 Sierra Bruno1,†Amelia Lamberty1,†Margaret McCoy1Zoe Mark1Nirav Daphtary2Minara Aliyeva2Kelly Butnor1
Sierra Bruno1,†Amelia Lamberty1,†Margaret McCoy1Zoe Mark1Nirav Daphtary2Minara Aliyeva2Kelly Butnor1 Matthew E. Poynter2
Matthew E. Poynter2 Vikas Anathy1
Vikas Anathy1 Brian Cunniff1*
Brian Cunniff1*
- 1Department of Pathology and Laboratory Medicine, University of Vermont, Burlington, VT, United States
- 2Department of Medicine, University of Vermont, Burlington, VT, United States
Mitochondria are multifaceted organelles necessary for numerous cellular signaling and regulatory processes. Mitochondria are dynamic organelles, trafficked and anchored to subcellular sites depending upon the cellular and tissue requirements. Precise localization of mitochondria to apical and basolateral membranes in lung epithelial cells is important for key mitochondrial processes. Miro1 is an outer mitochondrial membrane GTPase that associates with adapter proteins and microtubule motors to promote intracellular movement of mitochondria. We show that deletion of Miro1 in lung epithelial cells leads to perinuclear clustering of mitochondria. However, the role of Miro1 in epithelial cell response to allergic insults remains unknown. We generated a conditional mouse model to delete Miro1 in Club Cell Secretory Protein (CCSP) positive lung epithelial cells to examine the potential roles of Miro1 and mitochondrial trafficking in the lung epithelial response to the allergen, house dust mite (HDM). Our data show that Miro1 suppresses epithelial induction and maintenance of the inflammatory response to allergen, as Miro1 deletion modestly induces increases in pro-inflammatory signaling, specifically IL-6, IL-33, CCL20 and eotaxin levels, tissue reorganization, and airway hyperresponsiveness. Furthermore, loss of Miro1 in CCSP+ lung epithelial cells blocks resolution of the asthmatic insult. This study further demonstrates the important contribution of mitochondrial dynamic processes to the airway epithelial allergen response and the pathophysiology of allergic asthma.
Introduction
Allergic asthma is a complex, chronic inflammatory condition of the small airways that affects as many as 300 million people of all ages worldwide, including 25 million Americans (about 8.5% of the United States' population) (1–3). Generally, a combination of environmental and genetic factors can cause allergic asthma (4, 5) however, allergen exposure is the single most important cause. The main phenotypic drivers of acute episodes, or “attacks,” of airway inflammation are increased mucus production, and obstruction and narrowing of the airways (6). These disruptive changes to the airway epithelium, the first line of protection against external insults and pathogens, can lead to some of the classical symptoms associated with allergic asthma such as chest tightness, difficulty breathing, wheezing, and persistent coughing (7). In allergic asthma, the airway epithelium is frequently injured by allergens, leading to the secretion of pro-inflammatory cytokines and chemokines, as well as the recruitment and activation of innate and adaptive immune cells (8). Chronic activation of these pro-inflammatory molecules potentiates structural changes in the airways such as thickened basement membranes, shedding of the surface epithelium, subepithelial fibrosis, mucus hyperplasia and metaplasia, and increased angiogenesis surrounding the airways (9). These structural changes to the airways lead to functional changes, contributing to symptoms including difficulty breathing.
Currently, there is no cure for allergic asthma. Avoidance of triggers and the use of bronchodilators and anti-inflammatory drugs such as inhaled rapid-acting β-2 adrenergic agonists and oral corticosteroids are commonly recommended for the management of symptoms. However, these current treatment options, and even contemporary biological therapeutics, fail in upwards of 10% of patients or have serious side effects (10, 11), making it necessary to study novel molecular targets to better understand the pathophysiological nature of allergic asthma and to explore alternative therapeutic options.
Ample evidence suggests that mitochondria play a key role in lung health and pathophysiology (12–15). Mitochondria, the “powerhouses of the cell,” are the main producers of adenosine triphosphate (ATP). Moreover, mitochondria are a primary source of reactive oxygen species (ROS) (16), and can actively buffer calcium (Ca2+) (17). Following cellular stress, mitochondrial membranes can be damaged and may become dysfunctional. Mitochondrial dysfunction in respiratory diseases is characterized by ROS accumulation, loss of membrane potential, mitochondrial Ca2+ overload, mitochondrial DNA mutation or release, and mitophagy dysregulation (18). Increased production of ROS has been implicated in the development of chronic inflammatory lung conditions such as asthma (11, 19). As a result, these double-membraned organelles change in size, shape, and distribution depending on intracellular mitochondrial metabolite demands (20–22). Our previous work showed that mitochondrial fission is an early event following exposure of bronchial epithelial cells to house dust mite and that knockout of the pro-fission protein Dynamin Related Protein 1 (DRP1) exacerbates asthmatic phenotypes (23). Within the cell, mitochondria are transported on the microtubule-associated molecular motors kinesin and dynein (24–26), and tethered to these motors by a protein complex composed of trafficking kinesin protein 1 and 2 (TRAK1/2) and Mitochondrial Rho GTPase 1 and 2 (Miro1/2) (27–32). Miro1 shares approximately 60% homology with Miro2, but evidence shows Miro1 is the primary adaptor protein required for the subcellular positioning of mitochondria within differentiated cells (33, 34). The dynamic reorganization of mitochondrial networks is important in highly active cells, such as airway epithelial cells, due to their increased need in energy to maintain the functionality of the airways. Miro1 is critical for proper subcellular positioning of mitochondria in fibroblasts and distribution of mitochondrial derived molecules including ROS (35) and ATP (36). Evidence suggests that mitochondria are positioned at the apical and basolateral poles of polarized airway epithelial cells to constrict calcium signaling (37), and provide energy for mucus to be secreted and cleared (38–41), which becomes critical during high energy demand events such as stress or injury. Recent in vitro and in vivo models of COPD suggest that Miro1 plays a contributing role to this pathology (42, 43). Miro1 has been implicated in mesenchymal stem cell transfer of mitochondria to damaged lung epithelia following inhibition of the electron transport chain and allows for suppression of the allergic asthma phenotype in mouse models of the disease (17). Until now, the contribution of Miro1 in the context of allergic asthma remained unexplored.
Herein, we developed a novel mouse model to study Miro1 biology in the context of allergic asthma, in which we hypothesize that epithelial deletion of Miro1 leads to exacerbated inflammatory responses following chronic allergen exposure. We find that epithelial ablation of Miro1 in club cell secretory protein (CCSP) positive lung epithelial cells followed by exposure to the complex allergen house dust mite (HDM) leads to significant changes in tissue architecture and organization, inflammatory cell infiltrates, airway hyperresponsiveness, and forced pressure volume. Loss of Miro1 also disrupts the resolution phase following allergen exposure, providing evidence for the importance of Miro1 in mediating allergen-induced responses in lung epithelial cells.
Materials and methods
Study approval
All mouse studies were approved by the Institutional Animal Care and Use Committee of the University of Vermont, Burlington, VT, USA under protocol number PROTO202000216.
House dust Mite
HDM (XPB70D3A2.5, Stallergenes Greer, Lenoir, NC, USA) was suspended in Phosphate Buffered Saline (PBS). HDM concentration was determined by total protein concentration in HDM vial.
Epithelial tissue-specific transgenic mice
To achieve conditional airway epithelial specific deletion, C57BL/6 mice containing floxed Miro1 alleles (Miro1flx/flx) (34) were crossed with double transgenic mice containing a Club Cell secretory protein (CCSP) promoter fused to a reverse tetracycline trans activator (rTetA) and a Tet operon fused to Cre recombinase (TetOP-Cre), termed CCSP-rTetA/TetOP-Cre (44), to generate triple transgenic (CCSP-rTetA/TetOP-Cre/Miro1flx/flx) mice, referred to as ΔEpi-Miro1. Mice containing the three gene inserts were used to conditionally delete Miro1 in CCSP positive airway epithelial cells upon exposure to doxycycline-containing food (6 g/kg; Purina Diet Tech, St Louis, Mo) for 7 days. Littermates missing one of the three gene inserts (CCSP-rtTA, TetOP-Cre, or Miro1flx/flx) also on the doxycycline-containing food were used as control mice. Both female and male mice were used in this study and distributed equally among groups.
Mouse models of allergic asthma
An established model of allergic asthma was used for all experiments (45, 46). In this model, 25 µg of HDM or PBS was administered to anesthetized control and ΔEpi-Miro1 mice via nasopharyngeal aspiration. Mice were sensitized to the allergen 7 days after the start of the doxycycline-containing food. In the single model of allergen challenge, mice are sensitized on days 0 and 7 and challenged on day 14 with harvest 4 h later. In the multiple challenge model, the allergic response in mice was boosted 7 days after sensitization and challenged for five consecutive days 14 days after sensitization with HDM or PBS. Mice were studied 24 h following the last allergen challenge.
Airway hyperresponsiveness assessment
Mice were anesthetized using sodium pentobarbital (90 mg/kg) via intraperitoneal injection and tracheotomized using 18-guage cannulas. Mice were mechanically ventilated at a rate of 200 breaths/min using FlexiVent computer controlled small-animal ventilator (SCRIREQ, Montreal, QC, Canada). Airway hyperresponsiveness parameters including Newtonian resistance (Rn), tissue dampening (G), and tissue elastance (H) were measured in the mice after exposure to increasing concentrations (12.5 mg/ml, 25 mg/ml, and 50 mg/ml) of aerosolized methacholine. Measurement of lung mechanics presented are the average of three measurements encompassing the peak response.
Bronchoalveolar lavage fluid collection and processing
Bronchoalveolar lavage fluid (BALF) was collected by washing the airways with 1 ml of sterile PBS. Cells were isolated via centrifugation and total cell counts were determined using a hemocytometer (3110, Hausser Scientific, Horsham, PA, USA). Cytopsins were conducted and cells were stained using Hema3 stain reagents (Fisher Scientific, Waltham, MA, USA) to obtain differential cell counts from a minimum of 300 cells.
Tissue processing
Following euthanasia, left lung lobe tissue was collected, inflated, and fixed in 4% paraformaldehyde for 24–48 h at room temperature. Formaldehyde-fixed lung tissue was sent to the University of Vermont Medical Center for paraffin embedding. Tissue blocks were serially sectioned at a 5 μm thickness using a Leica 2030 manual paraffin microtome (Microscopy Imaging Center, University of Vermont). Tissue sections were mounted onto glass slides and dried in an oven at 52°C for 60 min. De-paraffinization and tissue rehydration for all staining procedures was achieved by immersing the glass slides through the following solutions: three 15 min xylene washes, two 5 min washes of 100% ethanol and 95% ethanol, and a 5 min wash of 70% ethanol, 50% ethanol, and dH2O.
Miro1 immunohistochemistry
Lung antigen retrieval was achieved by submersing slides in a DAKO antigen retrieval solution for 20 min at 95°C, slides were then allowed to cool down for 20 min at room temperature and rinsed in three 5 min PBS washes. Lung tissue slides were blocked in a 10% H2O2 in methanol solution for 15 min, followed by seven 5 min PBS washes. A 2.5% normal goat serum protein block (Vector Laboratories) was put on the slides for 15 min, followed by overnight incubation of primary α-Miro1 antibody (ABIN635090) diluted to a 1:200 concentration in PBS at 4°C. Following overnight incubation, slides were rinsed in seven 5 min PBS washes and a ImmPRESS polymer reagent (Vector Laboratories) was put on the tissue for 30 min at room temperature. Tissue was rinsed in seven 5 min PBS washes and exposed to a diaminobenzidine (DAB) peroxidase solution (Vector Laboratories) for immunohistochemical staining. Tissue was rinsed in dH2O and counterstained with hematoxylin and ammonium hydroxide. Images were captured at 20× magnification by Leica VERSA8 whole slide imager (Microscopy Imaging Center, University of Vermont).
Fluorescence staining and imaging
Antigen retrieval was done by heating slides for 20 min at 95°C in sodium citrate buffer with 0.05% TWEEN-20 then rinsed in dH2O. Sections were then blocked for 1 h in 1% BSA in PBS, followed by incubation using the following primary antibodies: TOMM20 (Millipore, MABT166), at 1:250 and CC10 (Santa Cruz Biotechnology, sc-390313), at 1:250; overnight at 4°C. Slides were then washed 3 × 5 min in PBS and incubated with species-specific Alexafluor-488- or Alexafluor-647-conjugated secondary antibodies, and counterstained with DAPI in PBS at 1:4,000 for nuclear localization. Sections were imaged using a Nikon A1R Confocal laser-scanning microscope. Images were captured at 20× and/or 40× magnification. The image files were converted to Tiff format. Brightness and contrast were adjusted equally in all images. The distance from the closest mitochondria to the apical membrane was measured using multiple line scans/cell and averaged using ImageJ.
Mucus metaplasia quantification
Periodic Acid Schiff (PAS) staining was conducted to assess mucus metaplasia. Lung tissue slides were immersed in 0.5% periodic acid for 10 min, followed by three 5 min rinses in dH2O, and 30 min in Schiff reagent. Next, two 1 min washes in 0.55% potassium metabisulfite, followed by a 10 min rinse under running water. Tissue was counterstained by immersing slides for 10 min in hematoxylin, rinsed under running water for 5 min, and 2 dips in 0.5% lithium carbonate. Images were captured at 20× magnification by Leica VERSA8 whole slide imager (Microscopy Imaging Center, University of Vermont). Mucus metaplasia was measured in the airways by quantitating the positively PAS-stained area using the Positive Pixel Count algorithm of Leica Aperio ImageScope Software (Aperio Technologies, San Diego, CA, USA). The Positive Pixel Count algorithm outputs were used to determine the positive PAS-stained area number of strong positive pixels normalized to lung tissue area.
Remodeling quantification
Masson's trichrome staining was conducted to assess remodeling changes. Lung tissue slides were immersed for 1 h in Bouin's solution at 56°C and cooled to room temperature. Tissue was rinsed under running water until stain disappeared. Tissue was stained for 10 min in Wiegert hematoxylin and washed under running water for 10 min. Next, tissue was stained for 2 min in Biebrich Scarlet-Acid Fuchsin solution, followed by a rinse in dH2O, and then immersed for 10–15 min in a phosphomolybdic/phosphotungstic acid solution. The last stain was 5 min in aniline blue, followed by a rinse in dH2O, and 3–5 min in a 1% acetic acid solution. Nuclei were stained in black; cytoplasm, keratin, and muscle fibers were stained in red; and collagen was stained in blue. Images were captured at 20× magnification by Leica VERSA8 whole slide imager (Microscopy Imaging Center, University of Vermont). Representative images of the small airways were captured for each experimental group. Images were de-identified and blindly scored using an arbitrary unit (A.U.) scale ranging from 1 to 8 by ∼6 separate individuals. The arbitrary unit scoring scale assessed changes in remodeling including collagen deposition, immune cell infiltration, epithelial layer thickening, and alveolar space changes. Lower scores signified little to no remodeling changes and higher scores signified increased remodeling changes. Mean A.U. scores per evaluator were obtained and these were averaged per experimental condition.
ELISAs
Right side lung lobes were flash frozen immediately after harvest and crushed to make lysates in buffer containing 137 mM Tris HCl (pH 8.0), 130 mM NaCl, and 1% NP-40. Samples were normalized to total lung protein and used to assess the abundance of IL-6, IL-33, CCL20, Eotaxin-1 (DuoSet ELISA Kits, R&D Systems, Minneapolis, MN, USA), IL-4, IL-13 (eBioscience Kits, Thermo Fisher Scientific, Waltham, MA, USA), and MUC5AC (Novus Biologicals, Littleton, CO, USA) per manufacturer's instructions.
Statistical analyses
Normal data were analyzed by Students t-test, one-way ANOVA and Tukey's multiple comparisons post-test using GraphPad Prism. A p-value <0.05 was considered significant. Data were averaged and expressed as the mean ± SEM.
Results
Epithelial Miro1 deletion restricts mitochondria to the basolateral side of the cell
To begin to understand the impact of Miro1 mediated mitochondrial trafficking on epithelial response to complex allergens, we generated an in vivo model system to conditionally delete Miro1 from CCSP + club cells in the mouse airway epithelium through the administration of doxycycline (Figure 1A). The CCSP-Cre mouse has been shown to restrict Cre expression to the lung (47, 48), although some studies indicate a possible subset of cells where CCSP is expressed during development (49) or in models of infection (50). Doxycycline-containing diet was provided to mice for 7 days prior to initiation of house dust mite (HDM) exposure to allow effective deletion. This diet was maintained throughout the duration of the dosing protocol until euthanasia. In an acute allergen recall challenge model, mice were sensitized on days 0 and 7 and challenged on day 14 with either 25 µg of HDM or phosphate buffered saline (PBS) control via intranasal inhalation and were harvested 4 h after the challenge (Figure 1B). Alteration of mitochondrial trafficking in club cells following Miro1 deletion were confirmed using immunofluorescence on fixed lung tissue sections with TOMM20 labeling the mitochondria and CC10 labeling club cells. Nuclei were stained with DAPI. Miro1 deletion induced mitochondrial constriction to the basolateral side of airway club cells and away from the apical surface (Figures 1C,D). We did not evaluate the morphology of mitochondria under these conditions due to limitations in resolution. Loss of Miro1 expression in lung epithelial cells of Miro1-deleted mice at time of harvest was observed in tissues stained with anti-Miro1 antibody by immunohistochemistry (Figure 1E).
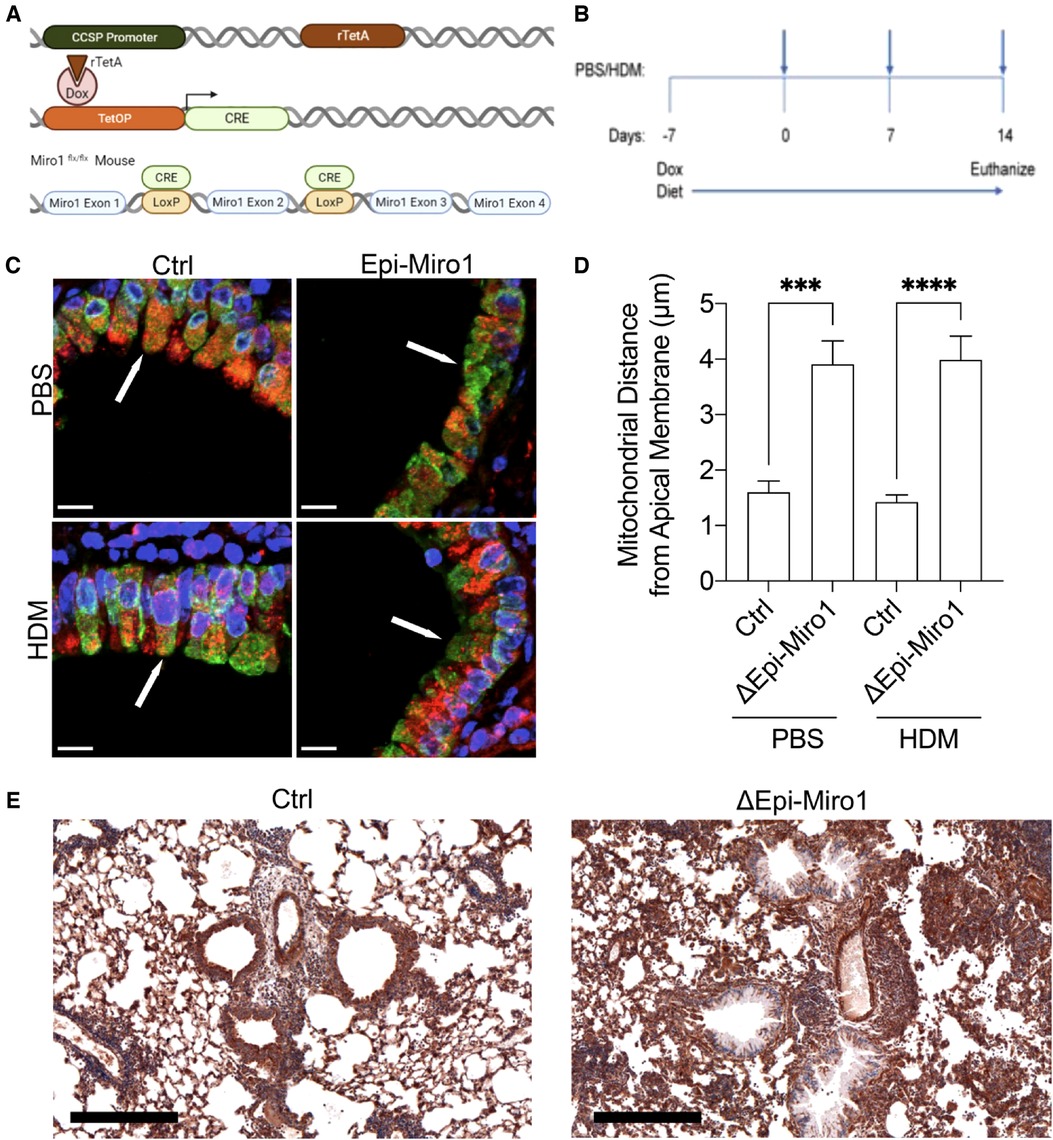
Figure 1. Miro1 deletion from club cells constricts mitochondria to the basolateral side of the cell. (A) Schematic of the doxycycline inducible deletion of Miro1 gene from CCSP positive epithelial cells. (B) Protocol schematic for the deletion of Miro1 via doxycycline containing food and single challenge HDM exposure protocol. Mice are sensitized on days 0 and 7 and challenged on day 14 with harvest 4 hours later. (C) Representative immunofluorescent images of TOMM20 (Red), CC10 (Green) and DAPI (Blue) in mouse lung tissue from representative mice. Arrows indicate areas of mitochondrial density in Ctrl mice that are lost at apical membranes in Epi-Miro1 mice. Scale Bar = 10 μm (D) Quantification of mitochondrial distance from the apical surface of club cells, n = 8–10 cells/airway from 5 mice per group; Two-way ANOVA with 2-stage linear set-up procedure. ***p < .001 vs. corresponding Ctrl group. Error bars represent mean ± SEM. (E) Immunohistochemical images of Miro1 expression in Ctrl and ΔEpi-Miro1 mice. Note loss of Miro1 IHC expression in the epithelial cell layer of ΔEpi-Miro1 mice. Scale bar = 300 μm
Epithelial Miro1 deletion does not alter initiation of inflammatory cell infiltration into the airways but does enhance pro-inflammatory cytokine abundance following single HDM challenge
Bronchoalveolar lavage fluid (BALF) was collected and used to assess inflammatory cell infiltration into the airways of the mice following HDM exposure. Total immune cell counts present in the BALF was marginally, though not significantly, enhanced in Miro1-deleted (ΔEpi-Miro1) mice exposed to HDM when compared to control (Ctrl) HDM-exposed mice (Figure 2A). This trending increase is attributed to slight increases in the numbers of eosinophils, neutrophils, and lymphocytes, with no discernable change in overall macrophage abundance in the airways (Figures 2B–E).
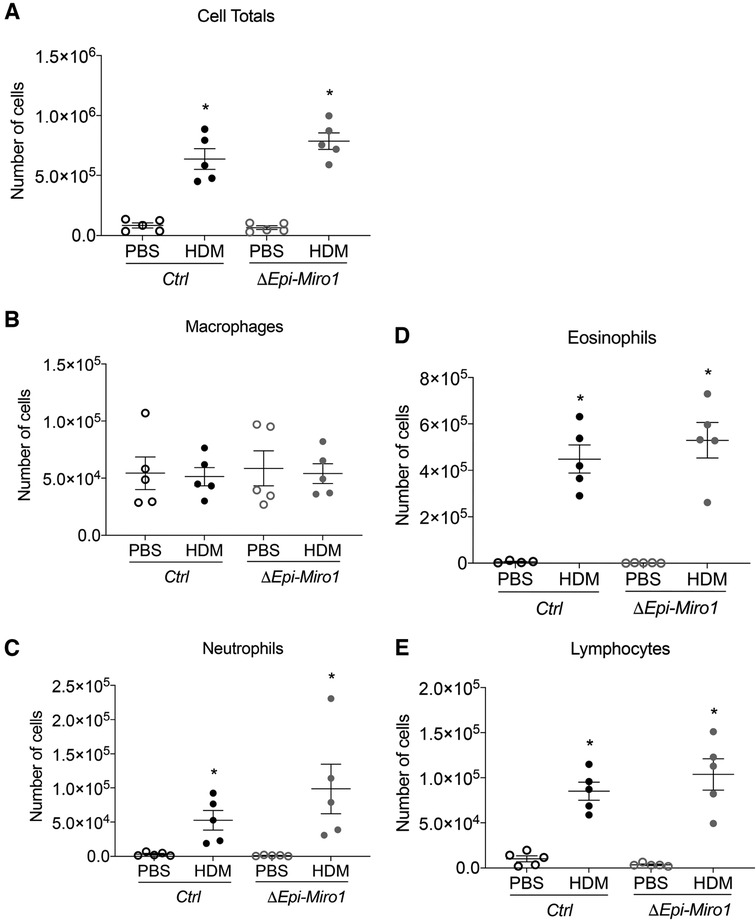
Figure 2. Epithelial Miro1 deletion does not significantly alter inflammatory cell recruitment following single HDM challenge. (A) Total cell infiltrates into the airways collected in the BALF, n = 5 mice per group: Two-way ANOVA with 2-stage linear set-up procedure, *p < 0.05 vs. corresponding PBS group. Error bars represent mean ± SEM. (B–E) Inflammatory cell-specific infiltrates present in the BALF, n = 5 mice per group; Two-way ANOVA followed by Sidak multiple comparisons test. *p < 0.05 vs. corresponding PBS group. Error bars represent mean ± SEM.
Though there were no significant cellular differences in the BALF between Ctrl and ΔEpi-Miro1 HDM-exposed mice, there were significant enhancements in several pro-inflammatory cytokines and chemokines in the lung tissue of Miro1 deleted HDM-exposed mice. More specifically, IL-6 and IL-33, two pro-inflammatory cytokines produced by airway epithelia known to play key roles in severity of asthma inflammation (20, 21), were modestly yet significantly upregulated in HDM-exposed mice following Miro1 deletion in club cells when compared to Ctrl HDM-challenged mice (Figures 3A,B). Further, the chemokine CCL20, produced by airway epithelial cells and known to play a significant role in mucus metaplasia, eosinophil recruitment, and IgE production in response to allergen (22, 23), was also significantly upregulated in HDM-exposed Miro1-deleted mice compared to Ctrls. (Figure 3C). Eotaxin, another key pro-inflammatory chemokine for eosinophil recruitment, did not differ between groups (Figure 3D). There were slight but not significant increases in production of the Th2 cytokines IL-4 and IL-13 as well (Figures 3E,F). These data suggest that Miro1 may help mediate the pro-inflammatory signaling response to allergen by airway epithelia.
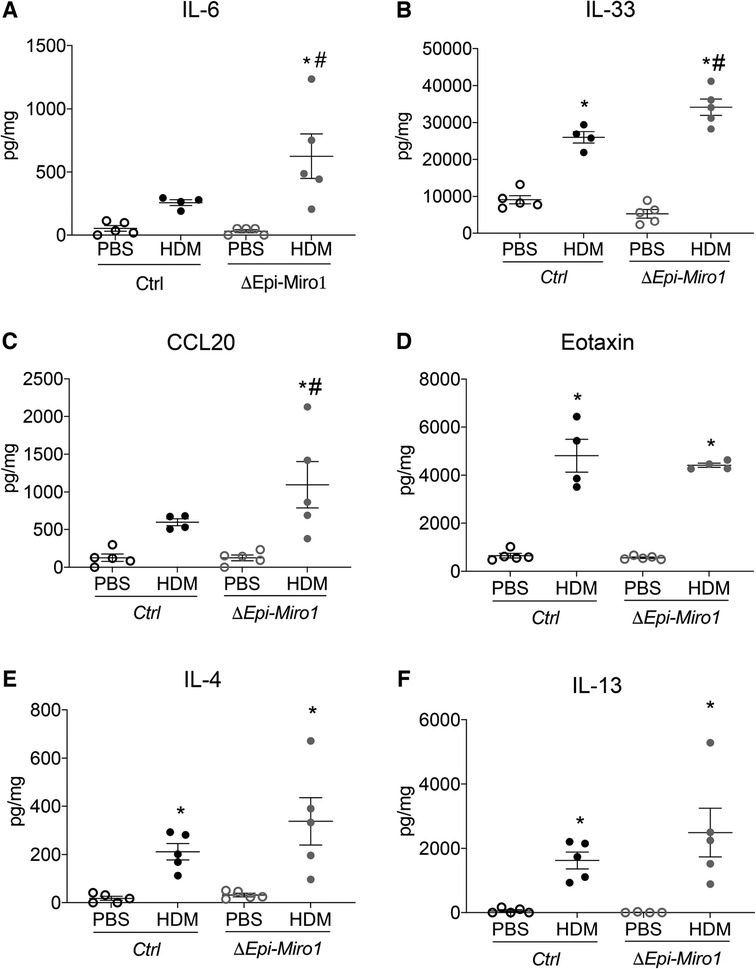
Figure 3. Epithelial Miro1 deletion enhances pro-inflammatory abundance following single HDM challenge. (A,B) ELISAs of pro-inflammatory cytokine abundance in lung tissue lysates, n = 5 mice per group; (C,D) ELISAs of pro-inflammatory chemokine abundance in lung tissue lysates, n = 5 mice per group; (E,F) ELISAs of Th2 cytokine abundance in lung tissue lysates, n = 5 mice per group: Two-way ANOVA followed by Sidak multiple comparisons test. *p < 0.05 vs. corresponding PBS group, #p < 0.05 vs. corresponding Ctrl group. Error bars represent mean ± SEM.
Epithelial Miro1 deletion enhances lymphocyte infiltration and pro-inflammatory signaling following multiple HDM challenges
We next aimed to determine whether these differences in initiation would translate to differences in progression and amplification of the inflammatory phenotype seen in a multi-challenge model of allergic asthma. We therefore utilized a challenge protocol in which we gave mice doxycycline 7 days before HDM sensitization. Mice where sensitized with 25μg HDM or PBS on days 0 and 7. Challenges occurred once a day on days 14 through 18, and mice were euthanized 24 h following the final challenge, on day 19. Mice were maintained on doxycycline-containing diet for the full duration of the experiment (Figure 4A). As with the single challenge protocol, there were no significant differences in total inflammatory cell infiltration into the BALF when comparing Ctrl and ΔEpi-Miro1 HDM-challenged mice (Figure 4B). Quantitation of inflammatory cell populations that enter the airspace following allergen exposure revealed no significant differences in macrophages, eosinophils, or neutrophils (Figures 4C–E). However, a significant enhancement in the number of lymphocytes present in the airways of Miro1-deleted HDM-challenged mice was observed compared to Ctrl mice (Figure 4F).
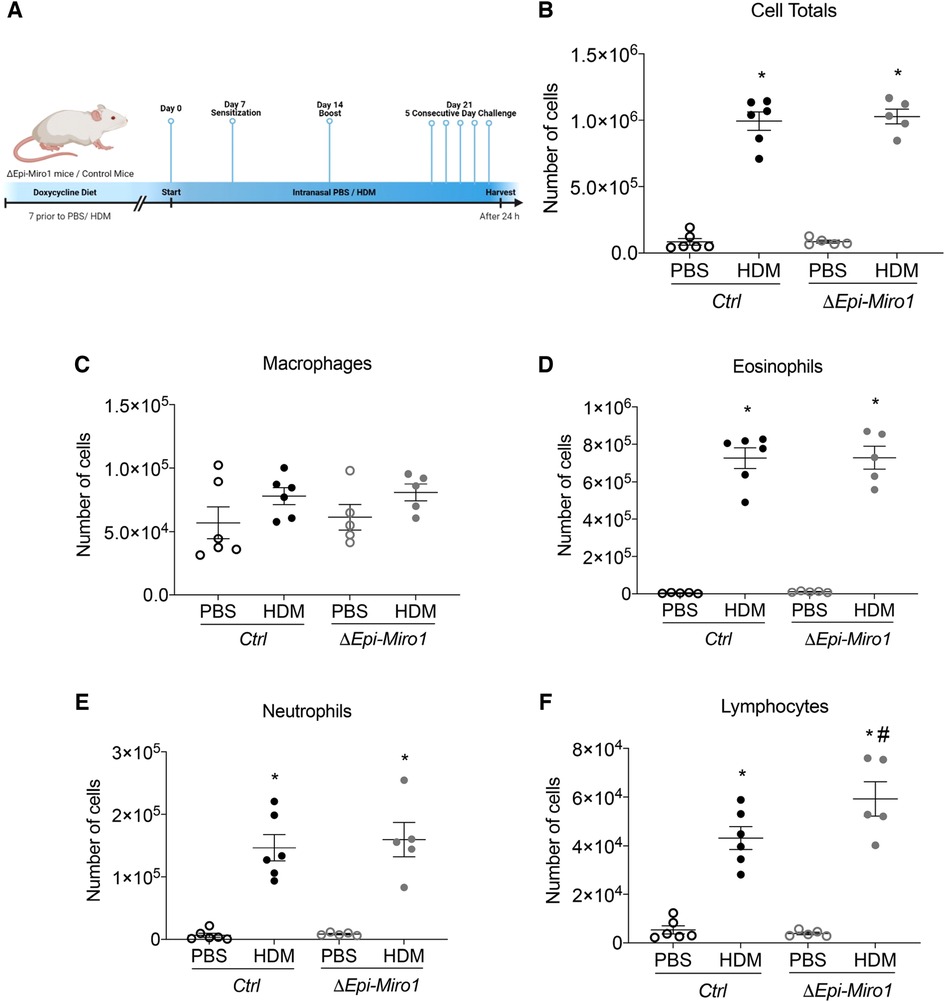
Figure 4. Epithelial Miro1 deletion significantly increases lymphocyte recruitment following multiple HDM challenges. (A) Schematic of deletion and multiple HDM challenge protocol; (B) Total cell infiltrates into the airways collected in the BALF, n = 5–6 mice per group: Two-way ANOVA with 2-stage linear set-up procedure. (C–F) Inflammatory cell-specific infiltrates present in the BALF, n = 5–6 mice per group: Two-way ANOVA followed by Sidak multiple comparisons test. *p < 0.05 vs. corresponding PBS group, #p < 0.05 vs. corresponding Ctrl group. Error bars represent mean ± SEM.
Pro-inflammatory cytokines and chemokines were also assessed, via ELISA, in lung tissue with this model. Though enhanced in the single challenge model of HDM exposure following Miro1 deletion, there were no discernable differences in expression of pro-inflammatory cytokines IL-6 and IL-33 between ΔEpi-Miro1 and Ctrl mice (Figures 5A,B). CCL20 and Eotaxin, however, were significantly enhanced in Miro1 deleted mice challenged multiple times with HDM when compared to Ctrl HDM mice (Figures 5C,D). And, as with the single challenge protocol, ΔEpi-Miro1 and Ctrl HDM mice had no significant differences in the abundance of Th2 cytokines IL-4 and IL-13, though Miro1 deleted HDM-challenged mice did trend higher than Ctrls (Figures 5E,F). These data together demonstrate a modest enhancement of inflammation in a more protracted model of allergic asthma following Miro1 epithelial deletion.
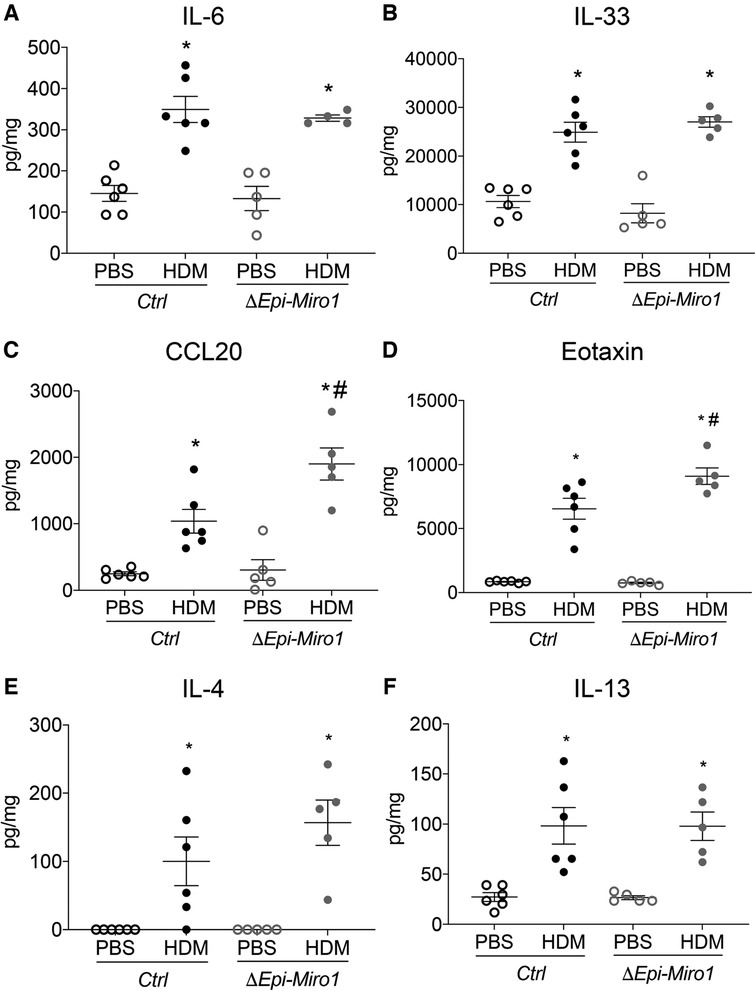
Figure 5. Epithelial Miro1 deletion significantly alters pro-inflammatory chemokine abundance following multiple HDM challenges. (A,B) ELISAs of pro-inflammatory cytokine abundance in lung tissue lysates, n = 5 mice per group; (C,D) ELISAs of pro-inflammatory chemokine abundance in lung tissue lysates, n = 5 mice per group; (E,F) ELISAs of Th2 cytokine abundance in lung tissue lysates, n = 5 mice per group; Two-way ANOVA followed by Sidak multiple comparisons test, *p < 0.05 vs. corresponding PBS group, #p < 0.05 vs. corresponding Ctrl group. Error bars represent mean ± SEM.
Conditional deletion of Miro1 in vivo augments mucus metaplasia and tissue remodeling
Mucus metaplasia is a hallmark of allergen-induced asthma and an indicator of its severity (51–53). Thus, we examined mucus metaplasia in lung tissue and MUC5AC abundance in the BALF after Miro1 deletion from the airway epithelium and HDM challenge. To assess mucus levels histologically, lung tissue was stained using the Periodic acid-Schiff stain (PAS) stain. Mucus staining was significantly increased in lung tissue of ΔEpi-Miro1 mice challenged with HDM, which exhibited extensive goblet cell hyperplasia when compared to the HDM-challenged Ctrls (Figures 6A,B). PAS staining was strong and concentrated in the epithelial cell layer in ΔEpi-Miro1 mice challenged with HDM (Figure 6A), suggesting epithelial Miro1 may regulate mucin secretion, further suppressing severity of the allergic response to HDM. Extensive immune cells were also observed in the surrounding lung tissue of ΔEpi-Miro1 mice challenged with HDM (Figure 6A). There were no statistically significant differences in the MUC5AC levels in the BALF between ΔEpi-Miro1 mice and Ctrls exposed to HDM in the BALF (Figure 6C), possibly due to cellular retention of mucus.
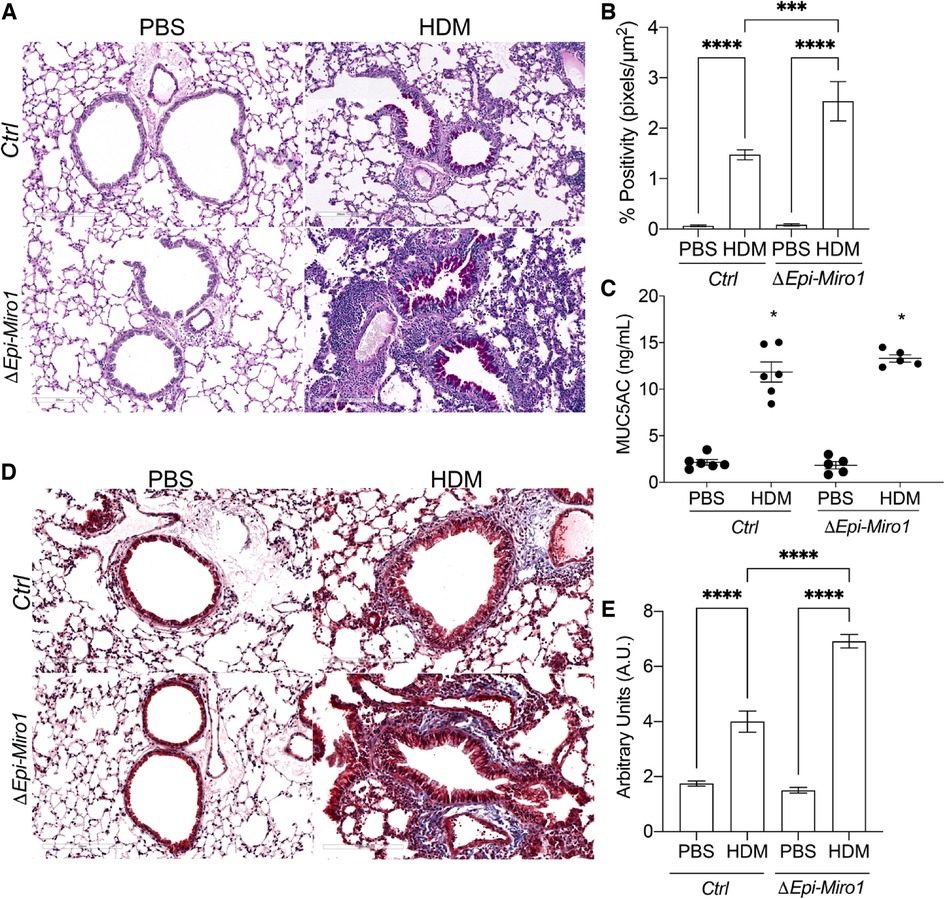
Figure 6. Conditional deletion of Miro1 in vivo augments mucus metaplasia and tissue remodeling (A) representative images of PAS staining in lung tissue sections from one experiment. (B) Quantification of PAS staining from multiple airways from n = 3–6 mice per group. (C) ELISA of MUC5AC in the BALF, n = 5–6 mice per group from one experiment: One-way ANOVA followed by Tukey multiple comparisons test. (D) Representative images of Masson's trichrome staining in lung tissue sections, n = 3–6. Scale bars are 200 µm. (E) Quantification of Masson's trichrome staining. The arbitrary unit scoring scale assessed changes in remodeling including collagen deposition, immune cell infiltration, epithelial layer thickening, and alveolar space changes. Lower scores signified little to no remodeling changes and higher scores signified increased remodeling changes. See materials and methods for scoring criteria. Error bars represent mean ± SEM. One-way ANOVA followed by Tukey multiple comparisons test, ***p = value < 0.001, ****p-value < 0.0001.
Next, we utilized Masson's trichrome (MT) staining to assess airway tissue remodeling, including collagen deposition, immune cell infiltration, epithelial hyperplasia, and alveolar space size in the lung tissue (Figure 6D). There were marked remodeling changes observed in the lung tissue of ΔEpi-Miro1 mice challenged with HDM when compared to control mice challenged with HDM (Figure 6D). ΔEpi-Miro1 exhibited more marked chronic small airway inflammation and reactive airway epithelial changes with more extensive peribronchiolar fibrosis (Figures 6D,E). These data suggest that Miro1 might help prevent severe remodeling changes in the airway epithelium following allergen exposure.
Conditional deletion of Miro1 in vivo enhances airway hyperresponsiveness following methacholine challenge
Methacholine-induced airway hyperresponsiveness (AHR) in the protracted HDM-induced allergic airway disease model was assessed. ΔEpi-Miro1 mice exposed to both PBS and HDM showed slightly increased, but significant, Newtonian resistance, tissue dampening, and tissue elastance compared to wild-type control animals (Figures 7A–C). These alterations in the AHR parameters, together with an increase in inflammation and mucus abundance are suggestive of increased airway constriction and collapse following exposure to HDM in airway epithelial Miro1-deleted mice. We also measured static compliance (Cst), which reflects the elastic properties of the respiratory system at rest. Cst was reduced in both Ctrl and ΔEpi-Miro1 mice exposed to HDM but only reached statistical significance in the ΔEpi-Miro1 group (Figure 7D). Lastly, we conducted a pressure-volume (Vpl and Ppl) curve analysis to calculate plateau pressure (Ppl) related to the total volume (Vpl) of methacholine delivered to the animals. HDM lowered both the Vpl and Ppl compared to PBS treated animals in both control and ΔEpi-Miro1 mice (Figure 7E).
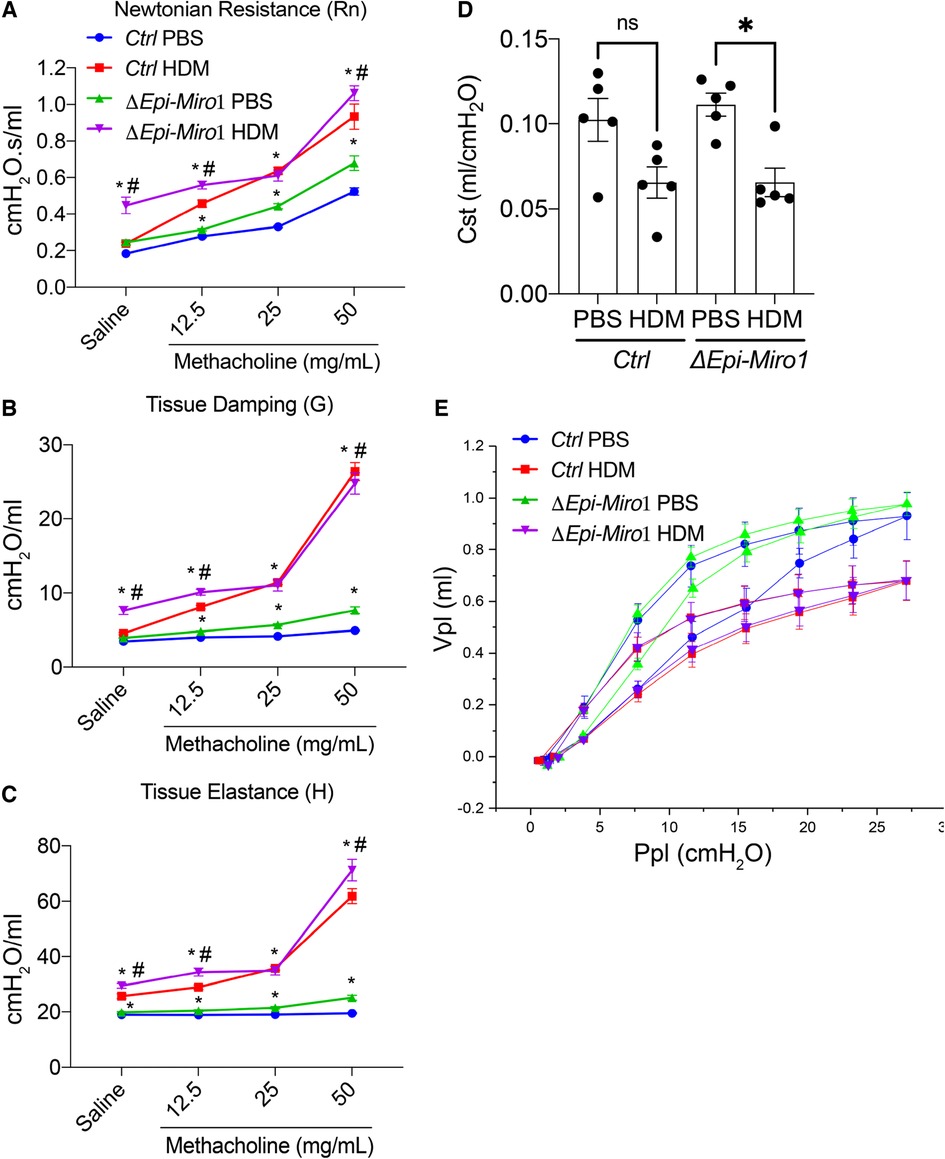
Figure 7. Epithelial Miro1 deletion significantly enhances AHR and cell death following multiple HDM challenges. (A) Newtonian Resistance (Rn) (B) Tissue Damping (G) and (C) Tissue Elastance (H) of control (Ctrl) and Epi-Miro1 mice challenged with PBS or HDM, n = 4–6 mice per group: Two-way ANOVA with 2-stage linear set-up procedure, *p < 0.05 vs. corresponding PBS group, #p < 0.05 vs. corresponding Ctrl group. Error bars represent mean ± SEM. (D) Static compliance (Cst) measurement in mice challenged with PBS or HDM, n = 4–6 mice per group: One-way ANOVA followed by Tukey multiple comparisons test, *p < 0.05. (E) Pressure volume curve analysis of indicated groups and treatments, n = 4–6 mice per group.
Resolution of AHR, pressure volume and static compliance is delayed in ΔEpi-Miro1
To evaluate resolution of the asthmatic phenotype induced by HDM exposure, we waited 2-weeks post HDM exposure in the multiple challenge protocol to evaluate cohorts of mice in Ctrl and ΔEpi-Miro1 groups. AHR parameters of tissue damping and elastance were still significantly increased in ΔEpi-Miro1 challenged compared to controls, whereas Newtonian resistance was comparable in both groups following methacholine inhalation (Figures 8A–C). Static compliance (Cst) measurements showed that Ctrl mice returned to control levels of approximately 0.10 ml/cmH2O while ΔEpi-Miro1 mice maintained a significantly lower Cst (Figure 8D), comparable to animals harvested 24 h post HDM exposure (Figure 7D). Lastly, pressure-volume loop analysis showed resolution in Ctrl mice while ΔEpi-Miro1 maintained a diminished pressure-volume loop (Figure 8E) comparable to animals harvested 24 h post-HDM (Figure 7E). Together these data indicate that deletion of Miro1 potentiates inflammatory responses and altered airway mechanics that fail to resolve following cessation of the HDM challenge.
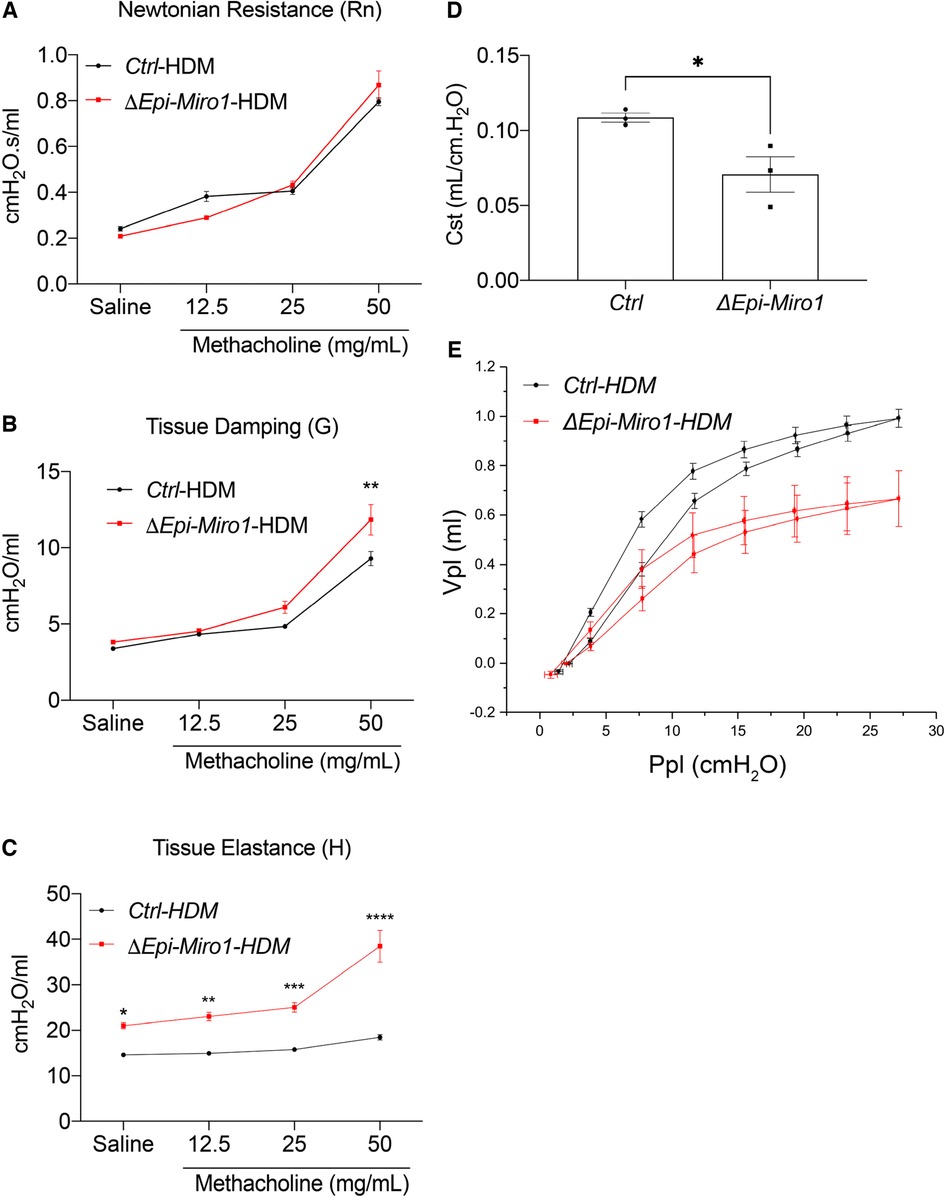
Figure 8. Resolution of AHR and pressure volume is delayed in epithelial Miro1 deleted mice compared to control mice. (A) Newtonian Resistance (Rn) (B) Tissue Damping (G) and (C) Tissue Elastance (H) of control (Ctrl) and Epi-Miro1 mice on the multiple challenge protocol and 2 weeks of no challenge, n = 3 mice per group, Two-way ANOVA with 2-stage linear set-up procedure. Error bars represent mean ± SEM. (D) Static compliance (Cst) measurement in mice challenged with PBS or HDM and allowed to resolve to 2 weeks, n = 3 mice per group, Students t-test, *p < 0.05. (E) Pressure volume curve analysis of indicated groups and treatments, n = 3 mice per group.
Discussion
The results from this study show that deletion of Miro1 from club cells leads to an increased inflammatory cell influx and enhanced secretion of pro-inflammatory cytokines in lung tissue following chronic HDM exposure. Miro1 epithelial deletion also leads to augmented mucus metaplasia and pronounced remodeling in the airways. Finally, Miro1 deletion alters lung mechanics following chronic allergen insult. The role of Miro1-mediated mitochondrial trafficking upon exposure to a complex allergen in airway epithelial cells, and their associated inflammatory responses, had not been demonstrated in previous literature. Our results provide compelling evidence for the role of Miro1 in airway epithelial cells by mediating inflammatory responses associated with HDM exposure and build upon our previous studies showing the importance of mitochondrial DRP1 to this process (23). However, the mechanistic details underlying the observed phenotype remain unclear, including additional aspects of Miro1 biology (54, 55) that where next explored in this study.
Chronic inflammation of the lungs caused by aeroallergens such as HDM is a hallmark of the pathophysiology associated with allergen-induced asthma (56). In this study, Miro1 was conditionally deleted from CCSP-positive airway epithelial cells in C57BL/6 mice. CCSP is predominantly expressed in club cells, a non-ciliated secretory epithelial cell subtype that is ubiquitously expressed in murine lungs (57, 58), although some studies indicate a possible subset of cells where CCSP is expressed during development (49) or in models of infection (50). ΔEpi-Miro1 and Ctrl mice were sensitized, boosted, and exposed for 5 consecutive days in a chronic model of allergen-induced inflammation to determine the contribution of Miro1 to HDM-induced lung inflammation. Our results show that epithelial deletion of Miro1 leads to an inflammatory cell influx in the BALF, especially lymphocytes, as well as an increase of pro-inflammatory mediators including CCL20 and eotaxin in the whole tissue lysate following HDM exposure. It is possible that the increase of CCL20, a lymphocyte and dendritic cell chemoattractant, and eotaxin, an eosinophil chemoattractant, promotes the increase in inflammatory cells following Miro1 deletion in airway epithelial cells. Specialized immunostainings on the lung tissue would be beneficial to determine the inflammatory cell profile in the observed sub-epithelial inflammation.
In agreement with our work, it was demonstrated that epithelial deletion of Miro1 leads to heightened pro-inflammatory responses following exposure to cigarette smoke (43). However, the inflammatory response in allergen-induced asthma is characterized by eosinophilia associated with an increased number of T lymphocytes and mast cell activation. CD4+ T lymphocytes regulate chronic inflammation in asthma via the release of Th2 adaptive cytokines (59, 60), which can lead to the observed changes in airway hyperresponsiveness, mucus production, and airway remodeling. Moreover, Miro1 plays a critical role in signaling for mitophagy and interacting with the Pink1/Parkin mitochondrial quality control system (61, 62), and compromised degradation of Parkin has been shown to promote inflammation and the release of mtDNA (63). However, mitophagy can activate apoptotic signaling pathways (64–66), an observation we made via the increase in the activity of caspases 3/7 (data not shown). Although we see non-significant increases in pro-inflammatory cytokines, Th2 cytokines, and inflammatory cell types in situ, future studies will measure these and other markers in the serum to assess their changes systemically. Our data suggest that Miro1 regulates inflammatory responses, primarily lymphocyte recruitment, following HDM exposure.
Airway remodeling has long been considered an important feature of asthma that results from longstanding inflammation and can lead to airway hyperresponsiveness (67). Structural changes within the airway wall such as epithelial membrane thickening, hypertrophy of smooth muscle cells, and peribronchial fibrosis result in the pathology associated with asthma (68). In this study, Miro1 deletion from epithelial cells led to prominent airway remodeling changes resulting in increased collagen deposition, heightened immune cell infiltration, and epithelial layer thickening, as demonstrated via Masson's trichrome staining. Miro1 deletion also led to increased smooth muscle surrounding the perimetry of the airways, as shown through immunohistochemical staining for α-smooth muscle actin (data not shown). The observed increase in eotaxin in the whole tissue lysate could be attributed to the increase in airway smooth muscle following Miro1 deletion, as airway smooth muscle has been shown to produce eotaxin and recruit eosinophils from systemic circulation (69). Moreover, other studies suggest that altered calcium homeostasis that leads to increased mitochondrial biogenesis results in increased bronchial smooth muscle mass (70). It is possible that lack of appropriate mitochondrial positioning in airway epithelial cells following Miro1 deletion could lead to calcium level dysregulation and increased airway smooth muscle. Together with our inflammatory cell profiles, these data are suggestive that Miro1 helps attenuate inflammation-associated remodeling changes.
Mucus is primarily produced and secreted by goblet cells, a specific subset of airway epithelial cells (71, 72). Goblet cell hyperplasia and metaplasia have been associated with asthma severity (52, 73), with mucus plugs being the primary cause of death in asthma due to asphyxiation from intraluminal airway obstruction (53). Our data suggest that club cell specific deletion of Miro1 leads to augmented mucus metaplasia as shown through the accumulation of mucus following exposure to HDM in the airway epithelium. However, there were no increases in mucin protein levels in the BALF after HDM exposure. Therefore, we hypothesize that mucins may remain in the secretory vesicles and not secreted into the intraluminal space following Miro1 deletion. Studies have shown that ATP is required to initiate a signaling cascade that results in a Ca2+ triggered fusion of the mucin granules to the membranes (38–41). Recent literature examining the role of Miro1 during alveolar formation observed a similar phenomenon where loss of epithelial Miro1 compromised the release of platelet-derived growth factor (74). Mitochondrial positioning via Miro1 also supports subcellular gradients of ATP and ROS towards the cell periphery in fibroblasts (35, 36) that may be also be lost in lung epithelial cells following Miro1 deletion, contributing to disrupted exocytic release. We suspect that Miro1 expression leading to the appropriate positioning of mitochondria in lung epithelial cells may be necessary for the secretion of mucins from airway epithelial cells, preventing more severe asthma phenotypes. However, it is unclear whether Miro1 deletion from club cells changes the functionality of mucus producing cells if transdifferentiated, as murine asthma models have shown a dramatic shift in cell phenotypes in the epithelium resulting from club cell differentiation to mucus cells (75).
The functional consequence of asthma is reversible airflow limitation (76). Airway inflammation alone may cause airflow limitation or through inflammatory mediators that act directly on airway smooth muscle (77). Enhanced tissue remodeling and mucus hyperplasia have been associated with increased airway hyperresponsiveness (78, 79). Our data show that Miro1 deletion from epithelial cells leads to changes in airway mechanics independent of HDM exposure, suggesting intrinsic alterations within the epithelium. The observed changes in airway hyperresponsiveness in this study could be attributed to the heightened immune cell infiltration, mucus obstruction as a result of goblet cell metaplasia, and enhanced smooth muscle levels and remodeling changes. Of striking importance is the inability for asthmatic phenotypes to be resolved in ΔEpi-Miro1 following a 2-week resolution phase. This information warrants investigation into Miro1 protein regulation in asthmatic models.
Results from this study suggest that Miro1-mediated mitochondrial trafficking contributes to the regulation of pro-inflammatory responses in the airway epithelium. Deletion of Miro1 was shown to augment chronic HDM-induced inflammatory responses in the lungs, associated with pronounced inflammatory cell infiltration and remodeling changes in the mouse lungs leading to altered lung mechanics. These results indicate a possible role for Miro1 in the development and progression of inflammatory responses and provide insights for the role of Miro1 in allergic airway diseases. Additional studies should be conducted to elucidate the mechanisms leading to disease. Altogether, these findings might have implications for the pharmacological targeting of Miro1 for the management and treatment of allergic airway diseases.
Data availability statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee of the University of Vermont.
Author contributions
SB, AL, MM, ZM, ND, MA and BC performed all experiments and contributed to data analysis and interpretation. KB, MEP, VA and BC contributed to data interpretation and manuscript editing. VA and BC conceived and designed the study. SB, AL and BC wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Support from the University of Vermont Department of Pathology and Laboratory Medicine (BC), Vermont Lung Center Pilot Project Grant (BC), Larner College of Medicine Pilot grant (BC). Supported by the National Institutes of Health R01HL133920 and R01HL142081 to MEP, HL 122383, HL141364, HL136917 to VA, and T32HL076122 to SB. Imaging work was performed at the Microscopy Imaging Center at the University of Vermont (RRID# SCR_018821). Confocal microscopy was performed on a Nikon A1R-ER point scanning confocal supported by NIH award number 1S10OD025030-01 from the Office of Research Infrastructure Programs.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet. (2017) 390(10100):1211–59. doi: 10.1016/S0140-6736(17)32154-2
2. Loftus PA, Wise SK. Epidemiology and economic burden of asthma. Int Forum Allergy Rhinol. (2015) 5(Suppl 1):S7–10. doi: 10.1002/alr.21547
3. GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: a systematic analysis for the global burden of disease study 2015. Lancet Respir Med. (2017) 5(9):691–706. doi: 10.1016/S2213-2600(17)30293-X
4. Toskala E, Kennedy DW. Asthma risk factors. Int Forum Allergy Rhinol. (2015) 5(Suppl 1):S11–6. doi: 10.1002/alr.21557
5. Subbarao P, Mandhane PJ, Sears MR. Asthma: epidemiology, etiology and risk factors. Cmaj. (2009) 181(9):E181–90. doi: 10.1503/cmaj.080612
6. Lemanske RF, Busse WW Jr. Asthma: clinical expression and molecular mechanisms. J Allergy Clin Immunol. (2010) 125(2 Suppl 2):S95–102. doi: 10.1016/j.jaci.2009.10.047
7. Patadia MO, Murrill LL, Corey J. Asthma: symptoms and presentation. Otolaryngol Clin North Am. (2014) 47(1):23–32. doi: 10.1016/j.otc.2013.10.001
8. Hammad H, Lambrecht BN. The basic immunology of asthma. Cell. (2021) 184(6):1469–85. doi: 10.1016/j.cell.2021.02.016
9. Tagaya E, Tamaoki J. Mechanisms of airway remodeling in asthma. Allergol Int. (2007) 56(4):331–40. doi: 10.2332/allergolint.R-07-152
10. Szefler SJ. Advancing asthma care: the glass is only half full!. J Allergy Clin Immunol. (2011) 128(3):485–94. doi: 10.1016/j.jaci.2011.07.010
11. Qu J, Li Y, Zhong W, Gao P, Hu C, et al. Recent developments in the role of reactive oxygen species in allergic asthma. J Thorac Dis. (2017) 9(1):E32–e43. doi: 10.21037/jtd.2017.01.05
12. Nam HS, Izumchenko E, Dasgupta S, Hoque MO, et al. Mitochondria in chronic obstructive pulmonary disease and lung cancer: where are we now? Biomark Med. (2017) 11(6):475–89. doi: 10.2217/bmm-2016-0373
13. Aghapour M, Remels AHV, Pouwels SD, Bruder D, Hiemstra PS, Cloonan SM, et al. Mitochondria: at the crossroads of regulating lung epithelial cell function in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. (2020) 318(1):L149–l164. doi: 10.1152/ajplung.00329.2019
14. Piantadosi CA, Suliman HB. Mitochondrial dysfunction in lung pathogenesis. Annu Rev Physiol. (2017) 79:495–515. doi: 10.1146/annurev-physiol-022516-034322
15. Larson-Casey JL, He C, Carter AB. Mitochondrial quality control in pulmonary fibrosis. Redox Biol. (2020) 33:101426. doi: 10.1016/j.redox.2020.101426
16. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. (2009) 417(1):1–13. doi: 10.1042/BJ20081386
17. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS, et al. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. (2004) 287(4):C817–33. doi: 10.1152/ajpcell.00139.2004
18. Zhou W, Qu J, Xie S, Sun Y, Yao H, et al. Mitochondrial dysfunction in chronic respiratory diseases: implications for the pathogenesis and potential therapeutics. Oxid Med Cell Longev. (2021) 2021:5188306. doi: 10.1155/2021/5188306
19. Comhair SA, Erzurum SC. Redox control of asthma: molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal. (2010) 12(1):93–124. doi: 10.1089/ars.2008.2425
20. McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. (2006) 16(14):R551–60. doi: 10.1016/j.cub.2006.06.054
21. Soubannier V, McBride HM. Positioning mitochondrial plasticity within cellular signaling cascades. Biochim Biophys Acta. (2009) 1793(1):154–70. doi: 10.1016/j.bbamcr.2008.07.008
22. Cunniff B, McKenzie AJ, Heintz NH, Howe AK, et al. AMPK Activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol Biol Cell. (2016) 27(17):2662–74. doi: 10.1091/mbc.e16-05-0286
23. Bruno SR, Kumar A, Mark ZF, Chandrasekaran R, Nakada E, Chamberlain N, et al. DRP1-Mediated Mitochondrial fission regulates lung epithelial response to allergen. Int J Mol Sci. (2021) 22(20). doi: 10.3390/ijms222011125
24. Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. (2005) 118(Pt 23):5411–9. doi: 10.1242/jcs.02745
25. Pilling AD, Horiuchi D, Lively CM, Saxton WM, et al. Kinesin-1 and dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. (2006) 17(4):2057–68. doi: 10.1091/mbc.e05-06-0526
26. Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, et al. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. (1998) 93(7):1147–58. doi: 10.1016/S0092-8674(00)81459-2
27. Fransson A, Ruusala A, Aspenström P. Atypical rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem. (2003) 278(8):6495–502. doi: 10.1074/jbc.M208609200
28. Fransson S, Ruusala A, Aspenström P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun. (2006) 344(2):500–10. doi: 10.1016/j.bbrc.2006.03.163
29. Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, et al. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. (2005) 47(3):379–93. doi: 10.1016/j.neuron.2005.06.027
30. Brickley K, Smith MJ, Beck M, Stephenson FA, et al. GRIF-1 and OIP106, members of a novel gene family of coiled-coil domain proteins: association in vivo and in vitro with kinesin. J Biol Chem. (2005) 280(15):14723–32. doi: 10.1074/jbc.M409095200
31. Glater EE, Megeath LJ, Stowers RS, Schwarz TL, et al. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. (2006) 173(4):545–57. doi: 10.1083/jcb.200601067
32. Stowers RS, Megeath LJ, Górska-Andrzejak J, Meinertzhagen IA, Schwarz TL, et al. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. (2002) 36(6):1063–77. doi: 10.1016/S0896-6273(02)01094-2
33. López-Doménech G, Covill-Cooke C, Ivankovic D, Halff EF, Sheehan DF, Norkett R, et al. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. (2018) 37(3):321–36. doi: 10.15252/embj.201696380
34. Nguyen TT, Oh SS, Weaver D, Lewandowska A, Maxfield D, Schuler MH, et al. Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease. Proc Natl Acad Sci U S A. (2014) 111(35):E3631–40. doi: 10.1073/pnas.1402449111
35. Alshaabi H, Shannon N, Gravelle R, Milczarek S, Messier T, Cunniff B, et al. Miro1-mediated mitochondrial positioning supports subcellular redox status. Redox Biol. (2021) 38:101818. doi: 10.1016/j.redox.2020.101818
36. Schuler MH, Lewandowska A, Caprio GD, Skillern W, Upadhyayula S, Kirchhausen T, et al. Miro1-mediated mitochondrial positioning shapes intracellular energy gradients required for cell migration. Mol Biol Cell. (2017) 28(16):2159–69. doi: 10.1091/mbc.e16-10-0741
37. Ribeiro CMP, Paradiso AM, Livraghi A, Boucher RC, et al. The mitochondrial barriers segregate agonist-induced calcium-dependent functions in human airway epithelia. J Gen Physiol. (2003) 122(4):377–87. doi: 10.1085/jgp.200308893
38. Evans CM, Williams OW, Tuvim MJ, Nigam R, Mixides GP, Blackburn MR, et al. Mucin is produced by clara cells in the proximal airways of antigen-challenged mice. Am J Respir Cell Mol Biol. (2004) 31(4):382–94. doi: 10.1165/rcmb.2004-0060OC
39. Davis CW, Dickey BF. Regulated airway goblet cell mucin secretion. Annu Rev Physiol. (2008) 70:487–512. doi: 10.1146/annurev.physiol.70.113006.100638
40. Davis CW, Lazarowski E. Coupling of airway ciliary activity and mucin secretion to mechanical stresses by purinergic signaling. Respir Physiol Neurobiol. (2008) 163(1-3):208–13. doi: 10.1016/j.resp.2008.05.015
41. Jaramillo AM, Azzegagh Z, Tuvim MJ, Dickey BF, et al. Airway mucin secretion. Ann Am Thorac Soc. (2018) 15(Suppl 3):S164–70. doi: 10.1513/AnnalsATS.201806-371AW
42. Sundar IK, Maremanda KP, Rahman I. Mitochondrial dysfunction is associated with Miro1 reduction in lung epithelial cells by cigarette smoke. Toxicol Lett. (2019) 317:92–101. doi: 10.1016/j.toxlet.2019.09.022
43. Sharma S, Wang Q, Muthumalage T, Rahman I, et al. Epithelial ablation of Miro1/Rhot1 GTPase augments lung inflammation by cigarette smoke. Pathophysiology. (2021) 28(4):501–12. doi: 10.3390/pathophysiology28040033
44. Perl AK, Tichelaar JW, Whitsett JA. Conditional gene expression in the respiratory epithelium of the mouse. Transgenic Res. (2002) 11(1):21–9. doi: 10.1023/A:1013986627504
45. van de Wetering C, Aboushousha R, Manuel AM, Chia SB, Erickson C, MacPherson MB, et al. Pyruvate kinase M2 promotes expression of proinflammatory mediators in house dust mite-induced allergic airways disease. J Immunol. (2020) 204(4):763–74. doi: 10.4049/jimmunol.1901086
46. Siddesha JM, Nakada EM, Mihavics BR, Hoffman SM, Rattu GK, Chamberlain N, et al. Effect of a chemical chaperone, tauroursodeoxycholic acid, on HDM-induced allergic airway disease. Am J Physiol Lung Cell Mol Physiol. (2016) 310(11):L1243–59. doi: 10.1152/ajplung.00396.2015
47. Li H, Cho SN, Evans CM, Dickey BF, Jeong JW, DeMayo FJ, et al. Cre-mediated recombination in mouse Clara cells. Genesis. (2008) 46(6):300–7. doi: 10.1002/dvg.20396
48. Perl AKT, Wert SE, Loudy DE, Shan Z, Blair PA, Whitsett JA, et al. Conditional recombination reveals distinct subsets of epithelial cells in trachea, bronchi, and alveoli. Am J Respir Cell Mol Biol. (2005) 33(5):455–62. doi: 10.1165/rcmb.2005-0180OC
49. Londhe VA, Maisonet TM, Lopez B, Jeng JM, Li C, Minoo P, et al. A subset of epithelial cells with CCSP promoter activity participates in alveolar development. Am J Respir Cell Mol Biol. (2011) 44(6):804–12. doi: 10.1165/rcmb.2009-0429OC
50. Bustos ML, Mura M, Hwang D, Ludkovski O, Wong AP, Keating A, et al. Depletion of bone marrow CCSP-expressing cells delays airway regeneration. Mol Ther. (2015) 23(3):561–9. doi: 10.1038/mt.2014.223
51. Dunican EM, Elicker BM, Gierada DS, Nagle SK, Schiebler ML, Newell JD, et al. Mucus plugs in patients with asthma linked to eosinophilia and airflow obstruction. J Clin Invest. (2018) 128(3):997–1009. doi: 10.1172/JCI95693
52. Aikawa T, Shimura S, Sasaki H, Ebina M, Takishima T, et al. Marked goblet cell hyperplasia with mucus accumulation in the airways of patients who died of severe acute asthma attack. Chest. (1992) 101(4):916–21. doi: 10.1378/chest.101.4.916
53. Kuyper LM, Paré PD, Hogg JC, Lambert RK, Ionescu D, Woods R, et al. Characterization of airway plugging in fatal asthma. Am J Med. (2003) 115(1):6–11. doi: 10.1016/S0002-9343(03)00241-9
54. Modi S, López-Doménech G, Halff EF, Covill-Cooke C, Ivankovic D, Melandri D, et al. Miro clusters regulate ER-mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nat Commun. (2019) 10(1):4399. doi: 10.1038/s41467-019-12382-4
55. Covill-Cooke C, Toncheva VS, Drew J, Birsa N, López-Doménech G, Kittler JT, et al. Peroxisomal fission is modulated by the mitochondrial Rho-GTPases, Miro1 and Miro2. EMBO Rep. (2020) 21(2):e49865. doi: 10.15252/embr.201949865
56. Murdoch JR, Lloyd CM. Chronic inflammation and asthma. Mutat Res. (2010) 690(1-2):24–39. doi: 10.1016/j.mrfmmm.2009.09.005
57. Singh G, Katyal SL. Clara cell proteins. Ann N Y Acad Sci. (2000) 923:43–58. doi: 10.1111/j.1749-6632.2000.tb05518.x
58. Singh G, Katyal S. Secretory proteins of Clara cells and type II cells. Comp Biol Norm Lung. (1992) 1:93–108.
59. Lloyd CM, Hessel EM. Functions of T cells in asthma: more than just T(H)2 cells. Nat Rev Immunol. (2010) 10(12):838–48. doi: 10.1038/nri2870
60. Robinson D, Hamid Q, Bentley A, Ying S, Kay A, Durham S, et al. Activation of CD4+ T cells, increased TH2-type cytokine mRNA expression, and eosinophil recruitment in bronchoalveolar lavage after allergen inhalation challenge in patients with atopic asthma. J Allergy Clin Immunol. (1993) 92(2):313–24. doi: 10.1016/0091-6749(93)90175-F
61. Bueno M, Lai YC, Romero Y, Brands J St, Croix CM, Kamga C, et al. PINK1 Deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest. (2015) 125(2):521–38. doi: 10.1172/JCI74942
62. Safiulina D, Kuum M, Choubey V, Gogichaishvili N, Liiv J, Hickey MA, et al. Miro proteins prime mitochondria for parkin translocation and mitophagy. Embo j. (2019) 38(2). doi: 10.15252/embj.201899384
63. Dimasuay KG, Schaunaman N, Martin RJ, Pavelka N, Kolakowski C, Gottlieb RA, et al. Parkin, an E3 ubiquitin ligase, enhances airway mitochondrial DNA release and inflammation. Thorax. (2020) 75(9):717–24. doi: 10.1136/thoraxjnl-2019-214158
64. Prakash YS, Pabelick CM, Sieck GC. Mitochondrial dysfunction in airway disease. Chest. (2017) 152(3):618–26. doi: 10.1016/j.chest.2017.03.020
65. Aggarwal S, Mannam P, Zhang J. Differential regulation of autophagy and mitophagy in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol. (2016) 311(2):L433–52. doi: 10.1152/ajplung.00128.2016
66. Mizumura K, Cloonan SM, Haspel JA, Choi AMK, et al. The emerging importance of autophagy in pulmonary diseases. Chest. (2012) 142(5):1289–99. doi: 10.1378/chest.12-0809
67. Fahy JV, Corry DB, Boushey HA. Airway inflammation and remodeling in asthma. Curr Opin Pulm Med. (2000) 6(1):15–20. doi: 10.1097/00063198-200001000-00004
68. Holgate ST. Pathogenesis of asthma. Clin Exp Allergy. (2008) 38(6):872–97. doi: 10.1111/j.1365-2222.2008.02971.x
69. Ghaffar O, Hamid Q, Renzi PM, Allakhverdi Z, Molet S, Hogg JC, et al. Constitutive and cytokine-stimulated expression of eotaxin by human airway smooth muscle cells. Am J Respir Crit Care Med. (1999) 159(6):1933–42. doi: 10.1164/ajrccm.159.6.9805039
70. Trian T, Benard G, Begueret H, Rossignol R, Girodet PO, Ghosh D, et al. Bronchial smooth muscle remodeling involves calcium-dependent enhanced mitochondrial biogenesis in asthma. J Exp Med. (2007) 204(13):3173–81. doi: 10.1084/jem.20070956
71. Ma J, Rubin BK, Voynow JA., mucins, mucus, and goblet cells. Chest. (2018) 154(1):169–76. doi: 10.1016/j.chest.2017.11.008
72. Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. (2010) 363(23):2233–47. doi: 10.1056/NEJMra0910061
73. Ordoñez CL, Khashayar R, Wong HH, Ferrando R, Wu R, Hyde DM, et al. Mild and moderate asthma is associated with airway goblet cell hyperplasia and abnormalities in mucin gene expression. Am J Respir Crit Care Med. (2001) 163(2):517–23. doi: 10.1164/ajrccm.163.2.2004039
74. Zhang K, Yao E, Chen B, Chuang E, Wong J, Seed RI, et al. Acquisition of cellular properties during alveolar formation requires differential activity and distribution of mitochondria. eLife. (2022) 11. doi: 10.7554/eLife.68598
75. Reader JR, Tepper JS, Schelegle ES, Aldrich MC, Putney LF, Pfeiffer JW, et al. Pathogenesis of mucous cell metaplasia in a murine asthma model. Am J Pathol. (2003) 162(6):2069–78. doi: 10.1016/S0002-9440(10)64338-6
76. Rogliani P, Ora J, Puxeddu E, Cazzola M, et al. Airflow obstruction: is it asthma or is it COPD? Int J Chron Obstruct Pulmon Dis. (2016) 11:3007–13. doi: 10.2147/COPD.S54927
77. Doeing DC, Solway J. Airway smooth muscle in the pathophysiology and treatment of asthma. J Appl Physiol. (2013) 114(7):834–43. doi: 10.1152/japplphysiol.00950.2012
78. Boulet LP, Turcotte H, Laviolette M, Naud F, Bernier MC, Martel S, et al. Airway hyperresponsiveness, inflammation, and subepithelial collagen deposition in recently diagnosed versus long-standing mild asthma. Influence of inhaled corticosteroids. Am J Respir Crit Care Med. (2000) 162(4 Pt 1):1308–13. doi: 10.1164/ajrccm.162.4.9910051
Keywords: Miro1, asthma, inflammation, mitochondria, house dust mite (HDM)
Citation: Bruno S, Lamberty A, McCoy M, Mark Z, Daphtary N, Aliyeva M, Butnor K, Poynter ME, Anathy V and Cunniff B (2023) Deletion of Miro1 in airway club cells potentiates allergic asthma phenotypes. Front. Allergy 4:1187945. doi: 10.3389/falgy.2023.1187945
Received: 16 March 2023; Accepted: 22 May 2023;
Published: 12 June 2023.
Edited by:
Xing Wang, The Affiliated Hospital of Southwest Medical University, ChinaReviewed by:
Deepak A. Deshpande, Thomas Jefferson University, United StatesRobert J. Lee, University of Pennsylvania, United States
© 2023 Bruno, Lamberty, McCoy, Mark, Daphtary, Aliyeva, Butnor, Poynter, Anathy and Cunniff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian Cunniff YmN1bm5pZmZAdXZtLmVkdQ==
†These authors have contributed equally to this work and share first authorship