 Toshiaki Kawakami
Toshiaki Kawakami Kazumi Kasakura1†
Kazumi Kasakura1†- 1Laboratory of Allergic Diseases, Center for Autoimmunity and Inflammation, La Jolla Institute for Immunology, La Jolla, CA, United States
- 2Department of Dermatology, School of Medicine, University of California, San Diego, La Jolla, CA, United States
- 3Atopy (Allergy) Research Center, Juntendo University Graduate School of Medicine, Tokyo, Japan
Acute exacerbation is the major cause of asthma morbidity, mortality, and health-care costs. Respiratory viral infections, particularly rhinovirus (RV) infections, are associated with the majority of asthma exacerbations. The risk for bronchoconstriction with RV is associated with allergic sensitization and type 2 airway inflammation. The efficacy of the humanized anti-IgE monoclonal antibody omalizumab in treating asthma and reducing the frequency and severity of RV-induced asthma exacerbation is well-known. Despite these clinical data, mechanistic details of omalizumab's effects on RV-induced asthma exacerbation have not been well-defined for years due to the lack of appropriate animal models. In this Perspective, we discuss potential IgE-dependent roles of mast cells and dendritic cells in asthma exacerbations.
Introduction
Asthma is a T helper cell 2 (Th2) cell-driven chronic inflammatory lung disease, characterized by airway inflammation, airway hyperresponsiveness (AHR), airway remodeling, and reversible airway obstruction (1, 2). The prevalence of asthma has been increasing for the last several decades (3). Five-10% of the patients have severe asthma, which is often difficult to treat (4). No treatment is curative, and existing drugs are often ineffective in controlling symptoms (5, 6). RV infection is associated with the majority of asthma exacerbations (7–12), whereas respiratory syncytial virus (RSV) is more associated with non-allergic asthma (11, 13). The risk for wheezing with RV is associated with allergic sensitization (e.g., HDM-specific IgE) and type 2 airway inflammation (11–17). Some, but not all, studies (18–21) suggest that impaired type I and III interferon (IFN) responses to RV infection may contribute to asthma exacerbation. RV infection also induces production of IL-25 (22), IL-33 (23), thymic stromal lymphopoietin (TSLP) (24), and granulocyte macrophage colony-stimulating factor (GM-CSF) (25) in lung epithelial cells, promoting type 2 airway inflammation (26–29). Clinical evidence supports the pathogenic role for IgE and mast cells in asthma and RV-induced asthma exacerbation: mast cells are increased in the airway epithelium (30) and within the smooth muscle layer (31–33) in allergic asthma, and in the alveolar parenchyma of uncontrolled allergic asthma (34). An increased percentage of degranulated mast cells are found in the mucous glands from fatal asthma (35). Unlike the Th2-low group, the Th2-high group of asthmatics highly express Th2 (IL4, IL5, IL13; IL-13-regulated genes, POSTN, CLCA1, SERPINB2) and mast cell (TPSB2, TPSAB1, and CPA3) genes in the airway epithelium (36–38), consistent with the recruitment of mast cells to the airway epithelium from the submucosa (39, 40). Mast cells are also recruited to the bronchial epithelium following RV infection (41). RV can replicate in mast cells and induce their activation (42–44). Importantly, omalizumab is indicated for moderate-to-severe asthma (45–48) and reduces RV-induced asthma exacerbation (47, 49, 50).
Despite these data supporting the pathogenic role for IgE and mast cells in asthma, the study of this topic has a twisted history. Studies in 1990s raised mixed results, with some supporting their role (51, 52), but others revealing the lack of their role in in vivo asthma models (51, 53–61). This confusion was resolved by two studies published in year 2000 showing that the contribution of mast cells to airway inflammation can be seen in mice sensitized with small amounts of a conveniently available antigen [i.e., ovalbumin (OVA)] without the strong Th2-skewing adjuvant alum (62, 63). However, the situation became murky again when clinically relevant allergens such as house dust mite (HDM) were increasingly used. Numerous studies have been conducted on HDM-induced airway inflammation in murine models. While most studies failed to mention on the role of IgE or mast cells (64–79), a recent study showed that HDM-induced airway inflammation is not dependent on IgE or FcεRI (80). In this Perspective, we will overview fundamental aspects of IgE, IgE receptors, and mechanisms of anti-IgE mAb's function. Then, we will describe cell types that are targeted by anti-IgE mAb-mediated protection against asthma exacerbations. Finally, we will discuss animal models of asthma and RV-induced asthma exacerbation, which will potentially solve the current enigma of how RV induces asthma exacerbations. Our discussion of IgE-related studies will be limited to those required for understanding the topic. Those who want to have an updated deeper understanding are referred to recent excellent reviews (81–87).
IgE, IgE Receptors, and Omalizumab
IgE is the least abundant immunoglobulin in serum. Multiple mechanisms control IgE levels from its synthesis to degradation: low efficiency of class-switch recombination to IgE, lower surface expression of membrane IgE (mIgE) than of mIgG1 on germinal center (GC) B cells, and increased apoptosis of IgE+ GC B cells. Recent studies on several reporter mice and GC reaction-mimicking B cell cultures (iGB cells) on 40LB feeder cells showed that IgE-producing B cells swiftly exit GCs and differentiate into plasma cells (PCs) and that IgE-producing GC cells die by apoptosis (88–91). A recent study found that IgE-BCR without antigen stimulation induces PI3K-mediated mTOR activation that increases IRF4 protein (not transcription), leading to IgE+ PC differentiation, and that chronic calcium signaling in IgE+ B cells and PCs culminates in apoptosis (92). Therefore, IgE+ memory B cells and IgE+ PCs are scarce. High-affinity IgE is produced by consecutive class-switch recombinations from IgM to IgG to IgE, whereas low-affinity IgE is produced by a direct switch from IgM to IgE (93). IgE production is induced by IL-4 and IL-13 (94, 95). T follicular helper (Tfh) cell-derived IL-4 is necessary for IgE production (96). Recently, a rare population of IL-13-producing Tfh cells (Tfh13) was shown to be required for production of high-affinity IgE (97). The half-life of infused IgE in the serum is very short (2 days) compared to IgG (18–23 days) (98). Several mechanisms likely contribute to the short half-life of IgE: rapid removal of free IgE from the circulation by binding to mast cells and basophils; degradation of IgE by extravascular and membrane-bound proteases; binding of IgE glycans to lectins leading to delivery for degradation; receptor-mediated endocytosis; and digestion of IgE in endolysosomes caused by lack of protection by FcRn.
There are two IgE receptors, the high-affinity receptor FcεRI and the low-affinity receptor CD23 (FcεRII). FcεRI expressed on mast cells and basophils is a heterotetramer of three subunits, an IgE-binding α subunit, a receptor-stabilizing and signal-amplifying β subunit, and signal-initiating disulfide-linked two γ subunits (99). IgE binds FcεRI at 1:1. While FcεRI expression is limited to mast cells and basophils in mice under homeostatic conditions, FcεRI in humans is additionally expressed by eosinophils, DCs and Langerhans cells. FcεRI is also expressed by bronchial epithelial cells in some asthmatics (100). The non-mast cell/basophil FcεRI consists of αγ2 heterotrimers. It has been known for long that levels of FcεRI expression on the cell surface correlates well-with serum levels of IgE (101, 102). This seems to be due to stabilization of FcεRI by bound IgE (103, 104). Thus, reduction in IgE levels (e.g., by omalizumab) will reduce the cell surface expression of FcεRI (105). Binding of a multivalent antigen to the mast cell and basophil surface IgE induces aggregation or crosslinking of FcεRI that triggers the activation of complex signaling events, eventually resulting in degranulation, eicosanoid synthesis and release, and cytokine production and secretion. These events cause allergic reactions ranging from local redness and itch to lethal systemic anaphylaxis.
CD23 is a type 2 transmembrane protein that forms a homotrimer composed of an IgE-binding C-type lectin head, a long α-helical coiled-coil stalk, a transmembrane domain and an N-terminal short cytoplasmic portion. CD23 is expressed on B cells, monocytes, DCs, Langerhans cells, eosinophils, and respiratory and gastrointestinal epithelial cells (106). Antigen presentation by CD23-bound IgE is known as IgE-mediated facilitated antigen presentation that amplifies Th2 responses upon re-exposure to the same antigen. In this phenomenon, IgE-antigen complexes are transported to the spleen by recirculating CD23+ B cells where they are delivered to CD8α− conventional DCs (cDCs) which induce proliferation of CD4+ T cells (107). CD23 expression by B cells is involved in regulation of IgE synthesis: engagement of CD23 with IgE suppresses IgE production and CD23-deficient mice exhibit stronger and long-lasting IgE response upon immunization (108). Conversely, CD23 transgenic mice exhibit decreased IgE production (109, 110). CD23-blocking mAb lumiliximab lowers IgE levels in humans (111). In contrast, soluble CD23 fragments promote IgE synthesis. Although CD23 plays a role in OVA-induced airway inflammation (112) by transcytosis of IgE immune complexes by lung epithelial cell CD23 (113), little is known about the role of CD23 in asthma exacerbations.
Omalizumab is a humanized IgG1κ monoclonal antibody (mAb) that binds to free human IgE and to membrane-bound form of IgE (mIgE) on the surface of B cells (114). Omalizumab inhibits IgE interactions with FcεRI and CD23 (115). As it binds to free IgE, omalizumab lowers free IgE levels and thus downregulates FcεRI levels on basophils and mast cells, limiting the degree of release of allergic mediators. Owing to these properties, omalizumab is indicated for the treatment of moderate-to-severe asthma (116), allergic rhinitis (117), and chronic idiopathic/spontaneous urticaria (CIU/CSU) (118–120). However, a newer anti-IgE mAb ligelizumab (121) failed to show its efficacy in the treatment of severe asthma (122), while it was more effective than omalizumab in the treatment of CIU/CSU (123). It was speculated that the differential efficiencies of omalizumab and ligelizumab in inhibiting FcεRI and CD23 bindings may contribute to the difference in the efficacies on severe asthma, although it requires further investigation.
Dendritic Cells
Children with severe asthma are susceptible to respiratory virus-induced asthma exacerbations, particularly those with high serum IgE levels (16, 124). IFNs secreted by plasmacytoid DCs (pDCs) are essential for the host defense to viral infections. pDCs express FcεRI and its expression is controlled by IgE levels. Enhanced FcεRI expression in asthma inhibits virus-induced IFN-α and IFN-λ1 responses of human pDCs (19, 125, 126). Omalizumab treatment of asthmatics reduces FcεRI expression on the pDC surface and increases RV-induced pDC IFN-α responses (19, 125–127). Studies demonstrated a counterregulatory mechanism between FcεRIα and TLR7, by which expression of these proteins is inversely proportional (19, 125). These observations can potentially explain the PROSE data that preventive administration of omalizumab could dampen the seasonal increase in asthma exacerbations among school children (50). Another DC subset might be involved in respiratory virus-induced asthma exacerbations. Sendai virus (SeV) infection in mice results in cysteinyl leukotrienes-induced recruitment and survival of CD49d+ neutrophils and subsequent enhancement of FcεRI expression on lung cDCs. Meanwhile anti-SeV IgE antibodies are produced, and these IgE antibodies stimulate the lung cDCs to produce CCL28 via FcεRI. CCL28 recruits IL-13-producing Th2 and type 2 innate lymphoid cells (ILC2) (11, 128). Thus, cDCs might also be targeted by omalizumab.
The clinical observations described above, particularly efficacy of omalizumab in reducing asthma exacerbations (49, 50, 116, 127, 129), strongly support the pathogenic role of IgE-bound mast cells, pDCs, and cDCs to RV-induced asthma exacerbation (Figure 1).
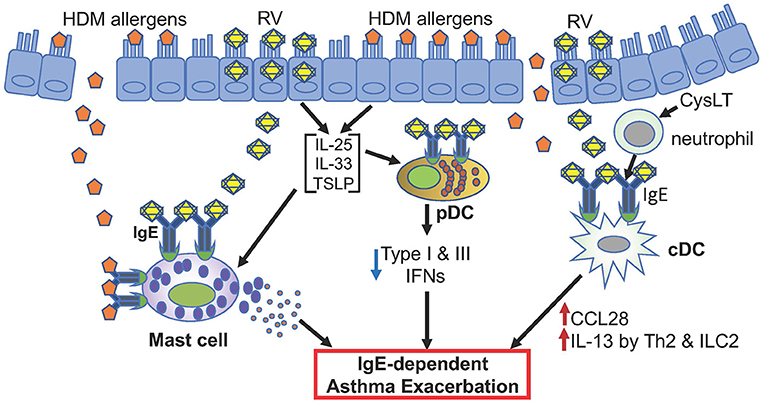
Figure 1. Hypothetical model of RV-induced asthma exacerbation. Only potential events involving IgE and FcεRI are depicted. Allergens such as HDM and rhinovirus antigens likely activate mast cells. pDCs highly expressing FcεRI in asthmatics may produce reduced levels of type I and type III IFNs when infected with rhinovirus. cDCs with high FcεRI expression may produce CCL28 that recruits IL-13-producing Th2 and ILC2 cells. These different cells likely contribute to RV-induced asthma exacerbation.
Mast Cells and IgE in HDM-Induced Models of Asthma
Early studies of animal models of asthma mostly used chicken OVA as a model allergen. More recently, numerous studies used HDM for sensitization and challenge purposes, as up to 90% of asthmatics are sensitized with HDM. When mice were repeatedly exposed to intranasal injections of a single allergen such as HDM, ragweed and Aspergillus, tolerance was induced (66). In contrast, sensitization to double and triple allergens broke through tolerance and caused AHR, eosinophilic inflammation, mast cell and smooth muscle hyperplasia, mucus production, and airway remodeling. Mucosal exposure to triple allergens in the absence of an adjuvant induced chronic airway inflammation. Anti-IL-5 and anti-IL-13 antibodies inhibited inflammation and AHR in the acute asthma model but not in the chronic triple-allergen model (66). A similar procedure of intranasal sensitization/challenge with the above triple allergens showed that airway epithelial cell-derived colony stimulating factor (CSF1/M-CSF) had a critical role in the production of allergen specific-IgE and regulated the recruitment of alveolar DCs and enhanced the migration of cDC2s to the draining lymph node, leading to Th2 cell-mediated allergic lung inflammation (69).
In typical airway inflammation experiments using HDM, mice are sensitized with a commercial HDM via the intranasal or intratracheal route and then challenged with the same allergen via the same route. One day or a few days after the last allergen challenge, bronchoalveolar lavage fluids (BALF) are collected. Leukocytes (e.g., increased eosinophils, neutrophils, macrophages, lymphocytes) and cytokines (e.g., increased mRNAs for and/or proteins of IL-4, IL-5, IL-13) in BALF and lungs are reported together with total or allergen-specific IgE and IgG and lung functions (usually showing methacholine-induced increase in lung resistance). Remodeling such as goblet cell metaplasia is also reported. Acute and chronic disease-mimicking procedures were developed. Involvement of specific molecules or cells in these airway inflammation models has been defined using pharmacological and immunological reagents as well as genetically engineered mice lacking these molecules or cell types. For example, antibodies against GM-CSF (64) or chemical antagonists against TLR4 (67) reduced Th2 phenotype and AHR. Using bone marrow chimeras, Hammad et al. showed that TLR4 expression on radioresistant lung structural cells, but not on DCs, is necessary and sufficient for DC activation in the lung and for priming of effector T helper responses to HDM (67). TLR4 triggering on structural cells caused production of the innate proallergic cytokines TSLP, GM-CSF, IL-25 and IL-33.
Intranasal administration of a high dose of HDM for 5 days/week over 3 weeks induced allergic airway inflammation with mast cell expansion and HDM-induced bronchoconstriction, which was abrogated in mast cell-deficient KitW−sh/W−sh mice (130). In another HDM model, WT mice showed allergic airway inflammation with increased tryptase in BALF, but KitW−sh/W−sh mice showed a selective impairment with reduced plasma IgE levels and BAL eosinophils (131). These KitW−sh/W−sh mice showing reduced AHR and inflammation compared to WT mice have the C57BL/6 genetic background. However, BALB/c-KitW−sh/W−sh mice showed as robust HDM-induced airway inflammatory phenotypes as did WT BALB/c mice (132). Allergic airway inflammation induced by repetitive HDM treatments was also reduced by cromoglygate, a mast cell stabilizer, before each HDM challenge (133). Interestingly, exacerbated AHR was induced by HDM in KitW−sh/W−sh mice engrafted with ST2-deficient mast cells, compared to KitW−sh/W−sh mice engrafted with WT mast cells (134), probably due to reduced prostaglandin E2 (PGE2) levels, which can inhibit IgE-mediated mast cell activation (135). IL-33/ST2-dependent mast cell induction of PGE2 could be responsible for the dampening effect on AHR (134). As researchers were concerned with abnormalities beyond mast cell deficiency in Kit mutant mast cell-deficient mice (e.g., KitW/Wv and KitW−sh/W−sh mice) and abnormal reconstitution of these mice with adoptively transferred mast cells (136, 137), non-Kit mutant mice have been used more often. Indeed, HDM-induced AHR was dependent on mast cells as demonstrated using KitW−sh/W−sh and Mas-TRECK mice (72).
Using wild-type and mutant BALB/c and FVB/N mice, McKnight et al. showed that neither IgE nor FcεRIα contributed to allergic airway disease, even in mice inoculated with the lowest dose of HDM, which readily induced detectable disease, but did not increase serum IgE or pulmonary mast cell levels (80). In contrast, high doses of HDM strikingly increased serum IgE and pulmonary mast cells, although both AHR and airway mast cell degranulation were equally elevated in wild-type and IgE-deficient mice. Thus, they concluded that IgE and FcεRIα-independent mechanisms are responsible for AHR and airway inflammation when low doses of HDM are inhaled repetitively. All the above HDM-induced airway inflammation data were obtained using soluble preparations of HDM. However, exacerbation of allergic asthma is also associated with an increase in ambient inhalable particulate matters (e.g., PM2.5 and PM10) from air pollutants. Furthermore, experimental allergic airway inflammation and/or AHR can be enhanced by particulate matters in an IL-33- or IL-1β-dependent manner (138–141). Importantly, Jin et al. compared AHR and pulmonary eosinophilia induced by soluble vs. particulate antigens (sAg vs. pAg) including HDM-conjugated polystyrene beads (142). They found that, compared with sAgs, pAgs triggered markedly heightened AHR and pulmonary eosinophilia in antigen-sensitized mice in a mast cell-dependent manner. pAgs mediated mast cell-dependent responses by enhancing retention of pAg/IgE/FcεRI complexes within lipid raft-enriched, CD63+ endocytic compartments, which prolonged IgE/FcεRI-triggered signaling and resulted in heightened cytokine responses. Animal models using particulate HDM are highly desirable to address the potential role of IgE/mast cells to mimic HDM-induced asthma.
Animal Models of Rhinovirus-Induced Asthma Exacerbation
RV is a member of the enterovirus genus in the Picornaviridae family of small, non-enveloped positive strand RNA viruses (143). There are three human RV genotypes (A, B, C). Ninety percent of RV-A and RV-B use human intercellular adhesion molecule-1 (ICAM-1) as their receptor (144), while the minor group uses the human and mouse proteins of low-density lipoprotein receptor family (145). RV-C viruses use cadherin-related family member 3 (CDHR3) as their receptor (146). RV-C and RV-A cause severe respiratory illness more often than RV-B (147). RV-induced lower respiratory illness is increased in asthmatics and correlates with virus load, augmented Th2, and/or impaired Th1 and IL-10 immunity (148). RV-1B (a member of the minor receptor group of RV-A) infection of BALB/c mice or RV-16 (RV-A, a member of the major receptor group) infection of mouse-human ICAM-1 transgenic mice induces viral replication in airway epithelial cells, airway inflammation (neutrophilia and lymphocytosis), mucin secretion, and increased IFNs response, similarly to human RV infection (149). Compared to RV-A (RV-1B) infection, RV-C (RV-C15) infection induced higher BALF eosinophilia, mRNA expression of IL-5, IL-13, IL-25, Muc5ac and Gob5/Clca, protein production of IL-5, IL-13, IL-25, IL-33 and TSLP, and expansion of ILC2 in naive and HDM-immunized BALB/c mice (150). Eosinophilic inflammation and mRNA expression of IL-13, Muc5ac and Muc5b were ILC2-dependent. It is well-known that RV infection causes increased IgE levels (151) and RV-specific IgE (152). However, unlike RSV infection (153), no studies have tested if RV-specific IgE could sensitize mast cells for RV antigen-mediated activation and if RV causes IgE-dependent airway inflammation and AHR in vivo. Such experiments remain to be conducted.
Synergistic interactions between RV infection and allergen sensitization and exposure increase the risk of asthma exacerbations (154, 155). Toussaint et al. found that experimental infection of asthmatics with RV-16 induces double-stranded DNA (dsDNA) release in nasal lavages, which is correlated with type 2 immune-mediated asthma exacerbation severity (156). Then, by comparing immune responses between PBS-sensitized/HDM-challenged and HDM-sensitized/HDM-challenged mice infected with live or UV-inactivated RV-1B 1 day after the last (and the second) HDM challenge, they found that live RV-induced exacerbation is associated with a more robust type 2 immune response (increased eosinophils, lymphocytes in BALF); higher serum IgE; enhanced airway inflammatory cells and mucus production; AHR; increased Th2 cells in lung; stronger production of Th2 cytokines from lung and mediastinal lymph node cells; increased recruitment of monocyte-derived DCs to lung and mediastinal lymph nodes and cDC2 to lung and higher dsDNA release in the airways than the other groups (156). These responses were reduced by DNase treatment and mostly recapitulated by exogenous dsDNA in place of RV infection. DNase treatment reduced the recruitment of monocyte-derived DCs to lung and mediastinal lymph nodes. dsDNA releases induced by RV-16 in human patients and by RV-1B in the mouse model were part of neutrophil extracellular traps (NETs). The pathogenic role of NETosis in type 2 inflammation was revealed by neutrophil depletion and NET inhibitor.
Discussion
Animal experiments have been the mainstay for mechanistic analysis of immune responses and disease pathogenesis, despite a wide recognition of differences between human and rodent immune systems (157–159). There is a discrepancy in clinical data showing omalizumab's efficacy in asthma and animal models lacking the effect of IgE or FcεRI deficiency. Clinical data indicate the presence of Th2-high and Th2-low asthmatic patients. Theoretically, Th2-high group may respond well to omalizumab. These different groups might be represented by animal models requiring IgE/FcεRI or not. However, things are not that simple. The discrepancy could be due to inappropriate animal models or simply lack of testing the role of IgE/FcεRI and mast cells. Most studies used a short course of less than a month. Careful analysis of acute vs. chronic models may reveal subtle differences in cellular and molecular requirements. Experiments with particulate HDM preparations or soluble HDM plus PM2.5 may be worth to test. Differences in HDM constituents between different lots could be a potential source of different results, as we experienced more than 10-fold differences in protein content per weight. Moreover, drastic differences in microbiota contained in HDM sources were recently noticed between different vendors (160). Thus, standardization of HDM preparations or use of single or combination of pure allergens is highly desirable. Incorporation of RV infection into HDM-induced airway inflammation adds another complexity. No systematic comparison between different RV types has been reported in airway disease or asthma exacerbation models, as experiments using RV-C has only recently begun. Since proliferation of human RV in mouse cells is severely limited, it may be useful to use a mouse cell-adapted RV variant (161). The landmark study by Toussaint et al. established an RV-induced asthma exacerbation model in HDM-allergic mice. Application or modification of this model to test effects of other RV species, mutant mice, and immunological/pharmacological agents will likely provide new insights into RV-induced asthma exacerbation in the future.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author Contributions
TK and YK wrote an initial manuscript. KK and TA edited it. All authors agree with the final version of the manuscript, contributed to the article, and approved the submitted version.
Funding
Work in the TK lab is supported by the US National Institutes of Health grants R01 AI146042, R21 AI153867, UMI AI109565. TA is supported by the Ministry of Education, Culture, Sports, Science and Technology, Japan (20K08808) and by a Grant-in-Aid for Special Research in Subsidies for ordinary expenses of private schools from the Promotion and Mutual Aid Corporation for Private Schools of Japan.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Holgate ST. Asthma: a simple concept but in reality a complex disease. Eur J Clin Invest. (2011) 41:1339–52. doi: 10.1111/j.1365-2362.2011.02534.x
2. Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. (2012) 18:716–25. doi: 10.1038/nm.2678
3. Eder W, Ege MJ, von Mutius E. The asthma epidemic. N Engl J Med. (2006) 355:2226–35. doi: 10.1056/NEJMra054308
4. Holgate ST, Polosa R. The mechanisms, diagnosis, and management of severe asthma in adults. Lancet. (2006) 368:780–93. doi: 10.1016/S0140-6736(06)69288-X
5. Holgate ST, Wenzel S, Postma DS, Weiss ST, Renz H, Sly PD. Asthma. Nat Rev Dis Primers. (2015) 1:15025. doi: 10.1038/nrdp.2015.25
6. Castillo JR, Peters SP, Busse WW. Asthma exacerbations: pathogenesis, prevention, and treatment. J Allergy Clin Immunol Pract. (2017) 5:918–27. doi: 10.1016/j.jaip.2017.05.001
7. Bizzintino J, Lee WM, Laing IA, Vang F, Pappas T, Zhang G, et al. Association between human rhinovirus C and severity of acute asthma in children. Eur Respir J. (2011) 37:1037–42. doi: 10.1183/09031936.00092410
8. Gern JE. How rhinovirus infections cause exacerbations of asthma. Clin Exp Allergy. (2015) 45:32–42. doi: 10.1111/cea.12428
9. Jackson DJ, Johnston SL. The role of viruses in acute exacerbations of asthma. J Allergy Clin Immunol. (2010) 125:1178–87; quiz 88–9. doi: 10.1016/j.jaci.2010.04.021
10. Khetsuriani N, Kazerouni NN, Erdman DD, Lu X, Redd SC, Anderson LJ, et al. Prevalence of viral respiratory tract infections in children with asthma. J Allergy Clin Immunol. (2007) 119:314–21. doi: 10.1016/j.jaci.2006.08.041
11. Mikhail I, Grayson MH. Asthma and viral infections: an intricate relationship. Ann Allergy Asthma Immunol. (2019) 123:352–8. doi: 10.1016/j.anai.2019.06.020
12. Kennedy JL, Pham S, Borish L. Rhinovirus and asthma exacerbations. Immunol Allergy Clin North Am. (2019) 39:335–44. doi: 10.1016/j.iac.2019.03.003
13. Jartti T, Gern JE. Role of viral infections in the development and exacerbation of asthma in children. The J Allergy Clin Immunol. (2017) 140:895–906. doi: 10.1016/j.jaci.2017.08.003
14. Friedlander SL, Busse WW. The role of rhinovirus in asthma exacerbations. J Allergy Clin Immunol. (2005) 116:267–73. doi: 10.1016/j.jaci.2005.06.003
15. Heymann PW, Kennedy JL. Rhinovirus-induced asthma exacerbations during childhood: the importance of understanding the atopic status of the host. J Allergy Clin Immunol. (2012) 130:1315–6. doi: 10.1016/j.jaci.2012.10.024
16. Kantor DB, Stenquist N, McDonald MC, Schultz BJ, Hauptman M, Smallwood CD, et al. Rhinovirus and serum IgE are associated with acute asthma exacerbation severity in children. J Allergy Clin Immunol. (2016) 138:1467–71 e9. doi: 10.1016/j.jaci.2016.04.044
17. Soto-Quiros M, Avila L, Platts-Mills TA, Hunt JF, Erdman DD, Carper H, et al. High titers of IgE antibody to dust mite allergen and risk for wheezing among asthmatic children infected with rhinovirus. J Allergy Clin Immunol. (2012) 129:1499–505 e5. doi: 10.1016/j.jaci.2012.03.040
18. Contoli M, Message SD, Laza-Stanca V, Edwards MR, Wark PA, Bartlett NW, et al. Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med. (2006) 12:1023–6. doi: 10.1038/nm1462
19. Durrani SR, Montville DJ, Pratt AS, Sahu S, DeVries MK, Rajamanickam V, et al. Innate immune responses to rhinovirus are reduced by the high-affinity IgE receptor in allergic asthmatic children. J Allergy Clin Immunol. (2012) 130:489–95. doi: 10.1016/j.jaci.2012.05.023
20. Edwards MR, Strong K, Cameron A, Walton RP, Jackson DJ, Johnston SL. Viral infections in allergy and immunology: how allergic inflammation influences viral infections and illness. J Allergy Clin Immunol. (2017) 140:909–20. doi: 10.1016/j.jaci.2017.07.025
21. Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. (2005) 201:937–47. doi: 10.1084/jem.20041901
22. Beale J, Jayaraman A, Jackson DJ, Macintyre JDR, Edwards MR, Walton RP, et al. Rhinovirus-induced IL-25 in asthma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci Transl Med. (2014) 6:256ra134. doi: 10.1126/scitranslmed.3009124
23. Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med. (2014) 190:1373–82. doi: 10.1164/rccm.201406-1039OC
24. Kato A, Favoreto Jr S, Avila PC, Schleimer RP. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J Immunol. (2007) 179:1080–7. doi: 10.4049/jimmunol.179.2.1080
25. Sanders SP, Kim J, Connolly KR, Porter JD, Siekierski ES, Proud D. Nitric oxide inhibits rhinovirus-induced granulocyte macrophage colony-stimulating factor production in bronchial epithelial cells. Am J Respir Cell Mol Biol. (2001) 24:317–25. doi: 10.1165/ajrcmb.24.3.4131
26. Divekar R, Kita H. Recent advances in epithelium-derived cytokines (IL-33, IL-25, and thymic stromal lymphopoietin) and allergic inflammation. Curr Opin Allergy Clin Immunol. (2015) 15:98–103. doi: 10.1097/ACI.0000000000000133
27. Roan F, Obata-Ninomiya K, Ziegler SF. Epithelial cell-derived cytokines: more than just signaling the alarm. J Clin Invest. (2019) 129:1441–51. doi: 10.1172/JCI124606
28. Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev. (2008) 226:172–90. doi: 10.1111/j.1600-065X.2008.00713.x
29. Werder RB, Zhang V, Lynch JP, Snape N, Upham JW, Spann K, et al. Chronic IL-33 expression predisposes to virus-induced asthma exacerbations by increasing type 2 inflammation and dampening antiviral immunity. J Allergy Clin Immunol. (2018) 141:1607–19 e9. doi: 10.1016/j.jaci.2017.07.051
30. Balzar S, Fajt ML, Comhair SA, Erzurum SC, Bleecker E, Busse WW, et al. Mast cell phenotype, location, and activation in severe asthma. Data from the severe asthma research program. Am J Respir Crit Care Med. (2011) 183:299–309. doi: 10.1164/rccm.201002-0295OC
31. Carroll NG, Mutavdzic S, James AL. Distribution and degranulation of airway mast cells in normal and asthmatic subjects. Eur Resp J. (2002) 19:879–85. doi: 10.1183/09031936.02.00275802
32. Amin K, Janson C, Boman G, Venge P. The extracellular deposition of mast cell products is increased in hypertrophic airways smooth muscles in allergic asthma but not in nonallergic asthma. Allergy. (2005) 60:1241–7. doi: 10.1111/j.1398-9995.2005.00823.x
33. Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. Mast-cell infiltration of airway smooth muscle in asthma. N Engl J Med. (2002) 346:1699–705. doi: 10.1056/NEJMoa012705
34. Andersson CK, Tufvesson E, Aronsson D, Bergqvist A, Mori M, Bjermer L, et al. Alveolar mast cells shift to an FcepsilonRI-expressing phenotype in mild atopic asthma: a novel feature in allergic asthma pathology. Allergy. (2011) 66:1590–7. doi: 10.1111/j.1398-9995.2011.02723.x
35. Carroll NG, Mutavdzic S, James AL. Increased mast cells and neutrophils in submucosal mucous glands and mucus plugging in patients with asthma. Thorax. (2002) 57:677–82. doi: 10.1136/thorax.57.8.677
36. Peters MC, Mekonnen ZK, Yuan S, Bhakta NR, Woodruff PG, Fahy JV. Measures of gene expression in sputum cells can identify TH2-high and TH2-low subtypes of asthma. J Allergy Clin Immunol. (2014) 133:388–94. doi: 10.1016/j.jaci.2013.07.036
37. Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci USA. (2007) 104:15858–63. doi: 10.1073/pnas.0707413104
38. Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. (2009) 180:388–95. doi: 10.1164/rccm.200903-0392OC
39. Altman MC, Lai Y, Nolin JD, Long S, Chen CC, Piliponsky AM, et al. Airway epithelium-shifted mast cell infiltration regulates asthmatic inflammation via IL-33 signaling. J Clin Invest. (2019) 129:4979–91. doi: 10.1172/JCI126402
40. Dougherty RH, Sidhu SS, Raman K, Solon M, Solberg OD, Caughey GH, et al. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2-high asthma. J Allergy Clin Immunol. (2010) 125:1046–53 e8. doi: 10.1016/j.jaci.2010.03.003
41. Zhu J, Message SD, Qiu Y, Mallia P, Kebadze T, Contoli M, et al. Airway inflammation and illness severity in response to experimental rhinovirus infection in asthma. Chest. (2014) 145:1219–29. doi: 10.1378/chest.13-1567
42. Akoto C, Davies DE, Swindle EJ. Mast cells are permissive for rhinovirus replication: potential implications for asthma exacerbations. Clini Exp Allergy. (2017) 47:351–60. doi: 10.1111/cea.12879
43. Hosoda M, Yamaya M, Suzuki T, Yamada N, Kamanaka M, Sekizawa K, et al. Effects of rhinovirus infection on histamine and cytokine production by cell lines from human mast cells and basophils. J Immunol. (2002) 169:1482–91. doi: 10.4049/jimmunol.169.3.1482
44. Liu H, Tan J, Liu J, Feng H, Pan D. Altered mast cell activity in response to rhinovirus infection provides novel insight into asthma. J Asthma. (2019) 2019:1–9. doi: 10.1080/02770903.2019.1585870
45. Holgate S, Buhl R, Bousquet J, Smith N, Panahloo Z, Jimenez P. The use of omalizumab in the treatment of severe allergic asthma: a clinical experience update. Respir Med. (2009) 103:1098–113. doi: 10.1016/j.rmed.2009.03.008
46. Holgate ST. New strategies with anti-IgE in allergic diseases. World Allergy Organ J. (2014) 7:17. doi: 10.1186/1939-4551-7-17
47. Busse WW, Morgan WJ, Gergen PJ, Mitchell HE, Gern JE, Liu AH, et al. Randomized trial of omalizumab (anti-IgE) for asthma in inner-city children. N Engl J Med. (2011) 364:1005–15. doi: 10.1056/NEJMoa1009705
48. Milgrom H, Berger W, Nayak A, Gupta N, Pollard S, McAlary M, et al. Treatment of childhood asthma with anti-immunoglobulin E antibody (omalizumab). Pediatrics. (2001) 108:E36. doi: 10.1542/peds.108.2.e36
49. Kantor DB, McDonald MC, Stenquist N, Schultz BJ, Smallwood CD, Nelson KA, et al. Omalizumab is associated with reduced acute severity of rhinovirus-triggered asthma exacerbation. Am J Resp Crit Care Med. (2016) 194:1552–5. doi: 10.1164/rccm.201606-1145LE
50. Teach SJ, Gill MA, Togias A, Sorkness CA, Arbes SJ, Calatroni A, et al. Preseasonal treatment with either omalizumab or an inhaled corticosteroid boost to prevent fall asthma exacerbations. J Allergy Clin Immunol. (2015) 136:1476–85. doi: 10.1016/j.jaci.2015.09.008
51. Hamelmann E, Tadeda K, Oshiba A, Gelfand EW. Role of IgE in the development of allergic airway inflammation and airway hyperresponsiveness–a murine model. Allergy. (1999) 54:297–305. doi: 10.1034/j.1398-9995.1999.00085.x
52. Martin TR, Takeishi T, Katz HR, Austen KF, Drazen JM, Galli SJ. Mast cell activation enhances airway responsiveness to methacholine in the mouse. J Clin Invest. (1993) 91:1176–82. doi: 10.1172/JCI116277
53. Brusselle GG, Kips JC, Tavernier JH, van der Heyden JG, Cuvelier CA, Pauwels RA, et al. Attenuation of allergic airway inflammation in IL-4 deficient mice. Clin Exp Allergy. (1994) 24:73–80. doi: 10.1111/j.1365-2222.1994.tb00920.x
54. Corry DB, Grunig G, Hadeiba H, Kurup VP, Warnock ML, Sheppard D, et al. Requirements for allergen-induced airway hyperreactivity in T and B cell-deficient mice. Mol Med. (1998) 4:344–55. doi: 10.1007/BF03401741
55. Korsgren M, Erjefalt JS, Korsgren O, Sundler F, Persson CG. Allergic eosinophil-rich inflammation develops in lungs and airways of B cell-deficient mice. J Exp Med. (1997) 185:885–92. doi: 10.1084/jem.185.5.885
56. Kung TT, Stelts D, Zurcher JA, Jones H, Umland SP, Kreutner W, et al. Mast cells modulate allergic pulmonary eosinophilia in mice. Am J Respir Cell Mol Biol. (1995) 12:404–9. doi: 10.1165/ajrcmb.12.4.7695919
57. MacLean JA, Sauty A, Luster AD, Drazen JM, De Sanctis GT. Antigen-induced airway hyperresponsiveness, pulmonary eosinophilia, and chemokine expression in B cell-deficient mice. Am J Resp Cell Mol Biol. (1999) 20:379–87. doi: 10.1165/ajrcmb.20.3.3291
58. Mehlhop PD, van de Rijn M, Goldberg AB, Brewer JP, Kurup VP, Martin TR, et al. Allergen-induced bronchial hyperreactivity and eosinophilic inflammation occur in the absence of IgE in a mouse model of asthma. Proc Natl Acad Sci USA. (1997) 94:1344–9. doi: 10.1073/pnas.94.4.1344
59. Nogami M, Suko M, Okudaira H, Miyamoto T, Shiga J, Ito M, et al. Experimental pulmonary eosinophilia in mice by Ascaris suum extract. Am Rev Resp Dis. (1990) 141:1289–95. doi: 10.1164/ajrccm/141.5_Pt_1.1289
60. Okudaira H, Nogami M, Matsuzaki G, Dohi M, Suko M, Kasuya S, et al. T-cell-dependent accumulation of eosinophils in the lung and its inhibition by monoclonal anti-interleukin-5. Int Arch Allergy Appl Immunol. (1991) 94:171–3. doi: 10.1159/000235354
61. Takeda K, Hamelmann E, Joetham A, Shultz LD, Larsen GL, Irvin CG, et al. Development of eosinophilic airway inflammation and airway hyperresponsiveness in mast cell-deficient mice. J Exp Med. (1997) 186:449–54. doi: 10.1084/jem.186.3.449
62. Kobayashi T, Miura T, Haba T, Sato M, Serizawa I, Nagai H, et al. An essential role of mast cells in the development of airway hyperresponsiveness in a murine asthma model. J Immunol. (2000) 164:3855–61. doi: 10.4049/jimmunol.164.7.3855
63. Williams CM, Galli SJ. Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. J Exp Med. (2000) 192:455–62. doi: 10.1084/jem.192.3.455
64. Cates EC, Fattouh R, Wattie J, Inman MD, Goncharova S, Coyle AJ, et al. Intranasal exposure of mice to house dust mite elicits allergic airway inflammation via a GM-CSF-mediated mechanism. J Immunol. (2004) 173:6384–92. doi: 10.4049/jimmunol.173.10.6384
65. Chan TK, Loh XY, Peh HY, Tan WNF, Tan WSD, Li N, et al. House dust mite-induced asthma causes oxidative damage and DNA double-strand breaks in the lungs. J Allergy Clin Immunol. (2016) 138:84–96 e1. doi: 10.1016/j.jaci.2016.02.017
66. Goplen N, Karim MZ, Liang Q, Gorska MM, Rozario S, Guo L, et al. Combined sensitization of mice to extracts of dust mite, ragweed, and aspergillus species breaks through tolerance and establishes chronic features of asthma. J Allergy Clin Immunol. (2009) 123:925–32 e11. doi: 10.1016/j.jaci.2009.02.009
67. Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via toll-like receptor 4 triggering of airway structural cells. Nat Med. (2009) 15:410–6. doi: 10.1038/nm.1946
68. Hammad H, Plantinga M, Deswarte K, Pouliot P, Willart MA, Kool M, et al. Inflammatory dendritic cells–not basophils–are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J Exp Med. (2010) 207:2097–111. doi: 10.1084/jem.20101563
69. Moon HG, Kim SJ, Jeong JJ, Han SS, Jarjour NN, Lee H, et al. Airway epithelial cell-derived colony stimulating factor-1 promotes allergen sensitization. Immunity. (2018) 49:275–87 e5. doi: 10.1016/j.immuni.2018.06.009
70. Fattouh R, Pouladi MA, Alvarez D, Johnson JR, Walker TD, Goncharova S, et al. House dust mite facilitates ovalbumin-specific allergic sensitization and airway inflammation. Am J Respir Crit Care Med. (2005) 172:314–21. doi: 10.1164/rccm.200502-198OC
71. Han Y, Chen L, Liu H, Jin Z, Wu Y, Wu Y, et al. Airway epithelial cGAS is critical for induction of experimental allergic airway inflammation. J Immunol. (2020) 204:1437–47. doi: 10.4049/jimmunol.1900869
72. Hitomi K, Tahara-Hanaoka S, Miki H, Iwata K, Shibayama S, Kubo M, et al. Allergin-1 on mast cells suppresses house dust mite-induced airway hyperresponsiveness in mice. Int Immunol. (2018) 30:429–34. doi: 10.1093/intimm/dxy025
73. Koyama D, Maruoka S, Gon Y, Shintani Y, Sekiyama T, Hiranuma H, et al. Myeloid differentiation-2 is a potential biomarker for the amplification process of allergic airway sensitization in mice. Allergol Int. (2015) 64:S37–45. doi: 10.1016/j.alit.2015.05.011
74. Miki H, Tahara-Hanaoka S, Almeida MS, Hitomi K, Shibagaki S, Kanemaru K, et al. Allergin-1 immunoreceptor suppresses house dust mite-induced allergic airway inflammation. J Immunol. (2020) 204:753–62. doi: 10.4049/jimmunol.1900180
75. Piyadasa H, Altieri A, Basu S, Schwartz J, Halayko AJ, Mookherjee N. Biosignature for airway inflammation in a house dust mite-challenged murine model of allergic asthma. Biol Open. (2016) 5:112–21. doi: 10.1242/bio.014464
76. Qian X, Aboushousha R, van de Wetering C, Chia SB, Amiel E, Schneider RW, et al. IL-1/inhibitory kappaB kinase epsilon-induced glycolysis augment epithelial effector function and promote allergic airways disease. J Allergy Clin Immunol. (2018) 142:435–50 e10. doi: 10.1016/j.jaci.2017.08.043
77. She L, Alanazi HH, Yan L, Zou Y, Sun Y, Dube PH, et al. Immune sensing of aeroallergen-associated double-stranded rna triggers an ifn response and modulates type 2 lung inflammation. J Immunol. (2019) 203:2520–31. doi: 10.4049/jimmunol.1900720
78. van de Wetering C, Aboushousha R, Manuel AM, Chia SB, Erickson C, MacPherson MB, et al. Pyruvate kinase M2 promotes expression of proinflammatory mediators in house dust mite-induced allergic airways disease. J Immunol. (2020) 204:763–74. doi: 10.4049/jimmunol.1901086
79. Willart MA, Deswarte K, Pouliot P, Braun H, Beyaert R, Lambrecht BN, et al. Interleukin-1alpha controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J Exp Med. (2012) 209:1505–17. doi: 10.1084/jem.20112691
80. McKnight CG, Jude JA, Zhu Z, Panettieri Jr RA, Finkelman FD. House dust mite-induced allergic airway disease is independent of ige and fcepsilonRIalpha. Am J Respir Cell Mol Biol. (2017) 57:674–82. doi: 10.1165/rcmb.2016-0356OC
81. Ando T, Kitaura J. Tuning IgE: IgE-associating molecules and their effects on IgE-dependent mast cell reactions. Cells. (2021) 10:1697. doi: 10.3390/cells10071697
82. Kanagaratham C, El Ansari YS, Lewis OL, Oettgen HC. IgE and IgG antibodies as regulators of mast cell and basophil functions in food allergy. Front Immunol. (2020) 11:603050. doi: 10.3389/fimmu.2020.603050
83. Shamji MH, Valenta R, Jardetzky T, Verhasselt V, Durham SR, Wurtzen PA, et al. The role of allergen-specific IgE, IgG and IgA in allergic disease. Allergy. (2021) 76:3627–41. doi: 10.1111/all.14908
84. Wade-Vallance AK, Allen CDC. Intrinsic and extrinsic regulation of IgE B cell responses. Curr Opin Immunol. (2021) 72:221–9. doi: 10.1016/j.coi.2021.06.005
85. He JS, Narayanan S, Subramaniam S, Ho WQ, Lafaille JJ, Curotto de Lafaille MA. Biology of IgE production: IgE cell differentiation and the memory of IgE responses. Curr Top Microbiol Immunol. (2015) 388:1–19. doi: 10.1007/978-3-319-13725-4_1
86. Engeroff P, Vogel M. The role of CD23 in the regulation of allergic responses. Allergy. (2021) 76:1981–9. doi: 10.1111/all.14724
87. Guntern P, Eggel A. Past, present, and future of anti-IgE biologics. Allergy. (2020) 75:2491–502. doi: 10.1111/all.14308
88. He JS, Meyer-Hermann M, Xiangying D, Zuan LY, Jones LA, Ramakrishna L, et al. The distinctive germinal center phase of IgE+ B lymphocytes limits their contribution to the classical memory response. J Exp Med. (2013) 210:2755–71. doi: 10.1084/jem.20131539
89. Laffleur B, Duchez S, Tarte K, Denis-Lagache N, Peron S, Carrion C, et al. Self-restrained B cells arise following membrane IgE expression. Cell Rep. (2015) 10:900–9. doi: 10.1016/j.celrep.2015.01.023
90. Yang Z, Sullivan BM, Allen CD. Fluorescent in vivo detection reveals that IgE(+) B cells are restrained by an intrinsic cell fate predisposition. Immunity. (2012) 36:857–72. doi: 10.1016/j.immuni.2012.02.009
91. Haniuda K, Fukao S, Kodama T, Hasegawa H, Kitamura D. Autonomous mIgE signaling through CD19 and BLNK prevents IgE-memory formation. Nat Immunol. (2016) 17:1109–17. doi: 10.1038/ni.3508
92. Newman R, Tolar P. Chronic calcium signaling in IgE(+) B cells limits plasma cell differentiation and survival. Immunity. (2021) 54:2756–71 e10. doi: 10.1016/j.immuni.2021.11.006
93. Xiong H, Dolpady J, Wabl M, Curotto de Lafaille MA, Lafaille JJ. Sequential class switching is required for the generation of high affinity IgE antibodies. J Exp Med. (2012) 209:353–64. doi: 10.1084/jem.20111941
94. Geha RS, Jabara HH, Brodeur SR. The regulation of immunoglobulin E class-switch recombination. Nat Rev Immunol. (2003) 3:721–32. doi: 10.1038/nri1181
95. Monticelli S, Vercelli D. Molecular regulation of class switch recombination to IgE through epsilon germline transcription. Allergy. (2001) 56:270–8. doi: 10.1034/j.1398-9995.2001.00129.x
96. Kobayashi T, Iijima K, Dent AL, Kita H. Follicular helper T cells mediate IgE antibody response to airborne allergens. J Allergy Clin Immunol. (2017) 139:300–13 e7. doi: 10.1016/j.jaci.2016.04.021
97. Gowthaman U, Chen JS, Zhang B, Flynn WF, Lu Y, Song W, et al. Identification of a T follicular helper cell subset that drives anaphylactic IgE. Science. (2019) 365:eaaw6433. doi: 10.1126/science.aaw6433
98. Lawrence MG, Woodfolk JA, Schuyler AJ, Stillman LC, Chapman MD, Platts-Mills TA. Half-life of IgE in serum and skin: Consequences for anti-IgE therapy in patients with allergic disease. J Allergy Clin Immunol. (2017) 139:422–8 e4. doi: 10.1016/j.jaci.2016.04.056
99. Turner H, Kinet JP. Signalling through the high-affinity IgE receptor Fc epsilonRI. Nature. (1999) 402(Suppl. 6760):B24–30. doi: 10.1038/35037021
100. Campbell AM, Vachier I, Chanez P, Vignola AM, Lebel B, Kochan J, et al. Expression of the high-affinity receptor for IgE on bronchial epithelial cells of asthmatics. Am J Respir Cell Mol Biol. (1998) 19:92–7. doi: 10.1165/ajrcmb.19.1.2648
101. Conroy MC, Adkinson Jr NF, lichtenstein LM. Measurement of IgE on human basophils: relation to serum IgE and anti-IgE-induced histamine release. J Immunol. (1977) 118:1317–21.
102. Malveaux FJ, Conroy MC, Adkinson Jr NF, Lichtenstein LM. IgE receptors on human basophils. Relationship to serum IgE concentration. J Clin Invest. (1978) 62:176–81. doi: 10.1172/JCI109103
103. Borkowski TA, Jouvin MH, Lin SY, Kinet JP. Minimal requirements for IgE-mediated regulation of surface Fc epsilon RI. J Immunol. (2001) 167:1290–6. doi: 10.4049/jimmunol.167.3.1290
104. Kubo S, Matsuoka K, Taya C, Kitamura F, Takai T, Yonekawa H, et al. Drastic up-regulation of fcepsilonri on mast cells is induced by IgE binding through stabilization and accumulation of fcepsilonri on the cell surface. J Immunol. (2001) 167:3427–34. doi: 10.4049/jimmunol.167.6.3427
105. MacGlashan Jr DW, Bochner BS, Adelman DC, Jardieu PM, Togias A, et al. Down-regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients with anti-IgE antibody. J Immunol. (1997) 158:1438–45.
106. Lee WT, Conrad DH. Murine B cell hybridomas bearing ligand-inducible Fc receptors for IgE. J Immunol. (1986) 136:4573–80.
107. Ding Z, Dahlin JS, Xu H, Heyman B. IgE-mediated enhancement of CD4(+) T cell responses requires antigen presentation by CD8alpha(-) conventional dendritic cells. Sci Rep. (2016) 6:28290. doi: 10.1038/srep28290
108. Yu P, Kosco-Vilbois M, Richards M, Kohler G, Lamers MC. Negative feedback regulation of IgE synthesis by murine CD23. Nature. (1994) 369:753–6. doi: 10.1038/369753a0
109. Payet ME, Woodward EC, Conrad DH. Humoral response suppression observed with CD23 transgenics. J Immunol. (1999) 163:217–23.
110. Payet-Jamroz M, Helm SL, Wu J, Kilmon M, Fakher M, Basalp A, et al. Suppression of IgE responses in CD23-transgenic animals is due to expression of CD23 on nonlymphoid cells. J Immunol. (2001) 166:4863–9. doi: 10.4049/jimmunol.166.8.4863
111. Rosenwasser LJ, Busse WW, Lizambri RG, Olejnik TA, Totoritis MC. Allergic asthma and an anti-CD23 mAb (IDEC-152): results of a phase I, single-dose, dose-escalating clinical trial. J Allergy Clin Immunol. (2003) 112:563–70. doi: 10.1016/S0091-6749(03)01861-X
112. Palaniyandi S, Liu X, Periasamy S, Ma A, Tang J, Jenkins M, et al. Inhibition of CD23-mediated IgE transcytosis suppresses the initiation and development of allergic airway inflammation. Mucosal Immunol. (2015) 8:1262–74. doi: 10.1038/mi.2015.16
113. Palaniyandi S, Tomei E, Li Z, Conrad DH, Zhu X. CD23-dependent transcytosis of IgE and immune complex across the polarized human respiratory epithelial cells. J Immunol. (2011) 186:3484–96. doi: 10.4049/jimmunol.1002146
114. Schulman ES. Development of a monoclonal anti-immunoglobulin E antibody (omalizumab) for the treatment of allergic respiratory disorders. Am J Respir Crit Care Med. (2001) 164:S6–11. doi: 10.1164/ajrccm.164.supplement_1.2103025
115. Pennington LF, Tarchevskaya S, Brigger D, Sathiyamoorthy K, Graham MT, Nadeau KC, et al. Structural basis of omalizumab therapy and omalizumab-mediated IgE exchange. Nat Commun. (2016) 7:11610. doi: 10.1038/ncomms11610
116. Busse W, Corren J, Lanier BQ, McAlary M, Fowler-Taylor A, Cioppa GD, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol. (2001) 108:184–90. doi: 10.1067/mai.2001.117880
117. Casale TB, Condemi J, LaForce C, Nayak A, Rowe M, Watrous M, et al. Effect of omalizumab on symptoms of seasonal allergic rhinitis: a randomized controlled trial. JAMA. (2001) 286:2956–67. doi: 10.1001/jama.286.23.2956
118. Maurer M, Rosen K, Hsieh HJ, Saini S, Grattan C, Gimenez-Arnau A, et al. Omalizumab for the treatment of chronic idiopathic or spontaneous urticaria. N Engl J Med. (2013) 368:924–35. doi: 10.1056/NEJMoa1215372
119. Kaplan AP, Gimenez-Arnau AM, Saini SS. Mechanisms of action that contribute to efficacy of omalizumab in chronic spontaneous urticaria. Allergy. (2017) 72:519–33. doi: 10.1111/all.13083
120. Cardet JC, Casale TB. New insights into the utility of omalizumab. J Allergy Clin Immunol. (2019) 143:923–6 e1. doi: 10.1016/j.jaci.2019.01.016
121. Gauvreau GM, Arm JP, Boulet LP, Leigh R, Cockcroft DW, Davis BE, et al. Efficacy and safety of multiple doses of QGE031 (ligelizumab) versus omalizumab and placebo in inhibiting allergen-induced early asthmatic responses. J Allergy Clin Immunol. (2016) 138:1051–9. doi: 10.1016/j.jaci.2016.02.027
122. Trischler J, Bottoli I, Janocha R, Heusser C, Jaumont X, Lowe P, et al. Ligelizumab treatment for severe asthma: learnings from the clinical development programme. Clin Transl Immunol. (2021) 10:e1255. doi: 10.1002/cti2.1255
123. Maurer M, Gimenez-Arnau AM, Sussman G, Metz M, Baker DR, Bauer A, et al. Ligelizumab for chronic spontaneous Urticaria. N Engl J Med. (2019) 381:1321–32. doi: 10.1056/NEJMoa1900408
124. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. (2015) 16:45–56. doi: 10.1038/ni.3049
125. Gill MA, Bajwa G, George TA, Dong CC, Dougherty II, et al. Counterregulation between the Fcepsilonri pathway and antiviral responses in human plasmacytoid dendritic cells. J Immunol. (2010) 184:5999–6006. doi: 10.4049/jimmunol.0901194
126. Schroeder JT, Bieneman AP, Xiao H, Chichester KL, Vasagar K, Saini S, et al. TLR9- and FcepsilonRI-mediated responses oppose one another in plasmacytoid dendritic cells by down-regulating receptor expression. J Immunol. (2005) 175:5724–31. doi: 10.4049/jimmunol.175.9.5724
127. Gill MA, Liu AH, Calatroni A, Krouse RZ, Shao B, Schiltz A, et al. Enhanced plasmacytoid dendritic cell antiviral responses after omalizumab. J Allergy Clin Immunol. (2018) 141:1735–43 e9. doi: 10.1016/j.jaci.2017.07.035
128. Hussain SA, Mejias A, Ramilo O, Peeples ME, Grayson MH. Post-viral atopic airway disease: pathogenesis and potential avenues for intervention. Expert Rev Clin Immunol. (2019) 15:49–58. doi: 10.1080/1744666X.2019.1541737
129. Esquivel A, Busse WW, Calatroni A, Togias AG, Grindle KG, Bochkov YA, et al. Effects of omalizumab on rhinovirus infections, illnesses, and exacerbations of asthma. Am J Respir Crit Care Med. (2017) 196:985–92. doi: 10.1164/rccm.201701-0120OC
130. Li S, Aliyeva M, Daphtary N, Martin RA, Poynter ME, Kostin SF, et al. Antigen-induced mast cell expansion and bronchoconstriction in a mouse model of asthma. Am J Physiol Lung Cell Mol Physiol. (2014) 306:L196–206. doi: 10.1152/ajplung.00055.2013
131. de Boer JD, Yang J, van den Boogaard FE, Hoogendijk AJ, de Beer R, van der Zee JS, et al. Mast cell-deficient kit mice develop house dust mite-induced lung inflammation despite impaired eosinophil recruitment. J Innate Immun. (2014) 6:219–26. doi: 10.1159/000354984
132. Hernandez JD, Yu M, Sibilano R, Tsai M, Galli SJ. Development of multiple features of antigen-induced asthma pathology in a new strain of mast cell deficient BALB/c-Kit(W-sh/W-sh) mice. Lab Invest. (2020) 100:516–26. doi: 10.1038/s41374-019-0354-2
133. Yu CK, Chen CL. Activation of mast cells is essential for development of house dust mite dermatophagoides farinae-induced allergic airway inflammation in mice. J Immunol. (2003) 171:3808–15. doi: 10.4049/jimmunol.171.7.3808
134. Zoltowska Nilsson AM, Lei Y, Adner M, Nilsson GP. Mast cell-dependent IL-33/ST2 signaling is protective against the development of airway hyperresponsiveness in a house dust mite mouse model of asthma. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L484–92. doi: 10.1152/ajplung.00270.2017
135. Plaza J, Torres R, Urbano A, Picado C, de Mora F. In Vitro and In Vivo validation of EP2-receptor agonism to selectively achieve inhibition of mast cell activity. Allergy Asthma Immunol Res. (2020) 12:712–28. doi: 10.4168/aair.2020.12.4.712
136. Reber LL, Marichal T, Galli SJ. New models for analyzing mast cell functions in vivo. Trends Immunol. (2012) 33:613–25. doi: 10.1016/j.it.2012.09.008
137. Rodewald HR, Feyerabend TB. Widespread immunological functions of mast cells: fact or fiction? Immunity. (2012) 37:13–24. doi: 10.1016/j.immuni.2012.07.007
138. Shadie AM, Herbert C, Kumar RK. Ambient particulate matter induces an exacerbation of airway inflammation in experimental asthma: role of interleukin-33. Clin Exp Immunol. (2014) 177:491–9. doi: 10.1111/cei.12348
139. Wang H, Song L, Ju W, Wang X, Dong L, Zhang Y, et al. The acute airway inflammation induced by PM2.5 exposure and the treatment of essential oils in Balb/c mice. Sci Rep. (2017) 7:44256. doi: 10.1038/srep44256
140. Shen Y, Zhang ZH, Hu D, Ke X, Gu Z, Zou QY, et al. The airway inflammation induced by nasal inoculation of PM2.5 and the treatment of bacterial lysates in rats. Sci Rep. (2018) 8:9816. doi: 10.1038/s41598-018-28156-9
141. Ogino K, Zhang R, Takahashi H, Takemoto K, Kubo M, Murakami I, et al. Allergic airway inflammation by nasal inoculation of particulate matter (PM2.5) in NC/Nga mice. PLoS ONE. (2014) 9:e92710. doi: 10.1371/journal.pone.0092710
142. Jin C, Shelburne CP, Li G, Potts EN, Riebe KJ, Sempowski GD, et al. Particulate allergens potentiate allergic asthma in mice through sustained IgE-mediated mast cell activation. J Clin Invest. (2011) 121:941–55. doi: 10.1172/JCI43584
143. Gern JE, Palmenberg AC. Rhinoviruses. 6th ed. In: Fields BN, Knipe DM, Howley PM, editors. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins (2013).
144. Greve JM, Davis G, Meyer AM, Forte CP, Yost SC, Marlor CW, et al. The major human rhinovirus receptor is ICAM-1. Cell. (1989) 56:839–47. doi: 10.1016/0092-8674(89)90688-0
145. Hofer F, Gruenberger M, Kowalski H, Machat H, Huettinger M, Kuechler E, et al. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc Natl Acad Sci USA. (1994) 91:1839–42. doi: 10.1073/pnas.91.5.1839
146. Bochkov YA, Watters K, Ashraf S, Griggs TF, Devries MK, Jackson DJ, et al. Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proc Natl Acad Sci USA. (2015) 112:5485–90. doi: 10.1073/pnas.1421178112
147. Lee WM, Lemanske Jr RF, Evans MD, Vang F, Pappas T, et al. Human rhinovirus species and season of infection determine illness severity. Am J Respir Crit Care Med. (2012) 186:886–91. doi: 10.1164/rccm.201202-0330OC
148. Message SD, Laza-Stanca V, Mallia P, Parker HL, Zhu J, Kebadze T, et al. Rhinovirus-induced lower respiratory illness is increased in asthma and related to virus load and Th1/2 cytokine and IL-10 production. Proc Natl Acad Sci USA. (2008) 105:13562–7. doi: 10.1073/pnas.0804181105
149. Bartlett NW, Walton RP, Edwards MR, Aniscenko J, Caramori G, Zhu J, et al. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat Med. (2008) 14:199–204. doi: 10.1038/nm1713
150. Rajput C, Han M, Ishikawa T, Lei J, Goldsmith AM, Jazaeri S, et al. Rhinovirus C infection induces type 2 innate lymphoid cell expansion and eosinophilic airway inflammation. Front Immunol. (2021) 12:649520. doi: 10.3389/fimmu.2021.649520
151. Zambrano JC, Carper HT, Rakes GP, Patrie J, Murphy DD, Platts-Mills TA, et al. Experimental rhinovirus challenges in adults with mild asthma: response to infection in relation to IgE. J Allergy Clin Immunol. (2003) 111:1008–16. doi: 10.1067/mai.2003.1396
152. Tam JS, Jackson WT, Hunter D, Proud D, Grayson MH. Rhinovirus specific IgE can be detected in human sera. J Allergy Clin Immunol. (2013) 132:1241–3. doi: 10.1016/j.jaci.2012.12.1091
153. Dakhama A, Park JW, Taube C, Chayama K, Balhorn A, Joetham A, et al. The role of virus-specific immunoglobulin E in airway hyperresponsiveness. Am J Respir Crit Care Med. (2004) 170:952–9. doi: 10.1164/rccm.200311-1610OC
154. Green RM, Custovic A, Sanderson G, Hunter J, Johnston SL, Woodcock A. Synergism between allergens and viruses and risk of hospital admission with asthma: case-control study. BMJ. (2002) 324:763. doi: 10.1136/bmj.324.7340.763
155. Murray CS, Poletti G, Kebadze T, Morris J, Woodcock A, Johnston SL, et al. Study of modifiable risk factors for asthma exacerbations: virus infection and allergen exposure increase the risk of asthma hospital admissions in children. Thorax. (2006) 61:376–82. doi: 10.1136/thx.2005.042523
156. Toussaint M, Jackson DJ, Swieboda D, Guedan A, Tsourouktsoglou TD, Ching YM, et al. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat Med. (2017) 23:681–91. doi: 10.1038/nm.4332
157. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
158. Zschaler J, Schlorke D, Arnhold J. Differences in innate immune response between man and mouse. Crit Rev Immunol. (2014) 34:433–54. doi: 10.1615/CritRevImmunol.2014011600
159. Gibertson SE, Weinmann AS. Conservation and divergence in gene regulation between mouse and human immune cells deserves equal emphasis. Trends Immunol. (2021) 42:1077–87. doi: 10.1016/j.it.2021.10.007
160. Teng F, Tacho-Pinot R, Sung B, Farber DL, Worgall S, Hammad H, et al. ILC3s control airway inflammation by limiting T cell responses to allergens and microbes. Cell Rep. (2021) 37:110051. doi: 10.1016/j.celrep.2021.110051
Keywords: asthma, rhinovirus, mast cells, dendritic cells, IgE, omalizumab
Citation: Kawakami T, Kasakura K, Kawakami Y and Ando T (2022) Immunoglobulin E-Dependent Activation of Immune Cells in Rhinovirus-Induced Asthma Exacerbation. Front. Allergy 3:835748. doi: 10.3389/falgy.2022.835748
Received: 14 December 2021; Accepted: 24 January 2022;
Published: 22 February 2022.
Edited by:
Tamara Haque, National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH), United StatesReviewed by:
Ulrich Matthias Zissler, Technical University of Munich, GermanyCopyright © 2022 Kawakami, Kasakura, Kawakami and Ando. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toshiaki Kawakami, dG9zaGlAbGppLm9yZw==
†Present address: Kazumi Kasakura, Janssen Pharmaceutical K.K., Tokyo, Japan