 Sifiso Vilakati
Sifiso Vilakati Giuliana Cortese
Giuliana Cortese
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Appl. Math. Stat. , 17 March 2025
Sec. Statistics and Probability
Volume 11 - 2025 | https://doi.org/10.3389/fams.2025.1519056
Ensuring patient safety is paramount in the development of new pharmaceutical products, which, while beneficial, can also pose potential risks. Traditional methods for analyzing safety data have been limited to single-stage randomization clinical trials. However, no existing methodology addresses the complexities of two-stage randomization designs with survival endpoints. This paper introduces a novel methodology utilizing inverse probability weights to analyze safety data in two-stage randomization designs. Our approach is applied to data from a leukemia clinical trial, the use of the weighted Aalen-Johansen estimator is recommended while the use of the weighted Kaplan-Meier is discouraged. This advancement provides a crucial tool for enhancing patient safety in complex clinical trial designs.
The safety of patients is an important aspect in the development of new pharmaceutical products. Any biologically active pharmaceutical product is meant to produce benefit to its users but can potentially cause harm as well. Of importance in the development of pharmaceutical products is the understanding of how the potential harms can manifest themselves and at what stage these potential harmful effects can be identified. Some pharmaceutical products fail at the development stage because of unanticipated safety issues. Some products pass through the development stage only to be called from the market place because of some undesired side effects that place the patients at serious health risks [1].
An adverse event is any untoward medical occurrence in a patient during the course of a clinical trial. An adverse event can be any unfavorable and unintended sign, symptom or disease temporally associated with the use of a medical product, whether it is related to the medical product or not. Adverse events can be classified into different categories, and in this study we shall focus on serious adverse events. A serious adverse event is defined as any untoward medical occurrence that; (1) may result in death, (2) is life threatening, (3) requires inpatient hospitalization or prolongation of existing hospitalization, (4) results in persistent or significant disability, and (5) is a congenital anomaly [2].
Although safety data are the most common and one of the most important types of data collected in clinical trials, in general more emphasis is given to the efficacy data. More methods are developed to analyze efficacy data but less attention is given to safety data, for example, more methodological developments have happened in the analysis of efficacy data for two-stage randomization designs [3–7] and to our knowledge no study has focused on the analysis of safety data from these designs. [8] advocate the use of survival analysis methods for analyzing safety data when the primary endpoint in a clinical trial is a time-to-event. In the sequel, we also advocate the use of survival analysis techniques suitable for two-stage randomization designs in the analysis of safety data from these designs.
Existing methods for analyzing safety data in clinical trials primarily focus on single-stage randomization designs [8]. These methods often fail to address the complexities and unique challenges posed by two-stage randomization designs, such as the need to account for multiple randomization points and the potential for varying patient responses at different stages. We propose a methodology to be used in the analysis of safety data from two-stage randomization designs. Our proposed methodology addresses these limitations by incorporating inverse probability weights, which allow for a more accurate and robust analysis of safety data in two-stage designs.
Consider a trial with two treatment arms, and let g = 1, 2 denote the treatment groups. We give a brief overview of the methods used in analyzing safety data when there is one randomization.
Analysis of safety data is often done using incidence proportions (IPs). These incidence proportions are only valid summaries under the assumption of similar exposure times in both treatment groups. In most cases this assumption is violated because in some trials the exposure times differ. The crude incidence proportion is defined as the number of patients experiencing the adverse event of interest divided by the total number of subjects in each study group. The IP is calculated as
where ag is the number of patients in treatment group g experiencing at least one serious AE and ng is the total number of patients in treatment group g. The IPs of two groups can be compared using the risk ratio, that is,
Another way of summarizing adverse events data is by using the incidence rate. The incidence rate (IR) is defined as
where ag is the number of patients in treatment group g experiencing at least one serious AE and (population-time at risk)g is the population time at risk of the first serious AE in treatment group g. The denominator in the above equation is the sum of all patients and the times at risk for the first serious AE. A patient who does not experience an AE contributes his/her follow-up time. The incidence rate ratio (IRR) is calculated as
with IRg being the incidence rate in group g to experience a serious AE.
To accommodate patient exposure times, the exposure adjusted incidence rates (EAIR) is defined as the number of subjects experiencing a serious AE divided by the total exposure time among the patients in the treatment group g;
where ag is the number of patients in treatment group g experiencing at least one serious AE and tig is the subject exposure time for individual i until the occurrence of first serious AE in treatment group g. For a subject with no AE, tig corresponds to the last follow-up time. This type of incidence rate is a valid statistic for treatment comparison when the incidence rate of a specific event is relatively constant over the study period. We interpret the EAIR as the number of serious AEs occurring in a population per unit time. The difference between the IR and the EAIR is that the denominator in the IR is the sum of all patients and the times at risk for the first serious AE. In the EAIR we sum the exposure times only.
Adverse events data are subject to competing risks. A patient that enters the study can either experience the AE of interest, die before experiencing the AE or be censored. Since patients may actually die before experiencing the AE, then death is a competing risk for the AE. After the patient has died, the AE cannot occur any more. With infinite follow-up and without censoring,
where dg is the number of deaths without an AE and ng is the total number of patients in treatment group g.
Consider the time interval [0, t]. Without censoring, the probability to experience the composite event (AE or death) is
For estimation, without censoring;
where T is the time to the first serious AE or death without an AE, is the number of patients in treatment group g experiencing at least one serious AE before or at time t and is the number of deaths in group g before or at time t.
The Kaplan-Meier (KM) estimator is sometimes used to estimate the cumulative incidence function (CIF), P(AE∈[0, t]), where death without experiencing an AE is treated as censored observation. In this case,
where ag(u) is the number of AEs in treatment group g at u and rg(u) denotes the number of patients with no AE before u, the so-called risk set, the product is over all AE times.
There has been a number of criticisms in using this approach in estimating Pg(AE ∈ [0, t]). Clearly, it ignores the competing risk set-up that exists in safety data. Another argument against this approach is that 1−KMg estimator aims at approximating a distribution function which approaches 1 as t becomes larger. On the other hand, Pg(AE ∈ [0, t])+Pg(Death ∈ [0, t]) tends to 1 as t becomes larger, hence the KM based estimator of Pg(AE ∈ [0, t]) is biased upwards [8]. Contrary to these arguments, the Kaplan-Meier estimator is still being used in some studies. In defense of this approach, in a response to Schmoor et al. [9], the authors of the paper Thanarajasingam et al. [10] argued that even though the Kaplan-Meier estimator tends to overestimate the Pg(AE ∈ [0, t]), the bias is minimal.
To estimate the Pg(AE ∈ [0, t]), the Aalen-Johansen estimator should be used in the competing risks situation [8]. The cumulative incidence function (CIF) of an AE is the expected proportion of patients experiencing an AE over the course of time. We note that
where is the KM estimator of the probability of not experiencing the composite event AE or death in treatment group g just before time u and dg(u) is the number of deaths in treatment group g at time u. The sum in (3) is the empirical probability to have an AE or death event in [0, t], that is, we are summing over all events times. Now, to get the probability of an adverse event in [0, t], we sum over the empirical probability of experiencing an AE, that is,
we sum over only event times for AEs. Without censoring, Equation 4 equals
this confirms that the incidence proportion is the correct estimate in the absence of censoring.
In the competing risks situation, a model for the cause-specific hazard function for an AE can be considered. First, we write the total hazard function;
where g denotes the treatment group. This can be decomposed into the sum of two cause-specific hazards, (D denotes death), which can be estimated by
Having decomposed the hazards in this manner, the Nelson-Aalen estimator of the cumulative hazard to experience an AE is given by
where the sum is over all AE times before t. Only AEs are counted in the numerator of Equation 5. In practice, death times are considered as right-censored times. Similarly, for estimating the cumulative hazard function for death, only death events are counted and AE events are censored.
Consider a two-stage randomization design in which patients are initially randomized to treatment A with levels A1 and A2. Those who respond and consent to further study are then randomized to a second treatment with levels B1 and B2. For simplicity, the term "response" will henceforth indicate both a response to the previous treatment and consent to the second randomization. The strategy AjBk, j, k = 1, 2 involves administering Aj followed by Bk if the patient responds to the initial treatment. We consider two-stage randomization designs where only responders proceed to the second stage, as seen in the CALGB 19,808 study [11]. Our goal is to estimate and compare survival distributions for the various treatment policies. For this purpose, we employ potential outcomes [12], not to focus on causal inference, but to use them as a framework for conceptualizing the problem.
In practice, each individual adheres to a single treatment strategy, resulting in only one observable outcome for that specific strategy. However, theoretically, individuals in the population can follow any treatment policy AjBk, meaning that one can envision a potential outcome for each possible strategy for every individual. Each person has their own set of potential outcomes, collectively known as their counterfactuals.
Here, we shall focus on data from patients who received induction therapy A1, since data from patients who received different induction therapies are independent. Data from patients who received A2 are analyzed in a similar manner. Interest is on estimating survival distributions for treatment policies A1B1 and A1B2. We assume that, associated with subject i is a set of random variables
where is the response status if patient i was assigned to A1. if patient i responds to treatment A1, otherwise. is the time from initial randomization to response for patient i defined only when ; T0i is the survival time for a patient who do not respond to first stage treatment. is the time from second randomization to death if patient i receives B1, and similarly is the time from second randomization to death if patient i receives B2 instead. If patient i is assigned to A1Bk, his/her survival time would be
We note that we can only observe T1i or T2i hence, Tki are potential outcomes. If then T1i = T2i = T0i.
Define Tk as the survival time for the population when all participants follow the treatment strategy A1Bk. Inferences about these distributions directly address the intent-to-treat question of interest. Using data from the two-stage design, we estimate the distribution of Tk.
Without right censoring, the observed data can be represented as a set of independent and identically distributed (iid) random vectors for i = 1, …, n, where Zi is an indicator for the B treatment defined only if We have Zi = 1 if patient i is assigned to B1 and Zi = 0 if assigned to B2. The observed survival time, Ti, is related to the potential outcomes as follows:
To incorporate right censoring, let Ci be the time to censoring for the ith patient. The observed data can then be represented as independent and identically distributed vectors , where Δi = I(Ti < Ci) is the failure indicator, Ui = min(Ti, Ci) is the observed time to either death or censoring. Ri = 0 if patient i is censored without having had a response to treatment A1 otherwise .
We assume that the second stage randomization is made independently of the other potential outcomes, that is
We note that πz, defined only if Ri = 1, is the probability of being randomized to the B treatment and it is typically fixed by design. In the analysis of safety data for two-stage randomization designs, we suggest the use of inverse probability weights since subjects who end up in B2 are considered missing under A1B1. We show how weighting can also be applied in the analysis of safety data for treatment policies. In the literature, two types of weights have been proposed [3, 4], in this paper we shall use time independent weights.
Let g = 1, 2, ... now denote the treatment policies. We make the following simplifying assumptions. We note that the events of interest can occur in both stages of the trial and we assume that the AEs occur after response for those who achieve complete remission. This makes the application of the inverse weights to be straight forward. Also, we assume that the states in the competing risk situation are absorbing. Let Wi1 = 1−Ri+RiZi/πz be the weight function for A1B1, that is, g = 1. For A1B2, let Wi2 = 1−Ri+RiZi/(1−πz). Similar weights are defined for the treatment policies A2B1 and A2B2.
For treatment policies, we define the incidence proportion as the weighted number of patients experiencing the adverse event divided by the weighted number of subjects in each study group. The weighting is done in such a way that the contribution of a non-responder is given a weight of 1 and a responder is given a weight of 1/πz or 1/(1−πz) where πz is the probability of being randomized to second stage treatment. With this definition,
where is the weighted number of patients in treatment policy g experiencing at least one serious AE, is the weighted number of patients in treatment policy g and Iig(event = AE) = 1 if patient i in treatment group g experiences at least one serious AE, it is zero otherwise. As an hypothetical example, we consider a trial where 100 patients are assigned to A1 and of these 100 patients, 80 respond to the A1 treatment and are equally randomized between B1 and B2. So about 40 patients are randomized to B1. Suppose that among the responders 15 develop serious AEs and among the non-responders 5 develop AEs. In calculating the WIP, the 5 patients receive a weight of 1 and the 15 patients receive a weight of 2, therefore we have 5+30 = 35. So, WIPA1B1 = 35/100 = 0.35. Without weighting: IPA1B1 = 20/60 = 0.33. In the theory of analyzing dynamic treatment regimes, patients who would have been randomized to B1 but end up in B2 are considered missing under the treatment policy A1B1. To deal with this “missingness,” inverse weights are used such that we still have 100 patients in the denominator in the above example.
To compare two treatment policies one can use the weighted risk ratio,
We define the weighted exposure adjusted incidence rate (WEAIR) as the weighted number of subjects experiencing at least one serious AE divided by the weighted exposure time among the subjects in a treatment policy, that is,
where is the weighted number of patients in treatment policy g experiencing at least one serious AE and tig is the subject exposure time until the occurrence of first serious AE in treatment policy g. For a subject with no AE, tig corresponds to the last follow-up time, and Wig is the inverse weight given to individual i in the treatment policy g. To compare two treatment policies one can use the weighted exposure adjusted incidence risk ratio,
Instead of using the usual Kaplan-Meier estimator, we suggest the use of the weighted Kaplan-Meier estimator in analyzing safety data from dynamic treatment regimes. To estimate the probability of an AE in some time interval [0, t], we can use 1−WKM, that is,
where w denotes that the event and the at risk processes are weighted. The numerator counts the AE events and the denominator gives the number at risk at time u. We weight these processes using the inverse probability weights depending on whether the individual is a responder or non-responder. Deaths before an AE are treated as censored observations. This estimator ignores the competing risks situation that exist in safety data. The most appropriate estimator is based on the Aalen-Johansen estimator.
The weighted Kaplan-Meier estimator ignores the competing risks situation that exists in AEs data. Death before an AE is a competing event. For the analysis of AEs data for dynamic treatment regimes, we propose the use of the weighted Aalen-Johansen estimator. The weighted Aalen-Johansen estimator of weighted cumulative incidence function is an appropriate method for estimating the probability of an AE in a competing risks situation:
where is the weighted Kaplan-Meier estimator of the probability of not experiencing the composite event AE or death just before time u. We sum over all events times (death or AE). Again, we weight the event processes with inverse weights. The probability of an AE in the time interval [0, t] is given
where here the sum is over all times of AE before t.
The all events (AE and death) weighted hazards is given by
This decomposes into the so-called cause specific weighted hazards, , which can be estimated by
where and are the weighted event processes. The quantity is the weighted at risk process for treatment policy g.
From the decomposition above, the Nelson-Aalen estimator for the weighted cumulative hazard to experience an AE is
In the numerator of Equation 14 we only count AEs, that is, we are summing over AEs times. We weight using the inverse probability of being in treatment policy g. In practice, we censor death events before an AE to estimate the weighted cumulative hazard for an AE. The procedure is similar for the weighted cumulative hazard for death without an AE.
We illustrate how this analysis can be done using the CALGB 19,808 toxicity dataset. In the CALGB 19,808 study, 302 patients were randomized between induction chemotherapy regimens consisting of cytosine arabinoside (Ara-C;A), daunorubicin (D), and etoposide (E) without (ADE) or with (ADEP) PSC-833 (P). The study was done to patients under the age of 60 with newly diagnosed acute myeloid leukemia. To be eligible, the patients should not have been previously treated for leukemia and be under the age of 60. For the first stage, the main objective of the trial was to determine whether the use of the Pgp-modulating agent PSC-833 in the ADEP regimen improved overall survival and disease free survival compared to ADE only. The randomization between ADE and ADEP was done at 1:1 ratio. The analysis of the first stage data is reported in [11]. In both treatment arms, 75% of the patients achieved complete remission (CR). Complete remission was defined using the National Cancer Institute Workshop criteria [13]. The 75% who achieved complete remission were further randomized to the second stage treatments namely recombinant interleukin-2 (rIL-2) and no rIL-2 (observation). We illustrate the proposed methodology on the toxicity dataset from the CALGB 19,808 study. Several variables were recorded in the toxicity dataset. The adverse events were graded in terms of their severity. The adverse event were graded as mild, moderate, severe, life-threatening, and fatal. The adverse event names and their categories are also given. In this illustration, we focus on the analysis of serious adverse events which are called life-threatening (serious AEs) in this dataset. Most of the analysis (other than the incidence proportions and ratios) will be based on the time to the first serious adverse event.
In the development of this methodology, we made some simplifying assumptions. One of them is that we assumed that for responders, the AE occurs after response to the first treatment. This makes it straightforward to apply the inverse probability weights. In this dataset, this assumption is not violated. The efficacy dataset has a variable named ind_crdays which gives the number of days from registration to when complete remission was reported. It can be seen that, for almost all the patients who responded, complete remission was achieved very early, for some as early as 24 days. We can then apply the methodology of this paper assuming the AE occurred after response to the responders to the induction treatments.
In the toxicity dataset, there is not an explicit time to the first serious adverse event. The time is given as an interval made up of two variables which are: AE starting day and AE ending day. The AE ending day refers to the number of days from registration to the end of AE reporting period. The AE occurred in the interval given by the two times. For purposes of this application, we used the AE ending day as our time variable. We could have used the middle value of the interval as our time variable. Interest is in comparing occurrences of AEs in different treatment policies. To achieve this, we merge the efficacy dataset and the toxicity dataset. The merging was done using the patient number which is present in both datasets.
There are four treatment policies embedded in the CALGB 19,808 study, namely; ADE - OBS, ADE - rIL-2, ADEP - OBS, and ADEP - rIL-2. For a detailed description of this study see In the second stage, some patients were randomized to observation. No active treatment was given to this group as patients were simply observed. There are no adverse events associated with the observation treatment option. In doing the analysis for the AEs, we only considered two treatment policies, which are ADE - rIL-2 and ADEP - rIL-2 for reasons given above. Other than the creation of the time to first serious AE variable, the data was analyzed without any further modifications.
Ignoring the censoring, we calculated the weighted incidence proportions for the treatment policies. The weighted incidence proportion for ADE - rIL-2 is 0.9797 and the weighted incidence proportion for the ADEP - rIL-2 is 0.9615. The probability of having a serious adverse event was slightly higher in the ADE - rIL-2 treatment policy. The weighted risk ratio, WRR = 0.9797/0.9615 = 1.015. The estimated risk of experiencing at least one serious AE is approximately the same in the two treatment policies.
To calculate the weighted exposure-adjusted incidence rate, we consider three scenarios a patient might be in during the trial. A patient who experiences a serious AE while still in the exposure time contributes to the time at risk his/her weighted time to the AE. A patient who dies without experiencing an AE contributes to the time at risk his/her weighted time to death. Lastly, a patient who does not experience a serious AE contributes to the time at risk for an AE his/her weighted time to the end of exposure. The weighted time at risk of exposure in the ADE - rIL-2 treatment policy is 9,498.593 days and for the ADEP - rIL-2 is 10,079.83 days. The WEAIR in the ADE - rIL-2 treatment policy is 0.0128 and WEAIR in the ADEP - rIL-2 is 0.0124. There is no major difference in the WEAIRs for the two treatment policies. This can be shown by calculating the weighted exposure-adjusted incidence risk ratio, WEAIRR = WEAIR1/WEAIR2 = 0.0128/0.0124 = 1.036. There is no difference in number of serious AEs occurring daily in the two treatment policies.
Figure 1 is obtained by treating death as censored and then taking 1−WKM. The probability of an AE is estimated by 1−WKM and this approach has been criticized as it ignores the competing risks situation. The graph shows no differences in the probabilities of experiencing an AE in the two treatment regimes.
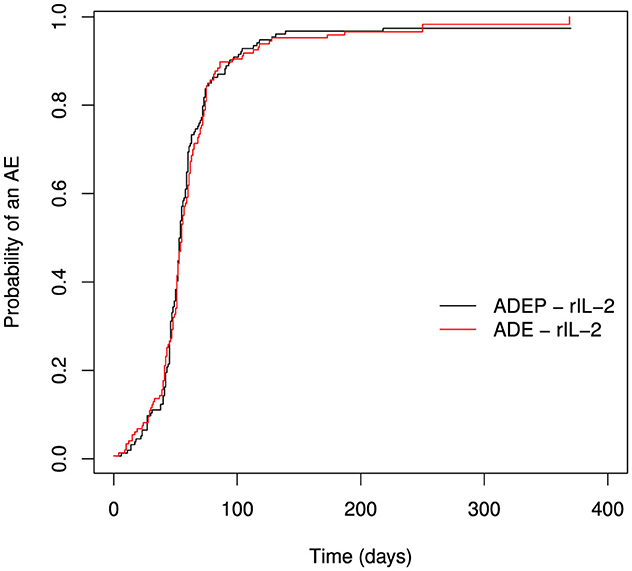
Figure 1. Estimating the probability of an AE using weighted Kaplan-Meier estimator.
The most appropriate approach of estimating the probability of an AE is the use of the weighted Aalen-Johansen estimator of the CIF. The graphs obtained from the weighted Kaplan-Meier looks similar to the ones from the weighted Aalen-Johansen estimator in Figure 2. This is not surprising since there were few deaths in the dataset. One should expect the two estimators to be similar if there are few deaths (competing risks). For this reason we do not show the graph for CIF for death events.
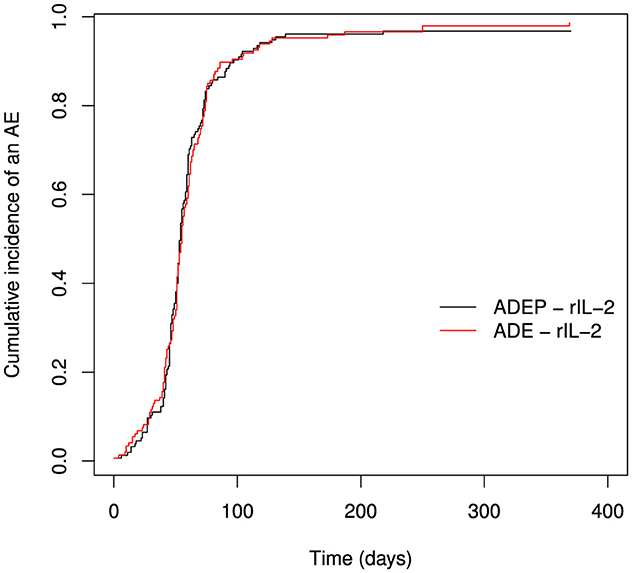
Figure 2. Estimating the probability of an AE using weighted Aalen-Johansen estimator.
The estimation of the probability of an AE by the weighted Kaplan-Meier tends to overestimate the probability. This could not be shown clearly in this analysis as there were few competing events (deaths). It can be seen though, that the graph of the weighted Kaplan-Meier is slightly above the graph from the Weighted Aalen-Johansen estimator in the tail of the distribution. This is depicted in Figure 3. In the reply to Schmoor et al. [9] by Thanarajasingam et al. [10] they argued that, even though the Kaplan-Meier estimator is biased upwards, its bias is minimal. This may not be true in all cases, but only in the case where the competing events are few. Since interest is in the probability of a competing event, that is, AE or death, if the count for deaths is close to zero, then the two estimators will be similar.
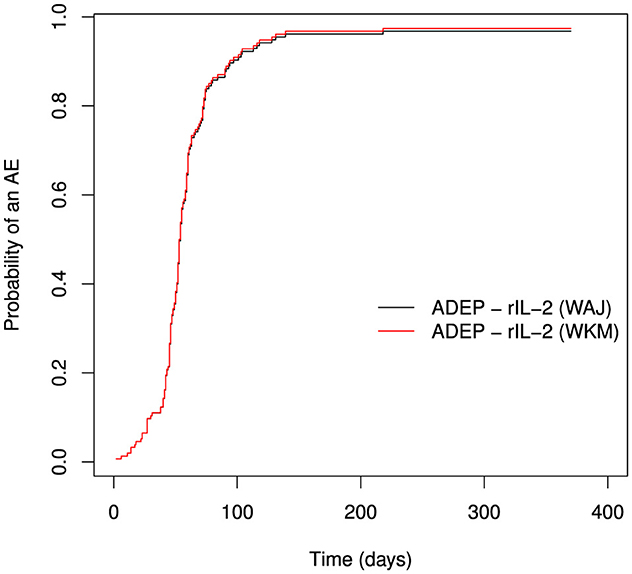
Figure 3. A comparison of weighted Aalen-Johansen and weighted Kaplan-Meier estimators.
In doing the analysis based on event-specific hazards, we only report the weighted cumulative hazards for an AE. Due to lower numbers of death before a serious AE, we did not include the graphs for death before an AE. When ignoring the competing event of death, we observe some differences between the cumulative hazards of ADEP - rIL-2 and ADEP - rIL-2. The cumulative hazard of experiencing an AE was higher in the ADEP - rIL-2 than in the ADE - rIL-2 treatment policy in the time period 100 days to about 180 days, thereafter the hazard of an AE is higher in the ADEP - rIL-2 treatment policy. For the earlier times, there is no much difference in the hazards of an AE between the two treatment policies. The cumulative hazard of experiencing an AE was equal in the first 100 days. This is shown in Figure 4 below.
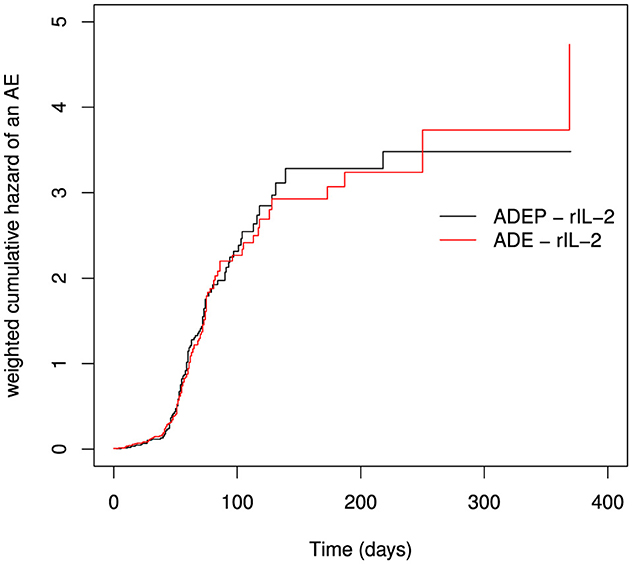
Figure 4. Weighted cumulative hazards for AEs using the Nelson-Aalen estimator.
There has been an acknowledgment that safety data does not receive the attention as efficacy data [1]. In most cases, the analysis of safety data has been done using crude incidence rates and this type of analysis may not be adequate. The use of time-to-event statistical methods is common practice for efficacy endpoints in clinical studies but such methods are rarely applied in the analysis of safety data. In this paper, we have given a general overview of the methods that are applicable to single stage study with a time-to-event endpoint. We then propose a methodology for analyzing safety data from two-stage randomization designs which uses inverse probability weights. The weighting is done in a similar way as in the analysis of efficacy data. We used time-independent inverse weights. A responder represents 1/πz patients who could have potentially been assigned to the treatment policy of interest. A non-responder only represents himself. In doing so, we have made the analysis of the safety data be in sync with the analysis of efficacy data from these study designs. We have focused on the time to the first serious AE.
We have made the assumption that adverse events (AEs) occur only after response for those achieving complete remission. This assumption makes the application of inverse weights straight forward. This assumption is only valid if response happens early in the first stage and this is the case in the CALGB 19,808 study. Future research is needed for cases where response is not observed early in the first stage. One suggestion would be to use time-dependent weights where an individual gets a weight depending on the time of occurrence of the AE. The problem will be that such methodology will not be in sync with the the methodology for analyzing efficacy data from these study designs.
The methodology proposed in this paper is descriptive in nature similar to the paper which motivated this study [8]. This is a limitation of this study. A further study would be to look at developing inferential procedures for our methodology.
The important aspect to note in safety data is the presence of competing risks situation. A patient who enters the study can experience the AE of interest, die before experiencing the AE or be censored. The use of the Kaplan-Meier estimator is not encouraged but if the competing events are few, then the bias is minimal.
The data analyzed in this study is subject to the following licenses/restrictions: data was requested from The Alliance for Clinical Trials in Oncology and cannot be shared without their consent. Requests to access these datasets should be directed to QWxsaWFuY2VzZXJ2aWNlY2VudGVyQGFsbGlhbmNlbmN0bi5vcmc=.
The studies involving humans were approved by the Alliance for Clinical Trials in Oncology. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
SV: Conceptualization, Writing – original draft, Formal analysis, Software. GC: Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that Gen AI was used in the creation of this manuscript. For paraphrazing.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gould AL. Statistical Methods for Evaluating Safety in Medical Product Development. London: John Wiley & Sons. (2015). doi: 10.1002/9781118763070
2. Chow SC, Liu JP. Design and Analysis of Clinical Trials: Concepts and Methodologies. London: John Wiley & Sons. (2008).
3. Guo X, Tsiatis A. A weighted risk set estimator for survival distributions in two-stage randomization designs with censored survival data. Int J Biostat. (2005) 1:1–15. doi: 10.2202/1557-4679.1000
4. Lunceford JK, Davidian M, Tsiatis AA. Estimation of survival distributions of treatment policies in two-stage randomization designs in clinical trials. Biometrics. (2002) 58:48–57. doi: 10.1111/j.0006-341X.2002.00048.x
5. Lokhnygina Y, Helterbrand JD. Cox regression methods for two-stage randomization designs. Biometrics. (2007) 63:422–8. doi: 10.1111/j.1541-0420.2007.00707.x
6. Wahed AS. Inference for two-stage adaptive treatment strategies using mixture distributions. J R Stat Soc Ser C (Appl Stat). (2010) 59:1–18. doi: 10.1111/j.1467-9876.2009.00679.x
7. Kidwell KM, Wahed AS. Weighted log-rank statistic to compare shared-path adaptive treatment strategies. Biostatistics. (2013) 14:299–314. doi: 10.1093/biostatistics/kxs042
8. Allignol A, Beyersmann J, Schmoor C. Statistical issues in the analysis of adverse events in time-to-event data. Pharm Stat. (2016) 15:297–305. doi: 10.1002/pst.1739
9. Schmoor C, Bender R, Beyersmann J, Kieser M, Schumacher M. Adverse event development in clinical oncology trials. Lancet Oncol. (2016) 17:e263–4. doi: 10.1016/S1470-2045(16)30223-6
10. Thanarajasingam G, Atherton PJ, Novotny PJ, Loprinzi CL, Sloan JA, Grothey A. Longitudinal adverse event assessment in oncology clinical trials: the Toxicity over Time (ToxT) analysis of Alliance trials NCCTG N9741 and 979254. Lancet Oncol. (2016) 17:663–70. doi: 10.1016/S1470-2045(16)00038-3
11. Kolitz JE, George SL, Marcucci G, Vij R, Powell BL, Allen SL, et al. P-glycoprotein inhibition using valspodar (PSC-833) does not improve outcomes for patients younger than age 60 years with newly diagnosed acute myeloid leukemia: Cancer and Leukemia Group B study 19808. Blood. (2010) 116:1413–21. doi: 10.1182/blood-2009-07-229492
12. Rubin DB. Estimating causal effects of treatments in randomized and nonrandomized studies. J Educ Psychol. (1974) 66:688. doi: 10.1037/h0037350
Keywords: adverse events, Aalen-Johansen estimator, inverse probability weights, toxicity, two-stage randomization designs
Citation: Vilakati S and Cortese G (2025) Analyzing safety data for two-stage randomization designs. Front. Appl. Math. Stat. 11:1519056. doi: 10.3389/fams.2025.1519056
Received: 29 October 2024; Accepted: 03 March 2025;
Published: 17 March 2025.
Edited by:
George Michailidis, University of Florida, United StatesReviewed by:
James Stamey, Baylor University, United StatesCopyright © 2025 Vilakati and Cortese. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuliana Cortese, Z2l1bGlhbmEuY29ydGVzZUB1bmlwZC5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.