
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Tuberc. , 15 October 2024
Sec. Therapeutic Advances in Tuberculosis and Non-Tuberculous Mycobacterial Disease
Volume 2 - 2024 | https://doi.org/10.3389/ftubr.2024.1417939
This article is part of the Research Topic Rising Stars in Tuberculosis and Mycobacterial Diseases: 2023 View all 5 articles
 Sasha E. Larsen1
Sasha E. Larsen1 Brittany D. Williams1,2
Brittany D. Williams1,2 Tiffany Pecor1Valerie A. Reese1Zhiyi Zhu1Dana E. Miller1Brendan K. Podell3
Tiffany Pecor1Valerie A. Reese1Zhiyi Zhu1Dana E. Miller1Brendan K. Podell3 Susan L. Baldwin1
Susan L. Baldwin1 Rhea N. Coler4*
Rhea N. Coler4*Mycobacterium bovis Bacille Calmette-Guérin (BCG) is the sole globally licensed vaccine against tuberculosis despite its relatively moderate protection of acute disease through adolescence. We hypothesize that vaccine efficacy from a mucosal BCG vaccination will be directly influenced by Mycobacterium tuberculosis (M.tb) strain and mouse background. Here we investigated the effectiveness of mucosal BCG vaccination via the intranasal route, in resistant and susceptible mouse strains, to protect against laboratory strain H37Rv and clinical strain HN878 M.tb aerosol challenge. We evaluated both pulmonary and disseminated CFU at 4-weeks post-infection in addition to survival endpoints in C57BL/6, SWR, and C3HeB/FeJ mice. Antigen specific T cell responses in the lung post-infection were also evaluated. We observed that in each case intranasal BCG afforded a significant reduction in pulmonary CFU at 4-weeks post-infection compared to matched untreated controls. However, only susceptible mouse strains, SWR and C3HeB/FeJ, demonstrated similarly robust control from bacterial dissemination when CFU in the spleen was evaluated at the same timepoint. In the case of both M.tb H37Rv and M.tb HN878 challenge, intranasal BCG significantly improved survival of each mouse cohort compared to unvaccinated controls. Together these data suggest that there is still much to be learned from the century old vaccine, BCG, and how it drives protection.
Mycobacterium bovis Bacille Calmette-Guérin (BCG) remains the sole licensed vaccine against Mycobacterium tuberculosis (M.tb), despite noted waning protection from disease (1–3). This is in part due to mixed efficacy reporting that may be due to factors like which BCG strains are used for vaccination (4, 5), composition of host genetics or prior exposures to other environmental mycobacteria (6–8) or other variables related to M.tb virulence (9, 10). Studies of outbreaks with known exposures to M.tb suggest BCG vaccination can result in reduced risk of test conversion in a gamma interferon release assay, T-SPOT®.TB-test, including in a school setting in the UK (11). Therefore, BCG is still deployed in nations with endemic M.tb. BCG alone has been unable to halt the population-level progression of tuberculosis disease (TB) that ravages low and middle income countries disproportionately (12, 13). Novel TB vaccine candidates have stalled through clinical and preclinical pipelines due to a lack of robust and tangible immune correlates for stage-gating advancement (14–16). Preceding novel vaccine candidates, the research community needs to leverage novel vaccine systems, routes of delivery and host genetics to identify the requirements for durable protection from TB disease.
While BCG does not provide durable immunity from TB disease or prevent infection, there are still ample ways in which immunologists and vaccine developers can leverage this century old vaccine to winnow down the most critical features of protective immunity, including in preclinical models. After the Lübeck disaster (17, 18), less effort was focused on mucosal BCG delivery as a mechanism for correlates of protection discovery. However, over the last two decades a trove of data has been shared in this regard. For example, Perdomo and colleagues demonstrated that lung resident memory T cells were induced by intratracheal immunization with BCG in a C57BL/6 mouse model (19). Intranasally (i.n.) delivered BCG is a standard control group for mycobacterial growth inhibition assays (MGIA) using ex vivo mouse samples for surrogate efficacy endpoints (20–22) and has been shown to protect BALB/c mice partially via accelerating IFNγ production at the pulmonary site (23). Others have shown that protection induced by mucosal BCG vaccination in susceptible DBA/2 mice is partially IL-17-dependent (24), and in other studies specifically drives critical cell homing to the lungs [reviewed here (25)]. The purpose of this study is to expand the understanding of the protective effect of i.n. vaccination with BCG against laboratory and clinical strains of M.tb across three different mouse strains with varying susceptibility to infection, C57BL/6, SWR, and C3HeB/FeJ.
Recent advancements in interrogating host genetics include the development and sharing of collaborative cross (CC) mice, which are genetically diverse but tractable, enabling identification of loci that correlate with protective responses to infection (26, 27). In CC and conventional mouse strains, a spectrum of sensitivity to M.tb infection is observed. In these studies, C57BL/6 mice are relatively resistant (low bacterial burden in tissues) to an intravenous (i.v.) challenge with M.tb H37Rv vs. other strains (28). However, when examining the protection afforded by subcutaneous (s.c.) BCG vaccination, male C57BL/6 were protected (>0.5 log10 CFU reduction) in both the lung and spleen 4 and 14 weeks post aerosol challenge with M.tb H37Rv [Figure 3 here (29)]. Despite their relative resistance to M.tb, C57BL/6 have been extensively used as a base challenge mouse strain when evaluating different M.tb strains, TB vaccines and immunity (30–32). Swiss inbred SWR mice, conversely, are well-known for being particularly sensitive to M.tb challenge which seems partially linked to defects in complement C5, inclusions in alveolar macrophages and worse disease progression due to accumulation of less functional effector T cells (30, 33–35). The C3HeB/FeJ mouse model is another canonically leveraged mouse strain given that they more faithfully recapitulate specific types of human-like necrotic TB lesions (36–39). C3HeB/FeJ mice (also referred to as Kramnik mice) have unique pulmonary pathology and are highly susceptible to M.tb challenge, making them well-utilized for vaccine and drug therapy studies (40–42) and immune cell influx evaluation to areas of infection (43). Using these three differentially susceptible mouse strains we executed a head-to-head evaluation of i.n. BCG vaccination on bacterial burden and survival after aerosol infection with a lab-adapted and clinical M.tb strains.
Cohorts of C57BL/6, SWR, and C3HeB/FeJ mice were either vaccinated with 5 × 105 CFU BCG intranasally or left untreated and 12 weeks later challenged with a low dose aerosol (LDA) using either laboratory M.tb H37Rv strain or clinical and highly virulent M.tb HN878 strain. Regardless of mouse strain or M.tb challenge strain, aerosol delivery of M.tb resulted in 20–67 CFU per mouse at 24 h (Table 1), in line with conventional LDA parameters.
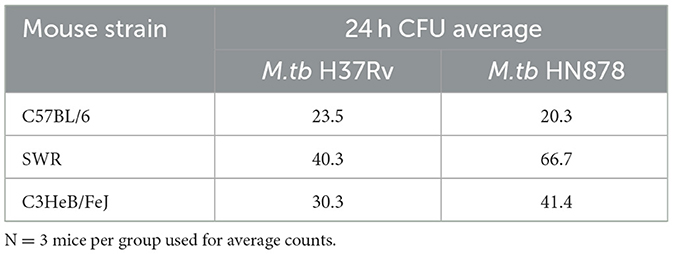
Table 1. Bacterial counts at 24 hours post M.tb infection.
At the 4-week post-infection timepoint we assessed cellular responses to M.tb H37Rv lysate (Figure 1). Antigen experienced (CD44+) CD4+ and CD8+ T cells were evaluated for CD154, CD107a, IL-2, IL-17, IL-21, IFNγ, and TNFα expression (gating strategy is presented in Supplementary Figure 1). We observed no significant differences between mouse strain or vaccination regimen in the cases of CD4+ T cells expressing IL-21, or TNFα after ex vivo stimulation with M.tb lysate (data not shown). Interestingly, by 4-weeks post-infection CD4+ T cells from SWR mice vaccinated with BCG demonstrated a significantly higher IL-17 response than unvaccinated controls (Figures 1G, H) and this pattern was similarly observed in the case of C3HeB/FeJ mice infected with M.tb HN878 (Figure 1H). When polyfunctional CD4+ T cells were evaluated for expression of two or more TH1 cytokines (IFNγ, IL-2, or TNFα), no statistically significant increases in BCG vaccinated cohorts were observed (Supplementary Figures 2A, B). However, in the case of mice infected with M.tb HN878, there was an increase in SWR BCG vaccinated mice with more CD4+ T cells co-expressing IL-17 and IL-2, and in BCG vaccinated C3HeB/FeJ mice having more CD4+ T cells co-expressing IL-17 with TNFα than matched unvaccinated cohorts (Supplementary Figures 3A, B). Otherwise, there are no notable trends of proinflammatory cytokine expression from CD4+ T cells across cohorts post-infection (Figure 1).
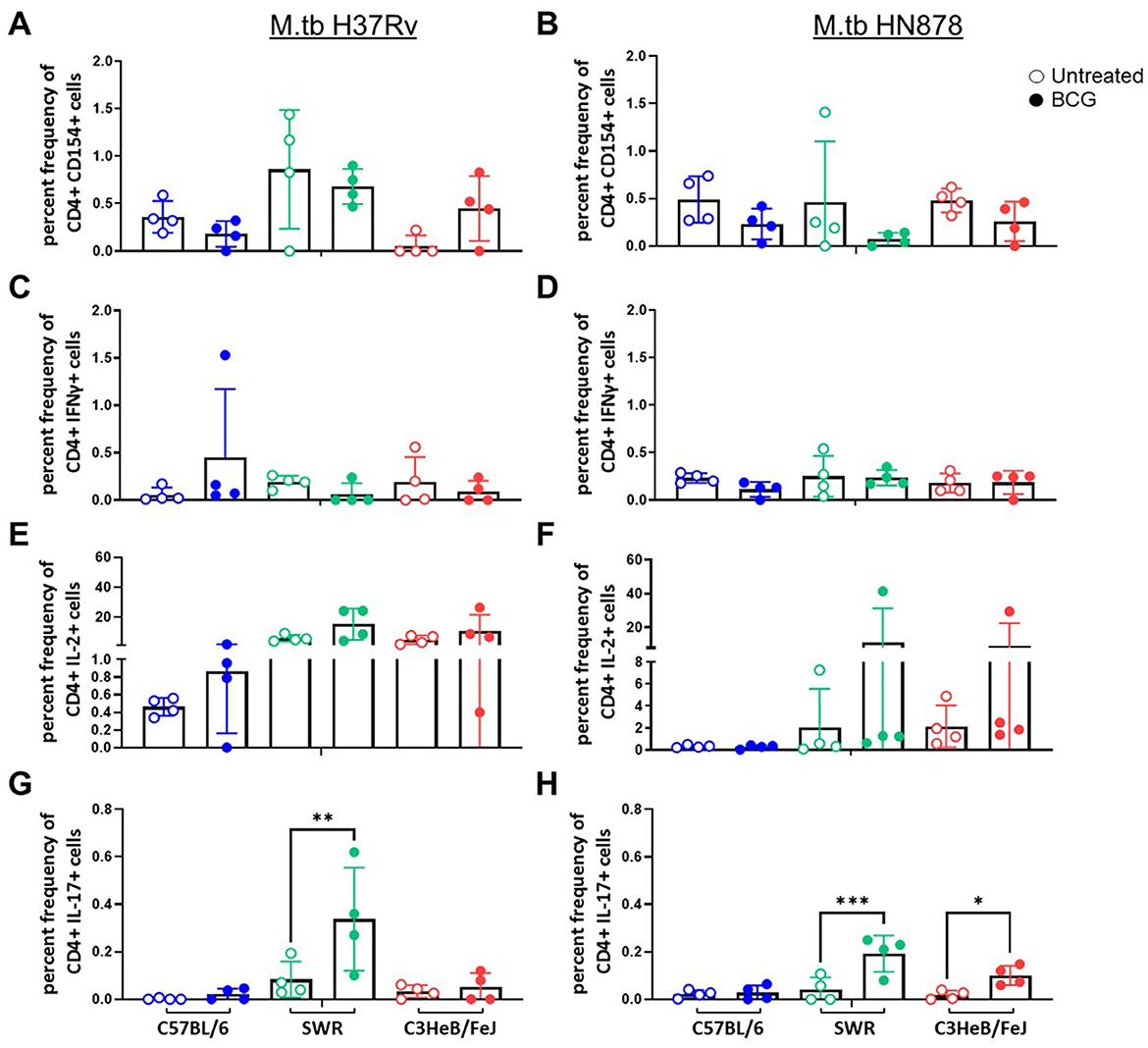
Figure 1. CD44+ CD4+ T cell proinflammatory cytokine expression after ex vivo M.tb H37Rv lysate stimulation. Percent frequency of CD44+ CD4+ cytokine expressing cells post stimulation with 10 μg/mL of M.tb H37Rv lysate from lung homogenates of n = 4 mice per group from either (A, C, E) M.tb H37Rv or (B, D, F) M.tb HN878 challenged cohorts 4 weeks post-infection. Untreated (open circles) cohorts were compared with i.n. BCG vaccinated (closed circles) cohorts of C57BL/6 (blue) SWR (green) or C3HeB/FeJ (red) mice. Expression of (A, B) CD154, (C, D) IFNγ, (E, F) IL-2, and (G, H) IL-17 were evaluated. One-Way ANOVA with multiple corrections for paired tests within mouse strain and between vaccination regimens used Šídák's multiple comparisons test. Significant differences noted in the figure where *p < 0.05, **p < 0.01, and ***p < 0.001.
We next evaluated CD8+ T cell cytotoxic activity and cytokine expression after ex vivo M.tb lysate stimulation by flow cytometry. We used CD107a as a marker of CD8+ T cell degranulation. Only in the case of C57BL/6 mice challenged with M.tb H37Rv did we observe a significant increase in CD107a staining in the BCG vaccinated group (Figure 2A), no significant differences were observed in M.tb HN878 challenged cohorts (Figure 2B). In the case of CD8+ T cells expressing proinflammatory cytokines we observed no major trends for IFNγ, where all cohorts had extremely low percent frequencies which did not change with M.tb strain or mouse strain (Figures 2C, D). Although not significant, there was a trend of higher CD8+ T cell IL-2 and TNFα expression in each mouse strain given i.n. BCG vaccination compared to untreated controls, 4-weeks after M.tb H37Rv challenge (Figures 2E, G). No similar trends were observed post-infection with M.tb HN878 for CD8+ T cell cytokines (Figures 2F, H).
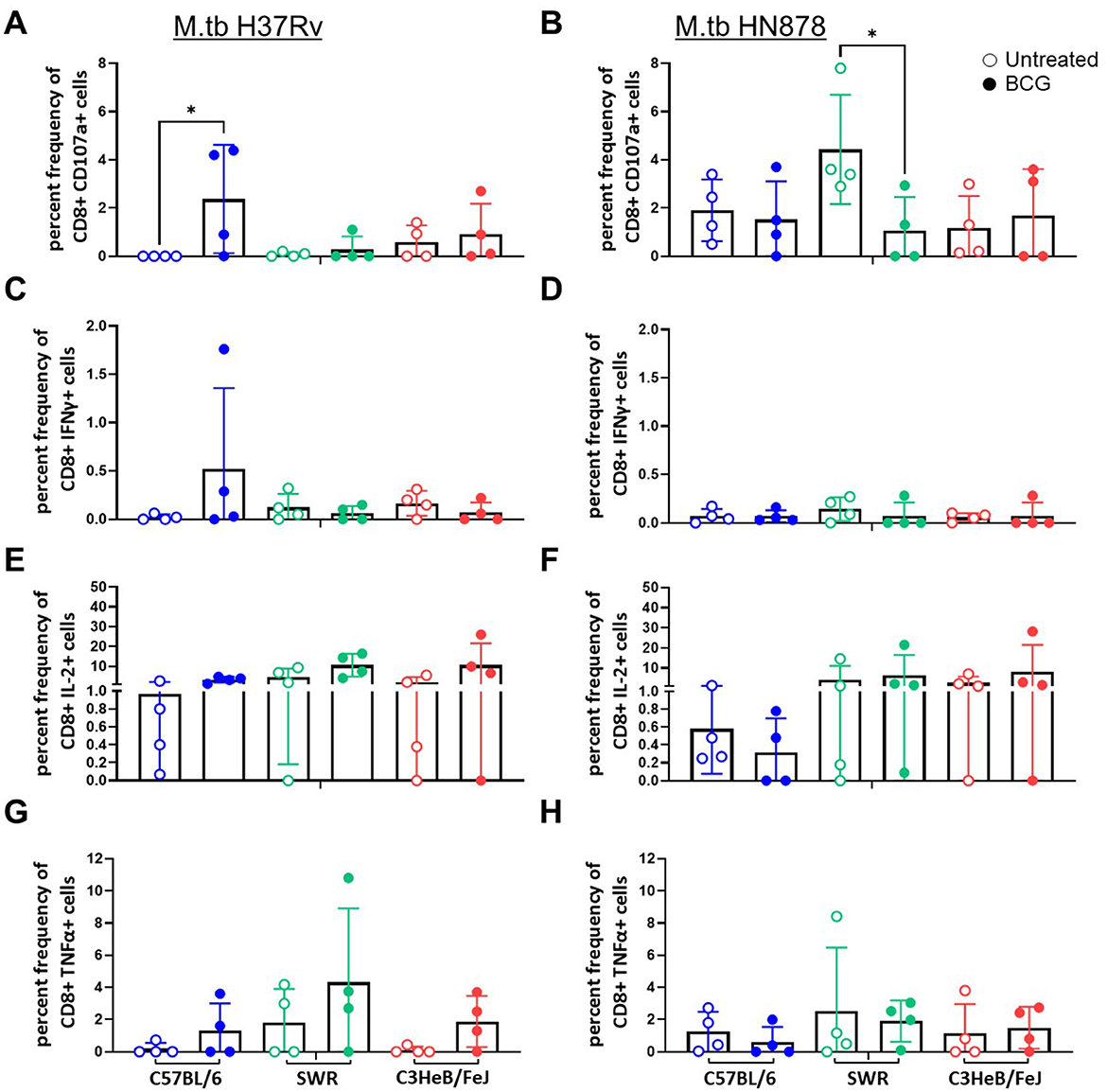
Figure 2. CD44+ CD8+ T cell proinflammatory cytokine expression after ex vivo M.tb H37Rv lysate stimulation. Percent frequency of CD8+ cytokine expressing cells post stimulation with 10 μg/mL of M.tb lysate from lung homogenates of n = 4 mice per group from either (A, C, E) M.tb H37Rv or (B, D, F) M.tb HN878 challenged cohorts 4 weeks post-infection. Untreated (open circles) cohorts were compared with i.n. BCG vaccinated (closed circles) cohorts of C57BL/6 (blue) SWR (green) or C3HeB/FeJ (red) mice. Expression of (A, B) CD107a, (C, D) IFNγ, (E, F) IL-2, and (G, H) TNFα were evaluated. One-Way ANOVA with multiple corrections for paired tests within mouse strain and between vaccination regimens used Šídák's multiple comparisons test. Significant differences noted in the figure where *p < 0.05.
In addition to total responses after ex vivo stimulation with M.tb lysate, antigen specific responses were evaluated. This included antigen Rv3874 (CFP-10) which is only expressed by M.tb as it is located in a region of deletion within BCG. At 4-weeks post-infection lung homogenate samples were assessed as described above for both CD4+ and CD8+ T cell cytokine expression. For CD4+ T cells, SWR mice with BCG vaccination consistently expressed significantly higher IL-2, IL-17 (Supplementary Figure 4) IL-21, and TNFα (data not shown) cytokines ex vivo compared with unvaccinated SWR controls following challenge with M.tb H37Rv, but this pattern was not observed in immunized SWR mice challenged with M.tb HN878. We observed BCG vaccinated SWR mice had a statistically higher percent frequency of CD4+ T cells co-expressing IL-2 and TNFα than unvaccinated SWR mice challenged with M.tb H37Rv (Supplementary Figure 2C). In addition, BCG vaccinated SWR cohorts demonstrated significantly higher CD4+ T cells co-expressing IL-17 with IL-2 than matched unvaccinated cohorts (Supplementary Figure 3C). C57BL/6 and C3HeB/FeJ mice were consistently low responders in each condition and no significant differences were observed between unvaccinated vs. BCG vaccinated comparisons for single or polyfunctional TH1 CD4+ T cells. Cohorts vaccinated with i.n. BCG and challenged with M.tb H37Rv generally trended higher in CD8+ T cell responses with ex vivo Rv3874 antigen stimulation. CD8+ T cells from SWR mice prophylactically immunized with i.n. BCG expressed significantly more IFNγ and IL-2 after ex vivo Rv3874 stimulation compared with unvaccinated controls (Supplementary Figure 5).
At 16-weeks post-vaccination and 4-weeks post-infection, bacterial burden was evaluated in each cohort. Lung Log10 CFU as well as spleen Log10 CFU were enumerated to evaluate the primary site of infection and representative organ for dissemination, respectively. Across each mouse and M.tb strain, prior i.n. vaccination with BCG resulted in significant protection in the lungs compared to untreated controls (Figures 3A, B). As expected, untreated susceptible mouse strains SWR and C3HeB/FeJ harbored significantly higher pulmonary bacterial burden than relatively resistant C57BL/6 in both infection schemes. Interestingly, unvaccinated C3HeB/FeJ mice had significantly higher lung bacterial burden than unvaccinated SWR mice at 4-weeks post-infection with M.tb HN878 but not M.tb H37Rv (Tables 2, 3). In the spleen, SWR mice with BCG vaccination were protected from both M.tb H37Rv and M.tb HN878 dissemination compared to unvaccinated controls, and C3HeB/FeJ mice showed significantly reduced bacterial burden compared to unvaccinated controls in the case of virulent M.tb HN878 infection (Figures 3C, D and Table 3). Unvaccinated cohorts infected with M.tb HN878 contained more heterogeneity and higher total percent lung lesion area, measured from accessory lobes stained with H&E, than unvaccinated controls infected with M.tb H37Rv by this 4-week post-infection timepoint (Figures 3E, F). There was a consistent trend of less lung lesion area induced by M.tb HN878 infection in BCG vaccinated groups, but this was only significantly lower in C3HeB/FeJ cohorts (Figure 3E). While C57BL/6 mice received the relatively lowest infection at the time of infection (Table 1), this cohort experienced nearly identical dissemination to the spleen across each M.tb challenge by 4-weeks post-infection (Table 3), suggesting the lack of protection in the BCG vaccinated animals from M.tb dissemination in this mouse strain was not due to initial M.tb deposition.
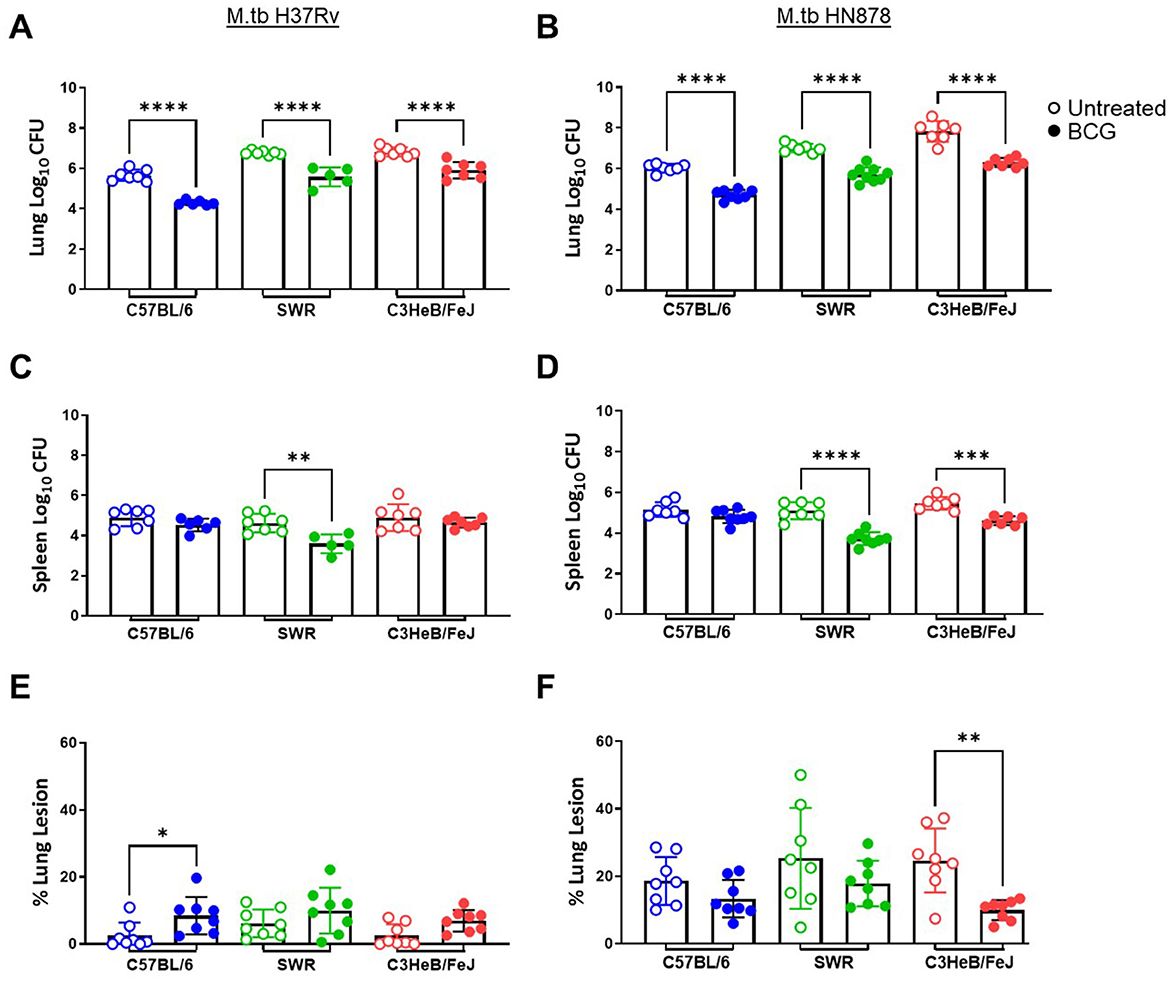
Figure 3. Pulmonary and disseminated bacterial burden and pathology 4 weeks post-infection. (A, B) Lung and (C, D) Spleen homogenate Log10 CFU counts and (E, F) percent lung lesion area from H&E stained accessory lobe slides evaluated 4 weeks post-infection with a LDA of (A, C, E) M.tb H37Rv or (B, D, F) M.tb HN878. Untreated groups are open circles and BCG vaccinated groups are noted as closed circles. One-Way ANOVA with multiple corrections for paired tests within mouse strain and between vaccination regimens used Šídák's multiple comparisons test. Significant differences noted in the figure where *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. n = 5–7 per group.
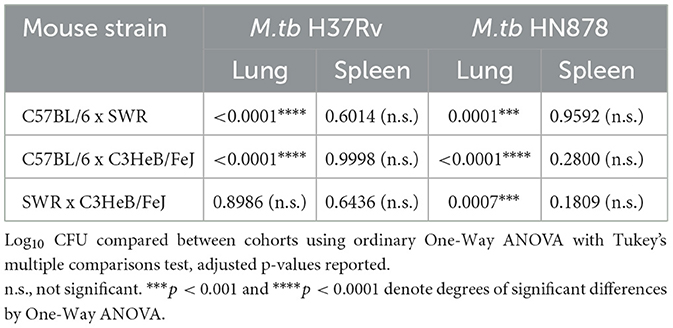
Table 2. Comparison of bacterial burden in unvaccinated mouse strains.

Table 3. Bacterial burden 4 weeks post-infection.
Lastly, durability of vaccine-mediated protection from morbidity was evaluated in n = 10 animals per group which were weighed weekly post-infection. Animals were observed for moribund characteristics or behaviors and met objective euthanasia criteria when weight loss exceeded 20% of maximum. We compared virulence of M.tb strains by median survival days across challenges as well as Log-rank hazard ratio evaluation of the full survival curves. For unvaccinated SWR and C3HeB/FeJ mice challenged with M.tb HN878 the median survival days was roughly half or worse of that for the same untreated cohorts challenged with M.tb H37Rv (Table 4). Overall, we observed that M.tb HN878 infection was more virulent over time by Log-rank Mantel-Cox test in unvaccinated SWR (p < 0.0001) and C3HeB/FeJ (p = 0.0001) mouse backgrounds compared to similarly challenged unvaccinated M.tb H37Rv cohorts, but not in unvaccinated C57BL/6 cohorts by the same test (p = 0.1477). In each mouse and M.tb challenge combination, i.n. BCG vaccination afforded greater cohort median survival days compared to untreated controls (Table 4) and provided durable protection from morbidity by probability of survival log-rank tests in each M.tb infection scheme within each mouse strain tested (Figure 4).
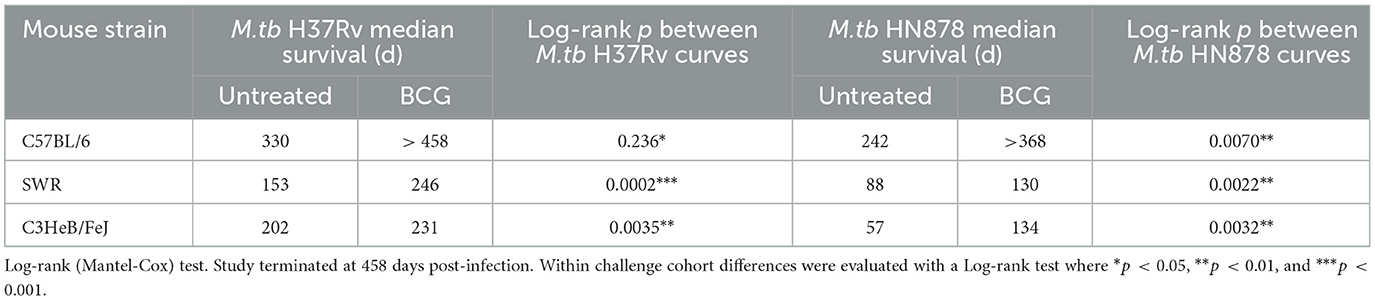
Table 4. Median survival days with and without mucosal BCG vaccination.
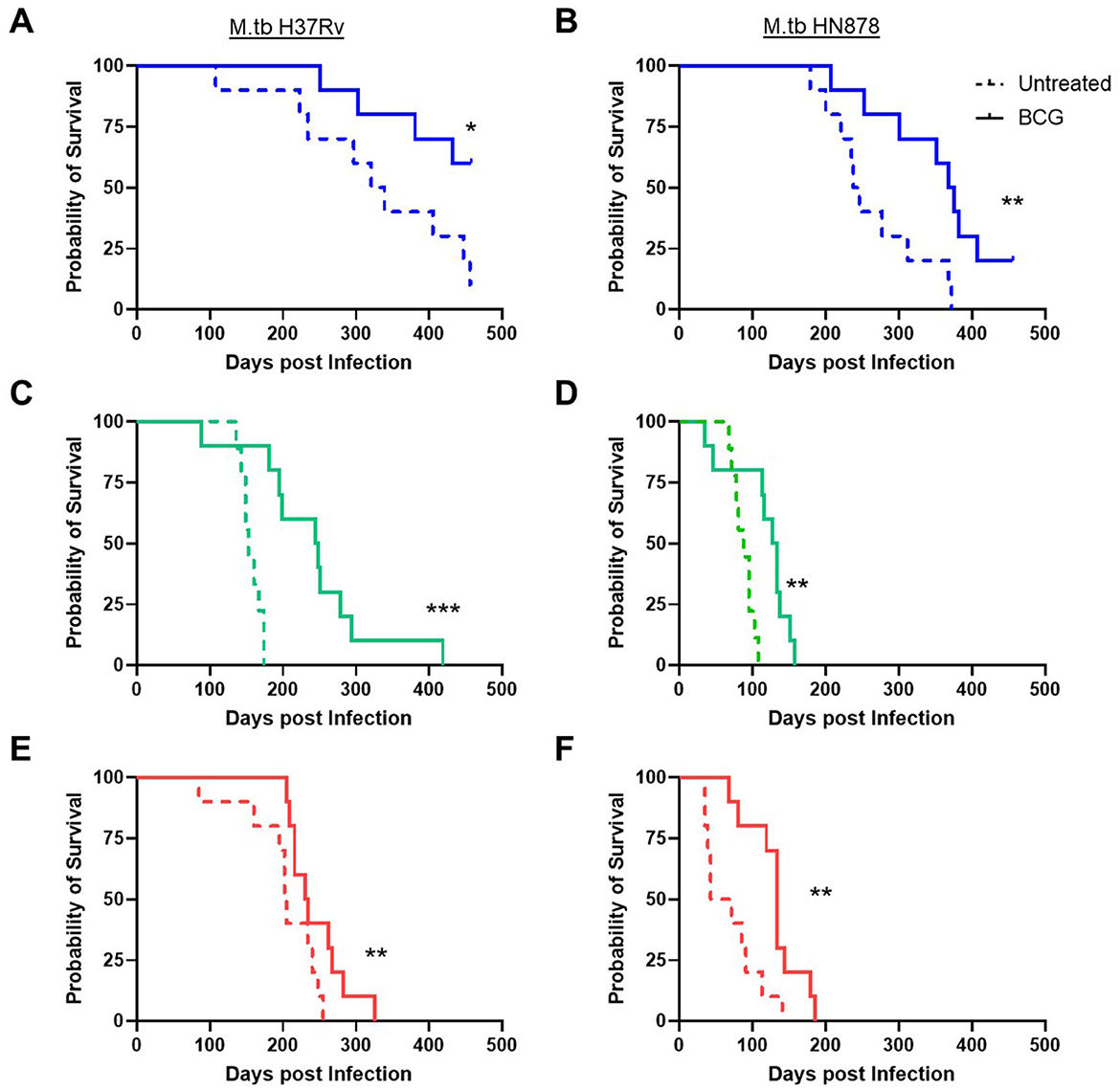
Figure 4. Survival of challenged cohorts and durable protection from mucosal BCG vaccination. Probability of survival, where n = 10 animals per group were weighed weekly and assessed for morbidity criteria post-infection with either (A, C, E) M.tb H37Rv or (B, D, F) M.tb HN878. Untreated (dashed line) cohorts were compared with i.n. BCG vaccinated (solid line) cohorts of (A, B) C57BL/6 (blue) (C, D) SWR (green) or (E, F) C3HeB/FeJ (red) mice. Comparisons with or without BCG vaccination within a mouse and M.tb challenge used a Log-rank (Mantel-Cox) test, significant differences are labeled accordingly *p < 0.05, **p < 0.01, and ***p < 0.001.
Here we demonstrate that mucosally delivered BCG affords significant protection from pulmonary bacterial burden and enhances survival in all mouse background and M.tb challenge settings evaluated in this study. However, protection from dissemination was variable as was the rate of decline with respect to survival outcomes. In the case of both SWR and C3HeB/FeJ mice challenged with M.tb HN878 there is a stark and rapid decline of the group, possibly suggesting a more uniform sensitivity than other combinations. These data serve as a reference for those needing survival as an endpoint, who may need to execute studies in shorter windows of time (SWR or C3HeB/FeJ x M.tb HN878) or examine the natural or vaccine-induced heterogeneity of an infection (C57BL/6 x M.tb H37Rv). Furthermore, we observed a significant reduction in percent lung lesion area in BCG-vaccinated C3HeB/FeJ mice compared to matched unvaccinated cohorts by 4-weeks post-infection with M.tb HN878 which is a fairly early time point for pathology assessments. These data are important given the power of translational pathological outcomes in C3HeB/FeJ mice.
Additionally, there is a significant and expected contribution of M.tb challenge strain on outcomes, where highly virulent M.tb HN878 infection resulted in higher bacterial burden, more immunopathology by 4-weeks post-infection and reduced survival and yet did not result in blatantly higher CD4+ or CD8+ T cell responses with ex vivo stimulation. While surprising, this is in line with prior observations where infection with M.tb Beijing strains, including M.tb HN878, over time induced an expansion of regulatory FoxP3+ T cells, downregulating effector-led immunity driven by BCG vaccination (44). The relatively weak and sometimes reduced expression of CD107a on CD8+ T cells derived from lung samples after ex vivo stimulation with M.tb lysate or recombinant antigen Rv3874 is congruent with human clinical studies which also observed reduced CD107a staining from lymph node samples compared to peripheral blood samples obtained from tuberculous lymphadenitis patients (45). Together these data may indicate a M.tb induced regulatory profile that is more acute closer to the site of infection.
Interestingly, BCG vaccinated SWR mice demonstrated a consistent increase in CD4+ T cells expressing IL-2 or IL-17 or both after ex vivo stimulation, which is notably lacking from the other two mouse strains. It is of interest in follow on studies to evaluate whether this contribution from IL-2 and IL-17 also contribute or correlate with reduced systemic and disseminated bacterial burden which was also observed in the SWR cohorts that received BCG vaccination. Aguilo et al. showed IL-17 dependent protection following pulmonary delivery of BCG, measured by a reduction in lung and spleen bacterial burden, and survival against M.tb H37Rv in the M.tb susceptible DBA/2 mouse strain (24). Interestingly, in this same study, BCG given by the i.d. route did not result in protection in the susceptible DBA/2 mice, but did protect wildtype C57BL/6 mice (24). Our group has also shown that mucosal delivery with a subunit vaccine is capable of inducing a skewed T helper 17 (TH17) immune response (46). Although we saw robust pulmonary M.tb protection following mucosal BCG immunization regardless of mouse background or M.tb strain in our current study, less consistent protection against M.tb was observed in the spleen. A limitation of all TB vaccine studies is that the field has not delivered discrete immune correlates of protection, and indeed we hypothesize that it is more likely that a suite of responses (47) are working together to limit M.tb in vivo and that many immune pathways together can garner similar results. It is unclear from these studies whether the CD4+ or CD8+ T cell responses from 4-weeks post-infection across mouse strains are meaningful toward protection and survival responses mediated by i.n. BCG vaccination or other immune parameters are driving those responses, such as T resident memory cells, humoral immune responses or specific trained innate immunity in the airway. The studies presented here are limited in that flow panels did not include markers that allow the discrimination of lung tissue-resident CD4+ and CD8+ T cells from those in the lung vasculature, which may further resolve differences in protection mediated by BCG vaccination. It has been observed that monocyte recruitment to the lungs after intradermal (i.d.) BCG vaccination correlated with protection in C57BL/6 mice, rather than T cell recruitment (48). These data together with CC models suggest that different host genetics in combination with BCG vaccination may pave different paths toward immune protection. All of these endpoints are worth pursuing.
Historically, oral BCG and i.d. BCG were evaluated for use in humans and ultimately i.d. was adopted. However, the oral route generates high-quality mucosal responses which has been observed in both mice and humans (49–51). Studying the unconventional administration of BCG (25) is making a resurgence in the TB vaccine community as a means to help define immune correlates of protection. This includes the notable use of intravenous (i.v.) delivery in non-human primate models that resulted in protection and robust immune endpoints across cohorts (52–55). In a diversity outbred mouse model, i.v. BCG enhanced survival but did not significantly reduce pulmonary M.tb burden compared to cohorts vaccinated via the i.d. route (56). Indeed, this may be due in part to the breadth of antigen-specific responses in i.v. vaccinated cohorts being larger than traditionally immunized i.d. mouse cohorts (57). While i.d. delivered BCG is controlled and cleared from the host overtime [in as little 12 weeks in mice as reported here (29)], it is unclear whether mucosal i.n. or systemic i.v. delivery of the vaccine would present novel niches for more long term persistence. This is part of ongoing discussions and preclinical studies about the safety of administering BCG i.v. clinically (25, 58). In addition to route, vaccine dose is known to be critical in meeting specific thresholds of protection against M.tb challenge in both mice (59) and other preclinical models (25, 53). Further work could compare routes and doses in mice, including i.n. and i.v. with traditional s.c./i.d. BCG, to obtain meaningful correlates that drive protection in the mouse model of TB. Cumulatively, these data suggest there is still much we can glean from BCG vaccination strategies if we vary the route or dose of delivery and carefully interrogate the interplay with different host or pathogen genetics.
Female C57BL/6, SWR, and C3HeB/FeJ mice were purchased from Charles River Laboratories at 5–8-weeks old. Mice were housed at (renamed post-receivership) the Infectious Disease Research Institutes (IDRI) biosafety level 3 (BSL-3) animal facility under pathogen-free conditions and were handled in accordance with approved protocols from the IDRI Institutional Animal Care and Use Committee (IACUC). The institute, formerly known as IDRI, operated under USDA Certificate # 91-R-0061 and PHS Assurance # A4337-01. All methods were carried out in accordance with animal welfare guidelines and regulations. For vaccination, mice were given an intranasal immunization of 5 × 105 colony forming units (CFU) of BCG Aeras Pasteur [as previously reported in (19)] in a total of 50 μl, 25 μl per nare, while under ketamine/xylazine anesthesia. The sequenced genome of BCG Aeras Pasteur propagated by our lab and maintained as vaccination stocks can be found at GenBank ID: SAMN38797945. Twelve weeks later, mice were challenged with a low dose aerosol (LDA, 25-100 CFU per mouse upon infection) of either M.tb H37Rv or M.tb HN878 (BEI Resources) using a Madison aerosol infection chamber. Delivery and deposition of M.tb dose was confirmed 24 h post-infection by evaluating bacterial burden from the lungs as described below. Ten mice per group were followed after M.tb infection for survival analysis. Animals with >20% weight loss, or moribund condition, were euthanized. The survival study was prematurely ended with C57BL/6 animals still not meeting endpoint criteria on day 458 post-infection, due to the Authors' prior institution (IDRI) abruptly entering receivership.
Intracellular staining and flow cytometry was performed on lung homogenates 4 weeks post-M.tb infection with either M.tb H37Rv or M.tb HN878. Samples were incubated in red blood cell lysis buffer (eBioscience), washed and resuspended in RPMI 1640 (Life Technologies) + 10% fetal bovine serum (FBS; BioWhittaker, Inc.) and subsequently aliquoted in 96-well round bottom plates. Cells were then stimulated with media alone, 10 μg/mL M.tb H37Rv whole cell lysate, 10 μg/mL of recombinant M.tb antigens Rv3478 (PPE60 Mtb39c), Rv3874 (CFP10), Rv3875 (ESTAT6), or 1 μg/mL phorbol myristate acetate (PMA; Calbiochem) + 1 μg/mL ionomycin (Sigma) and incubated at 37°C. During the stimulation fluorochrome-conjugated anti-mouse CD107a antibody (LAMP-1, clone 1D4B, BioLegend) was also added to the plates. After 1–2 h 1 μg/μl of GolgiPlug (BD Biosciences) was added and samples were incubated at 37°C for an additional 8 h. Samples then remained at 4°C until staining (<12 h). Samples were first surface stained with fluorochrome-conjugated antibodies against mouse CD4 (clone RM4-5, BioLegend), CD8 (clone 53-6.7, BioLegend), and 1 μg/mL of Fc receptor block anti-CD16/CD32 (clone 93, eBioscience) in PBS with 1% bovine serum albumin (BSA) for 10–15 min at room temperature (RT). Cells were then washed and fixed using BD Biosciences Fix/Perm reagent for 20 min at RT. Subsequently samples were washed with BD Perm/Wash followed by intracellular staining in Perm/Wash reagent with anti-mouse IFN-γ (clone XMG1.2, Invitrogen), IL-2 (clone JES6-5H4, eBioscience), IL-17A (clone Tc11-1BH10.1, BioLegend), TNF-α (clone MP6-XT22, eBioscience), CD44 (clone IM7, BioLegend), IL-21 (clone mhalx21, ThermoFisher Scientific), and CD154 (clone MR7, eBioscience) for 10 min at RT. All antibodies were used at 10 μl/mL. The samples were then incubated in 4% paraformaldehyde for 20 min to fix, decontaminate, and remove from the containment facility before washing and resuspension in PBS + 1% BSA. An LSRFortessa flow cytometer (BD Bioscience) was used for sample acquisition, and analysis was performed using FlowJo v10 (BD Biosciences).
To enumerate bacterial burden in infected animals, organs were homogenized at 24 h (n = 3, lung only) or 4 weeks (n = 5–8, lung and spleen) post-infection as previously described (60, 61). Lung and spleen tissue from infected animals was strain and homogenized in 2–5 mL of either RPMI + FBS (lung) or PBS + Tween-80 (Sigma-Aldrich) CFU buffer (spleen) using an Omni tissue homogenizer (Omni International, Kennesaw, GA). Homogenized tissue was plated neat or across a series of dilutions on Middlebrook 7H10 agar plates (Fisher Scientific, Waltham, MA) and subsequently incubated at 37°C and 5% CO2 for ~ 3 weeks before colonies were counted. For CFU plating, 5 μg/ml of thiophene-2-carboxylic acid hydrazide (TCH) was supplemented to 7H10 agar to inhibit the growth of BCG and to allow the growth of M.tb in duplicate plates. Bacterial burden as CFU/mL was calculated per organ and is presented here as Log10 values. Reduction in bacterial burden was calculated as the difference in mean Log10 values between groups assessed.
Lung accessory lobes from 4 weeks post M.tb infection were collected and temporarily stored in 10% normal buffered formalin. Hematoxylin and eosin (H&E) slides were generated at the Benaroya Research Institute Histology Core facility (Seattle, WA, USA). Blinded slides were analyzed by veterinary pathologist (Dr. Brendan Podell, Colorado State University). Image analysis and lesion area identification and percent lesion area was performed as previously described (35, 61).
For 4-week CFU, percent lesion area and ICS data comparing groups with or without BCG vaccination within a mouse and M.tb challenge, a One-Way ANOVA with multiple corrections for paired tests within mouse strain using Šídák's multiple comparisons test was used. The flow cytometry data were assessed using FlowJo v10 (BD), and SPICE (NIH, https://niaid.github.io/spice/). For survival comparisons with or without BCG vaccination within a mouse and M.tb challenge a Long-rank (Mantel-Cox) test was used and median days of survival for each group are reported in the text. Statistical analyses were performed using GraphPad Prism 10.2.2 software (GraphPad Software, San Diego, CA). Grubbs tests were run to identify potential outliers within cohorts. Significant differences are labeled accordingly in the figures where *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The animal study was approved by IDRI Institutional Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
SL: Conceptualization, Formal analysis, Investigation, Project administration, Resources, Visualization, Writing – original draft, Writing – review & editing. BW: Formal analysis, Investigation, Visualization, Writing – review & editing. TP: Formal analysis, Investigation, Visualization, Writing – review & editing. VR: Formal analysis, Investigation, Visualization, Writing – review & editing. DM: Visualization, Writing – review & editing. SB: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing. RC: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing. ZZ: Formal analysis, Investigation, Visualization, Writing – review & editing. BP: Data curation, Formal analysis, Investigation, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This project has been funded in whole or in part with Federal Funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. 75N93021C00029 (to RC) and Grant: R01AI125160 (to RC and SB) as well as Contract No: P30AI168023 as the Seattle Tuberculosis Research Advancement Center (to RC). BW was supported through National Institutes of Health Training grant to the University of Washington, Diseases of Public Health Importance No. AI00750922.
The authors would like to thank Winston Wicomb and the dedicated vivarium staff for their excellent animal care during this work. The following reagents were obtained through BEI Resources by the Coler Lab, NIAID, NIH: Mycobacterium tuberculosis, strain HN878, NR-13647 and strain H37Rv, and NR-13648.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/ftubr.2024.1417939/full#supplementary-material
1. Roy A, Eisenhut M, Harris RJ, Rodrigues LC, Sridhar S, Habermann S, et al. Effect of BCG vaccination against Mycobacterium tuberculosis infection in children: systematic review and meta-analysis. Br Med J. (2014) 349:g4643. doi: 10.1136/bmj.g4643
2. Mangtani P, Abubakar I, Ariti C, Beynon R, Pimpin L, Fine PE, et al. Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clin Infect Dis. (2014) 58:470–80. doi: 10.1093/cid/cit790
3. Abubakar I, Pimpin L, Ariti C, Beynon R, Mangtani P, Sterne JAC, et al. Systematic review and meta-analysis of the current evidence on the duration of protection by bacillus Calmette-Guérin vaccination against tuberculosis. Health Technol Assess. (2013) 17:1–372. doi: 10.3310/hta17370
4. Ritz N, Dutta B, Donath S, Casalaz D, Connell TG, Tebruegge M, et al. The influence of bacille Calmette-Guerin vaccine strain on the immune response against tuberculosis: a randomized trial. Am J Respir Crit Care Med. (2012) 185:213–22. doi: 10.1164/rccm.201104-0714OC
5. Zhang L, Ru H-W, Chen F-Z, Jin C-Y, Sun R-F, Fan X-Y, et al. Variable virulence and efficacy of BCG vaccine strains in mice and correlation with genome polymorphisms. Mol Ther. (2016) 24:398–405. doi: 10.1038/mt.2015.216
6. Poyntz HC, Stylianou E, Griffiths KL, Marsay L, Checkley AM, McShane H. Non-tuberculous mycobacteria have diverse effects on BCG efficacy against Mycobacterium tuberculosis. Tuberculosis. (2014) 94:226–37. doi: 10.1016/j.tube.2013.12.006
7. Ghasemi F, Kardan-Yamchi J, Heidary M, Karami-Zarandi M, Akrami S, Maleki A, et al. Effects of non-tuberculous mycobacteria on BCG vaccine efficacy: a narrative review. J Clin Tuberc Other Mycobact Dis. (2024) 36:100451. doi: 10.1016/j.jctube.2024.100451
8. Setiabudiawan TP, Reurink RK, Hill PC, Netea MG, van Crevel R, Koeken VACM. Protection against tuberculosis by bacillus Calmette-Guérin (BCG) vaccination: a historical perspective. Med. (2022) 3:6–24. doi: 10.1016/j.medj.2021.11.006
9. Hank JA, Chan JK, Edwards ML, Muller D, Smith DW. Influence of the virulence of Mycobacterium tuberculosis on protection induced by bacille Calmette-Guérin in guinea pigs. J Infect Dis. (1981) 143:734–8. doi: 10.1093/infdis/143.5.734
10. Smith I. Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin Microbiol Rev. (2003) 16:463–96. doi: 10.1128/CMR.16.3.463-496.2003
11. Eisenhut M, Paranjothy S, Abubakar I, Bracebridge S, Lilley M, Mulla R, et al. BCG vaccination reduces risk of infection with Mycobacterium tuberculosis as detected by gamma interferon release assay. Vaccine. (2009) 27:6116–20. doi: 10.1016/j.vaccine.2009.08.031
12. World Health Organization. Global Tuberculosis Report. Geneva: World Health Organization (2023).
13. World Health Organization. An investment case for new tuberulosis vaccines. Geneva: World Health Organization (2022).
14. Martin C, Aguilo N, Marinova D, Gonzalo-Asensio J. Update on TB vaccine pipeline. Appl Sci. (2020) 10:2632. doi: 10.3390/app10072632
15. Ottenhoff THM, Kaufmann SHE. Vaccines against tuberculosis: where are we and where do we need to go? PLoS Pathog. (2012) 8:e1002607. doi: 10.1371/journal.ppat.1002607
16. Zhuang L, Ye Z, Li L, Yang L, Gong W. Next-generation TB vaccines: progress, challenges, and prospects. Vaccines. (2023) 11:81304. doi: 10.3390/vaccines11081304
17. Fox GJ, Orlova M, Schurr E. Tuberculosis in newborns: the lessons of the “Lübeck Disaster” (1929–1933). PLoS Pathog. (2016) 12:e1005271. doi: 10.1371/journal.ppat.1005271
18. Donald P, Kaufmann S, Thee S, Mandalakas AM, Lange C. Pathogenesis of tuberculosis: the 1930 Lübeck disaster revisited. Eur Respir Rev. (2022) 31:2022. doi: 10.1183/16000617.0046-2022
19. Perdomo C, Zedler U, Kühl AA, Lozza L, Saikali P, Sander LE, et al. Mucosal BCG vaccination induces protective lung-resident memory T cell populations against tuberculosis. mBio. (2016) 7:16. doi: 10.1128/mBio.01686-16
20. Painter H, Willcocks S, Zelmer A, Reljic R, Tanner R, Fletcher H. Demonstrating the utility of the ex vivo murine mycobacterial growth inhibition assay (MGIA) for high-throughput screening of tuberculosis vaccine candidates against multiple Mycobacterium tuberculosis complex strains. Tuberculosis. (2024) 146:102494. doi: 10.1016/j.tube.2024.102494
21. Painter H, Harriss E, Fletcher HA, McShane H, Tanner R. Development and application of the direct mycobacterial growth inhibition assay: a systematic review. Front Immunol. (2024) 15:1355983. doi: 10.3389/fimmu.2024.1355983
22. Painter PSH, Cia F, Stockdale L, Tanner R, Willcocks S, Reljic R, et al. Adaption of the ex vivo mycobacterial growth inhibition assay for use with murine lung cells. Sci Rep. (2020) 20:60223. doi: 10.1038/s41598-020-60223-y
23. Lyadova IV, Vordermeier HM, Eruslanov EB, Khaidukov SV, Apt AS, Hewinson RG. Intranasal BCG vaccination protects BALB/c mice against virulent Mycobacterium bovis and accelerates production of IFN-gamma in their lungs. Clin Exp Immunol. (2001) 126:274–9. doi: 10.1046/j.1365-2249.2001.01667.x
24. Aguilo N, Alvarez-Arguedas S, Uranga S, Marinova D, Monzón M, Badiola J, et al. Pulmonary but not subcutaneous delivery of BCG vaccine confers protection to tuberculosis-susceptible mice by an interleukin 17-dependent mechanism. J Infect Dis. (2016) 213:831–9. doi: 10.1093/infdis/jiv503
25. Singh S, Saavedra-Avila NA, Tiwari S, Porcelli SA. A century of BCG vaccination: immune mechanisms, animal models, non-traditional routes and implications for COVID-19. Front Immunol. (2022) 13:959656. doi: 10.3389/fimmu.2022.959656
26. Smith CM, Proulx MK, Lai R, Kiritsy MC, Bell TA, Hock P, et al. Functionally overlapping variants control tuberculosis susceptibility in collaborative cross mice. MBio. (2019) 10:e02791. doi: 10.1128/mBio.02791-19
27. Noll KE, Ferris MT, Heise MT. The collaborative cross: a systems genetics resource for studying host-pathogen interactions. Cell Host Microbe. (2019) 25:484–98. doi: 10.1016/j.chom.2019.03.009
28. Smith CM, Baker RE, Proulx MK, Mishra BB, Long JE, Park SW, et al. Host-pathogen genetic interactions underlie tuberculosis susceptibility in genetically diverse mice. eLife. (2022) 11:e74419. doi: 10.7554/eLife.74419
29. Smith CM, Proulx MK, Olive AJ, Laddy D, Mishra BB, Moss C, et al. Tuberculosis susceptibility and vaccine protection are independently controlled by host genotype. MBio. (2016) 7:e01516. doi: 10.1128/mBio.01516-16
30. Turner OC, Keefe RG, Sugawara I, Yamada H, Orme IM. SWR mice are highly susceptible to pulmonary infection with Mycobacterium tuberculosis. Infect Immun. (2003) 71:5266–72. doi: 10.1128/IAI.71.9.5266-5272.2003
31. Medina E, North RJ. Resistance ranking of some common inbred mouse strains to Mycobacterium tuberculosis and relationship to major histocompatibility complex haplotype and Nramp1 genotype. Immunology. (1998) 93:270–4. doi: 10.1046/j.1365-2567.1998.00419.x
32. Jeon BY, Derrick SC, Lim J, Kolibab K, Dheenadhayalan V, Yang AL, et al. Mycobacterium bovis BCG immunization induces protective immunity against nine different Mycobacterium tuberculosis strains in mice. Infect Immun. (2008) 76:5173–80. doi: 10.1128/IAI.00019-08
33. Ooi YM, Colten HR. Genetic defect in secretion of complement C5 in mice. Nature. (1979) 282:207–8. doi: 10.1038/282207a0
34. Turner OC, Sugawara A, Yamada H, Cummings B, Orme IM. Crystalloid inclusions in the cytoplasm of alveolar macrophages of the SwR/J mouse a possible cause of susceptibility to Mycobacterium tuberculosis? J Submicrosc Cytol Pathol. (2001) 33:217–9.
35. Larsen SE, Baldwin SL, Orr MT, Reese VA, Pecor T, Granger B, et al. Enhanced anti-Mycobacterium tuberculosis immunity over time with combined drug and immunotherapy treatment. Vaccines. (2018) 6:20030. doi: 10.3390/vaccines6020030
36. Harper J, Skerry C, Davis SL, Tasneen R, Weir M, Kramnik I, et al. Mouse model of necrotic tuberculosis granulomas develops hypoxic lesions. J Infect Dis. (2012) 205:595–602. doi: 10.1093/infdis/jir786
37. Driver ER, Ryan GJ, Hoff DR, Irwin SM, Basaraba RJ, Kramnik I, et al. Evaluation of a mouse model of necrotic granuloma formation using C3HeB/FeJ mice for testing of drugs against Mycobacterium tuberculosis. Antimicrob Agents Chemother. (2012) 56:3181–95. doi: 10.1128/AAC.00217-12
38. Sissons J, Yan B-S, Pichugin AV, Kirby A, Daly MJ, Kramnik I. Multigenic control of tuberculosis resistance: analysis of a QTL on mouse chromosome 7 and its synergism with sst1. Genes Immun. (2009) 10:37–46. doi: 10.1038/gene.2008.68
39. Irwin SM, Driver E, Lyon E, Schrupp C, Ryan G, Gonzalez-Juarrero M, et al. Presence of multiple lesion types with vastly different microenvironments in C3HeB/FeJ mice following aerosol infection with Mycobacterium tuberculosis. Dis Model Mech. (2015) 8:591–602. doi: 10.1242/dmm.019570
40. Walter ND, Ernest JP, Dide-Agossou C, Bauman AA, Ramey ME, Rossmassler K, et al. Lung microenvironments harbor Mycobacterium tuberculosis phenotypes with distinct treatment responses. Antimicrob Agents Chemother. (2023) 67:e0028423. doi: 10.1128/aac.00284-23
41. Skerry C, Harper J, Klunk M, Bishai WR, Jain SK. Adjunctive TNF inhibition with standard treatment enhances bacterial clearance in a murine model of necrotic TB granulomas. PLoS ONE. (2012) 7:e39680. doi: 10.1371/journal.pone.0039680
42. Henao-Tamayo M, Obregón-Henao A, Creissen E, Shanley C, Orme I, Ordway DJ. Differential Mycobacterium bovis BCG vaccine-derived efficacy in C3Heb/FeJ and C3H/HeOuJ mice exposed to a clinical strain of Mycobacterium tuberculosis. Clin Vaccine Immunol. (2015) 22:91–8. doi: 10.1128/CVI.00466-14
43. Marzo E, Vilaplana C, Tapia G, Diaz J, Garcia V, Cardona P-J. Damaging role of neutrophilic infiltration in a mouse model of progressive tuberculosis. Tuberculosis. (2014) 94:55–64. doi: 10.1016/j.tube.2013.09.004
44. Ordway DJ, Shang S, Henao-Tamayo M, Obregon-Henao A, Nold L, Caraway M, et al. Mycobacterium bovis BCG-mediated protection against W-Beijing strains of Mycobacterium tuberculosis is diminished concomitant with the emergence of regulatory T cells. Clin Vaccine Immunol. (2011) 18:1527–35. doi: 10.1128/CVI.05127-11
45. Kathamuthu GR, Moideen K, Sridhar R, Baskaran D, Babu S. Diminished frequencies of cytotoxic marker expressing T- and NK cells at the site of Mycobacterium tuberculosis infection. Front Immunol. (2020) 11:585293. doi: 10.3389/fimmu.2020.585293
46. Orr MT, Beebe EA, Hudson TE, Argilla D, Huang P-WD, Reese VA, et al. Mucosal delivery switches the response to an adjuvanted tuberculosis vaccine from systemic TH1 to tissue-resident TH17 responses without impacting the protective efficacy. Vaccine. (2015) 33:6570–8. doi: 10.1016/j.vaccine.2015.10.115
47. Larsen SE, Williams BD, Rais M, Coler RN, Baldwin SL. It takes a village: the multifaceted immune response to Mycobacterium tuberculosis infection and vaccine-induced immunity. Front Immunol. (2022) 13:840225. doi: 10.3389/fimmu.2022.840225
48. Bickett TE, McLean J, Creissen E, Izzo L, Hagan C, Izzo AJ, et al. Characterizing the BCG induced macrophage and neutrophil mechanisms for defense against Mycobacterium tuberculosis. Front Immunol. (2020) 11:1202. doi: 10.3389/fimmu.2020.01202
49. R. Monteiro-Maia, Pinho RT. Oral bacillus Calmette-Guérin vaccine against tuberculosis: why not? Mem Inst Oswaldo Cruz. (2014) 109:838–45. doi: 10.1590/0074-0276140091
50. Lagranderie M, Chavarot P, Balazuc AM, Marchal G. Immunogenicity and protective capacity of Mycobacterium bovis BCG after oral or intragastric administration in mice. Vaccine. (2000) 18:1186–95. doi: 10.1016/S0264-410X(99)00386-2
51. Hoft DF, Xia M, Zhang GL, Blazevic A, Tennant J, Kaplan C, et al. PO and ID BCG vaccination in humans induce distinct mucosal and systemic immune responses and CD4(+) T cell transcriptomal molecular signatures. Mucosal Immunol. (2018) 11:486–95. doi: 10.1038/mi.2017.67
52. Irvine EB, O'Neil A, Darrah PA, Shin S, Choudhary A, Li W, et al. Robust IgM responses following intravenous vaccination with Bacille Calmette-Guérin associate with prevention of Mycobacterium tuberculosis infection in macaques. Nat Immunol. (2021) 22:1515–23. doi: 10.1038/s41590-021-01066-1
53. Darrah PA, Zeppa JJ, Maiello P, Hackney JA, Wadsworth MH 2nd, Hughes TK, et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature. (2020) 577:95–102. doi: 10.1038/s41586-019-1817-8
54. Sharpe S, White A, Sarfas C, Sibley L, Gleeson F, McIntyre A, et al. Alternative BCG delivery strategies improve protection against Mycobacterium tuberculosis in non-human primates: protection associated with mycobacterial antigen-specific CD4 effector memory T-cell populations. Tuberculosis. (2016) 101:174–90. doi: 10.1016/j.tube.2016.09.004
55. Morrison AL, Sarfas C, Sibley L, Williams J, Mabbutt A, Dennis MJ, et al. IV BCG vaccination and aerosol BCG revaccination induce mycobacteria-responsive γδ T cells associated with protective efficacy against M. tb challenge. Vaccines. (2023) 11:1604. doi: 10.3390/vaccines11101604
56. Kurtz SL, Mittereder LR, Lehman CC, Khan H, Gould VA, Elkins KL. Intravenous BCG vaccination of diversity outbred mice results in moderately enhanced protection against challenge with Mycobacterium tuberculosis compared to intradermal vaccination. Infect Immun. (2023) 91:e0016823. doi: 10.1128/iai.00168-23
57. Singh S, Bolz M, Cornelius A, Desvignes L. Intravenous BCG driven antigen recognition in a murine tuberculosis model. Comp Immunol Microbiol Infect Dis. (2022) 87:101838. doi: 10.1016/j.cimid.2022.101838
58. Lehman CC, King HA, Pokkali S, Bucsan AN, Seder R, Roederer M, et al. Repetitive vaccination with intravenous BCG is safe and immunogenic in naïve and SIV-infected macaques. J Immunol. (2023) 210:141. doi: 10.4049/jimmunol.210.Supp.141.16
59. Khatri B, Keeble J, Dagg B, Kaveh DA, Hogarth PJ, Ho MM. Efficacy and immunogenicity of different BCG doses in BALB/c and CB6F1 mice when challenged with H37Rv or Beijing HN878. Sci Rep. (2021) 11:23308. doi: 10.1038/s41598-021-02442-5
60. Larsen SE, Erasmus JH, Reese VA, Pecor T, Archer J, Kandahar A, et al. An RNA-based vaccine platform for use against Mycobacterium tuberculosis. Vaccines. (2023) 11:130. doi: 10.3390/vaccines11010130
Keywords: BCG, Mycobacterium tuberculosis, tuberculosis, lineages, mouse strain, vaccination
Citation: Larsen SE, Williams BD, Pecor T, Reese VA, Zhu Z, Miller DE, Podell BK, Baldwin SL and Coler RN (2024) Mucosal BCG delivery provides a spectrum of protection from different Mycobacterium tuberculosis strains across susceptible and resistant mouse backgrounds. Front. Tuberc. 2:1417939. doi: 10.3389/ftubr.2024.1417939
Received: 15 April 2024; Accepted: 24 September 2024;
Published: 15 October 2024.
Edited by:
Harald Gotten Wiker, University of Bergen, NorwayReviewed by:
Tim Lahey, Health Network, University of Vermont, United StatesCopyright © 2024 Larsen, Williams, Pecor, Reese, Zhu, Miller, Podell, Baldwin and Coler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rhea N. Coler, cmhlYS5jb2xlckBTZWF0dGxlY2hpbGRyZW5zLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.