 Anna Valerieva
Anna Valerieva Hilary J. Longhurst
Hilary J. Longhurst- 1Department of Allergology, Medical University of Sofia, Sofia, Bulgaria
- 2Department of Immunology, Auckland District Health Board, and Department of Medicine, University of Auckland, Auckland, New Zealand
Hereditary angioedema (HAE) is a rare disease caused by mutations in the SERPING1 gene. This results in deficient or dysfunctional C1 esterase inhibitor (C1-INH) and affects multiple proteases involved in the complement, contact-system, coagulation, and fibrinolytic pathways. Current options for the treatment and prevention of HAE attacks include treating all affected pathways via direct C1-INH replacement therapy; or specifically targeting components of the contact activation system, in particular by blocking the bradykinin B2 receptor (B2R) or inhibiting plasma kallikrein, to prevent bradykinin generation. Intravenously administered plasma-derived C1-INH (pdC1-INH) and recombinant human C1-INH have demonstrated efficacy and safety for treatment of HAE attacks, although time to onset of symptom relief varied among trials, specific agents, and dosing regimens. Data from retrospective and observational analyses support that short-term prophylaxis with intravenous C1-INH products can help prevent HAE attacks in patients undergoing medical or dental procedures. Long-term prophylaxis with intravenous or subcutaneous pdC1-INH significantly decreased the HAE attack rate vs. placebo, although breakthrough attacks were observed. Pathway-specific therapies for the management of HAE include the B2R antagonist icatibant and plasma kallikrein inhibitors ecallantide, lanadelumab, and berotralstat. Icatibant, administered for treatment of angioedema attacks, reduced B2R-mediated vascular permeability and, compared with placebo, reduced the time to initial symptom improvement. Plasma kallikrein inhibitors, such as ecallantide, block the binding site of kallikrein to prevent cleavage of high molecular weight kininogen and subsequent bradykinin generation. Ecallantide was shown to be efficacious for HAE attacks and is licensed for this indication in the United States, but the labeling recommends that only health care providers administer treatment because of the risk of anaphylaxis. In addition to C1-INH replacement therapy, the plasma kallikrein inhibitors lanadelumab and berotralstat are recommended as first-line options for long-term prophylaxis and have demonstrated marked reductions in HAE attack rates. Investigational therapies, including the activated factor XII inhibitor garadacimab and an antisense oligonucleotide targeting plasma prekallikrein messenger RNA (donidalorsen), have shown promise as long-term prophylaxis. Given the requirement of lifelong management for HAE, further research is needed to determine how best to individualize optimal treatments for each patient.
Introduction
Hereditary angioedema (HAE) is a rare and potentially life-threatening genetic disease that can cause recurrent episodes (attacks) of nonpruritic swelling of the skin, affecting the extremities (e.g., hands, feet), face, and genitals, as well as submucosal swelling of the gastrointestinal and upper respiratory tracts (1–3). Approximately 85% of patients have type I HAE (type I C1-INH-HAE), which is characterized by a deficiency in C1 esterase inhibitor (C1-INH) levels (2–4). Type II HAE (type II C1-INH-HAE) accounts for about 15% of cases and is associated with abnormal function of C1-INH in the presence of normal C1-INH levels. These 2 types of HAE are caused by mutations in the SERPING1 gene, which encodes C1-INH (2). A third type of HAE, in which both levels and function of C1-INH are normal (HAE-nl-C1INH) (2, 5), is associated with specific genetic mutations (i.e., F12, ANGPT1, HS3ST6, PLG, MYOF, and KNG1), although many patients with HAE-nl-C1INH have no currently identified genetic mutation (2, 6, 7). HAE substantially impairs patient health-related quality of life, disrupts daily activities, and adversely affects social and professional and/or academic functioning (3, 8–11). Additionally, HAE is associated with increased rates of anxiety and depression, likely related to the unpredictability of symptoms and the associated emotional and physical stress (8–11). Severe HAE attacks require immediate intervention and may necessitate an emergency department visit or hospitalization, adding to the disease burden (11, 12).
The ultimate goal of HAE treatment is to achieve complete disease control (i.e., prevent all HAE attacks) and normalize patients’ quality of life and daily functioning (12). For those patients who are unable to achieve complete disease control, therapy aims to reduce the number of HAE attacks and improve quality of life. Pharmacologic management of HAE consists of on-demand (acute) treatment of HAE attacks, short-term prophylaxis delivered prior to procedures or events anticipated to trigger HAE symptoms, and routine, long-term prophylaxis to prevent HAE attacks (2, 13). On-demand therapy aims to minimize morbidity and prevent mortality during an HAE attack, while long-term prophylaxis is broadly deemed valuable for helping to optimize patients’ quality of life and daily functioning. Minimizing the burden of treatment and associated adverse effects is also an important consideration, especially in patients receiving long-term prophylaxis (12, 13). In recent years, the range of pharmacologic treatment options for the management of HAE has expanded considerably to encompass agents with more convenient routes of administration that facilitate self-management, as well as medications with different mechanisms of action that target distinct components of the pathways involved in HAE (14). Multiple treatment options offer the opportunity to individualize therapy, provide an opportunity for both prophylaxis and on-demand therapy using synergistic mechanisms of action, and minimize the disease burden. This narrative review describes the biologic pathways of importance in HAE and provides an overview of therapies that target these pathways to prevent and treat HAE attacks.
Pathways of importance in HAE
The plasma contact system is composed of the enzymes factor XII and plasma prekallikrein and is involved in the generation of the inflammatory peptide bradykinin and in blood coagulation (15). This system, together with the nonenzymatic co-factor high molecular weight (HMW) kininogen, comprises the kallikrein–kinin pathway (15, 16). Factor XII is activated by a number of mechanisms to generate factor XIIa (activated factor XII), which then cleaves kallikrein from prekallikrein. Kallikrein further activates factor XII in a positive feedback loop and also cleaves the HMW kininogen, releasing bradykinin (Figure 1) (5, 16). Bradykinin plays a role in blood coagulation, fibrinolysis, and vasodilation and is the primary mediator of enhanced vascular permeability during an HAE attack (5, 15, 16). Additionally, cleavage of factor XIIa by kallikrein results in release of the active protease factor XIIf, which activates the classical complement pathway (16).
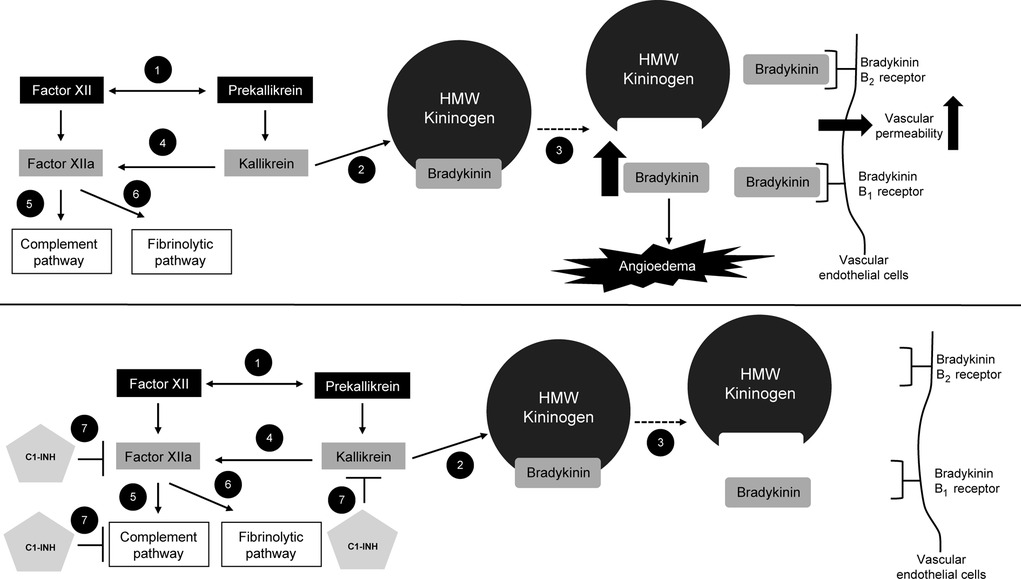
Figure 1. Dysregulation of signaling pathways in HAE. (1) When activated by trace amounts of factor XIIa, plasma prekallikrein and factor XII cleave each other to generate kallikrein and factor XIIa. (2) Kallikrein cleaves HMW plasma kininogen, leading to (3) the release of bradykinin. (4) Plasma kallikrein cleaves factor XIIa, leading to (5) activation of complement and (6) fibrinolytic pathways. In the top figure, the increase in bradykinin levels results in angioedema. Bradykinin binds bradykinin B1 and B2 receptors on vascular endothelial cells, leading to an increase in vascular permeability. In the bottom figure, (7) C1-INH inhibits factor XIIa, the complement pathway, and kallikrein, thus leading to a decrease in bradykinin production and reduced activation of bradykinin B1 and B2 receptors on vascular endothelial cells. C1-INH, C1 esterase inhibitor; HAE, hereditary angioedema; HMW, high molecular weight. Figure created with data from Cicardi M, et al. J Allergy Clin Immunol Pract. (2018) 6(4):1132–41; and Zuraw BL. N Engl J Med. (2008) 359(10):1027–36 (5, 16).
Under healthy conditions, C1-INH inhibits several proteases, including factors XIIa and XIIf and plasma kallikrein, as well as components of the early classical complement pathway (Figure 1) (5, 16). In types I and II HAE, C1-INH deficiency or dysfunction causes an increase in bradykinin levels because of dysregulation of the plasma contact system (5, 16). Unabated generation of bradykinin, resulting from insufficient C1-INH regulation of factor XIIa and kallikrein, leads to angioedema. Plasma kallikrein cleaves factor XIIa, leading to activation of the complement cascade; activation of complement results in the cleavage of C5 (to the anaphylatoxin C5a) and formation of the complement fragment C4a, which increases endothelial permeability (17). Cytokines can also affect endothelial permeability, with proinflammatory cytokines (e.g., interleukin [IL]-1, IL-4, IL-6, IL-8, IL-13) increasing permeability and anti-inflammatory cytokines (e.g., IL-1Ra, IL-10) inhibiting permeability (17). While evidence supports a key role of the bradykinin B2 (B2) receptor in angioedema, upregulation of the bradykinin B1 (B1) receptor during stress, trauma, or infection may influence susceptibility to angioedema, and prolonged B1 signaling may be involved in sustaining swelling during an HAE attack (16, 18). Thus, it is unclear whether blockade of both B1 and B2 receptors may be needed to completely prevent vascular leakage and associated swelling (18).
The lectin pathway is an activator of the complement cascade, which is mediated, in part, through mannose-binding lectin–associated serine proteases (MASPs) (19). MASP-1 is able to directly cleave HMW kininogen to generate and release bradykinin. C1-INH is an important regulator of the lectin pathway via inhibition of MASP-1 and MASP-2. Thus, dysregulation of MASPs may play a role in the pathophysiology of HAE by contributing to elevated bradykinin levels. The fibrinolytic pathway is also activated by factor XIIa (Figure 1), leading to conversion of plasminogen to plasmin; plasmin subsequently cleaves fibrin, leading to fibrin degradation (5). This pathway plays a greater role in some types of HAE-nl-C1INH pathophysiology, but also contributes to the endothelial dysfunction of types I and II HAE (20).
Pathways important in C1-inhibitor deficiency in HAE may also play a role in autoimmunity and neoplasms. A Swedish population-based cohort study reported that patients with HAE were at increased risk of autoimmune disease compared with individuals in the general population (odds ratio [OR], 1.6; 95% confidence interval [CI], 1.2–2.4); the risk of developing systemic lupus erythematosus was significantly greater in patients with HAE compared with individuals without HAE (OR, 71.9; 95% CI, 8.8–586.7) (21). While this study did not find an increased risk of cancer in patients with HAE vs. the general population (OR, 0.9; 95% CI, 0.6–1.4) (21), a retrospective review of medical records in Italy reported that neoplasms were the most common cause of mortality in patients with HAE, compared with cardiovascular disease as the most common cause of mortality in the general population (22). However, future studies are warranted to examine the role of HAE pathways in autoimmune disease and cancer.
Multiple pathway therapies
C1-INH replacement therapy
C1-INH products provide a direct replacement for the low levels or low functional activity of C1-INH in patients with type I or II HAE, respectively (13). Accordingly, administration of C1-INH replacement therapy during an HAE attack restores regulation of the cascade systems producing bradykinin by inhibiting the same targets as endogenous C1-INH (i.e., plasma kallikrein, factors XIIa and XIIf, and elements of the complement pathway, including MASP-1; Figure 1) (5, 13, 16).
On-demand therapy
The World Allergy Organization (WAO)/European Academy of Allergy and Clinical Immunology (EAACI) guidelines recommend treatment with intravenous (IV) C1-INH as soon as possible after onset of an HAE attack (13). Intravenously administered plasma-derived C1-INH (pdC1-INH; Berinert; CSL Behring LLC; Kankakee, IL) and recombinant human C1-INH (rhC1-INH; Ruconest; Pharming Healthcare Inc.; Warren, NJ; Table 1) (23–37) are efficacious and well tolerated as on-demand treatment of HAE attacks (Table 2) (38–41). In a phase 2/3, randomized, double-blind, placebo-controlled trial (n = 124), pdC1-INH (Berinert) 20 U/kg provided a significantly faster onset of symptom relief of abdominal or facial attacks compared with placebo (median, 0.5 vs. 1.5 h, respectively; P = 0.002) and a significantly shorter median time to complete resolution of symptoms (4.9 vs. 7.8 h, respectively; P = 0.02; Table 2) (38). These data were supported by an open-label extension trial (N = 57) of pdC1-INH 20 U/kg as on-demand treatment (median follow-up, 24 months; Table 2) (39). Another pdC1-INH product (Cinryze; ViroPharma Biologics LLC; Lexington, MA) administered as on-demand treatment also showed significantly faster onset of symptom relief compared with placebo (40) (Table 2) but is not approved for acute treatment of HAE in the United States or European Union (Table 1).
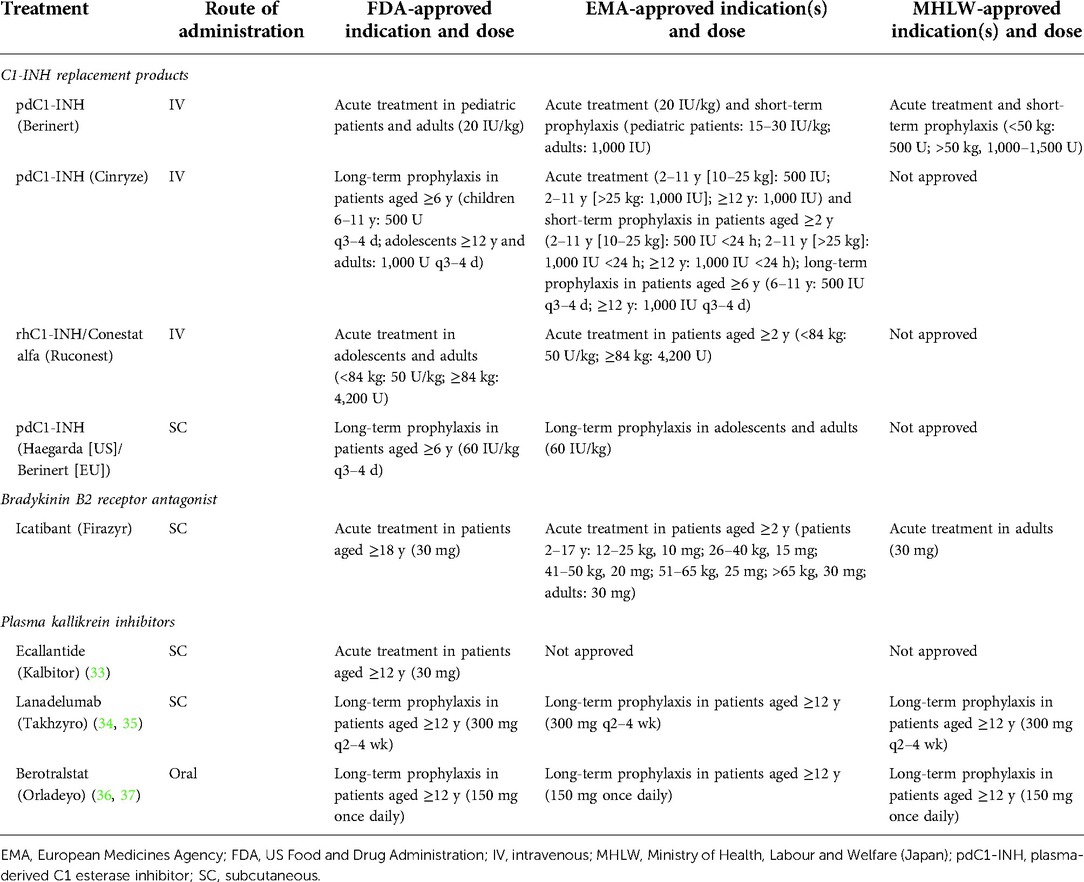
Table 1. Approved treatments for hereditary angioedema.

Table 2. Summary of clinical trials of C1-INH replacement therapy as on-demand treatment for HAE attacks.
Two similarly designed randomized, double-blind, placebo (saline)-controlled trials evaluated rhC1-INH 50 U/kg (n = 12) or 100 U/kg (n = 29) as on-demand treatment for HAE attacks (Table 2) (41). Compared with placebo, rhC1-INH significantly reduced the time to onset of symptom relief (50 U/kg: median, 122 vs. 495 min, respectively; P = 0.01; 100 U/kg: median, 66 vs. 495 min, respectively; P < 0.001), as well as the time to minimal symptoms (50 U/kg: median, 247 vs. 1,210 min, respectively; P = 0.001; 100 U/kg: median, 266 vs. 1,210 min, respectively; P < 0.001). The median time to onset of symptom relief with IV C1-INH replacement therapies has varied in relation to trial design, product formulation, and dosing, with a range of 0.4–1.2 h for pdC1-INH and approximately 1.0–2.0 h for rhC1-INH (Table 2) (38, 39, 41). Review of these trials suggests that there is a dose response, with higher doses providing earlier onset of relief, as well as relief in a greater percentage of patients (42).
Short-term prophylaxis
Surgical and dental procedures and other medical interventions (e.g., diagnostic procedures) may trigger an attack in patients with HAE (43–46). WAO/EAACI guidelines recommend short-term prophylactic therapy for patients with HAE undergoing these types of procedures, with C1-INH replacement therapies considered the first-line option for short-term prophylaxis (13). Patient-specific emotional triggers may also precipitate an HAE attack, so WAO/EAACI guidelines also suggest that short-term prophylaxis be considered prior to exposure to an anticipated stressful life event.
No C1-INH formulation is currently approved in the United States for short-term prophylaxis, although pdC1-INH (Berinert and/or Cinryze) is approved for this indication in the European Union and Japan (Table 1). Evidence for the efficacy and safety of C1-INH replacement therapy for short-term prophylaxis is limited to retrospective and observational analyses, including from patient registries (Table 3) (40, 46–54). Data from a retrospective study (N = 137) reported that patients ≥2 years of age with HAE who received short-term prophylaxis with pdC1-INH (Berinert) prior to medical procedures experienced a decrease in post-procedure HAE attacks compared with the number of attacks they experienced before being diagnosed with HAE (Table 3) (46). It is noteworthy that patients in this study received pdC1-INH 500 U, which is half the dose noted by WAO/EACCI guidelines (1,000 U or 20 U/kg) for short-term, pre-procedure prophylaxis with pdC1-INH (13, 46). Registry data also indicated a low cumulative HAE attack rate within 3 days of pdC1-INH (Berinert) administration as short-term prophylaxis (47). Numerical trends suggested greater efficacy with weight-based doses of ≥15 IU/kg or absolute doses ≥1,500 IU. A recombinant preparation of C1-INH has also proved effective. A retrospective study (N = 51; 92% with type I HAE) demonstrated that short-term prophylaxis with rhC1-INH was efficacious for increasing the percentage of medical and dental procedures that remained attack-free compared with procedures in which no short-term prophylaxis was administered (Table 3) (48). However, rhC1-INH is currently not approved for prophylaxis (Table 1) (28).

Table 3. Summary of clinical trials of C1-INH replacement therapy as short-term or long-term prophylaxis for HAE attacks.
Long-term prophylaxis
WAO/EAACI guidelines recommend consideration of long-term prophylaxis for all patients with type I or II HAE, taking into consideration patients’ preferences, disease activity, HAE impact on quality of life, availability of health care resources, and inability to achieve adequate control of symptoms with appropriate on-demand therapy (13). Patients with HAE should be evaluated for long-term prophylaxis at each office or telemedicine visit or at least annually because these factors can vary over time. C1-INH replacement (e.g., pdC1-INH) is recommended as a first-line, long-term prophylactic therapy (13). C1-INH products approved in the United States and the European Union for routine, long-term prophylaxis include IV pdC1-INH (Cinryze) and a subcutaneous (SC) formulation of pdC1-INH (Haegarda; CSL Behring LLC; Table 1).
A double-blind, placebo-controlled, crossover trial (N = 22) determined that pdC1-INH (Cinryze) 1,000 U IV administered every 3 to 4 days as long-term prophylaxis significantly reduced the HAE attack rate compared with placebo during a 12-week period (difference, 6.5 attacks; P < 0.001; Table 3) (40). In addition, HAE attack duration was significantly decreased for patients receiving pdC1-INH prophylaxis compared with those receiving placebo (2.1 vs. 3.4 days, respectively; P = 0.002; Table 3). An open-label prophylaxis trial (N = 146) of pdC1-INH 1,000 U (Cinryze) every 3–7 days for up to 2.6 years found a 90% decrease from baseline in the mean number of attacks [4.7 attacks/month (baseline) vs. 0.5 attacks/month] (49). During that trial, 140 patients underwent complement testing to assess C1-INH antigen levels and functional activity at baseline (immediately prior to treatment) and 1 h after treatment. Baseline functional activity was inversely correlated with HAE attack frequency (i.e., lower baseline C1-INH activity was predictive of an increased attack rate during treatment; R = 0.2; P = 0.01); however, no significant relationship was observed between postinjection C1-INH function and attack frequency (R = 0.09; P = 0.3) (49).
In a phase 3, randomized, double-blind, crossover trial (N = 90), a significant decrease in the mean frequency of attacks was observed with SC pdC1-INH 40 or 60 IU/kg, administered twice weekly for 16 weeks, compared with placebo (Table 3; P < 0.001 for both doses vs. placebo) (50). In that trial, 76% and 90% of patients were responders (i.e., ≥50% decrease in the number of attacks vs. placebo) with pdC1-INH 40 and 60 IU/kg, respectively. An open-label extension trial (N = 126) supported these results, with >90% of patients in the pdC1-INH 40- and 60-IU/kg treatment groups achieving a ≥50% reduction from baseline in HAE attacks (Table 3) (51). Pharmacokinetic modeling indicated that pdC1-INH administered SC exhibits a more consistent exposure and a lower peak-to-trough ratio compared with pdC1-INH given IV, which may account for the improved efficacy of SC pdC1-INH observed in clinical trials (50, 55).
A prospective, observational trial of long-term HAE prophylaxis with IV pdC1-INH (Berinert), which is not currently approved for this indication, reported that more than half of the patients (8/14) experienced a reduction in attack frequency during the last 12 months of treatment (mean duration, 9 years) compared with the attack frequency before initiation of long-term prophylactic therapy (Table 3) (52). However, 5 of the 14 patients experienced an increase in attack frequency, even with increased pdC1-INH dosing, and 1 patient discontinued participation in the trial after 5 years because of attack frequency (after receiving pdC1-INH 1,000 U daily for 1.5 years). One hypothesis for the increase in HAE attack rate during the trial was that frequent IV injections may be activating the plasma contact system, either directly or indirectly, leading to an increase in underlying disease activity that is otherwise masked by the effectiveness of pdC1-INH administration (52). Patients in this trial received pdC1-INH at least once weekly for a mean of 9 years (range, 4‒19 years). Cycling from increased C1-INH levels immediately after treatment to a decrease in C1-INH levels several days post-treatment may have lowered the threshold for activation of the plasma contact system (52). An observational registry trial (N = 47) of long-term HAE prophylaxis with pdC1-INH (Berinert; mean 9.2 months of treatment per patient) showed that more than two-thirds of patients (68.1%) experienced ≥1 attack within 7 days of receiving IV treatment (Table 3) (53).
rhC1-INH is also not approved as long-term prophylaxis of HAE (Table 1) but proved efficacious for this use in a phase 2, randomized, double-blind, placebo-controlled crossover trial (Table 3) (54). When administered once or twice weekly for 4 weeks, rhC1-INH reduced in a statistically significant manner the mean number of attacks, as well as the attack frequency, compared with placebo (54).
Single pathway therapies
Single pathway therapies that are currently available for the treatment of HAE include the SC administered B2 receptor antagonist icatibant (Firazyr; Takeda Pharmaceuticals America, Inc.; Lexington, MA), the SC administered plasma kallikrein inhibitors ecallantide (Kalbitor; Dyax Corp., a Takeda company; Lexington, MA) for attacks or lanadelumab (Takhzyro; Dyax Corp; Lexington, MA) for prophylaxis, as well as the oral plasma kallikrein inhibitor berotralstat (Orladeyo; BioCryst Pharmaceuticals, Inc.; Durham, NC; Table 1), also for prophylaxis. By blocking the effects of bradykinin at the B2 receptor (Figure 2), icatibant has been shown to reverse the vascular permeability observed in C1-INH gene knockout mice (56) and to decrease bradykinin-induced vasodilation in the forearms of healthy volunteers (57). Treatment with icatibant also decreased bradykinin levels in patients with HAE, but changes in plasma bradykinin levels were not directly related to the degree of symptom relief (58). Working upstream from icatibant, plasma kallikrein inhibitors block the binding site of kallikrein to prevent cleavage of HMW kininogen and subsequent bradykinin release and also reduce further activation of factor XIIa to disrupt the positive feedback loop that would otherwise lead to increased kallikrein production (Figure 2) (16, 33, 59, 60).
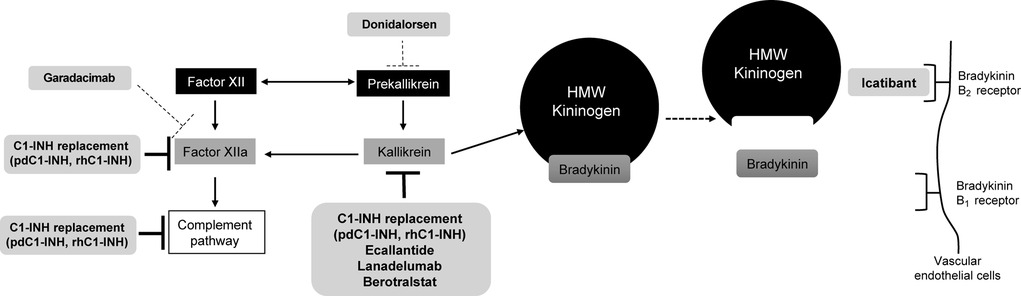
Figure 2. Therapeutic targeting of pathways in HAE. Inhibitors of the pathway are shown in light gray shaded boxes at their respective sites of action. Investigational therapies are shown with dashed lines at their target sites. C1-INH, C1 esterase inhibitor; HMW, high molecular weight; pdC1-INH, plasma-derived C1-INH; rhC1-INH, recombinant human C1-INH. Figure created with data from Cicardi M, et al. J Allergy Clin Immunol Pract. (2018) 6(4):1132–41; Zuraw BL. N Engl J Med. (2008) 359(10):1027–36; and Fijen LM, et al. Clin Rev Allergy Immunol. (2021) 61(1):66–7 (5, 14, 16).
On-demand therapy for HAE attacks
In addition to IV C1-INH replacement therapy, WAO/EAACI guidelines recommend IV ecallantide or icatibant as on-demand treatment of HAE attacks (13). Both agents are approved in the United States for this indication (Table 1), while icatibant is approved in the European Union (61) and Japan (62).
In two phase 3 randomized, double-blind trials (For Angioedema Subcutaneous Treatment [FAST]-1 [N = 56] and FAST-2 [N = 74]), icatibant 30 mg SC was administered for the treatment of acute HAE attacks. Icatibant reduced the median time to clinically significant relief of the patient’s index symptom compared with oral tranexamic acid 3 g daily for 2 days (FAST-2; 2.0 vs. 12.0 h, respectively; P < 0.001) and also reduced the median time compared with placebo, although the difference was not statistically significant (FAST-1; 2.5 vs. 4.6 h; P = 0.1; Table 4) (63–70). Furthermore, in both trials, icatibant significantly reduced the median time to initial symptom improvement by both patient assessment and investigator assessment. In a third phase 3 trial, icatibant 30 mg SC (n = 43) was significantly more efficacious than placebo (n = 45) for reducing the median time to onset of symptom relief for nonlaryngeal attacks (2.0 h vs. 19.8 h, respectively; P < 0.001) (65).
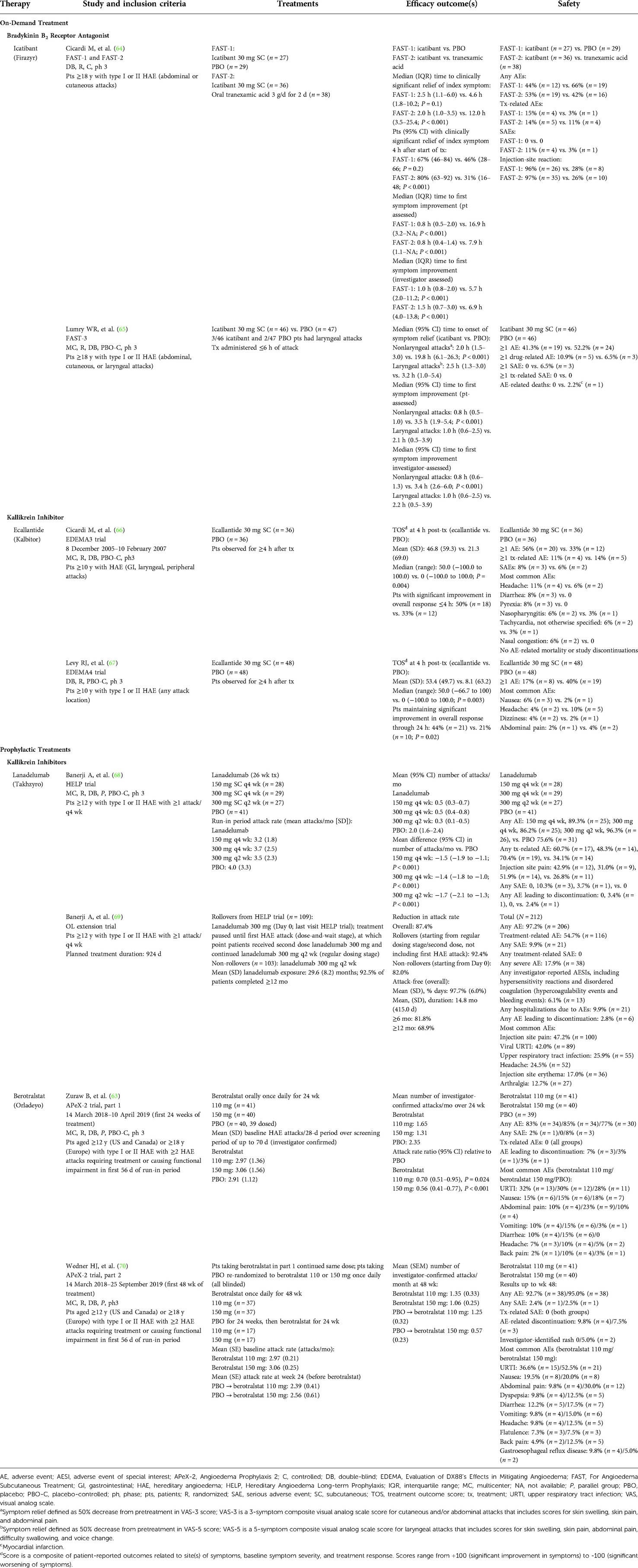
Table 4. Summary of clinical trials of bradykinin B2 receptor antagonists and kallikrein inhibitors as on-demand or prophylactic treatment of HAE attacks.
In a phase 3, randomized, double-blind trial (N = 72), on-demand treatment with ecallantide 30 mg SC significantly improved the median treatment outcome score (patient-reported composite comprising sites of symptoms, symptom severity, and response to treatment) compared with placebo (Table 4; P = 0.004) (66). Further, a greater percentage of patients who received ecallantide experienced significant improvement (“a lot better or resolved”) in overall response vs. placebo (50% vs. 33%, respectively) (66). Results of another phase 3, randomized, double-blind trial (N = 96) noted a significant improvement in treatment outcome score 4 h after dosing with ecallantide compared with placebo (P = 0.003) and an overall response maintained through 24 h (P = 0.02; Table 4) (67). Although anaphylaxis was not reported during these 2 trials (66, 67), a retrospective database analysis of ecallantide clinical trials identified 8 of 230 patients (3.5%) with hypersensitivity reactions consistent with anaphylaxis (71). All of these events occurred within 1 h of ecallantide administration, and no cases of anaphylaxis occurred after the first exposure (71). Accordingly, the US ecallantide prescribing information contains a boxed warning regarding the potential for anaphylaxis and states that only health care providers should administer the medication (33).
Long-term prophylaxis
The plasma kallikrein inhibitors lanadelumab and berotralstat are both recommended by WAO/EAACI guidelines as first-line options for long-term prophylaxis (13) and are approved in the United States, European Union, and Japan for this indication (Table 1). A phase 3 trial (N = 125) demonstrated that the 3 different lanadelumab SC dosing regimens examined (150 mg every 4 weeks, 300 mg every 4 weeks, or 300 mg every 2 weeks) all significantly reduced the mean number of attacks experienced by patients each month compared with placebo (P < 0.001 for all comparisons) over the 26-week treatment period (Table 4) (68). Patients who completed the trial (n = 109) or new patients (n = 103) were eligible for an open-label extension trial of lanadelumab 300 mg SC every 2 weeks (Table 4) (69). Long-term treatment with lanadelumab reduced the mean HAE attack rate by 87.4% overall. During treatment, patients were attack-free for 97.7% of days on average, and the mean duration of the attack-free period was >14 months. More than 80% of patients remained attack-free for ≥6 months and 69% were attack-free for ≥12 months.
In part 1 (a 24-week, placebo-controlled phase) of a phase 3, randomized, double-blind trial (N = 121), berotralstat 110 and 150 mg once daily significantly reduced the mean number of HAE attacks per month compared with placebo (P = 0.024 and P < 0.001, respectively; Table 4) (63). The attack rate ratio relative to placebo (95% CI) was 0.70 (0.51–0.95) for berotralstat 110 mg and 0.56 (0.41–0.77) for berotralstat 150 mg. Both doses of berotralstat significantly reduced the rate of HAE attacks in patients with ≥2 attacks per month at baseline (110 mg, P = 0.04; 150 mg, P = 0.005), but only berotralstat 150 mg significantly reduced the HAE attack rate among patients with <2 attacks per month at baseline (P = 0.009). In part 2 of this trial, berotralstat-treated patients in part 1 continued their assigned double-blind dose, and placebo-treated patients were re-randomized to double-blind berotralstat 110 mg or 150 mg once daily (Table 4) (70). Among patients who continued on berotralstat, the mean (SE) monthly attack rates declined from 2.97 (0.21) at baseline to 1.35 (0.33) at week 48 in the berotralstat 110-mg group and from 3.06 (0.25) at baseline to 1.06 (0.25) at week 48 in the berotralstat 150-mg group. For placebo-treated patients who switched to berotralstat, the decrease in HAE attack rates was similar to that observed in part 1 of the trial in the berotralstat treatment groups (Table 4) (70).
Investigational treatments
Garadacimab (CSL312) is a human recombinant monoclonal antibody that inhibits factor XIIa and thus acts on the complement pathway in addition to the kallikrein-kinin pathway (Figure 2) (72). It has been shown to potently inhibit bradykinin formation in plasma samples from patients with HAE and to reduce edema in animal models (72). Data have been published from a phase 2, randomized, double-blind trial (73). Patients were randomly assigned to receive an IV loading dose (placebo or garadacimab 40, 100, or 300 mg) followed by SC administration (placebo [n = 8], garadacimab 75 mg [n = 9], garadacimab 200 mg [n = 8], or garadacimab 600 mg [n = 7]) on day 6 and then every 4 weeks for 12 weeks. During the 12-week SC treatment period, the median number (interquartile range) of monthly attacks was 4.6 (3.1–5.0) in the placebo group, 0.0 (0.0–0.4) in the garadacimab 75-mg group, 0.0 (0.0–0.0) in the garadacimab 200-mg group, and 0.3 (0.0–0.7) in the garadacimab 600-mg group. Compared with placebo, garadacimab 75 mg, 200 mg, and 600 mg significantly reduced the median attack rate by 100% (P = 0.0002, post hoc analysis), 100% (P = 0.0002), and 93% (P = 0.0003), respectively. The most common adverse events were injection-site reactions (reported by 25%, 11%, 13%, and 57% of patients in the placebo and garadacimab 75-, 200-, and 600-mg groups, respectively). No serious adverse events, anaphylaxis, or thromboembolic events were reported. Phase 3 trials are ongoing.
Administration of an antisense oligonucleotide targeting plasma prekallikrein messenger RNA (Figure 2), which has been shown to prevent bradykinin formation (74), has been reported for 2 patients with severe bradykinin-mediated forms of HAE (75). The patients were initially treated with the parent antisense oligonucleotide IONIS-PKKRx SC for 12 to 16 weeks, followed by treatment with a ligand-conjugated form of the oligonucleotide, donidalorsen (formerly named IONIS-PKK-LRx) 80 mg SC every 3 to 4 weeks for 7 to 8 months. IONIS-PKKRx SC dosing was 200 mg once weekly, with optional dose loading in the first 2 weeks. If the 2 patients had breakthrough HAE attacks, dosing could be increased to 300 mg after 6 weeks and to 400 mg after 12 weeks. During IONIS-PKKRx treatment, the mean monthly attack rate decreased from 1.2 to 0.2 attacks (patient 1) and from 7.9 to 1.0 attacks (patient 2). When patients were switched to donidalorsen (IONIS-PKK-LRx), the mean monthly attack rate was 0 (patient 1) and 3.4 (patient 2). Plasma prekallikrein levels were reduced with both IONIS-PKKRx and donidalorsen (IONIS-PKK-LRx) treatment. Both patients experienced injection-site reactions.
A phase 2, randomized, double-blind, placebo-controlled trial was conducted to further examine the efficacy and safety of once monthly SC donidalorsen 80 mg in adults with HAE (76). During the ≤8-week study run-in period, the 20 patients (donidalorsen [n = 14]; placebo [n = 6]) included in the trial experienced a mean 2.7 HAE attacks/month (range, 1.0 to 5.6) (76). Donidalorsen significantly reduced the mean number of monthly attacks compared with placebo (0.2 vs. 2.2, respectively; 90% difference; P < 0.001) (76). Donidalorsen decreased plasma prekallikrein activity from baseline by 61% (76). The most commonly reported adverse events with donidalorsen were headache (14%) and nausea (7%); no patients in either treatment group experienced serious adverse events or discontinued the trial due to adverse events (76).
Discussion
Several options are available as on-demand treatment and as short-term or long-term prophylaxis of HAE attacks (2). Direct replacement with C1-INH products (e.g., pdC1-INH and rhC1-INH) treats all affected pathways, whereas single-target (pathway) therapies affect different components of the kallikrein-kinin system. Approved treatments for HAE vary in terms of indication(s), age limitations, route of administration, and frequency of dosing. C1-INH replacement therapies have a well-established efficacy and safety profile, given their long-term history of use in HAE (77), and newer agents are now being incorporated into HAE treatment regimens (78). Data for the C1-INH replacement therapies support dose-dependent efficacy, including the achievement of a zero-rate attack frequency (50, 54). Notably, the efficacy of C1-INH products for long-term prophylaxis was generally demonstrated in populations with a more severe HAE phenotype (on-placebo or pretreatment attack rate ∼4‒7 attacks/month) (40, 50, 54) than in similar trials of single-pathway treatment options (∼2–4 attacks/month) (Tables 3, 4) (63, 68). However, the phase 2 trial of garadacimab, an agent that targets both contact and fibrinolytic pathways, included patients with a more severe phenotype (mean 3.5–7.5 attacks/month) (73).
Although on-demand and prophylactic C1-INH treatments are effective for many patients with HAE (Tables 2–4), some patients may experience an increase in frequency of attacks while taking prophylactic therapy. Some authors have suggested that patients may develop tolerance to prophylactic therapy over time (52, 53). However, alternative explanations must be considered. For example, many patients lacking easy access to on-demand treatment do not treat every attack, and a percentage of attacks may remain untreated for other reasons (9, 79), so an increase in C1-INH usage may merely reflect better access to therapy and better patient education rather than tolerance induction. Prospectively collected observations based on patients receiving higher doses of prophylactic C1-INH do not suggest an increase in attack frequency and, if anything, show improved symptom control during 30 months of treatment, which disputes the theory of tolerance induction with C1-INH replacement therapy (51).
Long-term response to treatment may be dependent on baseline C1-INH levels in each patient (49). This is likely true for prophylaxis with C1-INH replacement therapies, but this hypothesis has not been specifically studied. Differences in study design, particularly in baseline or placebo HAE attack rates, treatment dosing, and route and frequency of administration, and a lack of head-to-head comparisons, make it difficult to evaluate any such relationship. For single-pathway therapies, genetic polymorphisms and perhaps environmental factors affecting other relevant pathways may be the most important predictors, along with factors affecting the degree of inhibition of the contact pathway target. Again, more research and better biomarkers are needed to determine the optimal approach for the individual patient. Given the lack of head-to-head clinical trials and differences among studies in patient populations, methods, and outcomes, which limit the ability to compare across trials, research is also needed to directly compare therapies in patients with HAE. Because both HAE therapies and a failure to control HAE symptoms have pharmacoeconomic implications (e.g., direct and indirect costs, including health care utilization), more data are needed about individual determinants of treatment response and the relative efficacy of HAE therapies (i.e., on-demand, short-term, and long-term prophylaxis) to enable delivery of an optimized treatment strategy for each patient.
In view of the wide variety of potentially relevant pathways regulated by C1-INH, it is perhaps surprising that several therapies that target only the contact pathway can produce a high level of control of angioedema (Figure 2). This reflects the central role of the contact pathway in angioedema generation and the multiple feedback loops between the contact pathway and other pathways implicated in angioedema generation. Further, new targeted agents in development for HAE have shown promise, albeit in small populations (73, 75).
In conclusion, some therapies for HAE target multiple pathways (i.e., C1-INH replacement therapy), while others target a single pathway (e.g., kallikrein inhibitors). Future research is needed to optimize therapeutic strategies and, in particular, to compare long-term outcomes both for angioedema control and for other consequences of C1-inhibitor deficiency, such as autoimmunity. In the meantime, health care providers and patients should establish an individualized management strategy that considers on-demand treatment and short-term prophylaxis, as well as long-term prophylaxis in appropriate patients, to minimize the disease burden of this condition.
Contribution to the field statement
Hereditary angioedema (HAE) is a rare genetic disease that causes unpredictable, recurrent episodes of skin/mucosal swelling (called attacks) that can affect multiple locations, such as the hands, feet, gastrointestinal tract, other intra-abdominal organs, face, and upper respiratory tract. This condition substantially impairs patient-related quality of life and interferes with their ability to function at home, work or school, or socially. Management of HAE aims to treat or, preferably, prevent attacks, improving quality of life. Swelling during HAE attacks is mediated by bradykinin. Different pathways contribute to bradykinin generation. Almost all cases of HAE are caused by deficiency or dysfunction of C1-esterase inhibitor (C1-INH), which under normal conditions regulates different pathways to inhibit bradykinin production. C1-INH replacement therapy has been used for decades to treat or prevent HAE attacks. While complete disease control with C1-INH replacement is possible, some patients experience breakthrough attacks. More recently available agents target a single pathway—the contact pathway—to prevent bradykinin generation or block the effects of bradykinin. We review the mechanism of action, efficacy, and safety of approved therapies, as well as investigational agents; consider the therapeutic potential of single-pathway treatment options vs. agents with broader effects; and identify gaps in research.
Author contributions
This work was developed from a poster presented at the 12th C1-Inhibitor Deficiency and Angioedema Workshop, June 3–6, 2021 (virtual). Both authors have contributed to the conception, development, drafting, and finalization of the manuscript. Both authors agree to be accountable for the content of the work. All authors contributed to the article and approved the submitted version.
Funding
Support for technical editorial and medical writing assistance was provided by Pharming Group NV, Leiden, the Netherlands. The authors did not receive any compensation for development of this manuscript. Pharming Group NV did not actively contribute to the content or have a role in the decision to submit but reviewed the article for scientific accuracy.
Acknowledgments
We thank Tobias M. Suiter, a former employee of Pharming Group NV, for thought-provoking discussions that inspired us to develop this manuscript. Technical editorial and medical writing assistance was provided, under the direction of the authors, by Synchrony Medical Communications, LLC, West Chester, PA.
Conflict of interest
AV has received consultancy/speaker honoraria/meeting sponsorship from, or collaborated in research with, Pharming Group NV, Takeda/Shire, Sobi, CSL Behring, and Pharvaris. HJL has consulted for, acted as speaker for, or collaborated in research with the following: Adverum, BioCryst, CSL Behring, GSK, Intellia, Ionis, KalVista, Pharming, Pharvaris, and Takeda.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. (2006) 119(3):267–74. doi: 10.1016/j.amjmed.2005.09.064
2. Busse PJ, Christiansen SC, Riedl MA, Banerji A, Bernstein JA, Castaldo AJ, et al. US HAEA Medical Advisory Board 2020 guidelines for the management of hereditary angioedema. J Allergy Clin Immunol Pract. (2021) 9(1):132–50. doi: 10.1016/j.jaip.2020.08.046
3. Longhurst HJ, Bork K. Hereditary angioedema: an update on causes, manifestations and treatment. Br J Hosp Med (Lond). (2019) 80(7):391–8. doi: 10.12968/hmed.2019.80.7.391
4. Bernstein JA. HAE Update: epidemiology and burden of disease. Allergy Asthma Proc. (2013) 34(1):3–6. doi: 10.2500/aap.2013.34.3623
5. Zuraw BL. Hereditary angioedema. N Engl J Med. (2008) 359(10):1027–36. doi: 10.1056/NEJMcp0803977
6. Bork K, Wulff K, Möhl BS, Steinmüller-Magin L, Witzke G, Hardt J, et al. Novel hereditary angioedema linked with a heparan sulfate 3-O-sulfotransferase 6 gene mutation. J Allergy Clin Immunol. (2021) 148(4):1041–8. doi: 10.1016/j.jaci.2021.01.011
7. Santacroce R, D’Andrea G, Maffione AB, Margaglione M, d’Apolito M. The genetics of hereditary angioedema: a review. J Clin Med. (2021) 10(9):2023. doi: 10.3390/jcm10092023
8. Hews-Girard J, Goodyear MD. Psychosocial burden of type 1 and 2 hereditary angioedema: a single-center Canadian cohort study. Allergy Asthma Clin Immunol. (2021) 17(1):61. doi: 10.1186/s13223-021-00563-0
9. Longhurst H, Bygum A. The humanistic, societal, and pharmaco-economic burden of angioedema. Clin Rev Allergy Immunol. (2016) 51(2):230–9. doi: 10.1007/s12016-016-8575-2
10. Lumry WR, Castaldo AJ, Vernon MK, Blaustein MB, Wilson DA, Horn PT. The humanistic burden of hereditary angioedema: impact on health-related quality of life, productivity, and depression. Allergy Asthma Proc. (2010) 31(5):407–14. doi: 10.2500/aap.2010.31.3394
11. Mendivil J, Murphy R, de la Cruz M, Janssen E, Boysen HB, Jain G, et al. Clinical characteristics and burden of illness in patients with hereditary angioedema: findings from a multinational patient survey. Orphanet J Rare Dis. (2021) 16(1):94. doi: 10.1186/s13023-021-01717-4
12. Maurer M, Aygören-Pürsün E, Banerji A, Bernstein JA, Balle Boysen H, Busse PJ, et al. Consensus on treatment goals in hereditary angioedema: a global Delphi initiative. J Allergy Clin Immunol. (2021) 148(6):1526–32. doi: 10.1016/j.jaci.2021.05.016
13. Maurer M, Magerl M, Betschel S, Aberer W, Ansotegui IJ, Aygören-Pürsün E, et al. The international WAO/EAACI guideline for the management of hereditary angioedema—the 2021 revision and update. Allergy. (2022) 77(7):1961–90. doi: 10.1111/all.15214
14. Fijen LM, Bork K, Cohn DM. Current and prospective targets of pharmacologic treatment of hereditary angioedema types 1 and 2. Clin Rev Allergy Immunol. (2021) 61(1):66–76. doi: 10.1007/s12016-021-08832-x
15. de Maat S, Joseph K, Maas C, Kaplan AP. Blood clotting and the pathogenesis of types I and II hereditary angioedema. Clin Rev Allergy Immunol. (2021) 60(3):348–56. doi: 10.1007/s12016-021-08837-6
16. Cicardi M, Zuraw BL. Angioedema due to bradykinin dysregulation. J Allergy Clin Immunol Pract. (2018) 6(4):1132–41. doi: 10.1016/j.jaip.2018.04.022
17. Debreczeni ML, Németh Z, Kajdácsi E, Farkas H, Cervenak L. Molecular dambusters: what is behind hyperpermeability in bradykinin-mediated angioedema? Clin Rev Allergy Immunol. (2021) 60(3):318–47. doi: 10.1007/s12016-021-08851-8
18. Hofman ZL, Relan A, Zeerleder S, Drouet C, Zuraw B, Hack CE. Angioedema attacks in patients with hereditary angioedema: local manifestations of a systemic activation process. J Allergy Clin Immunol. (2016) 138(2):359–66. doi: 10.1016/j.jaci.2016.02.041
19. Dobó J, Major B, Kékesi KA, Szabó I, Megyeri M, Hajela K, et al. Cleavage of kininogen and subsequent bradykinin release by the complement component: mannose-binding lectin-associated serine protease (MASP)-1. PLoS One. (2011) 6(5):e20036. doi: 10.1371/journal.pone.0020036
20. Kaplan AP, Joseph K. Pathogenesis of hereditary angioedema: the role of the bradykinin-forming cascade. Immunol Allergy Clin North Am. (2017) 37(3):513–25. doi: 10.1016/j.iac.2017.04.001
21. Björkman LS, Persson B, Aronsson D, Skattum L, Nordenfelt P, Egesten A. Comorbidities in hereditary angioedema-a population-based cohort study. Clin Transl Allergy. (2022) 12(3):e12135. doi: 10.1002/clt2.12135
22. Perego F, Gidaro A, Zanichelli A, Cancian M, Arcoleo F, Senter R, et al. Life expectancy in Italian patients with hereditary angioedema due to C1-inhibitor deficiency. J Allergy Clin Immunol Pract. (2020) 8(5):1772–4. doi: 10.1016/j.jaip.2020.01.007
23. CSL Behring GmbH. Berinert 2000 IU powder and solvent for solution for injection. Marburg, Germany: CSL Behring GmbH (2020).
24. CSL Behring LLC. Berinert (C1 esterase inhibitor [human]) for intravenous use. Freeze-dried powder for reconstitution. Kankakee, IL: CSL Behring LLC (2021).
25. Takeda Manufacturing Austria AG. Cinryze 500 IU powder and solvent for solution for injection: summary of product characteristics. Vienna, Austria: Takeda Manufacturing Austria AG (2016).
26. Takeda Pharmaceuticals U.S.A., Inc. Cinryze (C1 esterase inhibitor [human]) for intravenous use, freeze-dried powder for reconstitution. Lexington, MA: Takeda Pharmaceuticals U.S.A., Inc. (2021).
27. European Medicines Agency. Ruconest summary of product characteristics. Leiden: The Netherlands: Pharming Group N.V. (2020). [September 1, 2020]. Available from: https://www.ema.europa.eu/en/documents/product-information/ruconest-epar-product-information_en.pdf
28. Pharming Americas BV. Ruconest (C1 esterase inhibitor [recombinant]) for intravenous use, lyophilized powder for reconstitution. Leiden, The Netherlands: Pharming Americas BV (2020).
29. CSL Behring. Haegarda (C1 esterase inhibitor subcutaneous [human]) for subcutaneous injection, freeze-dried powder for reconstitution. Kankakee, IL: CSL Behring (2020).
30. CSL Behring UK Limited. Berinert 500 IU powder and solvent for solution for injection/infusion: summary of product characteristics. West Sussex, UK: CSL Behring UK Limited (2021).
31. Takeda Pharmaceuticals International AG. Firazyr 30 mg solution for injection in pre-filled syringe: summary of product characteristics. Dublin, Ireland: Takeda Pharmaceuticals International AG Ireland Branch (2013).
32. Takeda Pharmaceuticals America, Inc. Firazyr (icatibant) injection, for subcutaneous use. Lexington, MA: Takeda Pharmaceuticals America, Inc (2020).
33. Dyax Corp. Kalbitor (ecallantide) injection, for subcutaneous use. Lexington, MA: Dyax Corp (2020).
34. Takeda Pharmaceuticals International AG. Takhzyro 300 mg solution for injection: summary of product characteristics. Dublin, Ireland: Takeda Pharmaceuticals International AG Ireland Branch (2018).
35. Shire. Takhzyro (lanadelumab-flyo) injection, for subcutaneous use. Lexington, MA: Shire (2018).
36. Millmount Healthcare Limited. Orladeyo 150 mg hard capsules: summary of product characteristics. Meath, Ireland: Millmount Healthcare Limited (2021).
37. Millmount Healthcare Limited. Orladeyo (berotralstat) capsules, for oral use. Durham, NC: BioCryst Pharmaceuticals, Inc. (2020).
38. Craig TJ, Levy RJ, Wasserman RL, Bewtra AK, Hurewitz D, Obtułowicz K, et al. Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks. J Allergy Clin Immunol. (2009) 124(4):801–8. doi: 10.1016/j.jaci.2009.07.017
39. Craig TJ, Bewtra AK, Bahna SL, Hurewitz D, Schneider LC, Levy RJ, et al. C1 esterase inhibitor concentrate in 1085 hereditary angioedema attacks–final results of the I.M.P.A.C.T.2 study. Allergy. (2011) 66(12):1604–11. doi: 10.1111/j.1398-9995.2011.02702.x
40. Zuraw BL, Busse PJ, White M, Jacobs J, Lumry W, Baker J, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. (2010) 363(6):513–22. doi: 10.1056/NEJMoa0805538
41. Zuraw B, Cicardi M, Levy RJ, Nuijens JH, Relan A, Visscher S, et al. Recombinant human C1-inhibitor for the treatment of acute angioedema attacks in patients with hereditary angioedema. J Allergy Clin Immunol. (2010) 126(4):821–7. doi: 10.1016/j.jaci.2010.07.021
42. Hack CE, Relan A, van Amersfoort ES, Cicardi M. Target levels of functional C1-inhibitor in hereditary angioedema. Allergy. (2012) 67(1):123–30. doi: 10.1111/j.1398-9995.2011.02716.x
43. Aygören-Pürsün E, Martinez Saguer I, Kreuz W, Klingebiel T, Schwabe D. Risk of angioedema following invasive or surgical procedures in HAE type I and II–the natural history. Allergy. (2013) 68(8):1034–9. doi: 10.1111/all.12186
44. Singh U, Lumry WR, Busse P, Wedner HJ, Banerji A, Craig TJ, et al. Association between self-reported dental hygiene practices and dental procedure-related recurrent angioedema attacks in HAE subjects: a multicenter survey. J Allergy Clin Immunol Pract. (2020) 8(9):3162–9. doi: 10.1016/j.jaip.2020.05.041
45. Zanichelli A, Ghezzi M, Santicchia I, Vacchini R, Cicardi M, Sparaco A, et al. Short-term prophylaxis in patients with angioedema due to C1-inhibitor deficiency undergoing dental procedures: an observational study. PLoS One. (2020) 15(3):e0230128. doi: 10.1371/journal.pone.0230128
46. Farkas H, Zotter Z, Csuka D, Szabó E, Nébenführer Z, Temesszentandrási G, et al. Short-term prophylaxis in hereditary angioedema due to deficiency of the C1-inhibitor–a long-term survey. Allergy. (2012) 67(12):1586–93. doi: 10.1111/all.12032
47. Magerl M, Frank M, Lumry W, Bernstein J, Busse P, Craig T, et al. Short-term prophylactic use of C1-inhibitor concentrate in hereditary angioedema: findings from an international patient registry. Ann Allergy Asthma Immunol. (2017) 118(1):110–2. doi: 10.1016/j.anai.2016.10.006
48. Valerieva A, Staevska M, Jesenak M, Hrubiskova K, Sobotkova M, Zachova R, et al. Recombinant human C1 esterase inhibitor as short-term prophylaxis in patients with hereditary angioedema. J Allergy Clin Immunol Pract. (2020) 8(2):799–802. doi: 10.1016/j.jaip.2019.08.011
49. Zuraw BL, Kalfus I. Safety and efficacy of prophylactic nanofiltered C1-inhibitor in hereditary angioedema. Am J Med. (2012) 125(9):938.e1–e7. doi: 10.1016/j.amjmed.2012.02.020
50. Longhurst H, Cicardi M, Craig T, Bork K, Grattan C, Baker J, et al. Prevention of hereditary angioedema attacks with a subcutaneous C1 inhibitor. N Engl J Med. (2017) 376(12):1131–40. doi: 10.1056/NEJMoa1613627
51. Craig T, Zuraw B, Longhurst H, Cicardi M, Bork K, Grattan C, et al. Long-term outcomes with subcutaneous C1-inhibitor replacement therapy for prevention of hereditary angioedema attacks. J Allergy Clin Immunol Pract. (2019) 7(6):1793–802.e2. doi: 10.1016/j.jaip.2019.01.054
52. Bork K, Hardt J. Hereditary angioedema: long-term treatment with one or more injections of C1 inhibitor concentrate per week. Int Arch Allergy Immunol. (2011) 154(1):81–8. doi: 10.1159/000319213
53. Craig T, Shapiro R, Vegh A, Baker JW, Bernstein JA, Busse P, et al. Efficacy and safety of an intravenous C1-inhibitor concentrate for long-term prophylaxis in hereditary angioedema. Allergy Rhinol (Providence). (2017) 8(1):e13–e9. doi: 10.2500/ar.2017.8.0192
54. Riedl MA, Grivcheva-Panovska V, Moldovan D, Baker J, Yang WH, Giannetti BM, et al. Recombinant human C1 esterase inhibitor for prophylaxis of hereditary angio-oedema: a phase 2, multicentre, randomised, double-blind, placebo-controlled crossover trial. Lancet. (2017) 390(10102):1595–602. doi: 10.1016/S0140-6736(17)31963-3
55. Zuraw BL, Cicardi M, Longhurst HJ, Bernstein JA, Li HH, Magerl M, et al. Phase II study results of a replacement therapy for hereditary angioedema with subcutaneous C1-inhibitor concentrate. Allergy. (2015) 70(10):1319–28. doi: 10.1111/all.12658
56. Han ED, MacFarlane RC, Mulligan AN, Scafidi J, Davis AE 3rd. Increased vascular permeability in C1 inhibitor–deficient mice mediated by the bradykinin type 2 receptor. J Clin Invest. (2002) 109(8):1057–63. doi: 10.1172/JCI14211
57. Cockcroft JR, Chowienczyk PJ, Brett SE, Bender N, Ritter JM. Inhibition of bradykinin-induced vasodilation in human forearm vasculature by icatibant, a potent B2-receptor antagonist. Br J Clin Pharmacol. (1994) 38(4):317–21. doi: 10.1111/j.1365-2125.1994.tb04360.x
58. Bork K, Frank J, Grundt B, Schlattmann P, Nussberger J, Kreuz W. Treatment of acute edema attacks in hereditary angioedema with a bradykinin receptor-2 antagonist (icatibant). J Allergy Clin Immunol. (2007) 119(6):1497–503. doi: 10.1016/j.jaci.2007.02.012
59. Busse P, Kaplan A. Specific targeting of plasma kallikrein for treatment of hereditary angioedema: a revolutionary decade. J Allergy Clin Immunol Pract. (2022) 10:716–22. doi: 10.1016/j.jaip.2021.11.011
60. Duffey H, Firszt R. Management of acute attacks of hereditary angioedema: role of ecallantide. J Blood Med. (2015) 6:115–23. doi: 10.2147/JBM.S66825
61. European Medicines Agency. Kalbitor: withdrawal of the marketing authorisation application. [updated September 2, 2012March 17, 2022]. Available from: https://www.ema.europa.eu/en/medicines/human/withdrawn-applications/kalbitor#:∼:text=On%2011%20November%202011%2C%20Dyax,acute%20attacks%20of%20hereditary%20angioedema
62. Shire plc. Shire receives approval of Firazyr (icatibant injection) for the treatment of hereditary angioedema (HAE) attacks in Japan [press release]. (2018) [cited 2022 July 26]. Available from: https://www.globenewswire.com/news-release/2018/09/21/1574147/0/en/Shire-Receives-Approval-of-FIRAZYR-icatibant-injection-for-the-Treatment-of-Hereditary-Angioedema-HAE-Attacks-in-Japan.html
63. Zuraw B, Lumry WR, Johnston DT, Aygören-Pürsün E, Banerji A, Bernstein JA, et al. Oral once-daily berotralstat for the prevention of hereditary angioedema attacks: a randomized, double-blind, placebo-controlled phase 3 trial. J Allergy Clin Immunol. (2021) 148(1):164–72.e9. doi: 10.1016/j.jaci.2020.10.015
64. Cicardi M, Banerji A, Bracho F, Malbrán A, Rosenkranz B, Riedl M, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med. (2010) 363(6):532–41. doi: 10.1056/NEJMoa0906393
65. Lumry WR, Li HH, Levy RJ, Potter PC, Farkas H, Moldovan D, et al. Randomized placebo-controlled trial of the bradykinin B2 receptor antagonist icatibant for the treatment of acute attacks of hereditary angioedema: the FAST-3 trial. Ann Allergy Asthma Immunol. (2011) 107(6):529–37. doi: 10.1016/j.anai.2011.08.015
66. Cicardi M, Levy RJ, McNeil DL, Li HH, Sheffer AL, Campion M, et al. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. (2010) 363(6):523–31. doi: 10.1056/NEJMoa0905079
67. Levy RJ, Lumry WR, McNeil DL, Li HH, Campion M, Horn PT, et al. EDEMA4: a phase 3, double-blind study of subcutaneous ecallantide treatment for acute attacks of hereditary angioedema. Ann Allergy Asthma Immunol. (2010) 104(6):523–9. doi: 10.1016/j.anai.2010.04.012
68. Banerji A, Riedl MA, Bernstein JA, Cicardi M, Longhurst HJ, Zuraw BL, et al. Effect of lanadelumab compared with placebo on prevention of hereditary angioedema attacks: a randomized clinical trial. JAMA. (2018) 320(20):2108–21. doi: 10.1001/jama.2018.16773
69. Banerji A, Bernstein JA, Johnston DT, Lumry WR, Magerl M, Maurer M, et al. Long-term prevention of hereditary angioedema attacks with lanadelumab: the HELP OLE study. Allergy. (2022) 77(3):979–90. doi: 10.1111/all.15011
70. Wedner HJ, Aygören-Pürsün E, Bernstein J, Craig T, Gower R, Jacobs JS, et al. Randomized trial of the efficacy and safety of berotralstat (BCX7353) as an oral prophylactic therapy for hereditary angioedema: results of APeX-2 through 48 weeks (part 2). J Allergy Clin Immunol Pract. (2021) 9(6):2305–14.e4. doi: 10.1016/j.jaip.2021.03.057
71. Craig TJ, Li HH, Riedl M, Bernstein JA, Lumry WR, MacGinnitie AJ, et al. Characterization of anaphylaxis after ecallantide treatment of hereditary angioedema attacks. J Allergy Clin Immunol Pract. (2015) 3(2):206–12 e4. doi: 10.1016/j.jaip.2014.09.001
72. Cao H, Biondo M, Lioe H, Busfield S, Rayzman V, Nieswandt B, et al. Antibody-mediated inhibition of FXIIa blocks downstream bradykinin generation. J Allergy Clin Immunol. (2018) 142(4):1355–8. doi: 10.1016/j.jaci.2018.06.014
73. Craig T, Magerl M, Levy DS, Reshef A, Lumry WR, Martinez-Saguer I, et al. Prophylactic use of an anti-activated factor XII monoclonal antibody, garadacimab, for patients with C1-esterase inhibitor-deficient hereditary angioedema: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. (2022) 399:945–55. doi: 10.1016/s0140-6736(21)02225-x
74. Ferrone JD, Bhattacharjee G, Revenko AS, Zanardi TA, Warren MS, Derosier FJ, et al. IONIS-PKKRx a novel antisense inhibitor of prekallikrein and bradykinin production. Nucleic Acid Ther. (2019) 29(2):82–91. doi: 10.1089/nat.2018.0754
75. Cohn DM, Viney NJ, Fijen LM, Schneider E, Alexander VJ, Xia S, et al. Antisense inhibition of prekallikrein to control hereditary angioedema. N Engl J Med. (2020) 383(13):1242–7. doi: 10.1056/NEJMoa1915035
76. Fijen LM, Riedl MA, Bordone L, Bernstein JA, Raasch J, Tachdjian R, et al. Inhibition of prekallikrein for hereditary angioedema. N Engl J Med. (2022) 386(11):1026–33. doi: 10.1056/NEJMoa2109329
77. Zuraw BL. HAE Therapies: past present and future. Allergy Asthma Clin Immunol. (2010) 6(1):23. doi: 10.1186/1710-1492-6-23
78. Riedl MA, Banerji A, Gower R. Current medical management of hereditary angioedema: follow-up survey of US physicians. Ann Allergy Asthma Immunol. (2021) 126(3):264–72. doi: 10.1016/j.anai.2020.10.009
Keywords: bradykinin B2 receptor antagonist, complement C1 inhibitor protein, hereditary angioedema, kallikreins, prophylaxis
Citation: Valerieva A and Longhurst HJ (2022) Treatment of hereditary angioedema—single or multiple pathways to the rescue. Front. Allergy 3:952233. doi: 10.3389/falgy.2022.952233
Received: 24 May 2022; Accepted: 18 August 2022;
Published: 12 September 2022.
Edited by:
Richard L. Wasserman, Medical City Children’s Hospital, United StatesReviewed by:
Maureen Egan Bauer, Children’s Hospital Colorado, United StatesTakahiko Horiuchi, Kyushu University, Japan
© 2022 Valerieva and Longhurst. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Valerieva YW5uYS52YWxlcmlldmFAZ21haWwuY29t
†These authors have contributed equally to this work and share first authorship.
Specialty Section: This article was submitted to Skin Allergy, a section of the journal Frontiers in Allergy
Abbreviations AE, adverse event; B1, bradykinin B1; B2R, bradykinin B2 receptor; bw, body weight; CI, confidence interval; C1-INH; C1 esterase inhibitor; HAE, hereditary angioedema; EAACI, European Academy of Allergy and Clinical Immunology; FAST, For Angioedema Subcutaneous Treatment; HAE-nl-C1INH, hereditary angioedema with normal C1-INH; HMW, high molecular weight; IL, interleukin; IMPACT, International Multicenter Prospective Angioedema C1-INH Trial; IQR, interquartile range; IV, intravenous; LTP, long-term prophylaxis; MASP, mannose-binding lectin–associated serine proteases; OR, odds ratio; pdC1-INH, plasma-derived C1 esterase inhibitor; rhC1-INH, recombinant human C1 esterase inhibitor; SC, subcutaneous; SAE, serious adverse event; SE, standard error; WAO, World Allergy Organization.