 Lucas Farias Rodrigues1
Lucas Farias Rodrigues1 German Dario Ramírez-Zamudio2,3
German Dario Ramírez-Zamudio2,3 Guilherme Luis Pereira1,4Juliana Akamine Torrecilhas4Lucas Augustinho Trevisan4
Guilherme Luis Pereira1,4Juliana Akamine Torrecilhas4Lucas Augustinho Trevisan4 Otávio Rodrigues Machado Neto1,4
Otávio Rodrigues Machado Neto1,4 Luis Artur Loyola Chardulo1,4
Luis Artur Loyola Chardulo1,4 Welder Angelo Baldassini1,4
Welder Angelo Baldassini1,4 Rogério Abdallah Curi1,4*
Rogério Abdallah Curi1,4*- 1School of Agricultural and Veterinary Sciences (FCAV), São Paulo State University (UNESP), Jaboticabal, São Paulo, Brazil
- 2College of Animal Science and Food Engineering, University of São Paulo (USP), Pirassununga, São Paulo, Brazil
- 3Department of Animal Sciences, Center for Nutrition and Pregnancy, North Dakota State University, Fargo, ND, United States
- 4School of Veterinary Medicine and Animal Science (FMVZ), São Paulo State University (UNESP), Botucatu, São Paulo, Brazil
Introduction: This study investigated the impact of creep-feeding supplementation on the genome methylation of the Longissimus thoracis (LT) muscle in crossbred beef cattle (Bos taurus × Bos indicus).
Methods: The experiment involved 48 uncastrated F1 Angus-Nellore males (half-siblings), which were divided into two groups: NCF – no creep-feeding (n = 24) and CF – creep-feeding (n = 24). After weaning at 210 days, all animals were feedlot finished for 180 days under identical conditions. LT muscle biopsies were collected at weaning for genomic DNA methylation analysis by reduced representation bisulfite sequencing (RRBS).
Results and discussion: The groups differed significantly (CF > NCF: p < 0.05) to weaning weight (243.57±5.70 vs. 228.92±5.07kg), backfat thickness (12.96±0.86 vs. 10.61±0.42mm), LT muscle marbling score (366.11±12.39 vs. 321.50±13.65), and LT intramuscular fat content (5.80±0.23 vs. 4.95±0.20%). The weights at the beginning of the experiment and at slaughter (390 days) did not differ significantly. Mean methylation levels were higher in CF with 0.18% more CpG, 0.04% CHG, and 0.03% CHH. We identified 974 regions with differential methylation (DMRs: > 25% and q < 0.05), which overlapped with 241 differentially methylated genes (DMGs). Among these genes, 108 were hypermethylated and 133 were hypomethylated in CF group. Notably, 39 of these DMGs were previously identified as differentially expressed genes (DEGs: log2 fold change [0.5]) in the same animal groups. Over-representation analysis highlighted epigenetic regulations related to muscle growth, PPAR signaling, adipogenesis, insulin response, and lipid metabolism. Key DMGs/DEGs included: ACAA1, SORBS1, SMAD3, TRIM63, PRKCA, DNMT3A, RUNX1, NRG3, and SLC2A8. These epigenetic changes improved the performance of supplemented animals up to weaning and enhanced meat quality traits, particularly higher intramuscular fat. The results provided insights into the intricate interplay between nutrition, epigenetics, gene expression and phenotypes in beef cattle production.
1 Introduction
Beef cattle farming in Brazil has employed different strategies to increase animal performance and the quality of the meat produced. Particularly interesting strategies include the use of crossbred Bos taurus × Bos indicus cattle and creep-feeding throughout the cow-calf phase. Angus × Nellore animals are used to produce tender meat with a higher intramuscular fat (IMF) content to add value to the product and to meet the demands of consumers who are willing to pay more for higher quality (Lopes et al., 2020). Creep-feeding consists of supplementing calves during the suckling phase with a grain-based diet rich in energy and protein to obtain heavier individuals at weaning. This approach helps reduce the feeding time during the phase when the carcass is finished for slaughter (Dantas et al., 2010; Scheffler et al., 2014).
Although acetate is one of the main acetyl group donors for IMF deposition, glucose derived from concentrate-rich supplements is also considered an important substrate (Smith and Crouse, 1984). The authors of that study highlighted that the adequate intake of concentrates during the lactation period, especially those rich in starch, favors the proliferation and growth of fat cells. This fact is the result of increased glucose metabolism, which can enhance marbling without increasing visceral fat, thus contributing to improving meat quality (Baldassini et al., 2021). Furthermore, Ferreira et al. (2020) proposed that higher levels of dietary crude protein increase the digestion and intestinal absorption of starch and promote an increase in plasma insulin and glucose which, in turn, increase the deposition of IMF by favoring the synthesis of fatty acid precursors. According to Stewart (2010), several studies have demonstrated that energy supplementation throughout the cow-calf phase can favor IMF deposition; however, to maintain this increase until slaughter, it is necessary to continue feeding cattle grain-based diets.
Animals are able respond to different environmental/nutritional factors, exhibiting phenotypic plasticity because of changes in gene expression patterns (Zhang, 2015). Thus, phenotypes can be modified by nutritional modulations, i.e., dietary exposure can have consequences for growth and for animal health (McKay and Mathers, 2011). New evidence is constantly emerging that nutritional stimuli can modify DNA methylation and thus affect gene expression and the phenotypes of individuals (Day et al., 2015; Farias et al., 2015; Farkas et al., 2015). DNA methylation is a biological process that involves the addition of methyl groups (CH3) to the cytosine nucleotides of chromosomes. This process contributes to the epigenetic network that controls gene expression and, consequently, phenotypes (Zhang, 2015). Epigenetic marks in the genome, which can modify gene expression but not the DNA sequence, can be acquired and persist throughout an individual’s life (Stenz et al., 2018). These marks can also be transmitted from cells of one generation to another (Reik et al., 2001).
Based on the above, the aim of this study was to evaluate for the first time the effects of creep-feeding supplementation on genome methylation and, consequently, on gene expression in the Longissimus thoracis (LT) muscle of crossbred F1 Angus x Nellore cattle at weaning, as well as on performance, carcass, and meat quality traits.
2 Materials and methods
2.1 Animals and experimental treatments
All procedures involving animals were approved by the Animal Use Ethics Committee of the School of Agricultural and Veterinary Sciences (FCAV), UNESP, Jaboticabal, São Paulo, Brazil (Protocol number 013689/19).
Forty-eight uncastrated (intact) crossbred F1 Angus-Nellore bulls born to the same Aberdeen Angus (Bos taurus) sire (half-siblings) and their mothers (Nellore – Bos indicus) with a body weight of 414 ± 45 kg were stratified according to the date and number of births (primiparous or multiparous) and distributed in two 20-hectare Brachiaria grass paddocks. Calves were randomly assigned to two treatments from 30 days of age until weaning at 210 days. The following treatments were applied: no creep-feeding (NCF, n = 24) and creep-feeding (CF, n = 24). In the creep-feeding system, calves were kept with their mothers in the same paddock but had exclusive and free access to a supplement (ad libitum) corresponding to approximately 1% of their body weight. The adjustment of the supplement amount provided per animal was made weekly based on performance prediction using a diet evaluation and formulation system. The supplement offered to CF animals contained 22% crude protein and 65% total digestible nutrients on a dry matter basis and consisted of ground corn (44.8%), soybean meal (40.4%), and mineral mix (14.8%). After weaning, the animals of both treatments were taken to the feedlot area where they remained for 180 days in collective covered pens (three animals/pen with 10 m2 per animal). The two groups of animals received the same diet (Table 1), formulated with the maximum profit ration software RLM 3.3 (Lanna et al., 2011), using the ESALQ Tropicalized NRC system, ad libitum, divided into two equal parts (two times a day) at 8:00 am and 4:00 pm. The diet contained 12.6% forage and 87.4% corn-based concentrate.

Table 1. Ingredients and chemical-bromatological composition of the feedlot diet.
2.2 Collection of muscle tissue and performance, carcass and meat quality traits
At weaning, LT muscle samples were collected by biopsy from 12 animals randomly selected from each of the two treatments. For biopsy performed at the level of the 13th rib, the thoracic region was shaved and a local anesthetic (lidocaine 2%, 4mg/kg) was administered subcutaneously. After cleaning the site, a 1-cm incision was made and a sterilized Bergstrom biopsy needle was used to obtain 1 g of muscle tissue, which was immediately stored in liquid nitrogen and later in an ultrafreezer at -80°C. These muscle samples were used to extract total RNA for analysis of differential gene expression and DNA for analysis of differentiation of genomic methylation.
During the experiment, the animals were weighed at the beginning of the cow-calf phase (initial body weight – BWi), at weaning (weaning weight – WW), and at the end of the feedlot period (final body weight – BWf). BWi and WW were used to calculate average daily weight gain 1 (ADG1: beginning of the cow-calf phase to weaning), and BWf and WW were used to calculate ADG2 (weaning to the end of the feedlot period).
After 180 days of confinement, the 48 animals were transported to a commercial slaughterhouse and slaughtered after fasting from solids for 16 h by stunning and sectioning of the jugular vein. The carcasses were identified, washed, and divided into two halves. The half-carcasses were weighed individually to obtain the hot carcass weight (HCW) and chilled in the cold room for approximately 24 h at 1oC. After chilling, the carcasses were removed from the room and weighed. After weighing, the LT muscle of the left half-carcass was separated from the remaining carcass and backfat thickness (BFT) and ribeye area (REA: between the 12th and 13th thoracic vertebrae) were measured before deboning.
During deboning, LT samples (portion of approximately 12 cm) were collected between the 12th and 13th ribs of the left half-carcass, vacuum packed, and transported to the laboratory under refrigeration. In the laboratory, the samples were cut into 2.54-cm thick steaks, vacuum packed, and aged for 7 and 14 days. After these periods, physicochemical quality traits were analyzed, including marbling score (MS), IMF content, and Warner-Bratzler shear force (WBSF7 and WBSF14).
Additional information about the carcass and meat quality analyses used in this study can be found in Ramírez-Zamudio et al. (2023).
2.3 Statistical analysis of weight, weight gain, carcass and meat data
Performance data and carcass and meat quality traits of the 48 animals (n = 24/group) [BWi (kg), WW (kg), ADG1 (kg), BWf (kg), ADG2 (kg), HCW ( kg), REA (cm2), BFT (mm), MS (score), IMF (%), and WBSF7 and WBSF14 (kg)] were analyzed regarding the presence of discrepant information, homogeneity of variances, and normality of residuals. The data were expressed as means and their respective standard errors. Means were compared between the two treatments by the t-test.
2.4 Analysis of differential gene expression
Differential gene expression was analyzed by RNA sequencing (RNA-Seq) in 12 individuals from each treatment (NCF vs. CF). RNA extraction, preparation of the cDNA libraries, and sequencing and mapping of reads to the bovine reference genome (Bos taurus – ARS-UCD1.3) were performed following protocols and parameters recommended by the international scientific community. Differentially expressed genes (DEGs) were identified using the method implemented in the edgeR v.3.40.0 package (Robinson et al., 2010) of the R software, which considers pairwise comparison and assumes that the count data follow a negative binomial distribution. Detailed information on the differential gene expression analysis used can be found in Ramírez-Zamudio et al. (2023).
2.5 Analysis of DNA methylation
2.5.1 Genomic DNA extraction
DNA was extracted from LT muscle samples (50 mg) using the DNeasy Blood & Tissue kit (Qiagen, USA) according to the manufacturer’s instructions. The extracted DNA was quantified in a Qubit® 3.0 Fluorometer (Invitrogen, USA). DNA purity (260/280) and integrity were analyzed in a NanoDrop Lite microvolume spectrophotometer (Thermo Scientific, USA) and on 0.8% agarose gel, respectively. After dilution to the concentration necessary for methylation analysis (50 ng/µL), the DNA samples were stored in 1.7-mL minitubes at -20oC.
2.5.2 Library construction and reduced representation bisulfite sequencing
For analysis of differential DNA methylation, five DNA samples were randomly selected from the 12 animals of each treatment (NCF and CF) preliminarily submitted to RNA-Seq. The reduced representation bisulfite sequencing (RRBS) technique was applied using the Zymo-Seq RRBS Library kit (Zymo Research, USA) to sequence genome regions enriches for 5-methyl cytosines.
Digestion of genomic DNA aliquots, bisulfite conversions, and enrichment of the 10 libraries by PCR (one library/animal) were performed by the US company Zymo Research. The protocol consisted of digestion of 400 ng genomic DNA with 30 units of MspI (New England Biolab, USA) at 37oC, a restriction enzyme with a CCGG recognition site, followed by end repair of the fragments of approximately 150 bp and addition of adapters.
The fragments were ligated to adapters containing 5-methyl cytosine instead of cytosine as specified by Illumina (USA). Adapter-tagged fragments ≥ 50 bp were recovered using DNA Clean & Concentrator™-5 (Zymo Research, USA) and then treated with bisulfite using the EZ DNA Methylation-Lightning™ kit (Zymo Research, USA). The resulting PCR products for enrichment of the libraries were again purified using DNA Clean & Concentrator™-5. Clustering and sequencing of the libraries of the DNA fragments were performed by Zymo (USA) using HiSeq2500 v4 2x100bp (Illumina, USA) according to the manufacturer’s protocol on the NovaSeq 6000 platform (Illumina, USA).
2.5.3 Quality analysis of RRBS sequences and alignment to the reference genome
The FastQC 0.11.9 program (Andrews, 2010) was used to visualize the data obtained by RRBS, in which quality parameters were evaluated for the complete set of raw sequencing reads. Initial quality control was performed using TrimGalore v.0.6.4_dev (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), in which adapters and unknown or low-quality bases were removed from the ends of the sequences.
The Bismark v.0.22.3 program (Krueger and Andrews, 2011) was used to align the generated sequences previously treated with bisulfite to the bovine reference genome (Bos taurus – ARS-UCD1.2) using the -N1 option, which allows a single alignment error. The reference genome was first transformed into a bisulfite-converted version, with C-to-T conversion on the top original strand and G-to-A conversion on the bottom strand, and finally indexed using Bowtie2 (Langmead and Salzberg, 2012). The sequences that produced the best unique alignments in the two processes (top and bottom original strand alignment) were compared to the normal genomic sequence to confirm the presence of a C in the reference sequence. The methylation status of all cytosine positions in the sequences was inferred using the methylation_extractor function of the Bismark program. The rate or level of methylation ranged from 0% to 100% at each site (nucleotide), considering that tissues frequently comprise different populations of cells that can harbor different methylation signals.
2.5.4 Identification and characterization of cytosines, chromosome regions and differentially methylated genes
The metrics for cytosine methylation in the genome of the individuals analyzed were obtained with MethylDackel 0.5.0 (https://github.com/dpryan79/MethylDackel). This tool groups all cytosines into one of three sequence contexts – CpG, CHG and CHH, where H is the IUPAC ambiguity code for any nucleotide other than G.
To identify regions in the genome that are differentially methylated between NCF and CF individuals (differentially methylated regions – DMRs), i.e., regions that contain more than one methylated cytosine, the calculateDiffMeth function of the MethylKit 1.24.0 package was used (Akalin et al., 2012) in the R statistical environment (R Core Team: https://www.r-project.org) of the Bioconductor repository (https://bioconductor.org/packages/release/bioc/html/ methylKit.html). First, all cytosines were filtered for a minimum coverage of three reads and a maximum percentile of 99.9%. The methylKit function that normalizes coverage was applied using standard configurations. Next, the sliding window approach was used, with a window size of 1,000 bp and a step size of 500 bp. DMRs were defined when 1) the methylation difference was at least 25%; 2) the corrected p-value (q-value) was less than 0.05, and 3) sequence coverage was greater than 10-fold across all samples. Fisher’s exact test was implemented to calculate p-values, which were subsequently adjusted to q-values using the SLIM method (Wang et al., 2011).
Genes with ensemble id’s overlapping with DMRs shared between animals of the two groups were identified and termed differentially methylated genes (DMGs). The annotation of DMGs for identifying gene regions that harbor DMRs (promoter, transcription start site – TSS, gene body – exon and intron) was performed using the GenomationData package (version 3.8) through the methylKit library 1.24.0 (Akalin et al., 2012) of the R statistical software. The NCBI RefSeq annotation database file used was obtained from the UCSC Genome Browser. Promoters were defined 1,000 bp upstream and downstream of the gene’s TSS following the methylKit pattern. Information on CpG islands (CpGi) and shores was obtained from the UCSC Genome Browser.
2.5.5 Relationship between DMGs and DEGs
To identify genes exhibiting modified expression at weaning due to methylation, the list of genes identified as DMGs was contrasted with the list of DEGs, considering the filters of a 25% methylation difference and a log2 fold change (FC) of 0.5. In eukaryotes, CpGi are mainly located in gene promoter regions (regulatory regions), near the TSS. Methylation in these regions is associated with gene silencing; thus, in the absence of methylation of these islands, gene expression occurs (Oliveira et al., 2010; Portela and Esteller, 2010; Yuferov et al., 2010). CpG dinucleotides found outside these islands in regions known as “CpGi shores” are less methylated than the islands themselves (Tsankova et al., 2007; Robison and Nestler, 2011), but can also be associated with transcriptional inactivation. However, when methylation occurs in the body of genes (exons and introns), it may also be correlated with their expression (Doi et al., 2009; Irizarry et al., 2009; Portela and Esteller, 2010). CpGs in the gene body (exons/introns) and far from the TSS can inhibit or enhance gene expression (as long as the promoter is not methylated), depending on the genomic context. Methylation (CpG) of the gene body that occurs in a CpGi may be positively or negatively correlated with expression. On the other hand, if methylation does not occur in a CpGi, there will be a positive correlation with expression, i.e., increasing/decreasing methylation will lead to an increase/decrease in gene expression (Varley et al., 2013). These relationships were established and described by Enright et al (2003); Tost (2010), and Varley et al. (2013). Thus, the relationships found were classified as consistent (expected) or inconsistent (not expected), as shown in Figure 1.
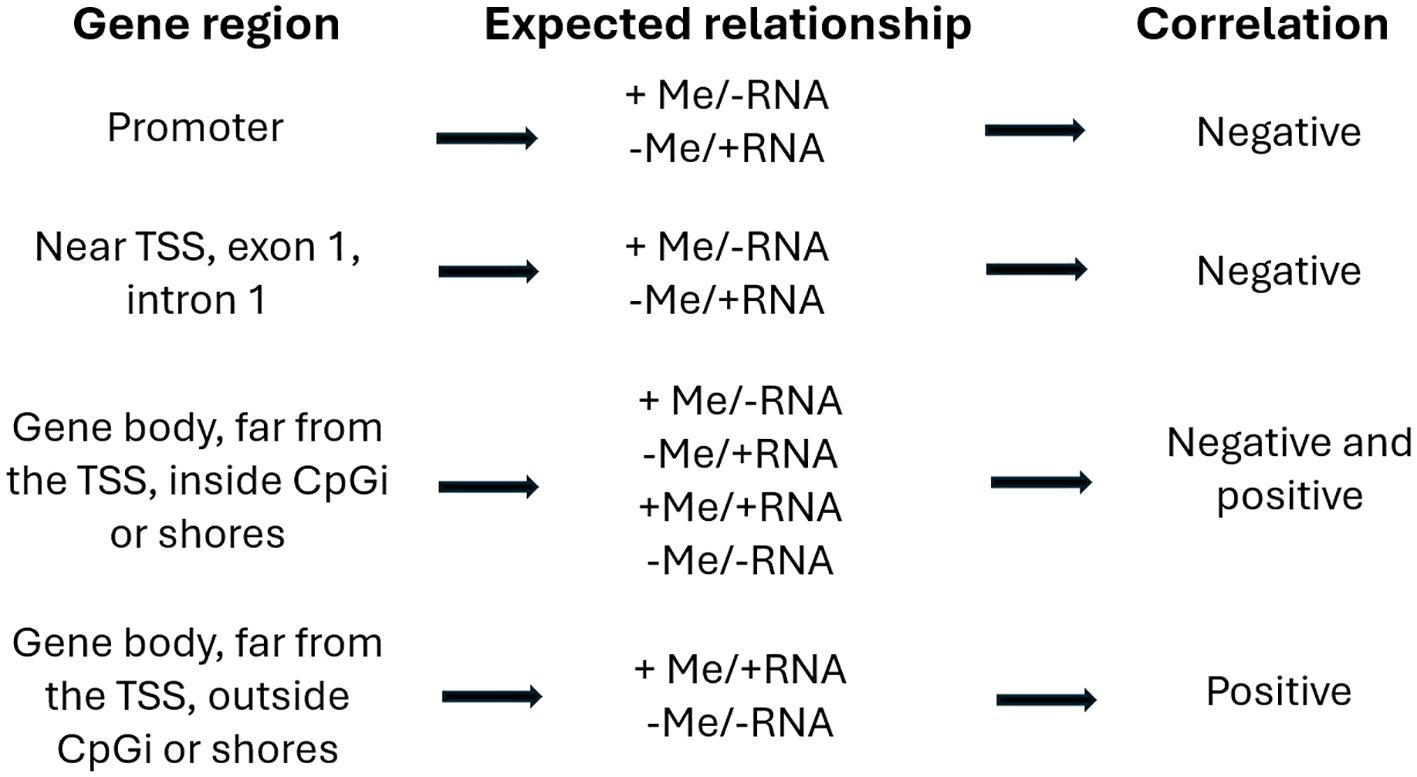
Figure 1. Expected relationships between the level of methylation in gene regions and expression in eukaryotic organisms.
2.5.6 Functional enrichment analysis
The set of genes identified as differentially methylated and differentially expressed – DMGs/DEGs – between NCF and CF individuals at weaning was used in over-representation analysis considering gene ontology (GO) terms (biological processes – BPs) and metabolic pathways (KEGG Pathway and WikiPathway databases). The WebGestalt tool, an online platform designed for functional analysis and interpretation of biological data (Wang et al., 2017), was used for this purpose. GO_BPs and pathways were enriched when p < 0.05.
3 Results
3.1 Performance, carcass and meat quality traits and differentially expressed genes at weaning
Regarding pre- and postweaning performance (Table 2), significant differences (p < 0.05) were observed between groups for WW and ADG1 during the cow-calf phase. For WW, CF animals (creep-feeding) were heavier than NCF animals (no creep-feeding) (p < 0.05). ADG1 was higher in CF compared to NCF (p < 0.05). No significant differences in BWi, BWf, ADG2, or HCW were observed between groups.
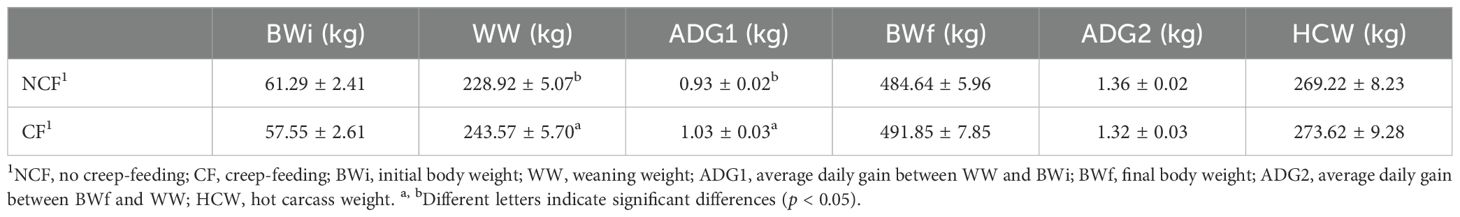
Table 2. Means and standard errors of animal weights at the beginning of the cow-calf phase, at weaning and at the end of the feedlot period, average daily gain during the cow-calf and finishing periods, and hot carcass weight obtained for the two treatments.
The meat quality and carcass traits are presented in Table 3. No significant differences between groups were observed for REA, WBSF7, or WBSF14. On the other hand, there were significant differences (p < 0.05) in BFT, IMF, and MS. For all three traits related to carcass fat deposition, CF animals (creep-feeding) had higher mean values than NCF animals (no creep-feeding).

Table 3. Means and standard errors of carcass backfat thickness and ribeye area, fat percentage, marbling score, and shear force after 7 and 14 days of aging obtained for the two treatments.
A total of 947 differentially expressed genes between NCF and CF (log2 FC [0.5]; FDR 5%) were identified; of these, 443 were downregulated and 504 were upregulated in CF (cow-calf phase with creep-feeding). Detailed information on the results of DEG analysis can be found in Ramírez-Zamudio et al. (2023).
3.2 Quality of extracted genomic DNA, sequencing and alignment of reads
The genomic DNA extracted from LT muscle samples obtained by biopsy at weaning had adequate quality for RRBS. The 260/280 ratio ranged from 1.87 to 2.01 and the 260/230 ratio from 1.48 to 1.97. The DNA concentration ranged from 83.4 to 752 ng/µL and agarose gel electrophoresis showed high molecular weight DNA samples, with no signs of degradation.
Sequencing of the bisulfite-converted DNA samples/libraries generated an average of approximately 32 million raw reads for samples of the two experimental groups. Approximately 33% of the reads generated per sample showed unique alignment, i.e., they aligned with only one region of the reference genome. About 45% of the reads were mapped to two or more regions and about 22% could not be aligned to the reference genome (Figure 2).
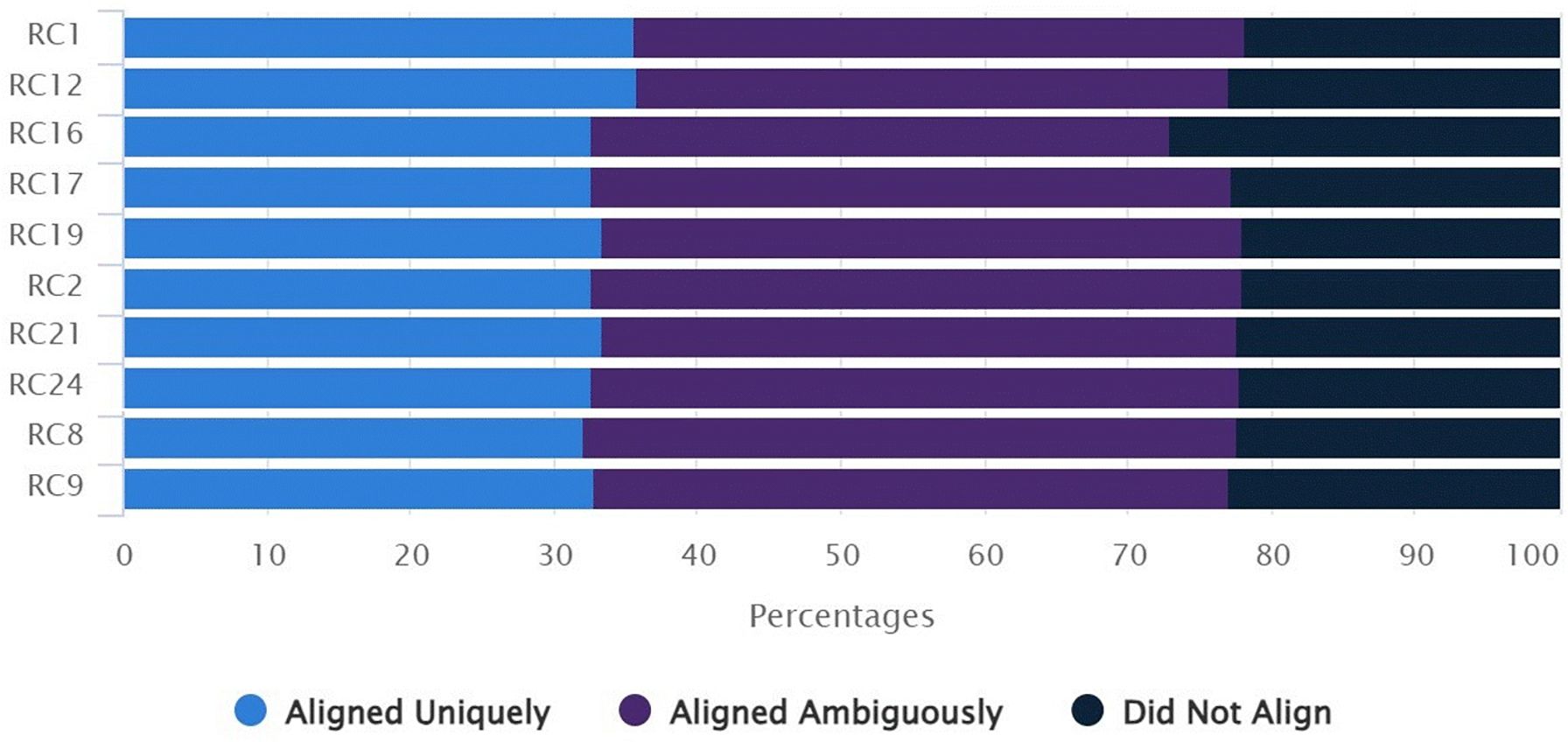
Figure 2. Percentage of reads generated by RRBS aligned or not to the bovine reference genome. RC1, RC2, RC8, RC9 and RC12: samples from NCF; RC16, RC17, RC19, RC21 and RC24: samples from CF. Blue bar: percentage of reads aligned to a unique region of the reference genome; purple bar: percentage of reads aligned to two or more regions of the reference genome; black bar: percentage of reads not aligned to regions of the reference genome.
The rate of bisulfite conversion of unmethylated cytosines exceeded 99% for CpG and non-CpG, indicating adequate bisulfitation of the samples.
Among reads mapped to a unique site in the genome, an average of seven million CpG cytosines were identified per sample, with a mean coverage of approximately 7X for CpG, as shown in Table 4.
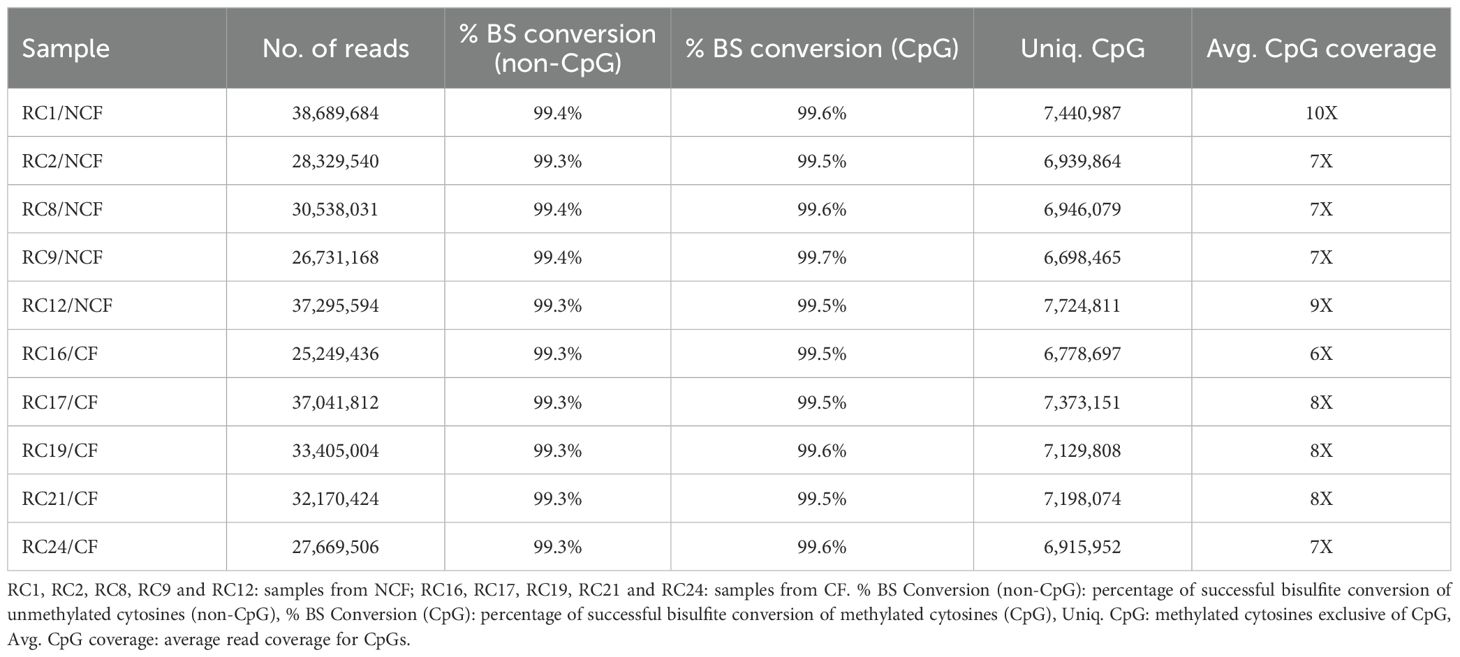
Table 4. Data generated by reduced representation bisulfite sequencing.
3.3 Identification and characterization of differentially methylated regions
For animals in CF, the mean genome-wide methylation level was 4.25% of CpG, 0.54% of CHG (where H = A, T or C), and 0.46% of CHH. The values for NCF animals were 4.07% of CpG, 0.50% of CHG and 0.43% of CHH, resulting in a higher mean methylation level (0.18% of CpG, 0.04% of CHG, and 0.03% of CHH) in animals of the supplemented group during the cow-calf phase.
Based on the contrast established between the groups of animals, 974 regions that differed in methylation levels at weaning were identified (DMRs: > 25% and q-value < 0.05). In CF, 431 regions showed hypermethylation, corresponding to 44.25% of all regions identified, while 543 showed hypomethylation, corresponding to 55.75%. Among DMRs, 54% occurred in intergenic regions of the genome, while the remaining regions were distributed across promoter and gene body regions (Figure 3A). About 22% of the DMRs were in CpGi, while 25% were present in island shores (Figure 3B).
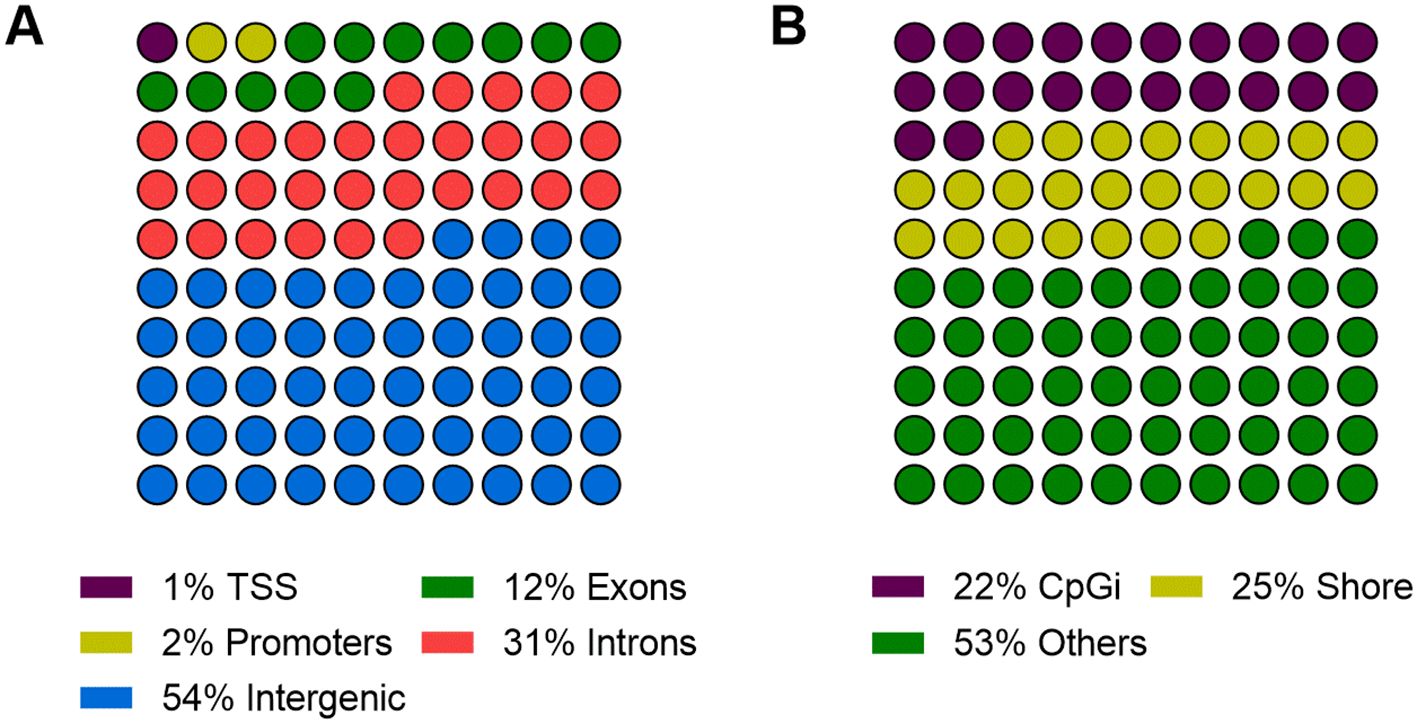
Figure 3. Proportion of DMRs among NCF and CF detected in different parts of the genome. (A) Overlap with intergenic and gene regions (promoter, transcription start site – TSS, exon and intron). (B) Overlap with CpG islands (CpGi), island shores, and others.
The 974 prospected DMRs overlapped with 241 genes which, as mentioned above, were called DMGs; among these genes, 108 were hypermethylated and 133 hypomethylated in CF. Of the 241 DMGs, 39 were correlated with DEGs, with a log2 FC ≥ 0.5 between the two experimental conditions. Part of the differentially expressed DMGs (DMGs/DEGs) exhibited more than one DMR, totaling 79 DMRs for 39 DMGs/DEGs (Table 5).
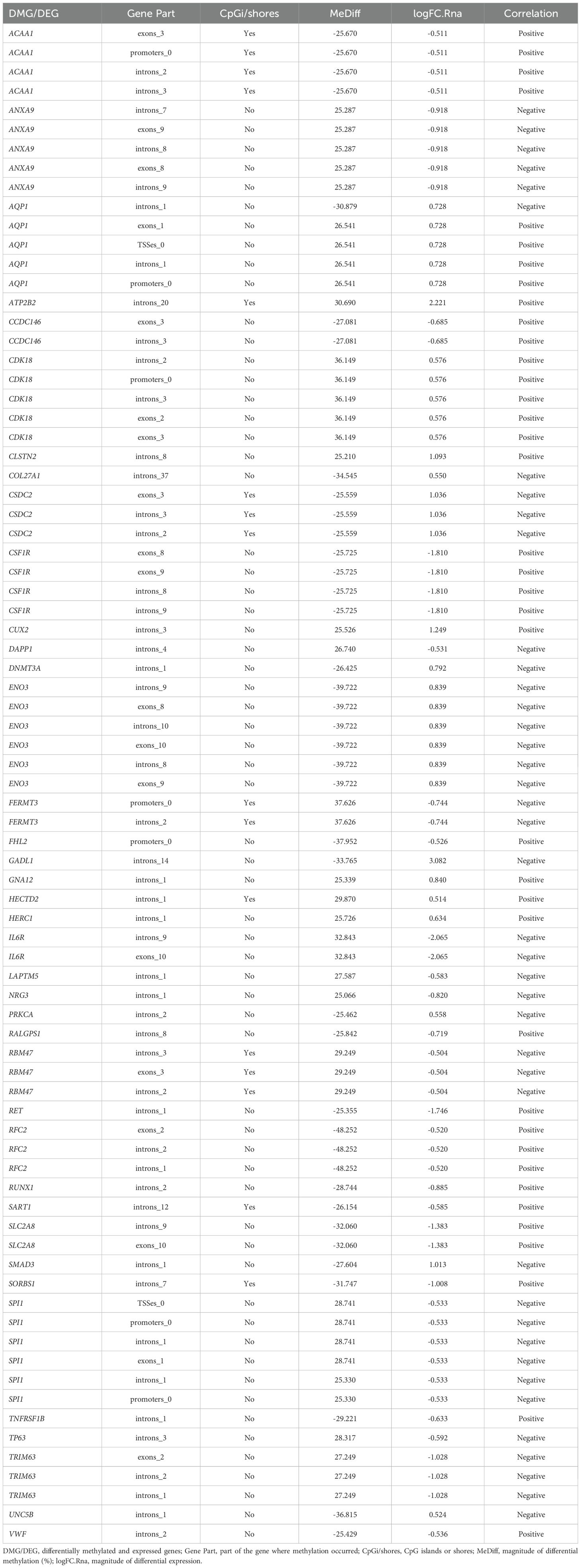
Table 5. Set of genes differentially methylated (DMGs) and differentially expressed (DEGs) between groups 1 and 2.
Twenty-one positively correlated DMGs/DEGs were identified; among these, nine [AQP1, ATP2B2, CDK18, CLSTN2, CSF1R, CUX2, GNA12, HECTD2, and HERC1] were hypermethylated and upregulated. Twelve genes [ACAA1, CCDC146, FHL2, RALGPS1, RET, RFC2, RUNX1, SART1, SLC2A8, SORBS1, TNFRSF1B, and VWF] exhibited hypomethylation associated with downregulation. On the other hand, 18 DMGs/DEGs showed a negative correlation. Among these, 10 [ANXA9, DAPP1, FERMT3, IL6R, LAPTM5, NRG3, RBM47, SPI1, TP63, and TRIM63] were identified as hypermethylated and downregulated, while eight [COL27A1, CSDC2, DNMT3A, ENO3, GADL1, PRKCA, SMAD3, and UNC5B] were characterized as hypomethylated and upregulated genes.
Thirty-three (85%) of the 39 DMGs/DEGs showed a methylation pattern compatible with the expression pattern (expected/consistent – according to Figure 1) and only six (15%) [ANXA9, ENO3, FHL2, GADL1, IL6R, and TP63] exhibited an unexpected/inconsistent pattern.
3.4 Functional enrichment analysis of DMGs/DEGs
Table 6 shows the BP-related GO terms enriched (p < 0.05) in the analysis involving the 39 DMGs/DEGs identified in animals of NCF and CF. Tables 7 and 8 list the enriched metabolic pathways (p < 0.05) using information from the KEGG and WikiPathways databases as a reference, respectively.
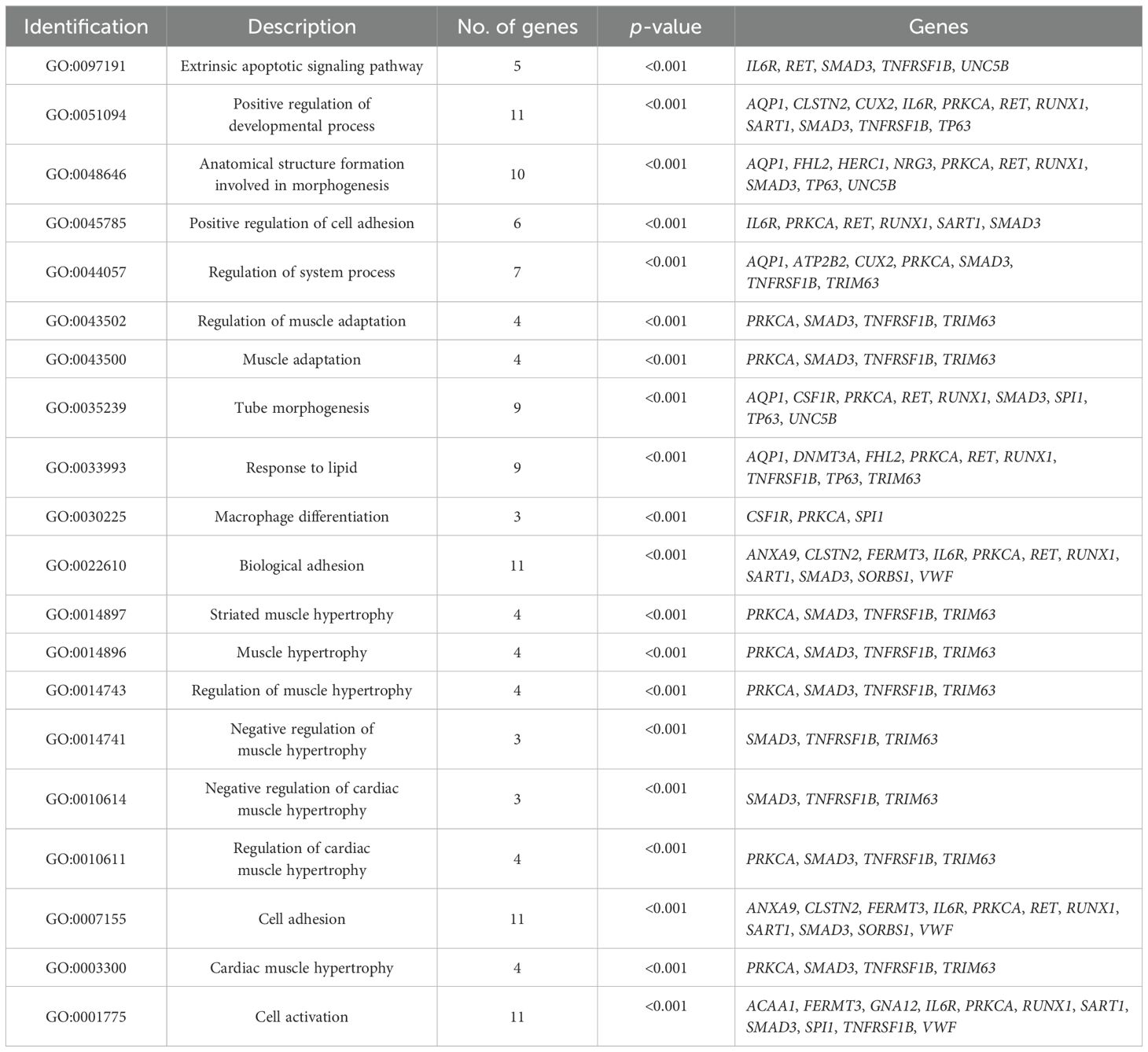
Table 6. Enriched biological processes (GO_BPs) considering genes differentially methylated and differentially expressed (DMGs/DEGs) between groups 1 and 2 at weaning.
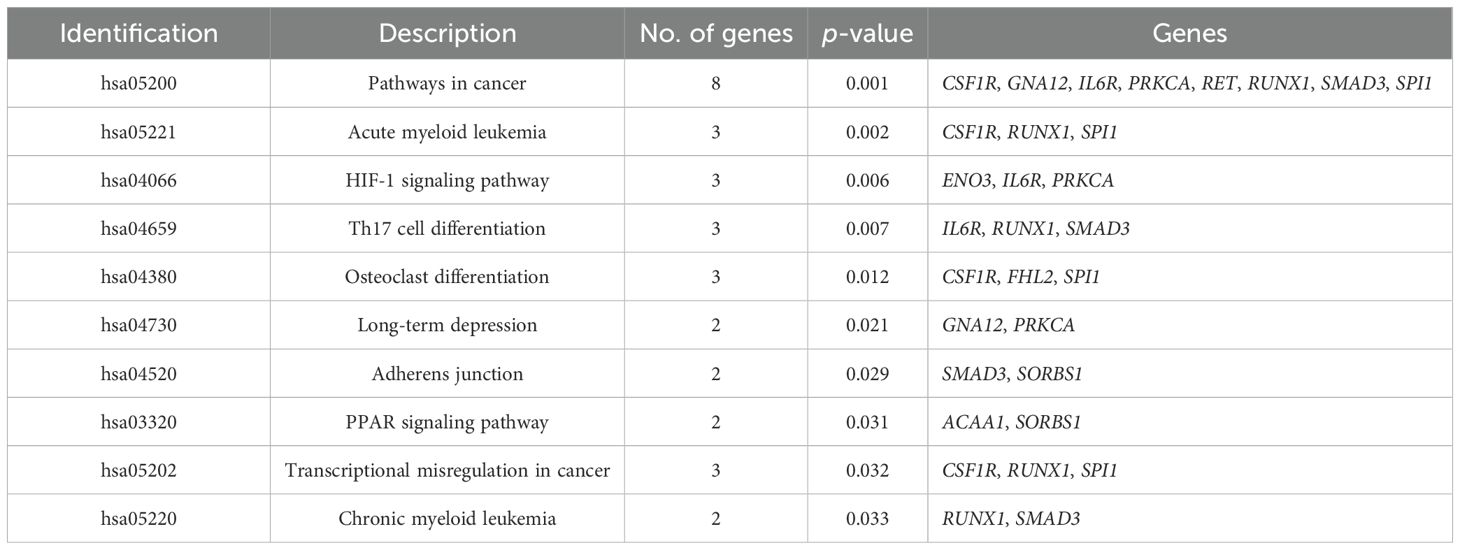
Table 7. Enriched metabolic pathways (KEGG Pathways) considering genes differentially methylated and differentially expressed (DMGs/DEGs) between groups 1 and 2 at weaning.
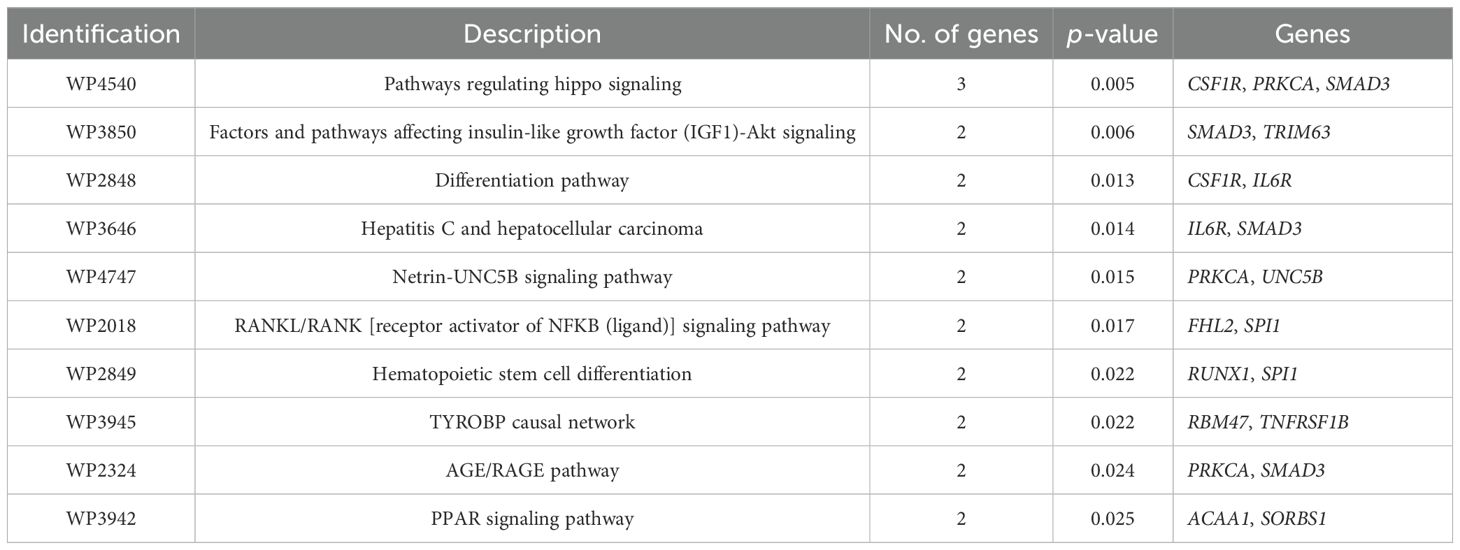
Table 8. Enriched metabolic pathways (WikiPathways) considering genes differentially methylated and differentially expressed (DMGs/DEGs) between groups 1 and 2 at weaning.
The higher WW, carcass fat coverage, and marbling (IMF and MS) observed in CF animals indicate that supplementation during the cow-calf phase favored muscle growth and development until weaning and lipid metabolism (adipogenesis and lipogenesis) until slaughter. Thus, BPs, metabolic pathways, and genes directly related to muscle development and fat deposition in the carcass were identified. The following BPs linked to muscle development can be highlighted: negative regulation of muscle hypertrophy (GO:0014741), regulation of muscle hypertrophy (GO:0014743), muscle hypertrophy (GO:0014896), striated muscle hypertrophy (GO:0014897), muscle adaptation (GO:0043500), and regulation of muscle adaptation (GO:0043502). These processes are related to the response of the muscle to different stimuli, not only for muscle growth but also for functional adaptations that improve the efficiency of this tissue. Another important process, response to lipid (GO:0033993), is related to the mechanisms of lipid metabolism. Regarding metabolic pathways, the following pathways can be highlighted as the most relevant: PPAR signalin pathway (hsa03320, WP3942) and factors and pathways affecting insulin-like growth factor (IGF1)-Akt signaling (WP3850).
Considering the enriched pathways and processes identified as the most important for the contrasting phenotypes between groups, the following DMGs/DEGs can be highlighted: ACAA1, SORBS1, SMAD3, TRIM63, PRKCA, DNMT3A, FHL2, NRG3, RUNX1, and TP63. The FHL2 and TP63 genes will not be included in the discussion since their methylation and expression patterns were incompatible with the expected. Although the SLC2A8 DMG/DEG is not part of any enriched BP or pathway, this gene will be discussed due to its high affinity for glucose in skeletal muscle and adipose tissues.
4 Discussion
4.1 Effect of supplementation on genomic methylation
Although subtle, the difference in the global methylation pattern between NCF and CF calves indicates changes in the genome in response to creep-feeding (increased methylation in CF). Andrés et al. (2021) demonstrated that diet can influence DNA methylation patterns by analyzing global blood methylation patterns, biochemical profiles, and metabolomes of ewe lambs born to mothers subjected to early food restriction or fed ad libitum. Results revealed significant enrichment in functional categories related to cellular processes, phosphorylation, nervous system, immune response, and reproductive function in lambs born to mothers with food restriction, attributed to differences in gene methylation within these categories. Nutrition can influence genome methylation patterns by providing substrates necessary for adequate DNA methylation or cofactors that modulate the enzymatic activity of DNA methyl transferases (DNMTs). As a universal methyl donor for methyltransferases, S-adenosylmethionine (SAM) is synthesized in the methionine cycle from different dietary precursors (McKay and Mathers, 2011; Feil and Fraga, 2012). All of these precursors enter the methionine pathway at different points and contribute to the net synthesis of SAM. Thus, increased availability of SAM results in global DNA hypermethylation and vice versa (Zhang, 2015). In addition to the indirect regulation of DNA methylation patterns by SAM modulation, several other dietary compounds can directly influence the expression or activity of DNMTs (Mukherjee et al., 2015).
Del Corvo et al. (2021) demonstrated an increase in methylation of approximately 0.54% in CpG, 0.06% in CHG, and 0.02% in CHH in Nellore cattle recovered after a period of heat stress. Values in Angus were 67.7% CG, 0.3% CHG, and 0.2% CHH methylation in the experimental period and 67.14% CG, 0.3% CHG, and 0.22% CHH methylation in the post challenge period. Even if they do not reach significance on a genomic scale, these changes can lead to distinct patterns of gene expression and, consequently, to divergent phenotypes (Ribeiro, 1994; Miller and Grant, 2012). Furthermore, analyzing the bovine methylome, De Souza et al. (2022) identified epigenetic mechanisms associated with meat tenderness in the Nellore breed, as well as genes with differential methylation in contrasting groups for meat tenderness. Taken together, these studies demonstrate the need to investigate genes that show differential methylation in contrasting groups of animals, either due to differences in a particular phenotype of interest or as a result of exposure to different experimental treatments, as well as to elucidate the metabolic pathways affected by these changes. Given that supplementation affects production costs, a limitation of this research was the absence of testing various supplementation levels. This could potentially reveal similar outcomes with lower supplementation levels or even more intriguing results with higher levels.
4.2 Metabolic pathways related to muscle and adipose tissue development
As practical implications, this study demonstrated that energy-protein supplementation of F1 Angus x Nellore calves during the rearing phase at 1% of their live weight was sufficient to promote, through changes in DNA methylation patterns and gene expression, increased muscle and fat deposition in the carcass, traits of interest to the market.
The insulin-like growth factor 1 (IGF1)-Akt signaling pathway plays a crucial role in muscle growth, influencing both protein synthesis and degradation by activating the mTOR pathway (Schiaffino and Mammucari, 2011; Jia et al., 2014; Yoshida and Delafontaine, 2020) and by inhibiting GSK3B (Cross et al., 1994, Cross et al., 1995) and FoxO transcription factors, thereby preventing muscle atrophy (Sartori et al., 2021). Within this context, calves that received a daily supplement of 1% of their body weight containing 22% crude protein and 65% total digestible nutrients, as well as inclusion of 44.8% ground corn as a glucose source, responded with a higher body weight at weaning. This improvement could be attributed to the activation of the IGF1-Akt pathway; although not measured in this study, this activation was possibly induced by the elevation of IGF-1 and insulin, thus facilitating protein synthesis via mTOR activation. However, supplementation during the cow-calf phase did not result in significant differences in body weight (BWf and HCW) after finishing or REA, an indicator of muscularity assessed during slaughter, although the IGF1-Akt pathway was active at weaning. This lack of a difference may indicate a change in gain composition (Asher et al., 2018), specifically a reduction in the rate of lean tissue deposition and an earlier onset of fat accumulation (Gerrits et al., 1997) after the supplementation period. This hypothesis is supported by the increase in BFT and marbling (IMF and MS) in animals supplemented during the cow-calf phase, possibly due to activation of the PPAR signaling pathway.
The PPAR signaling pathway involves PPAR nuclear receptors, which act as key transcription factors in the regulation of lipid and glucose metabolism (Gross et al., 2017). These receptors respond to environmental stimuli and modulate the expression of genes related to lipid metabolism and energy homeostasis (Lamichane et al., 2018; Attianese and Desvergne, 2015). During adipogenesis, specific activation of PPARγ, a key transcription factor of PPARs that is highly expressed in mature adipocytes, is fundamental for the commitment of mesenchymal stem cells to preadipocytes and their eventual differentiation into mature adipocytes (Queiroz et al., 2009; Wu et al., 1999). The inclusion of long-chain fatty acids in the diet has been associated with higher expression of PPAR-related genes in cattle and humans (Bionaz et al., 2012; Sampath and Ntambi, 2004), highlighting the importance of dietary sources for the stimulation of this pathway during adipocyte differentiation. Unsaturated fatty acids such as linoleic and linolenic acids are found in oilseeds such as soybeans (Ladeira et al., 2020). Although these acids undergo ruminal biohydrogenation, a fraction can escape to the duodenum and be absorbed (Duckett and Gillis, 2010). The present results of daily supplementation of calves with 1% of their body weight of creep feed, with the inclusion of 40.4% of soybean meal in the concentrate, suggest that a fraction of the unsaturated fatty acids in the supplement can reach skeletal muscle. This fact may contribute to the upregulation of genes associated with the PPAR pathway, which would explain the greater fat deposition in CF animals. The synergistic interaction between the IGF1-Akt and PPAR signaling pathways thus indicates a complex metabolic mechanism that regulates growth and body composition.
4.3 Main DMGs/DEGs related to muscle hypertrophy, adipogenesis and lipogenesis
In the present study, hypermethylation and downregulation of the neuregulin-3 (NRG3) gene at weaning were observed in calves supplemented during the cow-calf phase. This gene encodes peptide ligands for transmembrane tyrosine kinase receptors, which regulate cell growth and differentiation, affecting different cell types (Zhang et al., 1997). In humans, variants of NRG3 have been associated with the body mass index and biological processes that regulate body weight (Zhou et al., 2020).
Genes of the neuregulin family are important for the activation of skeletal muscle satellite cells, which are essential for repair processes (Gumà et al., 2010). Regarding muscle growth processes, satellite cells play a fundamental role in muscle fiber hypertrophy (Picard and Gagaoua, 2020). Although supplemented calves had higher WW, the downregulation of NRG3, as well as of RUNX family transcription factor 1 (RUNX1), in this case hypomethylated, may be a response to anticipated muscle hypertrophy. RUNX1 is known to cooperate with MyoD during the early stages of myogenesis (Zhou et al., 2022). Therefore, the downregulation of NRG3 and RUNX1 suggests that supplementation during the cow-calf phase triggered increased satellite cell activity, leading to the early downregulation of genes crucial to the anatomical structure formation involved in morphogenesis pathway, which is mainly active during the early periods of development such as the embryonic and fetal stages.
This hypothesis is strengthened by the hypomethylation and upregulation of the SMAD3 gene observed in calves supplemented during the cow-calf phase. This gene, which was enriched in the factors and pathways affecting insulin-like growth factor (IGF1)-Akt signaling pathway, plays an important role in the regulation of cellular responses to TGF-β (Shi et al., 2016). During fetal myogenesis, this gene is negatively regulated because of its repressive activity on myogenic regulatory factors such as those of the MyoD family (Liu et al., 2001). However, a study has shown that upregulation of SMAD3 significantly increases the number of satellite cells by self-renewal during postnatal myogenesis in mice, which may contribute to muscle hypertrophy (Ge et al., 2011). Additionally, these authors demonstrated that inactivation of SMAD3 in mice can upregulate MuRF1 and the ubiquitin-proteasome system responsible for muscle atrophy. Muscle ring finger 1, also known as TRIM63, was hypermethylated and downregulated in the supplemented calves studied here. Suppression of the expression of this gene that integrates the factors and pathways affecting insulin-like growth factor (IGF1)-Akt signaling pathway promotes muscle hypertrophy (Peris-Moreno et al., 2021), which would explain the higher WW observed in supplemented calves.
Intramuscular fat, a qualitative meat trait of high commercial value, is influenced not only by intense cell proliferation activity during fetal life but also by a specific interval within the first 250 days after birth. During this period, known as the “marbling window”, feeding a grain-rich diet specifically promotes the generation of new intramuscular adipocytes through hyperplasia, thereby establishing the sites for fat accumulation during the finishing phase (Du et al., 2013). This process paves the way for the activation of genes and other metabolic pathways associated with adipose tissue deposition. This hypothesis is strengthened by the observation of enrichment of the PPAR signaling pathway, accompanied by an increase in BFT and IMF at the end of the finishing period in supplemented calves. Genes of the PPAR family, particularly PPARγ, are key transcription factors that regulate preadipocyte differentiation and adipocyte metabolism (Duarte et al., 2013). Although the PPARγ gene was not directly demonstrated in this study, hypomethylation and upregulation of DNMT3A was observed in supplemented calves. Studies of subcutaneous adipose tissue in adult pigs revealed an increase in DNMT3A and PPARγ expression when compared to piglets (Qimuge et al., 2019). This increase in DNMT3A expression in adult animals suggests that the gene plays a fundamental role in the development and maturation of adipose tissue. Furthermore, the same authors found an increase in DNMT3A expression during the differentiation of intramuscular preadipocytes, indicating the involvement of this gene not only in the early development of adipose tissue but also in the maturation of adipocytes, a key process for the elevation of adipose tissue mass and for local metabolic regulation (Qimuge et al., 2019). The high expression of DNMT3A in supplemented calves observed at weaning may thus indicate that a high proportion of adipocytes were already mature and ready to start the intensive lipogenic process during the finishing period, which explains the greater marbling deposition in meat (IMF and MS) (Table 3).
The SORBS1 gene, a strong candidate associated with higher IMF content in cattle (Sasaki et al., 2006) that integrates the PPAR signaling pathway in the present study, was hypomethylated and downregulated at weaning in supplemented calves. SORBS1 is a member of the SORBS family and encodes proteins involved in insulin signaling and stimulation (Chang et al., 2016, Chang et al., 2018; Germain et al., 2015). In 3T3-L1 adipocytes, an established cell line commonly used to study adipocyte biology, SORBS1 plays an important role in the regulation of glucose uptake (Baumann et al., 2000). This gene was also found in C2C12 muscle cells (model of muscle cell line); in addition, its downregulation is related to insulin resistance due to AMPK activation in this cell type but not in 3T3-L1 adipocytes (Aye et al., 2023; Yang et al., 2024). This finding suggests that the downregulation of SORBS1 at weaning in supplemented calves is associated with a decrease in glucose utilization by muscle fibers, reducing competition between the latter and intramuscular adipocytes for this substrate for lipogenesis. The fact that skeletal muscle is a heterogeneous tissue that contains both muscle fibers and adipocytes (Ramírez-Zamudio et al., 2023) implies that the downregulation of SORBS1 cannot be necessarily attributed exclusively to adipocytes. Additionally, hypomethylation and upregulation of the PRKCA gene were detected in supplemented calves. Although PRKCA acts as a negative regulator of insulin receptor signaling, it is important to highlight that insulin is known for promoting adipose cell differentiation, IMF deposition, and glucose oxidation in muscle cells (Lim et al., 2011; Hausman et al., 2009). The results of this study suggest a complex role of PRKCA in the metabolism of supplemented calves, indicating that its upregulation may alter the typical metabolic functions of insulin in unpredicted manners. Insulin, when present in supraphysiological levels, can also induce adipogenic differentiation through the stimulation of IGF-1, a process known as transactivation (Fernyhough et al., 2007). These findings therefore reinforce our theory that muscle fibers of supplemented animals consume less glucose, increasing its availability for lipogenesis in adipocytes.
The reduced glucose utilization by muscle fibers in supplemented calves may also be supported by the observation of hypomethylation and downregulation of SLC2A8 at weaning. The SLC2A8 gene encodes GLUT8 transporters with high affinity for glucose in brain cells, as well as in skeletal muscle and adipose tissues (Gawlik et al., 2008). However, translocation of these transporters to the surface for insulin-stimulated glucose uptake has only been observed in murine muscle cells (Carayannopoulos et al., 2000) but not in adipocytes (Lisinski et al., 2001). Additionally, a study using cultured 3T3-L1 adipocytes suggested that GLUT8 expression is markedly increased during the phase of cell differentiation (Scheepers et al., 2001). This finding suggests that creep-feeding from day 30 of age until weaning of calves at 210 days, almost at the end of the “marbling window” (250 days) (Du et al., 2013), may have accelerated the differentiation of intramuscular adipose tissue cells when compared to the unsupplemented group. This anticipated adipocyte differentiation, facilitated by supplementation, may be associated with the downregulation of SLC2A8. This reflects complex metabolic dynamics involving various cell types and developmental stages within the muscle tissue that, in an integrated manner, affect glucose metabolism, energy homeostasis, and fat deposition.
The PPAR signaling pathway enriched in this study is essential for cell differentiation and lipid and carbohydrate metabolism (Feige et al., 2006). The ACAA1 gene associated with the PPAR signaling pathway was found to be hypomethylated and downregulated in animals submitted to creep-feeding. This gene plays an essential role in the synthesis and regulation of enzymes involved in the beta-oxidation of long-chain fatty acids (Wanders et al., 2010). Studies on sheep (Wang et al., 2021), birds (Li et al., 2019) and pigs (Yao et al., 2021) generally reported that ACAA1 expression is inversely associated with IMF deposition. These data are consistent with the results obtained here for supplemented calves, in which downregulation of ACAA1 was correlated with greater deposition of marbling fat. Thus, our theory is that the downregulation of ACAA1 in supplemented calves is due to the glucose-rich environment provided by the high dietary inclusion of corn (44.8%). Additionally, the decreased competition between muscle fibers and adipocytes for glucose, as demonstrated by regulation of the SORBS1 and PRKCA genes, which are members of the PPAR signaling pathway and regulators of glucose metabolism in muscle fibers, reduces the beta-oxidation of fatty acids in adipocytes.
5 Conclusion
The results of the present study support the hypothesis that the implementation of creep-feeding supplementation during the cow-calf phase induces changes in DNA methylation and gene expression levels, modifying the regulation of biological processes and metabolic pathways related to muscle growth and fat deposition (adipogenesis and lipogenesis) in beef cattle. Epigenetic regulations of genes that play key roles in muscle growth and development, PPAR signaling, adipogenesis, insulin response, and lipid metabolism were identified. These biological changes help explain the phenotypes observed in supplemented animals in terms of both performance until weaning (higher weight and weight gain until weaning) and traits related to meat quality (higher marbling).
Data availability statement
All relevant data are within the paper. RNA-Seq (RNA sequencing) and RRBS (reduced representation bisulfite sequencing) data may be made available by contacting the corresponding author (cm9nZXJpby5jdXJpQHVuZXNwLmJy).
Ethics statement
The animal studies were approved by Animal Use Ethics Committee of the School of Agricultural and Veterinary Sciences (FCAV), UNESP, Jaboticabal, São Paulo, Brazil (certificate number 013689/19). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
LR: Investigation, Software, Writing – original draft. GR-Z: Investigation, Writing – review & editing. GP: Investigation, Writing – review & editing, Conceptualization, Data curation, Formal Analysis, Methodology, Software, Supervision, Validation, Visualization, Writing – original draft. JT: Data curation, Methodology, Writing – review & editing. LT: Investigation, Software, Writing – original draft. OM: Conceptualization, Data curation, Funding acquisition, Methodology, Resources, Writing – review & editing. LC: Conceptualization, Data curation, Methodology, Writing – review & editing, Formal Analysis. WB: Conceptualization, Formal Analysis, Methodology, Writing – review & editing, Visualization. RC: Conceptualization, Formal Analysis, Methodology, Visualization, Writing – review & editing, Data curation, Funding acquisition, Investigation, Project administration, Resources, Software, Supervision, Validation, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Foundation of the State of São Paulo (Fundação de Amparo à Pesquisa do Estado de São Paulo – FAPESP) for funding (grants numbers 2019/ 22621-3 and 2023/05002-3). Coordination for the Improvement of Higher Education Personnel (Coordenação de Aperfeiçoamento de Pessoal de Nıvel ́Superior – CAPES), Finance code 001.
Acknowledgments
To the São Paulo State University (Universidade Estadual Paulista – UNESP) for providing the infrastructure for all the stages of this research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akalin A., Kormaksson M., Li S., Garrett-Bakelman F. E., Figueroa M. E., Melnick A., et al. (2012). methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13, 1–9. doi: 10.1186/gb-2012-13-10-r87
Andrés S., Madsen O., Montero O., Martín A., Giráldez F. J. (2021). The role of feed restriction on DNA methylation, feed efficiency, metabolome, biochemical profile, and progesterone patterns in the female filial generation (F1) obtained from early feed restricted ewes (F0). Front. Physiol. 12. doi: 10.3389/fphys.2021.779054
Andrews S. (2010). FastQC: a quality control analysis tool for high throughput sequencing data. Gitbub.
Asher A., Shabtay A., Cohen-Zinder M., Aharoni Y., Miron J., Agmon R., et al. (2018). Consistency of feed efficiency ranking and mechanisms associated with inter-animal variation among growing calves. J. Anim. Sci. 96, 990–1009. doi: 10.1093/jas/skx045
Attianese G. M. G., Desvergne B. (2015). Integrative and systemic approaches for evaluating PPARβ/δ (PPARD) function. Nucl. Receptor Signaling 13, 13001. doi: 10.1621/nrs.13001
Aye C. C., Hammond D. E., Rodriguez-Cuenca S., Doherty M. K., Whitfield P. D., Phelan M. M., et al. (2023). CBL/CAP is essential for mitochondria respiration complex I assembly and bioenergetics efficiency in muscle cells. Int. J. Mol. Sci. 24. doi: 10.3390/ijms24043399
Baldassini W. A., Ferreira M. S., Santiago B. M., Chardulo L. A. L., Curi R. A., Lanna D. P., et al. (2021). Intake, performance, meat quality and fatty acid profile of Nellore bulls finished in feedlot with diets containing dry corn gluten feed. Livestock Sci. 253, 104715. doi: 10.1016/j.livsci.2021.104715
Baumann C. A., Ribon V., Kanzaki M., Thurmond D. C., Mora S., Shigematsu S., et al. (2000). CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature 407 6801, 202–207. doi: 10.1038/35025089
Bionaz M., Thering B. J., Loor J. J. (2012). Fine metabolic regulation in ruminants via nutrient–gene interactions: saturated long-chain fatty acids increase expression of genes involved in lipid metabolism and immune response partly through PPAR-α activation. Br. J. Nutr. 107 2, 179–191. doi: 10.1017/S0007114511002777
Carayannopoulos M. O., Chi M. M., Cui Y., Pingsterhaus J. M., McKnight R. A., Mueckler M., et al. (2000). GLUT8 is a glucose transporter responsible for insulin-stimulated glucose uptake in the blastocyst. Proc. Natl. Acad. Sci. U.S.A. 97, 7313–7318. doi: 10.1073/pnas.97.13.7313
Chang T. J., Wang W. C., Hsiung C. A., He C. T., Lin M. W., Sheu W. H. H. S., et al. (2016). Genetic variation in the human SORBS1 gene is associated with blood pressure regulation and age at onset of hypertension: a SAPPHIRe Cohort Study. Medicine 95 10, e2970. doi: 10.1097/MD.0000000000002970
Chang T. J., Wang W. C., Hsiung C. A., He C. T., Lin M. W., Wayne H. S., et al. (2018). Genetic variation of SORBS1 gene is associated with glucose homeostasis and age at onset of diabetes: A SAPPHIRe Cohort Study. Sci. Rep. 8 1, 12. doi: 10.1038/s41598-018-28891-z
Cross D. A., Alessi D. R., Cohen P., Andjelkovich M., Hemmings B. A. (1995). Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378 6559, 785–789. doi: 10.1038/378785a0
Cross D. A., Alessi D. R., VanDenheede J. R., McDowell H. E., Hundal H. S., Cohen P. (1994). The inhibition of glycogen synthase kinase-3 by insulin or insulin-like growth factor 1 in the rat skeletal muscle cell line L6 is blocked by wortmannin, but not by rapamycin: evidence that wortmannin blocks activation of the mitogen-activated protein kinase pathway in L6 cells between Ras and Raf. Biochem. J. 303 1, 21–26. doi: 10.1042/bj3030021
Dantas C. C. O., De Mattos Negrão F., Geron L. J. V., Mexia A. A. (2010). O uso da técnica do Creep-feeding na suplementação de bezerros. PubVet 4, 899–904.
Day J. J., Kennedy A. J., Sweatt J. D. (2015). DNA methylation and its implications and accessibility for neuropsychiatric therapeutics. Annu. Rev. Pharmacol. Toxicol. 55, 591–611. doi: 10.1146/annurev-pharmtox-010814-124527
Del Corvo M., Lazzari B., Capra E., Zavarez L., Milanesi M., Utsunomiya Y. T., et al. (2021). Methylome patterns of cattle adaptation to heat stress. Front. Genet. 12. doi: 10.3389/fgene.2021.633132
De Souza M. M., Niciura S. C. M., Rocha M. I. P., Pan Z., Zhou H., Bruscadin J. J., et al. (2022). DNA methylation may affect beef tenderness through signal transduction in Bos indicus. Epigenet. Chromatin 15 15. doi: 10.1186/s13072-022-00449-4
Doi A., Park I. H., Wen B., Murakami P., Aryee M. J., Irizarry R., et al. (2009). Differential methylation of tissue-and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 41 12, 1350–1353. doi: 10.1038/ng.471
Du M., Huang Y., Das A., Yang Q., Duarte M., Dodson M., et al. (2013). Meat Science and Muscle Biology Symposium: Manipulating mesenchymal progenitor cell differentiation to optimize performance and carcass value of beef cattle. J. Anim. Sci. 91, 1419–1427. doi: 10.2527/jas.2012-5670
Duarte M. S., Paulino P. V. R., Das A. K., Wei S., Serrão N. V. L., Fu X., et al. (2013). Enhancement of adipogenesis and fibrogenesis in skeletal muscle of Wagyu compared with Angus cattle. J. Anim. Sci. 91, 2938–2946. doi: 10.2527/jas.2012-5892
Duckett S., Gillis M. (2010). Effects of oil source and fish oil addition on ruminal biohydrogenation of fatty acids and conjugated linoleic acid formation in beef steers fed finishing diets. J. Anim. Sci. 88, 2684–2691. doi: 10.2527/jas.2009-2375
Enright B. P., Jeong B. S., Yang X., Tian X. C. (2003). Epigenetic characteristics of bovine donor cells for nuclear transfer: levels of histone acetylation. Biol. Reprodution 69, 1525–1530. doi: 10.1095/biolreprod.103.019950
Farias N., Ho N., Butler S., Delaney L., Morrison J., Shahrzad S., et al. (2015). The effects of folic acid on global DNA methylation and colonosphere formation in colon cancer cell lines. J. Nutr. Biochem. 26 8, 818–826. doi: 10.1016/j.jnutbio.2015.02.002
Farkas S. A., Befekadu R., Hahn-Strömberg V., Nilsson T. K. (2015). DNA methylation and expression of the folate transporter genes in colorectal cancer. Tumor Biol. 36, 5581–5590. doi: 10.1007/s13277-015-3228-2
Feige J. N., Gelman L., Michalik L., Desvergne B., Wahli W. (2006). From molecular action to physiological outputs: peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 45, 120–159. doi: 10.1016/j.plipres.2005.12.002
Feil R., Fraga M. F. (2012). Epigenetics and the environment: emerging patterns and implications. Nat. Rev. Genet. 13 2, 97–109. doi: 10.1038/nrg3142
Fernyhough M. E., Okine E., Hausman G., Vierck J. L., Dodson M. V.. (2007). PPARγ and GLUT-4 expression as developmental regulators/markers for preadipocyte differentiation into an adipocyte. Domest. Anim. Endocrinol. 33 4, 367–378. doi: 10.1016/j.domaniend.2007.05.001
Ferreira M., Niehues M. B., Tomaz L. A., Baldassini W., Ladeira M., Arrigoni M., et al. (2020). Dry matter intake, performance, carcass traits and expression of genes of muscle protein metabolism in cattle fed increasing levels of de-oiled wet distillers grains. Anim. Feed Sci. Technol. 269, 114627. doi: 10.1016/j.anifeedsci.2020.114627
Gawlik V., Schmidt S., Scheepers A., Wennemuth G., Augustin R., Aumuller G., et al. (2008). Targeted disruption of Slc2a8 (GLUT8) reduces motility and mitochondrial potential of spermatozoa. Mol. Membrane Biol. 25, 224–235. doi: 10.1080/09687680701855405
Ge X., McFarlane C., Vajjala A., Lokireddy S., Ng Z. H., Tan C. K., et al. (2011). Smad3 signaling is required for satellite cell function and myogenic differentiation of myoblasts. Cell Res. 21, 1591–1604. doi: 10.1038/cr.2011.72
Germain M., Pezzolesi M. G., Sandholm N., McKnight A. J., Susztak K., LAJER M., et al. (2015). SORBS1 gene, a new candidate for diabetic nephropathy: results from a multi-stage genome-wide association study in patients with type 1 diabetes. Diabetologia 58, 543–548. doi: 10.1007/s00125-014-3459-6
Gerrits W. J., France J., Dijkstra J., Bosch M. W., Tolman G. H., Tamminga S. (1997). Evaluation of a model integrating protein and energy metabolism in preruminant calves. J. Nutr. 127, 1243–1252. doi: 10.1093/jn/127.6.1243
Gross B., Pawlak M., Lefebvre P., Staels B. (2017). PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 13 1, 36–49. doi: 10.1038/nrendo.2016.135
Gumà A., Martínez-Redondo V, López-Soldado I., Cantó C., Zorzano A.. (2010). Emerging role of neuregulin as a modulator of muscle metabolism. Am. J. Physiology-Endocrinology Metab. 298 4, E742–E750. doi: 10.1152/ajpendo.00541.2009
Hausman G. J., Dodson M. V., Ajuwon K., Azain M., Barnes K. M., Guan L. L., et al. (2009). Board-invited review: the biology and regulation of preadipocytes and adipocytes in meat animals. J. Anim. Science v. 87 N. 4 p. 1218-1246, 2009. doi: 10.2527/jas.2008-1427
Irizarry R. A., Ladd-Acosta C., Wen B., Wu Z., Montano C., Onyango P., et al. (2009). The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 41 2, 178–186. doi: 10.1038/ng.298
Jia L., Li Y. F., Wu G. F., Song Z. Y., Lu H. Z., Song C. C., et al. (2014). MiRNA-199a-3p regulates C2C12 myoblast differentiation through IGF-1/AKT/mTOR signal pathway. Int. J. Mol. Sci. 15 1, 296–308. doi: 10.3390/ijms15010296
Krueger F., Andrews S. R. (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27 11, 1571–1572. doi: 10.1093/bioinformatics/btr167
Ladeira M. M., de Oliveira D. M., Schoonmaker J. P., Chizzotti M. L., Barreto H. G., Paiva L. V., et al. (2020). Expression of lipogenic genes in the muscle of beef cattle fed oilseeds and vitamin E. Agri Gene 15, 100097. doi: 10.1016/j.aggene.2019.100097
Lamichane S., Dahal L. B., Kwon S. M. (2018). Pivotal roles of peroxisome proliferator-activated receptors (PPARs) and their signal cascade for cellular and whole-body energy homeostasis. Int. J. Mol. Sci. 19 4, 949. doi: 10.3390/ijms19040949
Langmead B., Salzberg S. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lanna D., Barioni L., Nepomuceno N., Almeida R., Tedeschi L. (2011). Ração de Lucro Máximo-RLM 3.2.1. Available online at: https://www.integrasoftware.com.br/rlm31/produto.php (Accessed October 12, 2020).
Li G., Fu S., Chen Y., Jin W., Zhai B., Li Y., et al. (2019). MicroRNA-15a regulates the differentiation of intramuscular preadipocytes by targeting ACAA1, ACOX1 and SCP2 in chickens. Int. J. Mol. Sci. 20, 16. doi: 10.3390/ijms20164063
Lim D., Kim N. K., Park H. S., Lee S. H., Cho Y. M., Oh S. J., et al. (2011). Identification of candidate genes related to bovine marbling using protein-protein interaction networks. Int. J. Biol. Sci. 7 7, 992. doi: 10.7150/ijbs.7.992
Lisinski I., Schurmann A., Joost H. G., Cushman S. W., Al-Hasani H. (2001). Targeting of GLUT6 (formerly GLUT9) and GLUT8 in rat adipose cells. Biochem. J. 358, 517–522. doi: 10.1042/bj3580517
Liu D., Black B. L., Derynck R. (2001). TGF-β inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. 15 22, 2950–2966. doi: 10.1101/gad.925901
Lopes L. S. F., Ferreira M. S., Baldassini W. A., Curi R. A., Pereira G. L., Machado Neto O. R., et al. (2020). Application of the principal component analysis, cluster analysis, and partial least square regression on crossbreed Angus-Nellore bulls feedlot finished. Trop. Anim. Health Production 52, 3655–3664. doi: 10.1007/s11250-020-02402-7
McKay J. A., Mathers J. C. (2011). Diet induced epigenetic changes and their implications for health. Acta Physiologica 202 2, 103–118. doi: 10.1111/j.1748-1716.2011.02278.x
Miller J. L., Grant P. A. (2012). The role of DNA methylation and histone modifications in transcriptional regulation in humans. Epigenetics: Dev. Dis. 61, 289–317. doi: 10.1007/978-94-007-4525-4_13
Mukherjee N., Kumar A. P., Ghosh R. (2015). DNA methylation and flavonoids in genitourinary cancers. Curr. Pharmacol. Rep. 1, 112–120. doi: 10.1007/s40495-014-0004-8
Oliveira N. F. P., Planello A. C. P., Andia D. C., Pardo A. P. S. (2010). Metilação de DNA e câncer. Rev. Bras. Cancerologia 56 4, 493–499. doi: 10.32635/2176-9745.RBC.2010v56n4.698
Peris-Moreno D., Cussonneau L., Combaret L., Polge C., Taillandier D. (2021). Ubiquitin Ligases at the heart of skeletal muscle atrophy control. Molecules 26 2, 407. doi: 10.3390/molecules26020407
Picard B., Gagaoua M. (2020). Muscle fiber properties in cattle and their relationships with meat qualities: An overview. J. Agric. Food Chem. 68 22, 6021–6039. doi: 10.1021/acs.jafc.0c02086
Portela A., Esteller M. (2010). Epigenetic modifications and human disease. Nat. Biotechnol. 28 10, 1057–1068. doi: 10.1038/nbt.1685
Qimuge N., He Z., Qin J., Sun Y., Wang X., Yu T., et al. (2019). Overexpression of DNMT3A promotes proliferation and inhibits differentiation of porcine intramuscular preadipocytes by methylating p21 and PPARg promoters. Gene 696, 54–62. doi: 10.1016/j.gene.2019.02.029
Queiroz J. C. F. D., Alonso-Vale M. I. C., Curi R., Lima F. B. (2009). Control of adipogenesis by fatty acids. Arquivos Brasileiros Endocrinologia Metabologia 53, 582–594. doi: 10.1590/S0004-27302009000500011
Ramírez-Zamudio G. D., Ganga M. J. G., Pereira G. L., Nociti R. P., Chiaratti M. R., Cooke R. F., et al. (2023). Effect of Cow-Calf Supplementation on Gene Expression, Processes, and Pathways Related to Adipogenesis and Lipogenesis in Longissimus thoracis Muscle of F1 Angus × Nellore Cattle at Weaning. Metabolites 13 2, 160–160. doi: 10.3390/metabo13020160
Reik W., Dean W., Walter J. (2001). Epigenetic reprogramming in mammalian development. Science 293 5532, 1089–1093. doi: 10.1126/science.10634
Ribeiro M. M. A. (1994). “Expressão gênica e metilação do DNA: causa ou consequência,” in Agroforum: Revista da Escola Superior Agrária de Castelo Branco, The University of Georgia, USA. vol. 15:19. , 9–14. Available at: http://hdl.handle.net/10400.11/676.
Robinson M. D., McCarthy D. J., Smyth G. K. (2010). EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26 1, 139–140. doi: 10.1093/bioinformatics/btp616
Robison A. J., Nestler E. J. (2011). Transcriptional and epigenetic mechanisms of addiction. Nat. Rev. Neurosci. 12 11, 623–637. doi: 10.1038/nrn3111
Sampath H., Ntambi J. M. (2004). Polyunsaturated fatty acid regulation of gene expression. Nutr. Rev. 62 9, 333–339. doi: 10.1111/j.1753-4887.2004.tb00058.x
Sartori R., Romanello V., Sandri M. (2021). Mechanisms of muscle atrophy and hypertrophy: implications in health and disease. Nat. Commun. 12 1, 330. doi: 10.1038/s41467-020-20123-1
Sasaki Y., Nagai K., Nagata Y., Doronbekov K., Nishimura S., Yoshioka S., et al. (2006). Exploration of genes showing intramuscular fat deposition-associated expression changes in musculus longissimus muscle. Anim. Genet. 37 1, 40–46. doi: 10.1111/j.1365-2052.2005.01380.x
Scheepers A., Doege H., Joost H. G., Schurmann A. (2001). Mouse GLUT8: genomic organization and regulation of expression in 3T3-L1 adipocytes by glucose. Biochem. Biophys. Res. Communication 288, 969–974. doi: 10.1006/bbrc.2001.5866
Scheffler J. M., McCann M. A., Greiner S. P., Jiang H., Hanigan M. D., Bridges G. A., et al. (2014). Early metabolic imprinting events increase marbling scores in fed cattle. J. Anim. Sci. 92 1, 320–324. doi: 10.2527/jas.2012-6209
Schiaffino S., Mammucari C. (2011). Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skeletal Muscle 1 1, 1–14. doi: 10.1186/2044-5040-1-4
Shi T., Peng W., Yan J., Cai H., Lan X., Lei C., et al. (2016). A novel 17 bp indel in the SMAD3 gene alters transcription level, contributing to phenotypic traits in Chinese cattle. Arch. Anim. Breed. 59 1, 151–157. doi: 10.5194/aab-59-151-2016
Smith S. B., Crouse D. J. (1984). Relative contributions of acetate, lactate and glucose to lipogenesis in bovine intramuscular and subcutaneous adipose tissue. J. Nutr. 114 4, 792–800. doi: 10.1093/jn/114.4.792
Stenz L., Schechter D. S., Serpa S. R., Paoloni-Giacobino A. (2018). Intergenerational transmission of DNA methylation signatures associated with early life stress. Curr. Genomics 19 8, 665–675. doi: 10.2174/1389202919666171229145656
Tost J. (2010). DNA methylation: an introduction to the biology and the disease-associated changes of a promising biomarker. Mol. Biotechnol. 44, 71–81. doi: 10.1007/s12033-009-9216-2
Tsankova N., Renthal W., Kumar A., Nestler E. J. (2007). Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 8 5, 355–367. doi: 10.1038/nrn2132
Varley K. E., Gertz J., Bowling K. M., Parker S. L., Reddy T. E., Pauli-Behn F., et al. (2013). Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 23, 555–585. doi: 10.1101/gr.147942.112
Wanders R. J. A., Ferdinandusse S., Brites P., Kemp S. (2010). Peroxisomes, lipid metabolism and lipotoxicity. Biochim. Biophys. Acta (BBA) - Mol. Cell Biol. Lipids 3, 272–280. doi: 10.1016/j.bbalip.2010.01.001
Wang Y., li X., Cao Y., Xiao C., Liu Y., Jin H., et al. (2021). Effect of the ACAA1 gene on preadipocyte differentiation in sheep. Front. Genet. 12. doi: 10.3389/fgene.2021.649140
Wang H., Tuominen L. K., Tsai C. (2011). SLIM: a sliding linear model for estimating the proportion of true null hypotheses in datasets with dependence structures. Bioinformatics 27 2, 225–231. doi: 10.1093/bioinformatics/btq650
Wang J., Vasaikar S., Shi Z., Greer M., Zhang B. (2017). WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 45, W1, W130–W137. doi: 10.1093/nar/gkx356
Wu Z., Rosen E. D., Brun R., Hauser S., Adelmant G., Troy A. E., et al. (1999). Cross-regulation of C/EBPα and PPARγ controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol. Cell 3 2, 151–158. doi: 10.1016/S1097-2765(00)80306-8
Yang Y. H., Fan X. X., Ye L., Huang W. J., Ko C. Y. (2024). Examining the molecular mechanisms of topiramate in alleviating insulin resistance: A study on C2C12 myocytes and 3T3L-1 adipocytes. Endocrine. 85, 168–180. doi: 10.1007/s12020-024-03706-6
Yao C., Pang D., Lu C., Xu A., Huang P., Ouyang H., et al. (2021). Investigation on the effect of two fat metabolism related pathways on intramuscular fat content in pigs. Pak. J. Zool 53, 1353–1366. doi: 10.17582/journal.pjz/20190407110454
Yoshida T., Delafontaine P. (2020). Mechanisms of IGF-1-mediated regulation of skeletal muscle hypertrophy and atrophy. Cells 9, 9. doi: 10.3390/cells9091970
Yuferov V., Levran O., Proudnikov D., Nielsen D. A., Kreek M. J. (2010). Search for genetic markers and functional variants involved in the development of opiate and cocaine addiction and treatment. Ann. New York Acad. Sci. 1, 184–207. doi: 10.1111/j.1749-6632.2009.05275.x
Zhang N. (2015). Epigenetic modulation of DNA methylation by nutrition and its mechanisms in animals. Anim. Nutr. 1 3, 144–151. doi: 10.1016/j.aninu.2015.09.002
Zhang D., Sliwkowski M. X., Mark M., Frantz G., Akita R., Sun Y., et al. (1997). Neuregulin-3 (NRG3): a novel neural tissue-enriched protein that binds and activates ErbB4. Proc. Natl. Acad. Sci. 94 18, 9562–9567. doi: 10.1073/pnas.94.18.9562
Zhou Y., Li Y., Meng Y., Wang J., Wu F., Ning Y., et al. (2020). Neuregulin 3 rs10748842 polymorphism contributes to the effect of body mass index on cognitive impairment in patients with schizophrenia. Trans. Psychiatry 10, 62. doi: 10.1038/s41398-020-0746-5
Keywords: Bos taurus x Bos indicus, methylome, nutrigenomics, RRBS, supplementation
Citation: Rodrigues LF, Ramírez-Zamudio GD, Pereira GL, Torrecilhas JA, Trevisan LA, Machado Neto OR, Chardulo LAL, Baldassini WA and Curi RA (2024) Epigenetic insights into creep-feeding: methylation profiling of Longissimus thoracis muscle at weaning in crossbred cattle. Front. Anim. Sci. 5:1476353. doi: 10.3389/fanim.2024.1476353
Received: 05 August 2024; Accepted: 11 October 2024;
Published: 31 October 2024.
Edited by:
Geoffrey E. Dahl, University of Florida, United StatesReviewed by:
Ravikanth Reddy Poonooru, University of Missouri, United StatesGiovanni Buonaiuto, University of Bologna, Italy
Copyright © 2024 Rodrigues, Ramírez-Zamudio, Pereira, Torrecilhas, Trevisan, Machado Neto, Chardulo, Baldassini and Curi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rogério Abdallah Curi, cm9nZXJpby5jdXJpQHVuZXNwLmJy