 Nadia V. Harerimana1,2,3
Nadia V. Harerimana1,2,3 Kathryn R. Bowles
Kathryn R. Bowles
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci. , 20 October 2022
Sec. Alzheimer's Disease and Related Dementias
Volume 14 - 2022 | https://doi.org/10.3389/fnagi.2022.1021918
This article is part of the Research Topic Genomics and Neurodegenerative Dementias View all 6 articles
Advances in genomic research over the last two decades have greatly enhanced our knowledge concerning the genetic landscape and pathophysiological processes involved in multiple neurodegenerative diseases. However, current insights arise almost exclusively from studies on individuals of European ancestry. Despite this, studies have revealed that genetic variation differentially impacts risk for, and clinical presentation of neurodegenerative disease in non-European populations, conveying the importance of ancestry in predicting disease risk and understanding the biological mechanisms contributing to neurodegeneration. We review the genetic influence of two important disease-associated loci, 17q21.31 (the “MAPT locus”) and APOE, to neurodegenerative disease risk in non-European populations, touching on global population differences and evolutionary genetics by ancestry that may underlie some of these differences. We conclude there is a need to increase representation of non-European ancestry individuals in genome-wide association studies (GWAS) and biomarker analyses in order to help resolve existing disparities in understanding risk for, diagnosis of, and treatment for neurodegenerative diseases in diverse populations.
The global prevalence of neurodegenerative diseases is likely to rise with the increasing life expectancy worldwide. Approximately 50 million people are currently affected by dementia (Nichols et al., 2019), which is estimated to increase to 130 million by 2050.1 Dementia is a major cause of disability, institutionalization, and mortality as well as a huge social and economic burden associated with the care of affected individuals (see text footnote 1). Among neurodegenerative diseases, Alzheimer’s disease (AD) is the most common and accounts for 60–70% of all cases (Seshadri and Wolf, 2007), whereas Parkinson’s disease (PD)/Lewy body dementia (LBD) is the second-most common neurodegenerative disorder, with a global prevalence of over 6 million (Nichols et al., 2019).
During the previous few decades, large-scale genome-wide association studies (GWAS) have succeeded in uncovering the genetic landscape and pathophysiological processes involved in neurodegenerative diseases. In particular, we have learned that apolipoprotein E (APOE) is the major susceptibility gene for late-onset AD (Lambert et al., 2013) and LBD (Chia et al., 2021). Similarly, the 17q21.31 locus, which is a 970 kb region of high linkage disequilibrium (LD) encoding two distinct haplotypes (H1 and H2) and encompassing the MAPT gene (Figure 1A), has been genetically associated with several primary tauopathies, as well as PD (Höglinger et al., 2011; Kouri et al., 2015; Jun et al., 2016; Bandres-Ciga et al., 2019; Nalls et al., 2019). Although this accumulated knowledge has greatly expanded our understanding of neurodegenerative diseases, these studies have focused on populations of European ancestry, thus it remains unclear how this knowledge extends to and is applicable to estimation of disease risk and understanding of pathogenic disease mechanisms in other global populations.
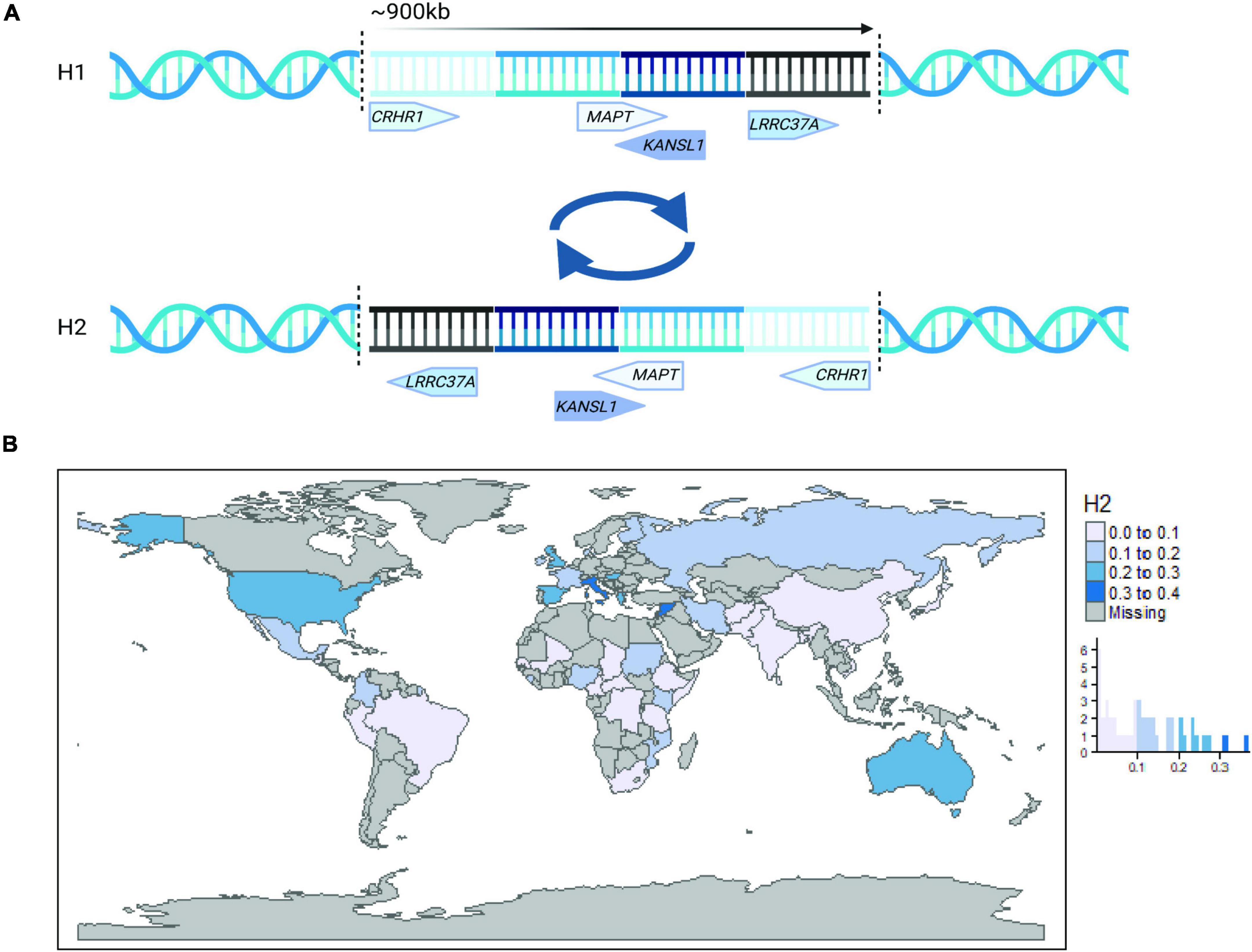
Figure 1. The 17q21.31 locus. (A) A simplified schematic illustration of the 17q21.31 inversion locus. The inversion breakpoints are represented by vertical dotted lines, and the approximate location and direction of four representative genes are presented for haplotype H1 (top) and H2 (bottom). (B) Worldwide 17q21.31 H2 allele frequency plotted by country and histogram. Haplotype frequency estimates were obtained from Donnelly et al. (2010), Steinberg et al. (2012), and publicly available 1000 Genomes Project data (www.internationalgenome.org).
To date, a large body of research has shown that genetic studies have not captured the level of diversity that exists globally, and neurodegenerative diseases are not an exception (Sirugo et al., 2019). Indeed, many European-based GWAS findings have not been replicated in other ancestral populations, thus making their findings less valuable and applicable across different populations. In this review, we discuss the evolutionary selection and differences in the genetic architecture of two important loci in neurodegenerative diseases, 17q21.31 and APOE, among diverse global populations. We also discuss the genetic associations of these loci with neurodegenerative diseases in non-European populations. Additionally, we discuss examples that illustrate why the inclusion of ethnically diverse populations in neurodegenerative genomic and biomarker studies will facilitate our understanding of the contribution of 17q21.31 and APOE to disease risk worldwide.
The 17q21.31 H2 haplotype occurs at strikingly different frequencies across global populations (Figure 1B), with the highest frequencies occurring in Southern Europe and Southwest Asia (Donnelly et al., 2010). In contrast, it occurs at variable frequencies across Africa and is practically absent in East Asian populations (Stefansson et al., 2005; Donnelly et al., 2010; Alves et al., 2015). The source of this apparent positive selection in Europeans has been under debate since the identification of the structural inversion in 2005 (Cruts et al., 2005; Stefansson et al., 2005).
The inversion at 17q21.31 is most likely a result of non-allelic homologous recombination, facilitated by the presence of repetitive, low copy number repeats at the distal ends of the locus (Stefansson et al., 2005; Zody et al., 2008; Itsara et al., 2012; Dennis and Eichler, 2016). By comparing sequence similarities across haplotypes, global populations, and non-human primates, several estimates place the origin of the inversion event at ∼3 million years ago (Cruts et al., 2005; Stefansson et al., 2005), pre-dating modern Homo sapiens. Indeed, the H2 inversion has been identified in non-human primates and is highly polymorphic, indicative of multiple, repetitive inversion events over the last 10 million years of evolution that have increased in their copy number complexity in humans (Zody et al., 2008; Watson et al., 2014). This hypothesis is also supported by a 30 kbp region of striking similarity between H1 and H2 haplotypes surrounding the CRHR1 gene at the 5′ end of the inversion, which is indicative of a possible double recombination event (Steinberg et al., 2012). However, another most common recent ancestor analysis based on haplotype structure rather than sequence similarity estimates a more recent inversion event occurring 16,400–108,400 years ago (Donnelly et al., 2010), which the authors argue correlates better with current global distributions of the H2 haplotype.
Interestingly, despite its low frequency, the inverted H2 allele has been proposed as the ancestral allele of African origin (Boettger et al., 2012; Steinberg et al., 2012), as evidenced by the presence of an H2 sub-haplotype lacking multiple duplication events (H2’) enriched in African hunter-gatherer populations compared to West African or European populations (Steinberg et al., 2012). Additionally, the H2 haplotype is much more homogeneous than H1, but greater diversity of H2 has been identified across African populations compared to Europeans (Stefansson et al., 2005; Alves et al., 2015). Given that the H2 inversion may have originated in Africa, it is curious why its frequency should be so low. The frequency of H2, combined with the striking homogeneity of H2 haplotypes in non-African populations, has led to the suggestion of a recent bottleneck and/or selective sweep following migration and gene flow out of Africa (Stefansson et al., 2005; Steinberg et al., 2012). However, more recent analyses suggest that there is little evidence for positive selection of H2 (Alves et al., 2015; Shimada and Nishida, 2020), and population differences may be explained by restricted recombination between haplotypes and demographic history, without the need for additional selective pressure (Steinberg et al., 2012; Alves et al., 2015).
Donnelly et al. (2010) highlighted that the high frequency of H2 in the Mediterranean raises the possibility of a Southwest Asian origin for the inversion, which would explain the comparatively high frequency of H2 in North African populations by gene flow back into Africa and would suggest that H2 haplotypes that originated in Africa could be in a non-inverted orientation. The inversion has been found to be highly polymorphic in Old World monkey species and Orangutan (Zody et al., 2008), and assessment of chromosome structure in H2 homozygote humans indicated heterozygosity of the inversion was present (Rao et al., 2010). However, this finding has not yet been replicated, possibly because studies confirming the H2 inversion status by fluorescent in situ hybridization or BAC cloning have utilized small samples from restricted ancestral populations (Stefansson et al., 2005; Gijselinck et al., 2006; Zody et al., 2008; Donnelly et al., 2010).
As well as variable H2 inversion frequencies across global populations, there are also highly variable frequencies of complex duplication events and copy number variants (CNVs) at the inversion breakpoints across populations on both haplotypes, but particularly on H1 (Boettger et al., 2012; Steinberg et al., 2012). Duplication events and CNVs are a major source of human genetic diversity by facilitating the creation of novel genes and regulatory elements (McElroy et al., 2009; Dennis and Eichler, 2016). It is thought that this is achieved by removing the ancestral selection restraint on a locus through the creation of genetic redundancy (Dennis and Eichler, 2016). It is therefore likely that variable CNVs on different H1 or H2 sub-haplotypes are likely to have altered functional effects, and that these are variable across different global populations. In 2012, two groups independently identified specific duplication events on the H1 haplotype that vary across populations (Boettger et al., 2012; Steinberg et al., 2012). Of note was the partial duplication of KANSL1 that is much more frequent in Europeans compared to Africans or East Asians and results in the production of novel transcripts of unknown functional effect (Boettger et al., 2012). In contrast, increased copy number of a region encompassing NSF, which is upstream of KANSL1, appears to be more frequent in East Asian populations compared to Europeans and Africans (Steinberg et al., 2012).
Surprisingly, since the identification of complex CNVs at the 17q21.31 locus, very little has been done to understand the functional effects of these variants and their relevance to disease risk. Similarly, while the distribution of H1 and H2 haplotypes and sub-haplotypes vary across global populations, the contribution of this locus to neurodegenerative disease risk in non-European populations remains largely unexplored. However, given the structural complexity of the locus and complex evolutionary history that results in distinct population-specific structural, and presumably functional, differences, it is important to expand our investigation of the 17q21.31 locus beyond European ancestry to truly understand its function and contribution to disease risk.
Similar to the 17q21.31 H2 haplotype, the distribution of different APOE alleles varies considerably across the world (Figure 2A). The ε3 allele is the most common isoform globally, with the highest allele frequency in Asia, Europe, and Africa in descending order (Singh et al., 2006). APOE ε2 is the least common isoform and is markedly higher in frequency in South African Zulu (19%) and Eurasian (18%) populations (Chikosi et al., 2000; Singh et al., 2006). Unlike in European populations and in contrast to the 17q21.31 H2 haplotype, the APOE ε4 allele is relatively highly abundant in central and southern regions of Africa, mainly in populations such as Pygmies (41%) and Khoisan (37%) (Corbo and Scacchi, 1999).
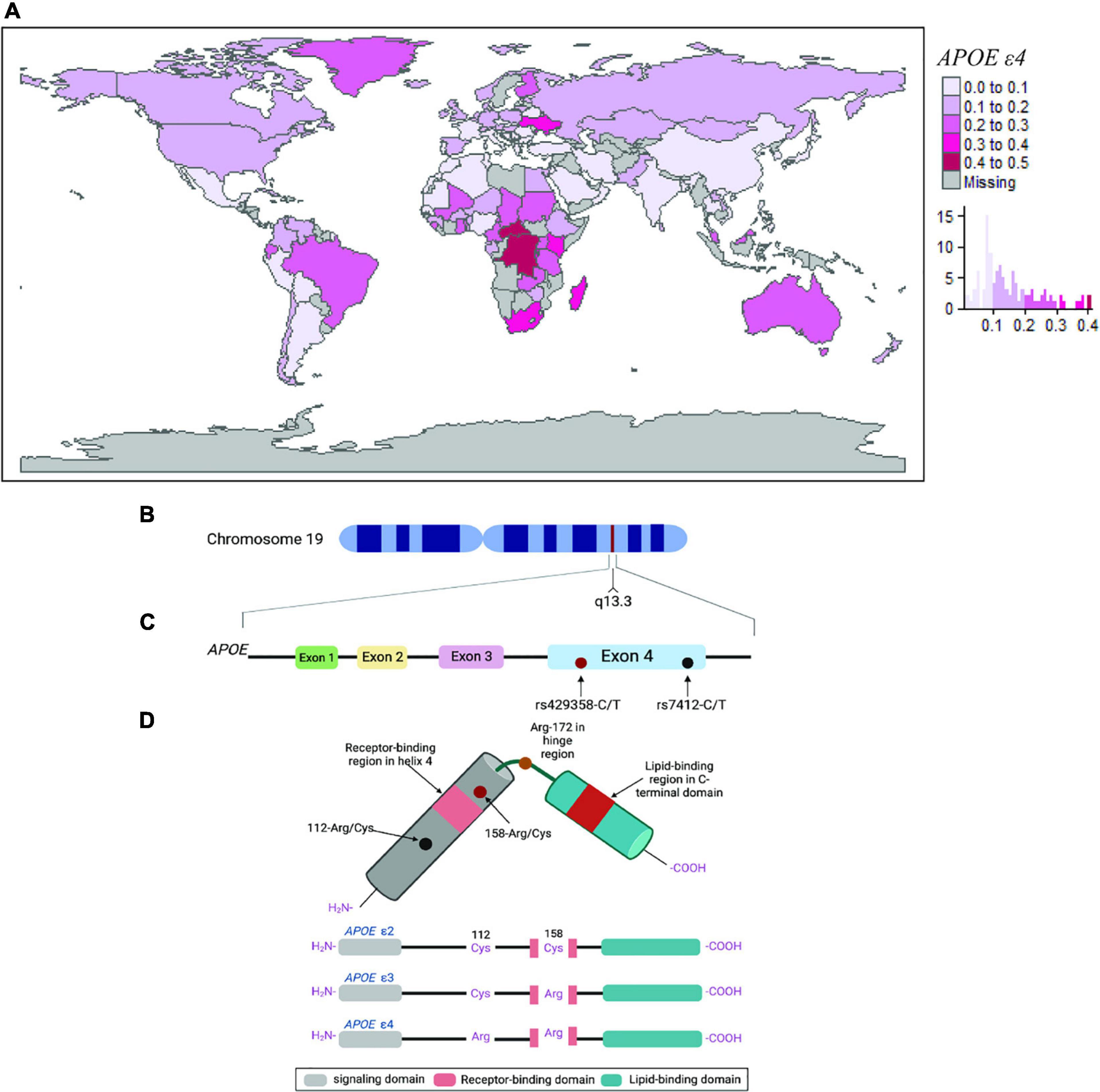
Figure 2. The APOE ε4 locus. (A) Worldwide APOE ε4 allele frequency plotted by country and by histogram. Allele frequency estimates were obtained from Singh et al. (2006) and other literature search (see Supplementary Information). (B–D) Schematic representation of the APOE ε4 locus. (B) The structure and location of the APOE gene on chromosome 19. (C) Estimated structure of the APOE protein. (D) The three main APOE isoforms ε2, ε3, and ε4, respectively, are the result of non-synonymous polymorphisms that cause amino acid changes at positions 112 and 158 of the APOE protein.
The APOE gene is widely expressed in all vertebrates (Duggan and Callard, 2001), but the ε4 allele has been only observed in chimpanzees and humans (Hanlon and Rubinsztein, 1995; Vamathevan et al., 2008). In early humans, it is thought that the origin of the ε4 allele correlates with the increased requirements for endurance in locomotion (i.e., physical exercise) around 1.8 million years ago during the development of a hunter-gatherer lifestyle, whereas the ε2 and ε3 alleles are estimated to date back to only 200,000–300,000 years ago (Raichlen and Alexander, 2014). APOE isoforms are thought to have originated in a South Asian subpopulation, followed by the westward migration of modern humans to Asia, Africa, Europe, and then North and South America. This hypothesis is supported by the relatively high frequency of ε3 in Asia (85%) and the near absence of ε2 in Amerindian populations from North and South America (4.6–4.9%) (Singh et al., 2006). The historical allelic divergence, combined with the relatively high frequency of the ε4 allele in populations where an economy of hunting still exists, or food supply has been sporadically available, indicates that ε4 is likely to be the ancestral allele (Hanlon and Rubinsztein, 1995; Seixas et al., 1999).
The human APOE gene is located on chromosome 19 at position q13.3 (Figures 2B,C) and encodes for a 299 amino acid protein (∼34 kDa) with multiple functions, particularly with cholesterol metabolism, lipid homeostasis, and innate immunity (Davignon et al., 1988), which are all likely to contribute to increased reproductive success and protection against infection load. Furthermore, APOE isoforms differ by a unique amino acid combination at positions 112 and 158: ε2 (Cys112, Cys158), ε3 (Cys112, Arg158), and ε4 (Arg112, Arg158), which modify their structure and function (Figure 2D). In regard to lipoprotein-binding preferences, APOE ε4 has a high affinity to very low-density lipoprotein (VLDL) particles (Mahley and Rall, 2000) while the ε2 has poor binding affinity to low-density lipoprotein (LDL), which are both associated with increased plasma cholesterol and triglycerides (Zhao et al., 2018). In addition, the ε4 allele has an exposed Arg-61 that is known to interact with Glu-255, which does not occur in the other isoforms, and has been suggested to represent another underlying factor to the adverse effects of the ε4 allele (Raffaï et al., 2001). Interestingly, mouse APOE ε4 lacks the equivalent of Arg-61 and resembles the ε3 allele in terms of lipoprotein-binding preferences (Raffaï et al., 2001).
Several hypotheses have been formulated about the functional and potential selective pressures contributing to the successful adaptation of the ε4 allele, including climate conditions, conservation practices, and infection load (Corbo and Scacchi, 1999). Consistent with the notion that meat eating (i.e., increased dietary fats) improves adaptive responses to pathogens and facilitates reproduction in populations where infections are highly prevalent, it is noted that fertility and fecundity were found to be higher in APOE ε4 allele carriers in the indigenous populations of South America (Corbo et al., 2004), West Africa (van Exel et al., 2017), and rural individuals of Western Europe (Jasienska et al., 2015). This hypothesis is also supported by the predominant presence of ε4 in indigenous people of Central Africa (40%), Australia (26%), Oceania (49%), and South America (27%) (Corbo and Scacchi, 1999; Singh et al., 2006).
In contrast, positive selection for the APOE ε4 allele in European populations is thought to reflect adaptations to extreme climate conditions (Eisenberg et al., 2010; Huebbe and Rimbach, 2017), such as high-latitude cold environments [e.g., Northern latitudes; (Lovegrove, 2003; Froehle and Schoeninger, 2006)] or low-latitude hot environments [e.g., near the equatorial level; (Eisenberg et al., 2010; Huebbe et al., 2011)], where human energy expenditure is known to be higher due to an increased requirement for thermoregulation. In Europe and Asia, ε4 allele distribution appears to follow a North-to-South latitudinal gradient, with a fourfold higher frequency in the North (e.g., >25% in Finland) than in the Mediterranean or South Asia area (e.g., <10% in Sardinia) (Egert et al., 2012). The most likely explanation is that high temperatures may influence demand for cholesterol indirectly via metabolism increase, thus promoting the accumulation of the ε4 allele. In this context, the APOE ε4 allele is also thought to protect against vitamin D deficiency via better absorption of fat-soluble vitamin D in geographical regions, such as northern Europe, subjected to diminished sunlight exposure (Gerdes, 2003; Huebbe and Rimbach, 2017).
Similar to the 17q21.31 locus, although there has been investigation with regard to the evolution and global distribution of APOE alleles, there are sparse data and inconsistent findings on the role of the ε4 allele in relation to neurodegenerative disease risk across global populations. However, understanding the contribution of APOE to disease risk in non-European populations is essential to uncover the biological mechanisms underlying global disease in order to develop appropriate and effective therapeutics for different populations.
The 17q21.31 locus has been genetically associated with 113 different traits across 176 studies (Buniello et al., 2019).2 Of these studies, only 12% included analysis of Asian, African, or Hispanic populations, and did not include assessment of any neurological, psychiatric, or neurodegenerative phenotypes. However, in European populations, the 17q21.31 locus has been genetically associated with several primary tauopathies, including progressive supranuclear palsy (Höglinger et al., 2011), corticobasal degeneration (CBD) (Kouri et al., 2015), frontotemporal dementia (FTD) (Ferrari et al., 2019), as well as the secondary tauopathy AD (Jun et al., 2016; Bellenguez et al., 2022) and Parkinson’s disease (Bandres-Ciga et al., 2019; Nalls et al., 2019), with the H2 haplotype consistently conferring protection against disease risk (Table 1). Given that Europeans account for an estimated 73% of all individuals included in genetic studies (Sirugo et al., 2019), it is therefore likely that the lack of association of the 17q21.31 locus with neurodegenerative disease in non-European populations is due to a paucity in data and analyses, especially for rarer diseases such as PSP.
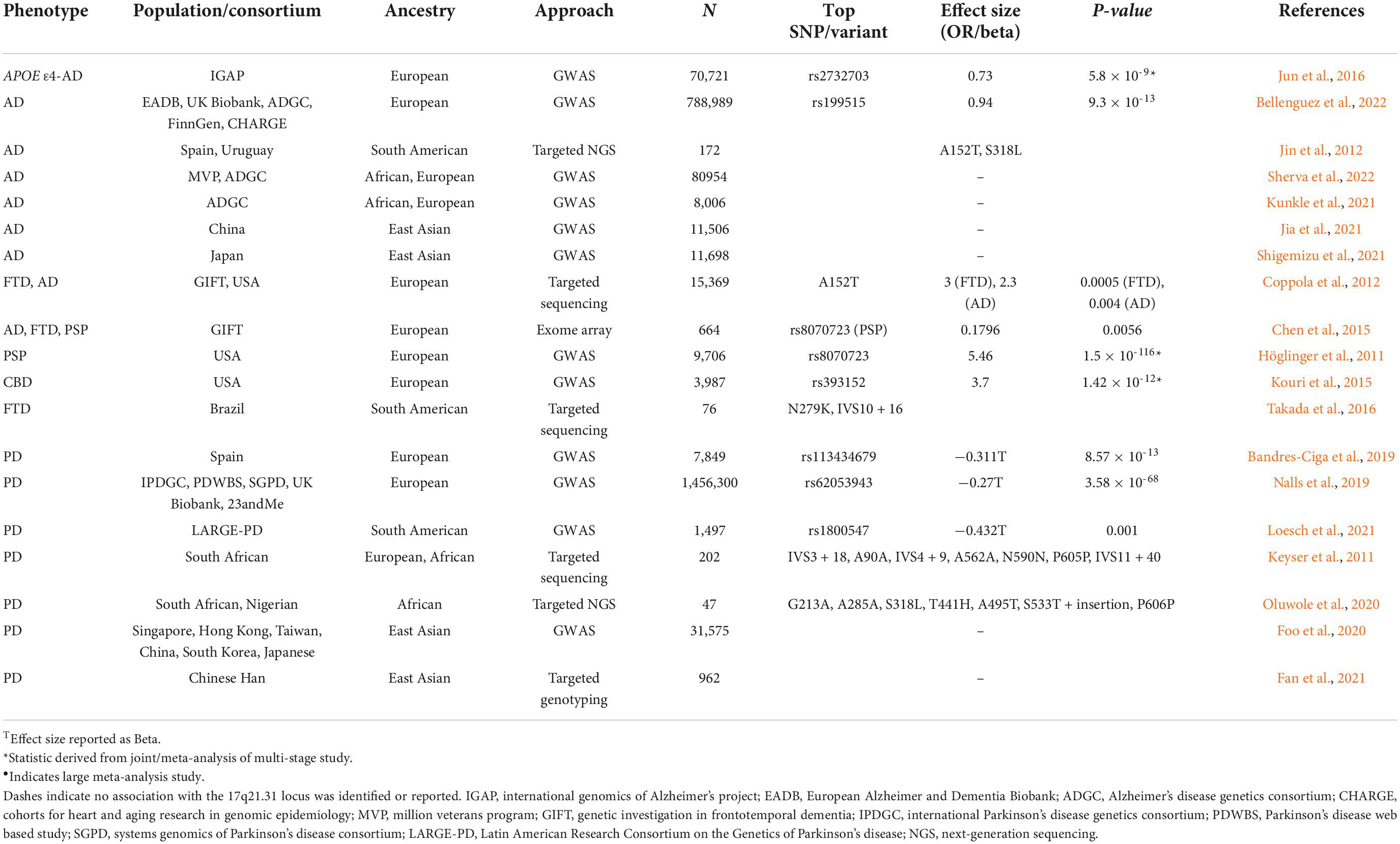
Table 1. Summary of 17q21.31 genetic association with neurodegenerative disease across global populations.
However, given global population differences in 17q21.31 structure, variation, and haplotype frequency, it is also possible that the lack of replication of genome-wide association signals at this locus for more common disorders, such as AD and PD, is a result of varied genetic architecture in non-European populations. For example, two recent PD GWAS conducted in Chinese Han and Japanese populations do not replicate the 17q21.31 association consistently observed in Europeans (Foo et al., 2020; Fan et al., 2021). This is not surprising, as the protective H2 haplotype is absent in these populations, and the relatively small sample sizes compared to European-ancestry consortia studies may preclude the identification of less common H1 sub-haplotypes that contribute to disease risk. To our knowledge, there are currently no PD GWAS that have been conducted in African ancestry populations; however the IPDGC-Africa consortium3 is addressing this gap in our understanding of PD genetics. Interestingly, African ancestry in South American Latino populations has been found to be protective against PD risk (Loesch et al., 2021), although this was not specific to the 17q21.31 locus. The same study also replicated the European effect size and direction of the 17q21.31 association with PD risk in South Americans, although due to a small sample size, the association did not reach significance (Loesch et al., 2021). Regardless, this is an indication of the relevance of the 17q21.31 locus to PD risk in non-European and admixed populations that warrants further investigation.
Despite the lack of PD GWAS, several groups have attempted to identify novel MAPT variants that may contribute to PD risk in African populations. For example, Keyser et al. (2011) identified two novel MAPT variants in South African individuals with PD (A91V and V635I), although these were predicted to be benign (Keyser et al., 2011). Similarly, Oluwole et al. (2020) identified an additional seven novel MAPT variants in black South African and Nigerian individuals that may be associated with PD risk, with unknown functional effects (Oluwole et al., 2020). While these data indicate the presence of ancestry-specific variants of MAPT, the association with PD risk is less convincing; this is unsurprising given that the GWAS association in European populations spans a ∼1 Mb region, which incorporates numerous genes other than MAPT. There is also limited evidence that the 17q21.31 signal implicates MAPT as the causative gene, but rather recent studies favor KANSL1, CRHR1, or LRRC37A2 as likely candidates contributing to PD risk (Loesch et al., 2021; Yao et al., 2021; Bowles et al., 2022). The contribution of rare variants in these specific genes has not yet been investigated in any population.
While Alzheimer’s disease is neuropathologically defined by the presence of neurofibrillary tau tangles, the genetic association of the 17q21.31 locus with AD risk is less clear. Until recently, an association signal within either MAPT or across the 17q21.31 locus has been notably absent, with the exception of in APOE ε4- populations specifically (Jun et al., 2016). However, it has just reached genome-wide significance in the most recent and largest European AD GWAS, independent of APOE genotype (Bellenguez et al., 2022). In addition, the rare MAPT A152T variant has been associated with the development of an AD-like dementia (Coppola et al., 2012), suggesting that the locus may play a role in modifying AD risk. Consistent with this, the MAPT A152T and S318L mutations were observed to occur 3× as frequently in AD cases compared to controls in an Ibero-American cohort, with Spanish and Uruguayan ancestry (Jin et al., 2012), with additional missense variants identified in exons 4a, 7, and 10 with unknown effects (Jin et al., 2012).
Several large GWAS have been conducted in non-European ancestry populations for AD, including the largest African-American AD GWAS to date, which utilized data derived from the Million Veterans Program (Sherva et al., 2022), as well as a recent publication from the ADGC (Kunkle et al., 2021). Large-scale studies have also been conducted in East Asian populations from China and Japan (Jia et al., 2021; Shigemizu et al., 2021). Similar to previous, smaller European ancestry studies, the 17q21.31 locus was not associated with AD risk in any of these GWAS. However, assessment of the 17q21.31 association in APOE ε4- individuals specifically may provide additional power in studies of non-European populations as they have done in Europeans (Jun et al., 2016), but this has been overlooked. Similarly, assessment of the 17q21.31 locus in fine-mapping and local ancestry analyses of known AD loci in non-European populations has been excluded for the same reason. As such, APOE genotype stratification analyses in these groups remain to be carried out in order to determine the contribution of the 17q21.31 locus and MAPT to AD risk.
Primary tauopathies, such as PSP and FTD, have been more challenging to investigate by GWAS due to their relative rarity compared to AD and PD. However, the effect size of the 17q21.31 locus is much larger for these disorders (e.g., odds ratio ∼4 compared to ∼1.5 for PD) (Höglinger et al., 2011; Nalls et al., 2019), thus necessitating smaller studies to observe a significant association in this region. Furthermore, rare autosomal dominant mutations in MAPT have demonstrated the direct relevance of this gene and locus to FTD/PSP pathogenesis.4 Given the relatively low frequency of PSP and FTD, little has been done to investigate the contribution of the locus in non-European populations. For example, Chen et al. (2015) embarked on a multi-ancestral exome array study of AD, FTD, and PSP, but there were not sufficient FTD or PSP cases available to carry out the multi-ancestry replication analyses (Chen et al., 2015).
Frontotemporal dementia has been reported as being less common in Asia than in Europe, and fewer individuals with FTD reported a positive family history of the disorder (9.5–20%) compared to Europeans (30–50%) (Ng, 2015), indicating that there may be a reduced genetic contribution of the 17q21.31 locus to disease risk in Asia. However, as epidemiological and clinical studies have also suggested there could be a slightly higher prevalence of PSP in Japan compared to European populations (Takigawa et al., 2016), the prevalence of primary tauopathies in East Asia requires additional investigation. In either case, given the absence of the protective H2 haplotype in these populations, understanding the genetic contribution of the 17q21.31 locus to FTD and PSP in Asia will likely provide valuable insight into the pathogenic mechanisms specific to H1 that underlie these diseases. Additionally, a common sub-type of PSP has been described that is defined by the presence of cerebellar ataxia, which appears to be specific to Asia (Krzosek et al., 2022), and suggests that there is likely to be ancestry-specific variation across the 17q21.31 locus H1 haplotype in East Asian populations that may differentially contribute to disease risk and clinical phenotypes.
Numerous rare MAPT variants associated with FTD have been identified in China and Japan (Kasuga et al., 2015; Ng, 2015). MAPT has been reported as the most common pathogenic FTD gene in China (Jiang et al., 2021), compared to the C9ORF72 expansion that is most prevalent in European familial FTD cases (Smith et al., 2013). Numerous autosomal dominant FTD MAPT mutations have also been identified in Colombia at a relatively high frequency compared to other populations (Takada et al., 2016; Zuluaga-Castaño et al., 2021; Acosta-Uribe et al., 2022), which is likely due to multiple founder events and population bottlenecks arising from Spanish invasion and African enslavement, followed by outbreaks of infectious disease (Acosta-Uribe et al., 2022). However, the relative abundance of MAPT variants identified in Colombia may also reflect the substantial efforts being conducted by large initiatives focused on identifying AD and FTD mutations in this population specifically. Regardless, given the frequency of pathogenic rare variants in this population, it would be of interest to assess the impact of this history and admixture on common 17q21.31 variants and structure and resulting function and contribution to disease risk that may be unique to this group.
The APOE locus is a well-established genetic risk factor for Alzheimer’s disease (AD) (Corder et al., 1993). Given the evolutionary shifts of this gene, the APOE alleles present a pronounced stepwise effect (ε2 < ε3 < ε4), where the ε2 allele confers substantial protection for AD and the ε4 allele increases risk in European ancestry populations [ε4/ε4 odds ratio (OR):12.5], but not conclusively in other ethnic groups (Farrer et al., 1997; Tang et al., 1998; Reiman et al., 2020; Table 2). In African populations, the genetic association of the ε4 allele with AD risk is weak or sometimes absent, but relatively present in African American individuals (ε4/ε4 OR: 5.7) (Farrer et al., 1997). The most recent and largest genome-wide association meta-analysis of African American individuals also confirmed APOE as the strongest risk factor of AD in this group (Sherva et al., 2022). This may be attributable to the presence of European admixture in African Americans, thus accounting for the population differences in the effect of the ε4 allele. Furthermore, African populations harbor the greatest diversity in the global population, yet only <2% of GWAS have included the African genome (Sirugo et al., 2019); thus, there may be additional protective variants that alleviate the effect of the ε4 allele in African populations yet to be identified. Additionally, the differential effect of the APOE ε4 allele in non-European admixed populations appears to be explained by the local ancestral background on which the allele lies; APOE ε4 alleles of African ancestry confer lower risk than those of European ancestry (Rajabli et al., 2018). In other words, individuals who have the APOE ε4 allele derived from an African ancestor have a lower AD risk as observed in the African population, while those who have inherited their APOE ε4 alleles from a European ancestor have the AD risk observed in European populations.
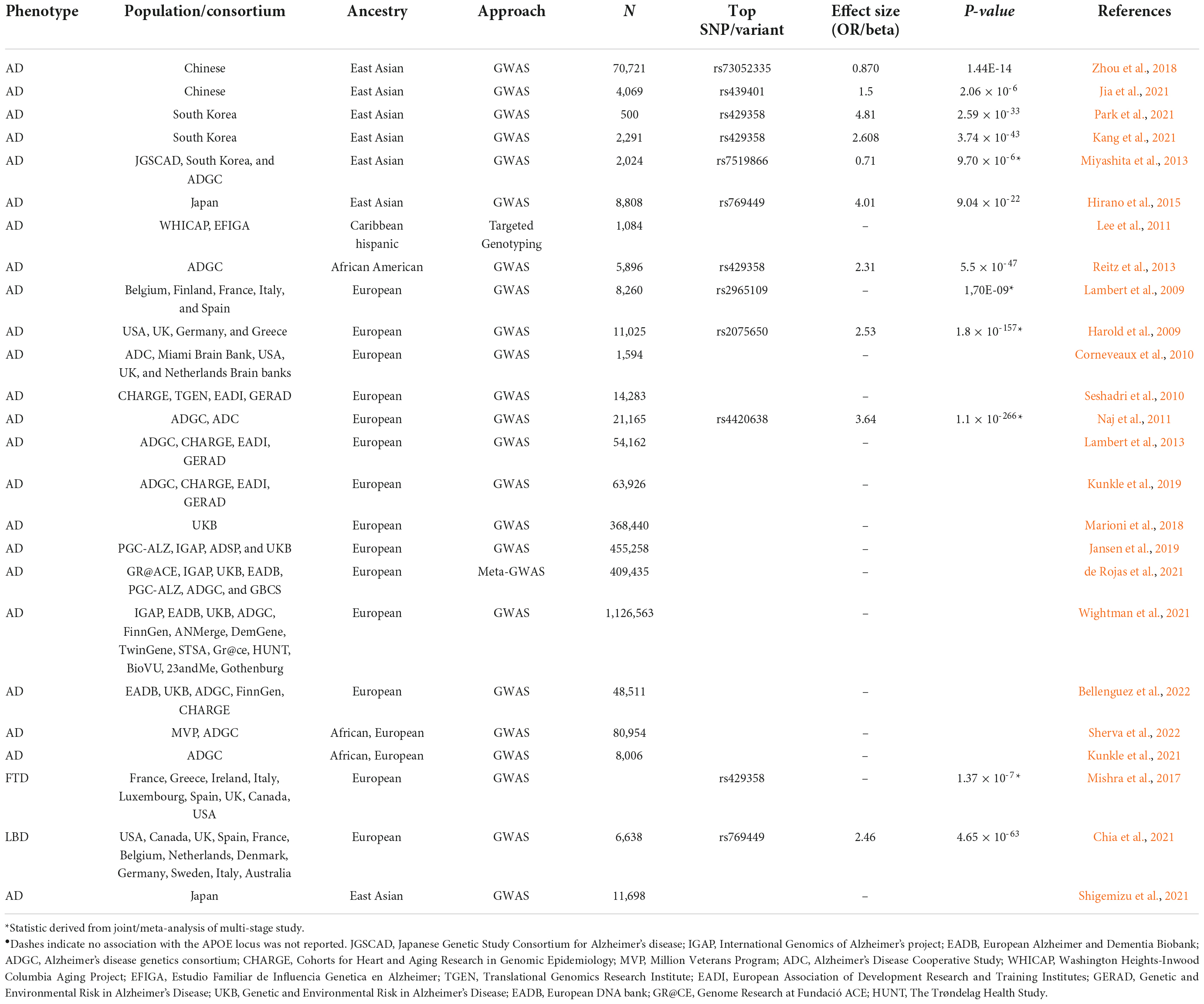
Table 2. Summary of APOE locus genetic association with neurodegenerative disease across global populations.
In contrast to the variable effect of APOE ε4 observed in African ancestry populations, East-Asian population seems to be the most susceptible to the effect of the ε4 allele on AD risk, with a higher odds ratio compared to European and other non-European populations (ε4/ε4 OR:33.12) (Farrer et al., 1997). A series of large-scale AD GWAS in East Asian individuals from Japanese (Shigemizu et al., 2021), Chinese (Jia et al., 2021), and South Korean cohorts (Kang et al., 2021) have all confirmed the APOE locus as the most significant contributor to AD risk, notably with higher odds ratios compared to European ancestry individuals. It is worth noting that the larger effect size in East Asians may be due to the differences in allele frequency such that the proportional of APOE ε4 frequency differs between cases and controls, thus resulting in a larger odds ratio even though the total difference in the allele frequency is similar across populations (Choi et al., 2019). This means that, compared to the European population, the effect of the ε4 allele on AD risk is stronger in East Asians who have a lower ε4 frequency and weaker in African populations who have a higher ε4 frequency. Alternatively, Choi et al. (2019) also point out the TT genotype within the APOE promoter, the SNP rs405509, is highly frequent in East Asians and possibly accounts for the observed high magnitude of the effect of ε4 on AD risk (Choi et al., 2019). Nonetheless, it would be interesting to assess different East Asian cohorts individually, as these cohorts are often conducted as part of large meta-analyses, which often fail to detect small effects loci that could be unique in each country.
The genetic association of APOE with AD risk in Hispanic\Latino populations is less clear. An association of the APOE ε4 allele and AD is remarkably absent in even the largest AD GWAS performed to date in Hispanic\Latino populations (Lee et al., 2011), which included admixed individuals from the Dominican Republic and Puerto Rico, indicating that the locus may not be a main genetic contributor. However, significant associations between APOE ε4 and AD risk have been observed in candidate genetic studies, but with smaller effect sizes relative to studies of European ancestry populations (ε4/ε4 OR: 2) (Farrer et al., 1997). Indeed, Hispanics/Latinos are highly admixed, with >90% of their ancestry derived from native Americans, African slave, and European invasion. Thus, disentangling the relationship between APOE and AD continues to be challenging in part due to the heterogeneity of the Hispanic\Latino population. As such, an Ameridian ancestry on APOE ε4 locus has been found to confer protection in Colombians (Moreno-Grau et al., 2018) and Brazilians (Benedet et al., 2012), while contributing to increased risk of AD in Peruvians (Marca-Ysabel et al., 2021). Interestingly, African ancestry APOE is thought to be protective in a Caribbean Hispanic cohort (Blue et al., 2019). Given the complex relationships between APOE genotype, ancestry, and AD risk across Hispanic\Latino populations, it is worth pointing out that local ancestry, allele frequencies, and patterns of LD should be considered when predicting the genetic risk of AD in populations with non-European ancestry.
APOE ε4 has been found to increase the risk of dementia with Lewy bodies (DLB) (Chia et al., 2021) and FTD (Mishra et al., 2017). However, the overlap with AD clinical and neuropathological symptoms has made it challenging to recruit patients with pure DLB and/or FTD, respectively. Only a few European studies have attempted to do so. The most recent and largest GWAS of DLB detected the APOE locus as the top GWAS association (Chia et al., 2021). Similarly, a gene-based association study led by Mishra et al. (2017) using GWAS summary files from the international FTD consortium confirmed that the APOE ε4 allele increases the risk of behavioral variant of FTD (bvFTD). Given that DLB and FTD clinically and pathologically resemble AD and PD, this has hindered the collection of large cohorts of cases whose diagnosis is certain, and as a consequence, could also be restraining the presence of large-scale GWAS in non-European populations (Orme et al., 2018). Furthermore, it is important to note that the GWAS of DLB and FTD are not as large as those for AD, and the heterogeneity of FTD subtypes further complicates these studies and reduces sample size and power. Despite this link with DLB and FTD, APOE has not clearly been associated with other neurodegenerative diseases, implying that APOE is not a major contributor to disease risk for these disorders. However, these associations remain to be conducted in non-European populations to allow the discovery of population-specific genetic factors.
Biomarkers for Alzheimer’s disease, including cerebrospinal fluid (CSF) and plasma levels of Aβ40, Aβ42, total tau (t-tau), and phosphorylated tau (p-tau-181/217), as well as neuroimaging measures such as PET and MRI, are important for an accurate clinical diagnosis of dementia, determination of disease progression, and serve as key outcome measures for clinical trials of novel therapeutics. Importantly, many of these biomarkers have been found to vary both by genetic variation at the APOE and 17q21.31 loci, as well as by race, thus implying a likely influence of genetic ancestry on disease pathogenesis and subsequent interpretation of biomarker results.
APOE ε4 has been associated with increased levels of both CSF and circulating t-tau and p-tau-181, as well as reduced plasma Aβ42 in European ancestry populations (Mattsson et al., 2018; Morris et al., 2019; Brickman et al., 2021; Deniz et al., 2021). However the relationship between APOE ε4 and markers of AD in African ancestry populations appears to be less well defined. Deniz et al. (2021) found that the APOEε4 allele was associated with reduced t-tau in plasma from African American participants, although consistent with European ancestry data, the levels increased with age and AD diagnosis (Deniz et al., 2021). Also consistent with European data, CSF and plasma Aβ42 appear to be reduced with APOEε4 genotype in both African and Asian ancestry populations (Cruchaga et al., 2013; Nakamura et al., 2018; Morris et al., 2019; Brickman et al., 2021; Deniz et al., 2021). However, a recent study of an admixed Brazilian cohort demonstrated not only that an increased proportion of African ancestry was associated with reduced neuritic plaque burden but that APOE ε4 was only associated with worse cognition and more severe neuropathology when it was of European origin (Naslavsky et al., 2022). Furthermore, a study of amyloid PET positivity in cognitively normal individuals revealed that self-reported black participants had reduced signal compared to whites, and this effect was larger in APOE ε4 carriers (Deters et al., 2021).
A race × APOE interaction for CSF t-tau and p-tau-181 has also been reported; while these markers were lower in African American participants compared to Europeans overall, this difference was driven only by APOE ε4 carriers (Howell et al., 2017; Garrett et al., 2019; Morris et al., 2019; Choudhury et al., 2021). These findings appear to be consistent with the GWAS data described above, in that there appears to be a reduced contribution of APOE ε4 to AD risk and pathogenesis in African ancestry populations compared to Europeans. In contrast, a recent multi-ethnic community study reported higher levels of p-tau-181 and p-tau-217 in plasma associated with the APOE ε4 genotype in White, Hispanic, and Black individuals, but that the accuracy of p-tau in classifying AD diagnosis was improved in Hispanic and Black participants compared to white participants (Brickman et al., 2021). These inconsistencies may be due to the relatively small sample sizes used to assess biomarker abundance in non-European ancestry populations, which limits the generalizability and reproducibility of results. Regardless, it is apparent that genetic ancestry at the APOE locus is likely influencing the interaction with amyloid and tau accumulation and phosphorylation, which has implications for the use of these biomarkers in diagnostic criteria of AD in diverse populations.
The contribution of genetic variation at the 17q21.31 locus to AD biomarker detection has received less attention than APOE genotype; however, a recent GWAS meta-analysis revealed that the locus was significantly associated with plasma t-tau in European Americans but not in African Americans (Sarnowski et al., 2022), which is suggestive that genetic architecture related to ancestry may influence tau pathology. Histopathological studies have reported no effect of race on Braak score postmortem (Sandberg et al., 2001; Naslavsky et al., 2022), although a more recent study identified an interaction between ancestry, tau burden, and dementia severity (Naslavsky et al., 2022). It is possible that this lack of effect of ancestry on tau pathology is a result of underpowered studies or may require a more comprehensive analysis applying modern neuropathological techniques combined with local ancestry analyses across the 17q21.31 locus to investigate this fully. While a more recent PET study identified higher 18F-Flortaucipir binding, indicative of increased tau pathology, in the choroid plexus and hippocampus of African American individuals compared to European ancestry individuals, it was hypothesized that the signal was likely due to increased tracer off-binding to melanin in the choroid plexus, which was also likely to have influenced the hippocampal signal (Lee et al., 2018). Therefore, the relationship between CSF and plasma tau to pathology in the brain in African ancestry individuals is unclear.
Interestingly, 17q21.31 variants associated with either CSF or plasma t-tau have not been H1/H2-defining tag SNPs, but rather have tagged specific sub-haplotypes of the major H1 allele (Laws et al., 2007; Kauwe et al., 2008; Sarnowski et al., 2022), which confers risk for AD (Bellenguez et al., 2022). The H1c tag SNP, rs242557, showed the strongest association with plasma t-tau in Europeans, possibly related to its association with increased MAPT expression (Sarnowski et al., 2022). The rs242557 minor allele occurs at a relatively high frequency in European populations (ALFA frequency = 0.36), is less common in African ancestry populations (∼0.32), and is the major allele in East Asian populations (∼0.57). To our knowledge, there are no reports comparing tau pathology or tau biomarkers between European, African, and Asian ancestry individuals stratified by 17q21.31 haplotype; however, given the lack of the H2 haplotype and increased frequency of tauopathy-associated H1 variants in East Asian populations, it may be expected that there will be a differential contribution of the 17q21.31 locus to neurodegenerative risk and tau biomarkers in this population. New efforts, such as the NCRAD-ACAD study5 are currently underway to characterize AD biomarkers in this understudied population and will provide valuable data and insight into the validity of current AD biomarkers in Asian cohorts.
Genetic studies of neurodegenerative diseases have historically focused on White, European ancestry populations. However, recent efforts to expand these analyses into globally diverse populations have revealed complex ancestry and population-specific associations that may reflect unique pathogenic mechanisms underlying disease susceptibility and progression. In this review, we have focused on two loci of interest; 17q21.31 and APOE. Both show distinct, complex evolutionary patterns of selection that likely underlie their differential disease risk across populations. However, a thorough assessment of their contributions to AD or other dementias in non-European populations is precluded by lack of data, resulting in part from lack of infrastructure and funding, availability and accessibility of neurologists and healthcare, as well as social stigma surrounding disease (Dekker et al., 2020).
The vast majority of genetic association and biomarker analyses in non-European ancestry individuals have been carried out in admixed American populations, further complicating our understanding of genetic ancestry to neurodegenerative disease risk. African Americans and Hispanic individuals in New York City were found to have an increased risk of AD, regardless of APOE genotype (Maestre et al., 1995), and African Americans are consistently reported as being more susceptible to AD risk even after correcting for relevant comorbidities such as cardiovascular health and diabetes, as well as socioeconomic factors (Barnes, 2022). However, the prevalence and incidence of dementia in Africa are among the lowest in the world (Akinyemi et al., 2022). It is therefore currently unclear to what extent these disparities are due to uncharacterized lifestyle, environmental and health effects resulting from racialization in the United States, or to complex and unique genetic interactions and variants resulting from admixture. Thus, increasing representation of non-European individuals as well as integrating risk factors that reflect lived experiences and biological data will allow us to thoroughly investigate the underlying mechanisms of neurodegenerative diseases and address the existent racial differences.
Biomarkers can assist in addressing the health disparities of AD in historically disadvantaged populations via establishing generalizable diagnostics and identifying individuals in the pre-clinical stages of AD (Gleason et al., 2022). Biomarker data for AD have been inconsistent in African ancestry populations, and entirely lacking in Asian populations, largely due to small sample sizes as a result of selection biases, stigmatization, and confounding due to social determinants of physical and cognitive health. Furthermore, the rate of participation of historically disadvantaged populations in biomarker research is lower than that of European ancestry individuals in the USA, due to historical trauma of lived experiences and a continued lack of trust in the healthcare system (Gleason et al., 2022). There is, undoubtedly, work to be done, but blood-based neurodegenerative biomarkers provide ways to improve recruitment and retention of marginalized ethnic individuals, and thus, advance the diversity of neurodegenerative genomic research.
Race and ancestry are inextricably intertwined, particularly in USA populations, and decoding genetic contributions of ancestry across the genome will likely prove to be challenging. Given the complexity of these loci and the frequency of admixed populations, the use of racial self-identification alone or global genetic ancestry estimates to stratify populations is unlikely to provide sufficient resolution. Additional local ancestry analyses of specific risk loci, integrated with biomarker data and environmental and socio-economic factors experienced as a consequence of race will therefore be crucial to understand the complex interaction between ancestry and neurodegenerative disease risk (Figure 3). Specific programs and studies of neurodegeneration, including frequency, genetic risk, and biomarker measurements, are currently underway in non-European ancestry countries, and initiatives that pair with, and support local research facilities are becoming increasingly common. These critical efforts hold great potential to reduce racial disparities in neurodegeneration research, and subsequently incidence and prevalence.
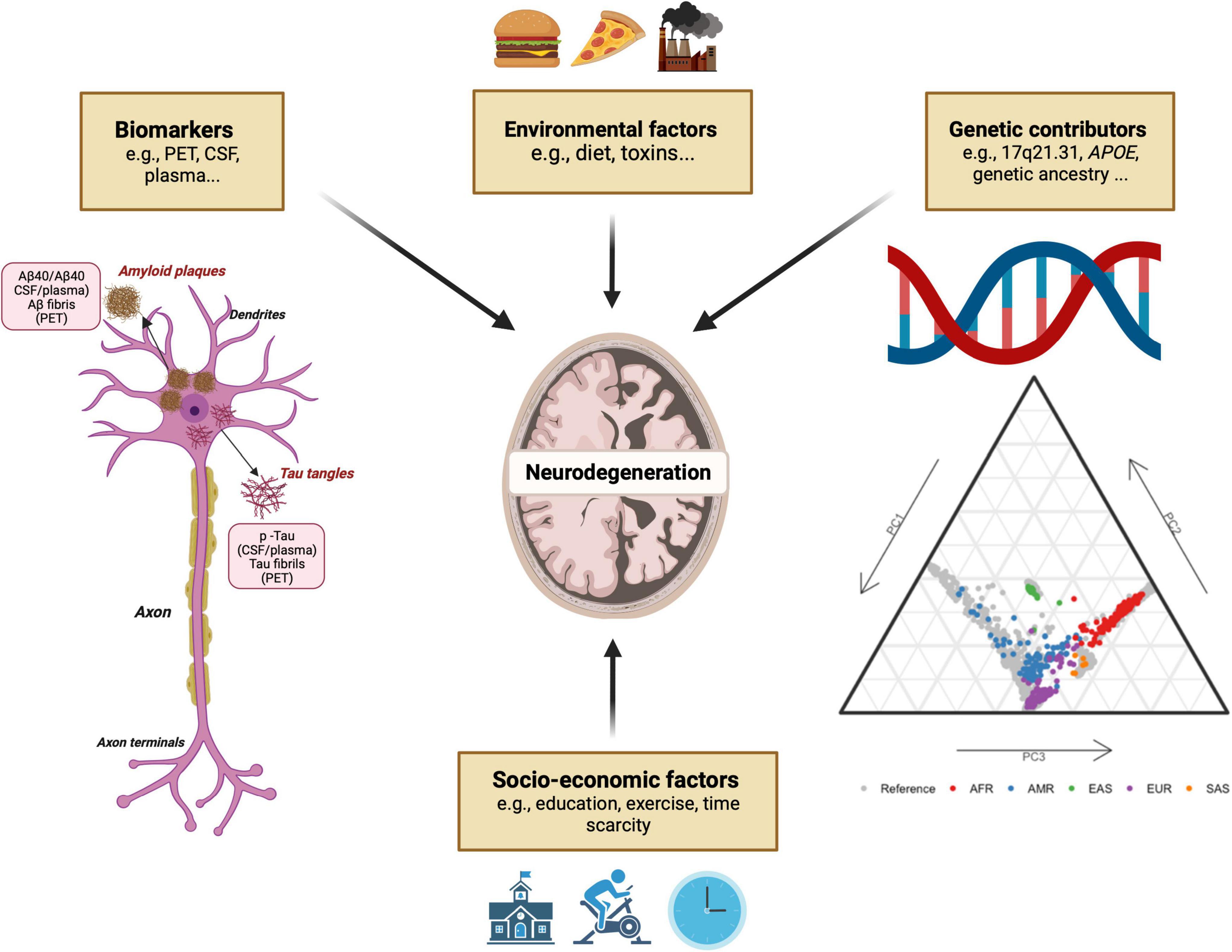
Figure 3. Main factors implicated in neurodegenerative disease and associated fluid biomarkers.
Incorporation of the amazing genetic diversity at the 17q21.31 locus and the clear differential association of APOE in different populations in studies of disease-associated pathways and mechanisms are required to fuel the needed interdisciplinary work necessary to promote health equity across the globe. We have discussed the complex interactions between evolutionary selection, genetic ancestry, and biomarker outcomes for two loci critical for neurodegenerative disease risk: 17q21.31 and APOE. Increasing representation of non-European individuals in genetic and biomarker studies will lead to an improved understanding of disease pathogenesis, and ultimately the development of therapeutic strategies that will be effective across diverse populations. We are optimistic for a future of improved inclusion of diverse populations in neurodegenerative research, such that we more accurately represent the level of diversity that exists globally. Indeed, many such important efforts are already underway.
KB organized and supervised the review with the active collaboration of the rest of the authors. NH wrote about APOE locus. KB wrote about the 17q21.31 locus. NH and KB made the figures. All authors provided sections of text covering their area of expertise, participated in the proofreading, discussion, read, and approved the final manuscript.
This work was supported by the Tau Consortium; the Rainwater Charitable Foundation (to AG); the JPB Foundation (to NH and AG); CurePSP #648-2017-04 (to KB); the Association for Frontotemporal Degeneration (AFTD) (to KB); the BrightFocus Foundation #A2021021S (to KB); and NIH U54 NS123746-01 (AG and KB).
Figures 1–3 were partially created using biorender.com.
Author AG is a member of the scientific advisory boards of Genentech and Muna Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.1021918/full#supplementary-material
Acosta-Uribe, J., Aguillón, D., Cochran, J. N., Giraldo, M., Madrigal, L., Killingsworth, B. W., et al. (2022). A neurodegenerative disease landscape of rare mutations in Colombia due to founder effects. Genome Med. 14, 1–22. doi: 10.1186/s13073-022-01035-9
Akinyemi, R. O., Yaria, J., Ojagbemi, A., Guerchet, M., Okubadejo, N., Njamnshi, A. K., et al. (2022). Dementia in Africa: Current evidence, knowledge gaps, and future directions. Alzheimers Dement. 18, 790–809. doi: 10.1002/alz.12432
Alves, J. M., Lima, A. C., Pais, I. A., Amir, N., Celestino, R., Piras, G., et al. (2015). Reassessing the evolutionary history of the 17q21 inversion polymorphism. Genome Biol. Evol. 7, 3239–3248. doi: 10.1093/gbe/evv214
Bandres-Ciga, S., Ahmed, S., Sabir, M. S., Blauwendraat, C., Adarmes-Gómez, A. D., Bernal-Bernal, I., et al. (2019). The genetic architecture of Parkinson disease in Spain: Characterizing population-specific risk, differential haplotype structures, and providing etiologic insight. Mov. Disord. 34, 1851–1863. doi: 10.1002/mds.27864
Barnes, L. L. (2022). Alzheimer disease in African American individuals: Increased incidence or not enough data? Nat. Rev. Neurol. 18, 56–62. doi: 10.1038/s41582-021-00589-3
Bellenguez, C., Küçükali, F., Jansen, I. E., Kleineidam, L., Moreno-Grau, S., Amin, N., et al. (2022). New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436. doi: 10.1038/s41588-022-01024-z
Benedet, A. L., Moraes, C. F., Camargos, E. F., Oliveira, L. F., Souza, V. C., Lins, T. C., et al. (2012). Amerindian genetic ancestry protects against Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 33, 311–317. doi: 10.1159/000339672
Blue, E. E., Horimoto, A. R. V. R., Mukherjee, S., Wijsman, E. M., and Thornton, T. A. (2019). Local ancestry at APOE modifies Alzheimer’s disease risk in Caribbean Hispanics. Alzheimers Dement. 15, 1524–1532. doi: 10.1016/j.jalz.2019.07.016
Boettger, L. M., Handsaker, R. E., Zody, M. C., and McCarroll, S. A. (2012). Structural haplotypes and recent evolution of the human 17q21.31 region. Nat. Genet. 44, 881–885. doi: 10.1038/ng.2334
Bowles, K. R., Pugh, D. A., Liu, Y., and Renton, A. E. (2022). 17q21.31 sub-haplotypes underlying H1-associated risk for Parkinson’s disease are associated with LRRC37A/2 expression in astrocytes. Mol. Neurodegener. 17, 1–21. doi: 10.1101/860668v2
Brickman, A. M., Manly, J. J., Honig, L. S., Sanchez, D., Reyes-Dumeyer, D., Lantigua, R. A., et al. (2021). Plasma p-tau181, p-tau217, and other blood-based Alzheimer’s disease biomarkers in a multi-ethnic, community study. Alzheimers Dement. 17, 1353–1364. doi: 10.1002/alz.12301
Buniello, A., Macarthur, J. A. L., Cerezo, M., Harris, L. W., Hayhurst, J., Malangone, C., et al. (2019). The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012. doi: 10.1093/nar/gky1120
Chen, J. A., Wang, Q., Davis-Turak, J., Li, Y., Karydas, A. M., Hsu, S. C., et al. (2015). A multiancestral genome-wide exome array study of Alzheimer disease, frontotemporal dementia, and progressive supranuclear palsy. JAMA Neurol. 72, 414–422. doi: 10.1001/jamaneurol.2014.4040
Chia, R., Sabir, M. S., Bandres-Ciga, S., Saez-Atienzar, S., Reynolds, R. H., Gustavsson, E., et al. (2021). Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat. Genet. 53, 294–303. doi: 10.1038/s41588-021-00785-3
Chikosi, A. B., Moodley, J., Pegoraro, R. J., Lanning, P. A., and Rom, L. (2000). Apolipoprotein E polymorphism in South African Zulu women with preeclampsia. Hypertens. Pregnancy 19, 309–314. doi: 10.1081/prg-100101992
Choi, K. Y., Lee, J. J., Gunasekaran, T. I., Kang, S., Lee, W., Jeong, J., et al. (2019). APOE promoter polymorphism-219T/G is an effect modifier of the influence of APOE ε4 on Alzheimer’s disease risk in a multiracial sample. J. Clin. Med. 8:1236. doi: 10.3390/jcm8081236
Choudhury, P., Ramanan, V. K., and Boeve, B. F. (2021). APOE ε4 Allele testing and risk of Alzheimer disease. JAMA 325, 484–485. doi: 10.1001/jama.2020.15085
Coppola, G., Chinnathambi, S., Lee, J. J., Dombroski, B. A., Baker, M. C., Soto-ortolaza, A. I., et al. (2012). Evidence for a role of the rare p.A152T variant in mapt in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum. Mol. Genet. 21, 3500–3512. doi: 10.1093/hmg/dds161
Corbo, R. M., and Scacchi, R. (1999). Apolipoprotein E (APOE) allele distribution in the world. Is APOE*4 a “thrifty” allele? Ann. Hum. Genet. 63, 301–310. doi: 10.1046/j.1469-1809.1999.6340301.x
Corbo, R. M., Ulizzi, L., Scacchi, R., Martínez-Labarga, C., and De Stefano, G. F. (2004). Apolipoprotein E polymorphism and fertility: A study in pre-industrial populations. Mol. Hum. Reprod. 10, 617–620. doi: 10.1093/molehr/gah082
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Corneveaux, J. J., Myers, A. J., Allen, A. N., Pruzin, J. J., Ramirez, M., Engel, A., et al. (2010). Association of CR1, CLU and PICALM with Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum. Mol. Genet. 19, 3295–3301. doi: 10.1093/hmg/ddq221
Cruchaga, C., Kauwe, J. S. K., Harari, O., Jin, S. C., Cai, Y., Karch, C. M., et al. (2013). GWAS of cerebrospinal fluid tau levels identifies novel risk variants for Alzheimer’s disease. Neuron 78, 256–268. doi: 10.1016/j.neuron.2013.02.026.GWAS
Cruts, M., Rademakers, R., Gijselinck, I., van der Zee, J., Dermaut, B., de Pooter, T., et al. (2005). Genomic architecture of human 17q21 linked to frontotemporal dementia uncovers a highly homologous family of low-copy repeats in the tau region. Hum. Mol. Genet. 14, 1753–1762. doi: 10.1093/hmg/ddi182
Davignon, J., Gregg, R. E., and Sing, C. F. (1988). Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis 8, 1–21. doi: 10.1161/01.atv.8.1.1
de Rojas, I., Moreno-Grau, S., Tesi, N., Grenier-Boley, B., Andrade, V., Jansen, I. E., et al. (2021). Common variants in Alzheimer’s disease and risk stratification by polygenic risk scores. Nat. Commun. 12:3417. doi: 10.1038/s41467-021-22491-8
Dekker, M. C. J., Coulibaly, T., Bardien, S., Ross, O. A., Carr, J., and Komolafe, M. (2020). Parkinson’s disease research on the African continent: Obstacles and opportunities. Front. Neurol. 11:512. doi: 10.3389/fneur.2020.00512
Deniz, K., Ho, C. C. G., Malphrus, K. G., Reddy, J. S., Nguyen, T., Carnwath, T. P., et al. (2021). Plasma biomarkers of Alzheimer’s disease in African Americans. J. Alzheimers Dis. 79, 323–334. doi: 10.3233/JAD-200828
Dennis, M. Y., and Eichler, E. E. (2016). Human adaptation and evolution by segmental duplication. Curr. Opin. Genet. Dev. 41, 44–52. doi: 10.1016/j.gde.2016.08.001
Deters, K., Napolioni, V., Sperling, R. A., Greicius, M. D., Mayeaux, R., Hohman, T., et al. (2021). Amyloid PET imaging in self-identified non-Hispanic black participants of the anti-amyloid in asymptomatic Alzheimer’s disease (A4) study. Neurology 96, e1491–e1500. doi: 10.1212/WNL.0000000000011599
Donnelly, M. P., Paschou, P., Grigorenko, E., Gurwitz, D., Mehdi, S. Q., Kajuna, S. L. B., et al. (2010). The distribution and most recent common ancestor of the 17q21 inversion in humans. Am. J. Hum. Genet. 86, 161–171. doi: 10.1016/j.ajhg.2010.01.007
Duggan, A. E., and Callard, I. P. (2001). Phylogenetic distribution of apolipoproteins A-I and E in vertebrates as determined by Western blot analysis. J. Exp. Zool. 290, 255–264. doi: 10.1002/jez.1056
Egert, S., Rimbach, G., and Huebbe, P. (2012). ApoE genotype: From geographic distribution to function and responsiveness to dietary factors. Proc. Nutr. Soc. 71, 410–424. doi: 10.1017/S0029665112000249
Eisenberg, D. T. A., Kuzawa, C. W., and Hayes, M. G. (2010). Worldwide allele frequencies of the human apolipoprotein E gene: Climate, local adaptations, and evolutionary history. Am. J. Phys. Anthropol. 143, 100–111. doi: 10.1002/ajpa.21298
Fan, L., Shi, C., Hu, X., Zhang, Z., Zheng, H., Luo, H., et al. (2021). Analysis of 12 GWAS-linked loci with Parkinson’s disease in the Chinese Han population. Front. Neurol. 12:623913. doi: 10.3389/fneur.2021.623913
Farrer, L. A., Cupples, L. A., Haines, J. L., Hyman, B., Kukull, W. A., Mayeux, R., et al. (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: A meta-analysis. J. Am. Med. Assoc. 278, 1349–1356. doi: 10.1001/jama.278.16.1349
Ferrari, R., Manzoni, C., and Hardy, J. (2019). Genetics and molecular mechanisms of frontotemporal lobar degeneration: An update and future avenues. Neurobiol. Aging 78, 98–110. doi: 10.1016/j.neurobiolaging.2019.02.006
Foo, J. N., Chew, E. G. Y., Chung, S. J., Peng, R., Blauwendraat, C., Nalls, M. A., et al. (2020). Identification of Risk Loci for Parkinson disease in Asians and comparison of risk between Asians and Europeans: A genome-wide association study. JAMA Neurol. 77, 746–754. doi: 10.1001/jamaneurol.2020.0428
Froehle, A. W., and Schoeninger, M. J. (2006). Intraspecies variation in BMR does not affect estimates of early hominin total daily energy expenditure. Am. J. Phys. Anthropol. 131, 552–559. doi: 10.1002/ajpa.20475
Garrett, S. L., McDaniel, D., Obideen, M., Trammell, A. R., Shaw, L. M., Goldstein, F. C., et al. (2019). Racial Disparity in cerebrospinal fluid amyloid and tau biomarkers and associated cutoffs for mild cognitive impairment. JAMA Netw. Open 2, 1–11. doi: 10.1001/jamanetworkopen.2019.17363
Gerdes, L. U. (2003). The common polymorphism of apolipoprotein E: Geographical aspects and new pathophysiological relations. Clin. Chem. Lab. Med. 41, 628–631. doi: 10.1515/CCLM.2003.094
Gijselinck, I., Bogaerts, V., Rademakers, R., van der Zee, J., Van Broeckhoven, C., and Cruts, M. (2006). Visualization of MAPT inversion on stretched chromosomes of tau-negative frontotemporal dementia patients. Hum. Mutat. 27, 1057–1059. doi: 10.1002/humu.20391
Gleason, C. E., Zuelsdorff, M., Gooding, D. C., Kind, A. J. H., Johnson, A. L., James, T. T., et al. (2022). Alzheimer’s disease biomarkers in black and nonhispanic white cohorts: A contextualized review of the evidence. Alzheimers Dement. 18, 1545–1564. doi: 10.1002/alz.12511
Hanlon, C. S., and Rubinsztein, D. C. (1995). Arginine residues at codons 112 and 158 in the apolipoprotein E gene correspond to the ancestral state in humans. Atherosclerosis 112, 85–90. doi: 10.1016/0021-9150(94)05402-5
Harold, D., Abraham, R., Hollingworth, P., Sims, R., Gerrish, A., Hamshere, M. L., et al. (2009). Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 41, 1088–1093. doi: 10.1038/ng.440
Hirano, A., Ohara, T., Takahashi, A., Aoki, M., Fuyuno, Y., Ashikawa, K., et al. (2015). A genome-wide association study of late-onset Alzheimer’s disease in a Japanese population. Psychiatr. Genet. 25, 139–146. doi: 10.1097/ypg.0000000000000090
Höglinger, G. U., Melhem, N. M., Dickson, D. W., Sleiman, P. M. A., Wang, L.-S., Klei, L., et al. (2011). Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet. 43, 699–705. doi: 10.1038/ng.859
Howell, J. C., Watts, K. D., Parker, M. W., Wu, J., Kollhoff, A., Wingo, T. S., et al. (2017). Race modifies the relationship between cognition and Alzheimer’s disease cerebrospinal fluid biomarkers. Alzheimers Res. Ther. 9, 1–10. doi: 10.1186/s13195-017-0315-1
Huebbe, P., and Rimbach, G. (2017). Evolution of human apolipoprotein E (APOE) isoforms: Gene structure, protein function and interaction with dietary factors. Ageing Res. Rev. 37, 146–161. doi: 10.1016/j.arr.2017.06.002
Huebbe, P., Nebel, A., Siegert, S., Moehring, J., Boesch-Saadatmandi, C., Most, E., et al. (2011). APOE ε4 is associated with higher vitamin D levels in targeted replacement mice and humans. FASEB J. 25, 3262–3270. doi: 10.1096/fj.11-180935
Itsara, A., Vissers, L. E. L. M., Steinberg, K. M., Meyer, K. J., Zody, M. C., Koolen, D. A., et al. (2012). Resolving the breakpoints of the 17q21.31 microdeletion syndrome with next-generation sequencing. Am. J. Hum. Genet. 90, 599–613. doi: 10.1016/j.ajhg.2012.02.013
Jansen, I. E., Savage, J. E., Watanabe, K., Bryois, J., Williams, D. M., Steinberg, S., et al. (2019). Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413. doi: 10.1038/s41588-018-0311-9
Jasienska, G., Ellison, P. T., Galbarczyk, A., Jasienski, M., Kalemba-Drozdz, M., Kapiszewska, M., et al. (2015). Apolipoprotein E (ApoE) polymorphism is related to differences in potential fertility in women: A case of antagonistic pleiotropy? Proc. Biol. Sci. 282:20142395. doi: 10.1098/rspb.2014.2395
Jia, L., Li, F., Wei, C., Zhu, M., Qu, Q., Qin, W., et al. (2021). Prediction of Alzheimer’s disease using multi-variants from a Chinese genome-wide association study. Brain 144, 924–937. doi: 10.1093/brain/awaa364
Jiang, Y., Jiao, B., Xiao, X., and Shen, L. (2021). Genetics of frontotemporal dementia in China. Amyotroph. Lateral Scler. Frontotemporal Degener. 22, 321–335. doi: 10.1080/21678421.2021.1880596
Jin, S. C., Pastor, P., Cooper, B., Cervantes, S., Benitez, B. A., Razquin, C., et al. (2012). Pooled-DNA sequencing identifies novel causative variants in PSEN1, GRN and MAPT in a clinical early-onset and familial Alzheimer’s disease Ibero-American cohort. Alzheimers Res. Ther. 4:34. doi: 10.1186/alzrt137
Jun, G., Ibrahim-Verbaas, C. A., Vronskaya, M., Lambert, J.-C., Chung, J., Naj, A. C., et al. (2016). A novel Alzheimer disease locus located near the gene encoding tau protein. Mol. Psychiatry 21, 108–117. doi: 10.1038/mp.2015.23
Kang, S., Gim, J., Lee, J., Gunasekaran, T. I., Choi, K. Y., Lee, J. J., et al. (2021). Potential novel genes for late-onset Alzheimer’s disease in East-Asian descent identified by APOE-stratified genome-wide association study. J. Alzheimers Dis. 82, 1451–1460. doi: 10.3233/JAD-210145
Kasuga, K., Kikuchi, M., Tokutake, T., Nakaya, A., Tezuka, T., Tsukie, T., et al. (2015). Systematic review and meta-analysis of Japanese familial Alzheimer’s disease and FTDP-17. J. Hum. Genet. 60, 281–283. doi: 10.1038/jhg.2015.15
Kauwe, J. S. K., Cruchaga, C., Mayo, K., Fenoglio, C., Bertelsen, S., Nowotny, P., et al. (2008). Variation in MAPT is associated with cerebrospinal fluid tau levels in the presence of amyloid-beta deposition. Proc. Natl. Acad. Sci. U.S.A. 105, 8050–8054. doi: 10.1073/pnas.0801227105
Keyser, R. J., Oppon, E., Carr, J. A., and Bardien, S. (2011). Identification of Parkinson’s disease candidate genes using CAESAR and screening of MAPT and SNCAIP in South African Parkinson’s disease patients. J. Neural Transm. 118, 889–897. doi: 10.1007/s00702-011-0591-z
Kouri, N., Ross, O. A., Dombroski, B., Younkin, C. S., Serie, D. J., Soto-ortolaza, A., et al. (2015). Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat. Commun. 6, 1–7. doi: 10.1038/ncomms8247
Krzosek, P., Madetko, N., Migda, A., Migda, B., Jaguś, D., and Alster, P. (2022). Differential Diagnosis of rare subtypes of progressive supranuclear palsy and PSP-like syndromes—infrequent manifestations of the most common form of atypical Parkinsonism. Front. Aging Neurosci. 14:804385. doi: 10.3389/fnagi.2022.804385
Kunkle, B. W., Grenier-Boley, B., Sims, R., Bis, J. C., Damotte, V., Naj, A. C., et al. (2019). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430. doi: 10.1038/s41588-019-0358-2
Kunkle, B. W., Schmidt, M., Klein, H.-U., Naj, A. C., Hamilton-Nelson, K. L., Larson, E. B., et al. (2021). Novel Alzheimer disease risk loci and pathways in African American individuals using the african genome resources panel: A meta-analysis. JAMA Neurol. 78, 102–113. doi: 10.1001/jamaneurol.2020.3536
Lambert, J. C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 41, 1094–1099. doi: 10.1038/ng.439
Lambert, J.-C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Laws, S. M., Perneczky, R., Drzezga, A., Diehl-Schmid, J., Ibach, B., Bäuml, J., et al. (2007). Association of the tau haplotype H2 with age at onset and functional alterations of glucose utilization in frontotemporal dementia. Am. J. Psychiatry 164, 1577–1584. doi: 10.1176/appi.ajp.2007.06091456
Lee, C. M., Jacobs, H. I. L., Marquié, M., Becker, J. A., Andrea, N. V., Jin, D. S., et al. (2018). 18F-flortaucipir binding in choroid plexus: Related to race and Hippocampus signal. J. Alzheimers Dis. 62, 1691–1702. doi: 10.3233/JAD-170840
Lee, J. H., Cheng, R., Barral, S., Reitz, C., Medrano, M., Lantigua, R., et al. (2011). Identification of novel loci for Alzheimer disease and replication of CLU, PICALM, and BIN1 in Caribbean Hispanic individuals. Arch. Neurol. 68, 320–328. doi: 10.1001/archneurol.2010.292
Loesch, D. P., Horimoto, A. R. V. R., Heilbron, K., Sarihan, E. I., Inca-Martinez, M., Mason, E., et al. (2021). Characterizing the genetic architecture of Parkinson’s disease in Latinos. Ann. Neurol. 90, 353–365. doi: 10.1002/ana.26153
Lovegrove, B. G. (2003). The influence of climate on the basal metabolic rate of small mammals: A slow-fast metabolic continuum. J. Comp. Physiol. B Biochem. Syst. Environ. Physiol. 173, 87–112. doi: 10.1007/s00360-002-0309-5
Maestre, G., Ottman, R., Stern, Y., Gurland, B., Chun, M., Tang, M. X., et al. (1995). Apolipoprotein E and Alzheimer’s disease: Ethnic variation in genotypic risks. Ann. Neurol. 37, 254–259. doi: 10.1002/ana.410370217
Mahley, R. W., and Rall, S. C. (2000). Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genom. Hum. Genet. 1, 507–537. doi: 10.1146/annurev.genom.1.1.507
Marca-Ysabel, M. V., Rajabli, F., Cornejo-Olivas, M., Whitehead, P. G., Hofmann, N. K., Illanes Manrique, M. Z., et al. (2021). Dissecting the role of Amerindian genetic ancestry and the ApoE ε4 allele on Alzheimer disease in an admixed Peruvian population. Neurobiol. Aging 101, 298.e11–298.e15. doi: 10.1016/j.neurobiolaging.2020.10.003
Marioni, R. E., Harris, S. E., Zhang, Q., McRae, A. F., Hagenaars, S. P., and Hill, W. D. (2018). GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 8:99. doi: 10.1038/s41398-018-0150-6
Mattsson, N., Ossenkoppele, R., Smith, R., Strandberg, O., Ohlsson, T., Jögi, J., et al. (2018). Greater tau load and reduced cortical thickness in APOE ε4-negative Alzheimer’s disease: A cohort study. Alzheimers Res. Ther. 10:77. doi: 10.1186/s13195-018-0403-x
McElroy, J. P., Nelson, M. R., Caillier, S. J., and Oksenberg, J. R. (2009). Copy number variation in African Americans. BMC Genet. 10:15. doi: 10.1186/1471-2156-10-15
Mishra, A., Ferrari, R., Heutink, P., Hardy, J., Pijnenburg, Y., and Posthuma, D. (2017). Gene-based association studies report genetic links for clinical subtypes of frontotemporal dementia. Brain 140, 1437–1446. doi: 10.1093/brain/awx066
Miyashita, A., Koike, A., Jun, G., Wang, L. S., Takahashi, S., Matsubara, E., et al. (2013). SORL1 is genetically associated with late-onset Alzheimer’s disease in Japanese, Koreans and Caucasians. PLoS One 8:e58618. doi: 10.1371/journal.pone.0058618
Moreno-Grau, S., Rodríguez-Gómez, O., Sanabria, Á, Pérez-Cordón, A., Sánchez-Ruiz, D., Abdelnour, C., et al. (2018). Exploring APOE genotype effects on Alzheimer’s disease risk and amyloid β burden in individuals with subjective cognitive decline: The FundacioACE healthy brain initiative (FACEHBI) study baseline results. Alzheimers Dement. 14, 634–643. doi: 10.1016/j.jalz.2017.10.005
Morris, J. C., Schindler, S. E., McCue, L. M., Moulder, K. L., Benzinger, T. L. S., Cruchaga, C., et al. (2019). Assessment of racial disparities in biomarkers for Alzheimer disease. JAMA Neurol. 76, 264–273. doi: 10.1001/jamaneurol.2018.4249
Naj, A. C., Jun, G., Beecham, G. W., Wang, L. S., Vardarajan, B. N., Buros, J., et al. (2011). Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 43, 436–441. doi: 10.1038/ng.801
Nakamura, A., Kaneko, N., Villemagne, V. L., Kato, T., Doecke, J., Doré, V., et al. (2018). High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 554, 249–254. doi: 10.1038/nature25456
Nalls, M. A., Blauwendraat, C., Vallerga, C. L., Heilbron, K., Bandres-ciga, S., Chang, D., et al. (2019). Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102. doi: 10.1016/S1474-4422(19)30320-5
Naslavsky, M. S., Suemoto, C. K., Brito, L. A., Scliar, M. O., Ferretti-Rebustini, R. E., Rodriguez, R. D., et al. (2022). Global and local ancestry modulate APOE association with Alzheimer’s neuropathology and cognitive outcomes in an admixed sample. Mol. Psychiatry 1–9. doi: 10.1038/s41380-022-01729-x [Epub ahead of print].
Ng, A. S. L. (2015). Genetics of frontotemporal dementia in Asia: Advancing knowledge through collaboration. Neurology 85, 2060–2062. doi: 10.1212/WNL.0000000000002045
Nichols, E., Szoeke, C. E. I., Vollset, S. E., Abbasi, N., Abd-Allah, F., Abdela, J., et al. (2019). Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the global burden of disease study 2016. Lancet Neurol. 18, 88–106. doi: 10.1016/S1474-4422(18)30403-4
Oluwole, O. G., Kuivaniemi, H., Abrahams, S., Haylett, W. L., Vorster, A. A., Van Heerden, C. J., et al. (2020). Targeted next-generation sequencing identifies novel variants in candidate genes for Parkinson’s disease in Black South African and Nigerian patients. BMC Med. Genet. 21:23. doi: 10.1186/s12881-020-0953-1
Orme, T., Guerreiro, R., and Bras, J. (2018). The genetics of dementia with lewy bodies: Current understanding and future directions. Curr. Neurol. Neurosci. Rep. 18:67. doi: 10.1007/s11910-018-0874-y
Park, J. H., Park, I., Youm, E. M., Lee, S., Park, J. H., Lee, J., et al. (2021). Novel Alzheimer’s disease risk variants identified based on whole-genome sequencing of APOE ε4 carriers. Transl. Psychiatry 11:296. doi: 10.1038/s41398-021-01412-9
Raffaï, R. L., Dong, L.-M., Farese, R. V., and Weisgraber, K. H. (2001). Introduction of human apolipoprotein E4 “domain interaction” into mouse apolipoprotein E. Proc. Natl. Acad. Sci. U.S.A. 98, 11587–11591. doi: 10.1073/pnas.201279298
Raichlen, D. A., and Alexander, G. E. (2014). Exercise, APOE genotype, and the evolution of the human lifespan. Trends Neurosci. 37, 247–255. doi: 10.1016/j.tins.2014.03.001
Rajabli, F., Feliciano, B. E., Celis, K., Hamilton-Nelson, K. L., Whitehead, P. L., Adams, L. D., et al. (2018). Ancestral origin of ApoE ε4 Alzheimer disease risk in Puerto Rican and African American populations. PLoS Genet. 14:e1007791. doi: 10.1371/journal.pgen.1007791
Rao, P. N., Li, W., Vissers, L. E. L. M., Veltman, J. A., and Ophoff, R. A. (2010). Recurrent inversion events at 17q21.31 microdeletion locus are linked to the MAPT H2 haplotype. Cytogenet. Genome Res. 129, 275–279. doi: 10.1159/000315901
Reiman, E. M., Arboleda-Velasquez, J. F., Quiroz, Y. T., Huentelman, M. J., Beach, T. G., Caselli, R. J., et al. (2020). Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat. Commun. 11:667. doi: 10.1038/s41467-019-14279-8
Reitz, C., Jun, G., Naj, A., Rajbhandary, R., Vardarajan, B. N., Wang, L. S., et al. (2013). Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ?4,and the risk of late-onset Alzheimer disease in African Americans. JAMA 309, 1483–1492. doi: 10.1001/jama.2013.2973
Sandberg, G., Stewart, W., Smialek, J., and Troncoso, J. C. (2001). The prevalence of the neuropathological lesions of Alzheimer’s disease is independent of race and gender. Neurobiol. Aging 22, 169–175. doi: 10.1016/S0197-4580(00)00236-0
Sarnowski, C., Ghanbari, M., Bis, J. C., Logue, M., Fornage, M., Mishra, A., et al. (2022). Meta-analysis of genome-wide association studies identifies ancestry-specific associations underlying circulating total tau levels. Commun. Biol. 5:336. doi: 10.1038/s42003-022-03287-y
Seixas, S., Trovoada, M. J., and Rocha, J. (1999). Haplotype analysis of the apolipoprotein E and apolipoprotein C1 loci in Portugal and São Tomé e Príncipe (Gulf of Guinea): Linkage disequilibrium evidence that APOE*4 is the ancestral APOE allele. Hum. Biol. 71, 1001–1008.
Seshadri, S., and Wolf, P. A. (2007). Lifetime risk of stroke and dementia: Current concepts, and estimates from the Framingham study. Lancet Neurol. 6, 1106–1114. doi: 10.1016/S1474-4422(07)70291-0
Seshadri, S., Fitzpatrick, A. L., Ikram, M. A., DeStefano, A. L., Gudnason, V., Boada, M., et al. (2010). Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303, 1832–1840. doi: 10.1001/jama.2010.574
Sherva, R., Zhang, R., Sahelijo, N., Jun, G., Anglin, T., Chanfreau, C., et al. (2022). African ancestry GWAS of dementia in a large military cohort identifies significant risk Loci Richard. medRxiv [Preprint]. doi: 10.1101/2022.05.25.22275553
Shigemizu, D., Mitsumori, R., Akiyama, S., Miyashita, A., Morizono, T., Higaki, S., et al. (2021). Ethnic and trans-ethnic genome-wide association studies identify new loci influencing Japanese Alzheimer’s disease risk. Transl. Psychiatry 11, 1–10. doi: 10.1038/s41398-021-01272-3
Shimada, M., and Nishida, T. (2020). Focusing on human haplotype diversity in numerous individual genomes demonstrates an evolutional feature of each locus. bioRxiv [Preprint]. doi: 10.1101/2020.03.28.012914
Singh, P. P., Singh, M., and Mastana, S. S. (2006). APOE distribution in world populations with new data from India and the UK. Ann. Hum. Biol. 33, 279–308. doi: 10.1080/03014460600594513
Sirugo, G., Williams, S., and Tishkoff, S. (2019). The missing diversity in human genetic studies. Cell 177, 26–31. doi: 10.1016/j.cell.2019.02.048.The
Smith, B. N., Newhouse, S., Shatunov, A., Vance, C., Topp, S., Johnson, L., et al. (2013). The C9ORF72 expansion mutation is a common cause of ALS+/-FTD in Europe and has a single founder. Eur. J. Hum. Genet. 21, 102–108. doi: 10.1038/ejhg.2012.98
Stefansson, H., Helgason, A., Thorleifsson, G., Steinthorsdottir, V., Masson, G., Barnard, J., et al. (2005). A common inversion under selection in Europeans. Nat. Genet. 37, 129–137. doi: 10.1038/ng1508
Steinberg, K. M., Antonacci, F., Sudmant, P. H., Kidd, J. M., Campbell, C. D., Vives, L., et al. (2012). Structural diversity and African origin of the 17q21. 31 inversion polymorphism. Nat. Publ. Gr. 44, 872–880. doi: 10.1038/ng.2335
Takada, L. T., Bahia, V. S., Guimarães, H. C., Costa, T. V. M. M., Vale, T. C., Rodriguez, R. D., et al. (2016). GRN and MAPT mutations in 2 frontotemporal dementia research centers in Brazil. Alzheimer Dis. Assoc. Disord. 30, 310–317. doi: 10.1097/WAD.0000000000000153
Takigawa, H., Kitayama, M., Wada-Isoe, K., Kowa, H., and Nakashima, K. (2016). Prevalence of progressive supranuclear palsy in Yonago: Change throughout a decade. Brain Behav. 6, 4–8. doi: 10.1002/brb3.557
Tang, M. X., Stern, Y., Marder, K., Bell, K., Gurland, B., Lantigua, R., et al. (1998). The APOE-epsilon4 allele and the risk of Alzheimer disease among African Americans, whites, and Hispanics. JAMA 279, 751–755. doi: 10.1001/jama.279.10.751
Vamathevan, J. J., Hasan, S., Emes, R. D., Amrine-Madsen, H., Rajagopalan, D., Topp, S. D., et al. (2008). The role of positive selection in determining the molecular cause of species differences in disease. BMC Evol. Biol. 8:273. doi: 10.1186/1471-2148-8-273
van Exel, E., Koopman, J. J. E., van Bodegom, D., Meij, J. J., de Knijff, P., Ziem, J. B., et al. (2017). Effect of APOE ε4 allele on survival and fertility in an adverse environment. PLoS One 12:e0179497. doi: 10.1371/journal.pone.0179497
Watson, C. T., Marques-Bonet, T., Sharp, A. J., and Mefford, H. C. (2014). The genetics of microdeletion and microduplication syndromes: An update. Annu. Rev. Genom. Hum. Genet. 15, 215–244. doi: 10.1146/annurev-genom-091212-153408
Wightman, D. P., Jansen, I. E., Savage, J. E., Shadrin, A. A., Bahrami, S., Holland, D., et al. (2021). A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282. doi: 10.1038/s41588-021-00921-z
Yao, S., Zhang, X., Zou, S. C., Zhu, Y., Li, B., Kuang, W. P., et al. (2021). A transcriptome-wide association study identifies susceptibility genes for Parkinson’s disease. NPJ Park. Dis. 7, 1–8. doi: 10.1038/s41531-021-00221-7
Zhao, N., Liu, C.-C., Qiao, W., and Bu, G. (2018). Apolipoprotein E, receptors, and modulation of Alzheimer’s disease. Biol. Psychiatry 83, 347–357. doi: 10.1016/j.biopsych.2017.03.003
Zhou, X., Chen, Y., Mok, K. Y., Zhao, Q., Chen, K., Chen, Y., et al. (2018). Identification of genetic risk factors in the Chinese population implicates a role of immune system in Alzheimer’s disease pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 115, 1697–1706. doi: 10.1073/pnas.1715554115
Zody, M. C., Jiang, Z., Fung, H.-C., Antonacci, F., Hillier, L. W., Cardone, M. F., et al. (2008). Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat. Genet. 40, 1076–1083. doi: 10.1038/ng.193
Keywords: APOE, 17q21.31, MAPT, haplotype, genetic ancestry, evolutionary genetics, non-European populations, neurodegeneration
Citation: Harerimana NV, Goate AM and Bowles KR (2022) The influence of 17q21.31 and APOE genetic ancestry on neurodegenerative disease risk. Front. Aging Neurosci. 14:1021918. doi: 10.3389/fnagi.2022.1021918
Received: 17 August 2022; Accepted: 26 September 2022;
Published: 20 October 2022.
Edited by:
Vivek Swarup, University of California, Irvine, United StatesReviewed by:
Chiara Fenoglio, University of Milan, ItalyCopyright © 2022 Harerimana, Goate and Bowles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathryn R. Bowles, a2Jvd2xlc0BlZC5hYy51aw==
†Present address: Kathryn R. Bowles, UK Dementia Research Institute, 49 Little France Crescent, Edinburgh BioQuarter, The University of Edinburgh, Edinburgh, United Kingdom
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.