
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Virol., 21 February 2022
Sec. Emerging and Reemerging Viruses
Volume 2 - 2022 | https://doi.org/10.3389/fviro.2022.834808
This article is part of the Research TopicEmerging and Re-emerging Viral DiseasesView all 37 articles
 Balamurali K. Ambati1
Balamurali K. Ambati1 Akhil Varshney2
Akhil Varshney2 Kenneth Lundstrom3*
Kenneth Lundstrom3* Giorgio Palú4
Giorgio Palú4 Bruce D. Uhal5
Bruce D. Uhal5 Vladimir N. Uversky6Adam M. Brufsky7
Vladimir N. Uversky6Adam M. Brufsky7Among numerous point mutation differences between the SARS-CoV-2 and the bat RaTG13 coronavirus, only the 12-nucleotide furin cleavage site (FCS) exceeds 3 nucleotides. A BLAST search revealed that a 19 nucleotide portion of the SARS-CoV-2 genome encompassing the furin cleavage site is a 100% complementary match to a codon-optimized proprietary sequence that is the reverse complement of the human mutS homolog (MSH3). The reverse complement sequence present in SARS-CoV-2 may occur randomly but other possibilities must be considered. Recombination in an intermediate host is an unlikely explanation. Single stranded RNA viruses such as SARS-CoV-2 utilize negative strand RNA templates in infected cells, which might lead through copy choice recombination with a negative sense SARS-CoV-2 RNA to the integration of the MSH3 negative strand, including the FCS, into the viral genome. In any case, the presence of the 19-nucleotide long RNA sequence including the FCS with 100% identity to the reverse complement of the MSH3 mRNA is highly unusual and requires further investigations.
Based on a recent publication describing insertion variants of SARS-CoV-2 (1) we would like to bring the attention to our recent findings related to the sequence of the furin cleavage site (FCS) in SARS-CoV-2 Spike (S) protein. The SARS-CoV-2 causing the COVID-19 pandemic (2) has 82.3% amino acid identity to bat coronavirus SL-CoVZC45, 77.2% amino acid identity to SARS-CoV, and 96.2% genome sequence identity to bat coronavirus RaTG13. While numerous point mutation differences exist between SARS-CoV-2 and RaTG13, only one insertion and dissimilarity exceeding 3 nucleotides (nt): a 12-nucleotide insertion coding for four amino acids (aa 681-684, PRRA) in the SARS-CoV-2 S protein has been discovered. This polybasic FCS differentiates SARS-CoV-2 from other b-lineage betacoronaviruses or any other sarbecovirus (3). An FCS addition enhanced the infectivity of SARS-CoV in 2009 (4). The absence of this FCS results in attenuated SARS-CoV-2 variants useful for animal vaccination, accentuating its relevance to human infection (5). This FCS is vital for human and ferret transmission (6), expands viral tropism to human cells (7), and is requisite for severe disease in two animal models of SARS-CoV-2 (5).
A peculiar feature of the nucleotide sequence encoding the PRRA furin cleavage site in the SARS-CoV-2 S protein is its two consecutive CGG codons. This arginine codon is rare in coronaviruses: relative synonymous codon usage (RSCU) of CGG in pangolin CoV is 0, in bat CoV 0.08, in SARS-CoV 0.19, in MERS-CoV 0.25, and in SARS-CoV-2 0.299 (8).
A BLAST search for the 12-nucleotide insertion led us to a 100% reverse match in a proprietary sequence (SEQ ID11652, nt 2751-2733) found in the US patent 9,587,003 filed on Feb. 4, 2016 (9) (Figure 1). Examination of SEQ ID11652 revealed that the match extends beyond the 12-nucleotide insertion to a 19-nucleotide sequence: 5′-CTACGTGCCCGCCGAGGAG-3′ (nt 2733-2751 of SEQ ID11652), such that the resulting mRNA would have 3′- GAUGCACGGGCGGCUCCUC-5′, or equivalently 5′- CU CCU CGG CGG GCA CGU AG-3′ (nucleotides 23547-23565 in the SARS-CoV-2 genome, in which the four bold codons yield PRRA, amino acids 681–684 of its spike protein). This is very rare in the NCBI BLAST database.
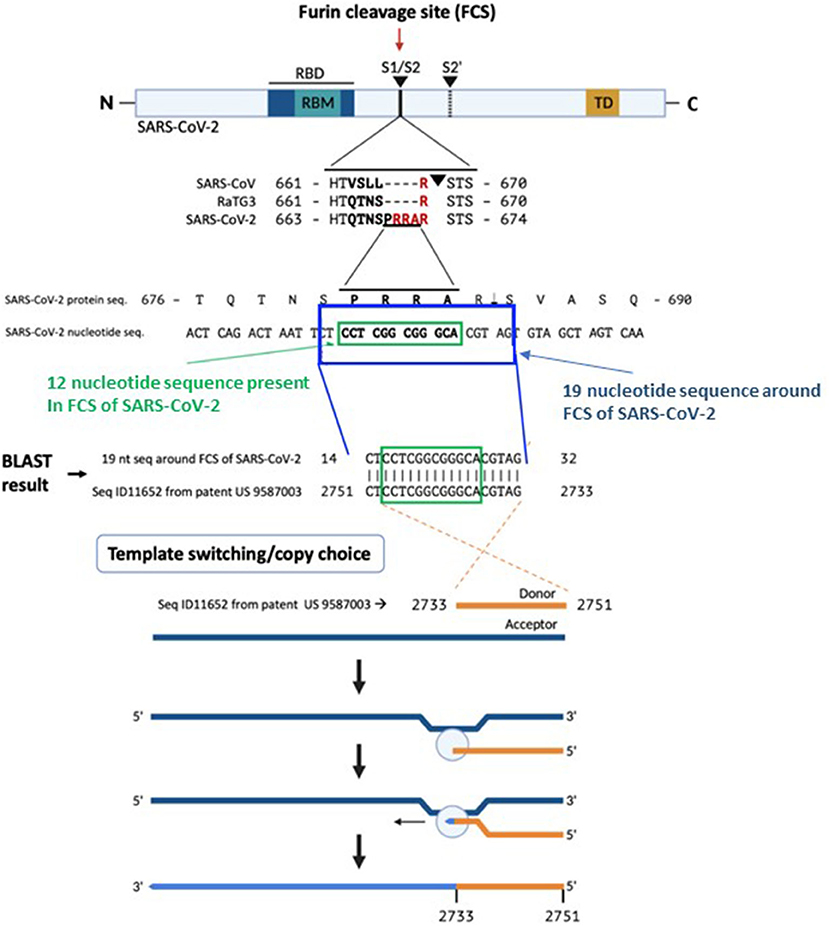
Figure 1. The origin of the furin sequence in SARS-CoV-2. Comparison of the protein sequences at the S1/S2 junction in SARS-CoV, RaTG13, and SARS-CoV-2 demonstrating the presence of the furin cleavage site (FCS) PRRA only in SARS-CoV-2. Based on a BLAST search of the 12-nucleotide stretch coding for the FCS PRRA, a 19-nucleotide long identical sequence was identified in the patented (US 958 7003) sequence Seq ID11652. SEQ ID11652 is transcribed to a MSH3 mRNA that appears to be codon optimized for humans. This 19-nucleotide sequence including 12 nucleotides coding for the FCS PRRA, present in the human MSH3 gene might have been introduced into the SARS-CoV-2 genome by the illustrated copy choice recombination mechanism in SARS-CoV-2 infected human cells overexpressing the MSH3 gene.
The correlation between this SARS-CoV-2 sequence and the reverse complement of a proprietary mRNA sequence is of uncertain origin. Conventional biostatistical analysis indicates that the probability of this sequence randomly being present in a 30,000-nucleotide viral genome is 3.21 ×10−11 (Figure 2).

Figure 2. Calculations of the probability of natural occurrence of the 19nt sequence under study. The SARS-CoV-2 genome is ~30,000 nucleotides long (P1). The patented sequence is ~3,300 nucleotides long (P2). The patented library encompasses 24'712 sequences of varying lengths with median length being in the range of 3,300 nucleotides. Conventional probability calculations are given of the probability of the presence of a 19-nucleotide sequence in the human genome and in one of the patented library sequences.
The proprietary sequence SEQ ID11652, read in the forward direction, encodes a 100% amino acid match to the human mut S homolog 3 (MSH3) (9, 10). MSH3 is a DNA mismatch repair protein (part of the MutS beta complex) (10). SEQ ID11652 is transcribed to a MSH3 mRNA that appears to be codon optimized for humans (11). We did not find the 19-nucleotide sequence CTCCTCGGCGGGCACGTAG in any mammalian or viral genomes except SARS-CoV-2 with 100% coverage and identity in the BLAST database (Supplementary Tables 1–3).
MSH3 replacement with a codon-optimized mRNA sequence for human expression likely has applications in cancers with mismatch repair deficiencies. While a portion of a reverse complement sequence being present in SARS-CoV-2 could be a random coincidence, other possibilities merit consideration.
Overexpression of MSH3 is known to interfere with mismatch repair (MSH2 sequestration from the MutS alpha complex comprising MSH2 and MSH6 results in MSH6 degradation and MutS alpha depletion) (12), which holds virologic importance. Induction of DNA mismatch repair deficiency results in permissiveness of influenza A virus (IAV) infection of human respiratory cells and increased pathogenicity (13). Mismatch repair deficiency may extend shedding of SARS-CoV-2 (14).
The absence of CTCCTCGGCGGGCACGTAG from any mammalian or viral genome in the BLAST database makes recombination in an intermediate host an unlikely explanation for its presence in SARS-CoV-2. A human-codon-optimized mRNA encoding a protein 100% homologous to human MSH3 could, during the course of viral research, inadvertently or intentionally induce mismatch repair deficiency in a human cell line, which would increase susceptibility to SARS-like viral infection. Infection of SEQ ID11652-MSH3-transduced human cells by a SARS-like virus could enable copy choice recombination (14). Replication of SARS-CoV-2 and other single stranded RNA viruses with an RNA genome of positive polarity is initiated by the synthesis of negative strand RNA in the cytoplasm of infected cells (15) (Figure 1). The negative strand RNA is a template for synthesis of positive stranded RNA utilized for translation of non-structural proteins, the replication and transcription complex, or new virion capsids. Coronaviruses generate double stranded RNA at an early stage of infection through genomic replication and mRNA transcription (16).
Acquisition of the reverse complement FCS sequence from an overexpressed positive sense MSH3 mRNA could occur through copy choice recombination with a negative sense SARS-CoV-2 RNA intermediate (14), involving jumping from one template to another (17) (Figure 1). The homology between SARS-CoV-2 and other known coronaviruses is discontinuous and most SARS-CoV-2 sequences derive from a relatively recent common ancestor with bat RaTG13. Moreover, similarity plots (SimPlots) have identified sudden changes in sequence identity between SARS-CoV-2 and RaTG13, signaling potential recombination events, which could explain the capability of SARS-CoV-2 binding to ACE2 through its RBD, which is not the case for the RaTG13 RBD (14).
A criticism of this hypothesis is that the identified sequence is on the opposite strand of the open reading frame in SEQ ID11652. However, cells transfected with MSH3, which induce mismatch repair deficiency could have targeted double-stranded cDNA encoding SEQ ID11652. Such cells co-transfected with a SARS-like virus expressing RdRp could attach to this 19-nucleotide sequence (14) and permit integration of a fragment from the negative strand into the viral genome, including the FCS, despite being on the opposite strand of the open reading frame. Mismatch repair mechanisms have enabled integration of short fragments from antisense strands in experimental models (18, 19). Microhomology can direct recombination between the MSH3 and a SARS-like virus, which could take place at the 19-nucleotide sequence of interest.
The presence in SARS-CoV-2 of a 19-nucleotide RNA sequence encoding an FCS at amino acid 681 of its spike protein with 100% identity to the reverse complement of a proprietary MSH3 mRNA sequence is highly unusual. Potential explanations for this correlation should be further investigated.
Publicly available datasets were analyzed in this study. This data can be found here: SEQ ID11652.
The investigation and original draft were initiated by BA, AV, and AB. The visualization was performed by BA, AV, AB, and GP. The writing and editing were done by BA, AV, KL, GP, BU, VU, and AB. All authors contributed to the article and approved the submitted version.
KL was employed by PanTherapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are thankful for the contribution of Dr. Jian Ying, University of Utah, Eureka, USA to the probability analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2022.834808/full#supplementary-material
1. Garushyants SK, Rogozin IB, Koonin EV. Template switching and duplications in SARS-CoV-2 genomes give rise to insertion variants that merit monitoring. Commun Biol. (2021) 4:1343. doi: 10.1038/s42003-021-02858-9
2. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature. (2020) 579:265–9. doi: 10.1038/s41586-020-2008-3
3. Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. (2020) 176:104742. doi: 10.1016/j.antiviral.2020.104742
4. Belouzard S, Chu V, Whittaker G. Activation of the SARS Coronavirus Spike Protein via Sequential Proteolytic Cleavage at 2 different Sites. Proc Natl Acad Sci USA. (2009) 106:5871–6.
5. Lau SY, Wang P, Mok BWY, Zhang AJ, Chu H, Lee ACY, et al. Attenuated SARS-CoV-2 variants with deletions at the S1/S2 junction. Emerg Microbes Infect. (2020) 9:837–42. doi: 10.1080/22221751.2020.1756700
6. Peacock TP, Goldhill DH, Zhou J, Baillon L, Frise R, Swann OC, et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat Microbiol. (2020) 6:899–909. doi: 10.1038/s41564-021-00908-w
7. Xia S, Lan Q, Su S, Wang X, Xu W, Liu Z, et al. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal Transd. Targeted Ther. (2020) 5:92. doi: 10.1038/s41392-020-0184-0
8. Kandeel M, Ibrahim A, Fayez M, Al-Nazawi M. From SARS and MERS CoVs to SARS-CoV-2: Moving toward more biased codon usage in viral structural and nonstructural genes. J Med Virol. (2020) 92:660–6. doi: 10.1002/jmv.25754
9. Bancel S, Chakraborty T, De Fougerolles A, Elbashir SM, John M, Roy A, et al. Modified Polynucleotides for the Production of Oncology-Related Proteins and Peptides. Cambridge, MA: United States Patent. (2016).
10. MacRae SL, McKnight Croken M, Calder RB, Aliper A, Milholland B, White RR, et al. DNA repair in species with extreme lifespan differences. Aging. (2015) 7:1171–84. doi: 10.18632/aging.100866
11. Mauro VP, Chappell SA. A critical analysis of codon optimization in human therapeutics. Trends Mol Med. (2014) 20:604–13. doi: 10.1016/j.molmed.2014.09.003
12. Marra G, Iaccarino I, Lettieri T, Roscilli G, Delmastro P, Jiricny J. Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proc Natl Acad Sci USA. (1998) 95:8568. doi: 10.1073/pnas.95.15.8568
13. Chambers BS, Heaton BE, Rausch K, Dumm RE, Hamilton JR, Cherry S, et al. DNA mismatch repair is required for the host innate response and controls cellular fate after influenza virus infection. Nat Microbiol. (2019) 4:1964–77. doi: 10.1038/s41564-019-0509-3
14. Gallaher WR. A palindromic RNA sequence as a common breakpoint contributor to copy-choice recombination in SARS-COV-2. Arch Virol. (2020) 165:2341–8. doi: 10.1007/s00705-020-04750-z
15. V'kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev. (2021) 19:155–70. doi: 10.1038/s41579-020-00468-6
16. Graham RL, Baric RS. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J Virol. (2010) 84:3146. doi: 10.1128/JVI.01394-09
17. Sola I, Almazan F, Zuniga S, Enjuanes L. Continuous and discontinuous RNA synthesis in coronaviruses. Annu Rev Virol. (2015) 2:265–88. doi: 10.1146/annurev-virology-100114-055218
18. Rakosy-Tican E, Lörincz-Besenyei E, Molnár I, Thieme R, Hartung F, Sprink T, et al. New Phenotypes of potato co-induced by mismatch repair deficiency and somatic hybridization. Front Plant Sci. (2019) 10:3. doi: 10.3389/fpls.2019.00003
Keywords: SARS-CoV-2 spike, furin cleavage site, MSH3 gene, sequence homology, recombinability
Citation: Ambati BK, Varshney A, Lundstrom K, Palú G, Uhal BD, Uversky VN and Brufsky AM (2022) MSH3 Homology and Potential Recombination Link to SARS-CoV-2 Furin Cleavage Site. Front. Virol. 2:834808. doi: 10.3389/fviro.2022.834808
Received: 13 December 2021; Accepted: 19 January 2022;
Published: 21 February 2022.
Edited by:
Xin Yin, Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences (CAAS), ChinaReviewed by:
Jitao Chang, Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences (CAAS), ChinaCopyright © 2022 Ambati, Varshney, Lundstrom, Palú, Uhal, Uversky and Brufsky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kenneth Lundstrom, bHVuZHN0cm9ta2VubmV0aEBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.