 Matteo Delucchi
Matteo Delucchi Paulina Näf
Paulina Näf Spencer Bliven
Spencer Bliven Maria Anisimova
Maria Anisimova
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
TECHNOLOGY AND CODE article
Front. Bioinform. , 25 June 2021
Sec. Protein Bioinformatics
Volume 1 - 2021 | https://doi.org/10.3389/fbinf.2021.691865
This article is part of the Research Topic Tandem Repeats: Methods and Roles in Protein Function and Evolution View all 4 articles
The Tandem Repeat Annotation Library (TRAL) focuses on analyzing tandem repeat units in genomic sequences. TRAL can integrate and harmonize tandem repeat annotations from a large number of external tools, and provides a statistical model for evaluating and filtering the detected repeats. TRAL version 2.0 includes new features such as a module for identifying repeats from circular profile hidden Markov models, a new repeat alignment method based on the progressive Poisson Indel Process, an improved installation procedure and a docker container. TRAL is an open-source Python 3 library and is available, together with documentation and tutorials viavital-it.ch/software/tral.
Recent years have seen an increased awareness of the importance of genomic tandem repeats (TRs) as functional features. TRs are adjacent repetitive units in genomic sequences, they are known for their associations with diseases and immune related functions and often play an important role in nucleic acid binding (Kajava, 2012; Delucchi et al., 2020; Gidley and Parmeggiani, 2021). TRs are found in abundance throughout the three domains of life (Marcotte et al., 1999; Delucchi et al., 2020). Many protein TRs fold in specific structures (Bassot and Elofsson, 2021). A significant fraction of TRs, however, remain unstructured. It is expected that new TRs can originate i. a. by replication slippage (Ellegren, 2004) or by duplication of intrinsically disordered regions (Delucchi et al., 2020).
The evolutionary conservation of many TRs supports their functional importance (Schaper et al., 2014; Delucchi et al., 2020; Chakrabarty and Parekh, 2021). For example, over 60% of mammalian TRs were estimated to be up to 300 Mya old, including well-studied repeats such as armadillo repeat proteins (ArmRP), leucine-rich repeats (LRR), HEAT and PHD-finger. Yet, despite strong conservation of some repeats, TR annotation and analysis remain challenging (Bahlo et al., 2018; Paladin et al., 2021; Chakrabarty and Parekh, 2021), particularly if TR units have diverged over time through indels and point mutations, duplication and loss of TR units, recombination, replication slippage and gene conversion e.g., Tørresen et al. (2019). In contrast, short TRs or microsatellites have been attracting attention as they are a rich source of genetic variability. For example, in the human genome these contribute about 3%, more than the entire protein coding sequences and are highly polymorphic [
Tandem Repeat Annotation Library (TRAL) has been developed in order to analyze a variety of in molecular sequences. TRAL implements a variety of tasks for analyzing tandem repeats (Schaper et al., 2015), both in DNA or amino acid sequences. These include: TR annotation using either sequence profiles or de novo TR detectors; identification and filtering of overlapping annotations; filtering by statistical significance; and retrieval of TR characteristics such as TR unit length, number, divergence, indel distribution and TR unit alignments (Schaper et al., 2012). TRAL was shown to be highly successful in the analyses of TRs, for example, relating structure and function of TR units to their evolutionary mode (Schaper et al., 2014). Unlike most other predictors, TRAL allows statistical validation of potential TR candidates based on the evolutionary definition of a tandem repeat. By TRAL’s definition, a TR region is statistically significant if its units share common ancestry (evaluated by testing for homology using a likelihood ratio test). For further details of the method we refer the reader to Anisimova et al. (2015), while Schaper et al. (2015) presents an extensive benchmarking of method’s performance in simulations with and without repeats as well as on real data.
Recently we conducted a large-scale survey of protein TRs, which relied on TRAL for TR annotation (Delucchi et al., 2020). This study highlighted the complexity of TR annotations in proteomic sequences and showed the diversity of their functional roles and origins.
The new release TRAL 2.0 brings new modules to annotate specific TRs across genomes and improved TR filtering by a new scoring function based on the realignment of TR units with an evolutionary aware indel model, the Poission Indel Process (Bouchard-Côté and Jordan, 2013).
Following the requests from our users, we provide many usability improvements, including easy installation and packaging. TRAL depends on numerous external software tools (Table 1). In the previous release, all these packages (with heterogeneous support and development status) had to be installed separately. The installation procedure could be time-consuming and difficult. Our new version provides an “easy setup” procedure, which enables installation of all required external software at once or individually on Linux. Additionally, fully configured TRAL environments are available as either a docker container (github.com/acg-team/tral/packages) or a Vagrant box.
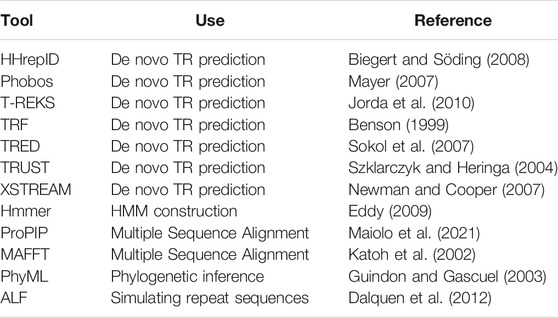
TABLE 1. External Software utilized by TRAL and installed using easy setup.
TRAL is written in Python version 3.6, and is available open-source under the GPL-2.0 license together with the much improved comprehensive documentation and tutorials on: github.com/acg-team/tral. External packages are governed by their own license terms and conditions.
We provide tutorials that explain how to interpret the output of TRAL. Examples are provided to illustrate de novo and cpHMM based TR annotation, statistical evaluation and filtering of the candidate repeats. Further, we provide explanations of the usage of command line tools to retrieve repeats from sequence databases and deal with large sequence data sets.
TRAL has traditionally focused on de novo repeat identification, i.e, without the prior knowledge about a potential TR unit (such as the TR unit length and its profile). This is useful for studying repeats in proteins that are expected to have repeated adjacent units but have not been annotated yet. The new search package provides methods for identifying and analyzing a specific repeat (defined by its amino acid or DNA profile) across one or more genomes. TR units in TRAL are represented using a circular profile hidden Markov model (cpHMM) (Figure 1) (Schaper et al., 2014). These can be generated from de novo searches or from other sources, such as Pfam alignments of known repeats.
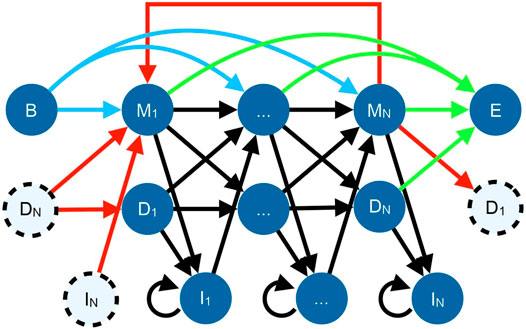
FIGURE 1. State diagram for the cpHMM (Schaper et al., 2014). Black arrows indicate standard profile HMM transitions within a repeat, with each amino acid corresponding to a match (
TRAL systematically detects the query cpHMM in a database of sequences, and allows filtering results by repeat length and statistical significance. A log-odds ratio is reported for each result, giving the cpHMM match probability normalized by the probability of a match to a random sequence. Searches of cpHMMs can be run either programmatically or using a command line tool.
The search module was used to identify members of the armadillo repeat protein family. This 42-residue repeat is well characterized and can be found across metazoan lineages. It forms a conserved alpha-barrel structure (Figure 2A). A cpHMM was constructed based on the alignment in Gul et al. (2017) (Figure 2B). TRAL was used to identify armadillo repeats across a selection of 94 metazoan species. Species were taken from the 15% Representative Proteome Group [2018_03 release; https://proteininformationresource.org/rps/; Chen et al. (2011)].
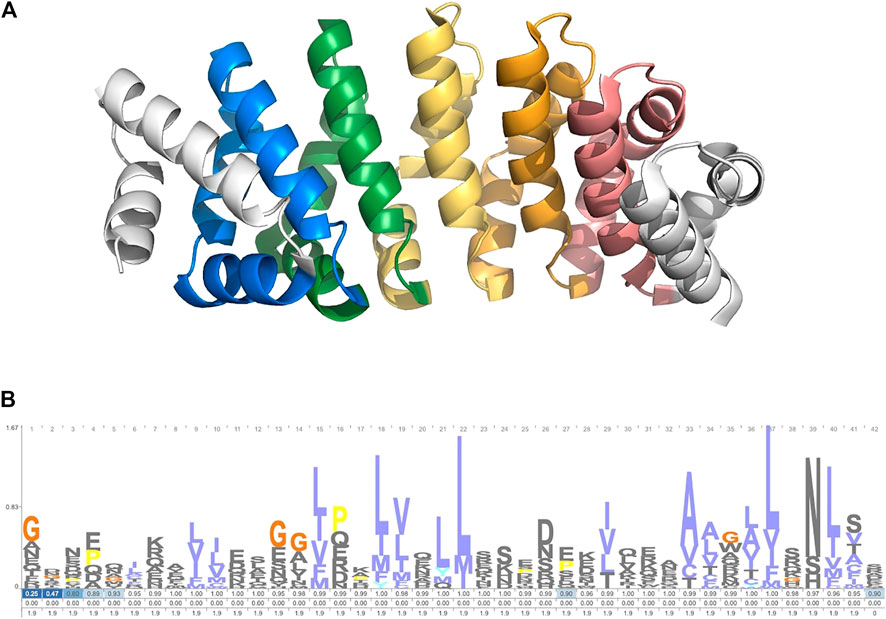
FIGURE 2. (A) Designed ArmRP YIIIM5AII (PDB: 5AEI) (Hansen et al., 2016). (B) Logo showing the cpHMM emmission and transmission probablities. Created with Skylign (Wheeler et al., 2014).
To filter out spurious annotations, TRAL uses scoring functions based on evolution models. Falsely filtering out a true TR may happen due to either unsuitability of the TR scoring function for TR classification, or inaccurate annotation of the predicted TR units, location, or unit alignment. Thus, better TR unit alignments should contribute to improving the TR prediction quality. Unit alignments are relatively short and are often dominated by short indels, making it difficult to accurately annotate TR unit borders. To improve the annotation of borders, we integrated a new alignment method for realigning TR units with an explicit evolutionary indel model - the progressive PIP (Maiolo et al., 2018) based on the PIP model (Bouchard-Côté and Jordan, 2013).
As modeling of indel evolution is computationally challenging, the dynamics of indels are typically not modeled explicitly, e.g., in the popular aligner MAFFT (Katoh et al., 2002), which was already integrated in TRAL to realign TR units.
The realignment of TR units can be performed after detecting TRs (de novo or based on an HMM) or for any given annotated TR. This allows for an iterative refinement of cpHMMs.
The realignment module allows the realignment of any given TR with MAFFT (default) or progressive PIP, ProPIP (Maiolo et al., 2021). When using progressive PIP, a TR unit phylogeny is inferred using PhyML (Guindon et al., 2010) from the initial unit alignment and used as a guide-tree when realigning with progressive PIP. The insertion and deletion rates (λ and μ) are estimated from the initial alignment. By default the indel rate is constant, and a variable indel rate option uses the γ-distribution with the shape parameter
TRAL was able to identify ArmRP members in all 94 species (Supplementary Figure S1). The number identified per species varied between 14 and 170, with a mean of 51 proteins per species (Supplementary Table S1). In humans, 107 ArmRP members were identified. Distinguishing ArmRP from related families such as HEAT proteins is difficult without structural information, but this result is comparable to other databases: Interpro annotates 127 human ArmRP in the “Armadillo” superfamily (IPR000225), while SMART includes 73 domains (SM00185).
To demonstrate the effect of the indel model on TRAL results, we used TRAL with default settings to realign the leucin-rich repeat units from the toll-like receptor TLR2 with MAFFT and ProPIP (Figure 3). The PIP-based alignment was slightly longer compared to MAFFT’s, which is consistent with the “phylogeny-aware” nature of this method preventing “overalignment”. The overall divergence was significantly lower, showing higher residue conservation in gap regions compared to MAFFT. This is consistent with (Maiolo et al., 2018), who showed that progressive PIP method infers gap patterns similar to those inferred by PRANK (Löytynoja, 2014), and also phylogenetically meaningful as supported by empirical studies e.g., Abram et al. (2010). While penalizing indels in aligners remains subjective, including the progressive PIP in TRAL allows an alternative method to be studied, particularly in TRs dominated by short indels.
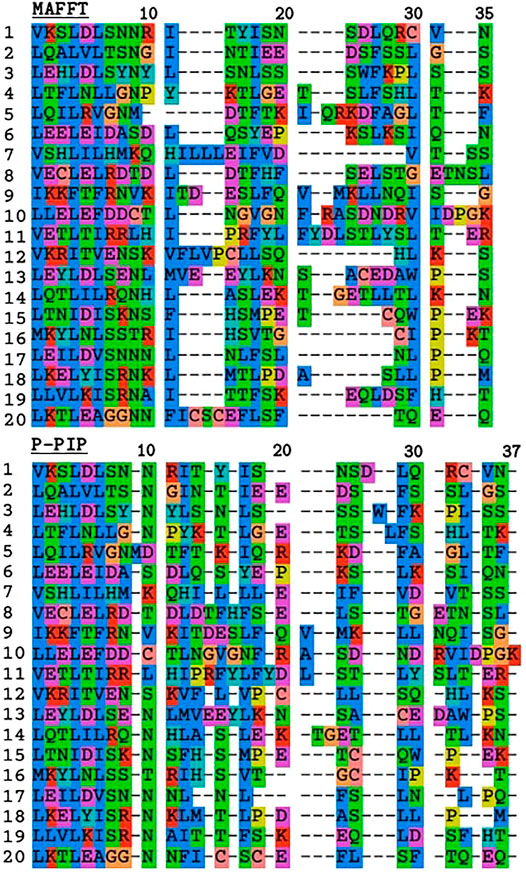
FIGURE 3. Comparison of TR unit alignments inferred with MAFFT and progressive PIP for leucine-rich repeats from TLR2. The first 11 residues correspond to the highly conserved segment with the motif LxxLxLxxNxL where “L” is Leu, Ile, Val or Phe, and “N” is Asn, Thr, Ser, or Cys (Matsushima et al., 2007).
With TRAL version 2.0 the tool became significantly easier to use and gained significant new features. Profile based TR detection is now easy and automated, allowing the investigation of TR defined by a specific query sequence or a provided profile model. The integration of TR unit alignment based on an explicit evolutionary aware indel model should help prevent unit over-alignment, contributing to the increased TR prediction quality. Moreover, the iterated refinement TR annotation allows to consider alternative views of indel evolution affecting the detected TRs.
Some limitations of TRAL remain. TRAL relies heavily on external software for identifying repeats, and many tools struggle with poorly conserved repeats. However, a plugin system exists to allow incorporating future advances in repeat detection into the TRAL framework. The statistical model used in TRAL was primarily used for protein alphabets. While TRAL can also be applied to polynucleotide sequences, the restricted alphabet limits the power such that significance thresholds may need to be adjusted for highly divergent sequences. For protein-coding sequences, in the future, we consider adding a codon alphabet to improve the power of detection. This may also bring additional advantages of being able to analyze protein coding repeats at the level of synonymous DNA changes [e.g., see section 4.3 of Kosiol and Anisimova (2019)].
Augmented with user-friendly documentation, tutorials, the simplified installation and independence of operating systems, TRAL 2.0 addresses a wider range of researchers.
Publicly available datasets were analyzed in this study. This data can be found here: https://github.com/acg-team/tral.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 823886 and SNSF grants CRSII5193832 and 31003A-176316.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Massimo Maiolo at the Zurich University of Applied Sciences for his help with the ProPIP aligner.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbinf.2021.691865/full#supplementary-material
Abram, M. E., Ferris, A. L., Shao, W., Alvord, W. G., and Hughes, S. H. (2010). Nature, Position, and Frequency of Mutations Made in a Single Cycle of HIV-1 Replication. J. Virol. 84, 9864–9878. Publisher: American Society for Microbiology Journals _eprint: https://jvi.asm.org/content/84/19/9864.full.pdf. doi:10.1128/JVI.00915-10
Anisimova, M., Pečerska, J., and Schaper, E. (2015). Statistical Approaches to Detecting and Analyzing Tandem Repeats in Genomic Sequences. Front. Bioeng. Biotechnol. 3, 1–6. doi:10.3389/fbioe.2015.00031
Bahlo, M., Bennett, M. F., Degorski, P., Tankard, R. M., Delatycki, M. B., and Lockhart, P. J. (2018). Recent Advances in the Detection of Repeat Expansions with Short-Read Next-Generation Sequencing. F1000Research 7. doi:10.12688/f1000research.13980.1
Bassot, C., and Elofsson, A. (2021). Accurate Contact-Based Modelling of Repeat Proteins Predicts the Structure of New Repeats Protein Families. PLOS Comput. Biol. 17, e1008798. doi:10.1371/journal.pcbi.1008798Publisher: Public Library of Science
Benson, G. (1999). Tandem Repeats Finder: a Program to Analyze DNA Sequences. Nucleic Acids Res. 27, 573–580. doi:10.1093/nar/27.2.573
Biegert, A., and Söding, J. (2008). De Novo identification of Highly Diverged Protein Repeats by Probabilistic Consistency. Bioinformatics 24, 807–814. doi:10.1093/bioinformatics/btn039
Bouchard-Côté, A., and Jordan, M. I. (2013). Evolutionary Inference via the Poisson Indel Process. Proc. Natl. Acad. Sci. 110, 1160–1166. National Academy of Sciences _eprint: https://www.pnas.org/content/110/4/1160.full.pdf. doi:10.1073/pnas.1220450110
Chakrabarty, B., and Parekh, N. (2021). DbStRiPs: Database of Structural Repeats in Proteins. Protein Sci. doi:10.1002/pro.4052 Available at: https://onlinelibrary.wiley.com/doi/pdf/10.1002/pro.4052
Chen, C., Natale, D. A., Finn, R. D., Huang, H., Zhang, J., Wu, C. H., et al. (2011). Representative Proteomes: A Stable, Scalable and Unbiased Proteome Set for Sequence Analysis and Functional Annotation. PLOS ONE 6, e18910. doi:10.1371/journal.pone.0018910 Publisher: Public Library of Science
Dalquen, D. A., Anisimova, M., Gonnet, G. H., and Dessimoz, C. (2012). ALF—A Simulation Framework for Genome Evolution. Mol. Biol. Evol. 29, 1115–1123. doi:10.1093/molbev/msr268
Delucchi, M., Schaper, E., Sachenkova, O., Elofsson, A., and Anisimova, M. (2020). A New Census of Protein Tandem Repeats and Their Relationship with Intrinsic Disorder. Genes 11, 407. doi:10.3390/genes11040407MDPI
Eddy, S. R. (2009). A New Generation of Homology Search Tools Based on Probabilistic Inference. Genome Informatics. Int. Conf. Genome Inform. 23, 205–211.
Ellegren, H. (2004). Microsatellites: Simple Sequences with Complex Evolution. Nat. Rev. Genet. 5, 435–445. doi:10.1038/nrg1348Nature Publishing Group
Gidley, F., and Parmeggiani, F. (2021). Repeat Proteins: Designing New Shapes and Functions for Solenoid Folds. Curr. Opin. Struct. Biol. 68, 208–214. doi:10.1016/j.sbi.2021.02.002
Giovannucci, E., Stampfer, M. J., Krithivas, K., Brown, M., Brufsky, A., Talcott, J., et al. (1997). The CAG Repeat within the Androgen Receptor Gene and its Relationship to Prostate Cancer. Proc. Natl. Acad. Sci. 94, 3320–3323. doi:10.1073/pnas.94.7.3320 Publisher: National Academy of Sciences Section: Biological Sciences
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 59, 307–321.
Guindon, S., and Gascuel, O. (2003). A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 52, 696–704. doi:10.1080/10635150390235520
Gul, I. S., Hulpiau, P., Saeys, Y., and van Roy, F. (2017). Metazoan Evolution of the Armadillo Repeat Superfamily. Cell Mol. Life Sci. 74, 525–541. doi:10.1007/s00018-016-2319-6
Hansen, S., Tremmel, D., Madhurantakam, C., Reichen, C., Mittl, P. R. E., and Plückthun, A. (2016). Structure and Energetic Contributions of a Designed Modular Peptide-Binding Protein with Picomolar Affinity. J. Am. Chem. Soc. 138, 3526–3532. doi:10.1021/jacs.6b00099 Publisher: American Chemical Society
Jorda, J., Xue, B., Uversky, V. N., and Kajava, A. V. (2010). Protein Tandem Repeats – the More Perfect, the Less Structured. FEBS J. 277, 2673–2682. eprint. https://febs.onlinelibrary.wiley.com/doi/pdf/10.1111/j.1742-4658.2010.07684.x. doi:10.1111/j.1742-4658.2010.07684.x
Kajava, A. V. (2012). Tandem Repeats in Proteins: From Sequence to Structure. J. Struct. Biol. 179, 279–288. doi:10.1016/j.jsb.2011.08.009
Katoh, K., Misawa, K., Kuma, K.-i., and Miyata, T. (2002). MAFFT: a Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Oxford Univ. Press 30, 3059–3066. doi:10.1093/nar/gkf436ISBN: 1362-4962 (Electronic) _eprint: journal.pone.0035671
Kosiol, C., and Anisimova, M. (2019). Selection Acting on Genomes, in Evolutionary Genomics: Statistical and Computational Methods. Editor M. Anisimova. Methods in Molecular Biology (New York, NY: Springer), 373–397. doi:10.1007/978-1-4939-9074-0_12
Löytynoja, A. (2014). Phylogeny-aware Alignment with PRANK. In Multiple Sequence Alignment Methods,. Editor D. J. Russell (Totowa, NJ: Humana Press), 155–170.
Maiolo, M., Gatti, L., Frei, D., Leidi, T., Gil, M., and Anisimova, M. (2021). ProPIP: a Tool for Progressive Multiple Sequence Alignment with Poisson Indel Process. BMC Bioinformatics Accepted pending revisions.
Maiolo, M., Zhang, X., Gil, M., and Anisimova, M. (2018). Progressive Multiple Sequence Alignment with Indel Evolution. BMC Bioinformatics 19, 1–8.
Marcotte, E. M., Pellegrini, M., Yeates, T. O., and Eisenberg, D. (1999). A Census of Protein repeats11Edited by. J. M. Thornton. J. Mol. Biol. 293, 151–160. doi:10.1006/jmbi.1999.3136
Matsushima, N., Tanaka, T., Enkhbayar, P., Mikami, T., Taga, M., Yamada, K., et al. (2007). Comparative Sequence Analysis of Leucine-Rich Repeats (LRRs) within Vertebrate Toll-like Receptors. BMC Genomics 8, 1–20. doi:10.1186/1471-2164-8-124
Mayer, C. (2007). Phobos: Highly Accurate Search for Perfect and Imperfect Tandem Repeats in Complete Genomes by Christoph Mayer Version: 3.3.11, 2006–2010.
Newman, A. M., and Cooper, J. B. (2007). XSTREAM: A Practical Algorithm for Identification and Architecture Modeling of Tandem Repeats in Protein Sequences. BMC Bioinformatics 8, 382. doi:10.1186/1471-2105-8-382
Paladin, L., Bevilacqua, M., Errigo, S., Piovesan, D., Mičetić, I., Necci, M., et al. (2021). RepeatsDB in 2021: Improved Data and Extended Classification for Protein Tandem Repeat Structures. Nucleic Acids Res. 49, D452–D457. doi:10.1093/nar/gkaa1097
Schaper, E., and Anisimova, M. (2015). The Evolution and Function of Protein Tandem Repeats in Plants. New Phytol. 206, 397–410. doi:10.1111/nph.13184 Available at: https://nph.onlinelibrary.wiley.com/doi/pdf/10.1111/nph.13184
Schaper, E., Gascuel, O., and Anisimova, M. (2014). Deep Conservation of Human Protein Tandem Repeats within the Eukaryotes. Mol. Biol. Evol. 31, 1132–1148. doi:10.1093/molbev/msu062
Schaper, E., Kajava, A. V., Hauser, A., Anisimova, M., and Zu, C. (2012). Repeat or Not Repeat ?— Statistical Validation of Tandem Repeat Prediction in Genomic Sequences. Mol. Biol. Evol. 40, 1–13. doi:10.1093/nar/gks726
Schaper, E., Korsunsky, A., Pečerska, J., Messina, A., Murri, R., Stockinger, H., et al. (2015). TRAL: Tandem Repeat Annotation Library. Bioinformatics 31, 3051–3053. doi:10.1093/bioinformatics/btv306
Sokol, D., Benson, G., and Tojeira, J. (2007). Tandem Repeats over the Edit Distance. Bioinformatics 23, e30–e35. doi:10.1093/bioinformatics/btl309
Szklarczyk, R., and Heringa, J. (2004). Tracking Repeats Using Significance and Transitivity. Bioinformatics 20, i311–i317. doi:10.1093/bioinformatics/bth911
Tørresen, O. K., Star, B., Mier, P., Andrade-Navarro, M. A., Bateman, A., Jarnot, P., et al. (2019). Tandem Repeats lead to Sequence Assembly Errors and Impose Multi-Level Challenges for Genome and Protein Databases. Nucleic Acids Res. 47, 10994–11006. doi:10.1093/nar/gkz841 Available at: https://academic.oup.com/nar/article-pdf/47/21/10994/31074469/gkz841.pdf
Vega, A., Sobrido, M. J., Ruiz-Ponte, C., Barros, F., and Carracedo, A. (2001). Rare HRAS1 Alleles Are a Risk Factor for the Development of Brain Tumors. Cancer 92, 2920–2926. CO;2-S. _eprint: https://acsjournals.onlinelibrary.wiley.com/doi/pdf/10.1002/1097-0142%2820011201%2992%3A11%3C2920%3A%3AAID-CNCR10110%3E3.0.CO%3B2-S. doi:10.1002/1097-0142(20011201)92:11⟨2920:AID-CNCR10110⟩3.0
Wheeler, T. J., Clements, J., and Finn, R. D. (2014). Skylign: a Tool for Creating Informative, Interactive Logos Representing Sequence Alignments and Profile Hidden Markov Models. BMC Bioinformatics 15, 7. doi:10.1186/1471-2105-15-7
Willems, T., Gymrek, M., Highnam, G., Consortium, T. G. P., Mittelman, D., and Erlich, Y. (2014). The Landscape of Human STR Variation. Genome Res. 24, 1894–1904. doi:10.1101/gr.177774.114 Cold Spring Harbor Laboratory Press Distributor: Cold Spring Harbor Laboratory Press Institution: Cold Spring Harbor Laboratory Press Label: Cold Spring Harbor Laboratory Press Publisher: Cold Spring Harbor Lab
Keywords: bioinformatics, computational biology, genome annotation, protein sequence analysis, tandem repeats, profile hidden markov models
Citation: Delucchi M, Näf P, Bliven S and Anisimova M (2021) TRAL 2.0: Tandem Repeat Detection With Circular Profile Hidden Markov Models and Evolutionary Aligner. Front. Bioinform. 1:691865. doi: 10.3389/fbinf.2021.691865
Received: 07 April 2021; Accepted: 11 June 2021;
Published: 25 June 2021.
Edited by:
Damiano Piovesan, University of Padua, ItalyReviewed by:
Tamas Lazar, Vrije University Brussel, BelgiumCopyright © 2021 Delucchi, Näf, Bliven and Anisimova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Anisimova, bWFyaWEuYW5pc2ltb3ZhQHpoYXcuY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.