 Jeremy P. McAleer
Jeremy P. McAleer- Department of Pharmaceutical Sciences, Marshall University School of Pharmacy, Huntington, WV, United States
Atopic dermatitis (AD) is an inflammatory skin disease characterized by epidermal barrier disruption, Th2 immune responses to skin allergens and microbial dysbiosis within affected lesions. Studies within the past decade have revealed genetic and environmental factors contributing to AD in children. Obesity is a metabolic disorder that often manifests early in life and is associated with reduced bacterial diversity, leading to skin colonization with lipophilic bacteria and intestinal colonization with pro-inflammatory species. These changes impair epithelial barriers and promote Th17 responses, which may worsen the severity of AD symptoms. While few studies have examined the contribution of microbiota in obesity-induced allergies, there is emerging evidence that PPAR-γ may be an effective therapeutic target. This review discusses the microbiome in pediatric AD, treatment with probiotics, how disease is altered by obesity and potential therapeutic effects of PPAR-γ agonists. While healthy skin contains diverse species adapted for specific niches, lesional skin is highly colonized with Staphylococcus aureus which perpetuates the inflammatory reaction. Treatments for AD should help to restore microbial diversity in the skin and intestine, as well as epithelial barrier function. Pre-clinical models have shown that PPAR-γ agonists can suppress Th17 responses, IgE production and mast cell function, while improving the epidermal barrier and microbial homeostasis. Overall, PPAR-γ agonists may be effective in a subset of patients with AD, and future studies should distinguish their metabolic and anti-inflammatory effects in order to inform the best therapies.
Introduction
Atopic march describes the successive development of allergic diseases beginning in infancy, including atopic dermatitis (AD), allergic rhinitis, asthma and food allergy (1). The first manifestation is usually AD, occurring in 85 percent of affected children before the age of 5 (2). In U.S. children, the prevalence of AD is 17 percent and is associated with a reduced quality of life due to anxiety and sleep disturbances. AD is typically caused by Th2 immune responses against skin allergens that lead to IgE production. Re-exposure to these allergens results in the degranulation of skin-resident mast cells that have been sensitized with IgE, leading to allergic manifestations including a rash, inflammation and pruritus. Longitudinal studies suggest that severe AD early in life increases the risk for allergic rhinitis or asthma in childhood or adulthood (2). These findings underscore the importance of identifying factors regulating the development of AD that may be exploited as therapeutic targets.
Epithelial surfaces of the body are colonized with microorganisms, collectively referred to as the microbiome. Many studies have characterized bacterial and fungal species present in the gastrointestinal (GI) tract, skin and lungs in healthy and diseased individuals, revealing several immunomodulatory functions. Nevertheless, our molecular understanding of how microbial colonization impacts the immune system is incomplete. Beneficial and detrimental roles for microbes have been identified in pediatric allergies, with protection associated with breastfeeding, vaginal delivery, having pets and avoiding antibiotics (3). Collectively, these studies suggest that dysbiosis, or imbalances in microbial species prevalence and diversity, contributes to atopy.
Obesity is a metabolic disorder that often manifests early in life and is associated with reduced bacterial diversity in the GI tract (4–6). Several lines of evidence suggest that obesity increases the severity of allergic diseases (7), including food allergies (8). While few studies have examined the contribution of microbiota in obesity-induced allergies, there is emerging evidence that PPAR-γ may be an effective therapeutic target. PPAR-γ is a lipid-sensing transcription factor that regulates genes involved in lipid metabolism, insulin sensitivity, adipogenesis and inflammation (9). Due to these functions, medications that stimulate PPAR-γ are approved for treating diabetes mellitus and inflammatory bowel diseases, underscoring its multi-functional roles. This review discusses the microbiome in pediatric AD, treatment with probiotics, how disease is altered by obesity and potential therapeutic effects of PPAR-γ agonists.
Atopic dermatitis
Pathophysiology
Atopic dermatitis (AD), or eczema, is a chronic, relapsing inflammatory skin disease with a prevalence of up to 25% in children and 7% in adults (10). Symptoms beginning in childhood may subside in adolescence or continue for years, involving periods of exacerbation and remission. Affected individuals have dry, cracked skin, intense pruritus and a erythematous rash due to Th2 immune responses against allergens (Figure 1). The skin barrier is impaired within crusted erythematous areas, associated with epidermal hyperplasia, scaling and lichenification (10). Some individuals have deficiencies in filaggrin or antimicrobial peptides, increasing permeability of the skin and susceptibility to opportunistic infections, respectively. Skin injury often precipitates AD, causing keratinocytes to produce cytokines that promote inflammation and immune activation (TSLP, IL-1, IL-6, IL-25, IL-33, TGF-b) (10). Th2 cells play a central role in driving pathogenesis, leading to allergen-specific IgE production and eosinophilia within affected lesions (Figure 1). Th17 cells also have a role, as lesional regions have increased expression of inflammatory genes including IL13, IL17A, IL17F, IL22, CCL17 and S100s (11). During infancy, atopic sensitization is also associated with IL9, IL33 and IL33R expression. Scratching affected regions leads to further impairment of the epidermal barrier, increasing susceptibility to opportunistic pathogens including Staphylococcus aureus.

Figure 1. Interactions between the immune system and microbiome in atopic dermatitis. In healthy individuals, the epidermal barrier is intact and maintained by filaggrin expression, antimicrobial peptides, and other factors. This is associated with a diverse microbiome that colonizes distinct niches on the skin surface. Atopic dermatitis (AD) patients have an impaired skin barrier leading to increased permeability within the keratinocyte layers. This is associated with reduced microbial diversity, including increased colonization with Staphylococcus aureus. These individuals are at increased risk of inflammatory skin injury, leading to the production of cytokines that facilitate Th2 cell differentiation. Allergen-specific Th2 cells then produce cytokines that promote eosinophil recruitment to the skin and IgE production by B cells. Mast cells that are sensitized with IgE release histamine and other inflammatory mediators following subsequent exposures to the skin allergen, while eosinophils mediate skin damage by releasing intracellular granules. Obesity is associated with increased lipid composition on the skin which facilitates dysbiosis, including colonization with lipophilic Corynebacterium species. The chronic inflammatory milieu in obesity promotes Th17 responses on epithelial surfaces including the skin. Cytokines produced by Th17 cells, including IL-17 and IL-22, impact keratinocyte differentiation, epithelial permeability and antimicrobial peptide production. In addition, diet-induced obesity is associated with increased production of IgE and mast cell accumulation in the skin. These effects of obesity are thought to increase the severity of AD in affected patients. PPAR-γ is a transcription factor with anti-inflammatory properties that also regulates lipid metabolism. Medications that target PPAR-γ may treat AD through multiple mechanisms including suppression of Th17 differentiation, mast cell accumulation, IgE production or pro-inflammatory cytokines. PPAR-γ agonists also improve insulin sensitivity, lipid metabolism, epidermal barrier function and microbial diversity, while pro-inflammatory effects include Th2 differentiation and IL-9 production. It is important to ascertain if the efficacy of PPAR-γ agonists in microbiota-dependent allergic diseases is influenced by body mass index, comorbidities or potential pro-inflammatory effects on the immune system in certain patient endotypes.
Obese patients with immunologic diseases including atopy and asthma have more severe disease than their lean counterparts (7, 12). Studies examining obesity in infancy and childhood also found a positive association with the prevalence of AD (13, 14). Gender differences have been identified, as only females with AD had higher abdominal obesity rates than healthy controls (15). Leptin deficiency is commonly associated with obesity; however, conflicting studies suggest it may not be directly involved in AD pathogenesis (16). A murine model investigating the mechanism of obesity-driven AD identified a role for Th17 cells (17). Although neutralization of Th2 cytokines (IL-4, IL-13) protected lean mice from AD, this treatment exacerbated disease in obese mice due to the lack of PPAR-γ expression in CD4 T cells (17). In addition, the PPAR-γ agonist rosiglitazone reduced AD severity in obese mice, demonstrating a protective anti-inflammatory function (17). While few studies have examined the role of microbiota in obesity-induced AD, it may contribute to the persistent low grade systemic inflammation that occurs in obese children (18).
Skin Microbiota
Healthy skin microbiota contains diverse species adapted for specific niches, including Cutibacterium, Malassezia, Staphylococcus and Corynebacterium (Figure 2) (19). The most prevalent bacteria in healthy skin include Cutibacterium acnes, Staphylococcus epidermidis, and Streptococcus mitis/oralis/pneumoniae/sanguinis (20). In addition, young children have increased colonization with Streptococcus, Granulicatella, Gemella, Rothia, Haemophilius and Candida spp., whereas Cutibacterium, Corynebacterium, Staphylococcus, Lactobacillus, Finegoldia and Anaerococcus are more abundant in adults. Over 100 fungal species have been identified on healthy skin, with most belonging to the phyla Ascomycota (21). Increased sebum production and structural changes after puberty may facilitate colonization with lipophilic microbes including Cutibacterium, Corynebacterium and Malassezia, replacing Streptococcus and Candida (20, 22). Thus, the skin microbiome in teenagers is more similar to adults than children. The overall diversity of skin microbes in children is due to specific environmental niches promoting colonization with certain species, which may change after the onset of puberty.
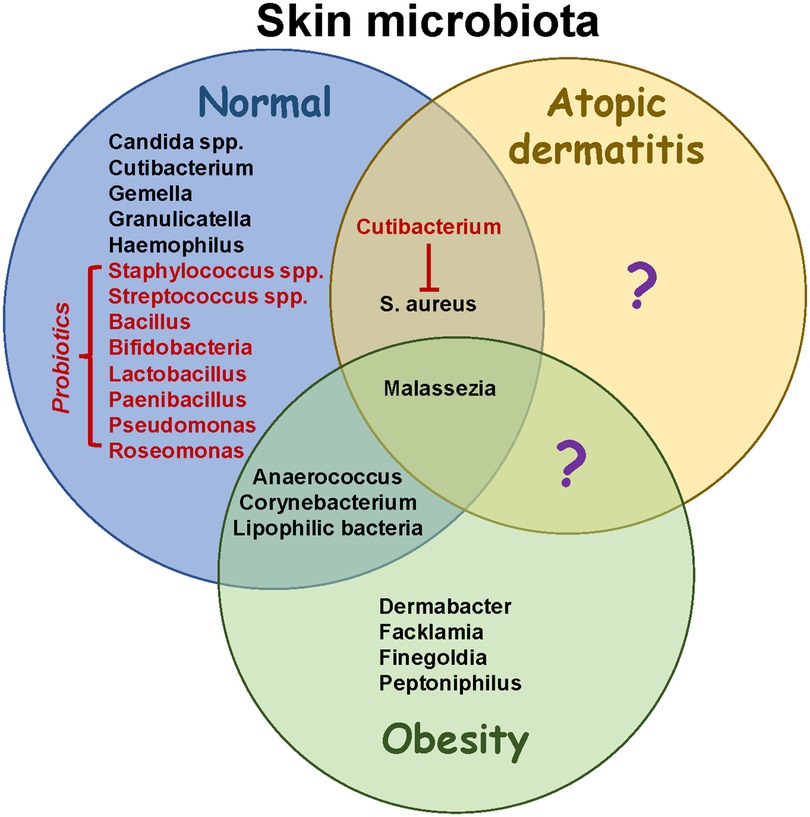
Figure 2. Microbiome changes in obesity and atopic dermatitis. Skin microbiota in healthy individuals has been well characterized, although more studies are necessary to elucidate developmental changes occurring throughout life. Structural changes associated with atopic dermatitis reduce bacterial diversity and increase colonization with Staphylococcus aureus. Potential probiotics (red font) help to restore diversity and have anti-inflammatory effects. In addition, they may suppress the growth of S. aureus. Obesity results in increased colonization with lipophilic species including Corynebacterium. While intestinal microbiota have been shown to contribute to AD severity in obesity (not shown), the role of skin microbiota in pathophysiology needs to be further studied. Identifying skin species that are unique to atopic dermatitis in the presence or absence of obesity (question marks) may help to guide therapeutic strategies to promote healthy microenvironments colonized with symbiotic microbiota.
Few studies have analyzed the impact of obesity on skin microbiota. Having a low body mass index (BMI) correlated with an increased Shannon Diversity Index compared to normal weight or obese individuals (23). Ten genera were enriched in underweight people, including Gordina, Lupinus and Prevotella, whereas seven were enriched in obesity including Anaerococcus, Finegoldia and Peptoniphilus (Figure 2). In addition, Corynebacterium colonization correlated with BMI (23). A mouse study found that skin Corynebacterium species and free fatty acids increased in response to a high fat diet (24). The authors speculated that increased adipogenesis created a microenvironment that favored colonization with lipophilic bacteria such as Corynebacterium. Dietary factors have also been shown to influence skin bacteria in humans (25). These data suggest that BMI and diet impact the composition of skin microbiota. Further studies are needed to analyze skin fungi in obesity, as Malassezia spp. are lipophilic and can induce Th17 responses (26), which may contribute to obesity-driven AD (17).
AD is characterized by reduced lipid content in skin, higher pH and increased transepidermal water loss, which may shape the composition of microbiota. Although children with AD had a more diverse microbiome in non-lesional skin compared to adults, dysbiosis occurred within skin lesions due to an impaired barrier (20). Dysbiosis was associated with reductions in Streptococcus, Cutibacterium and Malassezia, accompanied by increases in Staphylococcus aureus, suggesting an antagonistic relationship between skin commensals (20, 21). Streptococcus may inhibit S. aureus growth by producing hydrogen peroxide (27), whereas Cutibacterium and Corynebacterium are involved in porphyrin metabolism which may further suppress S. aureus colonization (20, 28). Dysbiosis might also be caused by a failure in antimicrobial peptide production which leads to increased colonization with S. aureus (29), immune activation in response to superantigens (30), and the development of AD (31). Patients are also sensitized to fungal antigens, including those from Malassezia spp., due to the disrupted epidermal barrier (21). Several lines of evidence implicate S. aureus in perpetuating AD. For instance, skin colonization at 3–6 months of age increases the risk of developing AD (32, 33), and both S. aureus and S. epidermidis increase during flares and decrease post-flare (34). Treating S. aureus-infected lesions with antibiotics reduces inflammation, demonstrating a critical role for microbiota in driving AD (35, 36). Topical treatments including corticosteroids, antibiotics and calcineurin inhibitors were associated with an increased diversity, including colonization with Streptococcus, Cutibacterium and Corynebacterium spp (34). Therefore, local inflammation during AD flares disrupts the microbiome by generating an environment that favors colonization with Staphylococcus spp. An adult study found that AD severity positively correlated with S. capitis and S. lugdunesis in lesional skin, and negatively correlated with S. hominis (37). Collectively, these observations suggest that factors influencing microbial colonization may impact an individual's susceptibility to AD.
Filaggrin (FLG)-deficiency contributes to dysbiosis by increasing skin pH, facilitating colonization with S. aureus (38). Virulence factors produced by S. aureus may then cause further breakdown of the skin barrier and stimulate immunity towards skin allergens (19). Skin microbiota are critical for the inflammation associated with FLG-deficiency. For instance, Flg−/− mice have spontaneous dermatitis and increased colonization with Staphylococcus spp. (39). In this model, dermatitis was dependent on IL-1β, but independent of Th2 cytokines. When raised germ-free, Flg−/− mice showed signs of dermatitis as neonates; however, this inflammation resolved in adulthood (39), suggesting that dysbiosis maintains the chronic inflammation in genetically susceptible individuals. Other genes associated with childhood AD include GRP1, CCL22, TTC27 (40), although their impact on skin colonization remains unclear.
Studies have explored if topically-applied probiotics can alleviate inflammation by restoring homeostasis. Ito, et al. demonstrated that skin inoculation with S. cohnii protected against spontaneous and chemical-induced AD by suppressing inflammation (41). Protection was attributed to the expression of glucocorticoid-inducible genes in skin, although S. cohnii strain-specific differences were observed. Roseomonas and Cutibacterium spp. may inhibit colonization with S. aureus and be suitable probiotics for AD skin (19); however, some Cutibacterium spp. (C. acnes) also facilitate S. aureus biofilm formation. A probiotic formulation containing Roseomonas mucosa, poly(vinyl pyrrolidione), poly(vinyl alcohol) and sodium alginate demonstrated antimicrobial activity against S. aureus (42). Other topical probiotics investigated for skin use include Bacillus, Bifidobacteria, Lactobacillus, Paenibacillus, Pseudomonas, Staphylococcus, Streptococcus, and others (43). These studies demonstrate a focus towards treating dysbiosis in order to reduce skin inflammation.
Gut microbiota in atopic dermatitis
The gut microbiome has been compared between infants with and without AD. Facultative anaerobes predominate during the first 6–12 months of life prior to colonization with obligate anaerobes (44). Notably, the anaerobe Akkermansia muciniphila was only detected in healthy infants and their mothers, suggesting it may correlate with protection against AD (45). Breast feeding may account for some of the microbiota differences between AD and non-AD infants (44). In the second year of life, moderate to severe AD was associated with a higher abundance of facultative anaerobes compared to healthy controls. This correlated with decreased production of the short chain fatty acid butyrate, and decreased expression of the butyrate receptor Gpr109a and Pparg in the colon of AD-induced mice (44). The authors speculated that low butyrate levels perturb the microbiome by decreasing oxygen consumption, promoting the growth of facultative anaerobes. In support, metabolic pathways responsive to oxidative stress are upregulated in the microbiome of AD patients (46). This was associated with increased colonization with Faecalibacterium prausnitzii and decreased levels of butyrate and propionate. In addition to Faecalibacterium, Bacteroides and Ruminococcus lactaris are increased in AD infants, whereas Bifidobacterium, Clostridium paraputrificum and Lachnospiraceae are decreased (45). Taken together, these data demonstrate that AD is associated with an altered microbial profile in the GI tract.
Several bacterial species are being tested for their therapeutic efficacy in AD. Oral administration of A. muciniphila or F. prausnitzii improved AD symptoms in mice, including dermatitis score, scratching behavior, serum IgE and TSLP (47). Treatment with these strains increased filaggrin in skin and ZO-1 expression in the intestine, demonstrating improved epithelial barriers. The mechanism of how A. muciniphila protects against AD may be multi-factorial, as monocolonization of germ-free mice upregulated genes involved in epithelial homeostasis, antigen presentation, immune activation and PPAR-α-dependent metabolism (48). Oral treatment with Pediococcus acidilactici decreased AD severity in mice, including erythema, hemorrhage, edema, excoriation, dryness and scratching behavior (49). In addition, P. acidilactici prevented the AD-induced decreases in Lactobacillales, Butyricicoccus, and Ruminococcus, demonstrating that it may help to restore intestinal homeostasis. Oral administration of Lactobacillus paracasei reduced AD-associated skin lesions, epidermal thickening, serum IgE and immune cell infiltration into skin lesions in a mouse model of AD (50). This was associated with decreased effector T cell cytokines and increased IL-10 and TGF-β. Similar results were found for L. plantarum, including increased colonization with butyrate-producing bacteria (51). In children with mild or moderate disease, L. plantarum supplementation resulted in a greater reduction in AD scores compared to placebo (52). A mouse model using Limosilactobacillus reuteri found that combining prenatal and postnatal treatment was better at improving AD and lowering serum IgE than postnatal treatment alone (53). This suggests maternal factors may influence the risk of developing AD in utero. Supplementation with L. reuteri increased microbial diversity in the GI tract, including colonization with Faecalibacterium, Bifidobacterium and Akkermansia (53). The anti-inflammatory effects of L. reuteri may have been due to PPAR-α signaling and retinol metabolism. These data demonstrate complex roles for microbiota and probiotics in protection against AD, including suppressing inflammation, improving microbial diversity, and increasing epithelial barrier function.
Gut microbiota and obesity
Few studies have examined the role of gut microbiota in obesity-induced allergies. Altering the microbiome with antibiotics during the first year leads to increased adiposity (54), demonstrating a role for bacteria in regulating metabolism. In support, human microbiota from obese donors increase body weight and adiposity when transferred to germ-free mice (55). Currently, a clinical trial is analyzing the microbial signature associated with obesity and AD (56). A mouse study demonstrated that diet-induced obesity aggravates contact hypersensitivity in an IL-17-dependent manner (57). This correlated with colonization of the GI tract by pro-inflammatory species including segmented filamentous bacteria, Clostridium type IV, and Enterococcus. Another study found that high fat diets increase IgE, small intestinal mast cells and gut permeability in response to food allergens (58). These data demonstrate that obesity-induced changes to the microbiome correlate with AD severity, and suggest that probiotics associated with leanness may protect against AD. Several species were shown to have lipid-lowering effects in human epidermal keratinocytes in vitro, including Bifidobacterium bifidum, Lactobacillus acidophilus, L. delbrueckii, L. casei, and L. gasseri (59), although their impact on obesity and atopy remain unknown. In vivo, Bifidobacterium breve persisted for at least 90 days after administration, whereas Lactobacillus salivarius colonization was transient (60), demonstrating that some probiotics may need more frequent administration than others.
PPAR-γ and childhood obesity
PPAR-γ is a master regulator of adipogenesis and functions as a transcription factor, improving insulin sensitivity (61). In addition, PPAR-γ has anti-inflammatory functions through its suppression of NF-κB and cyclooxygenase 2 in epithelial cells, granulocytes and T cells. High expression levels within adipose tissue, intestinal and immune cells contributes to the therapeutic efficacy of PPAR-γ agonists in type 2 diabetes and inflammatory bowel diseases (IBDs). Natural ligands for PPAR-γ include prostaglandins, medium to long chain fatty acids, foods and environmental pollutants (62). Genome studies have identified PPARG as one of the genes linked to childhood obesity (63, 64). While a dominant negative mutation is associated with severe insulin resistance, type 2 diabetes and hypertension (65), a gain of function is linked to extreme obesity (66). In mice, the dominant negative mutation exacerbates insulin resistance in the context of leptin-deficiency, demonstrating antagonistic roles for leptin and PPAR-γ in adipogenesis (67). PPAR-γ concentrations in obese children positively correlate with birth weight, but negatively correlate with waist circumference (68). Further, PPARG expression in immune cells and adipose tissue negatively correlate with obesity (69, 70), demonstrating complex functions for this transcription factor in regulating body weight. TMEM18 induces the expression of PPARG in adipose tissue and is critical for adipocyte differentiation (69). The inflammatory cytokine TNF suppresses both TMEM18 and PPARG1, leading to increased adipocyte size, decreased adiponectin, decreased insulin sensitivity and macrophage infiltration into adipose tissue. Some of the effects of PPAR-γ activity are mediated by adiponectin and ANGPTL4 (71, 72), with tissue-specific functions identified in the GI tract, skin, adipose tissue and immune cells (17, 73–79). Notably, microbiota have been found to regulate PPAR-γ signaling in the gut (75), although the role of microbiota on extra-intestinal functions of PPAR-γ are less clear.
PPAR-γ as a therapeutic target in atopic dermatitis
Due to its expression profile and anti-inflammatory effects, PPAR-γ may be a potential therapeutic target in microbiota-dependent diseases. The thiazolidinedione (TZD) rosiglitazone alleviated AD in response to a high fat diet, demonstrating a critical function for PPAR-γ (17). In this model, diet-induced obesity increased the severity of AD in an IL-17-depedendent manner. This was attributed to dietary fat downregulating Pparg within Th2 cells, allowing for the expansion of Th17 cells (17). A model of atopic march found the combination of rosiglitazone and dexamethasone suppressed allergic skin inflammation better than either medication alone, suggesting TZDs may synergize with glucocorticoids (76). Topical treatment also reduced subsequent lung inflammation following intranasal challenge. While it remains to be determined if PPAR-γ alters the microbiome in AD, Pparg expression is decreased in the colon of mice with AD, correlating with dysbiosis (44). Further, PPAR-γ agonists modulate intestinal microbes associated with Western diets or colitis (80–83). The mechanism through which PPAR-γ regulates GI microbiota involves suppressing lactate fermentation and promoting beta oxidation (84), facilitating colonization with anaerobes. Although PPAR-γ stimulation affects microbiota, therapeutic effects of agonists most likely arise from the suppression of Th17 cells (17), epidermal keratinocyte growth (85), mast cell development and differentiation (86, 87), and IgE production (88), demonstrating several anti-inflammatory functions that may benefit AD patients (Figure 1).
The etiology and pathophysiology of psoriasis bears some similarities with AD, including obesity, microbial dysbiosis and Th17-mediated inflammation (89). Clinical studies identified anti-psoriatic effects of TZDs (90), suggesting these medications may help in AD. Nevertheless, the skin microbiome is distinct in psoriasis and characterized by Corynebacteria spp. rather than S. aureus (91). Further, only AD patients have transcriptome signatures associated with epithelial barrier function and immune activation, suggesting that TZDs must be further studied before their use in pediatric AD patients. TZDs have a black box warning for congestive heart failure, a rare but serious side effect (92). Other adverse reactions include edema, weight gain, anemia and bone fractures. The use of mesalamine for IBD has been associated with renal impairment, hypersensitivity and photosensitivity. Further, PPAR-γ can exert pro-inflammatory effects through IL-9 which exacerbates dermatitis (93, 94). Although few studies have assessed the clinical use of PPAR-γ agonists in children, no serious adverse effects were reported in trials for IBD and autism (95, 96), suggesting an acceptable safety profile. Collectively, pre-clinical studies suggest that PPAR-γ agonists might be effective in a subset of patients with AD, and trials comparing their local vs. systemic administration may help to minimize the incidence of adverse reactions.
Conclusions
A surge of studies from the last decade helped to define the commensal microbiome in pediatric AD and how dysbiosis contributes to chronic diseases. Recent findings into the pathophysiology of obesity-driven AD have revealed PPAR-γ to be a potential therapeutic target. Future studies examining the efficacy of PPAR-γ modulators should distinguish their metabolic vs. anti-inflammatory effects in the gut, skin, adipose tissue and immune system, in order to inform the best therapies. The severity of AD and its role in initiating atopic march underscore the significance of utilizing effective treatments which may help to reduce the risk of asthma later in life.
Author contributions
This manuscript was written by JPM.
Funding
The publication charge of this manuscript was provided by the Marshall University School of Pharmacy.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hill DA, Spergel JM. The atopic march: critical evidence and clinical relevance. Ann Allergy Asthma Immunol. (2018) 120:131–7. doi: 10.1016/j.anai.2017.10.037
2. Spergel JM, Paller AS. Atopic dermatitis and the atopic march. J Allergy Clin Immunol. (2003) 112:S118–27. doi: 10.1016/j.jaci.2003.09.033
3. Musso P, Chiappini E, Bernardini R. Human microbiome and allergic diseases in children: pathogenetic role and therapeutic options. Curr Pediatr Rev. (2020) 16:89–94. doi: 10.2174/1573396315666191025110849
4. Houtman TA, Eckermann HA, Smidt H, de Weerth C. Gut microbiota and BMI throughout childhood: the role of firmicutes, bacteroidetes, and short-chain fatty acid producers. Sci Rep. (2022) 12:3140. doi: 10.1038/s41598-022-07176-6
5. Riva A, Borgo F, Lassandro C, Verduci E, Morace G, Borghi E, et al. Pediatric obesity is associated with an altered gut microbiota and discordant shifts in Firmicutes populations. Environ Microbiol. (2017) 19:95–105. doi: 10.1111/1462-2920.13463
6. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. (2009) 457:480–4. doi: 10.1038/nature07540
7. Hersoug LG, Linneberg A. The link between the epidemics of obesity and allergic diseases: does obesity induce decreased immune tolerance? Allergy. (2007) 62:1205–13. doi: 10.1111/j.1398-9995.2007.01506.x
8. Visness CM, London SJ, Daniels JL, Kaufman JS, Yeatts KB, Siega-Riz AM, et al. Association of obesity with IgE levels and allergy symptoms in children and adolescents: results from the national health and nutrition examination survey 2005–2006. J Allergy Clin Immunol. (2009) 123:1163–9. 1169 e1–4. doi: 10.1016/j.jaci.2008.12.1126
9. Decara J, Rivera P, Lopez-Gambero AJ, Serrano A, Pavon FJ, Baixeras E, et al. Peroxisome proliferator-activated receptors: experimental targeting for the treatment of inflammatory bowel diseases. Front Pharmacol. (2020) 11:730. doi: 10.3389/fphar.2020.00730
10. Mathias CB Inflammation of the skin and its therapeutic targets. In: Mathias CB, McAleer JP, Szollosi DE, editors. Pharmacology of immunotherapeutic drugs. London, UK: Springer Nature (2020). p. 141–74.doi: 10.1007/978-3-030-19922-7_5
11. Renert-Yuval Y, Del Duca E, Pavel AB, Fang M, Lefferdink R, Wu J, et al. The molecular features of normal and atopic dermatitis skin in infants, children, adolescents, and adults. J Allergy Clin Immunol. (2021) 148:148–63. doi: 10.1016/j.jaci.2021.01.001
12. Vehapoglu A, Cakin ZE, Kahraman FU, Nursoy MA, Toprak A. Is overweight/obesity a risk factor for atopic allergic disease in prepubertal children? A case-control study. J Pediatr Endocrinol Metab. (2021) 34:727–32. doi: 10.1515/jpem-2021-0051
13. Ali Z, Suppli Ulrik C, Agner T, Thomsen SF. Is atopic dermatitis associated with obesity? A systematic review of observational studies. J Eur Acad Dermatol Venereol. (2018) 32:1246–55. doi: 10.1111/jdv.14879
14. Nicholas MN, Keown-Stoneman CDG, Maguire JL, Drucker AM. Association between atopic dermatitis and height, body mass Index, and weight in children. JAMA Dermatol. (2022) 158:26–32. doi: 10.1001/jamadermatol.2021.4529
15. Iturriaga C, Bustos MF, Le Roy C, Rodriguez R, Cifuentes L, Silva-Valenzuela S, et al. Association between obesity and atopic dermatitis in children: a case-control study in a high obesity prevalence population. Pediatr Dermatol. (2022) 40(1):64–8. doi: 10.1111/pde.15110
16. Jimenez-Cortegana C, Ortiz-Garcia G, Serrano A, Moreno-Ramirez D, Sanchez-Margalet V. Possible role of leptin in atopic dermatitis: a literature review. Biomolecules. (2021) 11:1642–52. doi: 10.3390/biom11111642
17. Bapat SP, Whitty C, Mowery CT, Liang Y, Yoo A, Jiang Z, et al. Obesity alters pathology and treatment response in inflammatory disease. Nature. (2022) 604:337–42. doi: 10.1038/s41586-022-04536-0
18. Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Low-grade systemic inflammation in overweight children. Pediatrics. (2001) 107:E13. doi: 10.1542/peds.107.1.e13
19. Koh LF, Ong RY, Common JE. Skin microbiome of atopic dermatitis. Allergol Int. (2022) 71:31–9. doi: 10.1016/j.alit.2021.11.001
20. Shi B, Bangayan NJ, Curd E, Taylor PA, Gallo RL, Leung DYM, et al. The skin microbiome is different in pediatric versus adult atopic dermatitis. J Allergy Clin Immunol. (2016) 138:1233–6. doi: 10.1016/j.jaci.2016.04.053
21. Szczepanska M, Blicharz L, Nowaczyk J, Makowska K, Goldust M, Waskiel-Burnat A, et al. The role of the cutaneous mycobiome in atopic dermatitis. J Fungi (Basel). (2022) 8:1153–73. doi: 10.3390/jof8111153
22. Ward TL, Dominguez-Bello MG, Heisel T, Al-Ghalith G, Knights D, Gale CA. Development of the human mycobiome over the first month of life and across body sites. mSystems. (2018) 3(3):e00140–17. doi: 10.1128/mSystems.00140-17
23. Brandwein M, Katz I, Katz A, Kohen R. Beyond the gut: skin microbiome compositional changes are associated with BMI. Hum Microbiome J. (2019) 13:1–5. doi: 10.1016/j.humic.2019.100063
24. Moestrup KS, Chen Y, Schepeler T, Schweiger PJ, Jensen KB. Dietary control of skin lipid composition and microbiome. J Invest Dermatol. (2018) 138:1225–8. doi: 10.1016/j.jid.2017.12.005
25. Moitinho-Silva L, Boraczynski N, Emmert H, Baurecht H, Szymczak S, Schulz H, et al. Host traits, lifestyle and environment are associated with human skin bacteria. Br J Dermatol. (2021) 185:573–84. doi: 10.1111/bjd.20072
26. Sparber F, De Gregorio C, Steckholzer S, Ferreira FM, Dolowschiak T, Ruchti F, et al. The skin commensal yeast malassezia triggers a type 17 response that coordinates anti-fungal immunity and exacerbates skin inflammation. Cell Host Microbe. (2019) 25:389–403 e6. doi: 10.1016/j.chom.2019.02.002
27. Regev-Yochay G, Trzcinski K, Thompson CM, Malley R, Lipsitch M. Interference between Streptococcus pneumoniae and Staphylococcus aureus: in vitro hydrogen peroxide-mediated killing by Streptococcus pneumoniae. J Bacteriol. (2006) 188:4996–5001. doi: 10.1128/JB.00317-06
28. Orenstein A, Klein D, Kopolovic J, Winkler E, Malik Z, Keller N, et al. The use of porphyrins for eradication of Staphylococcus aureus in burn wound infections. FEMS Immunol Med Microbiol. (1997) 19:307–14. doi: 10.1111/j.1574-695X.1997.tb01101.x
29. Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. (2002) 347:1151–60. doi: 10.1056/NEJMoa021481
30. Leung DYM. Atopic dermatitis and the immune system: the role of superantigens and bacteria. J Am Acad Dermatol. (2001) 45:S13–6. doi: 10.1067/mjd.2001.117024
31. Bieber T, Paller AS, Kabashima K, Feely M, Rueda MJ, Ross Terres JA, et al. Atopic dermatitis: pathomechanisms and lessons learned from novel systemic therapeutic options. J Eur Acad Dermatol Venereol. (2022) 36:1432–49. doi: 10.1111/jdv.18225
32. Meylan P, Lang C, Mermoud S, Johannsen A, Norrenberg S, Hohl D, et al. Skin colonization by Staphylococcus aureus precedes the clinical diagnosis of atopic dermatitis in infancy. J Invest Dermatol. (2017) 137:2497–504. doi: 10.1016/j.jid.2017.07.834
33. Nakamura Y, Takahashi H, Takaya A, Inoue Y, Katayama Y, Kusuya Y, et al. Staphylococcus agr virulence is critical for epidermal colonization and associates with atopic dermatitis development. Sci Transl Med. (2020) 12(551):eaay4068. doi: 10.1126/scitranslmed.aay4068
34. Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. (2012) 22:850–9. doi: 10.1101/gr.131029.111
35. Kobayashi T, Glatz M, Horiuchi K, Kawasaki H, Akiyama H, Kaplan DH, et al. Dysbiosis and Staphylococcus aureus colonization drives inflammation in atopic dermatitis. Immunity. (2015) 42:756–66. doi: 10.1016/j.immuni.2015.03.014
36. Leyden JJ, Marples RR, Kligman AM. Staphylococcus aureus in the lesions of atopic dermatitis. Br J Dermatol. (1974) 90:525–30. doi: 10.1111/j.1365-2133.1974.tb06447.x
37. Edslev SM, Olesen CM, Norreslet LB, Ingham AC, Iversen S, Lilje B, et al. Staphylococcal communities on skin are associated with atopic dermatitis and disease severity. Microorganisms. (2021) 9:442–8. doi: 10.3390/microorganisms9020432
38. Rippke F, Schreiner V, Doering T, Maibach HI. Stratum corneum pH in atopic dermatitis: impact on skin barrier function and colonization with Staphylococcus Aureus. Am J Clin Dermatol. (2004) 5:217–23. doi: 10.2165/00128071-200405040-00002
39. Schwartz C, Moran T, Saunders SP, Kaszlikowska A, Floudas A, Bom J, et al. Spontaneous atopic dermatitis in mice with a defective skin barrier is independent of ILC2 and mediated by IL-1beta. Allergy. (2019) 74:1920–33. doi: 10.1111/all.13801
40. Jiang Z, Li J, Kong N, Kim JH, Kim BS, Lee MJ, et al. Accurate diagnosis of atopic dermatitis by combining transcriptome and microbiota data with supervised machine learning. Sci Rep. (2022) 12:290. doi: 10.1038/s41598-021-04373-7
41. Ito Y, Sasaki T, Li Y, Tanoue T, Sugiura Y, Skelly AN, et al. Staphylococcus cohnii is a potentially biotherapeutic skin commensal alleviating skin inflammation. Cell Rep. (2021) 35:109052. doi: 10.1016/j.celrep.2021.109052
42. Liu X, Qin Y, Dong L, Han Z, Liu T, Tang Y, et al. Living symbiotic bacteria-involved skin dressing to combat indigenous pathogens for microbiome-based biotherapy toward atopic dermatitis. Bioact Mater. (2023) 21:253–66. doi: 10.1016/j.bioactmat.2022.08.019
43. Habeebuddin M, Karnati RK, Shiroorkar PN, Nagaraja S, Asdaq SMB, Khalid Anwer M, et al. Topical probiotics: more than a skin deep. Pharmaceutics. (2022) 14:557–78. doi: 10.3390/pharmaceutics14030557
44. Lee MJ, Park YM, Kim B, Tae IH, Kim NE, Pranata M, et al. Disordered development of gut microbiome interferes with the establishment of the gut ecosystem during early childhood with atopic dermatitis. Gut Microbes. (2022) 14:2068366. doi: 10.1080/19490976.2022.2068366
45. Sung M, Choi Y, Park H, Huh CS. Gut microbiome characteristics in mothers and infants according to the presence of atopic dermatitis. Biomed Res Int. (2022) 2022:8145462. doi: 10.1155/2022/8145462
46. Song H, Yoo Y, Hwang J, Na YC, Kim HS. Faecalibacterium prausnitzii subspecies-level dysbiosis in the human gut microbiome underlying atopic dermatitis. J Allergy Clin Immunol. (2016) 137:852–60. doi: 10.1016/j.jaci.2015.08.021
47. Lee Y, Byeon HR, Jang SY, Hong MG, Kim D, Lee D, et al. Oral administration of Faecalibacterium prausnitzii and Akkermansia muciniphila strains from humans improves atopic dermatitis symptoms in DNCB induced NC/nga mice. Sci Rep. (2022) 12:7324. doi: 10.1038/s41598-022-11048-4
48. Derrien M, Van Baarlen P, Hooiveld G, Norin E, Muller M, de Vos WM. Modulation of mucosal immune response, tolerance, and proliferation in mice colonized by the mucin-degrader Akkermansia muciniphila. Front Microbiol. (2011) 2:166. doi: 10.3389/fmicb.2011.00166
49. Jeong DY, Ryu MS, Yang HJ, Jeong SY, Zhang T, Yang HJ, et al. Pediococcus acidilactici intake decreases the clinical severity of atopic dermatitis along with increasing mucin production and improving the gut microbiome in Nc/Nga mice. Biomed Pharmacother. (2020) 129:110488. doi: 10.1016/j.biopha.2020.110488
50. Kim WK, Jang YJ, Han DH, Jeon K, Lee C, Han HS, et al. Lactobacillus paracasei KBL382 administration attenuates atopic dermatitis by modulating immune response and gut microbiota. Gut Microbes. (2020) 12:1–14. doi: 10.1080/19490976.2020.1819156
51. Kim IS, Lee SH, Kwon YM, Adhikari B, Kim JA, Yu DY, et al. Oral administration of beta-glucan and Lactobacillus plantarum alleviates atopic dermatitis-like symptoms. J Microbiol Biotechnol. (2019) 29:1693–706. doi: 10.4014/jmb.1907.07011
52. Prakoeswa CRS, Herwanto N, Prameswari R, Astari L, Sawitri S, Hidayati AN, et al. Lactobacillus plantarum IS-10506 supplementation reduced SCORAD in children with atopic dermatitis. Benef Microbes. (2017) 8:833–40. doi: 10.3920/BM2017.0011
53. Zhou J, Xu G, Li X, Tu H, Li H, Chang H, et al. Limosilactobacillus reuteri FN041 prevents atopic dermatitis in pup mice by remodeling the ileal microbiota and regulating gene expression in peyer's patches after vertical transmission. Front Nutr. (2022) 9:987400. doi: 10.3389/fnut.2022.987400
54. Chen LW, Xu J, Soh SE, Aris IM, Tint MT, Gluckman PD, et al. Implication of gut microbiota in the association between infant antibiotic exposure and childhood obesity and adiposity accumulation. Int J Obes (Lond). (2020) 44:1508–20. doi: 10.1038/s41366-020-0572-0
55. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. (2013) 341:1241214. doi: 10.1126/science.1241214
56. Son MJ, Yang GJ, Jo EH, Shim YH, Kang SJ, Hong JE, et al. Association of atopic dermatitis with obesity via a multi-omics approach: a protocol for a case-control study. Medicine (Baltimore). (2019) 98:e16527. doi: 10.1097/MD.0000000000016527
57. Majewska-Szczepanik M, Kowalczyk P, Marcinska K, Strzepa A, Lis GJ, Wong FS, et al. Obesity aggravates contact hypersensitivity reaction in mice. Contact Dermatitis. (2022) 87:28–39. doi: 10.1111/cod.14088
58. Hussain M, Bonilla-Rosso G, Kwong Chung CKC, Bariswyl L, Rodriguez MP, Kim BS, et al. High dietary fat intake induces a microbiota signature that promotes food allergy. J Allergy Clin Immunol. (2019) 144:157–170 e8. doi: 10.1016/j.jaci.2019.01.043
59. Marras L, Caputo M, Bisicchia S, Soato M, Bertolino G, Vaccaro S, et al. The role of bifidobacteria in predictive and preventive medicine: a focus on eczema and hypercholesterolemia. Microorganisms. (2021) 9:836–56. doi: 10.3390/microorganisms9040836
60. Reddel S, Del Chierico F, Quagliariello A, Giancristoforo S, Vernocchi P, Russo A, et al. Gut microbiota profile in children affected by atopic dermatitis and evaluation of intestinal persistence of a probiotic mixture. Sci Rep. (2019) 9:4996. doi: 10.1038/s41598-019-41149-6
61. Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med. (2013) 19:557–66. doi: 10.1038/nm.3159
62. Stark JM, Coquet JM, Tibbitt CA. The role of PPAR-gamma in allergic disease. Curr Allergy Asthma Rep. (2021) 21:45. doi: 10.1007/s11882-021-01022-x
63. Akinci A, Kara A, Ozgur A, Turkkahraman D, Aksu S. Genomic analysis to screen potential genes and mutations in children with non-syndromic early onset severe obesity: a multicentre study in Turkey. Mol Biol Rep. (2022) 49:1883–93. doi: 10.1007/s11033-021-06999-2
64. Mateus Pellenz F, Crispim D, Silveira Assmann T. Systems biology approach identifies key genes and related pathways in childhood obesity. Gene. (2022) 830:146512. doi: 10.1016/j.gene.2022.146512
65. Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. (1999) 402:880–3. doi: 10.1038/47254
66. Ristow M, Muller-Wieland D, Pfeiffer A, Krone W, Kahn CR. Obesity associated with a mutation in a genetic regulator of adipocyte differentiation. N Engl J Med. (1998) 339:953–9. doi: 10.1056/NEJM199810013391403
67. Gray SL, Nora ED, Grosse J, Manieri M, Stoeger T, Medina-Gomez G, et al. Leptin deficiency unmasks the deleterious effects of impaired peroxisome proliferator-activated receptor gamma function (P465L PPARgamma) in mice. Diabetes. (2006) 55:2669–77. doi: 10.2337/db06-0389
68. Akyurek N, Aycan Z, Cetinkaya S, Akyurek O, Yilmaz Agladioglu S, Ertan U. Peroxisome proliferator activated receptor (PPAR)-gamma concentrations in childhood obesity. Scand J Clin Lab Invest. (2013) 73:355–60. doi: 10.3109/00365513.2013.786121
69. Landgraf K, Kloting N, Gericke M, Maixner N, Guiu-Jurado E, Scholz M, et al. The obesity-susceptibility gene TMEM18 promotes adipogenesis through activation of PPARG. Cell Rep. (2020) 33:108295. doi: 10.1016/j.celrep.2020.108295
70. Sadeghabadi ZA, Nourbakhsh M, Alaee M, Larijani B, Razzaghy-Azar M. Peroxisome proliferator-activated receptor gamma expression in peripheral blood mononuclear cells and angiopoietin-like protein 4 levels in obese children and adolescents. J Endocrinol Invest. (2018) 41:241–7. doi: 10.1007/s40618-017-0730-y
71. Stefan N, Stumvoll M. Adiponectin–its role in metabolism and beyond. Horm Metab Res. (2002) 34:469–74. doi: 10.1055/s-2002-34785
72. Yoon JC, Chickering TW, Rosen ED, Dussault B, Qin Y, Soukas A, et al. Peroxisome proliferator-activated receptor gamma target gene encoding a novel angiopoietin-related protein associated with adipose differentiation. Mol Cell Biol. (2000) 20:5343–9. doi: 10.1128/MCB.20.14.5343-5349.2000
73. Adachi M, Kurotani R, Morimura K, Shah Y, Sanford M, Madison BB, et al. Peroxisome proliferator activated receptor gamma in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut. (2006) 55:1104–13. doi: 10.1136/gut.2005.081745
74. Babaev VR, Yancey PG, Ryzhov SV, Kon V, Breyer MD, Magnuson MA, et al. Conditional knockout of macrophage PPARgamma increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. (2005) 25:1647–53. doi: 10.1161/01.ATV.0000173413.31789.1a
75. Byndloss MX, Olsan EE, Rivera-Chavez F, Tiffany CR, Cevallos SA, Lokken KL, et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science. (2017) 357:570–5. doi: 10.1126/science.aam9949
76. Deckers J, Bougarne N, Mylka V, Desmet S, Luypaert A, Devos M, et al. Co-activation of glucocorticoid receptor and peroxisome proliferator-activated receptor-gamma in murine skin prevents worsening of atopic march. J Invest Dermatol. (2018) 138:1360–70. doi: 10.1016/j.jid.2017.12.023
77. He W, Barak Y, Hevener A, Olson P, Liao D, Le J, et al. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A. (2003) 100:15712–7. doi: 10.1073/pnas.2536828100
78. Jones JR, Barrick C, Kim KA, Lindner J, Blondeau B, Fujimoto Y, et al. Deletion of PPARgamma in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc Natl Acad Sci U S A. (2005) 102:6207–12. doi: 10.1073/pnas.0306743102
79. Nobs SP, Natali S, Pohlmeier L, Okreglicka K, Schneider C, Kurrer M, et al. PPARgamma in dendritic cells and T cells drives pathogenic type-2 effector responses in lung inflammation. J Exp Med. (2017) 214:3015–35. doi: 10.1084/jem.20162069
80. Cevallos SA, Lee JY, Velazquez EM, Foegeding NJ, Shelton CD, Tiffany CR, et al. 5-Aminosalicylic acid ameliorates colitis and checks dysbiotic Escherichia coli expansion by activating PPAR-gamma signaling in the intestinal epithelium. mBio. (2021) 12(1):e03227–20. doi: 10.1128/mBio.03227-20
81. Lee JY, Cevallos SA, Byndloss MX, Tiffany CR, Olsan EE, Butler BP, et al. High-fat diet and antibiotics cooperatively impair mitochondrial bioenergetics to trigger dysbiosis that exacerbates Pre-inflammatory bowel disease. Cell Host Microbe. (2020) 28:273–284.e6. doi: 10.1016/j.chom.2020.06.001
82. Li JM, Yu R, Zhang LP, Wen SY, Wang SJ, Zhang XY, et al. Dietary fructose-induced gut dysbiosis promotes mouse hippocampal neuroinflammation: a benefit of short-chain fatty acids. Microbiome. (2019) 7:98. doi: 10.1186/s40168-019-0713-7
83. Xu P, Hong F, Wang J, Wang J, Zhao X, Wang S, et al. DBZ Is a putative PPARgamma agonist that prevents high fat diet-induced obesity, insulin resistance and gut dysbiosis. Biochim Biophys Acta Gen Subj. (2017) 1861:2690–701. doi: 10.1016/j.bbagen.2017.07.013
84. Gillis CC, Hughes ER, Spiga L, Winter MG, Zhu W, Furtado de Carvalho T, et al. Dysbiosis-associated change in host metabolism generates lactate to support Salmonella growth. Cell Host Microbe. (2018) 23:54–64 e6. doi: 10.1016/j.chom.2017.11.006
85. Ellis CN, Varani J, Fisher GJ, Zeigler ME, Pershadsingh HA, Benson SC, et al. Troglitazone improves psoriasis and normalizes models of proliferative skin disease: ligands for peroxisome proliferator-activated receptor-gamma inhibit keratinocyte proliferation. Arch Dermatol. (2000) 136:609–16. doi: 10.1001/archderm.136.5.609
86. Tachibana M, Wada K, Katayama K, Kamisaki Y, Maeyama K, Kadowaki T, et al. Activation of peroxisome proliferator-activated receptor gamma suppresses mast cell maturation involved in allergic diseases. Allergy. (2008) 63:1136–47. doi: 10.1111/j.1398-9995.2008.01677.x
87. Zhang Y, Li X, Fang S, Zhu Z, Yao M, Ying L, et al. Peroxisome proliferator-activated receptor gamma agonist suppresses mast cell maturation and induces apoptosis. Mol Med Rep. (2017) 16:1793–800. doi: 10.3892/mmr.2017.6802
88. Ruhl R, Dahten A, Schweigert FJ, Herz U, Worm M. Inhibition of IgE-production by peroxisome proliferator-activated receptor ligands. J Invest Dermatol. (2003) 121:757–64. doi: 10.1046/j.1523-1747.2003.12493.x
89. Barnas JL, Ritchlin CT. Etiology and pathogenesis of psoriatic arthritis. Rheum Dis Clin North Am. (2015) 41:643–63. doi: 10.1016/j.rdc.2015.07.006
90. Sobolev VV, Tchepourina E, Korsunskaya IM, Geppe NA, Chebysheva SN, Soboleva AG, et al. The role of transcription factor PPAR-gamma in the pathogenesis of psoriasis, skin cells, and immune cells. Int J Mol Sci. (2022) 23:9708–46. doi: 10.3390/ijms23179708
91. Fyhrquist N, Muirhead G, Prast-Nielsen S, Jeanmougin M, Olah P, Skoog T, et al. Microbe-host interplay in atopic dermatitis and psoriasis. Nat Commun. (2019) 10:4703. doi: 10.1038/s41467-019-12253-y
92. Xu J, Rajaratnam R. Cardiovascular safety of non-insulin pharmacotherapy for type 2 diabetes. Cardiovasc Diabetol. (2017) 16:18. doi: 10.1186/s12933-017-0499-5
93. Makita S, Takatori H, Matsuki A, Kawashima H, Iwata A, Tanaka S, et al. T-bet and STAT6 coordinately suppress the development of IL-9-mediated atopic dermatitis-like skin inflammation in mice. J Invest Dermatol. (2021) 141:1274–1285 e5. doi: 10.1016/j.jid.2020.08.029
94. Micosse C, von Meyenn L, Steck O, Kipfer E, Adam C, Simillion C, et al. Human “TH9” cells are a subpopulation of PPAR-gamma(+) TH2 cells. Sci Immunol. (2019) 4(31):eaat5943. doi: 10.1126/sciimmunol.aat5943
95. D'Agata I D, Vanounou T, Seidman E. Mesalamine in pediatric inflammatory bowel disease: a 10-year experience. Inflamm Bowel Dis. (1996) 2:229–35. doi: 10.1097/00054725-199612000-00001
Keywords: atopic dermatitis, microbiota, dysbiosis, obesity, PPAR-γ, probiotics, allergy
Citation: McAleer JP (2023) Obesity and the microbiome in atopic dermatitis: Therapeutic implications for PPAR-γ agonists. Front. Allergy 4:1167800. doi: 10.3389/falgy.2023.1167800
Received: 16 February 2023; Accepted: 13 March 2023;
Published: 27 March 2023.
Edited by:
Davide Leardini, University of Bologna, ItalyReviewed by:
Francesco Fabozzi, Bambino Gesù Pediatric Hospital (IRCCS), Italy© 2023 McAleer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeremy P. McAleer bWNhbGVlckBtYXJzaGFsbC5lZHU=
Specialty Section: This article was submitted to Infections and Microbiome, a section of the journal Frontiers in Allergy