 Yingzi Liu1Xuewen Xiao1Hui Liu1Xinxin Liao2,3,4,5,6Yafang Zhou2,3,4,5,6Ling Weng1,2,4,5,6Lu Zhou1Xixi Liu1Xiang-yun Bi1Tianyan Xu1Yuan Zhu1Qijie Yang1Sizhe Zhang1Xiaoli Hao1Weiwei Zhang2,4,5,6,7Junling Wang1,2,4,5,6Bin Jiao1,2,4,5,6*Lu Shen1,2,4,5,6,8*
Yingzi Liu1Xuewen Xiao1Hui Liu1Xinxin Liao2,3,4,5,6Yafang Zhou2,3,4,5,6Ling Weng1,2,4,5,6Lu Zhou1Xixi Liu1Xiang-yun Bi1Tianyan Xu1Yuan Zhu1Qijie Yang1Sizhe Zhang1Xiaoli Hao1Weiwei Zhang2,4,5,6,7Junling Wang1,2,4,5,6Bin Jiao1,2,4,5,6*Lu Shen1,2,4,5,6,8*- 1Department of Neurology, Xiangya Hospital, Central South University, Changsha, China
- 2National Clinical Research Center for Geriatric Disorders, Central South University, Changsha, China
- 3Department of Geriatrics, Xiangya Hospital, Central South University, Changsha, China
- 4Engineering Research Center of Hunan Province in Cognitive Impairment Disorders, Central South University, Changsha, China
- 5Hunan International Scientific and Technological Cooperation Base of Neurodegenerative and Neurogenetic Diseases, Changsha, China
- 6Key Laboratory of Hunan Province in Neurodegenerative Disorders, Central South University, Changsha, China
- 7Department of Radiology, Xiangya Hospital, Central South University, Changsha, China
- 8Key Laboratory of Organ Injury, Aging and Regenerative Medicine of Hunan Province, Changsha, China
Alzheimer’s disease (AD) is a progressive neurodegenerative disease associated with aging, environmental, and genetic factors. Amyloid protein precursor (APP) is a known pathogenic gene for familial Alzheimer’s disease (FAD), and now more than 70 APP mutations have been reported, but the genotype-phenotype correlation remains unclear. In this study, we collected clinical data from patients carrying APP mutations defined as pathogenic/likely pathogenic according to the American college of medical genetics and genomics (ACMG) guidelines. Then, we reanalyzed the clinical characteristics and identified genotype-phenotype correlations in APP mutations. Our results indicated that the clinical phenotypes of APP mutations are generally consistent with typical AD despite the fact that they show more non-demented symptoms and neurological symptoms. We also performed genotype-phenotype analysis according to the difference in APP processing caused by the mutations, and we found that there were indeed differences in onset age, behavioral and psychological disorders of dementia (BPSD) and myoclonus.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by progressive memory loss and cognitive decline (Sperling et al., 2011). According to the World Alzheimer Report 2018, 50 million people were living with dementia worldwide in 2018 (Alzheimer’s Disease International Consortium, 2018), and the number will more than triple to 152 million by 2050. The typical pathological feature of AD is extracellular deposits of amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles (Braak and Braak, 1996). Although the pathological changes of AD are relatively clear, the exact pathogenesis of this disease is still uncertain (Scheltens et al., 2016). AD is widely believed to be associated with aging, environmental and genetic factors (Farrer et al., 1997; Flicker, 2010; Barnes and Yaffe, 2011).
Alzheimer’s disease can be divided into familial Alzheimer’s disease (FAD) and sporadic Alzheimer’s disease (SAD), depending on whether there is a positive family history. Mutations in the amyloid protein precursor (APP), presenilin-1 (PSEN1), and presenilin-2 (PSEN2) genes can cause FAD. These three causative genes explained 5–10% of FAD (Loy et al., 2014; Cacace et al., 2016), and over 200 mutations in these genes have been described so far (Alzforum mutation database).1
Amyloid protein precursor mutations are the second most common pathogenic gene for AD, with an estimated mutation frequency of 1% (Cacace et al., 2016; Hinz and Geschwind, 2017). APP gene is positioned on chromosome 21q21.2–21q21.3 and has several different isoforms, of which the three most common isoforms are the 695 amino acid form, the 751, and the 770 amino acid forms (Bayer et al., 1999). APP695 is mainly produced by neurons, while APP751 and APP770 are primarily expressed on peripheral cells and platelets (Hardy, 1997; Guerreiro et al., 2012). All three isoforms consist of a single membrane-spanning domain, a large extracellular glycosylated N-terminus, and a shorter cytoplasmic C-terminus (Muresan and Ladescu Muresan, 2015) and can generate Aβ after sequential sequencing cleavages by β-secretase and γ-secretase (Nunan and Small, 2000). More than 70 APP mutations have been reported possibly associated with FAD since the first mutation V717I was discovered (Goate et al., 1991), and most of the mutations were found to increase the production of Aβ or alter the ratio of Aβ42/Aβ40 (Citron et al., 1992; De Jonghe et al., 2001; Kirkitadze et al., 2001; Nilsberth et al., 2001). Despite one research summarizing the APP missense mutations and their impacts on APP Processing (Theuns et al., 2006), the research on the genotype-phenotype of APP mutation is limited (Lindquist et al., 2009; Ryan et al., 2016; Jiang et al., 2019). Only a part of APP mutations targeted at specific populations was described, and there was no systematic summary of all APP mutations. However, to study the pathogenesis of AD better, it is imperative to fully understand its clinical characteristics and the correlations between genotype and phenotype.
In a previous study, we had systematically re-evaluated APP, PSEN1, and PSEN2 mutations according to the American college of medical genetics and genomics and the association for molecular pathology (ACMG-AMP) guidelines (Xiao et al., 2021). In this study, we collected detailed clinical data from FAD with APP mutations that were re-evaluated as pathogenic/likely pathogenic based on previous research. Then we reanalyzed the clinical characteristics and identified the genotype-phenotype correlations in AD caused by pathogenic/likely pathogenic APP mutations.
Materials and methods
Data sources and selection
We conducted a literature search using databases from the Alzforum mutation database (see text footnote 1) and PubMed with the keywords “APP” and “Alzheimer’s disease.” All the articles we included in this research described either clinical characteristics and/or neuropathological features. Almost all cases in these articles were diagnosed with AD according to the National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) (McKhann et al., 1984) criteria or the National Institute on Aging-Alzheimer’s Association (NIA-AA) (McKhann et al., 2011). APP mutations defined as variant of uncertain clinical significance (VUS) and benign/likely benign were excluded. Asymptomatic individuals and mild cognitive impairment (MCI) were also ruled out from the study.
Data extraction
The information collected directly from relevant manuscripts was related to the demographic data (origin, gender), age at onset (AAO), onset symptom, clinical feature, disease duration (calculated only for deceased patients), APOE allele, neuroimaging, electroencephalography (EEG), cerebrospinal fluid (CSF) biomarkers and the neuropathology for individually affected patients. Data were also extracted when the exact AAO and disease duration were unavailable in the article and the mean AAO, range, and number of patients.
Statistical analysis
The statistical analyses have been performed using the ANOVA test for continuous variables, chi-square for categorical variables, and correlation analysis for clinical phenotypes. All data were tested for normality and homogeneity of variance using the Shapiro-Wilk and Levene variance equality tests. All data were analyzed with IBM SPSS Statistics (version 23.0) and visualized using Prism 8 (GraphPad). Tests were considered statistically significant for P < 0.05.
Results
The overall clinical characteristics of pathogenic/likely pathogenic amyloid protein precursor mutations
A total of 31 APP mutations were re-evaluated as pathogenic/likely pathogenic based on the previous study, among which 28 mutations related to AD were included in this study, including 26 missense mutations, one double codon mutation, and only one mutation had a single base deletion. All the mutations were located in exon 16 (n = 7) and exon 17 (n = 21), near the splice site of α secretase, β secretase, and γ secretase. Several other mutations were near α, β, or γ secretase cleavage sites such as V669L or in the other region of APP than exon 16–17, but their pathogenic nature was questioned according to ACMG-AMP guidelines. Overall, 63 pedigrees exhibiting APP mutations were reported in this research, accounting for 180 affected subjects. Figure 1 shows the locations of APP gene mutations, the number of families and individuals affected by the mutations. The most frequently reported mutations were KM670/671NL (n = 43, near β cleavage site) and V717I (n = 69, near γ42 cleavage site).
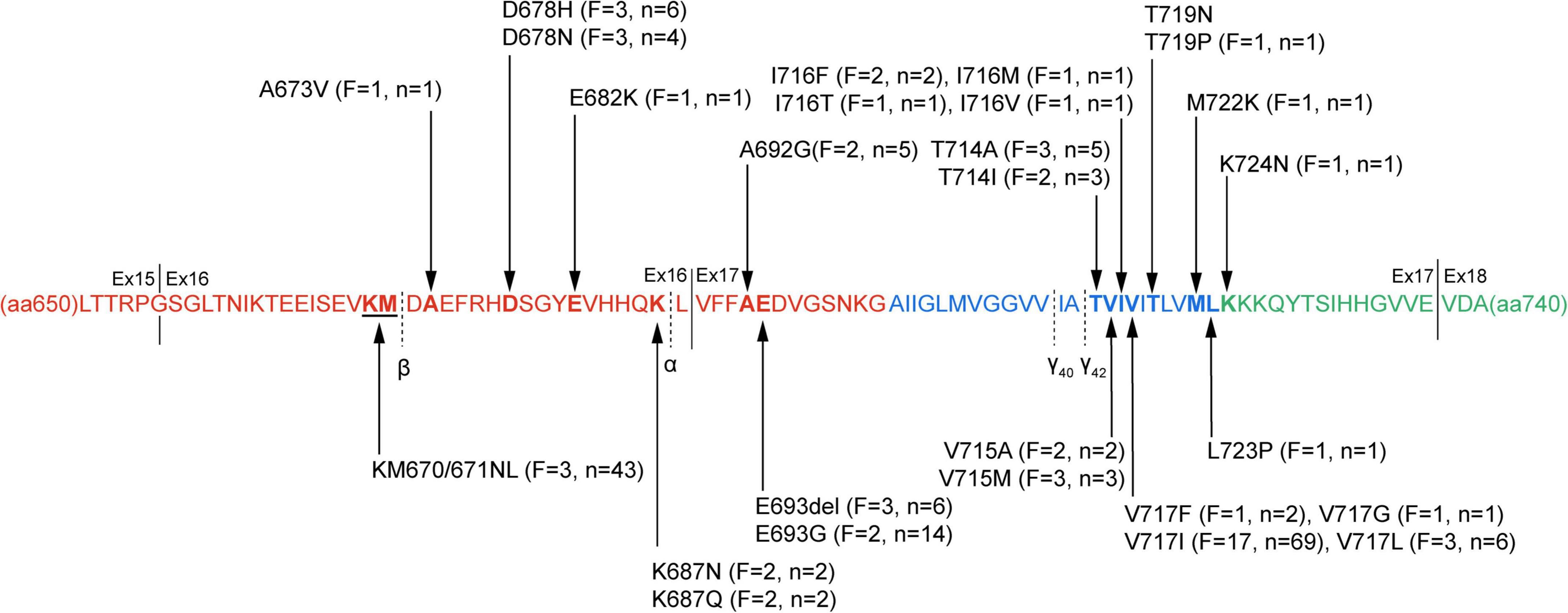
Figure 1. The pathogenic/likely pathogenic mutations in amyloid protein precursor (APP) protein. APP protein sequence from amino acid residue 650–740 is presented. The sequence in red depicts the extracellular domain, the transmembrane domain blue, and the intracellular domain green. Black arrow markers indicate pathogenic/likely pathogenic mutation sites, and the specific information on the mutation is described. The numbers of families and individuals affected are also shown in the figure. The cleavage sites of α-, β-, and γ secretases are marked with black dotted lines, and solid lines separate the different exons. F, the number of families affected in the mutations; n, the number of affected individuals; Ex, exon.
The clinical characteristics of APP gene mutations are summarized in Supplementary Table 1. The overall mean AAO in APP mutations was 50.7 years, ranging from 31 to 65 years. Gender information was available in 130 patients, including 55 male patients and 75 female patients (the ratio was 1:1.36). 74 cases reported the onset symptoms, and 170 described the clinical manifestations. Most cases had typical characteristics of memory loss (90.59%, 154/170), some patients also had non-amnestic clinical features including disorientation (39.41%, 67/170), visuospatial disorder (12.94%, 22/170) language impairment (27.65%, 47/170), apraxia/agnosia (49.41%, 84/170), dyscalculia (8.24%, 14/170), behavioral and psychological disorders of dementia (BPSD) (53.53%, 91/170). Some patients with APP mutations (e.g., A673V, A692G, and E693del) described neurological symptoms such as extrapyramidal symptoms (EPS), myoclonus, seizures, spastic paraplegia, and ataxia. And for those individuals who reported APOE allele, 70.49% (43/61) were APOE ε4 negative, 21.31% (13/61) had one APOE ε4, and 8.20% (5/61) had two copies of APOE ε4.
The age at onset and disease duration
Age at onset data were available in 123 subjects, in which only 9 (7.32%) patients had AAO <40 years old, and 12 (9.76%) had AAO >60 years old. The AAO of most patients was between 40 and 60 years old. Considering the different mutations, the lowest mean AAO was observed among the A673V and I716F mutations (36.0 and 38.0 years, respectively), while the highest was in the I716M mutation (64 years). We also noted that the AAO in some mutations (e.g., K687N and A692G) were roughly the same, while some mutations (e.g., D678N) showed wide variation. Age at onset could be significantly different among the affected individuals, even within the same family. In our research, the APOE allele did not affect the overall mean AAO of APP mutations, whereas, in the V717I mutation, carriers of the ε4 allele had an earlier AAO (p = 0.005).
The course of AD with APP mutations was slow. The mean duration of the disease ranged from 3 to 18 years, with an average of 8.7 years. A faster disease duration with a mean duration of 4 years was observed in the T714I carriers, while the patients with the V717I and V715M mutation experienced a more extended period of the disease (mean duration of 9.9 and 10.0 years, respectively). Similar to the AAO, the course of AD was found to vary widely in the same mutations, even within the same pedigrees.
The first symptoms and clinical presentation
Most of the patients reported amnesia as the first symptom (82.43%, 61/74), which was throughout almost all subjects and gradually aggravated. Disorientation, BPSD, or functional executive function impairment could be the initial symptoms in some individuals, accounting for 12.16% (9/74) in total. Besides, some patients (5.41%, 4/74) could also exhibit headache, vertigo, and other non-dementia symptoms as the first clinical manifestation. In comparison, no cases reported language impairment and dyscalculia at the onset.
With the progression of dementia, BPSD became the second most common clinical feature after amnesia. BPSD were classified into three subsyndromes, including psychotic syndrome (hallucinations and/or delusions), affective syndrome (agitation and/or depression and/or anxiety and/or irritability), and behavior syndrome (euphoria and/or apathy and/or disinhibition and/or aberrant motor behavior) (Garre-Olmo et al., 2010). Among the above three subsyndromes, the affective syndrome has the highest frequency (75.0%, 63/84) in carriers who reported BPSD, with depression and anxiety being the most common. Hallucinations and delusions of psychiatric symptoms were also frequent in APP mutations; Supplementary Table 1). Apraxia/agnosia was also a frequent clinical manifestation in AD, occurring in nearly half of the cases. Over 25% of subjects had disorientation and language impairment, while dyscalculia was rarely reported in dementia cases. We also found that individuals in the same pedigrees tended to be impaired in similar cognitive domains.
Neurological symptoms were concentrated in some of these APP mutations, with more than half of the cases clustered in the V717I mutation. In contrast, the A673V and D678N mutations only showed neurological symptoms in one and two patients, respectively. EPS was the most common atypical neurological feature in pathogenic/likely pathogenic APP mutations. Patients reported with EPS have at least one clinical manifestation of bradykinesia, rigidity, dystonia, stooped posture, and shuffling gait. Some patients also had other neurological symptoms besides EPS, such as myoclonus, seizures, spastic paraplegia, and ataxia. Pathological reflexes and frontal release signs were shown in APP mutations as well. Myoclonus, seizures, spastic paraplegia, and ataxia occur similarly in our research. However, myoclonus and seizures appeared in almost all mutations that reported neurological symptoms, whereas spastic paraplegia was only present in A673V, E693del, T714A, and V717I, ataxia only in E693del, I716F, and V717I. In addition to typical clinical features and neurological symptoms, a tiny percentage of individuals had headaches, vertigo, sleep disturbance, and other non-dementia symptoms (Supplementary Table 1).
The diagnostic findings
The neuroimaging, CSF, EEG, and neuropathology results were collected in Table 1. Most cases undergoing CT/MRI examination showed diffuse cerebral atrophy or a local involvement of parietal and temporal lobes or hippocampal region, accompanying with or without white matter lesion and other signs. All patients who received positron emission tomography (PET) and/or single-photon emission computed tomography (SPECT) were confirmed to have the following pattern, a temporal and parietal hypoperfusion/hypometabolism at first, then a progressive involvement in the frontal and occipital regions, even the precuneus, and finally the entire cerebral. However, two patients with E693del showed no difference in 11C-labeled Pittsburgh compound B (11C-PIB) imaging compared with non-demented people, and one harboring E693G mutation had no abnormal results in SPECT.
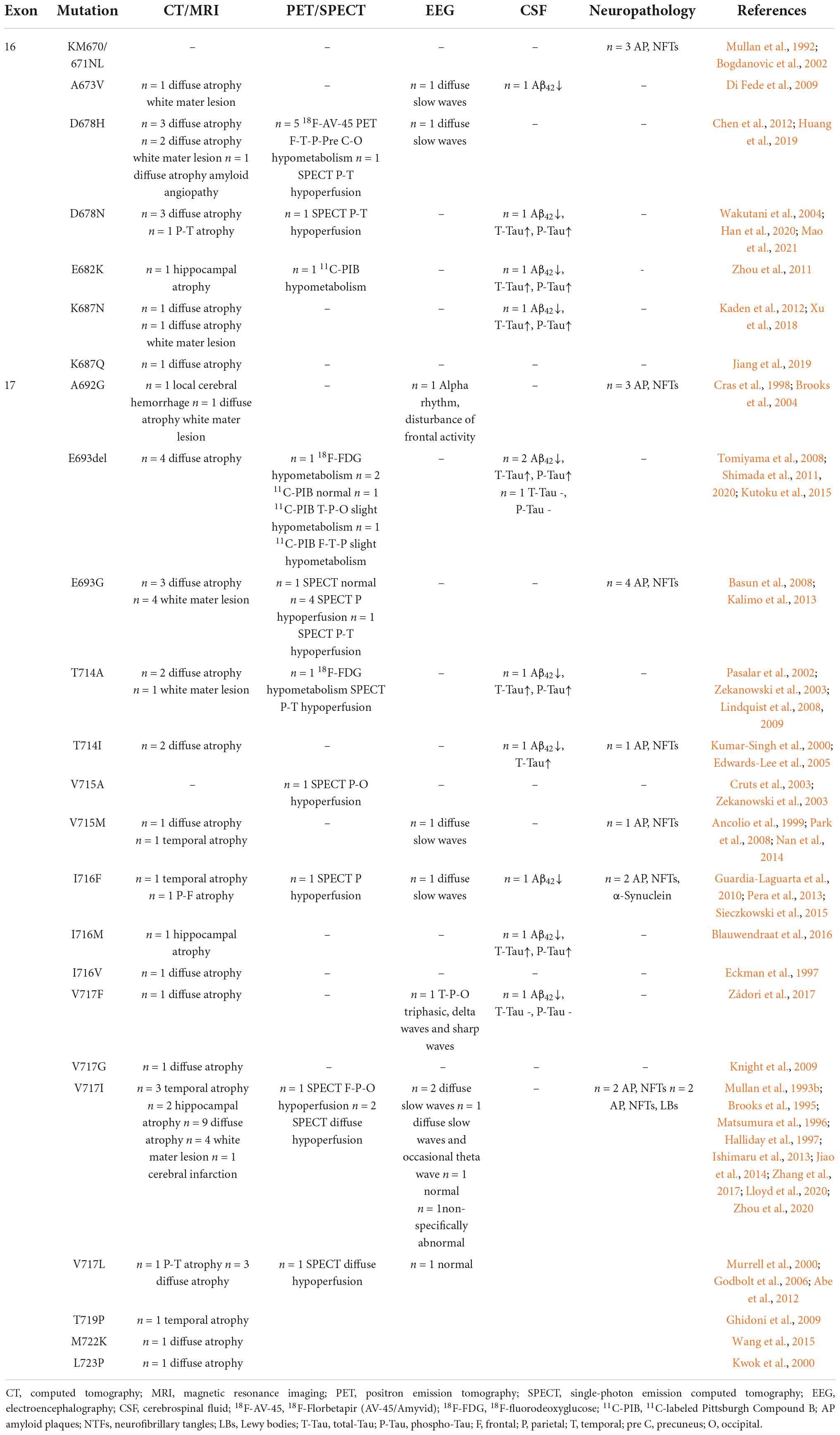
Table 1. Neuroimaging, electroencephalography, cerebrospinal fluid (CSF) analysis, and neuropathology findings in pathogenic/likely pathogenic amyloid protein precursor (APP) mutations.
Cerebrospinal fluid analysis was available in only 12 patients. Almost all cases matched the typical features of AD, the reduced Aβ42 levels and the increased levels of total-Tau (T-Tau) and phospho-Tau (P-Tau). However, one with E693G mutation and one with V717F mutation had normal levels of T-Tau and p-Tau. EEG was presented in 12 cases as well. And the results were as follows: 7 showed diffuse slow waves, two were normal, one was a non-specific abnormality, one had an alpha rhythm with disturbance of frontal activity, and one indicated T-P-O triphasic, delta waves, and sharp waves.
A total of 18 brain autopsies were performed in all studies. All the neuropathological findings fulfilled the Consortium to Establish a Registry for Alzheimer’s disease (CERAD) (Mirra et al., 1991) criteria, characterized by amyloid plaques and neurofibrillary tangles. In addition to amyloid plaques and neurofibrillary tangles, patients with I716F mutation have α-Synuclein in their brains, and Lewy bodies (LBs) were also present in patients with V717I mutation.
The genotype-phenotype correlations
Amyloid protein precursor mutations may alter APP processing, which may in turn diversify phenotypes. We collected the effects on APP processing in pathogenic/likely pathogenic APP mutations in Supplementary Table 2. The results showed that the mutation site adjacent to α/β secretase mainly increased the amount of total Aβ, while mutations near γ secretase alter the ratio of Aβ42/Aβ40. Based on the difference in biochemical results, we divided the pathogenic/likely pathogenic APP mutations into two groups: mutations close to α/β secretase and mutations near γ secretase, and compared their clinical features. Atypical mutations such as E693del were ruled out. A total of 130 cases (Complete data not available for all cases) were included in the study of genotype-phenotype correlation analyses, of which 38 were close to α/β secretase, and 92 belonged to the group near γ secretase. The clinical manifestations of the two groups were recorded in Table 2. The phenotype was mostly consistent (Figure 2). However, the AAO in the mutations near the α/β secretase site group was 52.2, a little later than 50.3 in group γ secretase (P = 0.049). The frequency of clinical BPSD was also less than that in group γ secretase (P = 0.008), and the incidence of myoclonus in the α/β secretase group was higher (P = 0.037).
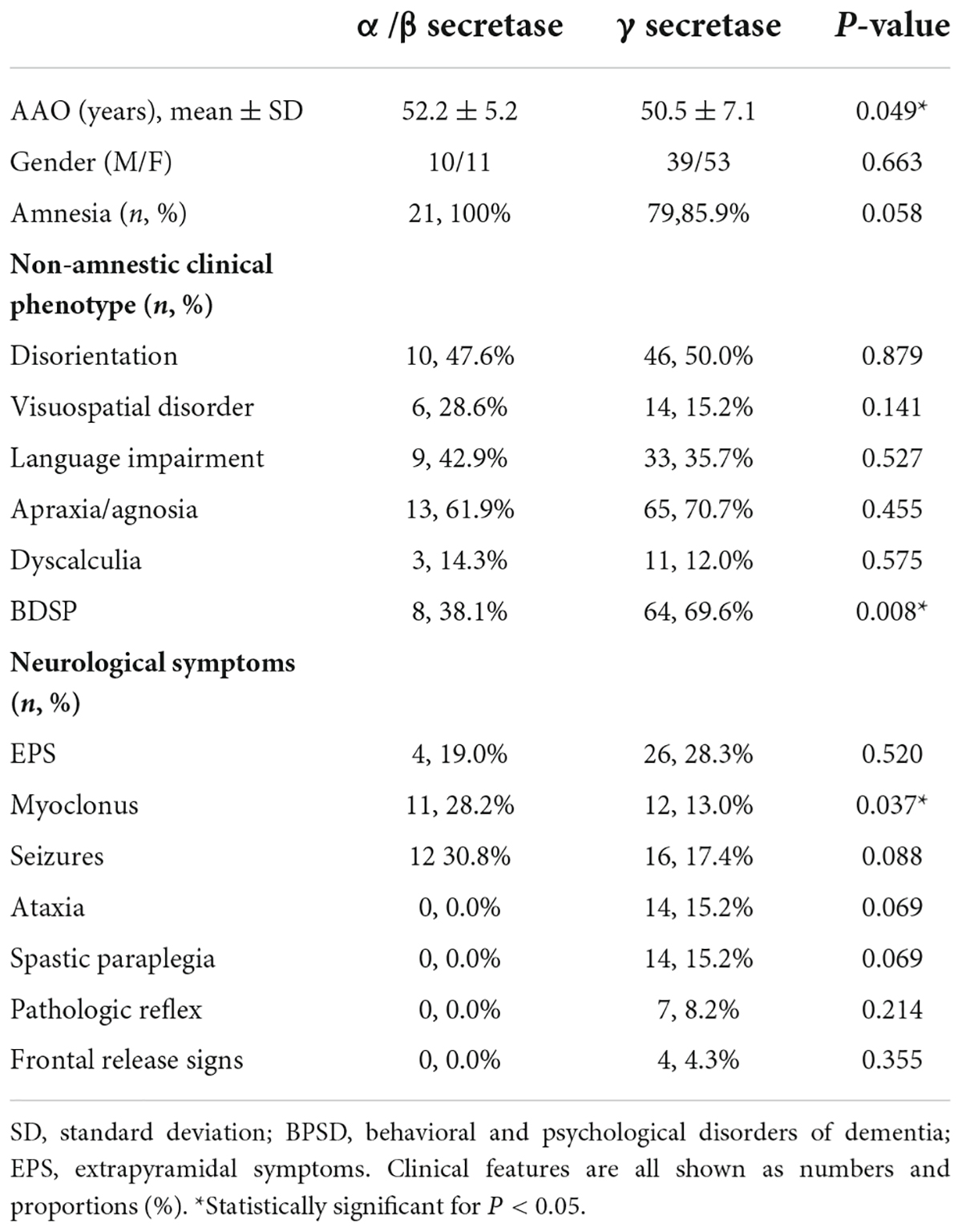
Table 2. The demographics and the frequencies of clinical features in mutations near α/β secretase and mutations near γ secretase.
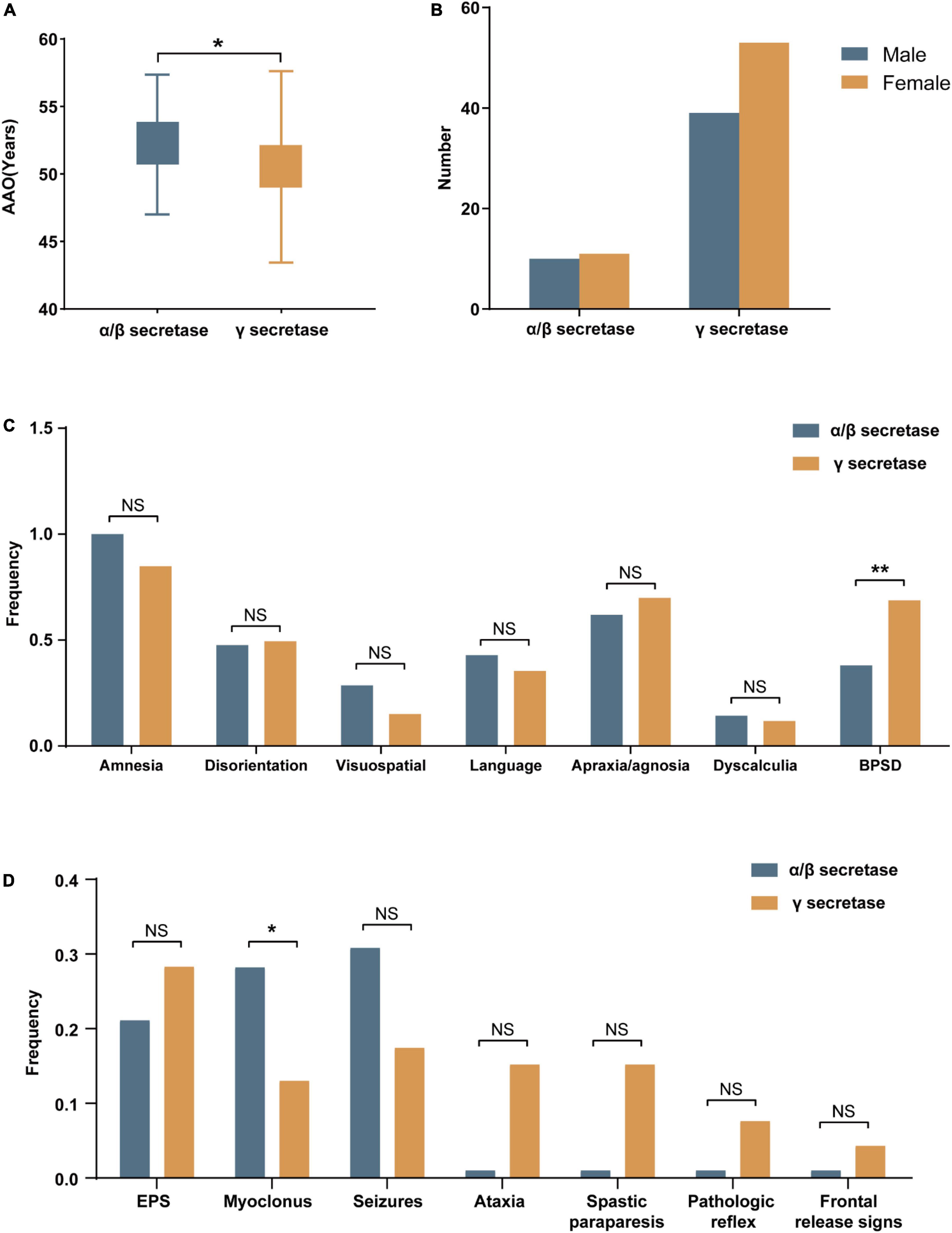
Figure 2. The demographics and the frequencies of clinical features in mutations near α/β secretase and near γ secretase. (A) The age at onset (AAO) and (B) The gender of mutations near α/β secretase and near γ secretase. (C) The frequencies of amnesia and non-amnestic clinical phenotype. (D) The frequencies of neurological symptoms (NS, no significance, *p < 0.05, **p < 0.01).
V717I mutation is the most frequently reported and clinically detailed APP mutation, and we also compared V717I mutation to all pathogenic/likely pathogenic APP mutations. A total of 147 patients were included in genotype-phenotype correlation analyses, with 60 carrying V717I mutation. There was little difference between the two groups except that the visuospatial impairment on V717I was very low, only 3.2% (Supplementary Figure 1). However, when comparing V717I alone with other APP mutations (Supplementary Tables 3, 4), we found that patients with the V717I mutation had a later onset and tended to have dyscalculia but had less damage in linguistic and visuospatial regions. V717I mutation carriers also had a higher prevalence of ataxia and spastic paraplegia regarding neurological symptoms (Supplementary Figure 2).
We also performed correlation analysis for clinical phenotypes. Apraxia/agnosia showed a weak positive correlation with language impairment (r = 0.308, p = 0.001), and BPS was negatively correlated with the visuospatial disorder (r = −0.301, p = 0.001). And the individuals with ataxia were more likely to have spastic paraplegia (r = 0.468, p = 1.711 × 10–7).
Discussion
This is the first study to collect clinical data on APP mutations defined as pathogenic/likely pathogenic according to ACMG and describe genotype-phenotype correlations of FAD cases with APP mutations. Although APP mutations are the second most common pathogenic gene for AD, the information on clinical manifestations of APP mutations was relatively limited. In general, APP mutations are consistent with the typical AD phenotype, even if there are some specific and heterogeneous features. And there are also some differences in clinical manifestations between APP mutations and PSEN1/PSEN2.
Previous studies verified that the pedigrees with APP mutations have an earlier mean AAO than those with PSEN2 mutations but later than families with PSEN1 mutations (Mullan et al., 1993a; Jayadev et al., 2010). A study of clinical phenotypic and genetic association analysis of autosomal dominant FAD in the UK demonstrated that the mean AAO of PSEN1 and APP mutations was 43.6 and 50.4, respectively (Ryan et al., 2016). The AAO of our study was 50.7, the same as the AAO of APP mutations in their research and later than that of PSEN1 mutations. APOE ε4 is a well-established risk factor for LOAD. APOE ε4 carriers had a significantly earlier AAO of AD than ε4 non-carriers. A study showed a decrease in 3.02 years in AAO for each unit increase in the number of ε4 alleles (Sorbi et al., 1995; Thambisetty et al., 2013). However, the APOE allele only affected the AAO of V717I mutation in our research. The limited data and confounding factors interfered with our ability to analyze the correlations between AAO and APOE alleles in other pathogenic/likely pathogenic APP mutations other than V717I. Moreover, since AAO measurement is usually retrospective and prone to recall bias, the accuracy of AAO itself remains to be determined.
Our results indicated that APP pathogenic/likely pathogenic mutations all have the following clinical characteristics. First, most cases start with amnesia. Second, the disease progresses relatively slowly. Third, patients rarely exhibit pure progressive amnesia and usually present with impairment in multiple cognitive domains. Fourth, BPSD frequently occurs in the progression of dementia and manifests in various forms, among which affective symptoms represented by depression and anxiety are the most common. Fifth, the neuroimaging, CSF biochemical, and neuropathological findings are typical in most cases. The clinical phenotype of APP mutations is similar to the SAD (Swearer et al., 1992), but some specificity and heterogeneity remain. For instance, patients with APP mutations are more likely to have apraxia/agnosia and perform worse than LOAD (Koedam et al., 2010; Smits et al., 2012).
Regarding BPSD, anxiety and depression are the most common in APP mutations, while depression and irritability are more frequent in LOAD (Gumus et al., 2021). The higher incidence of anxiety and depression with less irritability is also well represented in PSEN1 and PSEN2 (Kaiser et al., 2014; Panegyres and Chen, 2014), but they tend to have more hallucinations and delusions compared with APP mutations (Larner and Doran, 2006; Canevelli et al., 2014). The consistency and heterogeneity of the three pathogenic genes in BPSD may be related to both AAO and genes. In order to clarify whether there is a connection, it is necessary to pay more attention to the relationship between each subtype of BPSD in patients with dementia and AAO and genetics in future research. In addition, the disease progression of early-onset FAD is faster than that of late-onset SAD, with more extensive cognitive impairment and higher mortality (Jacobs et al., 1994), and this was broadly to our study. While in our study, the disease duration was not as short as previously reported, the average time from onset to death is only 6.6 years (Vermunt et al., 2019; Brück et al., 2021).
Similar to the non-amnestic phenotypes, neurological symptoms have higher morbidity in early-onset FAD than in late-onset SAD (Bateman et al., 2011), especially in pedigrees with AAO <40 years (Ryan and Rossor, 2010). Cases with APP, PSEN1, or PSEN2 mutations in FAD also differ in neurological symptoms. A study in autosomal dominant familial AD reported that apart from seizures and myoclonus, patients with PSEN1 mutations can present with other neurological symptoms while patients with APP mutations do not (Ryan et al., 2016). Differently, patients with APP mutation in this study also presented with EPS, spastic paraparesis, pathologic reflex, and ataxia. EPS is even more frequently in APP than in PSEN1, and the proportions of patients with myoclonus or seizures are the same. In the same study, Ryan et al. (2016) also pointed out that individuals with myoclonus tended to develop more seizures than those without myoclonus, yet this was not confirmed in our research. Instead, we found that patients with EPS or ataxia were more likely to have spastic paraplegia. Moreover, we also found that the severity of neurological symptoms would gradually increase as dementia progresses (Vöglein et al., 2019). Similarly, patients with neurological symptoms such as EPS experience a faster cognitive decline (Chui et al., 1994).
We conducted genotype-phenotype correlation analysis of APP mutations and found that biochemical differences due to mutations can lead to differences in clinical manifestations, which can occur in AAO, non-demented symptoms, and neurological symptoms. However, the clinical data of the patients we obtained were somewhat limited, which may have affected our interpretation of the results. Future studies need to focus more on the heterogeneity of clinical manifestations caused by differences in APP processing, Aβ amount, and the ratio of Aβ42/Aβ40, which may offer a better understanding of amyloid pathways in AD. Furthermore, the differences between V717I and all pathogenic/likely pathogenic APP mutations suggest that each APP mutation may have the diverse characteristic. For example, V715M and V717L have an earlier AAO than V717I. Some APP mutations had visuospatial and language impairments in addition to amnesia, while others had minor damage in these cognitive areas but showed more dyscalculia (e.g., V717I). And mutations in exon 16 are rare to show neurological symptoms compared to mutations in exon 17. Therefore, attention to the clinical manifestations of each mutation is highly warranted.
For the most reported V717I mutation, there was consistency in the pattern of symptoms between cases. There is a cognitive decline initially, with visuospatial impairment, disorientation, and language impairment. Dyscalculia, agnosia/apraxia and neurological symptoms occur as the disease progresses. However, there are differences in clinical manifestations among pedigrees (Mullan et al., 1993b). For instance, the APPV717I mutation in the Chinese population mainly manifests as affective symptoms, executive dysfunction and disorientation in the early stage, and spastic paraparesis and ataxia in the late stage are more common (Zhang et al., 2017). In addition, we also found differences in AAO and clinical manifestations within the same family. This may be due to variability in the expression of these mutations or related to other genetic or epigenetic factors (Román et al., 2019).
Although most pathogenic/likely pathogenic APP mutations are complete penetrance, some have been demonstrated to behave as incomplete dominant. A Caucasian woman with the homozygous mutation D678N had memory difficulties early in her third decade and developed full-blown clinical symptoms 10 years later. Her heterozygous affected siblings were generally diagnosed with dementia in their 60 s (Mastromoro et al., 2019). We also found asymptomatic carriers reported in some other pathogenic/likely pathogenic APP mutations, which may be due to incomplete dominant mutations or maybe just because of the individual differences as well as environmental factors that lead to a later onset in those carriers, and the researchers did not follow them up. And unlike other APP mutations (Zhou et al., 2011; Huang et al., 2019), A673V is a particular mutation caused by recessive homozygous mutation. It has a high amyloidosis effect in the homozygous state and an anti-amyloidosis effect in the heterozygous state (Di Fede et al., 2009). In addition, different mutations in the same site of APP can lead to different diseases. For example, E693K (Bugiani et al., 2010), E693Q (Wattendorff et al., 1982; Luyendijk and Bots, 1986) can lead to Cerebral Amyloid Angiopathy (CAA), E693G, and E693del lead to AD, D694N (Grabowski et al., 2001) mutation near 693 can lead to vascular dementia (VD). All of these suggest that there may be some discrepancy in the structure and toxicity of the mutant product Aβ, or other mechanisms related to other APP cleavage products besides Aβ such as APP intracellular domain (AICD). It is essential to use these atypical mutations as tools to study the pathogenesis of AD.
The limited clinical evidence makes it difficult to conduct further genotype-phenotype association analyses, especially to evaluate the clinical manifestations of different mutations at the same locus. And the reliability of the clinical data we collected remains to be determined. We are not able to be sure whether the specific manifestations unreported were because they were really absent or emerged after the study, or were just not mentioned in the articles. Besides, we only analyzed APP mutations and did not compare them with the other two pathogenic genes.
In conclusion, we collected the clinical data from patients with pathogenic/likely pathogenic APP mutations and performed an analysis of genotype-phenotypic association, which may help better understand the relationship between genotype and phenotype and may be beneficial for clinical practice prediction, diagnosis, and genetic counseling.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: https://www.alzforum.org/mutations.
Author contributions
YL and XX contributed to the conception and design of the study. YL collected the data, performed the statistical analysis, and wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
We are grateful to all subjects for participation in our study. This study was supported by the National Natural Science Foundation of China (Nos. 81671075 to LS, 81971029 to LS, 81701134 to BJ, 81901171 to XLu), the National Key R&D Program of China (Nos. 2017YFC0840100 and 2017YFC0840104 to LS, 2018YFC1312003 to JW), the Provincial Key Plan for Research and Development of Hunan (No. 2017SK2031 to LS), the Provincial Technology Innovation Guidance Plan Project of Hunan (No. 2018SK52601 to BJ).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.1013295/full#supplementary-material
Footnotes
References
Abe, M., Sonobe, N., Fukuhara, R., Mori, Y., Ochi, S., Matsumoto, T., et al. (2012). Phenotypical difference of amyloid precursor protein (APP) V717L mutation in Japanese family. BMC Neurol. 12:38. doi: 10.1186/1471-2377-12-38
Alzheimer’s Disease International Consortium (2018). World Alzheimer report 2018. London: Alzheimer’s Disease International.
Ancolio, K., Dumanchin, C., Barelli, H., Warter, J. M., Brice, A., Campion, D., et al. (1999). Unusual phenotypic alteration of beta amyloid precursor protein (betaAPP) maturation by a new Val-715 –> Met betaAPP-770 mutation responsible for probable early-onset Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 96, 4119–4124. doi: 10.1073/pnas.96.7.4119
Barnes, D. E., and Yaffe, K. (2011). The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 10, 819–828. doi: 10.1016/S1474-4422(11)70072-2
Basun, H., Bogdanovic, N., Ingelsson, M., Almkvist, O., Näslund, J., Axelman, K., et al. (2008). Clinical and neuropathological features of the arctic APP gene mutation causing early-onset Alzheimer disease. Arch. Neurol. 65, 499–505. doi: 10.1001/archneur.65.4.499
Bateman, R. J., Aisen, P. S., De Strooper, B., Fox, N. C., Lemere, C. A., Ringman, J. M., et al. (2011). Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res. Ther. 3:1. doi: 10.1186/alzrt59
Bayer, T. A., Cappai, R., Masters, C. L., Beyreuther, K., and Multhaup, G. (1999). It all sticks together–the APP-related family of proteins and Alzheimer’s disease. Mol. Psychiatry 4, 524–528. doi: 10.1038/sj.mp.4000552
Blauwendraat, C., Wilke, C., Jansen, I. E., Schulte, C., Simón-Sánchez, J., Metzger, F. G., et al. (2016). Pilot whole-exome sequencing of a German early-onset Alzheimer’s disease cohort reveals a substantial frequency of PSEN2 variants. Neurobiol. Aging 37, 208.e11–208.e17. doi: 10.1016/j.neurobiolaging.2015.09.016
Bogdanovic, N., Corder, E., Lannfelt, L., and Winblad, B. (2002). APOE polymorphism and clinical duration determine regional neuropathology in Swedish APP(670, 671) mutation carriers: implications for late-onset Alzheimer’s disease. J. Cell. Mol. Med. 6, 199–214. doi: 10.1111/j.1582-4934.2002.tb00187.x
Braak, H., and Braak, E. (1996). Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol. Scand. Suppl. 165, 3–12. doi: 10.1111/j.1600-0404.1996.tb05866.x
Brooks, W. S., Kwok, J. B., Halliday, G. M., Godbolt, A. K., Rossor, M. N., Creasey, H., et al. (2004). Hemorrhage is uncommon in new Alzheimer family with Flemish amyloid precursor protein mutation. Neurology 63, 1613–1617. doi: 10.1212/01.WNL.0000142965.10778.C7
Brooks, W. S., Martins, R. N., De Voecht, J., Nicholson, G. A., Schofield, P. R., Kwok, J. B., et al. (1995). A mutation in codon 717 of the amyloid precursor protein gene in an Australian family with Alzheimer’s disease. Neurosci. Lett. 199, 183–186. doi: 10.1016/0304-3940(95)12046-7
Brück, C. C., Wolters, F. J., Ikram, M. A., and de Kok, I. (2021). Heterogeneity in reports of dementia disease duration and severity: A review of the literature. J. Alzheimers Dis. 84, 1515–1522. doi: 10.3233/JAD-210544
Bugiani, O., Giaccone, G., Rossi, G., Mangieri, M., Capobianco, R., Morbin, M., et al. (2010). Hereditary cerebral hemorrhage with amyloidosis associated with the E693K mutation of APP. Arch. Neurol. 67, 987–995. doi: 10.1001/archneurol.2010.178
Cacace, R., Sleegers, K., and Van Broeckhoven, C. (2016). Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 12, 733–748. doi: 10.1016/j.jalz.2016.01.012
Canevelli, M., Piscopo, P., Talarico, G., Vanacore, N., Blasimme, A., Crestini, A., et al. (2014). Familial Alzheimer’s disease sustained by presenilin 2 mutations: systematic review of literature and genotype-phenotype correlation. Neurosci. Biobehav. Rev. 42, 170–179. doi: 10.1016/j.neubiorev.2014.02.010
Chen, W. T., Hong, C. J., Lin, Y. T., Chang, W. H., Huang, H. T., Liao, J. Y., et al. (2012). Amyloid-beta (Aβ) D7H mutation increases oligomeric Aβ42 and alters properties of Aβ-zinc/copper assemblies. PLoS One 7:e35807. doi: 10.1371/journal.pone.0035807
Chui, H. C., Lyness, S. A., Sobel, E., and Schneider, L. S. (1994). Extrapyramidal signs and psychiatric symptoms predict faster cognitive decline in Alzheimer’s disease. Arch. Neurol. 51, 676–681. doi: 10.1001/archneur.1994.00540190056015
Citron, M., Oltersdorf, T., Haass, C., McConlogue, L., Hung, A. Y., Seubert, P., et al. (1992). Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 360, 672–674. doi: 10.1038/360672a0
Cras, P., van Harskamp, F., Hendriks, L., Ceuterick, C., van Duijn, C. M., Stefanko, S. Z., et al. (1998). Presenile Alzheimer dementia characterized by amyloid angiopathy and large amyloid core type senile plaques in the APP 692Ala–>Gly mutation. Acta Neuropathol. 96, 253–260. doi: 10.1007/s004010050892
Cruts, M., Dermaut, B., Rademakers, R., Van den Broeck, M., Stögbauer, F., and Van Broeckhoven, C. (2003). Novel APP mutation V715A associated with presenile Alzheimer’s disease in a German family. J. Neurol. 250, 1374–1375. doi: 10.1007/s00415-003-0182-5
De Jonghe, C., Esselens, C., Kumar-Singh, S., Craessaerts, K., Serneels, S., Checler, F., et al. (2001). Pathogenic APP mutations near the gamma-secretase cleavage site differentially affect Abeta secretion and APP C-terminal fragment stability. Hum. Mol. Genet. 10, 1665–1671. doi: 10.1093/hmg/10.16.1665
Di Fede, G., Catania, M., Morbin, M., Rossi, G., Suardi, S., Mazzoleni, G., et al. (2009). A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 323, 1473–1477. doi: 10.1126/science.1168979
Eckman, C. B., Mehta, N. D., Crook, R., Perez-tur, J., Prihar, G., Pfeiffer, E., et al. (1997). A new pathogenic mutation in the APP gene (I716V) increases the relative proportion of A beta 42(43). Hum. Mol. Genet. 6, 2087–2089. doi: 10.1093/hmg/6.12.2087
Edwards-Lee, T., Ringman, J. M., Chung, J., Werner, J., Morgan, A., St George Hyslop, P., et al. (2005). An African American family with early-onset Alzheimer disease and an APP (T714I) mutation. Neurology 64, 377–379. doi: 10.1212/01.WNL.0000149761.70566.3E
Farrer, L. A., Cupples, L. A., Haines, J. L., Hyman, B., Kukull, W. A., Mayeux, R., et al. (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278, 1349–1356. doi: 10.1001/jama.1997.03550160069041
Flicker, L. (2010). Modifiable lifestyle risk factors for Alzheimer’s disease. J. Alzheimers Dis. 20, 803–811. doi: 10.3233/JAD-2010-091624
Garre-Olmo, J., López-Pousa, S., Vilalta-Franch, J., de Gracia Blanco, M., and Vilarrasa, A. B. (2010). Grouping and trajectories of the neuropsychiatric symptoms in patients with Alzheimer’s disease, part I: symptom clusters. J. Alzheimers Dis. 22, 1157–1167. doi: 10.3233/JAD-2010-101212
Ghidoni, R., Albertini, V., Squitti, R., Paterlini, A., Bruno, A., Bernardini, S., et al. (2009). Novel T719P AbetaPP mutation unbalances the relative proportion of amyloid-beta peptides. J. Alzheimers Dis. 18, 295–303. doi: 10.3233/JAD-2009-1142
Goate, A., Chartier-Harlin, M. C., Mullan, M., Brown, J., Crawford, F., Fidani, L., et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706. doi: 10.1038/349704a0
Godbolt, A. K., Beck, J. A., Collinge, J. C., Cipolotti, L., Fox, N. C., and Rossor, M. N. (2006). A second family with familial AD and the V717L APP mutation has a later age at onset. Neurology 66, 611–612. doi: 10.1212/01.WNL.0000197791.53828.2C
Grabowski, T. J., Cho, H. S., Vonsattel, J. P., Rebeck, G. W., and Greenberg, S. M. (2001). Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol. 49, 697–705. doi: 10.1002/ana.1009
Guardia-Laguarta, C., Pera, M., Clarimón, J., Molinuevo, J. L., Sánchez-Valle, R., Lladó, A., et al. (2010). Clinical, neuropathologic, and biochemical profile of the amyloid precursor protein I716F mutation. J. Neuropathol. Exp. Neurol. 69, 53–59. doi: 10.1097/NEN.0b013e3181c6b84d
Guerreiro, R. J., Gustafson, D. R., and Hardy, J. (2012). The genetic architecture of Alzheimer’s disease: beyond APP, PSENs and APOE. Neurobiol. Aging 33, 437–456. doi: 10.1016/j.neurobiolaging.2010.03.025
Gumus, M., Multani, N., Mack, M. L., and Tartaglia, M. C. (2021). Progression of neuropsychiatric symptoms in young-onset versus late-onset Alzheimer’s disease. Geroscience 43, 213–223. doi: 10.1007/s11357-020-00304-y
Halliday, G., Brooks, W., Arthur, H., Creasey, H., and Broe, G. A. (1997). Further evidence for an association between a mutation in the APP gene and Lewy body formation. Neurosci. Lett. 227, 49–52. doi: 10.1016/S0304-3940(97)00294-2
Han, L. H., Xue, Y. Y., Zheng, Y. C., Li, X. Y., Lin, R. R., Wu, Z. Y., et al. (2020). Genetic analysis of Chinese patients with early-onset dementia using next-generation sequencing. Clin. Interv. Aging 15, 1831–1839. doi: 10.2147/CIA.S271222
Hardy, J. (1997). Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 20, 154–159. doi: 10.1016/S0166-2236(96)01030-2
Hinz, F. I., and Geschwind, D. H. (2017). Molecular genetics of neurodegenerative dementias. Cold Spring Harb. Perspect. Biol. 9:a023705. doi: 10.1101/cshperspect.a023705
Huang, C. Y., Hsiao, I. T., Lin, K. J., Huang, K. L., Fung, H. C., Liu, C. H., et al. (2019). Amyloid PET pattern with dementia and amyloid angiopathy in Taiwan familial AD with D678H APP mutation. J. Neurol. Sci. 398, 107–116. doi: 10.1016/j.jns.2018.12.039
Ishimaru, T., Ochi, S., Matsumoto, T., Yoshida, T., Abe, M., Toyota, Y., et al. (2013). [A case report of early-onset Alzheimer’s disease with multiple psychotic symptoms, finally diagnosed as APPV717I mutation by genetic testing]. Seishin Shinkeigaku Zasshi 115, 1042–1050.
Jacobs, D., Sano, M., Marder, K., Bell, K., Bylsma, F., Lafleche, G., et al. (1994). Age at onset of Alzheimer’s disease: relation to pattern of cognitive dysfunction and rate of decline. Neurology 44, 1215–1220. doi: 10.1212/WNL.44.7.1215
Jayadev, S., Leverenz, J. B., Steinbart, E., Stahl, J., Klunk, W., Yu, C. E., et al. (2010). Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain 133(Pt 4), 1143–1154. doi: 10.1093/brain/awq033
Jiang, B., Zhou, J., Li, H. L., Chen, Y. G., Cheng, H. R., Ye, L. Q., et al. (2019). Mutation screening in Chinese patients with familial Alzheimer’s disease by whole-exome sequencing. Neurobiol. Aging 76, 215.e15–215.e21. doi: 10.1016/j.neurobiolaging.2018.11.024
Jiao, B., Tang, B., Liu, X., Xu, J., Wang, Y., Zhou, L., et al. (2014). Mutational analysis in early-onset familial Alzheimer’s disease in Mainland China. Neurobiol. Aging 35, 1957.e1–1957.e6. doi: 10.1016/j.neurobiolaging.2014.02.014
Kaden, D., Harmeier, A., Weise, C., Munter, L. M., Althoff, V., Rost, B. R., et al. (2012). Novel APP/Aβ mutation K16N produces highly toxic heteromeric Aβ oligomers. EMBO Mol. Med. 4, 647–659. doi: 10.1002/emmm.201200239
Kaiser, N. C., Liang, L. J., Melrose, R. J., Wilkins, S. S., Sultzer, D. L., and Mendez, M. F. (2014). Differences in anxiety among patients with early- versus late-onset Alzheimer’s disease. J. Neuropsychiatry Clin. Neurosci. 26, 73–80. doi: 10.1176/appi.neuropsych.12100240
Kalimo, H., Lalowski, M., Bogdanovic, N., Philipson, O., Bird, T. D., Nochlin, D., et al. (2013). The Arctic AβPP mutation leads to Alzheimer’s disease pathology with highly variable topographic deposition of differentially truncated Aβ. Acta Neuropathol. Commun. 1:60. doi: 10.1186/2051-5960-1-60
Kirkitadze, M. D., Condron, M. M., and Teplow, D. B. (2001). Identification and characterization of key kinetic intermediates in amyloid beta-protein fibrillogenesis. J. Mol. Biol. 312, 1103–1119. doi: 10.1006/jmbi.2001.4970
Knight, W. D., Ahsan, R. L., Jackson, J., Cipolotti, L., Warrington, E. K., Fox, N. C., et al. (2009). Pure progressive amnesia and the APPV717G mutation. Alzheimer Dis. Assoc. Disord. 23, 410–414. doi: 10.1097/WAD.0b013e31819cb7f3
Koedam, E. L., Lauffer, V., van der Vlies, A. E., van der Flier, W. M., Scheltens, P., and Pijnenburg, Y. A. (2010). Early-versus late-onset Alzheimer’s disease: more than age alone. J. Alzheimers Dis. 19, 1401–1408. doi: 10.3233/JAD-2010-1337
Kumar-Singh, S., De Jonghe, C., Cruts, M., Kleinert, R., Wang, R., Mercken, M., et al. (2000). Nonfibrillar diffuse amyloid deposition due to a gamma(42)-secretase site mutation points to an essential role for N-truncated A beta(42) in Alzheimer’s disease. Hum. Mol. Genet. 9, 2589–2598. doi: 10.1093/hmg/9.18.2589
Kutoku, Y., Ohsawa, Y., Kuwano, R., Ikeuchi, T., Inoue, H., Ataka, S., et al. (2015). A second pedigree with amyloid-less familial Alzheimer’s disease harboring an identical mutation in the amyloid precursor protein gene (E693delta). Intern. Med. 54, 205–208. doi: 10.2169/internalmedicine.54.3021
Kwok, J. B., Li, Q. X., Hallupp, M., Whyte, S., Ames, D., Beyreuther, K., et al. (2000). Novel Leu723Pro amyloid precursor protein mutation increases amyloid beta42(43) peptide levels and induces apoptosis. Ann. Neurol. 47, 249–253. doi: 10.1002/1531-8249(200002)47:2<249::AID-ANA18>3.0.CO;2-8
Larner, A. J., and Doran, M. (2006). Clinical phenotypic heterogeneity of Alzheimer’s disease associated with mutations of the presenilin-1 gene. J. Neurol. 253, 139–158. doi: 10.1007/s00415-005-0019-5
Lindquist, S. G., Nielsen, J. E., Stokholm, J., Schwartz, M., Batbayli, M., Ballegaard, M., et al. (2008). Atypical early-onset Alzheimer’s disease caused by the Iranian APP mutation. J. Neurol. Sci. 268, 124–130. doi: 10.1016/j.jns.2007.11.021
Lindquist, S. G., Schwartz, M., Batbayli, M., Waldemar, G., and Nielsen, J. E. (2009). Genetic testing in familial AD and FTD: mutation and phenotype spectrum in a Danish cohort. Clin. Genet. 76, 205–209. doi: 10.1111/j.1399-0004.2009.01191.x
Lloyd, G. M., Trejo-Lopez, J. A., Xia, Y., McFarland, K. N., Lincoln, S. J., Ertekin-Taner, N., et al. (2020). Prominent amyloid plaque pathology and cerebral amyloid angiopathy in APP V717I (London) carrier – phenotypic variability in autosomal dominant Alzheimer’s disease. Acta Neuropathol. Commun. 8:31. doi: 10.1186/s40478-020-0891-3
Loy, C. T., Schofield, P. R., Turner, A. M., and Kwok, J. B. (2014). Genetics of dementia. Lancet 383, 828–840. doi: 10.1016/S0140-6736(13)60630-3
Luyendijk, W., and Bots, G. T. (1986). Hereditary cerebral hemorrhage. Scand. J. Clin. Lab. Invest. 46:391. doi: 10.3109/00365518609083687
Mao, C., Li, J., Dong, L., Huang, X., Lei, D., Wang, J., et al. (2021). Clinical phenotype and mutation spectrum of Alzheimer’s disease with causative genetic mutation in a Chinese cohort. Curr. Alzheimer Res. 18, 265–272. doi: 10.2174/1567205018666210608120339
Mastromoro, G., Gambardella, S., Marchionni, E., Campopiano, R., Traversa, A., Di Bonaventura, C., et al. (2019). Unusual segregation of APP mutations in monogenic Alzheimer disease. Neuro Degener. Dis. 19, 96–100. doi: 10.1159/000502906
Matsumura, Y., Kitamura, E., Miyoshi, K., Yamamoto, Y., Furuyama, J., and Sugihara, T. (1996). Japanese siblings with missense mutation (717Val –> Ile) in amyloid precursor protein of early-onset Alzheimer’s disease. Neurology 46, 1721–1723. doi: 10.1212/WNL.46.6.1721
McKhann, G. M., Knopman, D. S., Chertkow, H., Hyman, B. T., Jack, C. R. Jr., Kawas, C. H., et al. (2011). The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269. doi: 10.1016/j.jalz.2011.03.005
McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D., and Stadlan, E. M. (1984). Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of department of health and human services task force on Alzheimer’s Disease. Neurology 34, 939–944. doi: 10.1212/WNL.34.7.939
Mirra, S. S., Heyman, A., McKeel, D., Sumi, S. M., Crain, B. J., Brownlee, L. M., et al. (1991). The consortium to establish a registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486. doi: 10.1212/WNL.41.4.479
Mullan, M., Crawford, F., Axelman, K., Houlden, H., Lilius, L., Winblad, B., et al. (1992). A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat. Genet. 1, 345–347. doi: 10.1038/ng0892-345
Mullan, M., Houlden, H., Crawford, F., Kennedy, A., Rogues, P., and Rossor, M. (1993a). Age of onset in familial early onset Alzheimer’s disease correlates with genetic aetiology. Am. J. Med. Genet. 48, 129–130. doi: 10.1002/ajmg.1320480303
Mullan, M., Tsuji, S., Miki, T., Katsuya, T., Naruse, S., Kaneko, K., et al. (1993b). Clinical comparison of Alzheimer’s disease in pedigrees with the codon 717 Val–>Ile mutation in the amyloid precursor protein gene. Neurobiol. Aging 14, 407–419. doi: 10.1016/0197-4580(93)90099-W
Muresan, V., and Ladescu Muresan, Z. (2015). Amyloid-β precursor protein: Multiple fragments, numerous transport routes and mechanisms. Exp. Cell Res. 334, 45–53. doi: 10.1016/j.yexcr.2014.12.014
Murrell, J. R., Hake, A. M., Quaid, K. A., Farlow, M. R., and Ghetti, B. (2000). Early-onset Alzheimer disease caused by a new mutation (V717L) in the amyloid precursor protein gene. Arch. Neurol. 57, 885–887. doi: 10.1001/archneur.57.6.885
Nan, S. J., Han, Y. Q., Fan, J., and Chen, Q. H. (2014). Blepharospasm in familial AD secondary to an APP mutation (V715M). Acta Neurol. Belg. 114, 333–334. doi: 10.1007/s13760-014-0291-1
Nilsberth, C., Westlind-Danielsson, A., Eckman, C. B., Condron, M. M., Axelman, K., Forsell, C., et al. (2001). The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat. Neurosci. 4, 887–893. doi: 10.1038/nn0901-887
Nunan, J., and Small, D. H. (2000). Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett. 483, 6–10. doi: 10.1016/S0014-5793(00)02076-7
Panegyres, P. K., and Chen, H. Y. (2014). Early-onset Alzheimer’s disease: a global cross-sectional analysis. Eur. J. Neurol. 21, 1149–54, e64–e65. doi: 10.1111/ene.12453
Park, H. K., Na, D. L., Lee, J. H., Kim, J. W., and Ki, C. S. (2008). Identification of PSEN1 and APP gene mutations in Korean patients with early-onset Alzheimer’s disease. J. Korean Med. Sci. 23, 213–217. doi: 10.3346/jkms.2008.23.2.213
Pasalar, P., Najmabadi, H., Noorian, A. R., Moghimi, B., Jannati, A., Soltanzadeh, A., et al. (2002). An Iranian family with Alzheimer’s disease caused by a novel APP mutation (Thr714Ala). Neurology 58, 1574–1575. doi: 10.1212/WNL.58.10.1574
Pera, M., Alcolea, D., Sánchez-Valle, R., Guardia-Laguarta, C., Colom-Cadena, M., Badiola, N., et al. (2013). Distinct patterns of APP processing in the CNS in autosomal-dominant and sporadic Alzheimer disease. Acta Neuropathol. 125, 201–213. doi: 10.1007/s00401-012-1062-9
Román, G. C., Mancera-Páez, O., and Bernal, C. (2019). Epigenetic factors in late-onset Alzheimer’s disease: MTHFR and CTH gene polymorphisms, metabolic transsulfuration and methylation pathways, and B vitamins. Int. J. Mol. Sci. 20:319. doi: 10.3390/ijms20020319
Ryan, N. S., and Rossor, M. N. (2010). Correlating familial Alzheimer’s disease gene mutations with clinical phenotype. Biomark. Med. 4, 99–112. doi: 10.2217/bmm.09.92
Ryan, N. S., Nicholas, J. M., Weston, P. S. J., Liang, Y., Lashley, T., Guerreiro, R., et al. (2016). Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer’s disease: a case series. Lancet Neurol. 15, 1326–1335. doi: 10.1016/S1474-4422(16)30193-4
Scheltens, P., Blennow, K., Breteler, M. M., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517. doi: 10.1016/S0140-6736(15)01124-1
Shimada, H., Ataka, S., Tomiyama, T., Takechi, H., Mori, H., and Miki, T. (2011). Clinical course of patients with familial early-onset Alzheimer’s disease potentially lacking senile plaques bearing the E693Δ mutation in amyloid precursor protein. Dement. Geriatr. Cogn. Disord. 32, 45–54. doi: 10.1159/000330017
Shimada, H., Minatani, S., Takeuchi, J., Takeda, A., Kawabe, J., Wada, Y., et al. (2020). Heavy tau burden with subtle amyloid β accumulation in the cerebral cortex and cerebellum in a case of familial Alzheimer’s disease with APP Osaka mutation. Int. J. Mol. Sci. 21:4443. doi: 10.3390/ijms21124443
Sieczkowski, E., Milenkovic, I., Venkataramani, V., Giera, R., Ströbel, T., Höftberger, R., et al. (2015). I716F AβPP mutation associates with the deposition of oligomeric pyroglutamate amyloid-β and α-synucleinopathy with Lewy bodies. J. Alzheimers Dis. 44, 103–114. doi: 10.3233/JAD-141524
Smits, L. L., Pijnenburg, Y. A., Koedam, E. L., van der Vlies, A. E., Reuling, I. E., Koene, T., et al. (2012). Early onset Alzheimer’s disease is associated with a distinct neuropsychological profile. J. Alzheimers Dis. 30, 101–108. doi: 10.3233/JAD-2012-111934
Sorbi, S., Nacmias, B., Forleo, P., Piacentini, S., Latorraca, S., and Amaducci, L. (1995). Epistatic effect of APP717 mutation and apolipoprotein E genotype in familial Alzheimer’s disease. Ann. Neurol. 38, 124–127. doi: 10.1002/ana.410380120
Sperling, R. A., Aisen, P. S., Beckett, L. A., Bennett, D. A., Craft, S., Fagan, A. M., et al. (2011). Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 280–292. doi: 10.1016/j.jalz.2011.03.003
Swearer, J. M., O’Donnell, B. F., Drachman, D. A., and Woodward, B. M. (1992). Neuropsychological features of familial Alzheimer’s disease. Ann. Neurol. 32, 687–694. doi: 10.1002/ana.410320513
Thambisetty, M., An, Y., and Tanaka, T. (2013). Alzheimer’s disease risk genes and the age-at-onset phenotype. Neurobiol. Aging 34, 2696.e1–2696.e5. doi: 10.1016/j.neurobiolaging.2013.05.028
Theuns, J., Marjaux, E., Vandenbulcke, M., Van Laere, K., Kumar-Singh, S., Bormans, G., et al. (2006). Alzheimer dementia caused by a novel mutation located in the APP C-terminal intracytosolic fragment. Hum. Mutat. 27, 888–896. doi: 10.1002/humu.20402
Tomiyama, T., Nagata, T., Shimada, H., Teraoka, R., Fukushima, A., Kanemitsu, H., et al. (2008). A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol. 63, 377–387. doi: 10.1002/ana.21321
Vermunt, L., Sikkes, S. A. M., van den Hout, A., Handels, R., Bos, I., van der Flier, W. M., et al. (2019). Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimers Dement. 15, 888–898. doi: 10.1016/j.jalz.2019.04.001
Vöglein, J., Paumier, K., Jucker, M., Preische, O., McDade, E., Hassenstab, J., et al. (2019). Clinical, pathophysiological and genetic features of motor symptoms in autosomal dominant Alzheimer’s disease. Brain 142, 1429–1440. doi: 10.1093/brain/awz050
Wakutani, Y., Watanabe, K., Adachi, Y., Wada-Isoe, K., Urakami, K., Ninomiya, H., et al. (2004). Novel amyloid precursor protein gene missense mutation (D678N) in probable familial Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 75, 1039–1042. doi: 10.1136/jnnp.2003.010611
Wang, Q., Jia, J., Qin, W., Wu, L., Li, D., Wang, Q., et al. (2015). A novel AβPP M722K mutation affects amyloid-β secretion and tau phosphorylation and may cause early-onset familial Alzheimer’s disease in Chinese individuals. J. Alzheimers Dis. 47, 157–165. doi: 10.3233/JAD-143231
Wattendorff, A. R., Bots, G. T., Went, L. N., and Endtz, L. J. (1982). Familial cerebral amyloid angiopathy presenting as recurrent cerebral haemorrhage. J. Neurol. Sci. 55, 121–135. doi: 10.1016/0022-510X(82)90094-6
Xiao, X., Liu, H., Liu, X., Zhang, W., Zhang, S., and Jiao, B. (2021). APP, PSEN1, and PSEN2 variants in Alzheimer’s disease: systematic re-evaluation according to ACMG guidelines. Front. Aging Neurosci. 13:695808. doi: 10.3389/fnagi.2021.695808
Xu, Y., Liu, X., Shen, J., Tian, W., Fang, R., Li, B., et al. (2018). The whole exome sequencing clarifies the genotype- phenotype correlations in patients with early-onset dementia. Aging Dis. 9:696–705. doi: 10.14336/AD.2018.0208
Zádori, D., Füvesi, J., Timár, E., Horváth, E., Bencsik, R., Szépfalusi, N., et al. (2017). The report of p.Val717Phe mutation in the APP gene in a Hungarian family with Alzheimer disease: a phenomenological study. Alzheimer Dis. Assoc. Disord. 31, 343–345. doi: 10.1097/WAD.0000000000000206
Zekanowski, C., Styczyńska, M., Pepłońska, B., Gabryelewicz, T., Religa, D., Ilkowski, J., et al. (2003). Mutations in presenilin 1, presenilin 2 and amyloid precursor protein genes in patients with early-onset Alzheimer’s disease in Poland. Exp. Neurol. 184, 991–996. doi: 10.1016/S0014-4886(03)00384-4
Zhang, G., Xie, Y., Wang, W., Feng, X., and Jia, J. (2017). Clinical characterization of an APP mutation (V717I) in five Han Chinese families with early-onset Alzheimer’s disease. J. Neurol. Sci. 372, 379–386. doi: 10.1016/j.jns.2016.10.039
Zhou, J., Chen, Y., Meng, F., Zhang, K., Liu, X., and Peng, G. (2020). Presenilin 1 and APP gene mutations in early-onset AD families from a Southeast region of China. Curr. Alzheimer Res. 17, 540–546. doi: 10.2174/1567205017666200624195809
Keywords: Alzheimer’s disease, APP mutations, clinical characteristics, genotype-phenotypic, non-dementia symptoms, neurological symptoms
Citation: Liu Y, Xiao X, Liu H, Liao X, Zhou Y, Weng L, Zhou L, Liu X, Bi X-y, Xu T, Zhu Y, Yang Q, Zhang S, Hao X, Zhang W, Wang J, Jiao B and Shen L (2022) Clinical characteristics and genotype-phenotype correlation analysis of familial Alzheimer’s disease patients with pathogenic/likely pathogenic amyloid protein precursor mutations. Front. Aging Neurosci. 14:1013295. doi: 10.3389/fnagi.2022.1013295
Received: 06 August 2022; Accepted: 12 September 2022;
Published: 14 October 2022.
Edited by:
John Wesson Ashford, United States Department of Veterans Affairs, United StatesReviewed by:
Eva Bagyinszky, Gachon University, South KoreaElena Milanesi, Victor Babes National Institute of Pathology (INCDVB), Romania
Shigeki Kawabata, Sompo Care Inc., Japan
Marc Tambini, Rutgers University, Newark, United States
Copyright © 2022 Liu, Xiao, Liu, Liao, Zhou, Weng, Zhou, Liu, Bi, Xu, Zhu, Yang, Zhang, Hao, Zhang, Wang, Jiao and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Jiao, 4011070@csu.edu.cn; Lu Shen, shenlu@csu.edu.cn