y., Krishnan, A., and Aluru, S. (2017). A greedy alignment-free distance estimator for phylogenetic inference. BMC Bioinform 18 (238), 238–8. doi:10.1186/s12859-017-1658-0
The Computational Pan-Genomics Consortium (2018). Computational pan-genomics: status, promises and challenges. Briefings Bioinforma. 19, 118–135. doi:10.1093/bib/bbw089
Keywords: elastic-degenerate string, intersection graph, pangenome comparison, matching statistics, SARS-CoV-2
Citation: Gabory E, Mwaniki MN, Pisanti N, Pissis SP, Radoszewski J, Sweering M and Zuba W (2024) Pangenome comparison via ED strings. Front. Bioinform. 4:1397036. doi: 10.3389/fbinf.2024.1397036
Received: 06 March 2024; Accepted: 23 August 2024;
Published: 26 September 2024.
Edited by:
Leena Salmela, University of Helsinki, FinlandReviewed by:
Jing Li, Integrated DNA Technologies, United StatesCinzia Pizzi, University of Padua, Italy
Copyright © 2024 Gabory, Mwaniki, Pisanti, Pissis, Radoszewski, Sweering and Zuba. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nadia Pisanti, bmFkaWEucGlzYW50aUB1bmlwaS5pdA==