 Xiushuang Chen1,2†Miao An3†Wenqian Zhang3†Kun Li1,2Muhammad Fakhar-e-Alam Kulyar3Kun Duan4Hui Zhou1Yu Wu1Xin Wan1Jianlong Li5Lingtong Quan6Zhanhai Mai5Wenxia Bai7Yi Wu1,2*
Xiushuang Chen1,2†Miao An3†Wenqian Zhang3†Kun Li1,2Muhammad Fakhar-e-Alam Kulyar3Kun Duan4Hui Zhou1Yu Wu1Xin Wan1Jianlong Li5Lingtong Quan6Zhanhai Mai5Wenxia Bai7Yi Wu1,2*- 1College of Veterinary Medicine, Nanjing Agricultural University, Nanjing, China
- 2MOE Joint International Research Laboratory of Animal Health and Food Safety, College of Veterinary Medicine, Nanjing Agricultural University, Nanjing, China
- 3College of Veterinary Medicine, Huazhong Agricultural University, Wuhan, China
- 4China Tobacco Henan Industrial Co. Ltd., Zhengzhou, China
- 5College of Veterinary Medicine, Xinjiang Agricultural University, Urumqi, China
- 6College of Life Sciences, Nanjing Agricultural University, Nanjing, China
- 7Nanjing Superbiotech Co. Ltd., Nanjing, China
The gut microbial community is closely related to mastitis, but studies regarding the influences of mastitis on gut microbiota in buffalo remain scarce. Herein, we characterized the differences in gut bacterial and fungal communities between mastitis-affected and healthy buffalos. Interestingly, although mastitis had no effect on gut bacterial and fungal diversities in the buffalos, some bacterial and fungal taxa were significantly altered. Bacterial and fungal taxonomic analysis showed that the preponderant bacterial phyla (Firmicutes and Bacteroidetes) and fungal phyla (Ascomycota and Basidiomycota) in buffalo were the same regardless of health status. At the level of genus, the changes in some gut bacterial and fungal abundances between both groups were gradually observed. Compared with healthy buffalos, the proportions of 3 bacterial genera (uncultured_bacterium_f_Muribaculaceae, Eubacterium_nodatum_group, and Lachnoclostridium_10) and 1 fungal genus (Pichia) in the mastitis-affected buffalo were significantly increased, whereas 4 bacterial genera (Ruminococcus_2, Candidatus_Stoquefichus, Turicibacter, and Cellulosilyticum) and 4 fungal genera (Cladosporium, Thermothelomyces, Ganoderma and Aspergillus) were significantly decreased. Taken together, this research revealed that there was significant difference in the compositions of the gut microbial community between the healthy and mastitis-affected buffalos. To our knowledge, this is the first insight into the characteristics of the gut microbiota in buffalos with mastitis, which is beneficial to understand the gut microbial information of buffalo in different health states and elucidate the pathogenesis of mastitis from the gut microbial perspective.
Introduction
Mastitis, an inflammatory response of the mammary parenchyma, affects almost all lactating mammals especially high-yield cows (1). It can lead to decreased milk production, severely restraining dairy industry development (2). Early investigation revealed that fecal microbiota transplantation from cows with mastitis to germ-free mice caused inflammations in multiple tissues, such as colon, spleen, and serum, as well as mastitis symptoms in the mammary gland (3). Moreover, probiotic administration has been demonstrated to effectively alleviate mastitis symptoms in some exploratory human clinical trials, indicating that the mechanism of mastitis protection may be mediated through the gut microbiota (4).
Growing evidence indicated that gut microbiota participated in multiple physiological and metabolic functions of the host, including nutrient acquisition, intestinal epithelium differentiation, and intestinal metabolism (5–8). Moreover, the gut microbiota has also been demonstrated to play role in intestinal mucosal barrier and immune system maturation, implying its contribution in disease prevention and immunologic functions (9, 10). However, multiple environmental-related factors, such as diet, nutritional deficiencies, antibiotic treatment, and exposure to contaminants, may affect gut microbial homeostasis or even induce gut microbial dysbiosis (11, 12). Stable gut microbiota enabled the intestines to function properly, whereas gut microbial dysbiosis may cause etiopathologic consequences (13, 14). Currently, gut microbial dysbiosis has been shown to be the core and critical factor of many gastrointestinal diseases, such as colonitis and diarrhea (15, 16). Additionally, disturbed gut microbiota and its metabolites could pass through the intestinal mucosal barrier and affect peripheral organ systems by blood circulation, causing physiological dysfunction and even disease, such as lipid disorders, diabetes, and non-alcoholic fatty liver (17, 18).
Recently, culture-independent techniques, mainly including metagenomic and 16S rDNA amplicon sequencing, have been successfully developed and widely applied to dissect the complicated gut microbial ecosystem, as well as investigate gut microbial alterations after suffering certain diseases (19, 20). By systematically investigating and analyzing the microbial information acquired, we can further understand the gut microbiota-host interaction and mechanisms contributing to ill-health, thereby formulating effective measures to minimize the collateral damage. Presently, high-throughput sequencing technologies have successfully dissected the gut microbiota of giraffes, yaks, goats, and dairy cattle, making considerable contributions to the etiological analysis, diagnosis, and treatment of multiple gastrointestinal and systemic diseases (5, 21, 22). As an important source of protein acquisition for humans, buffalo milk has increasingly attracted widespread attention due to its high fat, protein, mineral, and vitamin contents. However, mastitis dramatically decreases buffalo milk production and quality, causing significant health and economic burden in buffalo farming. Although the gut microbial importance in host health is widely acknowledged, scarce knowledge is known about the interaction between mastitis and gut microbiota in buffalo. Herein, we investigated the gut bacterial and fungal shifts of buffalo with mastitis.
Materials and Methods
Animals and Sample Collection
In this investigation, 10 buffalos (5 healthy and 5 with mastitis) in Jingzhou, China were used for sample acquisition, and all selected buffalos had similar characteristics, including age, weight, diet, immune background, and dwelling environment. Buffalo mastitis was diagnosed by the California mastitis test (CMT) using a commercial kit. Moreover, the confirmed cases did not receive any treatment prior to the sample collection. On the day of sample collection, all the selected buffalos were placed in separate areas to maximally decrease potential contamination among different samples of subjects. The sterilized fecal samplers were used for collecting the rectal feces of each buffalo. The collected samples were immediately placed intosterile plastic containers and transported to the laboratory and later stored at −80°C for further study.
16S rDNA and ITS Genes Amplicon Sequencing
Five fecal samples (~200 mg) from healthy and mastitis-affected buffaloes were unfrozen and homogenized before DNA extraction. Subsequently, the bacterial and fungal DNA of the processed fecal samples were extracted using QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany) based on the manufacturer's recommendations. The quality and quantity of the gDNA were evaluated via 0.8% (w/v) agarose gel electrophoresis and UV-Vis spectrophotometer (NanoDrop 2000, United States), respectively. To characterize the gut bacterial and fungal shifts, we amplified the V3/V4 and ITS2 regions utilizing bacterial (338F: ACTCCTACGGGAGGCAGCA and 806R: GGACTACHVGGGTWTCTAAT) and fungal (ITS5F: GGAAG TAAAAGTCGTAACAAGG and ITS2R: GCTGCGTTCTTCATCGA TGC) primers, respectively. PCR amplification was conducted as per the procedure previously described (5, 6). PCR products were subjected to target fragment recovery and quality evaluation and gel electrophoresis to obtain purified products. The recovered products were quantified by fluorescence using Quant-iT PicoGreen dsDNA Assay Kit on the Promega QuantiFluor fluorescent quantitative system and the libraries with a concentration above 2 nM and only one peak were considered qualified. The final purified products were applied for preparing the sequencing library using MiSeq Reagent Kit V3 (600 cycles) on the MiSeq sequencing machine.
Bioinformatics and Statistical Analysis
The raw data was requested to be preprocessed. Specifically, quality detection and primer removal were applied to initial data with some problematic sequences, including unqualified, short, or mismatched sequences, to acquire clean reads through the Trimmomatic (v0.33) and Cutadapt software (1.9.1). The collected clean reads were subjected to splice and secondary filter as per the length range of different regions using Usearch software (v10). Afterward, recognition and removal of chimera sequences were conducted to achieve effective reads. The effective reads with 97 similarities were clustered into the same operational taxonomic unit (OTU). Moreover, Venn graphs were also generated to visualize the OTUs abundance and distribution in each group. To further dissect the influence of mastitis on gut microbial diversity and abundance, we computed multiple alpha diversity indexes according to the OTUs' distribution. On the other hand, beta diversity analysis was used for characterizing the differences between gut bacterial and fungal principal components. The assessment of sequencing depth for each sample was based on rank abundance and rarefaction curves. Differential bacterial and fungal taxa were determined by the LEfSe and Metastats analysis. Data analysis was performed by SPSS statistical program (v20.0) and P-values (means ± SD) <0.05 were recognized as statistically significant.
Results
Sequence Analysis
In this study, we collected a total of 10 fecal samples for amplicon sequencing and 535,789 (CB = 264,271, MB = 271,518), and 707,929 (CB = 353,169, MB = 354,760) original sequences were achieved from the gut bacterial and fungal communities, respectively (Table 1). After quality assessment, 906,588 (CB = 372,157, MB = 534,431) eligible sequences were identified, with a median read count of 372,15 (ranging from 306,42 to 452,06) and 534,43 (ranging from 462,29 to 603,82) reads from bacterial V3/4 and fungal ITS2 regions from each sample, respectively (Table 2). The qualified sequences were clustered into 671 bacterial OTUs and 142 fungal OTUs as per 97% sequence similarity (Figures 1A–C,G–I). Additionally, the amounts of unique bacterial OTUs in CB and MB were 16 and 2 and 653 OTUs were shared in both groups, accounting for approximately 97.31% of the total bacterial OTUs. Meanwhile, we also observed 142 common OTUs in CB and MB, which consisted of more than 100% of the overall fungal OTUs. The results of accumulation and rarefaction curves demonstrated that almost all species can be detected (Figures 1D–F,J–L).
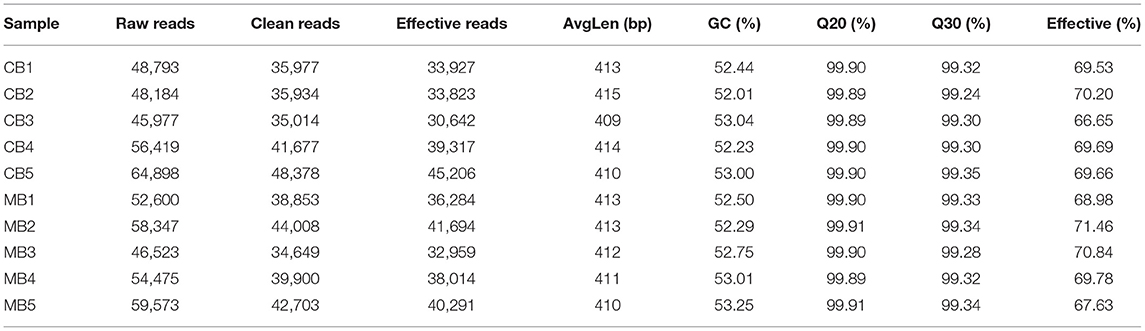
Table 1. Bacterial sequence information from amplicon sequencing.
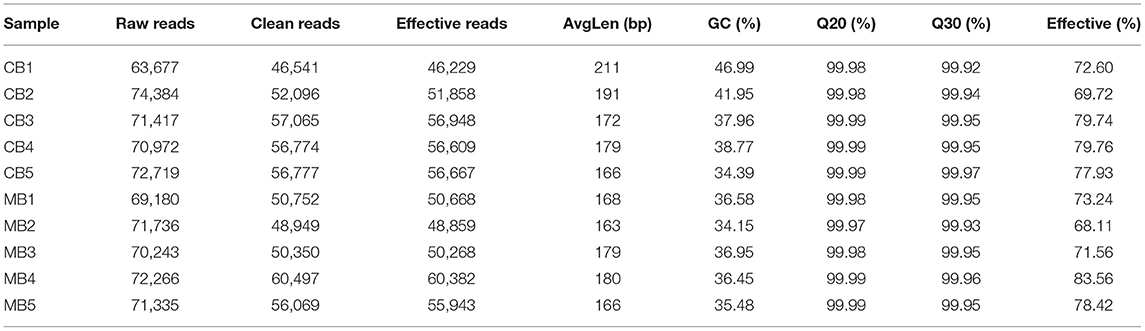
Table 2. Fungal sequence information from amplicon sequencing.
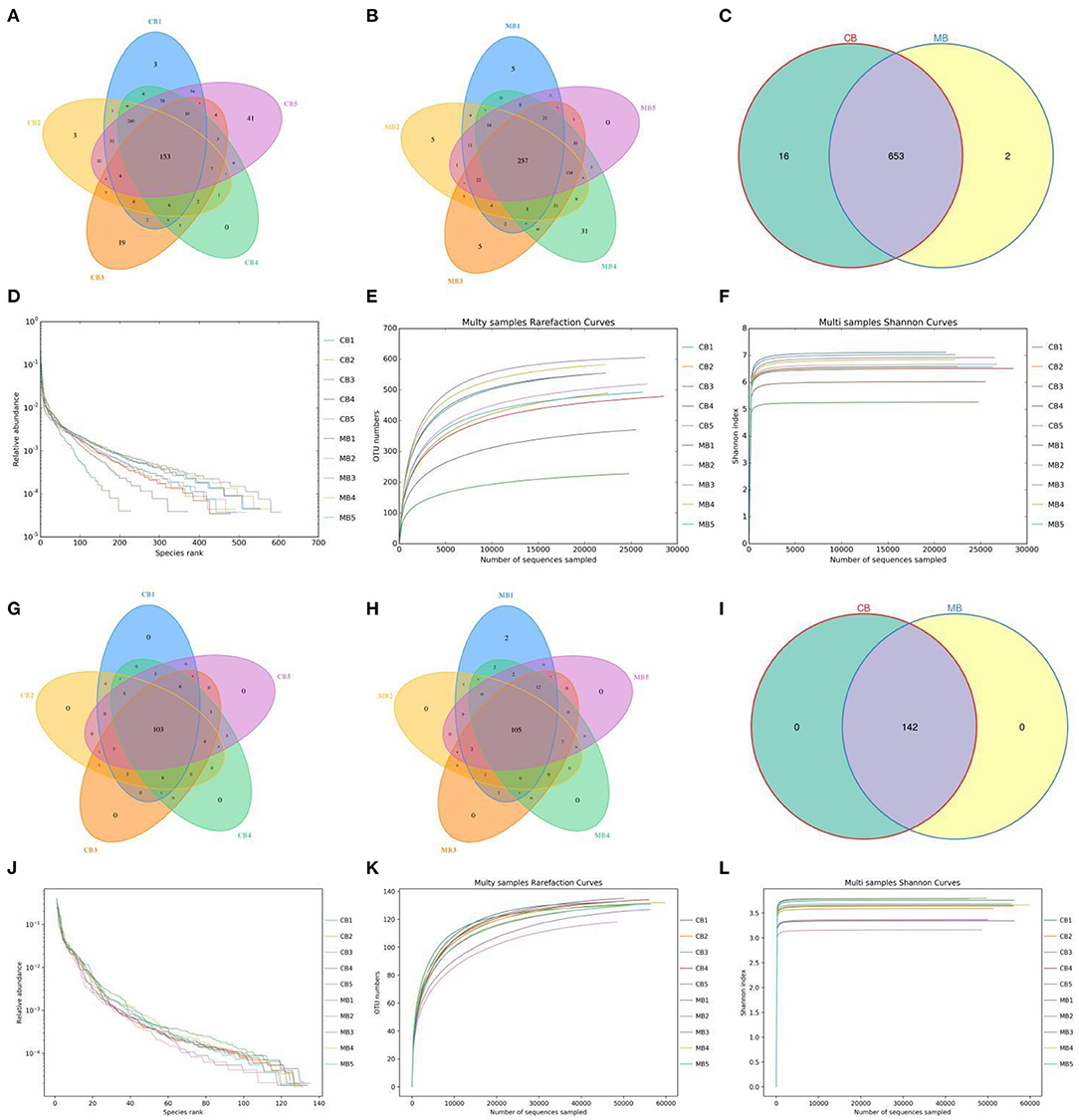
Figure 1. Operational taxonomic units (OTUs) distribution and sequencing data analysis. Venn diagrams for gut bacterial (A–C) and fungal (G–I) OTUs distribution. Gut bacterial (D–F) and fungal (J–L) sequencing depth and evenness were assessed by rank abundance and rarefaction curves.
Microbial Diversities Analysis Associated With Mastitis
To further explore the influence of mastitis on the gut microbiota of a buffalo, we calculated the alpha and beta diversity indices that could reflect gut microbial diversity. Bacterial and fungal Good's coverage estimates in each sample of CB and MB were almost 100%, implying excellent coverage. Furthermore, there were no significant differences in the bacterial and fungal Chao1 (496.79 ± 144.06 vs. 534.90 ± 79.22, P = 0.62; 134.77 ± 3.85 vs. 133.91 ± 7.35, P = 0.82), ACE (495.99 ± 143.39 vs. 529.78 ± 77.53, P = 0.65; 134.01 ± 2.01 vs. 134.20 ± 5.87, P = 0.94), Simpson (0.96 ± 0.011 vs. 0.96 ± 0.010, P =0.69; 0.83 ± 0.025 vs. 0.82 ± 0.030, P = 0.44) and Shannon (6.47 ± 0.72 vs. 6.62 ± 0.37, P = 0.70; 3.62 ± 0.17 vs. 3.53 ± 0.26, P = 0.53) indices between CB and MB (Figures 2A–H). Alpha-diversity analysis indicated that mastitis had no distinct effect on the gut bacterial and fungal diversity and abundance in buffalos. Principal coordinate analysis (PCoA) plots that reflect the similarities and differences among different samples were used to assess the gut bacterial and fungal beta-diversity. Results of the beta-diversity analysis showed that the samples in CB and MB were clustered together, implying similar gut microbial principal components (Figures 2I–L).
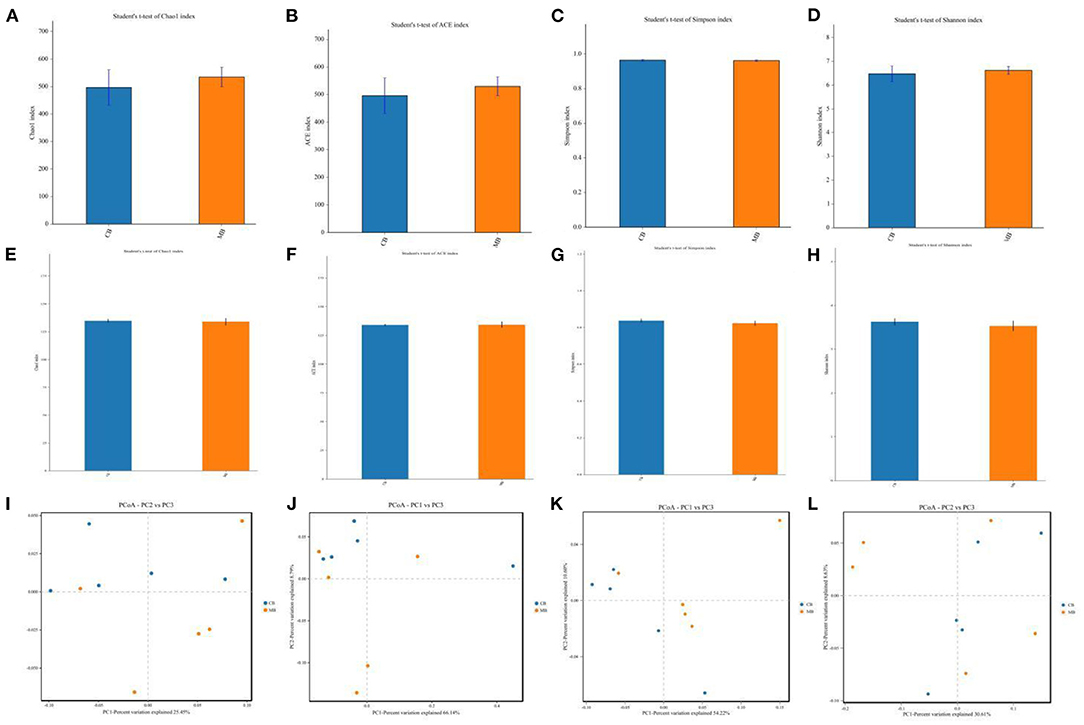
Figure 2. Changes of gut bacterial and fungal diversities associated with mastitis in buffalos. (A–D) Represent gut bacterial Chao, ACE, Simpson, and Shannon indices, respectively. (E–H) Represent gut fungal Chao, ACE, Simpson, and Shannon indices, respectively. (I,J) Represent gut bacterial PCoA maps, whereas (K,L) Represent gut fungal PCoA maps.
Comparative Analysis of Bacterial Taxonomic Composition
In this microbiome investigation, a total of 9 bacterial phyla and 155 genera were recognized in CB and MB, ranging from 7 to 9 phyla and 103 to 134 genera per sample, respectively. The phyla Firmicutes (70.31, 68.19%), Bacteroidetes (27.88, 27.50%), Spirochaetes (0.53, 1.84%), and Proteobacteria (0.56, 1.46%) were the four most preponderant bacterial phyla in samples of CB and MB regardless of health condition, which accounted for approximately 99.00% of the total composition (Figure 3A). Other phyla, such as Patescibacteria (0.32, 0.45%), Tenericutes (0.25, 0.33%), Actinobacteria (0.091, 0.10%), Verrucomicrobia (0.028, 0.10%), and Cyanobacteria (0.024, 0.020%), in CB and MB were identified in lower abundances. At the genus level, the most dominant bacterial genera in the CB were Ruminococcaceae_UCG-005 (13.79%) followed by the Bacteroides (6.88%) and Rikenellaceae_RC9_gut_group (6.42%). However, Ruminococcaceae_UCG-005 (19.25%), Rikenellaceae_RC9_gut_group (7.06%) and uncultured_bacterium_f_Lachnospiraceae (6.70%) were abundantly present in the MB (Figure 3B). Bacterial distribution, as well as correlation of both groups during mastitis could also be observed by the clustering heatmap and network diagram, respectively (Figures 3E, 4).
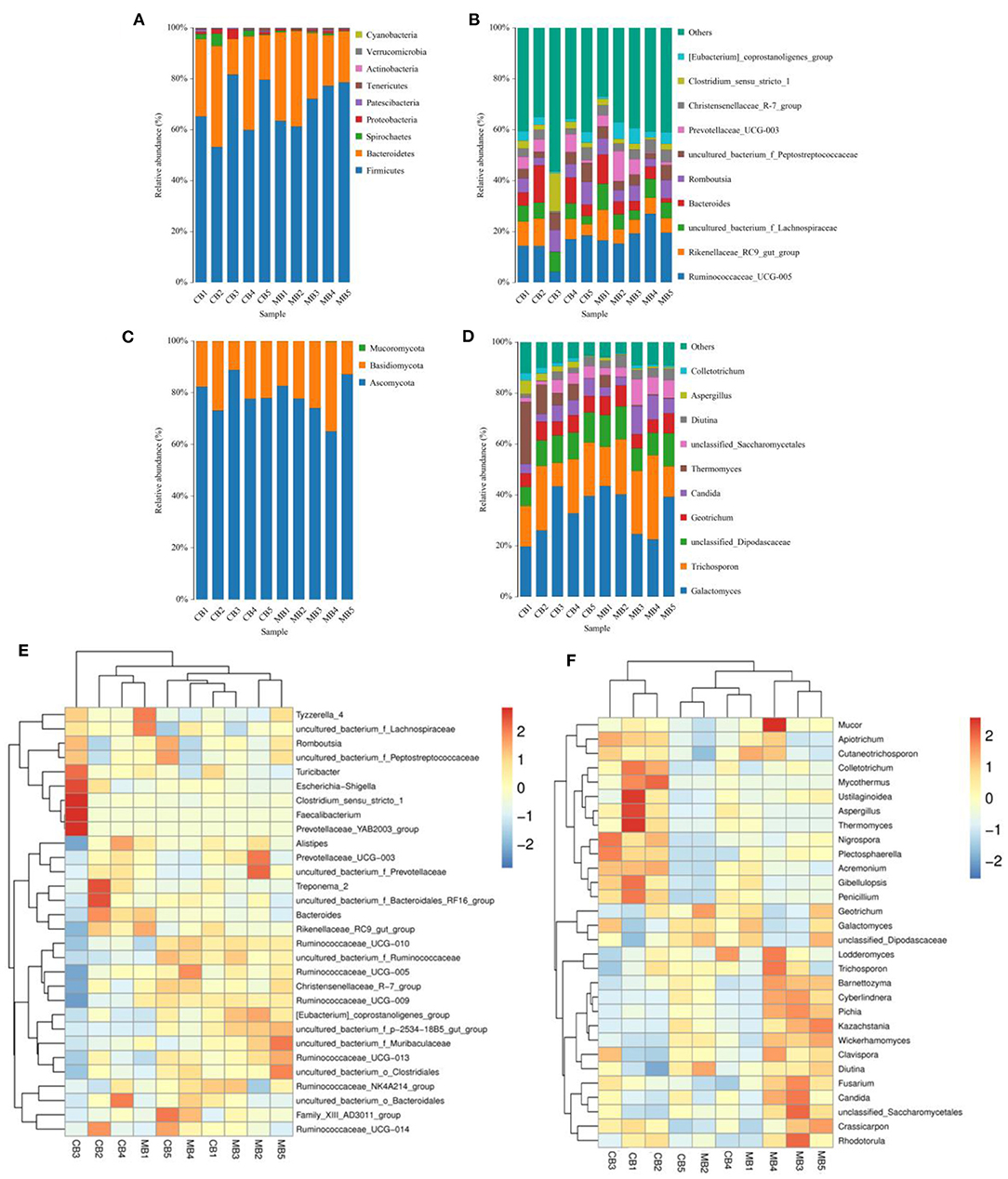
Figure 3. The proportions of preponderant bacterial (A,B) and fungal (C,D) taxa at the level of phylum and genus associated with mastitis in buffalos. The color-block in the heatmap indicates the normalized relative richness of each bacterial (E) and fungal (F) genera in healthy and mastitis-affected buffalos.
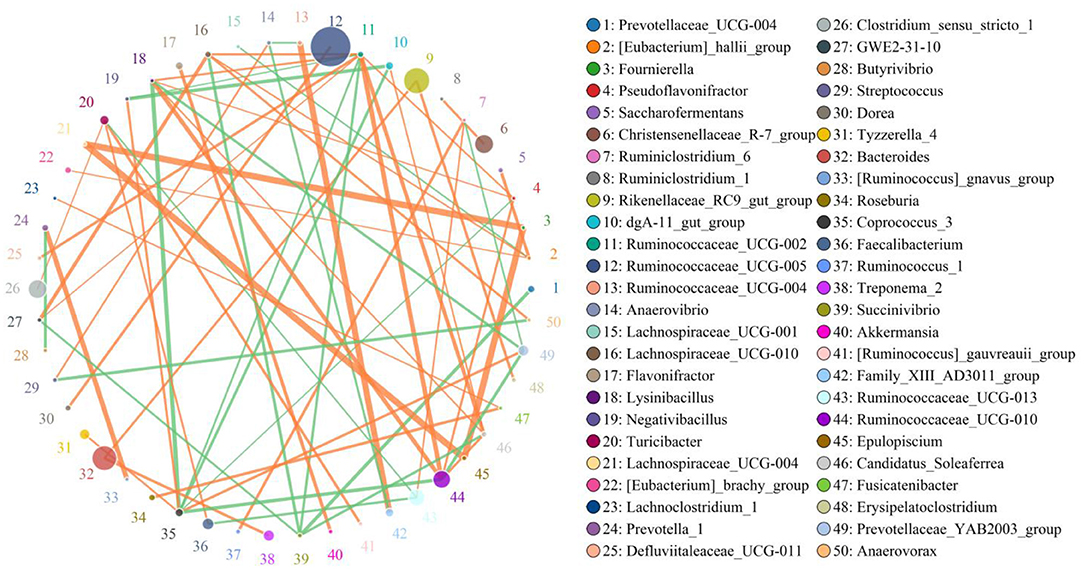
Figure 4. The network diagram visualizes correlations between different bacterial genera. The orange lines indicate a positive correlation and the green lines indicate a negative correlation.
To further assess the influences of mastitis on the gut microbiota in buffalos, we performed Metastats analysis on different classification levels. At the genus level, uncultured_bacterium_f_Muribaculaceae, Eubacterium_nodatum_group, and Lachnoclostridium_10 were significantly more dominant in the MB group than in the CB group, whereas the Ruminococcus_2 was lower (Figure 5A). Besides the above-mentioned differential taxa, the MB group also showed dramatically lower richness of Candidatus_Stoquefichus, Turicibacter, and Cellulosilyticum (Figures 6A,B).
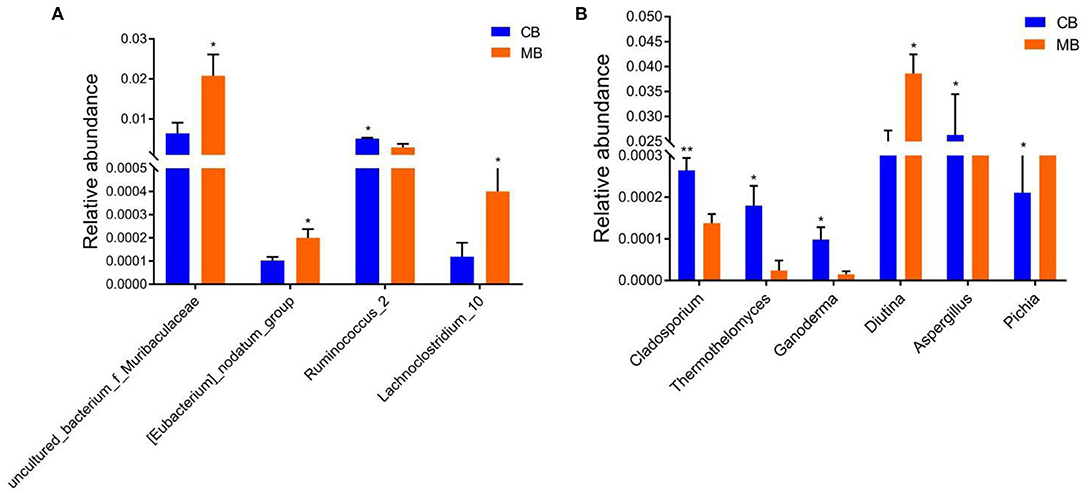
Figure 5. Significant changes in the intestinal bacteria (A) and fungi (B) associated with mastitis in buffalos. Data was indicated as mean ± SD. *p < 0.05, **p < 0.01.
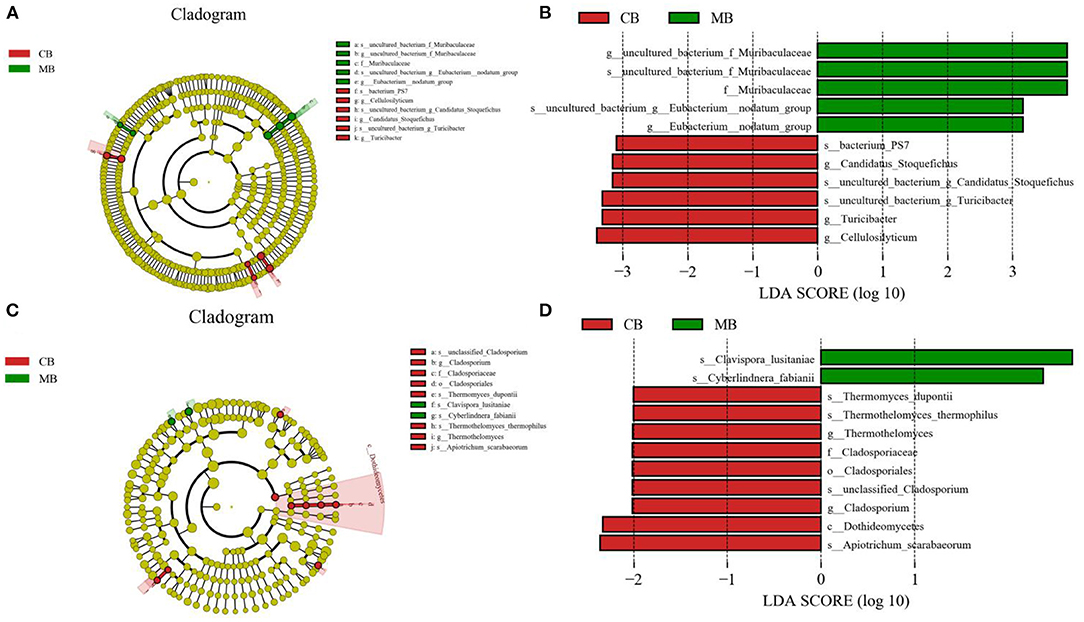
Figure 6. Recognition of differential taxa associated with mastitis in buffalo. Phylogenetic distribution of bacterial (A) and fungal (C) taxa with obvious differences were visualized through the cladogram. The criterion of bacterial (B) and fungal (D) significance was performed at LDA scores > 2.
Comparative Analysis of Fungal Taxonomic Composition
There were 3 phyla and 66 genera recognized in the gut fungal community of CB and MB and the dominant phyla and genera were presented in Figures 3C,D. The Ascomycota (77.11, 80.05%) and Basidiomycota (22.83, 19.91%) were the most prevalent fungal phyla in both groups, making up approximately 99.00% of the overall fungal composition. The Ascomycota (77.11, 80.05%) and Basidiomycota (22.83, 19.91%) were the most prevalent fungal phyla in both groups, making up approximately 99.00% of the overall fungal composition. At the genus level, the Galactomyces (33.66, 32.91%), Trichosporon (21.62, 18.51%), and unclassified_Dipodascaceae (11.13, 10.24%) were the most prevalent fungal genera in both groups, which accounted for over 60.00% of the total taxonomical group identified. The clustering heatmap also showed the distribution and changes of gut fungal community in buffalo with mastitis (Figure 3F).
Metastats analysis was used to compare the differences in the gut fungal community of both groups (Figure 5B). At the genus level, Cladosporium, Thermothelomyces, Ganoderma, and Aspergillus were all significantly overrepresented in the CB group, whereas Pichia was the most abundant fungus in MB. Similar results were also observed in Figures 6C,D.
Discussion
Mastitis is a common disease in buffalos that seriously affects milk production and animal health, causing enormous economic loss (23). However, multiple factors, including pathogen infection, unclean environment, nutritional deficiency, and stress reaction, cause mastitis to be difficult to control (23–25). A gut microbial community containing trillions of microorganisms has been demonstrated to be a complex and interactive ecosystem that participates in the positive regulation of host metabolism and health (22, 26). Although, these microorganisms, including bacteria and fungi, colonize the intestine, they can expand their negative impact beyond the gastrointestinal tract and thus cause the development of other diseases (27–29). Recent investigations about gut microbiota have also indicated its key role in the development of mastitis in dairy cows (30, 31). Presently, the study of the relationship between gut microbiota and mastitis has covered many species, but research regarding the gut bacterial and fungal communities in buffalo with mastitis remains scarce. Here, we systematically dissected the gut bacterial and fungal changes in buffalo associated with mastitis and indicated distinct changes in gut bacterial and fungal taxa in buffalo during mastitis.
Growing evidence indicated that the gut microbiota was a dynamic system that was inevitably influenced by multiple intrinsic and extrinsic factors, including diet, age, and sex environment (32–34). Generally, the physiological fluctuations of gut microbiota caused by the above-mentioned factors cannot affect intestinal normal function and homeostasis. However, intestinal-related diseases, such as diarrhea, colitis, and colorectal cancer, have been demonstrated to perturb intestinal homeostasis, resulting in dysbiosis (35, 36). Moreover, recent studies have also shown that diabetes, high blood pressure, and obesity could also cause significant changes in the gut microbiota (37–39). In this study, we selected feces to explore the gut microbial changes in buffalos with mastitis in consideration of the samples' availability and subjects' particularity. Results indicated that there were no significant differences in the gut microbial alpha-diversity between healthy and mastitis-affected buffalo, suggesting that mastitis had no effect on the gut microbial diversity and abundance of the buffalos. Consistent with this study, Ma et al. (3) also demonstrated that there were no obvious differences in the gut microbiota alpha-diversity between healthy and mastitis-affected cows. Notably, although the differences in gut bacterial and fungal diversities between controls and mastitis-affected subjects were not significant, the percentages of some bacteria and fungi altered markedly, implying that these intestinal bacteria and fungi are constantly self-adjusting to the current intestinal environment.
In this microbiome investigation, we observed that Firmicutes and Bacteroidetes were the most preponderant bacteria, whereas Basidiomycota and Ascomycota were the most dominant fungi in buffalos, regardless of health status (40–42). Notably, these microbial phyla have also been demonstrated to be widespread in other ruminants, such as goats, cows, giraffes, and yaks, indicating their key roles in intestinal ecology and function (43, 44). Earlier studies indicated that most members of Firmicutes were intestinal beneficial bacteria involved in the regulation of the immune system, gut microbial homeostasis, and intestinal barrier function (45). Moreover, its members contribute to the degradation of cellulose in ruminants (40). As the dominant bacteria in the gut, Bacteroidetes was responsible for degrading carbohydrates and proteins, showing the great potential for promoting the maturation of the gastrointestinal immune system (43). At the genus level, Bacteroides were abundantly present in the healthy buffalo, which was inconsistent with the findings of mastitis-affected buffalo. As intestinal anaerobion, Bacteroides can decompose polysaccharides and play a key role in the intestinal ecosystem (46). As intestinal beneficial bacteria, Rikenellaceae has been demonstrated to possess multiple probiotic properties and control the development of colitis by regulating T-regulatory cell differentiation (47). Ruminococcaceae can degrade cellulose and starch, displaying positive regulation in growing development and feed efficiency (48). Moreover, Ruminococcaceae, a potential probiotic candidate, plays active roles in the secretion of short-chain fatty acids (SCFAs), intestinal homeostasis, and host health (49).
The shifts of some specific bacteria and fungi could dissect the potential relationship between gut microbial community and mastitis, thus we further investigated the gut bacterial and fungal changes associated with mastitis. Results showed a significant decrease in 4 bacterial genera (Ruminococcus_2, Candidatus_Stoquefichus, Turicibacter, and Cellulosilyticum) and 4 fungal genera (Cladosporium, Thermothelomyces, Ganoderma, and Aspergillus), as well as an increase in 3 bacterial genera (uncultured_bacterium_f_Muribaculaceae, Eubacterium_nodatum_group and Lachnoclostridium_10) and 1 fungal genus (Pichia) in mastitis-affected buffalo. These bacteria and fungi may play an important role in intestinal homeostasis and functions, as well as the development of buffalo with mastitis. Ruminococcus and Cellulosilyticum have been shown to possess the characteristics of decomposing cellulose and starch (50–52). Notably, Ruminococcus is also a potential producer of SCFAs (53). Numerous pieces of evidence indicated that SCFAs play a fundamental role in gut microbial homeostasis and host metabolism (54, 55). Furthermore, SCFAs were also involved in the regulation of intestinal permeability, immunologic function, and cell proliferation (56, 57). Therefore, the higher proportions of Ruminococcus and Cellulosilyticum in the ruminant are beneficial to maintain energy intake and intestinal function.
Taken together, this study first compared and analyzed the differences in gut microbiota between healthy and mastitis-affected buffalos. Results showed that mastitis did not alter the gut bacterial and fungal diversity, but the proportions of some bacterial and fungal taxa altered significantly. This study also contributes to understanding the gut microbial information of buffalos and shows that the changes in gut bacterial and fungal communities may be an important factor of mastitis. Notably, this research is also beneficial to prevent and treat mastitis in buffalos from the gut microbial perspective.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by Ethics Committee of the Nanjing Agricultural University. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author Contributions
XC, MA, and WZ conceived and designed the experiments. MA and WZ contributed sample collection and reagents preparation. HZ, YiW, and XW analyzed the data. XC wrote the manuscript. YuW, MFK, KD, JL, LQ, ZM, and WB revised the manuscript. All authors reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was financially supported by the National Natural Science Foundation of China (NSFC, Grant Nos. 31872514 and 32172900).
Conflict of Interest
KD was employed by China Tobacco Henan Industrial Co. Ltd. WB was employed by Nanjing Superbiotech Co. Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Omaleki L, Browning GF, Allen JL, Markham PF, Barber SR. Molecular epidemiology of an outbreak of clinical mastitis in sheep caused by Mannheimia haemolytica. Vet Microbiol. (2016) 191:82–7. doi: 10.1016/j.vetmic.2016.06.005
2. Swinkels JM, Hilkens A, Zoche-Golob V, Kromker V, Buddiger M, Jansen J, et al. Social influences on the duration of antibiotic treatment of clinical mastitis in dairy cows. J Dairy Sci. (2015) 98:2369–80. doi: 10.3168/jds.2014-8488
3. Ma C, Sun Z, Zeng B, Huang S, Zhao J, Zhang Y, et al. Cow-to-mouse fecal transplantations suggest intestinal microbiome as one cause of mastitis. Microbiome. (2018) 6:200. doi: 10.1186/s40168-018-0578-1
4. Hu X, Guo J, Zhao C, Jiang P, Maimai T, Yanyi L, et al. The gut microbiota contributes to the development of Staphylococcus aureus-induced mastitis in mice. ISME J. (2020) 14:1897–910. doi: 10.1038/s41396-020-0651-1
5. Li A, Liu B, Li F, He Y, Wang L, Fakhar-E-Alam KM, et al. Integrated bacterial and fungal diversity analysis reveals the gut microbial alterations in diarrheic giraffes. Front Microbiol. (2021) 12:712092. doi: 10.3389/fmicb.2021.712092
6. Hu J, Nie Y, Chen J, Zhang Y, Wang Z, Fan Q, et al. Gradual changes of gut microbiota in weaned miniature piglets. Front Microbiol. (2016) 7:1727. doi: 10.3389/fmicb.2016.01727
7. Liu X, Nagy P, Bonfini A, Houtz P, Bing XL, Yang X, et al. Microbes affect gut epithelial cell composition through immune-dependent regulation of intestinal stem cell differentiation. Cell Rep. (2022) 38:110572. doi: 10.1016/j.celrep.2022.110572
8. Guo X, Sha Y, Lv W, Pu X, Liu X, Luo Y, et al. Sex differences in rumen fermentation and microbiota of Tibetan goat. Microb Cell Fact. (2022) 21:55. doi: 10.1186/s12934-022-01783-8
9. Xiong Y, Ji L, Zhao Y, Liu A, Wu D, Qian J. Sodium butyrate attenuates taurocholate-induced acute pancreatitis by maintaining colonic barrier and regulating gut microorganisms in mice. Front Physiol. (2022) 13:813735. doi: 10.3389/fphys.2022.813735
10. Li MX, Li MY, Lei JX, Wu YZ, Li ZH, Chen LM, et al. Huangqin decoction ameliorates DSS-induced ulcerative colitis: role of gut microbiota and amino acid metabolism, mTOR pathway and intestinal epithelial barrier. Phytomedicine. (2022) 100:154052. doi: 10.1016/j.phymed.2022.154052
11. Li A, Wang Y, He Y, Liu B, Iqbal M, Mehmood K, et al. Environmental fluoride exposure disrupts the intestinal structure and gut microbial composition in ducks. Chemosphere. (2021) 277:130222. doi: 10.1016/j.chemosphere.2021.130222
12. Liao J, Liu Y, Yi J, Li Y, Li Q, Li Y, et al. Gut microbiota disturbance exaggerates battery wastewater-induced hepatotoxicity through a gut-liver axis. Sci Total Environ. (2022) 809:152188. doi: 10.1016/j.scitotenv.2021.152188
13. Li A, Ding J, Shen T, Han Z, Zhang J, Abadeen ZU, et al. Environmental hexavalent chromium exposure induces gut microbial dysbiosis in chickens. Ecotoxicol Environ Saf. (2021) 227:112871. doi: 10.1016/j.ecoenv.2021.112871
14. Zhu L, Xu F, Wan W, Yu B, Tang L, Yang Y, et al. Correction to: gut microbial characteristics of adult patients with allergy rhinitis. Microb Cell Fact. (2020) 19:192. doi: 10.1186/s12934-020-01441-x
15. Wang Y, Zhang H, Zhu L, Xu Y, Liu N, Sun X, et al. Dynamic distribution of gut microbiota in goats at different ages and health states. Front Microbiol. (2018) 9:2509. doi: 10.3389/fmicb.2018.02509
16. Xi L, Qin X, Song Y, Han J, Li Z, Zhang J. Gut microbial alterations in diarrheal baer's pochards (Aythya baeri). Front Vet Sci. (2021) 8:756486. doi: 10.3389/fvets.2021.756486
17. Sun CY, Zheng ZL, Chen CW, Lu BW, Liu D. Targeting gut microbiota with natural polysaccharides: effective interventions against high-fat diet-induced metabolic diseases. Front microbiol. (2022) 13:859206. doi: 10.3389/fmicb.2022.859206
18. Bauer KC, Littlejohn PT, Ayala V, Creus-Cuadros A, Finlay BB. Nonalcoholic fatty liver disease and the gut-liver axis: exploring an undernutrition perspective. Gastroenterology. (2022) 162:1858-75. doi: 10.1053/j.gastro.2022.01.058
19. Ning Y, Qi J, Dobbins MT, Liang X, Wang J, Chen S, et al. Comparative analysis of microbial community structure and function in the gut of wild and captive amur tiger. Front microbiol. (2020) 11:1665. doi: 10.3389/fmicb.2020.01665
20. Hu L, Geng S, Li Y, Cheng S, Fu X, Yue X, et al. Exogenous fecal microbiota transplantation from local adult pigs to crossbred newborn piglets. Front Microbiol. (2017) 8:2663. doi: 10.3389/fmicb.2017.02663
21. Xin J, Chai Z, Zhang C, Zhang Q, Zhu Y, Cao H, et al. Comparing the microbial community in four stomach of dairy cattle, yellow cattle and three yak herds in qinghai-tibetan plateau. Front Microbiol. (2019) 10:1547. doi: 10.3389/fmicb.2019.01547
22. Li A, Yang Y, Qin S, Lv S, Jin T, Li K, et al. Microbiome analysis reveals gut microbiota alteration of early-weaned Yimeng black goats with the effect of milk replacer and age. Microb Cell Fact. (2021) 20:78. doi: 10.1186/s12934-021-01568-5
23. Singha S, Ericsson CD, Chowdhury S, Nath SC, Paul OB, Hoque MA, et al. Occurrence and aetiology of subclinical mastitis in water buffalo in Bangladesh. J Dairy Res. (2021) 88:314–20. doi: 10.1017/S0022029921000698
24. Krishnamoorthy P, Suresh KP, Jayamma KS, Shome BR, Patil SS, Amachawadi RG. An Understanding of the global status of major bacterial pathogens of milk concerning bovine mastitis: a systematic review and meta-analysis (Scientometrics). Pathogens. (2021) 10:545. doi: 10.3390/pathogens10050545
25. Zaatout N. An overview on mastitis-associated Escherichia coli: Pathogenicity, host immunity and the use of alternative therapies. Microbiol Res. (2022) 256:126960. doi: 10.1016/j.micres.2021.126960
26. Podar PT, Yang Z, Bjornsdottir SH, Podar M. Comparative analysis of microbial diversity across temperature gradients in hot springs from yellowstone and iceland. Front Microbiol. (2020) 11:1625. doi: 10.3389/fmicb.2020.01625
27. Gao Y, Liu Y, Ma F, Sun M, Song Y, Xu D, et al. Lactobacillus plantarum Y44 alleviates oxidative stress by regulating gut microbiota and colonic barrier function in Balb/C mice with subcutaneous d-galactose injection. Food Funct. (2021) 12:373–86. doi: 10.1039/D0FO02794D
28. Zhong G, Wan F, Lan J, Jiang X, Wu S, Pan J, et al. Arsenic exposure induces intestinal barrier damage and consequent activation of gut-liver axis leading to inflammation and pyroptosis of liver in ducks. Sci Total Environ. (2021) 788:147780. doi: 10.1016/j.scitotenv.2021.147780
29. Xu R, Aruhan Xiu L, Sheng S, Liang Y, Zhang H, et al. Exopolysaccharides from lactobacillus buchneri TCP016 attenuate LPS- and d-GalN-Induced liver injury by modulating the gut microbiota. J Agric Food Chem. (2019) 67:11627–37. doi: 10.1021/acs.jafc.9b04323
30. Pang M, Xie X, Bao H, Sun L, He T, Zhao H, et al. Insights into the bovine milk microbiota in dairy farms with different incidence rates of subclinical mastitis. Front Microbiol. (2018) 9:2379. doi: 10.3389/fmicb.2018.02379
31. Hu X, Li S, Fu Y, Zhang N. Targeting gut microbiota as a possible therapy for mastitis. Eur J Clin Microbiol Infect Dis. (2019) 38:1409–23. doi: 10.1007/s10096-019-03549-4
32. Yuan X, Chen R, Zhang Y, Lin X, Yang X. Sexual dimorphism of gut microbiota at different pubertal status. Microb Cell Fact. (2020) 19:152. doi: 10.1186/s12934-020-01412-2
33. Chen L, Li S, Xiao Q, Lin Y, Li X, Qu Y, et al. Composition and diversity of gut microbiota in Pomacea canaliculata in sexes and between developmental stages. BMC Microbiol. (2021) 21:200. doi: 10.1186/s12866-021-02259-2
34. Chang J, Yao X, Zuo C, Qi Y, Chen D, Ma W. The gut bacterial diversity of sheep associated with different breeds in Qinghai province. BMC VET RES. (2020) 16:254. doi: 10.1186/s12917-020-02477-2
35. Chen F, Dai X, Zhou CC, Li KX, Zhang YJ, Lou XY, et al. Integrated analysis of the faecal metagenome and serum metabolome reveals the role of gut microbiome-associated metabolites in the detection of colorectal cancer and adenoma. Gut. (2021) 71:1315–25. doi: 10.1136/gutjnl-2020-323476
36. Yang Y, Misra BB, Liang L, Bi D, Weng W, Wu W, et al. Integrated microbiome and metabolome analysis reveals a novel interplay between commensal bacteria and metabolites in colorectal cancer. Theranostics. (2019) 9:4101–14. doi: 10.7150/thno.35186
37. Calabrese CM, Valentini A, Calabrese G. Gut microbiota and type 1 diabetes mellitus: the effect of mediterranean diet. Front Nutr. (2020) 7:612773. doi: 10.3389/fnut.2020.612773
38. Li X, Wei T, Li J, Yuan Y, Wu M, Chen F, et al. Tyrosol ameliorates the symptoms of obesity, promotes adipose thermogenesis, and modulates the composition of gut microbiota in HFD fed mice. Mol Nutr Food Res. (2022) e2101015. doi: 10.1002/mnfr.202101015
39. Ye Y, Xu H, Xie Z, Wang L, Sun Y, Yang H, et al. Time-restricted feeding reduces the detrimental effects of a high-fat diet, possibly by modulating the circadian rhythm of hepatic lipid metabolism and gut microbiota. Front Nutr. (2020) 7:596285. doi: 10.3389/fnut.2020.596285
40. Zhang L, Jiang X, Li A, Waqas M, Gao X, Li K, et al. Characterization of the microbial community structure in intestinal segments of yak (Bos grunniens). Anaerobe. (2020) 61:102115. doi: 10.1016/j.anaerobe.2019.102115
41. Zhuang Y, Chai J, Cui K, Bi Y, Diao Q, Huang W, et al. Longitudinal Investigation of the Gut Microbiota in Goat Kids from Birth to Postweaning. Microorganisms. (2020) 8:1111. doi: 10.3390/microorganisms8081111
42. Li K, Mehmood K, Zhang H, Jiang X, Shahzad M, Dong X, et al. Characterization of fungus microbial diversity in healthy and diarrheal yaks in Gannan region of Tibet Autonomous Prefecture. Acta Trop. (2018) 182:14–26. doi: 10.1016/j.actatropica.2018.02.017
43. Sun B, Wang X, Bernstein S, Huffman MA, Xia DP, Gu Z, et al. Marked variation between winter and spring gut microbiota in free-ranging Tibetan Macaques (Macaca thibetana). Sci Rep. (2016) 6:26035. doi: 10.1038/srep26035
44. Garneau JE, Tremblay DM, Moineau S. Characterization of 1706, a virulent phage from Lactococcus lactis with similarities to prophages from other Firmicutes. Virology. (2008) 373:298–309. doi: 10.1016/j.virol.2007.12.002
45. Wang Y, Li A, Liu J, Mehmood K, Wangdui B, Shi H, et al. L pseudomesenteroides and L johnsonii isolated from yaks in Tibet modulate gut microbiota in mice to ameliorate enteroinvasive Escherichia coli-induced diarrhea. Microb Pathog. (2019) 132:1–9. doi: 10.1016/j.micpath.2019.04.020
46. Spence C, Wells WG, Smith CJ. Characterization of the primary starch utilization operon in the obligate anaerobe bacteroides fragilis: regulation by carbon source and oxygen. J Bacteriol. (2006) 188:4663–72. doi: 10.1128/JB.00125-06
47. Seshadri R, Leahy SC, Attwood GT, Teh KH, Lambie SC, Cookson AL, et al. Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection. Nat Biotechnol. (2018) 36:359–67. doi: 10.1038/nbt.4110
48. Kong F, Hua Y, Zeng B, Ning R, Li Y, Zhao J. Gut microbiota signatures of longevity. Curr Biol. (2016) 26:R832–3. doi: 10.1016/j.cub.2016.08.015
49. Reddivari L, Veeramachaneni D, Walters WA, Lozupone C, Palmer J, Hewage M, et al. Perinatal Bisphenol A Exposure Induces Chronic Inflammation in Rabbit Offspring via Modulation of Gut Bacteria and Their Metabolites. mSystems. (2017) 2:e00093–17. doi: 10.1128/mSystems.00093-17
50. Miller TL, Currenti E, Wolin MJ. Anaerobic bioconversion of cellulose by Ruminococcus albus, Methanobrevibacter smithii, and Methanosarcina barkeri. Appl Microbiol Biotechnol. (2000) 54:494–8. doi: 10.1007/s002530000430
51. Cai S, Li J, Hu FZ, Zhang K, Luo Y, Janto B, et al. Cellulosilyticum ruminicola, a newly described rumen bacterium that possesses redundant fibrolytic-protein-encoding genes and degrades lignocellulose with multiple carbohydrate- borne fibrolytic enzymes. Appl Environ Microbiol. (2010) 76:3818–24. doi: 10.1128/AEM.03124-09
52. De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A. (2010) 107:14691–6. doi: 10.1073/pnas.1005963107
53. Juste CGM, Stampini DMH, Tako E. Effects of iron and zinc biofortified foods on gut microbiota in vivo (Gallus gallus): a systematic review. Nutrients. (2021) 13:189. doi: 10.3390/nu13010189
54. Cherta-Murillo A, Pugh JE, Alaraj-Alshehhi S, Hajjar D, Chambers ES, Frost GS. The effect of short-chain fatty acids on glycemic control in humans: a systematic review and Meta-analysis. Am J Clin Nutr. (2022). doi: 10.1093/ajcn/nqac085. [Epub ahead of print].
55. Zhen Y, Ge L, Xu Q, Hu L, Wei W, Huang J, et al. Normal light-dark and short-light cycles regulate intestinal inflammation, circulating short-chain fatty acids and gut microbiota in period2 gene knockout mice. Front Immunol. (2022) 13:848248. doi: 10.3389/fimmu.2022.848248
56. Feng Y, Wang Y, Wang P, Huang Y, Wang F. Short-chain fatty acids manifest stimulative and protective effects on intestinal barrier function through the inhibition of nlrp3 inflammasome and autophagy. Cell physiol biochem. (2018) 49:190–205. doi: 10.1159/000492853
Keywords: gut microbiota, bacterial, fungal, buffalo, mastitis
Citation: Chen X, An M, Zhang W, Li K, Kulyar MF-e-A, Duan K, Zhou H, Wu Y, Wan X, Li J, Quan L, Mai Z, Bai W and Wu Y (2022) Integrated Bacteria-Fungi Diversity Analysis Reveals the Gut Microbial Changes in Buffalo With Mastitis. Front. Vet. Sci. 9:918541. doi: 10.3389/fvets.2022.918541
Received: 12 April 2022; Accepted: 24 May 2022;
Published: 27 June 2022.
Edited by:
Fazul Nabi, Lasbela University of Agriculture, Water and Marine Sciences, PakistanReviewed by:
Tariq Jamil, Friedrich Loeffler Institut, GermanyPrerona Boruah, DY Patil Deemed to be University, India
Copyright © 2022 Chen, An, Zhang, Li, Kulyar, Duan, Zhou, Wu, Wan, Li, Quan, Mai, Bai and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Wu, wuyi2001cn@163.com
†These authors have contributed equally to this work