 Eistine Boateng1*Draginja Kovacevic2Vladimira Oldenburg1Madeleine Rådinger3Susanne Krauss-Etschmann1,2,4
Eistine Boateng1*Draginja Kovacevic2Vladimira Oldenburg1Madeleine Rådinger3Susanne Krauss-Etschmann1,2,4
- 1Early Life Origins of Chronic Lung Disease, Research Center Borstel, Leibniz Lung Center, Airway Research Center North (ARCN), German Center for Lung Research (DZL), Borstel, Germany
- 2DZL Laboratory for Experimental Microbiome Research, Research Center Borstel, Leibniz Lung Center, Airway Research Center North (ARCN), German Center for Lung Research (DZL), Borstel, Germany
- 3Krefting Research Centre, Department of Internal Medicine and Clinical Nutrition, Institute of Medicine, University of Gothenburg, Gothenburg, Sweden
- 4Institute for Experimental Medicine, Christian-Albrechts-Universität zu Kiel, Kiel, Germany
The airway epithelial cells and overlying layer of mucus are the first point of contact for particles entering the lung. The severity of environmental contributions to pulmonary disease initiation, progression, and exacerbation is largely determined by engagement with the airway epithelium. Despite the cellular cross-talk and cargo exchange in the microenvironment, epithelial cells produce miRNAs associated with the regulation of airway features in asthma. In line with this, there is evidence indicating miRNA alterations related to their multifunctional regulation of asthma features in the conducting airways. In this review, we discuss the cellular components and functions of the airway epithelium in asthma, miRNAs derived from epithelial cells in disease pathogenesis, and the cellular exchange of miRNA-bearing cargo in the airways.
Introduction
The lung is constantly exposed to particles with diverse aerodynamic properties that determine where they are deposited in the airways. The airway epithelial cells exhibit a barrier function to protect the submucosal layer from pathogens, allergens, and pollutants in inhaled air. Furthermore, the airway epithelium coordinates with cells of the innate and adaptive immune systems to maintain host defense. In allergic disease, airway epithelial cells produce cytokines e.g. TSLP, IL-25, and IL-33 which drives dendritic cells toward Th2 response, activates eosinophils, and stimulates the production of type 2 cytokines in mast cells, respectively (1–4). Thus, epithelial cells support the initiation of airway inflammation and the pathogenesis of allergy related diseases e.g. allergic asthma.
Asthma is a complex, heterogenous disease characterized by symptoms such as recurrent episodes of wheezing, breathlessness, chest tightness, cough, and expiratory airflow fluctuation. Asthma is often dichotomized into endotypes distinguished by pathophysiological mechanisms. The most studied endotypes are type 2 and non-type 2, which are defined by various phenotypes (5). Emerging studies are still unraveling the pathophysiological mechanisms of asthma (5–8) though it is known that the disease is marked by chronic airway inflammation (9) which subsequently leads to hypersecretion of mucus, bronchial hyperresponsiveness, and airway edema and remodeling (7, 10, 11). Airway epithelial remodeling in asthma is demonstrated by subepithelial fibrosis, thickening of the sub-basement membrane, increase in smooth muscle mass, mucus gland hypertrophy, and vascular congestion, culminating in airway congestion and bronchoconstriction (6, 12). These prominent pathological features highlight the involvement of the airway epithelium in asthma [reviewed in (13, 14)]. Therefore, the epithelial biology of the conducting airways fundamentally provides insight to the understanding of the mechanistic development of asthma.
Goblet cell hyperplasia and mucus hypersecretion are striking pathophysiological features reported during asthma exacerbation (15). To define the molecular mechanisms leading to these manifestations, some studies have explained the role of EGFR in mucus cell metaplasia and mucus production in bronchial epithelial cells (16–18). Furthermore, a study done in patients with asthma demonstrated that epithelial cells increase the levels of GAD65/67 and GABAAR subunits in response to allergen challenge (19). The authors showed that the inhibition of GABA signaling decreased ovalbumin or IL-13-induced goblet cell hyperplasia and mucin production in mice.
It is reported that epithelial cells express several asthma susceptibility genes e.g. IL-1RL1, IL-33, TSLP, CDHR3, PCDH1, MUC5AC, KIF3A, EFHC1 and GSDMB (11, 20–22). Moreover, epithelial gene expression can be modulated by miRNAs, small noncoding RNAs that bind to target mRNAs leading to degradation or even translational repression (23–25). miRNAs are ∼22 nucleotides in length (23), and processed by two RNase III proteins, Drosha and Dicer. After transcription, the miRNA, known as primary or pri-miRNA, is processed into a precursor miRNA (pre-miRNA) and further into mature miRNA. It is widely known that miRNAs bind to sequence-specific recognition sites at the 3′ UTR of their target to repress gene expression. Nonetheless, miRNAs also bind to regions such as the 5′ UTR, coding sequence, and promoters (26). The molecules not only suppress genes, but also stimulate gene expression (27). miRNAs are involved in numerous biological processes, including the pathogenesis of many diseases. Therefore, miRNAs could serve as valuable biomolecules for disease diagnosis, progression, exacerbation and treatment response. For instance, as miRNAs are dysregulation during the pathogenesis of allergy and asthma, their levels can be quantitatively explored in patient samples from the skin, lung, and nose, and in sputum, exhaled breath condensates, and specific cell types (28).
miRNAs have been shown to be differentially expressed in bronchial epithelial cells following diesel exhaust particle, tobacco smoke, and virus exposures (28). Indeed, several miRNAs modulate genes involved in epithelial barrier function, proliferation, response to viral infection and mucus production (29–33). It is however suggest that the impact of miRNAs on the airway epithelium might differ with disease severity (31).
The thickening of the epithelium (34) and induction of goblet cell hyperplasia in asthma suggest a role for a dysregulated network of intracellular molecules in response to alterations in the milieu of the lung. We have earlier discussed the engagement of miRNAs in multiple gene regulation during the pathogenesis of asthma (35). miRNAs are altered in bronchial epithelial cells in patients with asthma (32), and some evidence suggests their contribution to the modulation of genes controlling cellular phenotypes. For example, a study showed that in humans, the expression of miR-4423 was restricted in the airway epithelium, while the overexpression of this miRNA in normal human bronchial epithelial cells (HBECs) contributed to an increase in cells expressing ciliated cell markers (36). Additionally, there was defective multiciliogenesis in the trachea, bronchus, and bronchiole of miR-34/449 “triple knockout” embryos (37), suggesting that the miR-34/449 family is essential for normal ciliation in the respiratory epithelium. Another study also used normal HBECs to demonstrate that miRNAs are differentially expressed in differentiated and undifferentiated cells (38). Other biological processes have been shown to be regulated by miRNAs in the lung epithelium, including the disruption of intercellular adhesion proteins and release of cytokines (39).
The protective function of epithelial cells in the airway is periodically challenged, as they are the first point of contact from the environment. The epithelia of the proximal and distal air spaces are variously vulnerable depending on the velocity of air entering the lungs. Hence, the airway epithelium is a primary site for the commencement and aggravation of respiratory diseases. In this review, we summarize the cellular components of the airway epithelium and their roles in the pathogenesis of asthma. Importantly, we focus on airway epithelial cell function in asthma, the connection between deregulated miRNAs and epithelial cell function, and finally, cross-talk in the microenvironment of the airway epithelium.
The airway epithelium
The microscopic aspects of lung development and signaling mechanisms have been exquisitely explained (40). As this review is about asthma, our focus is particularly on the lower respiratory system, which consists of the conducting and respiratory zones. The trachea, bronchi, and terminal bronchioles make up the conducting zone. The respiratory bronchioles, alveolar duct and alveoli form the respiratory zone, consisting of different epithelial cell types. The conducting zone transports respiratory gases and the respiratory zone functions in gas exchange. The respiratory epithelium comprises pseudostratified ciliated columnar epithelium with other functionally heterogeneous cells. The diverse cellular components include goblet, ciliated columnar, basal, brush, neuroendocrine cells, and pulmonary ionocytes. The airway epithelium merges with the pulmonary alveolar epithelium which consists of type I and type II cells. The type I cells make up the majority of the alveolar surface area and are essential for the air-blood barrier in the lungs (41, 42). The type II cells differentiate into type I cells in response to injury repair (43, 44).
In response to injury and stress, the lung is capable of regenerating the airway epithelium. The multipotent capacity of a specific cell type in the neighborhood of the basal lamina defines the cellular lineage hierarchy in the bronchial airway. These cells, known as basal cells, act as progenitor cells that maintain airway homeostasis through self-renewal and differentiation into specialized cell types such as ciliated and goblet cells (45, 46). Though it is unclear which signaling pathways primarily orchestrate the process, basal cells are capable of differentiating into tuft cells, pulmonary ionocytes, and neuroendocrine cells (47). In contrast, it is well documented that the fate of basal cell differentiation into other cell types is mediated by transcriptional networks of the Notch signaling pathway (48). Despite the multipotent progenitor characteristics of basal cells, their abundance is diverse in the conducting airways. In the human lung, basal cells are higher in the large airways as compared to the smaller airways (49). In addition to basal cells, maintenance of the airway epithelium is also supported by some subsets of club cells that terminally differentiate into ciliated cells in response to epithelial damage (50, 51). Additionally, pulmonary neuroendocrine cells in the bronchial epithelium proliferate and repair the neighboring epithelium (52), and rare clusters are shown to function as stem cells (53).
The ciliated cells of the epithelium have numerous motile cilia protruding from basal bodies on the apical part of the cells. These cilia are made of structural and motor proteins that orchestrate coordinated ciliary movement in the airway. Thus, the protective layer of mucus lining the epithelium is biomechanically beaten by the cilia in the direction leading outside the lung. This function supports the removal of inhaled particles and pathogens from the airways. It is suggested that motile cilia in the respiratory epithelium possess various forms of sensory functions such as mechanoreceptive and chemoreceptive properties [reviewed in (54)]. In contrast to their biomechanical role, ciliated cells of the airway are also targets for invading bacteria and viruses (55). Additionally, it has been demonstrated that ciliated cells transdifferentiate into specialized epithelial cells in response to injury (56).
Club cells are cuboidal non-ciliated, non-mucus secreting, and microvilli studded, with dense cytoplasmic granules (57). These cells are dome-shaped, facing the bronchiolar lumen. In humans, club cells are heterogeneous, and one subtype expresses high levels of the basal cell marker KRT5 (58). Like basal cells, this population of club cells is multipotent (58). There are variants of functional club cells located in the airways. For example, at the bronchioalveolar-duct junction and the microenvironment of neuroendocrine cells, club cells can assume progenitor properties to support epithelial regeneration in response to injury (59–61). Other functional features of club cells include their critical role in inflammation, biotransformation of environmental toxicants, mitigation of oxidative stress, and maintenance of the barrier function of the airway epithelium (62–64). Participation in these mechanisms involve the secretion of club cell protein 10 (CC10), KL-6 protein, lipids, glycoproteins, and surfactants (surfactant proteins A, B, and D).
Goblet cells are located throughout the conducting airway of the respiratory epithelium. Ultrastructural analysis of these cells shows that they are polarized, with nuclei located on the basal side and secretary granules located on the apical side. Along with club cells, goblet cells are also formed from epithelial cells under the triggering influence of IL-13 (65). In addition to IL-13, some inflammatory mediators, irritant gases, proteinases, bacterial products, and cigarette smoke exposures increase the number of goblet cells in the airways (66). Goblet cells contribute mucins to the mucus component in the airways, supporting the viscoelastic property of the mucus in the airway.
Since their discovery, the physiological and mechanical roles of pulmonary ionocytes were associated with mucus viscosity and clearance in cystic fibrosis (47, 67). Pulmonary ionocytes are differentiated from basal cells (47, 67) and transcriptionally distinguished by the expression of FOXI1 and the highest level of CFTR among the cells in the airway epithelium (68). Apart from basal cell differentiation into ionocytes, a possible cell lineage from tuft-like cells has been shown (69). In humans, FOXI1+ ionocytes are found in reduced amounts in the small, compared to large airway epithelium (68). Also, antibody inhibition of the Notch receptors in HBECs has been shown to decrease the number of pulmonary ionocytes (67), suggesting the influence of the signaling pathway in their production.
Epithelial cells in asthma
As a first line of defense, the mucus lining of the epithelium traps foreign particles for further clearance. Usually after an invasion by particles from the environment, epithelial cells react by increasing mucus secretion and ciliary beating in an attempt to cleanse the airway of harmful and insoluble particles. Disruptions in the mucus barrier expose the epithelial cells to allergens and noxious agents which could lead to severe damages. However, epithelial cells are strongly linked together by tight and adherens junctions to form a protective physical barrier over the underlying stromal region of the airway. Thus, the integrity of the airway epithelial landscape and the junctional complexes is essential to cushion against excessive injury from particles and invading pathogens.
Epithelial cells are often frail in asthmatic airways. This is marked by apoptosis and shedding of epithelial cells in the bronchial mucosa of asthma patients compared to healthy controls (70). In an in vitro experiment, allergen exposed airway epithelial cells released IL-25 (71). In another study on patients with mild atopic asthma, the epithelium and submucosa of bronchial biopsies showed increased immunoreactivity to IL-25, IL-33, and TSLP after allergen challenge (72). These findings highlight the role of airway epithelial cells in supporting inflammation by synthesizing lipid mediators and cytokines, which contribute to some of the pathological features of asthma (1, 73). As part of the initial steps in allergic response to inhaled particles and pathogens, the airway epithelial cells release chemokines and cytokines to attract and activate dendritic cells. These cytokines also activate the group 2 innate lymphoid cells (ILC2s) known to support type 2 inflammation in allergic diseases (74). Altogether, the interplay between airway epithelial cells and antigen-presenting cells in the milieu of inflammatory mediators stimulate key features of asthma.
Airway epithelial cells express various proportions of molecules that support the pathogenesis of asthma and indicate susceptibility to the disease. In an experimental mouse model, the Toll-like receptor (TLR) 4 was found to be highly expressed in the conducting airway epithelial cells (75). In this study, inhibition of TLR4 reduced airway lymphocytosis and eosinophilia, IL-5 and IL-13 in bronchoalveolar lavage fluid (BALF), peri-bronchovascular infiltrates, goblet cell hyperplasia, and airway hyperresponsiveness in HDM treated mice. There is also evidence that the cadherin-related family member 3 (CDHR3) expressed in airway epithelial cells is associated with exacerbation susceptibility in children with severe asthma (76). Another molecule, protocadherin 1 (PCDH1), expressed in bronchial epithelial cells, has emerged as a susceptibility gene for bronchial hyperresponsiveness and asthma in children (77, 78). In addition, these genes which are expressed in epithelial cells also indicate asthma susceptibility: ADAM33, GPRA, SPINK5, IRAKM, DPP10, and HLA-G (79).
The specialized functions of the cells in the airway epithelium suggest their independent response in allergy and asthma conditions. These cells encounter various levels of molecular-specific alterations detrimental to their functional performance. In a recent study, the level of TRPV4 was demonstrated to be increased in the apical membrane of bronchiolar cells in biopsies from asthma patients (80). Elevated levels of TRPV4 in club cells were moreover shown to act as signals for allergic inflammation. Previous studies have also demonstrated significantly reduced CC10 in the epithelial cells of small airways and in the plasma of asthma patients compared to controls (81, 82). A decrease in serum levels of CC10 has also been reported in patients with overlapping features of asthma and COPD (83). In mouse model of OVA-induced asthma, the mRNA and protein levels of CC10 were reduced in lung tissues and the airway, respectively (84), and CC10 was further associated with exacerbation of asthmatic phenotypes. These studies show that the immunomodulatory, anti-inflammatory, and anti-phospholipase A2 properties of CC10 are threatened in asthma conditions, suggesting a functional role of club cells in the disease.
A characteristic feature of airway epithelial remodeling is goblet cell metaplasia. In a mouse model with conditionally expressed spdef in the non-ciliated epithelial cells, increases were found in goblet cell metaplasia, eosinophil infiltration, and airway hyperresponsiveness (85). Again, there were marked increase in the mRNA levels of Th2 cytokines (Il-13 and Ccl17), eosinophilic chemoattractants (Ccl24 and Ccl20) and targets that signify dendritic cell activation (Il-33, Il-25, and Csf2) (85). Noteworthy, goblet cell metaplasia is coordinated by a wide range of molecules and signaling pathways e.g. Notch2, IL-4, IL-13, TNF-α, and EGF (86–89). In both mice and cultured epithelial cells, inhibition of IL-13 and the STAT6 signaling attenuated goblet cell metaplasia (90). In the mice, there was also evidence of decreased goblet cell metaplasia and prevention of HDM-induced airway hyperreactivity and inflammation. In another mouse study, inhibition of the CREB binding protein (CBP)/β-catenin pathway mitigated goblet cell metaplasia and mucus production in an HDM-induced model of allergic airway disease (91). Elevated FOXM1 has been shown in goblet and club cells of an HDM asthma mouse model and in patients with severe asthma (92). Deletion of FOXM1 in club cells decreased inflammation and airway resistance in an HDM-induced mouse model, and also reduced goblet cell metaplasia in the airway epithelium (92). Hyperplasia of goblet cells is also associated with chronic hypersecretion of mucus in asthma. The mechanisms of goblet cell hyperplasia and contribution to the apical mucus layer of the epithelium in humans and asthma mouse models have been described elsewhere (15, 93–96).
Ciliary function remains an important component of the respiratory airway epithelium. Despite the chemical barrier functions elicited by the airway epithelial cells the biomechanical action of ciliated cells is relevant for the clearance of foreign particles. Airway epithelial cell cilia respond to chemical stimuli and the physical properties of liquid components overlying them (97–99). Epithelial cell loss is usually marked in asthmatic airways – instigated by disruption and decreased formation of cellular tight junctions (100). These epithelial changes increase permeability to allergens (100, 101), mucus hypersecretion, and depletion of ciliated cells in the airway (102). Progenitor cells expressing FOXJ1, a marker for ciliated cells, are capable of differentiating into goblet cells under the influence of IL-13 (103). This underscores the role of ciliated cells in epithelial regeneration in response to injury. In bronchial biopsies from children with chronic onset of asthma, some cells co-expressed mucus (MUC5AC and CEACAM5) and ciliated cell genes (FOXJ1 and PIFO), and were named as mucus ciliated cells (104). It was further demonstrated that the cells exhibited elevated levels of IL-4/IL-13-induced genes, as well as asthma genes such as CSTI and POSTN, suggesting the involvement of these types of cells in mucus cell metaplasia (104).
Epithelial cell miRNAs in asthma
The role of airway epithelial cells in the pathogenesis of asthma merit the discussion of intracellular molecules that are functionally active in disease progression or symptom alleviation. miRNAs are dysregulated in respiratory diseases, and capable of modulating the expression of various genes implicated in airway biology and diseases (35). Murine models support investigations into miRNAs and their role in asthma and allergic airway disease. Using HDM treatment as a model for allergic airway disease in mice, data from one study showed greater than 5-fold increase in miR-145, miR-21 and let-7b in dissected airways compared to samples from saline-treated mice (105). Notably, inhibition of miR-145 reduced HDM-induced mucus hypersecretion in the airway epithelial cells and recruitment of eosinophils to the airway (105). Eosinophil recruitment was further demonstrated by a reduced number of eosinophils in BALF. However, the authors neither identified specific targets nor described the molecular mechanisms leading to reduced mucus hypersecretion after miR-145 antagonism.
In response to HDM-induced allergic asthma, one study found elevated levels of miR-16, miR-21, and miR-126 in the airway walls of mice (106). On the contrary, HDM-induced TLR-4 and MyD88 deficient mice demonstrated decreased levels of one of the miRNAs i.e. miR-126. Inhibition of miR-126 in the airway wall of WT mice suppressed HDM-induced mucus hypersecretion and other allergy-related features (106). Moreover, the MyD88 signaling pathway was found to regulate the expression of miR-126 and HDM-induced allergic inflammation. Altogether, transcriptomes of the airway walls of HDM challenged mice treated with ant-miR-126 showed differential regulation of genes that encode for proteins making up certain regions of the Ig kappa, Ig lambda and heavy chains. OBF.1 [Oct binding factor 1 a.k.a. BOB.1 (B-cell Oct binding protein 1)] and PU.1 were significantly upregulated, whereas GATA3 was downregulated. OBF.1 regulated the production of antibodies and PU.1 negatively regulated TLR4 and Th2 response.
Anti-mmu-mir-106a attenuated goblet cell metaplasia and mucin content in the airways of mice with OVA-induced allergic asthma (107). However, the molecular events leading to reduced goblet cell metaplasia require further investigation, as this was not demonstrated in the study. In contrast, some studies have demonstrated the involvement of miR-125b and miR-330 in the inhibition of goblet cell differentiation in allergic airway inflammation and in IL-13-induced MUC5AC secretion in bronchial epithelial cells, respectively (108, 109). Intranasal administration of miR-145–5p antagomir to mice reversed HDM-induced decrease in KIF3A (expressed in airway epithelial cells), reduced inflammatory response in the lung, and ultimately, modulated asthma phenotypes (39).
Analyses of samples from human subjects have been instrumental in studying airway biology and miRNAs in asthma. Dysregulated levels of miRNAs are reported in diverse human samples. For instance, decreased levels of miR-34b-5p, miR-34c-5p, miR-449a and miR-449b-5p were demonstrated in bronchial epithelial brushings from both steroid-naive and steroid-using asthma cohorts (110). As already described, miR-449 is important for the differentiation of ciliated cells (111). This was demonstrated by overexpressing miR-449 in proliferating human airway epithelial cells (HAECs), which eventually decreased the levels of NOTCH1 and DLL1. Additionally, inhibition of miR-449 in differentiating HAECs increased the protein levels of NOTCH1 and DLL1. Altogether, lower protein levels of NOTCH1 and DLL1 were found in multiciliated cells compared to undifferentiated and non-ciliated differentiated cells (111). Since the pathogenesis of asthma is impacted by multiple genetic factors, the reduction of miR-34b-5p and miR-34c-5p in asthma should be further investigated to determine their molecular influence in the disease.
Microarray analyses of bronchial epithelial cells from asthmatic patients have also shown decreased levels of miR-18a, miR-27a, miR-128, and miR-155 (32). Inhibition of all of these miRNAs together increased the release of IL-6 from bronchial epithelial cells compared to inhibition of each miRNA individually. Importantly, these findings potentially caution the selection of specific dysregulated miRNAs to provide mechanistic explanations of phenotypes and pathological observations in experimental disease conditions.
Bronchial epithelial cells from mild asthmatic patients indicated strong downregulation of miR-203 and significant increases in miR-487b, miR-181c, and miR-let-7f levels (112). Higher expression of the aquaporin gene AQP4, a predicted target for miR-203, was found in bronchial epithelial cells from mild asthma patients. AQP4 controls water transport across the cell membrane and we suggest that the level is possibly upregulated in the bronchial epithelium to support the clearance of obstructive fluids in asthmatic airways. Hence, miR-203 is decreased to support this process in asthma pathogenesis.
Among the numerous miRNAs differentially expressed in the bronchial epithelium of healthy and severe asthma patients, the levels of miR-19b-3p, miR-20a-5p, and miR-135b-5p were shown to be decreased while those of miR-574-3p and miR-625-3p were increased in asthma patients after validation in an expanded cohort (113). Using the Ingenuity Pathway Analysis, the authors ascertained that the numerous miRNAs dysregulated in severe asthma condition relate to several biological processes including epithelial repair and inflammation (113).
In a recent study, miR-141-3p was increased in primary HBECs and as well induced following allergic stimulation in patients with asthma (33). Knockdown of miR-141-3p in IL-13-treated HBECs led to decreased goblet cell genes and mucus-producing goblet cells (33). Also, endobronchial biopsy samples from patients with severe asthma displayed increased epithelial thickness, and bronchial epithelial cells showed an elevated rate of cell proliferation compared to samples from patients with mild asthma (30). Corresponding with the observation, the level of miR-19a was increased in bronchial epithelial cells in the severe asthma condition. Further evidence revealed that the transfection of miR-19a into bronchial epithelial cells promoted cell proliferation (30). Airway biopsies of patients with asthma demonstrated a decrease in miR-146a compared to the level in non-asthmatic controls (114). Additionally, stimulation of HBECs with cytokines associated with asthma (TNF-α, IL-17A, and IL-4) resulted in increased miR-146a. Thus, miR-146a response to pro-inflammatory cytokines implicates its relevance in neutrophilic asthma (114). Moreover, in this study, patients with neutrophilic asthma showed significant increase in the mRNA levels of IL-8 and CXCL1 in airway epithelial cells. Importantly, a negative association between miR-146a level (in bronchial brushing) and neutrophil counts (in BALF) was reported in asthmatic patients (114). The authors showed that overexpression of miR-146a in HBECs controlled inflammatory response by reducing the mRNA level and release of IL-8 and CXCL1, respectively. Additionally, the mRNA levels of the interferon response genes, IFITM1 and IRF1, were moderately increased in miR-146a transfected HBECs. Nevertheless, the regulation of these proteins and genes in this study only indicates a causal effect from miR-146a transfection into HBECs and not necessarily the regulation of putative targets.
While miRNAs regulate various genes to modulate phenotypes in the airway epithelium of asthma patients and experimental conditions, a single miRNA can also regulate multiple genes and several miRNAs can regulate the same gene. The epithelium of patients with asthma display several structural and functional anomalies linked to dysregulated miRNAs. miRNA candidates profiled in the airway epithelium may work synergistically or elicit additive effects to other subsets in the microenvironment of the epithelium to modulate these phenotypes. Thus, we posit that the study of specific miRNAs among a dysregulated pool may not adequately disentangle the mechanistic involvement of epithelial miRNAs in the pathogenesis of asthma. Because miRNA dysregulation in the asthmatic airway epithelium may be molecularly programmed in response to triggers, miRNAs should be collectively studied in relation to phenotypes in asthmatic airways and the pathogenesis of the disease in general. Figure 1 shows airway epithelial cell miRNAs and the features of asthma they are mechanistically involved with.
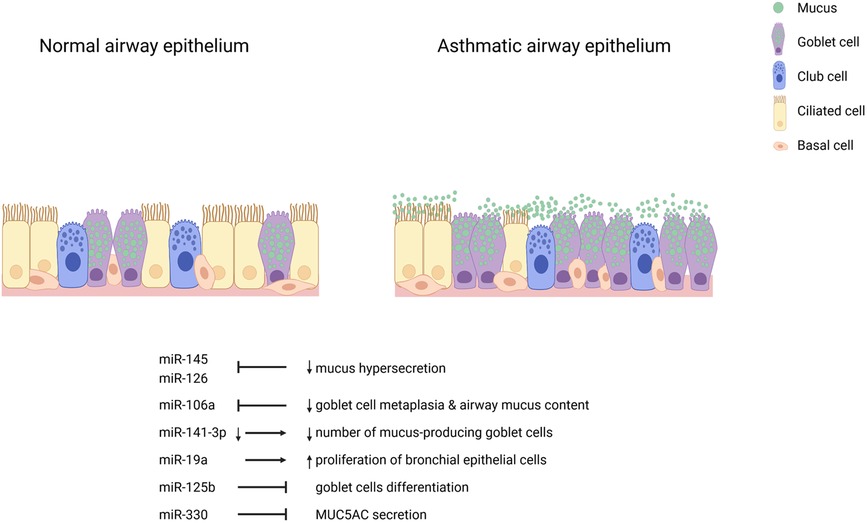
Figure 1. Airway epithelial cell miRNAs in asthma. Inhibition of miR-145 and miR-126 decrease mucus hypersecretion. Inhibition of miR-106a reduces goblet cell metaplasia and airway mucus content. Decreased miR-141-3p leads to fewer mucus-producing goblet cells. miR-19a induces the proliferation of bronchial epithelial cells. miR-125b and miR-330 inhibit goblet cell differentiation and IL-13-induced MUC5AC secretion, respectively.
Cellular cross-talk in the vicinity of the airway epithelium
Cells release extracellular vesicles (EVs) containing bioactive cargoes that influence cellular functions in asthma (115, 116). Thus, the exchange of biomolecules in the vicinity of the airway epithelium may be supported by EVs. In a recent publication, it was demonstrated that epithelial cells exchange cargoes to alter mucin hypersecretion and miRNA content of exosomes in target cells (117).
EV miRNAs are believed to support the infiltration of inflammatory cells to the lungs, regulate Th2 inflammation, modulate mast cell degranulation, regulate cytokine production, and function in airway remodeling and hyperresponsiveness (118). Under physiological and pathological conditions, the number and content of EVs is altered. For example, treatment of HBECs with T2 (IL-4 + IL-13) and T17 (IL-17A + TNFα) cytokines induced the release of EVs (119). In an in-vitro model of experimental asthma, dysregulated miRNAs were found in the EV cargoes of normal HBECs challenged with IL-13 (120). These findings suggest a link between miR-34a, miR-92b and miR-210 to early stage Th2 response in the airway and asthma. In another study, HDM-exposed mice showed increased BALF EVs bearing miRNAs suggested to be involved in allergic airway inflammation (121).
Dysregulated intracellular miRNAs in airway epithelial cells determine cellular response and phenotypes. The cross-talk orchestrated through cargo exchange between the cells is therefore a key component in asthma pathogenesis. Moreover, cell signaling mechanisms and pathways crucial for the progression and mitigation of asthma require the unraveling of miRNAs generated by specific cells in the airways. This could support the understanding of the pathomechanisms of goblet cell hyperplasia and metaplasia, and mucus hypersecretion in asthma.
Conclusion
Epithelial cells are the first cellular contact of environmental materials entering the lung. miRNA regulation of targets in the airway support the maintenance of epithelial barrier integrity or dysfunction in asthma. Communications established between epithelial cells in the airways depict the relevance of miRNAs in response to alterations in the cellular milieu in asthmatic conditions. A gap of knowledge remains when it comes to understanding the role of epithelial cell miRNAs in asthma. Further studies require the use of single cell small RNA sequencing combined with functional assays, including cell lines with genetically modified miRNAs and target genes to expand this research field. Moreover, defining the possible vesicular cargo exchanges between the epithelial cells in the airways and dysregulated miRNAs in each specific cell type is essential to broaden understanding of signaling pathways connected to the pathophysiology of asthma.
Author contributions
E.B wrote the manuscript. S.K.-E, M.R., D.K and V.O. reviewed and edited the manuscript. D.K. designed the figure. All authors contributed to the article and approved the submitted version.
Acknowledgments
The authors thank Reghan Borer for editing support and the Leibniz Lung Center, member of the German Center for Lung Research (DZL), Borstel, Germany.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ying S, O'Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, et al. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J Immunol. (2005) 174(12):8183–90. doi: 10.4049/jimmunol.174.12.8183
2. Zhou B, Comeau MR, De Smedt T, Liggitt HD, Dahl ME, Lewis DB, et al. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol. (2005) 6(10):1047–53. doi: 10.1038/ni1247
3. Altman MC, Lai Y, Nolin JD, Long S, Chen CC, Piliponsky AM, et al. Airway epithelium-shifted mast cell infiltration regulates asthmatic inflammation via Il-33 signaling. J Clin Invest. (2019) 129(11):4979–91. doi: 10.1172/JCI126402
4. Suzukawa M, Morita H, Nambu A, Arae K, Shimura E, Shibui A, et al. Epithelial cell-derived Il-25, but not Th17 cell-derived Il-17 or Il-17f, is crucial for murine asthma. J Immunol. (2012) 189(7):3641–52. doi: 10.4049/jimmunol.1200461
5. Kuruvilla ME, Lee FE, Lee GB. Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy Immunol. (2019) 56(2):219–33. doi: 10.1007/s12016-018-8712-1
6. Gans MD, Gavrilova T. Understanding the immunology of asthma: pathophysiology, biomarkers, and treatments for asthma endotypes. Paediatr Respir Rev. (2020) 36:118–27. doi: 10.1016/j.prrv.2019.08.002
7. Saetta M, Turato G. Airway pathology in asthma. Eur Respir J Suppl. (2001) 34:18s–23s. doi: 10.1183/09031936.01.00229501
8. Suraya R, Nagano T, Katsurada M, Sekiya R, Kobayashi K, Nishimura Y. Molecular mechanism of asthma and its novel molecular target therapeutic agent. Respir Investig. (2021) 59(3):291–301. doi: 10.1016/j.resinv.2020.12.007
9. Reddel HK, Bateman ED, Becker A, Boulet LP, Cruz AA, Drazen JM, et al. A summary of the new gina strategy: a roadmap to asthma control. Eur Respir J. (2015) 46(3):622–39. doi: 10.1183/13993003.00853-2015
10. Bergeron C, Tulic MK, Hamid Q. Airway remodelling in asthma: from benchside to clinical practice. Can Respir J. (2010) 17(4):e85–e93. doi: 10.1155/2010/318029
11. Heijink IH, Kuchibhotla VNS, Roffel MP, Maes T, Knight DA, Sayers I, et al. Epithelial cell dysfunction, a major driver of asthma development. Allergy. (2020) 75(8):1902–17. doi: 10.1111/all.14421
12. Mims JW. Asthma: definitions and pathophysiology. Int Forum Allergy Rhinol. (2015) 5(Suppl 1):S2–S6. doi: 10.1002/alr.21609
13. Calven J, Ax E, Radinger M. The airway epithelium-a central player in asthma pathogenesis. Int J Mol Sci. (2020) 21(23):8907. doi: 10.3390/ijms21238907
14. Wang Y, Bai C, Li K, Adler KB, Wang X. Role of airway epithelial cells in development of asthma and allergic rhinitis. Respir Med. (2008) 102(7):949–55. doi: 10.1016/j.rmed.2008.01.017
15. Rogers DF. Airway goblet cell hyperplasia in asthma: hypersecretory and anti-inflammatory? Clin Exp Allergy. (2002) 32(8):1124–7. doi: 10.1046/j.1365-2745.2002.01474.x
16. Casalino-Matsuda SM, Monzon ME, Forteza RM. Epidermal growth factor receptor activation by epidermal growth factor mediates oxidant-induced goblet cell metaplasia in human airway epithelium. Am J Respir Cell Mol Biol. (2006) 34(5):581–91. doi: 10.1165/rcmb.2005-0386OC
17. Enomoto Y, Orihara K, Takamasu T, Matsuda A, Gon Y, Saito H, et al. Tissue remodeling induced by hypersecreted epidermal growth factor and amphiregulin in the airway after an acute asthma attack. J Allergy Clin Immunol. (2009) 124(5):913–20 e1-7. doi: 10.1016/j.jaci.2009.08.044
18. Jia Z, Bao K, Wei P, Yu X, Zhang Y, Wang X, et al. Egfr activation-induced decreases in Claudin1 promote Muc5ac expression and exacerbate asthma in mice. Mucosal Immunol. (2021) 14(1):125–34. doi: 10.1038/s41385-020-0272-z
19. Xiang YY, Wang S, Liu M, Hirota JA, Li J, Ju W, et al. A gabaergic system in airway epithelium is essential for mucus overproduction in asthma. Nat Med. (2007) 13(7):862–7. doi: 10.1038/nm1604
20. Bhaker S, Portelli MA, Rakkar K, Shaw D, Johnson S, Brightling C, et al. Human bronchial epithelial cells from patients with asthma have an altered gene expression profile. ERJ Open Res. (2022) 8(1):00625–2021. doi: 10.1183/23120541.00625-2021
21. Tsai YH, Parker JS, Yang IV, Kelada SNP. Meta-analysis of airway epithelium gene expression in asthma. Eur Respir J. (2018) 51(5):1701962. doi: 10.1183/13993003.01962-2017
22. Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U S A. (2007) 104(40):15858–63. doi: 10.1073/pnas.0707413104
24. Johansson K, Weidner J, Radinger M. Micrornas in type 2 immunity. Cancer Lett. (2018) 425:116–24. doi: 10.1016/j.canlet.2018.03.036
25. Mehta A, Baltimore D. Micrornas as regulatory elements in immune system logic. Nat Rev Immunol. (2016) 16(5):279–94. doi: 10.1038/nri.2016.40
26. Broughton JP, Lovci MT, Huang JL, Yeo GW, Pasquinelli AE. Pairing beyond the seed supports microrna targeting specificity. Mol Cell. (2016) 64(2):320–33. doi: 10.1016/j.molcel.2016.09.004
27. Vasudevan S. Posttranscriptional upregulation by micrornas. Wiley Interdiscip Rev RNA. (2012) 3(3):311–30. doi: 10.1002/wrna.121
28. Weidner J, Bartel S, Kilic A, Zissler UM, Renz H, Schwarze J, et al. Spotlight on micrornas in allergy and asthma. Allergy. (2021) 76(6):1661–78. doi: 10.1111/all.14646
29. Bondanese VP, Francisco-Garcia A, Bedke N, Davies DE, Sanchez-Elsner T. Identification of host mirnas that may limit human rhinovirus replication. World J Biol Chem. (2014) 5(4):437–56. doi: 10.4331/wjbc.v5.i4.437
30. Haj-Salem I, Fakhfakh R, Berube JC, Jacques E, Plante S, Simard MJ, et al. Microrna-19a enhances proliferation of bronchial epithelial cells by targeting Tgfbetar2 gene in severe asthma. Allergy. (2015) 70(2):212–9. doi: 10.1111/all.12551
31. Huang H, Lu H, Liang L, Zhi Y, Huo B, Wu L, et al. Microrna-744 inhibits proliferation of bronchial epithelial cells by regulating Smad3 pathway via targeting transforming growth factor-Beta1 (Tgf-Beta1) in severe asthma. Med Sci Monit. (2019) 25:2159–68. doi: 10.12659/MSM.912412
32. Martinez-Nunez RT, Bondanese VP, Louafi F, Francisco-Garcia AS, Rupani H, Bedke N, et al. A microrna network dysregulated in asthma controls Il-6 production in bronchial epithelial cells. PLoS One. (2014) 9(10):e111659. doi: 10.1371/journal.pone.0111659
33. Siddiqui S, Johansson K, Joo A, Bonser LR, Koh KD, Le Tonqueze O, et al. Epithelial Mir-141 regulates Il-13-induced airway mucus production. JCI Insight. (2021) 6(5):e139019. doi: 10.1172/jci.insight.139019
34. Cohen LEX, Tarsi J, Ramkumar T, Horiuchi TK, Cochran R, DeMartino S. Epithelial cell proliferation contributes to airway remodeling in severe asthma. Am J Respir Crit Care Med. (2007) 176(2):138–45. doi: 10.1164/rccm.200607-1062OC
35. Boateng E, Krauss-Etschmann S. miRNAs in lung development and diseases. Int J Mol Sci. (2020) 21(8):2765. doi: 10.3390/ijms21082765
36. Perdomo C, Campbell JD, Gerrein J, Tellez CS, Garrison CB, Walser TC, et al. Microrna 4423 is a primate-specific regulator of airway epithelial cell differentiation and lung carcinogenesis. Proc Natl Acad Sci USA. (2013) 110(47):18946–51. doi: 10.1073/pnas.1220319110
37. Otto T, Candido SV, Pilarz MS, Sicinska E, Bronson RT, Bowden M, et al. Cell cycle-targeting micrornas promote differentiation by enforcing cell-cycle exit. Proc Natl Acad Sci USA. (2017) 114(40):10660–5. doi: 10.1073/pnas.1702914114
38. Martinez-Anton A, Sokolowska M, Kern S, Davis AS, Alsaaty S, Taubenberger JK, et al. Changes in microrna and mrna expression with differentiation of human bronchial epithelial cells. Am J Respir Cell Mol Biol. (2013) 49(3):384–95. doi: 10.1165/rcmb.2012-0368OC
39. Xiong T, Du Y, Fu Z, Geng G. Microrna-145-5p promotes asthma pathogenesis by inhibiting kinesin family member 3a expression in mouse airway epithelial cells. J Int Med Res. (2019) 47(7):3307–19. doi: 10.1177/0300060518789819
40. Morrisey EE, Hogan BL. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell. (2010) 18(1):8–23. doi: 10.1016/j.devcel.2009.12.010
41. Haies DM, Gil J, Weibel ER. Morphometric study of rat lung cells. I. Numerical and dimensional characteristics of parenchymal cell population. Am Rev Respir Dis. (1981) 123(5):533–41. doi: 10.1164/arrd.1981.123.5.533
42. Wang Y, Tang Z, Huang H, Li J, Wang Z, Yu Y, et al. Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc Natl Acad Sci U S A. (2018) 115(10):2407–12. doi: 10.1073/pnas.1719474115
43. Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. (2013) 123(7):3025–36. doi: 10.1172/JCI68782
44. Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature. (2014) 507(7491):190–4. doi: 10.1038/nature12930
45. Rock JR, Randell SH, Hogan BL. Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis Model Mech. (2010) 3(9–10):545–56. doi: 10.1242/dmm.006031
46. Whitsett JA. Airway epithelial differentiation and mucociliary clearance. Ann Am Thorac Soc. (2018) 15(Suppl 3):S143–8. doi: 10.1513/AnnalsATS.201802-128AW
47. Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, et al. A revised airway epithelial hierarchy includes cftr-expressing ionocytes. Nature. (2018) 560(7718):319–24. doi: 10.1038/s41586-018-0393-7
48. Rock JR, Gao X, Xue Y, Randell SH, Kong YY, Hogan BL. Notch-dependent differentiation of adult airway basal stem cells. Cell Stem Cell. (2011) 8(6):639–48. doi: 10.1016/j.stem.2011.04.003
49. Boers JE, Ambergen AW, Thunnissen FB. Number and proliferation of basal and parabasal cells in normal human airway epithelium. Am J Respir Crit Care Med. (1998) 157(6 Pt 1):2000–6. doi: 10.1164/ajrccm.157.6.9707011
50. Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology. (2003) 8(4):432–46. doi: 10.1046/j.1440-1843.2003.00493.x
51. Rawlins EL, Okubo T, Xue Y, Brass DM, Auten RL, Hasegawa H, et al. The role of Scgb1a1+ clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell. (2009) 4(6):525–34. doi: 10.1016/j.stem.2009.04.002
52. Song H, Yao E, Lin C, Gacayan R, Chen MH, Chuang PT. Functional characterization of pulmonary neuroendocrine cells in lung development, injury, and tumorigenesis. Proc Natl Acad Sci USA. (2012) 109(43):17531–6. doi: 10.1073/pnas.1207238109
53. Ouadah Y, Rojas ER, Riordan DP, Capostagno S, Kuo CS, Krasnow MA. Rare pulmonary neuroendocrine cells are stem cells regulated by Rb, P53, and notch. Cell. (2019) 179(2):403–16 e23. doi: 10.1016/j.cell.2019.09.010
54. Bloodgood RA. Sensory reception is an attribute of both primary cilia and motile cilia. J Cell Sci. (2010) 123(Pt 4):505–9. doi: 10.1242/jcs.066308
55. Kuek LE, Lee RJ. First contact: the role of respiratory cilia in host-pathogen interactions in the airways. Am J Physiol Lung Cell Mol Physiol. (2020) 319(4):L603–19. doi: 10.1152/ajplung.00283.2020
56. Park KS, Wells JM, Zorn AM, Wert SE, Laubach VE, Fernandez LG, et al. Transdifferentiation of ciliated cells during repair of the respiratory epithelium. Am J Respir Cell Mol Biol. (2006) 34(2):151–7. doi: 10.1165/rcmb.2005-0332OC
57. Singh G, Katyal SL. Clara cell proteins. Ann N Y Acad Sci. (2000) 923:43–58. doi: 10.1111/j.1749-6632.2000.tb05518.x
58. Rostami MR, LeBlanc MG, Strulovici-Barel Y, Zuo W, Mezey JG, O'Beirne SL, et al. Smoking shifts human small airway epithelium club cells toward a lesser differentiated population. NPJ Genom Med. (2021) 6(1):73. doi: 10.1038/s41525-021-00237-1
59. Giangreco A, Reynolds SD, Stripp BR. Terminal bronchioles harbor a unique airway stem cell population that localizes to the bronchoalveolar duct junction. Am J Pathol. (2002) 161(1):173–82. doi: 10.1016/S0002-9440(10)64169-7
60. Hong KU, Reynolds SD, Giangreco A, Hurley CM, Stripp BR. Clara cell secretory protein-expressing cells of the airway neuroepithelial body microenvironment include a label-retaining subset and are critical for epithelial renewal after progenitor cell depletion. Am J Respir Cell Mol Biol. (2001) 24(6):671–81. doi: 10.1165/ajrcmb.24.6.4498
61. Reynolds SD, Giangreco A, Power JH, Stripp BR. Neuroepithelial bodies of pulmonary airways serve as a reservoir of progenitor cells capable of epithelial regeneration. Am J Pathol. (2000) 156(1):269–78. doi: 10.1016/S0002-9440(10)64727-X
62. Gamez AS, Gras D, Petit A, Knabe L, Molinari N, Vachier I, et al. Supplementing defect in club cell secretory protein attenuates airway inflammation in copd. Chest. (2015) 147(6):1467–76. doi: 10.1378/chest.14-1174
63. Mango GW, Johnston CJ, Reynolds SD, Finkelstein JN, Plopper CG, Stripp BR. Clara cell secretory protein deficiency increases oxidant stress response in conducting airways. Am J Physiol. (1998) 275(2):L348–56. doi: 10.1152/ajplung.1998.275.2.L348
64. Tokita E, Tanabe T, Asano K, Suzaki H, Rubin BK. Club cell 10-Kda protein attenuates airway mucus hypersecretion and inflammation. Eur Respir J. (2014) 44(4):1002–10. doi: 10.1183/09031936.00080913
65. Laoukili J, Perret E, Willems T, Minty A, Parthoens E, Houcine O, et al. Il-13 alters mucociliary differentiation and ciliary beating of human respiratory epithelial cells. J Clin Invest. (2001) 108(12):1817–24. doi: 10.1172/JCI13557
66. Rogers DF. Airway goblet cells: responsive and adaptable front-line defenders. Eur Respir J. (1994) 7(9):1690–706. doi: 10.1183/09031936.94.07091678
67. Plasschaert LW, Zilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, et al. A single-cell atlas of the airway epithelium reveals the Cftr-rich pulmonary ionocyte. Nature. (2018) 560(7718):377–81. doi: 10.1038/s41586-018-0394-6
68. Okuda K, Dang H, Kobayashi Y, Carraro G, Nakano S, Chen G, et al. Secretory cells dominate airway cftr expression and function in human airway superficial epithelia. Am J Respir Crit Care Med. (2021) 203(10):1275–89. doi: 10.1164/rccm.202008-3198OC
69. Goldfarbmuren KC, Jackson ND, Sajuthi SP, Dyjack N, Li KS, Rios CL, et al. Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat Commun. (2020) 11(1):2485. doi: 10.1038/s41467-020-16239-z
70. Prefontaine D, Hamid Q. Airway epithelial cells in asthma. J Allergy Clin Immunol. (2007) 120(6):1475–8. doi: 10.1016/j.jaci.2007.09.041
71. Kouzaki H, Tojima I, Kita H, Shimizu T. Transcription of interleukin-25 and extracellular release of the protein is regulated by allergen proteases in airway epithelial cells. Am J Respir Cell Mol Biol. (2013) 49(5):741–50. doi: 10.1165/rcmb.2012-0304OC
72. Wang W, Li Y, Lv Z, Chen Y, Li Y, Huang K, et al. Bronchial allergen challenge of patients with atopic asthma triggers an alarmin (Il-33, Tslp, and Il-25) response in the airways epithelium and submucosa. J Immunol. (2018) 201(8):2221–31. doi: 10.4049/jimmunol.1800709
73. Whetstone CE, Ranjbar M, Omer H, Cusack RP, Gauvreau GM. The role of airway epithelial cell alarmins in asthma. Cells. (2022) 11(7):1105. doi: 10.3390/cells11071105
74. Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med. (2013) 5(174):174ra26. doi: 10.1126/scitranslmed.3004812
75. Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via toll-like receptor 4 triggering of airway structural cells. Nat Med. (2009) 15(4):410–6. doi: 10.1038/nm.1946
76. Bonnelykke K, Sleiman P, Nielsen K, Kreiner-Moller E, Mercader JM, Belgrave D, et al. A genome-wide association study identifies Cdhr3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet. (2014) 46(1):51–5. doi: 10.1038/ng.2830
77. Mortensen LJ, Kreiner-Moller E, Hakonarson H, Bonnelykke K, Bisgaard H. The Pcdh1 gene and asthma in early childhood. Eur Respir J. (2014) 43(3):792–800. doi: 10.1183/09031936.00021613
78. Toncheva AA, Suttner K, Michel S, Klopp N, Illig T, Balschun T, et al. Genetic variants in protocadherin-1, bronchial hyper-responsiveness, and asthma subphenotypes in German children. Pediatr Allergy Immunol. (2012) 23(7):636–41. doi: 10.1111/j.1399-3038.2012.01334.x
79. Moheimani F, Hsu AC, Reid AT, Williams T, Kicic A, Stick SM, et al. The genetic and epigenetic landscapes of the epithelium in asthma. Respir Res. (2016) 17(1):119. doi: 10.1186/s12931-016-0434-4
80. Wiesner DL, Merkhofer RM, Ober C, Kujoth GC, Niu M, Keller NP, et al. Club cell Trpv4 serves as a damage sensor driving lung allergic inflammation. Cell Host Microbe. (2020) 27(4):614–28 e6. doi: 10.1016/j.chom.2020.02.006
81. Laing IA, Hermans C, Bernard A, Burton PR, Goldblatt J, Le Souef PN. Association between plasma Cc16 levels, the A38g polymorphism, and asthma. Am J Respir Crit Care Med. (2000) 161(1):124–7. doi: 10.1164/ajrccm.161.1.9904073
82. Shijubo N, Itoh Y, Yamaguchi T, Imada A, Hirasawa M, Yamada T, et al. Clara cell protein-positive epithelial cells are reduced in small airways of asthmatics. Am J Respir Crit Care Med. (1999) 160(3):930–3. doi: 10.1164/ajrccm.160.3.9803113
83. Oh JY, Lee YS, Min KH, Hur GY, Lee SY, Kang KH, et al. Decreased serum club cell secretory protein in asthma and chronic obstructive pulmonary disease overlap: a pilot study. Int J Chron Obstruct Pulmon Dis. (2018) 13:3411–7. doi: 10.2147/COPD.S174545
84. Zhu L, An L, Ran D, Lizarraga R, Bondy C, Zhou X, et al. The club cell marker Scgb1a1 downstream of Foxa2 is reduced in asthma. Am J Respir Cell Mol Biol. (2019) 60(6):695–704. doi: 10.1165/rcmb.2018-0199OC
85. Rajavelu P, Chen G, Xu Y, Kitzmiller JA, Korfhagen TR, Whitsett JA. Airway epithelial Spdef integrates goblet cell differentiation and pulmonary Th2 inflammation. J Clin Invest. (2015) 125(5):2021–31. doi: 10.1172/JCI79422
86. Dabbagh K, Takeyama K, Lee HM, Ueki IF, Lausier JA, Nadel JA. Il-4 induces mucin gene expression and goblet cell metaplasia in vitro and in vivo. J Immunol. (1999) 162(10):6233–7.10229869
87. Danahay H, Pessotti AD, Coote J, Montgomery BE, Xia D, Wilson A, et al. Notch2 is required for inflammatory cytokine-driven goblet cell metaplasia in the lung. Cell Rep. (2015) 10(2):239–52. doi: 10.1016/j.celrep.2014.12.017
88. Lora JM, Zhang DM, Liao SM, Burwell T, King AM, Barker PA, et al. Tumor necrosis factor-alpha triggers mucus production in airway epithelium through an Ikappab kinase beta-dependent mechanism. J Biol Chem. (2005) 280(43):36510–7. doi: 10.1074/jbc.M507977200
89. Tyner JW, Kim EY, Ide K, Pelletier MR, Roswit WT, Morton JD, et al. Blocking airway mucous cell metaplasia by inhibiting Egfr antiapoptosis and Il-13 transdifferentiation signals. J Clin Invest. (2006) 116(2):309–21. doi: 10.1172/JCI25167
90. Sun L, Ren X, Wang IC, Pradhan A, Zhang Y, Flood HM, et al. The Foxm1 inhibitor Rcm-1 suppresses goblet cell metaplasia and prevents Il-13 and Stat6 signaling in allergen-exposed mice. Sci Signal. (2017) 10(475):eaai8583. doi: 10.1126/scisignal.aai8583
91. Kuchibhotla VNS, Starkey MR, Reid AT, Heijink IH, Nawijn MC, Hansbro PM, et al. Inhibition of beta-catenin/creb binding protein signaling attenuates house dust mite-induced goblet cell metaplasia in mice. Front Physiol. (2021) 12:690531. doi: 10.3389/fphys.2021.690531
92. Ren X, Shah TA, Ustiyan V, Zhang Y, Shinn J, Chen G, et al. Foxm1 promotes allergen-induced goblet cell metaplasia and pulmonary inflammation. Mol Cell Biol. (2013) 33(2):371–86. doi: 10.1128/MCB.00934-12
93. Alagha K, Bourdin A, Vernisse C, Garulli C, Tummino C, Charriot J, et al. Goblet cell hyperplasia as a feature of neutrophilic asthma. Clin Exp Allergy. (2019) 49(6):781–8. doi: 10.1111/cea.13359
94. Zeki AA, Bratt JM, Rabowsky M, Last JA, Kenyon NJ. Simvastatin inhibits goblet cell hyperplasia and lung arginase in a mouse model of allergic asthma: a novel treatment for airway remodeling? Transl Res. (2010) 156(6):335–49. doi: 10.1016/j.trsl.2010.09.003
95. Chen M, Lv Z, Zhang W, Huang L, Lin X, Shi J, et al. Triptolide suppresses airway goblet cell hyperplasia and Muc5ac expression via Nf-Kappab in a murine model of asthma. Mol Immunol. (2015) 64(1):99–105. doi: 10.1016/j.molimm.2014.11.001
96. Mishina K, Shinkai M, Shimokawaji T, Nagashima A, Hashimoto Y, Inoue Y, et al. Ho-1 inhibits Il-13-induced goblet cell hyperplasia associated with Clca1 suppression in normal human bronchial epithelial cells. Int Immunopharmacol. (2015) 29(2):448–53. doi: 10.1016/j.intimp.2015.10.016
97. Horvath G, Sorscher EJ. Luminal fluid tonicity regulates airway ciliary beating by altering membrane stretch and intracellular calcium. Cell Motil Cytoskeleton. (2008) 65(6):469–75. doi: 10.1002/cm.20273
98. Luk CK, Dulfano MJ. Effect of Ph, viscosity and ionic-strength changes on ciliary beating frequency of human bronchial explants. Clin Sci (Lond). (1983) 64(4):449–51. doi: 10.1042/cs0640449
99. Shah AS, Ben-Shahar Y, Moninger TO, Kline JN, Welsh MJ. Motile cilia of human airway epithelia are chemosensory. Science. (2009) 325(5944):1131–4. doi: 10.1126/science.1173869
100. Xiao C, Puddicombe SM, Field S, Haywood J, Broughton-Head V, Puxeddu I, et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol. (2011) 128(3):549–56 e1-12. doi: 10.1016/j.jaci.2011.05.038
101. Hackett TL, Singhera GK, Shaheen F, Hayden P, Jackson GR, Hegele RG, et al. Intrinsic phenotypic differences of asthmatic epithelium and its inflammatory responses to respiratory syncytial virus and air pollution. Am J Respir Cell Mol Biol. (2011) 45(5):1090–100. doi: 10.1165/rcmb.2011-0031OC
102. Post S, Heijink IH, Hesse L, Koo HK, Shaheen F, Fouadi M, et al. Characterization of a lung epithelium specific E-cadherin knock-out model: implications for obstructive lung pathology. Sci Rep. (2018) 8(1):13275. doi: 10.1038/s41598-018-31500-8
103. Turner J, Roger J, Fitau J, Combe D, Giddings J, Heeke GV, et al. Goblet cells are derived from a Foxj1-expressing progenitor in a human airway epithelium. Am J Respir Cell Mol Biol. (2011) 44(3):276–84. doi: 10.1165/rcmb.2009-0304OC
104. Vieira Braga FA, Kar G, Berg M, Carpaij OA, Polanski K, Simon LM, et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med. (2019) 25(7):1153–63. doi: 10.1038/s41591-019-0468-5
105. Collison A, Mattes J, Plank M, Foster PS. Inhibition of house dust mite-induced allergic airways disease by antagonism of microrna-145 is comparable to glucocorticoid treatment. J Allergy Clin Immunol. (2011) 128(1):160–7 e4. doi: 10.1016/j.jaci.2011.04.005
106. Mattes J, Collison A, Plank M, Phipps S, Foster PS. Antagonism of microrna-126 suppresses the effector function of Th2 cells and the development of allergic airways disease. Proc Natl Acad Sci USA. (2009) 106(44):18704–9. doi: 10.1073/pnas.0905063106
107. Sharma A, Kumar M, Ahmad T, Mabalirajan U, Aich J, Agrawal A, et al. Antagonism of mmu-Mir-106a attenuates asthma features in allergic murine model. J Appl Physiol (1985). (2012) 113(3):459–64. doi: 10.1152/japplphysiol.00001.2012
108. Liu Z, Chen X, Wu Q, Song J, Wang L, Li G. Mir-125b inhibits goblet cell differentiation in allergic airway inflammation by targeting Spdef. Eur J Pharmacol. (2016) 782:14–20. doi: 10.1016/j.ejphar.2016.04.044
109. Su Y, Wang J, Zou J, Han W, Li S. Mir-330 regulates interleukin-13-induced Muc5ac secretion by targeting Munc18b in human bronchial epithelial cells. Int J Clin Exp Pathol. (2018) 11(7):3463–70.31949724
110. Solberg OD, Ostrin EJ, Love MI, Peng JC, Bhakta NR, Hou L, et al. Airway epithelial mirna expression is altered in asthma. Am J Respir Crit Care Med. (2012) 186(10):965–74. doi: 10.1164/rccm.201201-0027OC
111. Marcet B, Chevalier B, Luxardi G, Coraux C, Zaragosi LE, Cibois M, et al. Control of vertebrate multiciliogenesis by Mir-449 through direct repression of the Delta/notch pathway. Nat Cell Biol. (2011) 13(6):693–9. doi: 10.1038/ncb2241
112. Jardim MJ, Dailey L, Silbajoris R, Diaz-Sanchez D. Distinct microrna expression in human airway cells of asthmatic donors identifies a novel asthma-associated gene. Am J Respir Cell Mol Biol. (2012) 47(4):536–42. doi: 10.1165/rcmb.2011-0160OC
113. Martinez-Nunez RT, Rupani H, Plate M, Niranjan M, Chambers RC, Howarth PH, et al. Genome-wide posttranscriptional dysregulation by micrornas in human asthma as revealed by Frac-Seq. J Immunol. (2018) 201(1):251–63. doi: 10.4049/jimmunol.1701798
114. Kivihall A, Aab A, Soja J, Sladek K, Sanak M, Altraja A, et al. Reduced expression of Mir-146a in human bronchial epithelial cells alters neutrophil migration. Clin Transl Allergy. (2019) 9:62. doi: 10.1186/s13601-019-0301-8
115. Mortaz E, Alipoor SD, Varahram M, Jamaati H, Garssen J, Mumby SE, et al. Exosomes in severe asthma: update in their roles and potential in therapy. Biomed Res Int. (2018) 2018:2862187. doi: 10.1155/2018/2862187
116. Srinivasan A, Sundar IK. Recent updates on the role of extracellular vesicles in the pathogenesis of allergic asthma. Extracell Vesicles Circ Nucl Acids. (2021) 2:127–47. doi: 10.20517/evcna.2021.03
117. Gupta R, Radicioni G, Abdelwahab S, Dang H, Carpenter J, Chua M, et al. Intercellular communication between airway epithelial cells is mediated by exosome-like vesicles. Am J Respir Cell Mol Biol. (2019) 60(2):209–20. doi: 10.1165/rcmb.2018-0156OC
118. Nagano T, Katsurada M, Dokuni R, Hazama D, Kiriu T, Umezawa K, et al. Crucial role of extracellular vesicles in bronchial asthma. Int J Mol Sci. (2019) 20(10):2589. doi: 10.3390/ijms20102589
119. Ax E, Jevnikar Z, Cvjetkovic A, Malmhall C, Olsson H, Radinger M, et al. T2 and T17 cytokines alter the cargo and function of airway epithelium-derived extracellular vesicles. Respir Res. (2020) 21(1):155. doi: 10.1186/s12931-020-01402-3
120. Bartel S, La Grutta S, Cilluffo G, Perconti G, Bongiovanni A, Giallongo A, et al. Human airway epithelial extracellular vesicle mirna signature is altered upon asthma development. Allergy. (2020) 75(2):346–56. doi: 10.1111/all.14008
Keywords: asthma, miRNA, signaling, epithelial cells, lung
Citation: Boateng E, Kovacevic D, Oldenburg V, Rådinger M and Krauss-Etschmann S (2022) Role of airway epithelial cell miRNAs in asthma. Front. Allergy 3:962693. doi: 10.3389/falgy.2022.962693
Received: 6 June 2022; Accepted: 1 September 2022;
Published: 20 September 2022.
Edited by:
Isaac Kirubakaran Sundar, University of Kansas Medical Center, United StatesReviewed by:
Bogdan Jakiela, Jagiellonian University Medical College, PolandTilman Sanchez-Elsner, University of Southampton, United Kingdom
© 2022 Boateng, Kovacevic, Oldenburg, Rådinger and Krauss-Etschmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eistine Boateng eboateng@fz-borstel.de
Specialty Section: This article was submitted to Asthma, a section of the journal Frontiers in Allergy