Department of Physiology, Feinberg School of Medicine, Northwestern University, Chicago, IL, USA
A goal of many laboratories that study aging is to find a key cellular change(s) that can be manipulated and restored to a young-like state, and thus, reverse the age-related cognitive deficits. We have chosen to focus our efforts on the alteration of intrinsic excitability (as reflected by the postburst afterhyperpolarization, AHP) during the learning process in hippocampal pyramidal neurons. We have consistently found that the postburst AHP is significantly reduced in hippocampal pyramidal neurons from young adults that have successfully learned a hippocampus-dependent task. In the context of aging, the baseline intrinsic excitability of hippocampal neurons is decreased and therefore cognitive learning is impaired. In aging animals that are able to learn, neuron changes in excitability similar to those seen in young neurons during learning occur. Our challenge, then, is to understand how and why excitability changes occur in neurons from aging brains and cause age-associated learning impairments. After understanding the changes, we should be able to formulate strategies for reversing them, thus making old neurons function more as they did when they were young. Such a reversal should rescue the age-related cognitive deficits.
There is little question that learning new tasks becomes more difficult with normal aging; especially those tasks that rely on the hippocampus and related medial temporal lobe structures for forming episodic, or declarative, memories. It has even been suggested that age-related cognitive deficits starts as early as 20-years old in humans (Salthouse, 2009
). As this special issue of Frontiers in Aging Neuroscience can attest, there are many potential contributing factors and targets to systematically evaluate in order to understand the cellular alterations that accompany normal aging and the aging-related learning and memory impairments. Given the importance of the hippocampus in forming new memories, the hippocampus has been, and continues to be, a brain region that is extensively studied.
In this review, we will focus on data gathered from both in vitro and in vivo experiments that demonstrate changes in the neural circuitry of hippocampal CA1 region that occurs with learning and normal aging. Specifically, we will relate the alterations in intrinsic excitability (as reflected by the postburst afterhyperpolarization, AHP) measured in vitro to the dynamic in vivo neuronal changes observed in CA1 pyramidal neurons that are necessary for successful learning. We will discuss other brain regions that also undergo similar intrinsic excitability changes (in the postburst AHP) with learning and aging. In addition, we will review potential cellular mechanisms that are involved in establishing the learning-related postburst AHP alteration observed in hippocampal pyramidal neurons as well as how these cellular mechanisms may be altered with normal aging and lead to learning and memory impairments.
In pyramidal neurons, the postburst AHP is evoked by a burst of action potentials and is defined by its three phases: fast, medium and slow. As there are many excellent in depth reviews regarding the postburst AHP (e.g.: Nicoll, 1988
; Storm, 1990
; Sah, 1996
; Sah and Faber, 2002
; Wu et al., 2002
; Faber and Sah, 2003
; Disterhoft and Oh, 2006a
, 2007
), we will focus on the learning and the aging-related alterations for each of the phases in this review.
Fast Afterhyperpolarization (fAHP)
The fAHP, which lasts 2–5 ms, is a significant part of the repolarization process following an action potential in neurons (Storm, 1987
). In hippocampal pyramidal neurons, the fAHP measured at the soma is mainly a voltage and calcium-dependent potassium current, IC, mediated by BK channels (Poolos and Johnston, 1999
). Recently, Elizabeth Matthews in our laboratory demonstrated that the BK mediated fAHP is significantly reduced in CA1 pyramidal neurons following successful trace eyeblink conditioning task in young adult (Matthews et al., 2008
) and in aging rats (Matthews et al., 2009
) (Figure 1
B). However, there was no significant age-related difference in the fAHP in these neurons (Matthews et al., 2009
) (Figure 1
D).
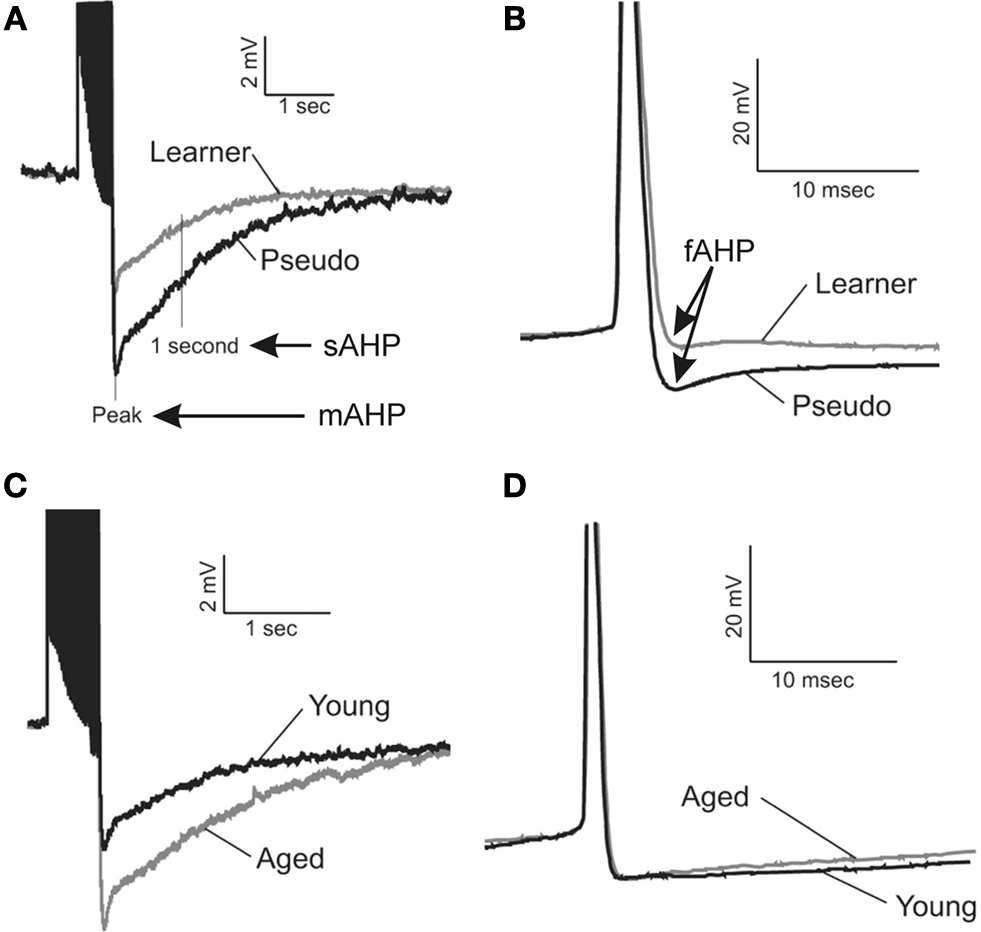
Figure 1. Learning and aging-related alterations in the fast, medium and slow afterhyperpolarizations. Illustrated are examples of the postburst AHPs evoked with a 50Hz train of action potentials from young adult (3–4 mo) and aging (29–31 mo) rats. The action potentials are truncated for illustrative purposes. The fast AHP was measured from individual action potentials that were elicited by an 800 ms depolarizing current step sufficient to elicit a minimum of 4 action potentials. The fast (B), medium and slow (A) afterhyperpolarizations (AHP) are significantly reduced in CA1 pyramidal neurons from both young and aging rats that successfully learned the hippocampus-dependent trace eyeblink conditioning task. (C) The medium and slow AHPs are significantly larger in CA1 neurons from behaviorally naïve aged rats as compared to those from young adults. (D) However, the fast AHP is not altered with aging in behaviorally naïve rats. These data suggest potentially different cellular mechanisms are involved in modulating learning-related AHP reductions from those that are involved in aging-related alterations in the medium and slow AHPs. Reprinted with permission from Matthews et al. (2009)
© by the Society for Neuroscience.
Medium and Slow Postburst Afterhyperpolarization (mAHP; sAHP)
The burst of action potentials in pyramidal neurons is followed by the medium and slow postburst afterhyperpolarization. In most pyramidal neurons, the two phases are identified by their sensitivity to the bee venom, apamin (Sah, 1996
; Sah and Faber, 2002
; Stocker, 2004
; Disterhoft and Oh, 2006a
). The mAHP typically lasts for hundreds of milliseconds (Sah and Faber, 2002
) and is blocked by apamin (Sah, 1996
; Stocker et al., 1999
; Oh et al., 2000
; Sah and Faber, 2002
; Sailer et al., 2002
; Stocker, 2004
). Thus, the channel that underlies the mAHP is generally considered to be the apamin-sensitive SK channel which mediates this calcium-dependent potassium current (Sah and Faber, 2002
; Stocker, 2004
). However, there is also evidence that IM and Ih, and not apamin-sensitive SK channels, may mediate the mAHP in CA1 pyramidal neurons (Gu et al., 2005
). The sAHP is not affected by apamin and is mediated by a calcium-dependent potassium current that lasts for seconds, but the channel that underlies this current has yet to be identified (Sah and Faber, 2002
; Disterhoft and Oh, 2006a
). Although the channel identity of the sAHP is unknown, the sAHP channels have been shown to be localized on the proximal dendrites and/or soma of CA1 neurons (Sah and Bekkers, 1996
; Bekkers, 2000
). Since both the mAHP and sAHP are observed after a burst of action potentials and undergo similar learning and aging-related changes, we will refer to both of them throughout our manuscript as the postburst AHP, unless specified.
We and others have repeatedly demonstrated that the postburst AHP is significantly reduced in hippocampal pyramidal neurons from animals that have successfully learned an associative task. For example, the postburst AHP is significantly reduced in CA1 and CA3 pyramidal neurons following trace eyeblink conditioning (Figure 1
A) (Moyer et al., 1996
, 2000
; Thompson et al., 1996a
; Kuo et al., 2008
; Matthews et al., 2008
, 2009
; Oh et al., 2009
), spatial water maze learning (Oh et al., 2003
), and olfactory discrimination learning (Zelcer et al., 2006
). Recently, the postburst AHP has been shown to be significantly reduced in CA1 pyramidal neurons following contextual and trace fear conditioning (Kaczorowski and Disterhoft, 2009
; McKay et al., 2009
). Additionally, successful olfactory rule learning has been shown to significantly reduce the postburst AHP of piriform cortical neurons (Saar et al., 1998
; Saar and Barkai, 2003
). The functional consequence of the learning-related postburst AHP reduction in these hippocampal pyramidal and piriform cortical neurons is that these neurons are able to fire significantly more action potentials to a prolonged depolarizing stimulus: i.e., the intrinsic neuronal excitability of these neurons is increased by the reduced postburst AHP.
In accordance with the role of the hippocampus for memory formation, the learning-related postburst AHP reduction in hippocampal pyramidal neurons is also transient. Numerous lesion studies have demonstrated that an intact hippocampus is necessary for successful learning, but the hippocampus is not the storage site of memory. For example, an elegant study by Richard Thompson and colleagues (Kim et al., 1995
) demonstrated that bilateral hippocampal lesions 1 day after rabbits had learned the trace eyeblink conditioning task abolished the memory of the learned conditioned eyeblink responses in these rabbits. However, if the same bilateral hippocampal lesions were performed a month after the rabbits had learned the trace eyeblink conditioning task, there was no deficit in recall of the learned conditioned response. This effect was presumably due to the fact that the memory of the learned association had been transferred to the neocortex from the hippocampus. These data are in full agreement with the in vitro biophysical studies by Disterhoft and colleagues (Moyer et al., 1996
; Thompson et al., 1996a
) who demonstrated that the learning-related postburst AHP reduction in hippocampal pyramidal neurons are maximal 1 day after the rabbits have learned the trace eyeblink conditioning task and subsequently returned to the basal, naïve state within a week after the rabbits have learned the task. Thus, these studies together suggest that the learning-related increased intrinsic excitability of hippocampal pyramidal neurons (via a postburst AHP reduction) observed immediately after successful learning is essential for transfer of the learned memory to neocortical regions where consolidation of the learned association takes place.
The postburst AHP has been demonstrated to be significantly larger in CA1 pyramidal neurons with normal aging (Landfield and Pitler, 1984
; Kumar and Foster, 2002
, 2007
; Disterhoft and Oh, 2007
; Gant and Thibault, 2009
). The functional consequence of this age-related postburst AHP enlargement is that these CA1 neurons fire fewer action potentials to a prolonged depolarizing stimulus: i.e., the intrinsic neuronal excitability of these neurons is reduced by the larger postburst AHP. This age-related postburst AHP increase in CA1 pyramidal neurons has been postulated to be a factor in the age-related learning impairments (Disterhoft and Oh, 2006a
, 2007
; Foster, 2007
) as nearly half of the aging animals failed to learn the trace eyeblink conditioning task (Thompson et al., 1996b
; Knuttinen et al., 2001
) and compounds (e.g., cholinesterase inhibitors) that reduced the postburst AHP in CA1 pyramidal neurons from aged animals ameliorated the age-related learning deficit (Disterhoft and Oh, 2006b
). An important finding is that those aging animals that successfully learn the trace eyeblink conditioning task have neurons with significantly reduced postburst AHPs that are nearly identical to those in CA1 neurons from young adult animals that learned (Moyer et al., 2000
) (Figure 1
C). Furthermore, those aged rats with smaller, young-like postburst AHP measures comprised the cognitively aged-unimpaired group that were able to successfully learn the spatial water maze task (Tombaugh et al., 2005
). Taken as a whole, these data suggest that the large postburst AHP in aging CA1 neurons prevent these neurons from participating in the neural network necessary for successful learning of hippocampus-dependent tasks. However, if these aging CA1 neurons are restored to a young-like state with pharmacological compounds (Disterhoft and Oh, 2006b
, 2007
) or remain in a young-like state (Tombaugh et al., 2005
), then these aging CA1 neurons can successful be integrated into the neural network that is required for hippocampus-dependent learning to occur.
The in vitro AHP data strongly suggest that the intrinsic neuronal excitability change is a critical factor for successful learning. However, they do not address causality or the relationship between the learning-related AHP changes and successful learning. To shed light on this question, we have extended the original experiments of Berger et al. (1976)
[who demonstrated hippocampal unit changes during delay (a non-hippocampus dependent) eyeblink conditioning in rabbits] by performing a series of in vivo recordings in the CA1 region as subjects were trained on the hippocampus-dependent trace eyeblink conditioning task (reviewed in: Disterhoft and Oh, 2006a
).
The activity of CA1 pyramidal neurons in vivo is dramatically increased prior to initial expression of a conditioned eyeblink response (McEchron and Disterhoft, 1997
), or the “ah-ha” moment. After the learned response becomes consistent and even overlearned, the neuronal activity of many CA1 pyramidal neurons is suppressed/inhibited during training trials (McEchron and Disterhoft, 1997
). These data suggest that initially acquiring a hippocampus-dependent task requires an increase in neuronal activity in the CA1 region which can be provided by the learning-related reduction of the postburst AHP. But during overtraining, active inhibition may be necessary for persistence of memory and to insure that only stimuli relevant to expression of the learned response pass through the hippocampal circuit. This active inhibition may also be a protective cellular mechanism to prevent over excitation of the neural circuit that may lead to pathological conditions, such as inflammation and apoptosis. But how is this inhibition achieved? Recent in vitro data gathered by Bridget McKay in our laboratory indicate that synaptically mediated inhibition onto CA1 pyramidal neurons is increased while the synaptic and somatically evoked postburst AHP is reduced in hippocampal slices from rats that have successfully learned the trace eyeblink conditioning task (McKay and Disterhoft, 2008
). Thus, these congruent in vivo and in vitro data suggest that the learning-related postburst AHP reduction may well precede the expression of the learned response.
In addition to the learning-related alterations in firing rate in vivo, the basal firing rate of CA1 pyramidal neurons may be directly correlated with the postburst AHP. As stated previously, there are two distinct populations in the aging group: those that are able to successfully learn and those that fail (Thompson et al., 1996b
; Knuttinen et al., 2001
; Tombaugh et al., 2005
). Notably, aging animals that are able to learn have postburst AHPs that are similar to that observed in young adults (Moyer et al., 2000
; Tombaugh et al., 2005
) and have in vivo basal firing rates that are also similar to that observed in young adults (McEchron et al., 2001
). Those aging animals that fail to learn have significantly larger postburst AHPs (Moyer et al., 2000
; Tombaugh et al., 2005
) and have significantly lower in vivo basal firing rates than young adults (McEchron et al., 2001
). However, the in vivo basal firing rate of CA1 neurons from aging animals can be elevated by pharmacological compounds that also reduce the postburst AHP in vitro and ameliorate the age-related learning impairments (Thompson et al., 1990
) (Figure 2
). Therefore, modulation of intrinsic neuronal excitability of hippocampal pyramidal neurons has a direct impact on the neuronal network activity.
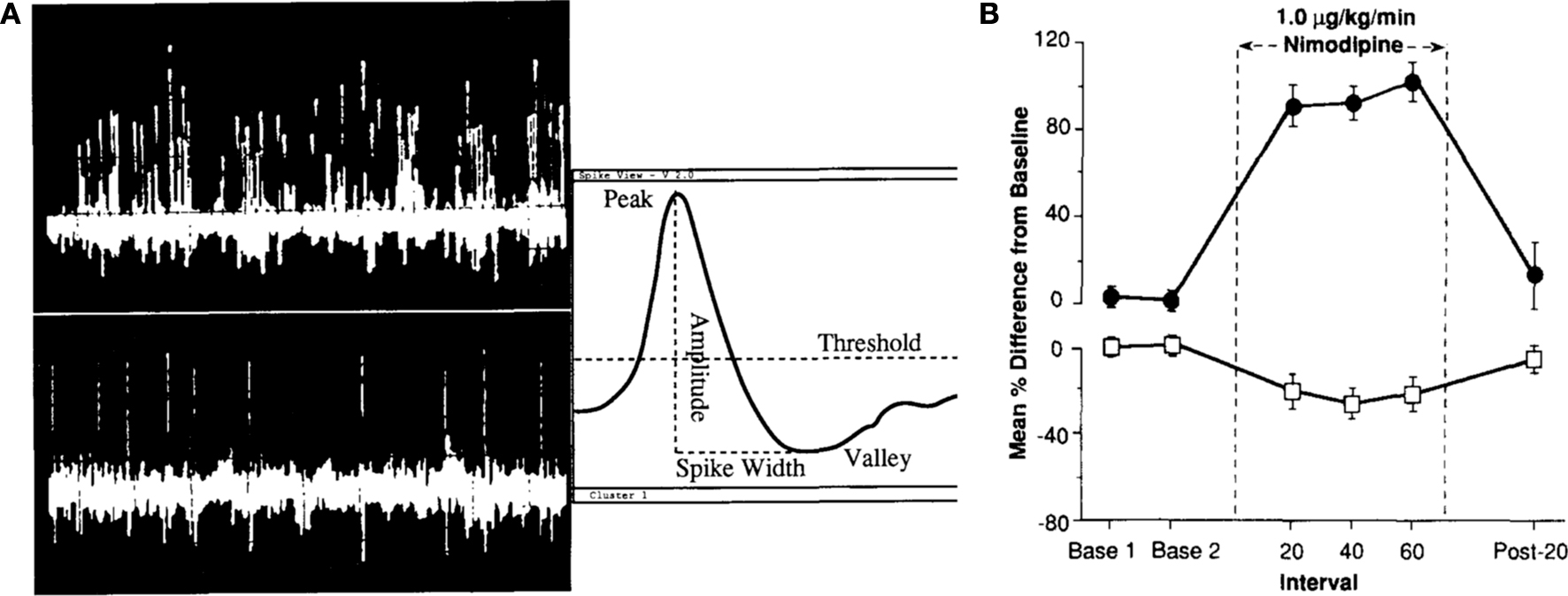
Figure 2. Baseline in vivo firing rate of CA1 pyramidal neurons is significantly enhanced with L-type calcium channel blocker nimodipine. (A) Single-unit discrimination (firing neurons) using the spike separation algorithms is illustrated. The top left trace shows 1 s of a typical multi-unit signal recorded midway through stratum pyramidale of field CAl in the dorsal hippocampus with clear theta frequency (4–8 Hz) modulation of the firing of many cells. The lower left trace shows 5 s of a classic “single-unit” signal (pyramidal neuron) recorded in the same rabbit. Simple window discrimination of this “unitary” signal (possessing fairly uniform peak-to-peak spike amplitude) would yield a frequency estimation of 2.0 ± 0.3 Hz. (B) Intravenous infusion of nimodipine significantly increased the spontaneous activity of CA1 pyramidal neurons (filled circles) while reducing the spontaneous activity of theta (interneuron) cells (open box) recorded from the dorsal hippocampus of aging rabbits. Reprinted with permission from Thompson et al. (1990)
© Elsevier Science Publishers B. V. (Biomedical Division).
Work by Edi Barkai and colleagues has demonstrated that piriform cortical neurons undergo similar learning-related postburst AHP reductions as those observed in hippocampal pyramidal neurons (Saar et al., 1998
; Saar and Barkai, 2003
). Given the similarities in the learning-related postburst AHP reductions between the piriform cortical neurons and the hippocampal pyramidal neurons, it would seem logical to assume that reduction in the postburst AHP is a cellular mechanism of learning and even perhaps memory in all pyramidal neurons. However, this is apparently not the case. Rather, it is the proper modulation of the postburst AHP of the pyramidal neurons in a particular brain region that is the key.
Recent work by Porter and colleagues demonstrate the point regarding optimal modulation, for neurons in prefrontal cortex (Santini et al., 2008
). The infralimbic prefrontal pyramidal neurons have been shown to be involved in learning to extinguish the learned fear response (Quirk et al., 2003
; Pare et al., 2004
). However, it has also been suggested that tonic activity of these infralimbic neurons contributes to the suppression of the fear response (Santini et al., 2008
). Thus, the intrinsic excitability of these infralimbic neurons is reduced (via increase in the postburst AHP, resulting in stronger spike-frequency adaptation) after a rat has learned fear conditioning (Santini et al., 2008
). Furthermore, the postburst AHPs of these infralimbic neurons are restored to the pre-fear conditioned state after extinction training sessions (Santini et al., 2008
). Thus, the proper modulation of the postburst AHP in these infralimbic neurons is the converse of the changes seen in hippocampal and piriform cortical pyramidal neurons and is an essential part of the neural network to allow both the formation and extinction of the fear memory.
In addition to the learning-related postburst AHP modulation, the pharmacological modulation of the postburst AHP also needs to be appropriate for the relevant brain region. In the hippocampal pyramidal neurons, pharmacological compounds that reduce the postburst AHP have been shown to be beneficial in reversing the age-related learning impairments (reviewed in: Wu et al., 2002
; Disterhoft and Oh, 2006b
, 2007
). In the infralimbic pyramidal neurons, activation of PKA has been shown to reduce the postburst AHP and increase the excitability of these neurons (Mueller et al., 2008
) which is an essential component of extinction learning (Santini et al., 2008
). However, for normal working memory tasks, over excitation of prefrontal pyramidal neurons (i.e., postburst AHP reductions) with pharmacological compounds that activate protein kinases has been shown to be detrimental in both young and aging subjects (Taylor et al., 1999
; Ramos et al., 2003
; Birnbaum et al., 2004
; Arnsten et al., 2005
; Brennan et al., 2009
). Luebke and colleagues found that increased firing in layer 2–3 prefrontal pyramidal neurons is correlated with poor performance on the delayed non-matching to sample task (Chang et al., 2005
). Therefore, successful learning is reliant on the proper modulation of the intrinsic excitability of pyramidal neurons (via modulating the postburst AHP) within the particular cortical region that is engaged during learning.
Protein kinase A (PKA) is not the sole protein kinase that impacts the postburst AHP. Other kinases, such as protein kinase C and α-calcium/calmodulin-dependent kinase II, are also capable of modulating the postburst AHP (Nicoll, 1988
; Wu et al., 2002
; Disterhoft and Oh, 2006a
). There is also evidence that the postburst AHP is maintained by a balance in the activity of protein kinases and phosphatases (Pedarzani et al., 1998
). However, the importance of PKA in contributing to the learning-related postburst AHP reduction has recently been demonstrated in rat CA1 pyramidal neurons after trace eyeblink conditioning (Oh et al., 2009
). Activation of PKA has been shown to significantly reduce the postburst AHP in hippocampal pyramidal neurons from behaviorally naïve (Pedarzani and Storm, 1993
, 1996
; Wu et al., 2002
; Disterhoft and Oh, 2006a
; Oh et al., 2009
) and pseudoconditioned (Oh et al., 2009
) subjects. However, the postburst AHP in CA1 neurons from animals that successfully learned the trace eyeblink conditioning task was not reduced by PKA activation: i.e., the effect of PKA on the postburst AHP from trained animals was occluded (Oh et al., 2009
) (Figure 3
). PKA has been extensively studied in regards to its activity in long-term synaptic potentiation and in the CREB signaling cascade (Impey et al., 1999
; Bailey et al., 2000
; Kandel, 2001
). It has also been shown that activation of CREB can significantly reduce the postburst AHP in CA1 pyramidal neurons (Lopez de Armentia et al., 2007
). Thus, the cellular signaling molecule that mediates long-term synaptic changes and activates the protein synthesis cascade (PKA) is also involved in mediating the learning-related postburst AHP reduction. Studies are underway in our laboratory to determine if the same signaling molecules and pathways are activated to maintain the learning-related intrinsic excitability changes.
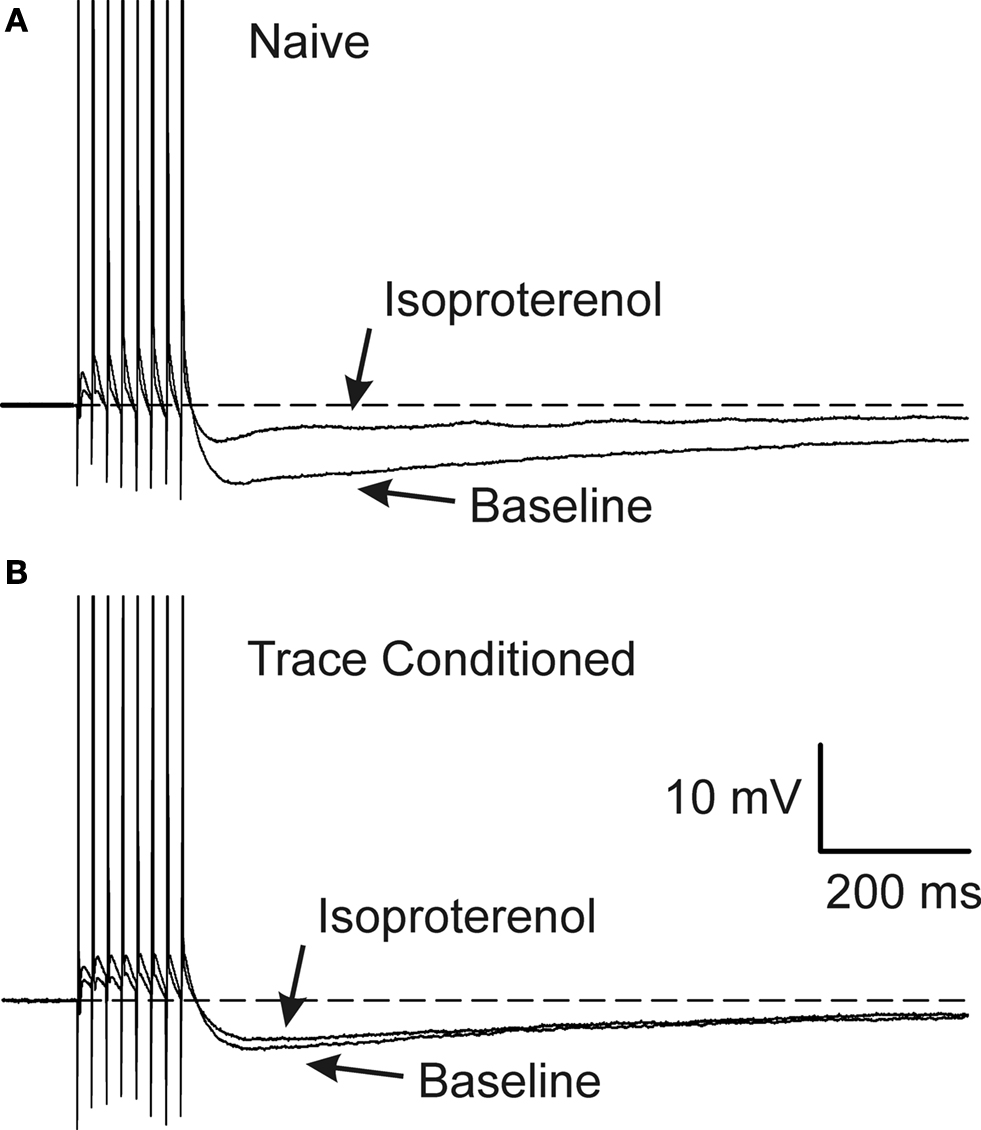
Figure 3. Learning occluded the PKA mediated AHP reduction. Examples of 1μM isoproterenol’s effect on a CA1 neuron from a naïve (A) and a trace EBC (B) rat are illustrated. Note that isoproterenol reduced the postburst AHP without abolishing it in the CA1 neuron from the naïve animal (A), whereas, isoproterenol had minimal effect on the CA1 neuron from a trained rat (B). The APs have been truncated for illustration purposes. Reprinted with permission from Oh et al. (2009)
© by the National Academy of Sciences.
Alteration in PKA activity, in addition to the age-related enlargement of the postburst AHP in hippocampal pyramidal neurons, may contribute to the normal aging-related learning impairments (Disterhoft and Oh, 2006a
, 2007
; Gant and Thibault, 2009
). Age-related adenylyl cyclase (AC) 1 and 9 mRNA reductions in the hippocampus have been suggested to be a factor in the age-related spatial learning impairments (Mons et al., 2004
). Notably, compounds that enhance cAMP/PKA signaling in the hippocampus have been shown to ameliorate this age-related spatial learning deficit (Bach et al., 1999
). Furthermore, alterations in PKA activity have been linked to Alzheimer’s disease; as β-amyloid has been shown to inactivate PKA in hippocampal neurons (Vitolo et al., 2002
) and AC type 1 has also been shown to be reduced in hippocampus of Alzheimer’s disease patients (Yamamoto et al., 2000
). It is also been demonstrated that the activity of calcineurin (a protein phosphatase) is increased in the CA1 region with normal aging (Foster et al., 2001
; Monti et al., 2005
). Calcineurin activity has been shown to reduce CREB activation (Bito et al., 1996
; Winder and Sweatt, 2001
). Notably, the increased calcineurin activity has been correlated with learning deficits on the spatial water maze task and reduced CREB activity in aging subjects (Foster et al., 2001
). Thus, the cellular mechanism that is important for successful learning and protein synthesis may be compromised with normal aging that leads to learning impairments and subsequent pathology of aging.
As the postburst AHP is mainly mediated by calcium-dependent potassium currents, compounds that block voltage-gated calcium channels (VGCCs) have been suggested to facilitate learning. Specifically, L-type VGCCs have received much attention since these channels have been linked to the slow AHP current (Marrion and Tavalin, 1998
), are found predominantly on the soma and proximal dendrites (Westenbroek et al., 1990
; Hell et al., 1993
; Christie et al., 1995
; Magee and Johnston, 1995a
), and contribute significantly to the slow AHP (Norris et al., 1998
; Thibault et al., 2001
; Power et al., 2002
). L-type VGCC blockers have been shown to facilitate reversal learning (McMonagle-Strucko and Fanelli, 1993
) and enhance inhibitory avoidance learning (Quevedo et al., 1998
) in normal young adults. Recently, we have found that protein levels of L-type VGCC subunits (CaV1.2 and CaV1.3) are significantly reduced in the CA1 region from young adult rats that have successfully learned the trace eyeblink conditioning task (Oh et al., 2008
; Nunez-Santana et al., 2009
). Thus, alteration of VGCCs may contribute to the learning-related postburst AHP reduction in CA1 pyramidal neurons and to successful learning.
The “Calcium Hypothesis of Brain Aging” (Gibson and Peterson, 1987
; Landfield, 1987
; Khachaturian, 1989
, 1994
; Khachaturian et al., 1989
; Disterhoft et al., 1994
) has been a major inspiration of research into examining alterations of calcium with normal aging. The L-type VGCCs have been a focus of attention as numerous studies have shown age-related increases in L-type VGCCs in CA1 hippocampal neurons resulted in enhanced calcium current and postburst AHP (Landfield and Pitler, 1984
; Moyer et al., 1992
; Moyer and Disterhoft, 1994
; Thibault and Landfield, 1996
; Norris et al., 1998
; Power et al., 2002
; Veng and Browning, 2002
). Blockade of L-type VGCCs with nimodipine has been shown to be beneficial in reversing the age-related learning impairments in trace eyeblink conditioning (Deyo et al., 1989
), in increasing the basal firing rate of CA1 neurons in vivo (Thompson et al., 1990
), and in significantly increasing intrinsic excitability of CA1 neurons in vitro by reducing the postburst AHP (Moyer et al., 1992
; Campbell et al., 1996
; Norris et al., 1998
; Kumar and Foster, 2002
; Power et al., 2002
). However, the postburst AHP can still be evoked in young and aging neurons after complete L-type VGCC blockade, indicating that other calcium sources and mechanisms contribute to the postburst AHP (Power et al., 2002
). This notion is further supported by experiments that utilized knockout mice which lacked either the L-type VGCC CaV1.2 or CaV1.3 and found that only deletion of CaV1.3 reduced the slow AHP by approximately 40% (Gamelli et al., 2009
). Importantly, the relative inhibition by the L-type blocker nimodipine reduced the currents underlying the AHP by a similar degree in young and aged neurons, suggesting that the L-type VGCC may not be the sole determinant of the AHP difference during the aging process (Power et al., 2002
).
Other VGCCs can potentially contribute to the age-related postburst AHP enhancement. Studies have shown that VGCCs are non-uniformly distributed on CA1 pyramidal cells: L and N-type VGCCs are found predominantly on the soma and proximal dendrites; T and R-type VGCCs are found predominantly on the apical dendrites (Westenbroek et al., 1990
, 1992
, 1995
; Christie et al., 1995
, 1996
; Magee et al., 1995
; Magee and Johnston, 1995a
,b
; Magee and Carruth, 1999
). This pattern of distribution could differentially impact the stimulus arriving on the soma and proximal dendrites from distal dendrites, given that VGCCs have dissimilar biophysical kinetic properties (Hille, 2001
). Takahashi et al. (1989)
showed that low voltage-activated calcium currents are not different from neonatal, young and aged CA1 neurons; however these measurements were taken in the somatic region and T-type VGCCs are found predominantly on the dendrites. On the other hand, Hemond and Jaffe (2005)
have reported age-related increase in calcium transients evoked with a train of action potentials in CA1 pyramidal dendrites, using calcium imaging techniques. The distribution of VGCCs still needs to be studied in normal aging. Nonetheless, these data strongly suggest that calcium signaling is different from soma to dendrites and this may result in physiological events being modulated in many diverse ways; e.g., the potential source(s) of calcium for the postburst AHP may be different in neurons from aging as compared to young animals based on changes in the distribution of VGCCs.
There are other potential sources that can impact the calcium levels with aging. Calcium released from calcium-induced calcium release (CICR) mechanisms (via ryanodine receptors on the endoplasmic reticulum, ER) has been shown to contribute to the postburst AHP (Torres et al., 1995
, 1996
; Shah and Haylett, 2000
) and has been demonstrated to be a significant source of calcium for the postburst AHP in CA1 neurons from aging animals (Kumar and Foster, 2004
; Gant et al., 2006
). In addition to the ryanodine mediated CICR, intracellular calcium level can be elevated via activation of inositol-triphosphate receptors (IP3R) on the ER (Ross et al., 2005
; Watanabe et al., 2006
; Hong and Ross, 2007
; Power and Sah, 2007
). Notably, binding experiments revealed elevation in IP3R in several areas, including the CA1 region, in aging animals (Araki et al., 1994
). Therefore, alterations in VGCCs are not the only route that can directly impact the calcium levels in the cytosol of neurons with normal aging.
Calcium buffering and clearance mechanisms have also been implicated to be altered with normal aging. An increase in calcium buffering capacity, without a change in resting [Ca2+], has been reported in dissociated basal forebrain neurons from aged animals, using calcium imaging techniques (Murchison and Griffith, 1998
, 2007
; Murchison et al., 2009
). Recently, using calcium imaging techniques with both CCD and two-photon laser-scanning microscopy, we have found that calcium buffering capacity is increased and the resting [Ca2+] is reduced in CA1 pyramidal neurons from hippocampal slices of aging as compared to young animals (Oliveira et al., 2009
). There are numerous calcium binding proteins of which calbindin-D28K, hippocalcin, parvalbumin and calretinin have been most studied. Among them, calbindin-D28K and hippocalcin have been shown to be reduced in hippocampus of aged animals (Villa et al., 1994
; de Jong et al., 1996
; Krzywkowski et al., 1996
; Kishimoto et al., 1998
; Furuta et al., 1999
), while no changes in parvalbumin and calretinin were found (de Jong et al., 1996
; Krzywkowski et al., 1996
; Kishimoto et al., 1998
); thus, excluding these calcium binding proteins as the source of the increase in calcium buffering capacity with aging.
There are multiple pathways to remove free calcium from the cytosol. On the plasma membrane, the Na+–Ca2+ exchanger and the Ca2+-ATPase are the major proteins extruding calcium (Hille, 2001
; Nicholls, 2009
), and both mechanisms have been demonstrated to be decreased with aging (Martinez-Serrano et al., 1992
; Zaidi et al., 1998
). Calcium can also be stored within the mitochondria (via Ca2+-uniporter) and the ER (via sarcoplasmic endoplasmic reticulum Ca2+-ATPase) (Hille, 2001
; Nicholls, 2009
). It has been suggested that the alteration in the Ca2+ uniporter activity is a source of reduced mitochondrial function with normal aging (Satrustegui et al., 1996
). Concurrently, the loading of the ER was shown to be drastically reduced in aging neurons, suggesting impairments in SERCA activity (Verkhratsky et al., 1994
). These data strongly suggest that the altered calcium clearance mechanisms during aging may have significant negative impact on neuronal function.
Collectively, these findings clearly show that altered calcium signaling can negatively impact neuronal function, in addition to impacting the postburst AHPs, with aging. Notably, L-type VGCC blockers, like nimodipine, have been shown to reverse the age-related impairments in synaptic plasticity (Foster, , 2007
, 1999
), to reduce the postburst AHP (Moyer et al., 1992
; Campbell et al., 1996
; Norris et al., 1998
; Power et al., 2002
), to reduce calcium action potentials (Moyer and Disterhoft, 1994
), to increase baseline activity of hippocampal pyramidal neurons (Thompson et al., 1990
), and to rescue learning in age-impaired subjects (Deyo et al., 1989
). Furthermore, nimodipine has been shown to have clinical benefits in aging human studies (Ban et al., 1990
). Therefore, pharmacological approaches that are designed to counteract the age-related calcium handling changes (e.g., CICR proteins) that also restore the calcium-dependent postburst AHPs to a young-like state may ultimately be beneficial in restoring learning and memory capacity of aging individuals.
Since the “calcium hypothesis of brain aging” was formulated nearly 30 years ago, numerous laboratories have uncovered important pieces of information about the workings of the neuron and the neural network. We have chosen to focus our efforts on one of the few consistent discoveries, the relationship of a cellular biomarker of learning and of aging in the hippocampus, the postburst AHP, to cognition in aging (Figure 4
). The postburst AHP is reduced during the learning process in young adults. With normal aging, the postburst AHP is increased resulting in a reduced basal activity of the hippocampal pyramidal neurons. It is our hypothesis that this reduction in the intrinsic excitability is a cause of cognitive impairment with normal aging (Disterhoft and Oh, 2006a
). However, in those aging animals that are able to successfully learn, the neuronal changes in excitability are similar to those seen in young adults. Our challenge, then, is to understand how and why the excitability changes occur in neurons from aging brains and cause age-associated learning impairments. After understanding the changes, we may be able to formulate strategies for reversing them, thus making old neurons function more as they did when they were young. The lack of channel identity that underlies the slow postburst AHP limits direct examination of the cellular signaling pathway that leads to the learning and aging-related alterations of the slow postburst AHP. However, given its calcium dependence for activation, studies that have examined alterations in sources of calcium with learning (Nunez-Santana et al., 2009
) and aging (Thibault and Landfield, 1996
; Veng and Browning, 2002
; Oliveira et al., 2009
) have proved to be fruitful. Additionally, studies that have examined the signaling cascades involved in gene transcription have identified numerous potential signaling molecules that may also be involved in regulating intrinsic excitability (i.e., postburst AHP) of neurons. Thus, the future challenge is to use new cutting-edge technologies [e.g., multiphoton calcium imaging and gene silencing (siRNA) techniques] to identify the molecular components and the signaling cascades that are engaged for the learning-related postburst AHP reductions, and to determine if they are the source of the age-related learning impairments and the age-related postburst AHP enlargements.
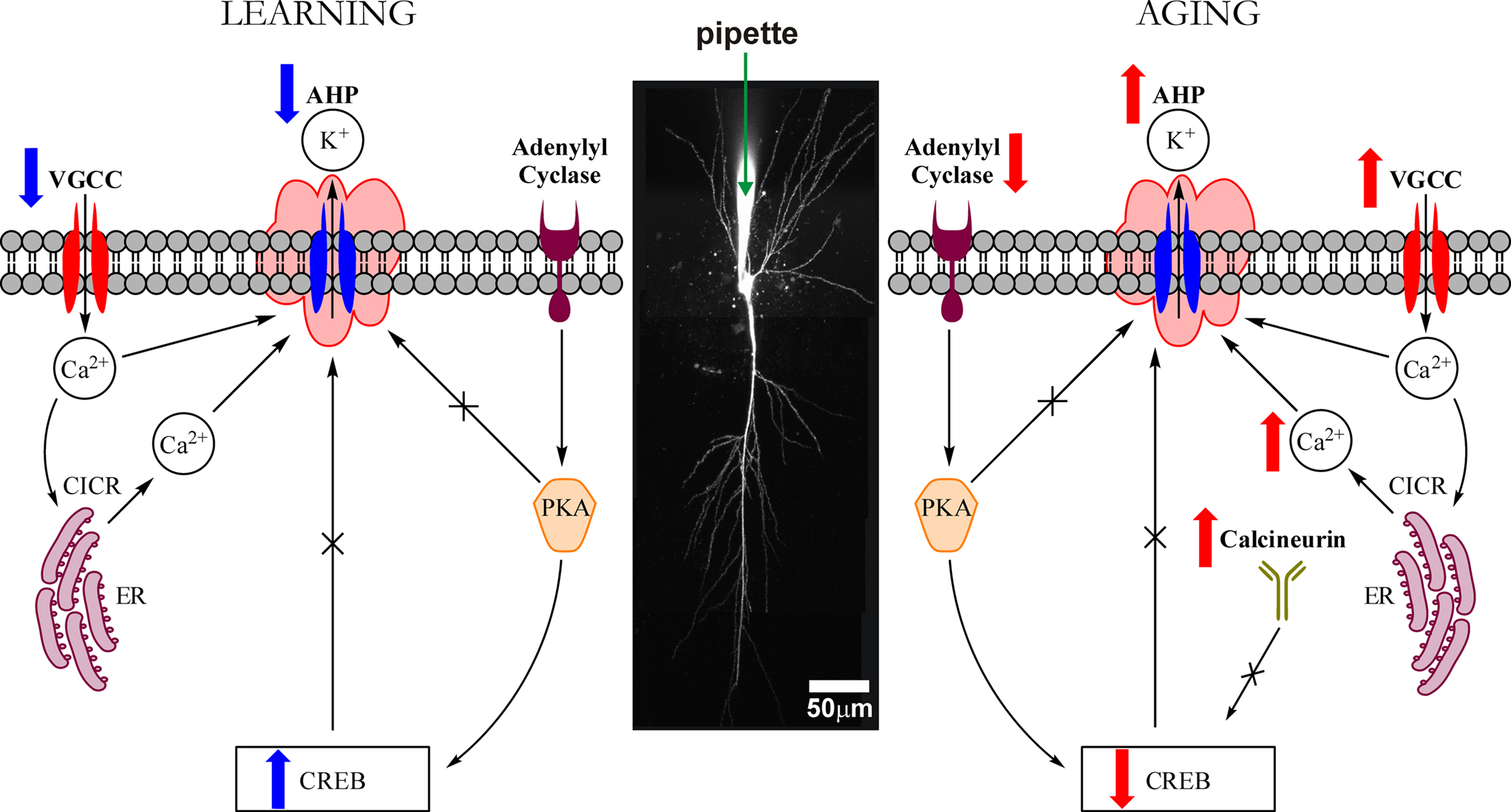
Figure 4. Highly simplified schematic of cellular signaling mechanisms that modulate the intrinsic excitability (i. e., the postburst AHP) of hippocampal pyramidal neurons during learning and with aging. The postburst AHP is a cellular response mechanism to a burst of action potentials that reduces the membrane potential (makes it more hyperpolarized) and to prevent the neuron from firing more action potentials. The postburst AHP has been consistently found to be altered during learning and with aging, making it an ideal cellular “biomarker” of intrinsic excitability change. The postburst AHP is activated by Ca2+ ions that enter the cell via VGCCs and those that are released from the ER via Ca2+-induced Ca2+ release (CICR) mechanisms. We have recent evidence that suggests that the protein levels of L-type VGCCs are significantly reduced in CA1 region after successful learning (Nunez-Santana et al., 2009
) which could contribute to the learning-related postburst AHP reduction. We also have shown that PKA activity is involved in mediating the learning-related postburst AHP reduction (Oh et al., 2009
). The postburst AHP has also been shown to be reduced by CREB activation (Lopez de Armentia et al., 2007
). Given that protein synthesis is essential for learning trace eyeblink conditioning (Inda et al., 2005
) and that CREB activation is necessary for protein synthesis (Kandel, 2001
), the cellular mediators of PKA and CREB activation should also be impacted by learning and modulate the postburst AHP. The postburst AHP is significantly increased with normal aging (Disterhoft and Oh, 2006a
, 2007
). There are numerous potential factors for this increase, but a few of the major factors are (1) increased L-type VGCCs (Moyer et al., 1992
; Thibault and Landfield, 1996
; Norris et al., 1998
; Power et al., 2002
) (2) increased CICR (Kumar and Foster, 2004
; Gant et al., 2006
), and (3) increased calcineurin activity and expression (Foster et al., 2001
) that leads to reduction in CREB activation (Bito et al., 1996
; Bach et al., 1999
; Winder and Sweatt, 2001
; Mons et al., 2004
). All of these factors can also lead to cognitive deficits in aging. VGCC: voltage-gated calcium channel. AHP: afterhyperpolarization. K+: potassium ion. Ca2+: calcium ion. CICR: Ca2+-induced Ca2+ release. ER: endoplasmic reticulum. PKA: protein kinase A. CREB: cAMP response element binding.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supported by NIH grants R37AG008796 and R01AG017139 (John F. Disterhoft).
Bach, M. E., Barad, M., Son, H., Zhuo, M., Lu, Y. F., Shih, R., Mansuy, I., Hawkins, R. D., and Kandel, E. R. (1999). Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 5280–5285.