 Yahan Sun
Yahan Sun Qinghu Yang
Qinghu Yang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci. , 27 August 2021
Sec. Alzheimer's Disease and Related Dementias
Volume 13 - 2021 | https://doi.org/10.3389/fnagi.2021.707466
Alzheimer’s disease (AD) is a neurodegenerative disease with complex pathological characteristics, whose etiology and pathogenesis are still unclear. Over the past few decades, the role of the extracellular matrix (ECM) has gained importance in neurodegenerative disease. In this review, we describe the role of the ECM in AD, focusing on the aspects of synaptic transmission, amyloid-β-plaque generation and degradation, Tau-protein production, oxidative-stress response, and inflammatory response. The function of ECM in the pathological process of AD will inform future research on the etiology and pathogenesis of AD.
Alzheimer’s disease (AD) is the most common cause of dementia and the fourth leading cause of death after cardiovascular disease, cancer, and acquired immune deficiency syndrome (AIDS). AD symptoms usually manifest as memory loss, cognitive decline, personality changes, and other neurological indications including depression, restlessness, anxiety, and aggressive behaviors. The typical pathological characteristics of AD are amyloid-beta (Aβ) deposits, neurofibrillary tangles (NFTs), synapse and neuron loss, glial activation, and disorganization of the extracellular matrix (ECM). Numerous hypotheses, such as the Aβ hypothesis (Hardy and Selkoe, 2002), the oxidative stress hypothesis (Maccioni et al., 2010), and the Tau protein abnormal phosphorylation hypothesis (Terry and Buccafusco, 2003), have attempted to explain the etiology of AD and have targeted both genetic and environmental factors (Wenk, 2003), yet the causative mechanisms of this disease remain elusive.
The ECM is a structural network composed of various macromolecules secreted by cells, namely collagen, elastin, fibronectin, laminin, glycoproteins like tenascin-R (TNR), and tenascin-C (TNC), glycosaminoglycans (GAGs), and proteoglycans (Frantz et al., 2010). In the central nervous system (CNS), neural ECM molecules produced and released by neurons and surrounding cells (astrocytes, oligodendrocytes, etc.) are extensively accumulated in the extracellular space (Testa et al., 2019). Proteoglycans consist of a core protein covalently attached to one of five GAG chains (Yanagishita, 1993), namely chondroitin sulfate, keratan sulfate, dermatan sulfate, heparan sulfate, and the non-sulfated GAG hyaluronan. The carboxyl group of uronic acid on the surface of hyaluronic acid (HA) has a large amount of negative charge, and its repulsive effect causes the whole molecule to stretch and swell; the hydrophilic group can also combine with a large number of water molecules to make the matrix isotonic and edema.
These form a viscous colloid and generate swelling pressure, giving the tissue good elasticity and resistance to pressure, as well as interfere indirectly with parameters of neuronal excitability by means of their influence on extracellular space volume (Arranz et al., 2014). Chondroitin sulfate proteoglycan (CSPG) usually acts as a barrier molecule and plays an important role in embryonic development and plasticity of the central nervous system in adulthood (Miyata and Kitagawa, 2016). Astrocytes promote the growth and development of the brain by releasing factors such as TNC (Pollen et al., 2015), which regulates neuronal development. TNR is a membrane-bound connexin that is closely related to the formation and stability of the perineural network (Suttkus et al., 2014). The ECM serves to maintain cell morphology and structure (Blumenthal et al., 2014) and is involved in the survival, differentiation, development, and migration of nerve cells. There are three main types of ECM in the brain and spinal cord: the ubiquitously present “loose” ECM; cell membrane-bound molecules such as TNR; and perineuronal nets (PNNs) that wrap around specific neurons (Celio et al., 1998; Deepa et al., 2006; Soleman et al., 2013). As a critical component of the extracellular neural space, the ECM is closely related to several neurodegenerative diseases, especially AD. Many studies have reported alterations in the expression profile of ECM proteins in early-onset AD. Table 1 lists changes in ECM protein expression during the development of AD. Such changes before the occurrence of AD could disturb the homeostasis of the nervous system and promote the onset of AD.
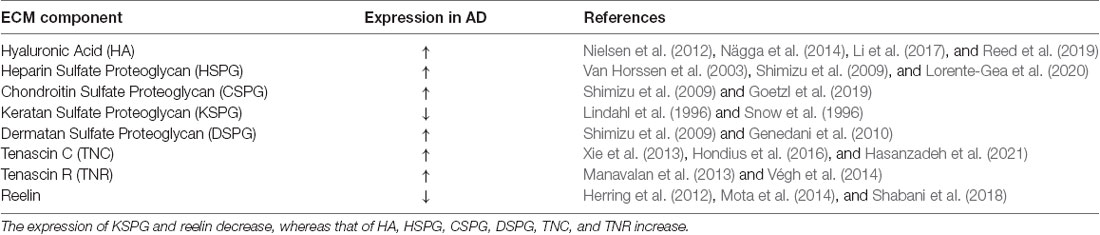
Table 1. Expression of extracellular matrix (ECM) component changes in the pathological state of AD.
As we communicate in this review, different types of ECM may participate in the pathology of AD via different pathways (Figure 1). Here, we focus on the role of the glycosaminoglycans and proteoglycans in the occurrence of AD from the aspects of synaptic signal transmission, Aβ plaque generation and degradation, Tau protein production, oxidative stress response, and the inflammatory response. Taken together, these pieces of evidence strongly support that ECM dysregulation is closely associated with AD, and future research can better inform efforts to study the etiology and pathogenesis of AD.
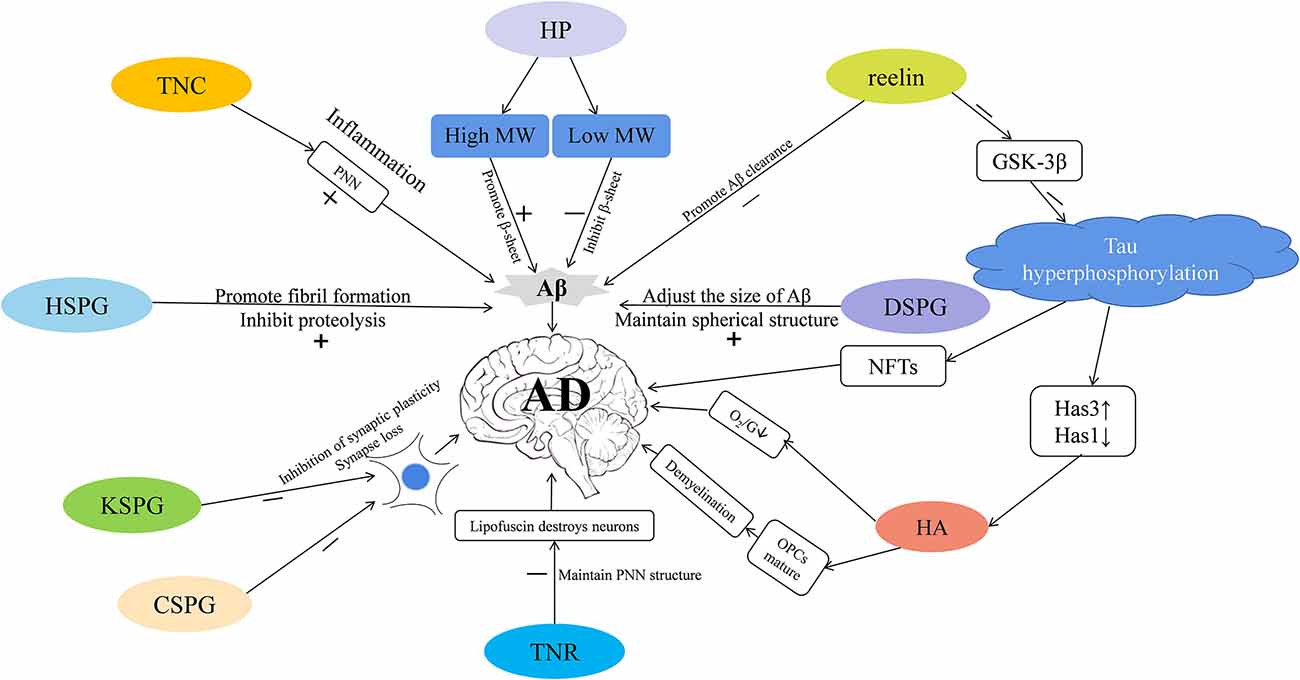
Figure 1. The extracellular matrix (ECM) participates in the progression of Alzheimer’s disease (AD) via different mechanisms. Tenascin-C (TNC) is increased in AD—it increases the stability of perineuronal nets (PNNs) to reduce the clearance of amyloid-β (Aβ), and participates in inflammatory pathways that leads to the occurrence of AD. PNNs also wrap around neurons and protect them from the neurotoxic effects of Aβ. Heparin (HP) is increased in AD—high molecular-weight (MW) HP promotes the formation of β-sheet secondary structure, and low molecular-weight (MW) HP inhibits it, which in turn affects the production of Aβ. Heparin sulfate proteoglycans (HSPGs) are increased in AD; they promote the formation of Aβ fibrils, inhibit amyloid hydrolysis, and promote the production of Aβ. Dermatan sulfate proteoglycans (DSPGs) are increased in AD—they may modulate the size of Aβ plaques, maintain the spherical structure of Aβ, thereby regulating the occurrence of AD. Reelin is decreased in AD—it can promote the clearance of Aβ, and inhibit the expression of GSK-3β that phosphorylates the Tau protein and promotes neurofibrillary tangle (NFT) formation, thus mediating the occurrence of AD. Tau hyperphosphorylation reduces hyaluronan synthase 1 (Has1), increases hyaluronan synthase 3 (Has3), and up-regulates short-chain Hyaluronic acid (HA). HA is increased in AD; it inhibits the maturation of OPCs, causes demyelination, reduces the supply of brain oxygen and glucose (O2/G), and may promote the occurrence of AD. TENASCIN-R (TNR) is increased in AD; it increases PNNs stability and may prevent lipofuscin from destroying neurons. Keratan sulfate proteoglycan (KSPG) is decreased in AD and chondroitin sulfate proteoglycan (CSPG) is increased; both these macromolecules may inhibit synaptic plasticity, cause synapse loss, and promote pathological damage in AD.
Synaptic changes are a frequent occurrence in neurodegenerative diseases and are likewise observed in the early stages of AD. The loss of synapses in the AD brain is closely associated with cognitive dysfunction and the decline of learning and memory. Many dendritic spine abnormalities and a decrease in synapse number have been observed in cognition-related brain areas, like the prefrontal cortex and hippocampus, in the early stages of AD (Vertes et al., 1999).
AD is also called a “synaptic degeneration disease” (Selkoe, 2002). The critical balance between excitatory and inhibitory neurotransmission, which is key to normal cognitive function, is completely disrupted in the AD state (Boyce et al., 2016). Some reports implicate keratan sulfate proteoglycan (KSPG) in the regulation of synaptic function (Snow et al., 1996). The level of KSPGs in the cerebral cortices of AD patients is much less than in healthy individuals (Table 1, Lindahl et al., 1996). KSPGs have been reported as primarily located at synapses and dystrophic neurites within neuritic plaques of AD and the normal, aged brain (Snow et al., 1996). Interestingly, the core protein of KSPG, SV2Proteoglycan (SV2PG), is mainly located on the synaptic vesicle membrane (Buckley and Kelly, 1985), indicating that this position may be a potential site where the keratan sulfate chain attaches to carry out the functions of the KSPG (Scranton et al., 1993). In addition to being present in most synapses, the SV2PG amino-acid sequence has homology with other proteins identified as transporters (Bajjalieh et al., 1992; Feany et al., 1992; Gingrich et al., 1992), suggesting a potential function in synaptic vesicle transport. The localization of SV2PG on dystrophic neurites indicates that changes in neurotransmission may mainly involve abnormal neurites in AD (Buckley and Kelly, 1985; Snow et al., 1996). The lack of highly sulfated KSPGs in AD may affect neurotransmission and weaken communication between neurons, thereby impairing the learning and memory of AD patients (Figure 1; Lindahl et al., 1996).
In addition to neurotransmission, synapse number and neuronal plasticity are also affected in AD (Vertes et al., 1999). Hyaluronic acid (HA) is one of the main components of the ECM (De La Motte and Drazba, 2011) that is linked to AD pathology via its effects on neuronal function and plasticity. The expression of HA increases with the progression of AD (Table 1, Reed et al., 2019). HA can inhibit the maturation of oligodendrocyte progenitors (Back et al., 2005). Abnormal expression of HA will demyelinate neurons, impair the transmission of nerve signals, limit remyelination, and cause white matter lesions (Figure 1; Montine et al., 2012). In human magnetic resonance imaging studies (Back et al., 2011), it was observed that white matter lesions in the medial prefrontal cortex (mPFC) were significantly associated with vascular injury and co-localized with areas rich in HA. The atypical increase of HA content in the brains of AD patients may cause vascular injury, a decrease of cerebral blood flow, decrease of oxygen and glucose supply to the brain, synapse loss, decline of neuronal function, and ultimately aggravate the cognitive deficits of AD patients (Figure 1; Park et al., 2014). Finally, HA is also present in the core of PNNs (Figure 2), which wrap around neurons and inhibit neuronal plasticity during aging and disease (Sorg et al., 2016).
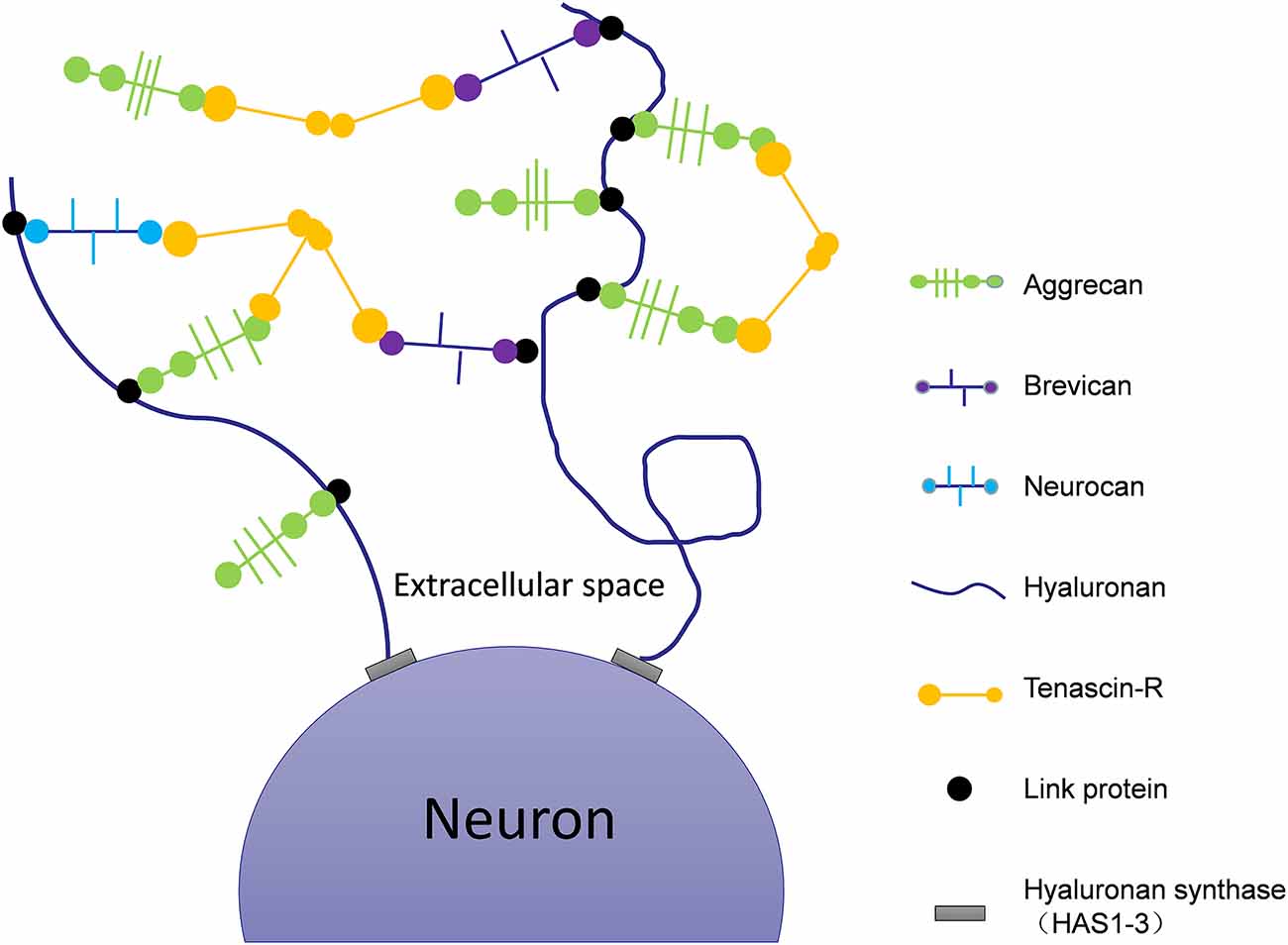
Figure 2. Schematic of the molecular composition of PNNs. Hyaluronan synthases are located at the neuronal cell membrane, synthesizing hyaluronan and secreting it into the perineuronal area. Members of the lectican family (aggrecan, neurocan, and brevican) bind to the hyaluronic backbone. Link proteins stabilize the binding of CSPGs to hyaluronan. Tenascin-R (TNR) crosslinks the lecticans to form stable PNNs.
It was reported that Aβ plaques produced in the brains of AD patients can cause changes in synaptic plasticity (Abramov et al., 2009). According to these reports, the chondroitin sulfate proteoglycan (CSPG) content in the brains of AD patients is higher than that of healthy individuals (Table 1, Goetzl et al., 2019). Chondroitin sulfate 4 (C-4) is expressed in the core of senile plaques (SPs) and neurofibrillary tangles (NTFs), non-chondroitin sulfate (C-0) is expressed in intracellular NTFs and dystrophic neurites of SPs, and chondroitin sulfate 6 (C-6) is expressed in the core of NTFs and around SPs (Dewitt et al., 1993). The combination of CSPGs and the inhibitory receptor protein tyrosine phosphatase σ (PTPσ) inhibits neuronal plasticity (Paveliev et al., 2016). Injecting chondroitinase ABC (ChABC) into the hippocampus of AD mice cuts off the CSPG network, thereby reducing Aβ plaques in the molecular layer, restoring synaptic density around Aβ plaques, enhancing synaptic plasticity, and finally improving the long-term memory capabilities of AD mice (Figure 1; Howell et al., 2015). Interestingly, the injection of ChABC into the secondary visual cortex (V2L) to relieve CSPG load can reduce long-term spatial memory in rats without affecting short-term memory, suggesting that the mechanisms of long-term and short-term memory are distinct. Moreover, rats’ recall of remote, but not recent, visual fear memories is dependent on intact PNNs in V2L (Thompson et al., 2018).
The PNNs are highly stable structures relieved from constant renewal as they are not exposed to the catabolic intracellular environment. Therefore, it has been proposed that PNN may continue to be a material framework for stabilizing long-term memory (Tsien, 2013). Moreover, the PNNs promote the fast-spiking activity of Parvalbumin-positive (PV+) interneurons, and consequently the excitatory–inhibitory balance of neural networks required for cognitive functions (Lensjø et al., 2017), which is essential for consolidation and retrieval of memories (Xia et al., 2017). The efficacy of ChABC in AD model and non-AD model animals may be caused by differences in CSPG content (Goetzl et al., 2019). In non-AD model animals, CSPG expression is relatively normal and sufficient to maintain the balance of the PNN network. Using ChABC to break up CSPG affects the stability of PNN and the material framework of long-term memory (Tsien, 2013), resulting in a decrease in long-term memory. Indeed, in AD model animals, the content of CSPG increases (Goetzl et al., 2019), and the normal structure of PNN can be restored by injection of ChABC so as to restore long-term memory.
In the brains of AD animal models, PNNs surround Aβ plaques, blocking the degradation of amyloid and inhibiting the growth of neurites (Hockfield et al., 1990; Pizzorusso et al., 2002; Mcrae et al., 2007). This restricts synaptic plasticity and affects the transfer of information between neurons. Tenascin-R (TNR), which is involved in the formation of PNNs (Figure 2), is increased in the brains of AD patients (Végh et al., 2014). The main components of PNNs are lecticans (aggrecan, brevican, neurocan, and versican), phosphacan, hyaluronan, TNR, and link proteins (cartilage link protein Crtl-1/HAPLN-1 and brain link protein Bral2/HAPLN-4; Figure 2). They interact with each other to provide the molecular backbone of the PNN, thereby creating a stable scaffold around the cell bodies and proximal dendrites of neurons (Figure 1; Suttkus et al., 2014). PNNs inhibit the outgrowth of neurites (Hockfield et al., 1990; Pizzorusso et al., 2002; Mcrae et al., 2007), which may lead to restricted synaptic plasticity and defective neurotransmission over the course of AD, aggravating the disease’s pathological damage.
When Aβ oligomers form, they become anchored to the cell membrane and result in the breakdown of the phospholipid bilayer (Friedman et al., 2009). They also interact with the hydrophobic region of the cell membrane to form transmembrane pores, which leads to the outflow of cell contents and an imbalance of ion homeostasis (Österlund et al., 2019). High molecular-weight heparin (HP) can bind to Aβ and promote the conversion of Aβ peptides from random coils to β sheets (Bruinsma et al., 2010), thereby promoting amyloid aggregation and fibrosis and stabilizing the formed senile plaques. On the contrary, low molecular-weight HP can reverse the process of amyloidosis by hindering the formation of β sheets (Walzer et al., 2002), and thus prevent the neuropathic process induced by Aβ (Figure 1). This fact indicates that HP proteins of different molecular weights play different roles in the course of AD. Studies have shown that removing the O-sulfuric acid group in HP weakens its role in promoting Aβ aggregation. Moreover, when the sulfuric acid group is completely removed, its promotion of Aβ aggregation disappears completely (Ariga et al., 2010), indicating that the sulfuric acid group is very important for the aggregation of Aβ.
The ECM-component HSPG is also increased in the brains of AD patients (Table 1, Van Horssen et al., 2003). Agrin is an HSPG containing nine protease-inhibiting regions (Stone and Nikolics, 1995; Castillo et al., 1999) and prevents proteases from degrading the Aβ protein. Agrin can enhance the formation of Aβ fibrils in vitro and can protect fibrils against proteolysis (Gupta-Bansal et al., 1995). As AD progresses, immature, non-fibrillar Aβ plaques eventually develop into mature, fibrillar Aβ plaques (Figure 1; Selkoe, 1991). This finding supports the contribution of HSPG to the formation of mature Aβ fibrils in AD. The pathogenic mechanism of this process operates as follows: Agrin prevents proteases from degrading the Aβ protein, the continuous deposition of which leads to the disease. Studies have shown that the degree of sulfation of the HSPG is essential to promote the formation of fibrils of Aβ (Cohen et al., 1997) or other amyloid peptides (Castillo et al., 1998). The sulfation pattern of HSPG does not occur randomly (Dennissen et al., 2002). Five anti-HS antibodies obtained through phage-display technology were used to label the topological structure of HSPG epitopes, and it was found that each antibody recognized a unique epitope structure of sulfate groups. This shows that the position of the sulfate group is very important for the function of the HSPG (Ten Dam et al., 2003, 2006; Kurup et al., 2007; Wijnhoven et al., 2008). Highly N-sulfated and O-sulfated HS may participate in the initiation of Aβ aggregation (Hileman et al., 1998) or stabilize previously formed Aβ (Holm Nielsen et al., 2000). The His-His-Gln-Lys amino-acid sequence on the Aβ peptide chain is considered the binding site for the interaction between glycosaminoglycans and Aβ, among which His13 may be the main site for the function of the sulfate group.
There are different structures of Aβ aggregates (namely, monomers, oligomers, and fibers) in the AD brain, indicating that there may be a connection between the progression of AD and the structure of Aβ (Gremer et al., 2017). Therefore, understanding what maintains the spherical structure of Aβ aggregates will provide a new perspective for the treatment of AD. Dermatan sulfate proteoglycans (DSPGs) are upregulated in the brains of AD rats (Table 1, Genedani et al., 2010). Using a variety of antibodies to identify decorin, the core protein of DSPG, in the brains of AD patients, positive staining was found in the filamentous structure of amyloid deposits and NFTs. Unlike HSPG, which tends to be evenly distributed in neuritic plaques (NPs) containing amyloid, decorin is mainly distributed around spherical amyloid plaques and the edges of amyloid fiber bundles. The lack of decorin in the center of certain amyloid plaques indicates that the distribution of DSPGs is spatially restricted (Snow et al., 1992). The abnormal distribution of decorin around amyloid plaques suggests that DSPGs may help regulate the size of the amyloid plaques in NPs or maintain its spherical structure (Figure 1; Obrink, 1973; Flint et al., 1984; Vogel et al., 1984; Scott, 1988). Moreover, the close relationship between blood vessels and amyloid plaques (Nortley et al., 2019) suggests that vascular-wall proteoglycans may be associated with Aβ deposits or NFTs in nearby plaques. Different DSPGs were also shown to have different binding affinities for amyloid (Buée et al., 1993). Taken together, this evidence supports that DSPGs play an important role in the formation of amyloid plaques and the pathogenesis of AD. After the induction of β-amyloid fibrils in the rat striatum, the DSPG content and the total charge density were shown to increase significantly (Genedani et al., 2010), indicating that the production of Aβ in the AD brain may trigger a responsive change in DSPG expression.
It has been reported that the expression of reelin and its glycosylation pattern is altered in the cerebrospinal fluid of AD patients (Botella-López et al., 2006). Specifically, there is evidence to show that the depletion of reelin is an early event in AD pathology (Table 1). A recent study showed that reelin and its downstream signaling members APOER2, VLDLR, and DAB1 are all affected in AD. Since reelin depletion occurs before the onset of Aβ pathology, the decline of reelin may play a role in the pathological precipitation of Aβ (Herring et al., 2012). Consistent with the above, the expression of reelin in the entorhinal cortex of transgenic mice and humans with AD is reduced (Chin et al., 2007). Further, reelin has been shown to inhibit Aβ production, promote Aβ clearance, and prevent Tau protein hyperphosphorylation (Figure 1; Dulabon et al., 2000; Brich et al., 2003; Deane et al., 2008).
Tau belongs to the microtubule-associated protein (MAP) family, which can effectively stabilize microtubules and promote retrograde (periphery to the nucleus) and anterograde (nucleus to outer periphery) axonal transport (Kadavath et al., 2015; Wang and Mandelkow, 2016). Therefore, the normal function of tau is essential for neuronal transport and synaptic structure (Avila et al., 2016; Guo et al., 2017). Tau hyperphosphorylation can lead to the deterioration of the dendritic structure and axonal transport, as well as the depolymerization of microtubules (Baudier and Cole, 1987).
HA synthases (HAS), including Has1, Has2, and Has3, are widely expressed in the central nervous system of mice. All types of HAS are located in the cell bodies of neurons, but only Has1 is found in axons. In TauP301S transgenic mice where tau protein is overexpressed, it was found that Has1 is no longer localized to axons, similar to the redistribution of Has1 expression observed in the brains of AD patients. Additionally, in the brains of TauP301S transgenic mice, Has1 expression decreases, whereas Has3 expression increases, leading to the upregulation of short-chain HA in the ECM (Figure 1; Li et al., 2017). The localization of Has1 depends on intact microtubules, and its mislocalization caused by the hyperphosphorylation of Tau disrupts the balance of ECM components, promotes ECM recombination, and inhibits the formation of PNNs. Under such conditions, neurons become more susceptible to the invasion of neurotoxic substances such as Aβ, which inhibit synaptic remodeling and aggravate AD.
Reelin is activated through the SFK/PI3K/Akt pathway to inhibit the expression of GSK-3β and prevent Tau phosphorylation, another pathological occurrence in AD. Recent studies have found that GSK-3β is the most effective Tau protein kinase and can promote an abnormal increase in Tau phosphorylation (Figure 1; Ohkubo et al., 2003). Consistently, studies have shown that the expression of GSK-3β colocalizes with NFTs, and both its expression and activity are significantly increased in the brains of AD patients.
Under healthy physiological conditions, the reactive oxygen species (ROS) metabolized by the body is maintained in a steady state of redox by the body’s natural antioxidant system. The hypothesis of free radical damage in AD postulates that (Cabungcal et al., 2013) the body’s defense systems are weakened with disease progression, causing the accumulation of a pathologically high level of free radicals, which react with unsaturated fatty acids in the cell membrane to form lipid peroxides such as malondialdehyde. Malondialdehyde combines with proteins, nucleic acids, and other biological macromolecules to form insoluble lipofuscin deposits in cells (Kun et al., 2018). Lipofuscin can damage cell structure, disrupt cell metabolism, accelerate cell senescence and death, as well as affect learning and memory (Giaccone et al., 2011; Kwon et al., 2016). As shown in Table 1, as the course of AD progresses, the expression of tenascin-R increases (Végh et al., 2014). Neurons in the cortex and subcortex are surrounded by PNNs, which protects them from neurofibrillary degeneration (Brückner et al., 1999; Morawski et al., 2012). Studies have shown that compared with unprotected neurons, those surrounded by PNNs are less affected by the accumulation of lipofuscin present in AD (Morawski et al., 2004). Since lipofuscin is an indicator of oxidative stress and aging (Sohal and Brunk, 1989), this finding indicates that the PNN may have a protective function against oxidative stress-induced neurodegeneration in AD.
Aβ-induced cerebrovascular (CV) deficits are mediated by ROS (Iadecola et al., 2009). Application of exogenous, soluble Aβ (Aβ1–40 and Aβ1–42 monomers) onto isolated mouse cerebral arterioles leads to significant oxidative stress and vasomotor dysfunction, and anti-ROS strategies markedly improve these CV deficits (Dietrich et al., 2010). Tg2576 mice in age from 2–3 months with elevated levels of endogenous, soluble Aβ species display substantial oxidative stress and CV deficits (Park et al., 2005). HSPGs are an attractive upstream candidate for Aβ-induced ROS production and CV dysfunction in AD. HSPGs bind Aβ with high affinity and promote their intracellular uptake in multiple cell types (Sandwall et al., 2010), including human cerebral vascular smooth muscle cells (VSMC; Kanekiyo and Bu, 2009). Therefore, HSPGs could be key mediators of Aβ1–42-induced oxidative stress and Aβ1–40-induced VSMC dysfunction. Aβ could interact with the cell surface or extracellular-matrix HSPGs, leading to intracellular calcium influx and ROS production. Toxic ROS species could directly damage the VSMC contractile machinery, leading to a hypercontractile phenotype that would reduce the supply of oxygen and glucose to the brain, thereby aggravating the pathology of AD (Reynolds et al., 2016).
Chondroitin sulfate (CS) oligosaccharides have also been tested as a therapeutic strategy in AD. They block Aβ-induced oxidative stress in SH-SY5Y cells and mitochondrial dysfunction (Zhao et al., 2020) in AD mice. They also inhibit oxidative stress, production of pro-inflammatory cytokines, and activation of the toll-like receptor pathway in Aβ-injured BV2 microglia (Zhao et al., 2020). CS oligosaccharides were reported to significantly suppress Aβ-induced oxidative stress by increasing the activity of antioxidant enzymes, including SOD and GSH-Px. It has been suggested that CS oligosaccharides may bind Aβ fibrils and inhibit them from interacting with cell and mitochondrial membranes; this process may be associated with the molecular weight of the CS oligosaccharides (Zhang et al., 2018). As such, modulation of ROS and identification of the upstream inducers of Aβ-mediated ROS production will be instrumental in designing novel therapies to prevent Aβ-induced CV dysfunction and to ameliorate the effects that these vascular deficits have on AD dementia.
Neuroinflammation is a result of the biological response of microglia and invading immune cells to harmful substances (Gendelman, 2002). Studies have shown that the pathology of AD (including Aβ accumulation and tau hyperphosphorylation) can cause inflammation in susceptible areas (Akiyama et al., 2000; Hamelin et al., 2016). The low molecular-weight HA fragments synthesized by Has3 or degraded by hyaluronidase can cause inflammation (Simpson et al., 2015). In severe AD, tumor necrosis factor (TNF)-stimulated gene-6 (TSG-6) was shown to be significantly increased and was found in NeuN-positive neurons and microglia (Hanger et al., 2014). TSG-6 is believed to reduce inflammation and provide tissue protection for various organs (Day and Milner, 2019). It is expressed in the brains of adult rodents and may become cross-linked with HA during the formation of glial scars in the central nervous system (Coulson-Thomas et al., 2016), thereby changing the structure of HA (Baranova et al., 2011) to one that enhances interaction with its receptors (Lesley et al., 2004; Lawrance et al., 2016; Richter et al., 2018). In this way, TSG-6 may help HA to alleviate neuroinflammation during AD (Day and Milner, 2019).
In AD, it has been reported that microglia (Krstic and Knuesel, 2013; Niraula et al., 2017) and astrocytes (Batarseh et al., 2016) can enhance the phagocytosis of harmful substances to resist the damage caused by AD; however, other studies suggest they secrete pro-inflammatory cytokines, which can damage brain cells and aggravate the process of AD. Studies have shown that a tenascin-C (TNC) deficiency reduces pro- but enhances anti-inflammatory activation in the mutated APP-transgenic mouse brain, associated with a reduced cerebral Aβ load and higher levels of postsynaptic density protein 95 (PSD-95; Xie et al., 2013). In other words, TNC transforms the neuroinflammatory process from pro- to anti-inflammatory (Xie et al., 2013). Reducing the level of TNC in the brains of AD mice significantly increased the number of microglia and macrophages, and decreased the activity of β-secretase and γ-secretase in the hippocampus and cortex (Heneka et al., 2015), which can effectively reduce the course of synapse damage in AD. This finding indicates that the pathogenesis of AD is not limited to neurons, but also includes interactions with the immune mechanisms of the brain. Misfolded and aggregated proteins bind to pattern-recognition receptors on microglia and astrocytes, which trigger the innate immune response characterized by the release of inflammatory mediators, thereby aggravating the progression of AD (Heneka et al., 2015). As an endogenous activator, TNC can accumulate in the AD brain and cause chronic inflammation via the action of pro-inflammatory cytokines (Figure 1).
The latest genomic and epidemiological studies have shown that inflammation and immune responses in the brain are key factors in the pathogenesis and progression of AD (Vanitallie, 2017). Therefore, using anti-inflammatory factors, such as those in the ECM, to relieve pro-inflammatory responses is a viable strategy for the treatment of AD.
Fibrillar Aβ plays a central role in neurotoxicity in AD brains (Mucke and Selkoe, 2012); it was shown to induce oxidative damage and destroy the cytosolic Ca2+ homeostasis of hippocampal neurons (Resende et al., 2007). Studies have shown that Aβ promotes an increase in the concentration of Ca2+ in the cytoplasm of neurons (Kuchibhotla et al., 2008), which increases the excitability of neurons and causes an increase in the frequency of action potentials (AP; Scarnati et al., 2020). The increase in neuronal AP frequency may be related to the shortened refractory period and lower threshold potential (Tamagnini et al., 2015), which is a common cause of neuronal death in most degenerative diseases. Despite being the main component of senile plaques in AD, Aβ protein does not show neurotoxicity to CSPG-containing neurons. However, when CSPG is removed with ChABC, Aβ1–42 becomes neurotoxic to neurons (Miyata et al., 2007), suggesting that the neuroprotective properties of PNNs could be harnessed as an effective treatment in AD. Furthermore, DSPGs in the ECM prevent the neurotoxic factors within and around senile plaques from interacting with the nearby neurons or astrocytes, which restricts the further spread of neurotoxic effects. Accordingly, neurons wrapped in DSPG-rich ECM are not susceptible to Aβ’s toxic effects (Díaz-Nido et al., 2002). Therefore, an increase in DSPGs may play a key role in reducing amyloid neurotoxicity. The “two-sidedness” of the effects of CSPG and DSPG in AD allows researchers to selectively eliminate or enhance the expression of CSPG or DSPG.
The pathogenesis of AD is complex. In this article, we found that the same ECM may participate in the initiation and development of AD through a variety of ways, while different ECMs may participate in the pathogenesis of AD via the same pathway. This review thus provides a broader basis for further understanding AD pathogenesis.
In summary, different components of the ECM have different roles in the neuropathology of AD, and regulating the expression of individual components is an important step to stabilize or improve the course of the disease. For example, as shown in Figure 2, HA, CSPG, TNR, and other components make up PNNs, which together maintain the stability of the extracellular environment. There may be potential interactions between the different components of the ECM, so that the ECM may participate in a variety of ways. Although the ECM component has various functions, it is also specific in different pathologies of AD. Therefore, this special ECM component will be a potential target and biomarker for the development and treatment of AD. This review describes the possible relevant role of some components of the ECM in AD, but the molecular mechanisms underlying the pathogenesis of AD remain unclear. Therefore, further elucidating the contributions of other ECM components in the pathogenesis of AD will be of great significance for its treatment and that of other AD-related neurodegenerative diseases.
QY and YS made substantial contributions to the conception and design of the review and gave final approval of the version to be published. SX, MJ, LY, XL, and ZB participated in writing the particular sections of the manuscript and approved the final version. QY and YS prepared figures. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant No. 31760275, 31860270, and 31960174), the PhD research startup foundation of Yan’an University (Grant No. YDBK2019-41), the Yan’an High-level Talent Project (Grant No. 203010108), Key Research and Development Program of Shaanxi (Program No. 2018ZDXM-NY-038 and 2018ZDCXL-SF-01-04), Shaanxi Province Innovative Talent Promotion Plan—Key Science and Technology Innovation Team (2017KCT-35), and the Department of Science and Technology of Shaanxi Province (2020NY-007 and 2020JM-555).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abramov, E., Dolev, I., Fogel, H., Ciccotosto, G. D., Ruff, E., and Slutsky, I. (2009). Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat. Neurosci. 12, 1567–1576. doi: 10.1038/nn.2433
Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G. M., et al. (2000). Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421. doi: 10.1016/s0197-4580(00)00124-x
Ariga, T., Miyatake, T., and Yu, R. K. (2010). Role of proteoglycans and glycosaminoglycans in the pathogenesis of Alzheimer’s disease and related disorders: amyloidogenesis and therapeutic strategies—a review. J. Neurosci. Res. 88, 2303–2315. doi: 10.1002/jnr.22393
Arranz, A. M., Perkins, K. L., Irie, F., Lewis, D. P., Hrabe, J., Xiao, F., et al. (2014). Hyaluronan deficiency due to Has3 knock-out causes altered neuronal activity and seizures via reduction in brain extracellular space. J. Neurosci. 34, 6164–6176. doi: 10.1523/JNEUROSCI.3458-13.2014
Avila, J., Jiménez, J. S., Sayas, C. L., Bolós, M., Zabala, J. C., Rivas, G., et al. (2016). Tau structures. Front. Aging Neurosci. 8:262. doi: 10.3389/fnagi.2016.00262
Back, S. A., Kroenke, C. D., Sherman, L. S., Lawrence, G., Gong, X., Taber, E. N., et al. (2011). White matter lesions defined by diffusion tensor imaging in older adults. Ann. Neurol. 70, 465–476. doi: 10.1002/ana.22484
Back, S. A., Tuohy, T. M., Chen, H., Wallingford, N., Craig, A., Struve, J., et al. (2005). Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat. Med. 11, 966–972. doi: 10.1038/nm1279
Bajjalieh, S. M., Peterson, K., Shinghal, R., and Scheller, R. H. (1992). SV2, a brain synaptic vesicle protein homologous to bacterial transporters. Science 257, 1271–1273. doi: 10.1126/science.1519064
Baranova, N. S., Nilebäck, E., Haller, F. M., Briggs, D. C., Svedhem, S., Day, A. J., et al. (2011). The inflammation-associated protein TSG-6 cross-links hyaluronan via hyaluronan-induced TSG-6 oligomers. J. Biol. Chem. 286, 25675–25686. doi: 10.1074/jbc.M111.247395
Batarseh, Y.S., Duong, Q.V., Mousa, Y.M., Al Rihani, S.B., Elfakhri, K., and Kaddoumi, A. (2016). Amyloid-β and astrocytes interplay in amyloid-β related disorders. Int. J. Mol. Sci. 17:338. doi: 10.3390/ijms17030338
Baudier, J., and Cole, R. D. (1987). Phosphorylation of tau proteins to a state like that in Alzheimer’s brain is catalyzed by a calcium/calmodulin-dependent kinase and modulated by phospholipids. J. Biol. Chem. 262, 17577–17583.
Blumenthal, N. R., Hermanson, O., Heimrich, B., and Shastri, V. P. (2014). Stochastic nanoroughness modulates neuron-astrocyte interactions and function via mechanosensing cation channels. Proc. Natl. Acad. Sci. U S A 111, 16124–16129. doi: 10.1073/pnas.1412740111
Botella-López, A., Burgaya, F., Gavín, R., García-Ayllón, M. S., Gómez-Tortosa, E., Peña-Casanova, J., et al. (2006). Reelin expression and glycosylation patterns are altered in Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 103, 5573–5578. doi: 10.1073/pnas.0601279103
Boyce, R., Glasgow, S. D., Williams, S., and Adamantidis, A. (2016). Causal evidence for the role of REM sleep theta rhythm in contextual memory consolidation. Science 352, 812–816. doi: 10.1126/science.aad5252
Brich, J., Shie, F. S., Howell, B. W., Li, R., Tus, K., Wakeland, E. K., et al. (2003). Genetic modulation of tau phosphorylation in the mouse. J. Neurosci. 23, 187–192. doi: 10.1523/JNEUROSCI.23-01-00187.2003
Brückner, G., Hausen, D., Härtig, W., Drlicek, M., Arendt, T., and Brauer, K. (1999). Cortical areas abundant in extracellular matrix chondroitin sulphate proteoglycans are less affected by cytoskeletal changes in Alzheimer’s disease. Neuroscience 92, 791–805. doi: 10.1016/j.foodchem.2021.130679
Bruinsma, I. B., Te Riet, L., Gevers, T., Ten Dam, G. B., Van Kuppevelt, T. H., David, G., et al. (2010). Sulfation of heparan sulfate associated with amyloid-beta plaques in patients with Alzheimer’s disease. Acta Neuropathol. 119, 211–220. doi: 10.1007/s00401-009-0577-1
Buckley, K., and Kelly, R. B. (1985). Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. J. Cell Biol. 100, 1284–1294. doi: 10.1083/jcb.100.4.1284
Buée, L., Ding, W., Anderson, J. P., Narindrasorasak, S., Kisilevsky, R., Boyle, N. J., et al. (1993). Binding of vascular heparan sulfate proteoglycan to Alzheimer’s amyloid precursor protein is mediated in part by the N-terminal region of A4 peptide. Brain Res. 627, 199–204. doi: 10.1001/jamaoncol.2021.2705
Cabungcal, J. H., Steullet, P., Morishita, H., Kraftsik, R., Cuenod, M., Hensch, T. K., et al. (2013). Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc. Natl. Acad. Sci. U S A 110, 9130–9135. doi: 10.1073/pnas.1300454110
Castillo, G. M., Cummings, J. A., Yang, W., Judge, M. E., Sheardown, M. J., Rimvall, K., et al. (1998). Sulfate content and specific glycosaminoglycan backbone of perlecan are critical for perlecan’s enhancement of islet amyloid polypeptide (amylin) fibril formation. Diabetes 47, 612–620. doi: 10.2337/diabetes.47.4.612
Castillo, G. M., Lukito, W., Wight, T. N., and Snow, A. D. (1999). The sulfate moieties of glycosaminoglycans are critical for the enhancement of beta-amyloid protein fibril formation. J. Neurochem. 72, 1681–1687. doi: 10.1046/j.1471-4159.1999.721681.x
Celio, M. R., Spreafico, R., De Biasi, S., and Vitellaro-Zuccarello, L. (1998). Perineuronal nets: past and present. Trends Neurosci. 21, 510–515. doi: 10.1016/s0166-2236(98)01298-3
Chin, J., Massaro, C. M., Palop, J. J., Thwin, M. T., Yu, G. Q., Bien-Ly, N., et al. (2007). Reelin depletion in the entorhinal cortex of human amyloid precursor protein transgenic mice and humans with Alzheimer’s disease. J. Neurosci. 27, 2727–2733. doi: 10.1523/JNEUROSCI.3758-06.2007
Cohen, N. A., Kaufmann, W. E., Worley, P. F., and Rupp, F. (1997). Expression of agrin in the developing and adult rat brain. Neuroscience 76, 581–596. doi: 10.1016/s0306-4522(96)00345-4
Coulson-Thomas, V. J., Lauer, M. E., Soleman, S., Zhao, C., Hascall, V. C., Day, A. J., et al. (2016). Tumor necrosis factor-stimulated gene-6 (TSG-6) is constitutively expressed in adult central nervous system (CNS) and associated with astrocyte-mediated glial scar formation following spinal cord injury. J. Biol. Chem. 291, 19939–19952. doi: 10.1074/jbc.M115.710673
Day, A. J., and Milner, C. M. (2019). TSG-6: a multifunctional protein with anti-inflammatory and tissue-protective properties. Matrix Biol. 78, 60–83. doi: 10.1016/j.matbio.2018.01.011
De La Motte, C. A., and Drazba, J. A. (2011). Viewing hyaluronan: imaging contributes to imagining new roles for this amazing matrix polymer. J. Histochem. Cytochem. 59, 252–257. doi: 10.1369/0022155410397760
Deane, R., Sagare, A., Hamm, K., Parisi, M., Lane, S., Finn, M. B., et al. (2008). apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Invest. 118, 4002–4013. doi: 10.1172/JCI36663
Deepa, S. S., Carulli, D., Galtrey, C., Rhodes, K., Fukuda, J., Mikami, T., et al. (2006). Composition of perineuronal net extracellular matrix in rat brain: a different disaccharide composition for the net-associated proteoglycans. J. Biol. Chem. 281, 17789–17800. doi: 10.1074/jbc.M600544200
Dennissen, M. A., Jenniskens, G. J., Pieffers, M., Versteeg, E. M., Petitou, M., Veerkamp, J. H., et al. (2002). Large, tissue-regulated domain diversity of heparan sulfates demonstrated by phage display antibodies. J. Biol. Chem. 277, 10982–10986. doi: 10.1074/jbc.M104852200
Dewitt, D. A., Silver, J., Canning, D. R., and Perry, G. (1993). Chondroitin sulfate proteoglycans are associated with the lesions of Alzheimer’s disease. Exp. Neurol. 121, 149–152. doi: 10.1006/exnr.1993.1081
Díaz-Nido, J., Wandosell, F., and Avila, J. (2002). Glycosaminoglycans and beta-amyloid, prion and tau peptides in neurodegenerative diseases. Peptides 23, 1323–1332. doi: 10.1055/s-0041-1733782
Dietrich, H. H., Xiang, C., Han, B. H., Zipfel, G. J., and Holtzman, D. M. (2010). Soluble amyloid-beta, effect on cerebral arteriolar regulation and vascular cells. Mol. Neurodegener. 5:15. doi: 10.1186/1750-1326-5-15
Dulabon, L., Olson, E. C., Taglienti, M. G., Eisenhuth, S., Mcgrath, B., Walsh, C. A., et al. (2000). Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron 27, 33–44. doi: 10.1016/s0896-6273(00)00007-6
Feany, M. B., Lee, S., Edwards, R. H., and Buckley, K. M. (1992). The synaptic vesicle protein SV2 is a novel type of transmembrane transporter. Cell 70, 861–867. doi: 10.1016/0092-8674(92)90319-8
Flint, M. H., Craig, A. S., Reilly, H. C., Gillard, G. C., and Parry, D. A. (1984). Collagen fibril diameters and glycosaminoglycan content of skins–indices of tissue maturity and function. Connect Tissue Res. 13, 69–81. doi: 10.3109/03008208409152144
Frantz, C., Stewart, K. M., and Weaver, V. M. (2010). The extracellular matrix at a glance. J. Cell Sci. 123, 4195–4200. doi: 10.1242/jcs.023820
Friedman, R., Pellarin, R., and Caflisch, A. (2009). Amyloid aggregation on lipid bilayers and its impact on membrane permeability. J. Mol. Biol. 387, 407–415. doi: 10.1016/j.jmb.2008.12.036
Gendelman, H. E. (2002). Neural immunity: friend or foe. J. Neurovirol. 8, 474–479. doi: 10.1080/13550280290168631
Genedani, S., Agnati, L. F., Leo, G., Buzzega, D., Maccari, F., Carone, C., et al. (2010). Beta-Amyloid fibrillation and/or hyperhomocysteinemia modify striatal patterns of hyaluronic acid and dermatan sulfate: possible role in the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 7, 150–157. doi: 10.2174/156720510790691074
Giaccone, G., Orsi, L., Cupidi, C., and Tagliavini, F. (2011). Lipofuscin hypothesis of Alzheimer’s disease. Dement. Geriatr. Cogn. Dis. Extra 1, 292–296. doi: 10.1159/000329544
Gingrich, J. A., Andersen, P. H., Tiberi, M., El Mestikawy, S., Jorgensen, P. N., Fremeau, R. T., et al. (1992). Identification, characterization and molecular cloning of a novel transporter-like protein localized to the central nervous system. FEBS Lett. 312, 115–122. doi: 10.1016/0014-5793(92)80917-6
Goetzl, E. J., Nogueras-Ortiz, C., Mustapic, M., Mullins, R. J., Abner, E. L., Schwartz, J. B., et al. (2019). Deficient neurotrophic factors of CSPG4-type neural cell exosomes in Alzheimer disease. FASEB J. 33, 231–238. doi: 10.1096/fj.201801001
Gremer, L., Schölzel, D., Schenk, C., Reinartz, E., Labahn, J., Ravelli, R. B. G., et al. (2017). Fibril structure of amyloid-β(1–42) by cryo-electron microscopy. Science 358, 116–119. doi: 10.1126/science.aao2825
Guo, T., Noble, W., and Hanger, D. P. (2017). Roles of tau protein in health and disease. Acta Neuropathol. 133, 665–704. doi: 10.1007/s00401-017-1707-9
Gupta-Bansal, R., Frederickson, R. C., and Brunden, K. R. (1995). Proteoglycan-mediated inhibition of A beta proteolysis. A potential cause of senile plaque accumulation. J. Biol. Chem. 270, 18666–18671. doi: 10.1074/jbc.270.31.18666
Hamelin, L., Lagarde, J., Dorothée, G., Leroy, C., Labit, M., Comley, R. A., et al. (2016). Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain 139, 1252–1264. doi: 10.1093/brain/aww017
Hanger, D. P., Lau, D. H., Phillips, E. C., Bondulich, M. K., Guo, T., Woodward, B. W., et al. (2014). Intracellular and extracellular roles for tau in neurodegenerative disease. J. Alzheimers Dis. 40, S37–45. doi: 10.3233/JAD-132054
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hasanzadeh, Z., Nourazarian, A., Nikanfar, M., Laghousi, D., Vatankhah, A. M., and Sadrirad, S. (2021). Evaluation of the serum Dkk-1, Tenascin-C, oxidative stress markers levels and Wnt signaling pathway genes expression in patients with Alzheimer’s disease. J. Mol. Neurosci. 71, 879–887. doi: 10.1007/s12031-020-01710-9
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Herring, A., Donath, A., Steiner, K. M., Widera, M. P., Hamzehian, S., Kanakis, D., et al. (2012). Reelin depletion is an early phenomenon of Alzheimer’s pathology. J. Alzheimers Dis. 30, 963–979. doi: 10.3233/JAD-2012-112069
Hileman, R. E., Fromm, J. R., Weiler, J. M., and Linhardt, R. J. (1998). Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. Bioessays 20, 156–167. doi: 10.1002/(SICI)1521-1878(199802)20:2<156::AID-BIES8>3.0.CO;2-R
Hockfield, S., Kalb, R. G., Zaremba, S., and Fryer, H. (1990). Expression of neural proteoglycans correlates with the acquisition of mature neuronal properties in the mammalian brain. Cold Spring Harb. Symp. Quant. Biol. 55, 505–514. doi: 10.1101/sQ16.1990.055.01.049
Holm Nielsen, E., Nybo, M., Junker, K., Toftedal Hansen, P., Rasmussen, I. M., and Svehag, S. E. (2000). Localization of human serum amyloid P component and heparan sulfate proteoglycan in in vitro-formed Abeta fibrils. Scand. J. Immunol. 52, 110–112. doi: 10.1046/j.1365-3083.2000.00775.x
Hondius, D. C., Van Nierop, P., Li, K. W., Hoozemans, J. J., Van Der Schors, R. C., Van Haastert, E. S., et al. (2016). Profiling the human hippocampal proteome at all pathologic stages of Alzheimer’s disease. Alzheimers Dement. 12, 654–668. doi: 10.1016/j.jalz.2015.11.002
Howell, M. D., Bailey, L. A., Cozart, M. A., Gannon, B. M., and Gottschall, P. E. (2015). Hippocampal administration of chondroitinase ABC increases plaque-adjacent synaptic marker and diminishes amyloid burden in aged APPswe/PS1dE9 mice. Acta Neuropathol. Commun. 3:54. doi: 10.1186/s40478-015-0233-z
Iadecola, C., Park, L., and Capone, C. (2009). Threats to the mind: aging, amyloid and hypertension. Stroke 40, S40–S44. doi: 10.1161/STROKEAHA.108.533638
Kadavath, H., Hofele, R. V., Biernat, J., Kumar, S., Tepper, K., Urlaub, H., et al. (2015). Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. U S A 112, 7501–7506. doi: 10.1073/pnas.1504081112
Kanekiyo, T., and Bu, G. (2009). Receptor-associated protein interacts with amyloid-beta peptide and promotes its cellular uptake. J. Biol. Chem. 284, 33352–33359. doi: 10.1074/jbc.M109.015032
Krstic, D., and Knuesel, I. (2013). Deciphering the mechanism underlying late-onset Alzheimer disease. Nat. Rev. Neurol. 9, 25–34. doi: 10.1038/nrneurol.2012.236
Kuchibhotla, K. V., Goldman, S. T., Lattarulo, C. R., Wu, H. Y., Hyman, B. T., and Bacskai, B. J. (2008). Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59, 214–225. doi: 10.1016/j.neuron.2008.06.008
Kun, A., González-Camacho, F., Hernández, S., Moreno-García, A., Calero, O., and Calero, M. (2018). Characterization of amyloid-β plaques and autofluorescent lipofuscin aggregates in alzheimer’s disease brain: a confocal microscopy approach. Methods Mol. Biol. 1779, 497–512. doi: 10.1007/978-1-4939-7816-8_31
Kurup, S., Wijnhoven, T. J., Jenniskens, G. J., Kimata, K., Habuchi, H., Li, J. P., et al. (2007). Characterization of anti-heparan sulfate phage display antibodies AO4B08 and HS4E4. J. Biol. Chem. 282, 21032–21042. doi: 10.1074/jbc.M702073200
Kwon, H. J., Cha, M. Y., Kim, D., Kim, D. K., Soh, M., Shin, K., et al. (2016). Mitochondria-targeting ceria nanoparticles as antioxidants for Alzheimer’s disease. ACS Nano 10, 2860–2870. doi: 10.1021/acsnano.5b08045
Lawrance, W., Banerji, S., Day, A. J., Bhattacharjee, S., and Jackson, D. G. (2016). Binding of hyaluronan to the native lymphatic vessel endothelial receptor LYVE-1 is critically dependent on receptor clustering and hyaluronan organization. J. Biol. Chem. 291, 8014–8030. doi: 10.1074/jbc.M115.708305
Lensjø, K. K., Lepperød, M. E., Dick, G., Hafting, T., and Fyhn, M. (2017). Removal of perineuronal nets unlocks juvenile plasticity through network mechanisms of decreased inhibition and increased gamma activity. J. Neurosci. 37, 1269–1283. doi: 10.1523/JNEUROSCI.2504-16.2016
Lesley, J., Gál, I., Mahoney, D. J., Cordell, M. R., Rugg, M. S., Hyman, R., et al. (2004). TSG-6 modulates the interaction between hyaluronan and cell surface CD44. J. Biol. Chem. 279, 25745–25754. doi: 10.1074/jbc.M313319200
Li, Y., Li, Z. X., Jin, T., Wang, Z. Y., and Zhao, P. (2017). Tau pathology promotes the reorganization of the extracellular matrix and inhibits the formation of perineuronal nets by regulating the expression and the distribution of hyaluronic acid synthases. J. Alzheimers Dis. 57, 395–409. doi: 10.3233/JAD-160804
Lindahl, B., Eriksson, L., Spillmann, D., Caterson, B., and Lindahl, U. (1996). Selective loss of cerebral keratan sulfate in Alzheimer’s disease. J. Biol. Chem. 271, 16991–16994. doi: 10.1074/jbc.271.29.16991
Lorente-Gea, L., García, B., Martín, C., Ordiales, H., García-Suárez, O., Piña-Batista, K. M., et al. (2020). Heparan sulfate proteoglycans undergo differential expression alterations in Alzheimer disease brains. J. Neuropathol. Exp. Neurol. 79, 474–483. doi: 10.1093/jnen/nlaa016
Maccioni, R. B., Farías, G., Morales, I., and Navarrete, L. (2010). The revitalized tau hypothesis on Alzheimer’s disease. Arch. Med. Res. 41, 226–231. doi: 10.1016/j.arcmed.2010.03.007
Manavalan, A., Mishra, M., Feng, L., Sze, S. K., Akatsu, H., and Heese, K. (2013). Brain site-specific proteome changes in aging-related dementia. Exp. Mol. Med. 45:e39. doi: 10.1038/emm.2013.76
Mcrae, P. A., Rocco, M. M., Kelly, G., Brumberg, J. C., and Matthews, R. T. (2007). Sensory deprivation alters aggrecan and perineuronal net expression in the mouse barrel cortex. J. Neurosci. 27, 5405–5413. doi: 10.1523/JNEUROSCI.5425-06.2007
Miyata, S., and Kitagawa, H. (2016). Chondroitin sulfate and neuronal disorders. Front. Biosci. (Landmark Ed) 21, 1330–1340. doi: 10.2741/4460
Miyata, S., Nishimura, Y., and Nakashima, T. (2007). Perineuronal nets protect against amyloid beta-protein neurotoxicity in cultured cortical neurons. Brain Res. 1150, 200–206. doi: 10.1016/j.brainres.2007.02.066
Montine, T. J., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Dickson, D. W., et al. (2012). National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123, 1–11. doi: 10.1007/s00401-011-0910-3
Morawski, M., Brückner, G., Jäger, C., Seeger, G., Matthews, R. T., and Arendt, T. (2012). Involvement of perineuronal and perisynaptic extracellular matrix in Alzheimer’s disease neuropathology. Brain Pathol. 22, 547–561. doi: 10.1111/j.1750-3639.2011.00557.x
Morawski, M., Brückner, M. K., Riederer, P., Brückner, G., and Arendt, T. (2004). Perineuronal nets potentially protect against oxidative stress. Exp. Neurol. 188, 309–315. doi: 10.1016/j.expneurol.2004.04.017
Mota, S. I., Ferreira, I. L., Valero, J., Ferreiro, E., Carvalho, A. L., Oliveira, C. R., et al. (2014). Impaired Src signaling and post-synaptic actin polymerization in Alzheimer’s disease mice hippocampus—linking NMDA receptors and the reelin pathway. Exp. Neurol. 261, 698–709. doi: 10.1016/j.expneurol.2014.07.023
Mucke, L., and Selkoe, D. J. (2012). Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2:a006338. doi: 10.1101/cshperspect.a006338
Nägga, K., Hansson, O., Van Westen, D., Minthon, L., and Wennström, M. (2014). Increased levels of hyaluronic acid in cerebrospinal fluid in patients with vascular dementia. J. Alzheimers Dis. 42, 1435–1441. doi: 10.5999/aps.2020.02257
Nielsen, H. M., Palmqvist, S., Minthon, L., Londos, E., and Wennström, M. (2012). Gender-dependent levels of hyaluronic acid in cerebrospinal fluid of patients with neurodegenerative dementia. Curr. Alzheimer Res. 9, 257–266. doi: 10.2174/156720512800107537
Niraula, A., Sheridan, J. F., and Godbout, J. P. (2017). Microglia priming with aging and stress. Neuropsychopharmacology 42, 318–333. doi: 10.1038/npp.2016.185
Nortley, R., Korte, N., Izquierdo, P., Hirunpattarasilp, C., Mishra, A., Jaunmuktane, Z., et al. (2019). Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 365:eaav9518. doi: 10.1126/science.aav9518
Obrink, B. (1973). The influence of glycosaminoglycans on the formation of fibers from monomeric tropocollagen in vitro. Eur. J. Biochem. 34, 129–137. doi: 10.1111/j.1432-1033.1973.tb02739.x
Ohkubo, N., Lee, Y. D., Morishima, A., Terashima, T., Kikkawa, S., Tohyama, M., et al. (2003). Apolipoprotein E and reelin ligands modulate tau phosphorylation through an apolipoprotein E receptor/disabled-1/glycogen synthase kinase-3beta cascade. FASEB J. 17, 295–297. doi: 10.1096/fj.02-0434fje
Österlund, N., Moons, R., Ilag, L. L., Sobott, F., and Gräslund, A. (2019). Native ion mobility-mass spectrometry reveals the formation of β-barrel shaped amyloid-β hexamers in a membrane-mimicking environment. J. Am. Chem. Soc. 141, 10440–10450. doi: 10.1021/jacs.9b04596
Park, L., Anrather, J., Zhou, P., Frys, K., Pitstick, R., Younkin, S., et al. (2005). NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J. Neurosci. 25, 1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005
Park, L., Koizumi, K., El Jamal, S., Zhou, P., Previti, M. L., Van Nostrand, W. E., et al. (2014). Age-dependent neurovascular dysfunction and damage in a mouse model of cerebral amyloid angiopathy. Stroke 45, 1815–1821. doi: 10.1161/STROKEAHA.114.005179
Paveliev, M., Fenrich, K. K., Kislin, M., Kuja-Panula, J., Kulesskiy, E., Varjosalo, M., et al. (2016). HB-GAM (pleiotrophin) reverses inhibition of neural regeneration by the CNS extracellular matrix. Sci. Rep. 6:33916. doi: 10.1038/srep33916
Pizzorusso, T., Medini, P., Berardi, N., Chierzi, S., Fawcett, J. W., and Maffei, L. (2002). Reactivation of ocular dominance plasticity in the adult visual cortex. Science 298, 1248–1251. doi: 10.1126/science.1072699
Pollen, A. A., Nowakowski, T. J., Chen, J., Retallack, H., Sandoval-Espinosa, C., Nicholas, C. R., et al. (2015). Molecular identity of human outer radial glia during cortical development. Cell 163, 55–67. doi: 10.1016/j.cell.2015.09.004
Reed, M. J., Damodarasamy, M., Pathan, J. L., Chan, C. K., Spiekerman, C., Wight, T. N., et al. (2019). Increased hyaluronan and TSG-6 in association with neuropathologic changes of Alzheimer’s disease. J. Alzheimers Dis. 67, 91–102. doi: 10.3233/JAD-180797
Resende, R., Pereira, C., Agostinho, P., Vieira, A. P., Malva, J. O., and Oliveira, C. R. (2007). Susceptibility of hippocampal neurons to Abeta peptide toxicity is associated with perturbation of Ca2+ homeostasis. Brain Res. 1143, 11–21. doi: 10.1016/j.brainres.2007.01.071
Reynolds, M. R., Singh, I., Azad, T. D., Holmes, B. B., Verghese, P. B., Dietrich, H. H., et al. (2016). Heparan sulfate proteoglycans mediate Aβ-induced oxidative stress and hypercontractility in cultured vascular smooth muscle cells. Mol. Neurodegener. 11:9. doi: 10.1186/s13024-016-0073-8
Richter, R. P., Baranova, N. S., Day, A. J., and Kwok, J. C. (2018). Glycosaminoglycans in extracellular matrix organisation: are concepts from soft matter physics key to understanding the formation of perineuronal nets? Curr. Opin. Struct. Biol. 50, 65–74. doi: 10.1016/j.sbi.2017.12.002
Sandwall, E., O’callaghan, P., Zhang, X., Lindahl, U., Lannfelt, L., and Li, J. P. (2010). Heparan sulfate mediates amyloid-beta internalization and cytotoxicity. Glycobiology 20, 533–541. doi: 10.1093/glycob/cwp205
Scarnati, M. S., Clarke, S. G., Pang, Z. P., and Paradiso, K. G. (2020). Presynaptic calcium channel open probability and changes in calcium influx throughout the action potential determined using AP-waveforms. Front. Synaptic Neurosci. 12:17. doi: 10.3389/fnsyn.2020.00017
Scott, J. E. (1988). Proteoglycan-fibrillar collagen interactions. Biochem. J. 252, 313–323. doi: 10.1042/bj2520313
Scranton, T. W., Iwata, M., and Carlson, S. S. (1993). The SV2 protein of synaptic vesicles is a keratan sulfate proteoglycan. J. Neurochem. 61, 29–44. doi: 10.1111/j.1471-4159.1993.tb03535.x
Selkoe, D. J. (1991). The molecular pathology of Alzheimer’s disease. Neuron 6, 487–498. doi: 10.1016/0896-6273(91)90052-2
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Shabani, S., Farbood, Y., Mard, S. A., Sarkaki, A., Ahangarpour, A., and Khorsandi, L. (2018). The regulation of pituitary-thyroid abnormalities by peripheral administration of levothyroxine increased brain-derived neurotrophic factor and reelin protein expression in an animal model of Alzheimer’s disease. Can. J. Physiol. Pharmacol. 96, 275–280. doi: 10.1139/cjpp-2016-0434
Shimizu, H., Ghazizadeh, M., Sato, S., Oguro, T., and Kawanami, O. (2009). Interaction between beta-amyloid protein and heparan sulfate proteoglycans from the cerebral capillary basement membrane in Alzheimer’s disease. J. Clin. Neurosci. 16, 277–282. doi: 10.1016/j.jocn.2008.04.009
Simpson, M. A., De La Motte, C., Sherman, L. S., and Weigel, P. H. (2015). Advances in hyaluronan biology: signaling, regulation and disease mechanisms. Int. J. Cell Biol. 2015:690572. doi: 10.1155/2015/690572
Snow, A. D., Mar, H., Nochlin, D., Kresse, H., and Wight, T. N. (1992). Peripheral distribution of dermatan sulfate proteoglycans (decorin) in amyloid-containing plaques and their presence in neurofibrillary tangles of Alzheimer’s disease. J. Histochem. Cytochem. 40, 105–113. doi: 10.1177/40.1.1370306
Snow, A. D., Nochlin, D., Sekiguichi, R., and Carlson, S. S. (1996). Identification in immunolocalization of a new class of proteoglycan (keratan sulfate) to the neuritic plaques of Alzheimer’s disease. Exp. Neurol. 138, 305–317. doi: 10.1006/exnr.1996.0069
Sohal, R. S., and Brunk, U. T. (1989). Lipofuscin as an indicator of oxidative stress and aging. Adv. Exp. Med. Biol. 266, 17–26. doi: 10.1007/978-1-4899-5339-1_2
Soleman, S., Filippov, M. A., Dityatev, A., and Fawcett, J. W. (2013). Targeting the neural extracellular matrix in neurological disorders. Neuroscience 253, 194–213. doi: 10.1016/j.neuroscience.2013.08.050
Sorg, B. A., Berretta, S., Blacktop, J. M., Fawcett, J. W., Kitagawa, H., Kwok, J. C., et al. (2016). Casting a wide net: role of perineuronal nets in neural plasticity. J. Neurosci. 36, 11459–11468. doi: 10.1523/JNEUROSCI.2351-16.2016
Stone, D. M., and Nikolics, K. (1995). Tissue- and age-specific expression patterns of alternatively spliced agrin mRNA transcripts in embryonic rat suggest novel developmental roles. J. Neurosci. 15, 6767–6778. doi: 10.1523/JNEUROSCI.15-10-06767.1995
Suttkus, A., Rohn, S., Weigel, S., Glöckner, P., Arendt, T., and Morawski, M. (2014). Aggrecan, link protein and tenascin-R are essential components of the perineuronal net to protect neurons against iron-induced oxidative stress. Cell Death Dis. 5:e1119. doi: 10.1038/cddis.2014.25
Tamagnini, F., Novelia, J., Kerrigan, T. L., Brown, J. T., Tsaneva-Atanasova, K., and Randall, A. D. (2015). Altered intrinsic excitability of hippocampal CA1 pyramidal neurons in aged PDAPP mice. Front. Cell Neurosci. 9:372. doi: 10.3389/fncel.2015.00372
Ten Dam, G. B., Hafmans, T., Veerkamp, J. H., and Van Kuppevelt, T. H. (2003). Differential expression of heparan sulfate domains in rat spleen. J. Histochem. Cytochem. 51, 727–739. doi: 10.1177/002215540305100604
Ten Dam, G. B., Kurup, S., Van De Westerlo, E. M., Versteeg, E. M., Lindahl, U., Spillmann, D., et al. (2006). 3-O-sulfated oligosaccharide structures are recognized by anti-heparan sulfate antibody HS4C3. J. Biol. Chem. 281, 4654–4662. doi: 10.1074/jbc.M506357200
Terry, A. V. Jr., and Buccafusco, J. J. (2003). The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 306, 821–827. doi: 10.1124/jpet.102.041616
Testa, D., Prochiantz, A., and Di Nardo, A. A. (2019). Perineuronal nets in brain physiology and disease. Semin. Cell Dev. Biol. 89, 125–135. doi: 10.1016/j.semcdb.2018.09.011
Thompson, E. H., Lensjø, K. K., Wigestrand, M. B., Malthe-Sørenssen, A., Hafting, T., and Fyhn, M. (2018). Removal of perineuronal nets disrupts recall of a remote fear memory. Proc. Natl. Acad. Sci. U S A 115, 607–612. doi: 10.1073/pnas.1713530115
Tsien, R. Y. (2013). Very long-term memories may be stored in the pattern of holes in the perineuronal net. Proc. Natl. Acad. Sci. U S A 110, 12456–12461. doi: 10.1073/pnas.1310158110
Van Horssen, J., Wesseling, P., Van Den Heuvel, L. P., De Waal, R. M., and Verbeek, M. M. (2003). Heparan sulphate proteoglycans in Alzheimer’s disease and amyloid-related disorders. Lancet Neurol. 2, 482–492. doi: 10.1016/s1474-4422(03)00484-8
Vanitallie, T. B. (2017). Alzheimer’s disease: Innate immunity gone awry? Metabolism 69, S41–S49. doi: 10.1016/j.metabol.2017.01.014
Végh, M. J., Heldring, C. M., Kamphuis, W., Hijazi, S., Timmerman, A. J., Li, K. W., et al. (2014). Reducing hippocampal extracellular matrix reverses early memory deficits in a mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 2:76. doi: 10.1186/s40478-014-0076-z
Vertes, R. P., Fortin, W. J., and Crane, A. M. (1999). Projections of the median raphe nucleus in the rat. J. Comp. Neurol. 407, 555–582.
Vogel, K. G., Paulsson, M., and Heinegård, D. (1984). Specific inhibition of type I and type II collagen fibrillogenesis by the small proteoglycan of tendon. Biochem. J. 223, 587–597. doi: 10.1042/bj2230587
Walzer, M., Lorens, S., Hejna, M., Fareed, J., Hanin, I., Cornelli, U., et al. (2002). Low molecular weight glycosaminoglycan blockade of beta-amyloid induced neuropathology. Eur. J. Pharmacol. 445, 211–220. doi: 10.1016/s0014-2999(02)01759-4
Wang, Y., and Mandelkow, E. (2016). Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21. doi: 10.1038/nrn.2015.1
Wenk, G. L. (2003). Neuropathologic changes in Alzheimer’s disease. J. Clin. Psychiatry 64, 7–10. Available online at: https://www.psychiatrist.com/read-pdf/12701/.
Wijnhoven, T. J., Van De Westerlo, E. M., Smits, N. C., Lensen, J. F., Rops, A. L., Van Der Vlag, J., et al. (2008). Characterization of anticoagulant heparinoids by immunoprofiling. Glycoconj. J. 25, 177–185. doi: 10.1007/s10719-007-9070-z
Xia, F., Richards, B. A., Tran, M. M., Josselyn, S. A., Takehara-Nishiuchi, K., and Frankland, P. W. (2017). Parvalbumin-positive interneurons mediate neocortical-hippocampal interactions that are necessary for memory consolidation. eLife 6:e27868. doi: 10.7554/eLife.27868
Xie, K., Liu, Y., Hao, W., Walter, S., Penke, B., Hartmann, T., et al. (2013). Tenascin-C deficiency ameliorates Alzheimer’s disease-related pathology in mice. Neurobiol. Aging 34, 2389–2398. doi: 10.1016/j.neurobiolaging.2013.04.013
Yanagishita, M. (1993). Function of proteoglycans in the extracellular matrix. Acta Pathol. Jpn. 43, 283–293. doi: 10.1111/j.1440-1827.1993.tb02569.x
Zhang, Q., Na, Z., Cheng, Y., and Wang, F. (2018). Low-molecular-weight chondroitin sulfate attenuated injury by inhibiting oxidative stress in amyloid β-treated SH-SY5Y cells. Neuroreport 29, 1174–1179. doi: 10.1097/WNR.0000000000001092
Keywords: Alzheimer’s disease, extracellular matrix, synaptic transmission, amyloid-β plaque, tau-protein, oxidative stress, inflammation
Citation: Sun Y, Xu S, Jiang M, Liu X, Yang L, Bai Z and Yang Q (2021) Role of the Extracellular Matrix in Alzheimer’s Disease. Front. Aging Neurosci. 13:707466. doi: 10.3389/fnagi.2021.707466
Received: 10 May 2021; Accepted: 04 August 2021;
Published: 27 August 2021.
Edited by:
Harry Wilhelm Steinbusch, Maastricht University, NetherlandsReviewed by:
Karen Schmitt, Central Institute of Mental Health (ZI), GermanyCopyright © 2021 Sun, Xu, Jiang, Liu, Yang, Bai and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qinghu Yang, aHF5YW5nQHlhdS5lZHUuY24=; Zhantao Bai, enRiYWlAeWF1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.