
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Anal. Sci. , 13 February 2023
Sec. Omics
Volume 3 - 2023 | https://doi.org/10.3389/frans.2023.1118494
This article is part of the Research Topic Perspectives in Omics 2022 View all 7 articles
 Valentina Calabrese1*
Valentina Calabrese1* Arnaud Salvador1
Arnaud Salvador1 Yohann Clément1Thomas Alexandre Brunet1Anabelle Espeyte2Arnaud Chaumot2Olivier Geffard2
Yohann Clément1Thomas Alexandre Brunet1Anabelle Espeyte2Arnaud Chaumot2Olivier Geffard2 Davide Degli-Esposti2
Davide Degli-Esposti2 Sophie Ayciriex1*
Sophie Ayciriex1*The aquatic environment is one of the most complex biosystems, as organism at all trophic levels may be exposed to a multitude of pollutants. As major goals, ecotoxicology typically investigates the impact of toxic pollutants on the ecosystems through the study of sentinel organisms. Over the past decades, Mass Spectrometry (MS)-based omics approaches have been extended to sentinel species both in laboratory and field exposure conditions. Single-omics approaches enable the discovery of biomarkers mirroring the health status of an organism. By covering a restricted set of the molecular cascade, they turn out to only partially satisfy the understanding of complex ecotoxicological effects. In contrast, a more complete understanding of the ecotoxicity pathways can be accessed through multi-omics approaches. In this perspective, we provide a state-of-the-art and a critical evaluation on further developments in MS-based single and multi-omics studies in aquatic ecotoxicology. As case example, literature regarding Gammarids freshwater amphipods, non-model sentinel organisms sensitive to pollutants and environmental changes and crucial species for downstream ecosystems, will be reviewed.
Since the advent of industrialization, human activities have increased as a result of human population growth, leading to the release of novel entities into the environment (Persson et al., 2022). Water, an essential substance on Earth for living organisms, is also one of the most heavily polluted environments. In the biosphere, freshwater ecosystems represent one of the most delicate compartments, hosting around 10% of the animal kingdom (Balian et al., 2008; Brondizio et al., 2019). The toxicological effects on freshwater organisms caused by exposure to pollutants may include altered reproduction, changes in nutrition habits, physiological or morphological anomalies, migration, death, and extinction (Ahmed et al., 2022). Adverse effects on living organisms are related to modifications at different biological levels, including modulation of gene expression, protein synthesis or metabolic pathways. During the past decades, ecotoxicological research has relied on the use of robust analytical techniques and modern bioinformatics approaches for the discovery of genes, proteins, metabolites, lipids involved in stress responses. Among these technologies, mass spectrometry (MS)-based omics represents a gold standard for both structural and quantitative analysis of thousands of compounds down to ultra-trace levels (Girolamo et al., 2013; Groh and Suter, 2020). The past years of research have been mainly based on the use of single omics (proteomics, lipidomics, or metabolomics, to name a few) leading to a limited view of the investigated system. More recently, multi-omics approaches, based on the integration of multiple omics data on a same sample have been implemented in ecotoxicology studies leading to a more performant elucidation of complex processes (Nam et al., 2022; Faugere et al., 2023).
Gammarids represent the most abundant macroinvertebrate species, in terms of biomass, in freshwater environments. More importantly, they represent keystone species being involved in the detritus cycle, providing prey for secondary consumers, and intervening in the food web. Furthermore, Gammarids are very sensitive to diverse chemical compounds as metals, organics, or oil spills, and may be subjected to bioaccumulation of toxic compounds. Since almost a century, Gammarids have been used as bioindicator species in freshwater ecotoxicological assessment. Considering these peculiarities and drawing on our own experience in the field, we focus on Gammarids to present the advancements and perspective on the use of MS-based omics in ecotoxicology research.
Proteomics, i.e., the large-scale study of proteins expressed by an organism, has been largely investigated in ecotoxicology for biomarker discovery. Protein sequence identification has been historically performed through shotgun approaches in which peptide proxies, obtained after proteins have undergone tryptic digestion, are analyzed through liquid chromatography tandem mass spectrometry (LC-MS/MS). The first studies performed on model organisms were conducted by comparing experimental and in silico fragmentation spectra of peptides derived from nucleoside sequences, for protein annotation. Proteogenomics, based on high resolution MS (i.e., shotgun proteomics), have enabled protein identification in the absence of genomic data from unfinished genome or from species-specific RNA sequences, also for non-model organisms (Armengaud et al., 2014). In the case of Gammarus fossarum, proteogenomics has enabled the identification of 1,873 proteins involved in reproductive pathway (Trapp et al., 2014), to characterize the female core-proteome (Trapp et al., 2016), and to identify proteins related to endocrine perturbation caused by exposure to xenobiotics (Trapp et al., 2015, Koenig et al., 2021). Even if proteogenomics has in part gained prevalence in ecotoxicology research, the technique remains costly, suffers from poor reproducibility and reduced sensitivity (Aggarwal et al., 2022).
Indeed, for quantification purposes, improvements in both sensitivity and selectivity have been reached using targeted MS employing the multiple reaction monitoring (MRM) acquisition mode (Liebler and Zimmerman, 2013). This MS-based technique relies on the use of low-resolution mass spectrometers (triple quadrupole or hybrid quadrupole-linear ion trap) which enables three stages of analyses constituted by precursor ion selection, fragmentation, and fragments ions selection, namely, MRM transition. Only ions satisfying both m/z criteria (precursor and fragment ions) are detected, allowing increasing specificity and signal to noise ratio in complex samples analysis. Ecotoxicological research has made profit of MRM for the discovery and quantification of vitellogenin-related potential ecotoxicological biomarkers in Gammarus fossarum (Simon et al., 2010). However, classic MRM mode limits the number of monitored transitions as a compromise between dwell time transition and the total duty cycle (Rodriguez-Aller et al., 2013). While dwell time corresponds to the time necessary for the acquisition of an MRM transition, the duty cycle is the time spent monitoring an analyte. Importantly, the higher the duty cycle, the greater number of acquired points for chromatographic peak analysis which results in better quality data. In opposition, an increase of monitored transitions for a single analysis (multiplexing ability) may lead to poor reproducibility due to lower duty cycles, especially in coupling with Ultra High-Pressure Liquid Chromatography (UHPLC) which implies narrower peak widths. The MRM3 approach enables increased sensitivity and selectivity by monitoring in addition to the classic approach, second generation of product ions without any further improvement in the multiplexing ability (Jaffuel et al., 2013). A first step towards increased multiplexing has been reached out through the scheduled MRM algorithm. In this operational mode, narrow retention time windows are set up to acquire the MRM transitions only during the expected analyte chromatographic elution (Bertsch et al., 2010). While a prior knowledge of the retention time is required, algorithms adapt dwell times maintaining optimal duty cycles. This allows the monitoring of hundreds of compounds in a single analysis without sacrificing signal-to-noise ratio and reproducibility (Leprêtre et al., 2022). This multiplexing approach is mainly based on the reliance of measured retention times and suffers of matrix effects which can cause time-window shifts. Despite tedious complications in terms of use for consumables, instrument time and operator work, this may represent a problem both for intra-laboratory reproducibility and method transferability among different analytical platforms. More recently, Scout-MRM (renamed scout-triggered MRM or stMRM) has enabled a more reliable multiplexing method for both quantification and identification (Rougemont et al., 2017; Ayciriex et al., 2020; Salvador et al., 2020). Monitoring of concurrent MRM transitions is triggered by marker transitions of known/exogenous compounds (scout compounds), instead of retention time windows. When spiked into the biological sample, scout compounds ideally distribute uniformly along the chromatogram, allowing the monitoring of concomitant MRM transitions of analytes during an acquisition window spanning from the first scout triggering transition to a second one exceeding a chosen intensity threshold. Faugere et al. (2020) optimized and applied for the first time in aquatic ecotoxicology, a method based on Scout-MRM mode for broad and multiplex analysis of proteins in adult Gammarids. Based on preliminary optimization of 44 labelled peptides (Gouveia et al., 2017), Scout-MRM method enabled 157-protein multiplex quantification and identification. The study represents a good example in which low resolution mass spectrometers serves both for robust quantification (increased signal to noise ratio and specificity) and identification based on the simultaneous monitoring of 4 MRM transitions per analyte. The relevance of optimized method was demonstrated through its application on adult Gammarids exposed to pesticides contamination, and on the modulation of proteins involved in key physiological pathways.
Similarly to proteomics, metabolomics and lipidomics give access to an in-depth perception of stress responses acting on metabolites and lipids in exposed organisms. Importantly, metabolites and lipids present extremely diverse chemical compounds with different polarity and a great structural diversity resulting in the presence of different isobaric and isomeric forms. MS-based metabolomics and lipidomics present many advantages over historical NMR-based analyses, principally relying on the greater sensitivity and the possibility to combine hyphenated techniques (i.e., liquid chromatography) for the detection and characterization of compounds in complex biological samples. Moreover, MS enables faster analyses and reduces needed sample quantity (Letertre et al., 2021). While the use of MRM based techniques is mainly used in for quantitative analysis, the advent of high-resolution mass spectrometry (HRMS) has enabled ions exact mass measurements and the acquisition of fragmentation mass spectra at high precision for the identification of potential biomarkers (Rampler et al., 2020; Heiles, 2021). Coupling separative techniques (liquid or gas chromatography) to MS in a targeted or non-targeted approach is routinely used to investigate respectively a known subset of compounds or to obtain undiscriminating information on the whole range of analytes present in the examined sample (Bletsou et al., 2015). Modern MS platforms providing high resolving power, mass accuracy and sensitivity have shed light on alternative approaches, such as shotgun metabolomics, lipidomics, glycomics (Hu et al., 2020; Bui et al., 2022). These techniques enable fast characterization and quantification of metabolites and lipids in crude extracts ionized through direct injection into an electrospray (ESI) source and resolved through the utilization of high-resolution analyzers (Han and Gross, 2005; García-Sevillano et al., 2015). In this extent, the terms diverge from the expression “shotgun proteomics”, which indicates bottom-up techniques for the digestion of crude protein extracts and analysis through LC-MS/MS (Wu and Maccoss, 2002). While proteomics has been widely applied to study stress responses on Gammarids, metabolomics and lipidomics applications remain limited. Targeted metabolomics has been applied to measure changes in the concentration of 29 selected metabolites in G. pulex exposed to xenobiotics (Gómez-Canela et al., 2016), while combination of non-targeted metabolomics and chemometrics have highlighted putative metabolic biomarkers of G. fossarum male and female organisms exposed to pharmaceuticals (Bonnefoy et al., 2019). Similar approaches have been applied to the discovery of lipid biomarkers involved in female G. fossarum reproductive function perturbation caused by the exposition to fenoxycarb, a carbamate insecticide (Arambourou et al., 2018). Despite these studies being relatively recent, biomarker discovery has not been always followed by identification or structural characterization. A single study performed on G. pulex has allowed to get a hint on metabolites identity through molecular formula attribution on exact mass measurements and KEGG database screening (Sheikholeslami et al., 2020).
“Old is the new black”: what is the contribution of other MS and bioinformatics tools in ecotoxicology studies?
The literature reviewed previously focuses on Gammarids species. It is only a case study and reflects the need for ecotoxicology to move towards the application of high throughput analytical methods that are well established in other application areas such as phytochemistry, pharmacology or medicine. To reiterate an already expressed concept, the identification of compounds in the absence of isolated and characterized compounds is a challenge in omics sciences. Data dependent analysis (DDA) and data independent analysis (DIA) are nowadays routinely used to acquire high-resolution fragmentation spectra for thousands of compounds in one single measurement (Fernández-Costa et al., 2020). While DDA-MS performs fragmentation only for selected precursor ions above a certain intensity threshold defined by the operator, DIA-MS enables an indiscriminate fragmentation of all precursor ions. Sequential window acquisition of all theoretical mass spectra (SWATH-MS) is a modified version of DIA-MS in which ions fragmentation is triggered by specific isolation windows (Anjo et al., 2017). In this acquisition mode, all precursors which fall in the m/z mass detection range are fragmented without prior detection, assuring in-depth acquisition for a broad range of compounds in complex samples. The method has been specifically optimized for the identification and quantification of non-labelled protein, but it has recently shown potential applications in metabolomics and lipidomics (Raetz et al., 2020). In all cases, the annotation of the compounds depends strictly on the possibility to compare them with reference fragmentation spectra. Molecular networking (available on GNPS, MetGem and MS-DIAL platform), is a bioinformatic tool permitting the grouping of structurally correlated compounds which fragment similarly (Tsugawa et al., 2015; Olivon et al., 2018; Nothias et al., 2020). These tools allow faster compound annotation as match with database fragmentation spectra.
The use of ultrahigh-resolution analyzers as Fourier Transform Cyclotron Ion Resonance (FTICR) and Orbitrap can offer unrivaled resolution (∼106 at m/z 200 for FTICR in direct infusion mode), mass precision typically below 1 ppm, high sensitivity and good dynamic range enabling the detection of thousands of peaks in a few-minutes run, isobar resolution and access to the fine isotopic peak distribution (Hernández et al., 2012). Unique formula assignment for thousand peaks allows global molecular profiling through the use of van Krevelen diagrams (Laszakovits and MacKay, 2021).
In addition, ion mobility mass spectrometry (IMS) enables separation of isomers based on their tridimensional conformation in gas phase, expressed by their collision cross section (CCS) (Kanu et al., 2008). IMS has been successfully integrated into classic LC-MS set up offering more confident identification of potential biomarkers (either protein, peptides, or small molecules) based on the comparison of experimental and database/theoretical CCS values (Zhou et al., 2022).
While these MS tools are nowadays routinely applied in metabolomics for the characterization of natural compounds in plants (Calabrese et al., 2022, 2023) or human biofluids (Zhu et al., 2021), applications in ecotoxicological research remains still very limited (Taylor et al., 2009; Duarte, et al., 2022) and totally unexplored in Gammarids.
In all the aforementioned techniques, spatial information of interesting compounds in biological tissue is missing. MS imaging is an emerging field in life science allowing real label-free molecular imaging of biological tissue sections. In this approach, information on the spatial distribution of the molecules within a tissue or whole organism can be attained, giving another level of omics information and hints on metabolic pathway (Amstalden van Hove et al., 2010). Despite the fact that this technique has been widely used in biology, MS imaging is relatively new in the ecotoxicology field (Fu et al., 2021). Nano-Secondary Ion Mass Spectrometry (nanoSIMS) imaging has been used to study the selective distribution of silver and gold toxic nanoparticles respectively in the cuticle and the gut area of G. fossarum tissue sections (Mehennaoui et al., 2018) and Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS) has been employed to assess the dynamic changes in lipid composition during the maturation of oocytes in G. fossarum (Fu et al., 2021). IMS has been also integrated in MSI offering in situ separation and mapping of isobaric and isomeric lipids in the muscle and oocytes of females of G. fossarum and disclosing differential lipid composition and abundance in the different organs (Fu et al., 2020).
The integration of all the aforementioned approaches can lead to improvements in the optic of both high throughput MS-based single layer and multiple layers-omics approaches (Figure 1).
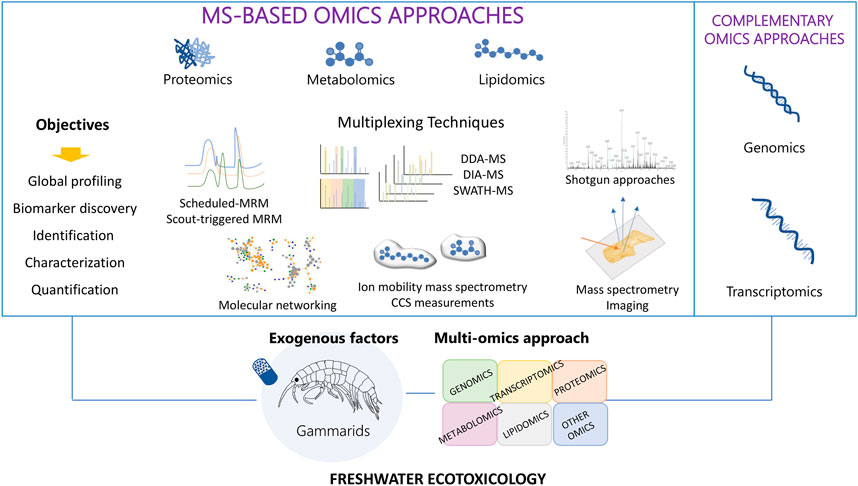
FIGURE 1. Summary of MS-based techniques for a high multiplexing and throughput multi-omics approach applied to freshwater sentinel organism such as Gammarids in ecotoxicology.
Omics approaches pave the way for an initial understanding of ecotoxicological adverse responses but lack of the ability to predict complex mechanisms underlying stressed organisms. There is nowadays a growing consciousness for the need to apply multi-omics approaches based on the use of multi-layers analysis to obtain high-value integrative information. Although multi-omics approaches are increasingly spreading, some scientific challenges have still to be faced. Beside from large investments in terms of diverse analytical platforms, qualified operators, time and resources, multi-omics need optimized analytical protocols and modern tools for data analysis and integration. For example, an effort should be made towards the reduction of biases among the different omics layers, upstream the analysis. In ecotoxicology, different organs or organisms are used to obtain pertinent samples specific for each single-omics layer, introducing biological variability in the analysis. On the other hand, additional analytical variability could arise from multiple sample extraction steps and instrumental runs. Thus, reduction of variability can be reached either by getting multi-omics information from a unique sample and in the ideal case, from a single organism, down to a single cell (Li et al., 2021). In these conditions it is possible to obtain optimized correlation of results for a better understanding of ecotoxicological effects. Recently, Faugere et al. developed a procedure based on a liquid-liquid extraction step (MTBE/MeOH) for the simultaneous extraction of proteins, lipids, and metabolites from a unique sample of G. fossarum, which turned out to improve compound recovery and repeatability, with respect to classic independent fractions sample preparation (Faugere et al., 2023). In the study, MS-based multi-omics in combination with multivariate data analysis revealed specific proteomics, metabolomics and lipidomics signatures of the different female reproductive stages. Besides from this illustration, rare examples of multi-omics development for ecotoxicology application have been published and most of the present literature on MS-based multi-omics approaches in freshwater ecotoxicology is restrained to the study of model organisms, such as zebrafish (Danio rerio) or organisms belonging to the Daphniidae family (Huang et al., 2017; Wang et al., 2019; Jia et al., 2022; Marana et al., 2022).
On another side, there is the question concerning multi-omics data integration and the development of predictive models for highly complex datasets with intrinsic variability. In the last years, progressively sophisticated and appealing approaches have been developed based on the expansion of classic chemometrics, artificial intelligence and machine learning. Among these, “data integration analysis for biomarker discovery using latent components” (DIABLO) and “mining interesting numerical pattern sets” (MINT) enable respectively N-integration (same biological N samples measured on different ‘omics platforms) and the P-integration (several independent data sets or studies measured on the same predictors) of multi-omics datasets (Rohart et al., 2017). These methods consider complex factors as heterogeneity between omics platforms and give adequate weight to different-omics layers. In addition, weighted correlation network analysis (WGCNA) has been extended to MS-based proteomics and metabolomics to find clusters of highly correlated proteins and metabolites (lipids, sugars, and so on), and to highlight connection between specific pollutants and adverse outcomes (Pei et al., 2017; Degli Esposti et al., 2019). Efforts are being made also towards the analysis of complex multi-omics datasets sampled frequently over time in longitudinal studies (Bodein et al., 2022).
Among the available platforms, MetaboAnalyst 5.0 and Workflow4Metabolomics (within the Galaxy framework) are distinguished thanks to their user-friendly and MS-driven workflows, from single-omics data exploration and analysis to comprehensive integration and visualization of multi-omics datasets. Table 1 gathers the principal platforms for MS data integration in single and multi-omics applications, together with a summarized description and advantages.
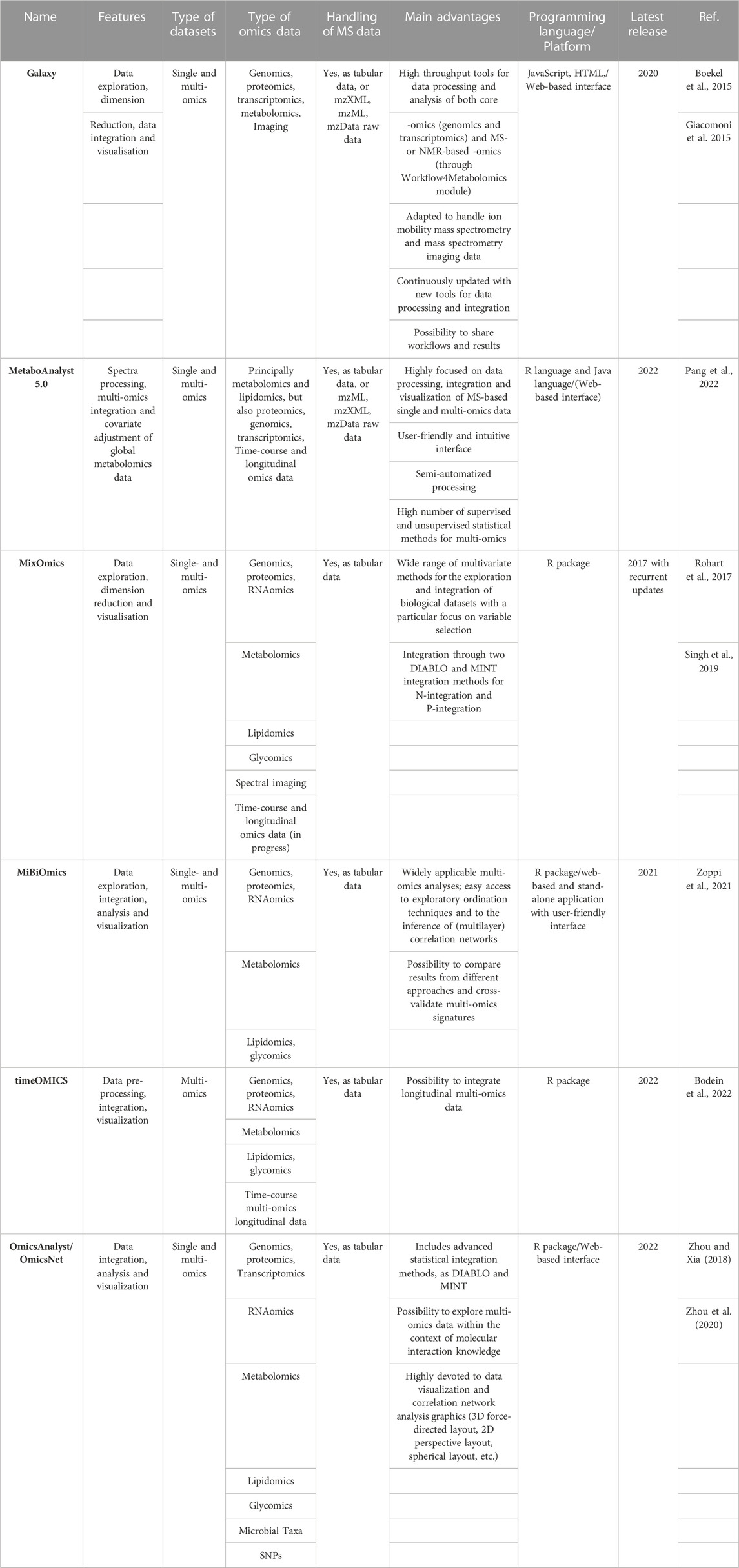
TABLE 1. Principal platforms for MS data exploration, integration and visualization in single and multi-omics applications.
The journey to a better understanding of freshwater ecosystems and gammarid species has just begun. In the near future, the main efforts in ecotoxicology studies should be addressed to the use of alternative mass spectrometry platforms, for identification and quantification of entire metabolome, lipidome, and proteome. From another perspective, the optimization and validation of universal sample treatments compatible with high-throughput multi-omics analyses have still to be developed, with the highest expectative in single-cell analysis. To go much further, the use of novel and progressively complete integration methods of large datasets based on MS-omics (and other non-MS-based omics) data can fill the gap between molecular response, toxicity pathways and apical physiological effects in exposed organisms and provide a more holistic view in ecotoxicology.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
VC and SA designed the concept and wrote the article, with contributions from AS, YC, TAB, AE, AC, OG, and DD-E. VC prepared the figure. All authors contributed to manuscript revision, read and approved the submitted version.
VC was supported by a post-doctoral fellowship of the SENS research funding of the Université Claude Bernard Lyon 1. This work was also supported by the French National Research Agency (ANR) (young investigator grant, ANR-18-CE34-0008 PLAN-TOX and ANR-18-CE34-0013 APPROve), and the French GDR “Aquatic Ecotoxicology” framework which aims at fostering stimulating discussions and collaborations for more integrative approaches.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aggarwal, S., Raj, A., Kumar, D., Dash, D., and Yadav, A. K. False discovery rate: The achilles’ heel of proteogenomics. Brief. Bioinform (2022) 23:bbac163. doi:10.1093/bib/bbac163
Ahmed, S. F., Kumar, P. S., Kabir, M., Zuhara, F. T., Mehjabin, A., Tasannum, N., et al. Threats, challenges and sustainable conservation strategies for freshwater biodiversity. Environ. Res. (2022) 214:113808. doi:10.1016/j.envres.2022.113808
Amstalden van Hove, E. R., Smith, D. F., and Heeren, R. M. A. A concise review of mass spectrometry imaging. J. Chromatogr. A (2010) 1217:3946–3954. doi:10.1016/j.chroma.2010.01.033
Anjo, S. I., Santa, C., and Manadas, B. SWATH-MS as a tool for biomarker discovery: From basic research to clinical applications. Proteomics (2017) 17:1600278. doi:10.1002/pmic.201600278
Arambourou, H., Fuertes, I., Vulliet, E., Daniele, G., Noury, P., Delorme, N., et al. Fenoxycarb exposure disrupted the reproductive success of the amphipod Gammarus fossarum with limited effects on the lipid profile. PLoS One (2018) 13:e0196461. doi:10.1371/journal.pone.0196461
Armengaud, J., Trapp, J., Pible, O., Geffard, O., Chaumot, A., and Hartmann, E. M. Non-model organisms, a species endangered by proteogenomics. J. proteomics (2014) 105:5–18. doi:10.1016/j.jprot.2014.01.007
Ayciriex, S., Carrière, R., Bardet, C., Blanc, J. Y. L., Salvador, A., Fortin, T., et al. Streamlined development of targeted mass spectrometry-based method combining scout-MRM and a web-based tool indexed with scout peptides. Proteomics (2020) 20(2):1900254. doi:10.1002/pmic.201900254
Balian, E. v., Segers, H., Lévèque, C., and Martens, K. The freshwater animal diversity assessment: An overview of the results. Hydrobiologia (2008) 595:627–637. doi:10.1007/s10750-007-9246-3
Bertsch, A., Jung, S., Zerck, A., Pfeifer, N., Nahnsen, S., Henneges, C., et al. Optimal de novo design of MRM experiments for rapid assay development in targeted proteomics. J. Proteome Res. (2010) 9:2696–2704. doi:10.1021/pr1001803
Bletsou, A. A., Jeon, J., Hollender, J., Archontaki, E., and Thomaidis, N. S. Targeted and non-targeted liquid chromatography-mass spectrometric workflows for identification of transformation products of emerging pollutants in the aquatic environment. TrAC - Trends Anal. Chem. (2015) 66:32–44. doi:10.1016/j.trac.2014.11.009
Bodein, A., Scott-Boyer, M. P., Perin, O., Lê Cao, K. A., and Droit, A. timeOmics: an R package for longitudinal multi-omics data integration. Bioinformatics (2022) 38(2):577–579. doi:10.1093/bioinformatics/btab664
Boekel, J., Chilton, J. M., Cooke, I. R., Horvatovich, P. L., Jagtap, P. D., Käll, L., et al. Multi-omic data analysis using Galaxy. Nat. Biotechnol. (2015) 33(2):137–139. doi:10.1038/nbt.3134
Bonnefoy, C., Fildier, A., Buleté, A., Bordes, C., Garric, J., Vulliet, E., et al. Untargeted analysis of nanoLC-HRMS data by ANOVA-PCA to highlight metabolites in Gammarus fossarum after in vivo exposure to pharmaceuticals. Talanta (2019) 202:221–229. doi:10.1016/j.talanta.2019.04.028
Brondizio, E. S., Settele, J., Díaz, S., and Ngo, H. T. Global assessment report on biodiversity and ecosystem services of the intergovernmental science-policy platform on biodiversity and ecosystem services. Bonn, Germany: IPBES (2019). doi:10.5281/zenodo.3831673
Bui, D. T., Jung, J., Kitova, E. N., Li, Z., Willows, S. D., Boddington, M. E., et al. Mass spectrometry-based shotgun glycomics using labeled glycan libraries. Anal. Chem. (2022) 94(12):4997–5005. doi:10.1021/acs.analchem.1c04779
Calabrese, V., Schmitz-Afonso, I., Prevost, C., Afonso, C., and Elomri, A. Molecular networking and collision cross section prediction for structural isomer and unknown compound identification in plant metabolomics: A case study applied to Zhanthoxylum heitzii extracts. Anal. Bioanal. Chem. (2022) 414(14):4103–4118. doi:10.1007/s00216-022-04059-7
Calabrese, V., Schmitz-Afonso, I., Riah-Anglet, W., Trinsoutrot-Gattin, I., Pawlak, B., and Afonso, C. Direct introduction MALDI FTICR MS based on dried droplet deposition applied to non-targeted metabolomics on Pisum Sativum root exudates. Talanta (2023) 253:123901. doi:10.1016/j.talanta.2022.123901
Degli Esposti, D., Almunia, C., Guery, M. A., Koenig, N., Armengaud, J., Chaumot, A., et al. Co-expression network analysis identifies gonad-and embryo-associated protein modules in the sentinel species Gammarus fossarum. Sci. Rep. (2019) 9(1):7862. doi:10.1038/s41598-019-44203-5
Duarte, B., Feijão, E., Cruz de Carvalho, R., Duarte, I. A., Marques, A. P., Maia, M., et al. Untargeted metabolomics reveals antidepressant effects in a marine photosynthetic organism: The diatom Phaeodactylum tricornutum as a case study. Biology (2022) 11(12):1770. doi:10.3390/biology11121770
Faugere, J., Brunet, T. A., Clément, Y., Espeyte, A., Geffard, O., Lemoine, J., et al. Development of a multi-omics extraction method for ecotoxicology: Investigation of the reproductive cycle of Gammarus fossarum. Talanta (2023) 253:123806. doi:10.1016/j.talanta.2022.123806
Faugere, J., Gouveia, D., Ayciriex, S., Chaumot, A., Almunia, C., François, A., et al. High-multiplexed monitoring of protein biomarkers in the sentinel Gammarus fossarum by targeted scout-MRM assay, a new vision for ecotoxicoproteomics. J. Proteomics (2020) 226:103901. doi:10.1016/j.jprot.2020.103901
Fernández-Costa, C., Martínez-Bartolomé, S., McClatchy, D. B., Saviola, A. J., Yu, N. K., and Yates, J. R. Impact of the identification strategy on the reproducibility of the DDA and DIA results. J. Proteome Res. (2020) 19:3153–3161. doi:10.1021/acs.jproteome.0c00153
Fu, T., Knittelfelder, O., Geffard, O., Clément, Y., Testet, E., Elie, N., et al. Shotgun lipidomics and mass spectrometry imaging unveil diversity and dynamics in Gammarus fossarum lipid composition. iScience (2021) 24:102115. doi:10.1016/j.isci.2021.102115
Fu, T., Oetjen, J., Chapelle, M., Verdu, A., Szesny, M., Chaumot, A., et al. In situ isobaric lipid mapping by MALDI–ion mobility separation–mass spectrometry imaging. J. Mass Spectrom. (2020) 55:e4531. doi:10.1002/jms.4531
García-Sevillano, M. A., García-Barrera, T., Navarro, F., Montero-Lobato, Z., and Gómez-Ariza, J. L. Shotgun metabolomic approach based on mass spectrometry for hepatic mitochondria of mice under arsenic exposure. BioMetals (2015) 28:341–351. doi:10.1007/s10534-015-9837-9
Giacomoni, F., Le Corguille, G., Monsoor, M., Landi, M., Pericard, P., Petera, M., et al. Workflow4Metabolomics: A collaborative research infrastructure for computational metabolomics. Bioinformatics (2015) 31(9):1493–1495. doi:10.1093/bioinformatics/btu813
Girolamo, F. D., Lante, I., Muraca, M., and Putignani, L. The role of mass spectrometry in the “omics” era. Curr. Org. Chem. (2013) 17(23):2891–2905. doi:10.2174/1385272817888131118162725
Gómez-Canela, C., Miller, T. H., Bury, N. R., Tauler, R., and Barron, L. P. Targeted metabolomics of Gammarus pulex following controlled exposures to selected pharmaceuticals in water. Sci. Total Environ. (2016) 562:777–788. doi:10.1016/j.scitotenv.2016.03.181
Gouveia, D., Chaumot, A., Charnot, A., Almunia, C., François, A., Navarro, L., et al. Ecotoxico-proteomics for aquatic environmental monitoring: First in situ application of a new proteomics-based multibiomarker assay using caged amphipods. Environ. Sci. Technol. (2017) 51(22):13417–13426. doi:10.1021/acs.est.7b03736
Han, X., and Gross, R. W. Shotgun lipidomics: Multidimensional MS analysis of cellular lipidomes. Expert Rev. Proteomics (2005) 2:253–264. doi:10.1586/14789450.2.2.253
Heiles, S. Advanced tandem mass spectrometry in metabolomics and lipidomics—Methods and applications. Anal. Bioanal. Chem. (2021) 413(24):5927–5948. doi:10.1007/s00216-021-03425-1
Hernández, F., Sancho, J. v., Ibáñez, M., Abad, E., Portolés, T., and Mattioli, L. Current use of high-resolution mass spectrometry in the environmental sciences. Anal. Bioanal. Chem. (2012) 403:1251–1264. doi:10.1007/s00216-012-5844-7
Hu, C., Duan, Q., and Han, X. Strategies to improve/eliminate the limitations in shotgun lipidomics. Proteomics (2020) 20(11):1900070. doi:10.1002/pmic.201900070
Huang, S. S. Y., Benskin, J. P., Veldhoen, N., Chandramouli, B., Butler, H., Helbing, C. C., et al. A multi-omic approach to elucidate low-dose effects of xenobiotics in zebrafish (Danio rerio) larvae. Aquat. Toxicol. (2017) 182:102–112. doi:10.1016/j.aquatox.2016.11.016
Jia, J., Dong, C., Han, M., Ma, S., Chen, W., Dou, J., et al. Multi-omics perspective on studying reproductive biology in Daphnia sinensis. Genomics (2022) 114:110309. doi:10.1016/j.ygeno.2022.110309
Kanu, A. B., Dwivedi, P., Tam, M., Matz, L., and Hill, H. H. Ion mobility-mass spectrometry. J. Mass Spectrom. (2008) 43:1–22. doi:10.1002/jms.1383
Laszakovits, J. R., and MacKay, A. A. Data-based chemical class regions for van krevelen diagrams. J. Am. Soc. Mass Spectrom. (2021) 33(1):198–202. doi:10.1021/jasms.1c00230
Leprêtre, M., Geffard, O., Espeyte, A., Faugere, J., Ayciriex, S., Salvador, A., et al. Multiple reaction monitoring mass spectrometry for the discovery of environmentally modulated proteins in an aquatic invertebrate sentinel species, Gammarus fossarum. Gammarus Fossarum. Environ. Pollut. (2022) 315:120393. doi:10.1016/j.envpol.2022.120393
Letertre, M. P., Giraudeau, P., and De Tullio, P. Nuclear magnetic resonance spectroscopy in clinical metabolomics and personalized medicine: Current challenges and perspectives. Front. Mol. Biosci. (2021) 8:698337. doi:10.3389/fmolb.2021.698337
Li, Y., Li, H., Xie, Y., Chen, S., Qin, R., Dong, H., et al. An integrated strategy for mass spectrometry-based multiomics analysis of single cells. Anal. Chem. (2021) 93:14059–14067. doi:10.1021/acs.analchem.0c05209
Liebler, D. C., and Zimmerman, L. J. Targeted quantitation of proteins by mass spectrometry. Biochemistry (2013) 52:3797–3806. doi:10.1021/bi400110b
Marana, M. H., Poulsen, R., Thormar, E. A., Clausen, C. G., Thit, A., Mathiessen, H., et al. Plastic nanoparticles cause mild inflammation, disrupt metabolic pathways, change the gut microbiota and affect reproduction in zebrafish: A full generation multi-omics study. J. Hazard Mater (2022) 424:127705. doi:10.1016/j.jhazmat.2021.127705
Mehennaoui, K., Cambier, S., Serchi, T., Ziebel, J., Lentzen, E., Valle, N., et al. Do the pristine physico-chemical properties of silver and gold nanoparticles influence uptake and molecular effects on Gammarus fossarum (Crustacea Amphipoda)? Sci. Total Environ. (2018) 643:1200–1215. doi:10.1016/j.scitotenv.2018.06.208
Nam, S.-E., Bae, D.-Y., Ki, J.-S., Ahn, C.-Y., and Rhee, J.-S. The importance of multi-omics approaches for the health assessment of freshwater ecosystems. Mol. Cell Toxicol. (2022). doi:10.1007/s13273-022-00286-2
Nothias, L.-F., Petras, D., Schmid, R., Dührkop, K., Rainer, J., Sarvepalli, A., et al. Feature-based molecular networking in the GNPS analysis environment. Nat. Methods (2020) 17:905–908. doi:10.1038/s41592-020-0933-6
Olivon, F., Elie, N., Grelier, G., Roussi, F., Litaudon, M., and Touboul, D. MetGem software for the generation of molecular networks based on the t-SNE algorithm. Anal. Chem. (2018) 90:13900–13908. doi:10.1021/acs.analchem.8b03099
Pang, Z., Zhou, G., Ewald, J., Chang, L., Hacariz, O., Basu, N., et al. Using MetaboAnalyst 5.0 for LC–HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat. Protoc. (2022) 17(8):1735–1761. doi:10.1038/s41596-022-00710-w
Pei, G., Chen, L., and Zhang, W., WGCNA application to proteomic and metabolomic data analysisMethods Enzym. 585. Academic Press (2017). p. 135–158. doi:10.1016/bs.mie.2016.09.016
Persson, L., Carney Almroth, B. M., Collins, C. D., Cornell, S., de Wit, C. A., Diamond, M. L., et al. Outside the safe operating space of the planetary boundary for novel entities. Environ. Sci. Technol. (2022) 56(3):1510–1521. doi:10.1021/acs.est.1c04158
Raetz, M., Bonner, R., and Hopfgartner, G. SWATH-MS for metabolomics and lipidomics: Critical aspects of qualitative and quantitative analysis. Metabolomics (2020) 16:71. doi:10.1007/s11306-020-01692-0
Rampler, E., Abiead, Y. E., Schoeny, H., Rusz, M., Hildebrand, F., Fitz, V., et al. Recurrent topics in mass spectrometry-based metabolomics and lipidomics—Standardization, coverage, and throughput. Anal. Chem. (2020) 93(1):519–545. doi:10.1021/acs.analchem.0c04698
Rodriguez-Aller, M., Gurny, R., Veuthey, J. L., and Guillarme, D. Coupling ultra-high-pressure liquid chromatography with mass spectrometry: Constraints and possible applications. J. Chromatogr. A (2013) 1292:2–18. doi:10.1016/j.chroma.2012.09.061
Rohart, F., Gautier, B., Singh, A., and Lê Cao, K. A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. (2017) 13(11):e1005752. doi:10.1371/journal.pcbi.1005752
Rougemont, B., Bontemps Gallo, S., Ayciriex, S., Carrière, R., Hondermarck, H., Lacroix, J. M., et al. Scout-MRM: Multiplexed targeted mass spectrometry-based assay without retention time scheduling exemplified by Dickeya dadantii proteomic analysis during plant infection. Anal. Chem. (2017) 89(3):1421–1426. doi:10.1021/acs.analchem.6b03201
Salvador, A., Carrière, R., Ayciriex, S., Margoum, C., Leblanc, Y., and Lemoine, J. Scout-multiple reaction monitoring: A liquid chromatography tandem mass spectrometry approach for multi-residue pesticide analysis without time scheduling. J. Chromatogr. A (2020) 1621:461046. doi:10.1016/j.chroma.2020.461046
Sheikholeslami, M. N., Gómez-Canela, C., Barron, L. P., Barata, C., Vosough, M., and Tauler, R. Untargeted metabolomics changes on Gammarus pulex induced by propranolol, triclosan, and nimesulide pharmaceutical drugs. Chemosphere (2020) 260:127479. doi:10.1016/j.chemosphere.2020.127479
Simon, R., Jubeaux, G., Chaumot, A., Lemoine, J., Geffard, O., and Salvador, A. Mass spectrometry assay as an alternative to the enzyme-linked immunosorbent assay test for biomarker quantitation in ecotoxicology: Application to vitellogenin in Crustacea (Gammarus fossarum). J. Chromatogr. A (2010) 1217:5109–5115. doi:10.1016/j.chroma.2010.06.015
Singh, A., Shannon, C. P., Gautier, B., Rohart, F., Vacher, M., Tebbutt, S. J., et al. Diablo: An integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics (2019) 35(17):3055–3062. doi:10.1093/bioinformatics/bty1054
Taylor, N. S., Weber, R. J. M., Southam, A. D., Payne, T. G., Hrydziuszko, O., Arvanitis, T. N., et al. A new approach to toxicity testing in Daphnia magna: Application of high throughput FT-ICR mass spectrometry metabolomics. Metabolomics (2009) 5(1):44–58. doi:10.1007/s11306-008-0133-3
Trapp, J., Almunia, C., Gaillard, J. C., Pible, O., Chaumot, A., Geffard, O., et al. Proteogenomic insights into the core-proteome of female reproductive tissues from crustacean amphipods. J. Proteomics (2016) 135:51–61. doi:10.1016/j.jprot.2015.06.017
Trapp, J., Armengaud, J., Pible, O., Gaillard, J. C., Abbaci, K., Habtoul, Y., et al. Proteomic investigation of male Gammarus fossarum, a freshwater crustacean, in response to endocrine disruptors. J. Proteome Res. (2015) 14:292–303. doi:10.1021/pr500984z
Trapp, J., Geffard, O., Imbert, G., Gaillard, J. C., Davin, A. H., Chaumot, A., et al. Proteogenomics of Gammarus fossarum to document the reproductive system of amphipods. Mol. Cell. Proteomics (2014) 13:3612–3625. doi:10.1074/mcp.M114.038851
Tsugawa, H., Cajka, T., Kind, T., Ma, Y., Higgins, B., Ikeda, K., et al. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. methods (2015) 12(6):523–526. doi:10.1038/nmeth.3393
Wang, P., Ng, Q., Zhang, B., Wei, Z., Hassan, M., He, Y., et al. Employing multi-omics to elucidate the hormetic response against oxidative stress exerted by nC60 on Daphnia pulex. Environ. Pollut. (2019) 251:22–29. doi:10.1016/j.envpol.2019.04.097
Wu, C. C., and Maccoss, M. J. Shotgun proteomics: Tools for the analysis of complex biological systems. Curr. Opin. Mol. Ther. (2002) 4:242–250.
Zhou, G., Pang, Z., Lu, Y., Ewald, J., and Xia, J. OmicsNet 2.0: A web-based platform for multi-omics integration and network visual analytics. Nucleic Acids Res. (2022) 50:W527–W533. doi:10.1093/nar/gkac376
Zhou, G., and Xia, J. OmicsNet: A web-based tool for creation and visual analysis of biological networks in 3D space. Nucleic acids Res. (2018) 46(W1):W514–W522. doi:10.1093/nar/gky510
Zhou, Z., Luo, M., Chen, X., Yin, Y., Xiong, X., Wang, R., et al. Ion mobility collision cross-section atlas for known and unknown metabolite annotation in untargeted metabolomics. Nat. Commun. (2020) 11(1):4334. doi:10.1038/s41467-020-18171-8
Zhu, Y., Wancewicz, B., Schaid, M., Tiambeng, T. N., Wenger, K., Jin, Y., et al. Ultrahigh-resolution mass spectrometry-based platform for plasma metabolomics applied to type 2 diabetes research. J. Proteome Res. (2021) 20(1):463–473. doi:10.1021/acs.jproteome.0c00510
Keywords: ecotoxicology, Gammarids, mass spectrometry, omics, MS imaging
Citation: Calabrese V, Salvador A, Clément Y, Brunet TA, Espeyte A, Chaumot A, Geffard O, Degli-Esposti D and Ayciriex S (2023) Challenges and perspectives in MS-based omics approaches for ecotoxicology studies: An insight on Gammarids sentinel amphipods. Front. Anal. Sci. 3:1118494. doi: 10.3389/frans.2023.1118494
Received: 07 December 2022; Accepted: 03 February 2023;
Published: 13 February 2023.
Edited by:
Martin Giera, Leiden University Medical Center (LUMC), NetherlandsReviewed by:
Carmen Bedia, Institute of Environmental Assessment and Water Research (CSIC), SpainCopyright © 2023 Calabrese, Salvador, Clément, Brunet, Espeyte, Chaumot, Geffard, Degli-Esposti and Ayciriex. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valentina Calabrese, dmFsZW50aW5hLmNhbGFicmVzZUB1bml2LWx5b24xLmZy; Sophie Ayciriex, c29waGllLmF5Y2lyaWV4QHVuaXYtbHlvbjEuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.