 Allison M. Schwalb1,†
Allison M. Schwalb1,† Imran Anwar1,†
Imran Anwar1,† Isabel DeLaura1,†
Isabel DeLaura1,† Joseph M. Ladowski1
Joseph M. Ladowski1 Janghoon Yoon1Rafaela Belloni1
Janghoon Yoon1Rafaela Belloni1 Mingqing Song1Carolyn Glass2Jun Wang1
Mingqing Song1Carolyn Glass2Jun Wang1 Stuart Knechtle1
Stuart Knechtle1 Jean Kwun1*
Jean Kwun1*
- 1Duke Transplant Center, Duke University School of Medicine, Durham, NC, United States
- 2Department of Pathology, Duke University School of Medicine, Durham, NC, United States
Objective: Cardiac Allograft Vasculopathy (CAV), a process of vascular damage accelerated by antibody-mediated rejection (AMR), is one of the leading causes of cardiac transplant failure. Proteasome inhibitors (PIs) are utilized to treat AMR, however PI-associated toxicity limits their therapeutic utility. Novel immunoproteasome inhibitors (IPIs) have higher specificity for immune cells and have not been investigated for AMR in cardiac transplant patients. We sought to evaluate IPI effect on AMR in a murine cardiac transplant model.
Methods: Fully MHC mismatched C57BL/6 to huCD52Tg heterotopic heart transplantations were performed. Recipients were treated with alemtuzumab (10 µg, IP) on days −2, −1, 2, and 4 and anti-CD25mAb (PC61, 100 µg, IP) on day 7 to accelerate AMR with or without IPI (ONX-0914,15 mg/kg, SQ), administered on transplant day and three times a week thereafter.
Results: Animals without IPI gradually developed post-transplant donor-specific antibody (DSA) and showed a significantly elevated DSA level compared to animals receiving IPI. (TFXM 48.86 vs. 14.17; p = 0.0291, BFXM 43.53 vs. 6.114; p = 0.0031). Accordingly, H&E staining of allograft showed reduced evidence of AMR with IPI compared to controls (P = 0.0410). Notably, increased mortality was observed in the IPI treated group.
Conclusion: This study demonstrated the ability of ONYX-0914, an IPI, to control post-transplant DSA production and the AMR development in a heart transplant model. However, IPI-resistant DSA production was also observed and increased mortality with IPI therapy raises concerns about potential toxicity. Further investigation is warranted to assess the utility and potential risk associated with the use of IPI as a post-transplant maintenance immunosuppression.
Introduction
Heart transplants provide life-saving treatment for patients with end-stage heart failure. Advances in management of heart transplantation have significantly improved overall survival in the first-year post-transplant; however, there has been little change to the risk of death after 1-year post-transplant (1). The leading cause of heart transplant failure is cardiac allograft vasculopathy (CAV), the development of intimal hyperplasia and vascular fibrosis within the vasculature of transplanted cardiac tissue (2, 3). CAV is a consequence of endothelial damage by both immune and nonimmune factors. and prevention of hyperlipidemia, hypertension, and other non-immune mediated CAV risk factors have been shown to reduce mortality in heart transplant recipients (4). Antibody-mediated rejection (AMR) has been identified as a primary immune-mediated catalyst in the development of CAV (5, 6). Furthermore, episodes of acute AMR and donor-specific antibody (DSA) production have been specifically associated with increased likelihood of CAV development (5, 7) and cardiovascular mortality (8). Preventing and managing AMR is thus paramount to improving graft survival and reducing morbidity and mortality in heart transplant recipients.
Plasma cells secreting antibodies including DSA are important therapeutic targets to address AMR. Currently, most treatment options for AMR are focused either on antibody (plasmapheresis or IVIg) or B cell removal (via rituximab) (9). Proteasome inhibitors (PIs), through their inhibition of the 20s subunit of the proteasome and accumulation of intracellular proteins, preferentially lead to the apoptosis of plasma cells and prevention of antibody production (10, 11). To this effect, PIs such as bortezomib have been utilized clinically to both prevent and treat AMR in solid organ transplant recipients (12). Unfortunately, use of PIs is limited due to their nonspecific inhibition of proteasomes in all cells, resulting in significant toxicity which limits therapeutic dosing and long-term utilization in transplant recipients (13–15).
Novel immunoproteasome inhibitors (IPIs) present an attractive alternative to PIs to manage and prevent CAV. Immunoproteasomes, the target of IPIs, are more highly expressed in cells of hematopoietic origin or those which have been exposed to inflammatory mediators such as IFN-γ and TNF-α (16, 17). IPIs' selective inhibition has been shown to reduce toxicity profiles without compromising plasma cell depletion activity in multiple myeloma (18) and pre-clinical models of solid organ transplant (19). However, the efficacy of immunoproteasome inhibition in preventing AMR has not been evaluated in cardiac transplantation models. Early studies in murine models have suggested that immunoproteasomes are up regulated during acute and chronic AMR in heart transplantation, which validates the potential utility of immunoproteasome inhibition in preventing AMR, DSA production, and CAV (20). The goal of the present study is to determine whether post-transplant IPI treatment can prevent the development of DSA, reduce AMR, and prevent CAV in a murine heart transplant model (21).
Materials and methods
Animals
Homozygous huCD52Tg (H-2k) mice originally provided by Herman Waldman (22). HuCD52Tg (H-2k) mice were bred as homozygotes and maintained at Duke Laboratory Animal Resources. C57BL/6 (H-2b) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). The mice were housed in a pathogen-free barrier facility. The study was approved by the Duke University Animal Care and Use Committee (IACUC#A055-21-03).
Heterotopic heart transplantation
At approximately 6–12 weeks of age, the C57BL/6 donor hearts were transplanted into the huCD52Tg recipients using a technique like that described previously (21, 23). The recipients were treated with 10 µg of alemtuzumab (i.p.) at days −2, −1, +2, and +4 relative to transplantation. The recipients also received 100 µg of anti-CD25 mAb (PC61, i.p.) at POD 7 to deplete T regulatory cells and accelerate AMR (24). A total of 30 mice were assigned to one of two treatment protocols: immunoproteasome inhibitor (IPI group, ONX-0914, IV, three times weekly at 15 mg/kg) and control (no injection). Mice in the IPI group were administered IPI between post-transplant days 7 and 50 (Figure 1). All animals were sacrificed at 7 weeks post-transplant.
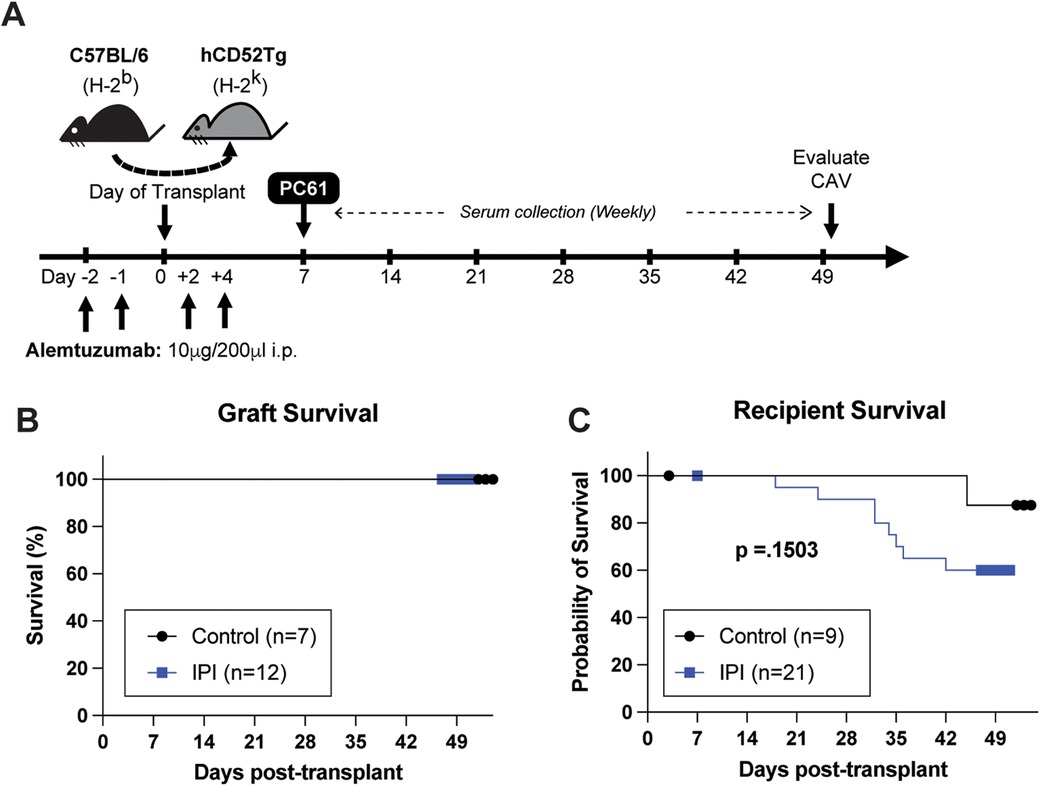
Figure 1. Schematic representation of experimental design and overall survival comparisons between IPI and control groups. (A) Visual representation of drug administration and data collection timepoints during the 7-week study period. (B) Comparison of heterotopic allograft survival in control group (black) and IPI group (blue) recipients that survived to study completion. (C) Comparison of animal survival between recipients assigned to the control and IPI groups during the study period.
DSA detection
A flow cytometry crossmatch was performed to measure DSA as described previously (21, 24). Recipient blood was obtained from the recipients via submandibular bleeding at POD 7, 14, 28, 42, and at the time of sacrifice. Donor splenocytes were prepared from C57BL/6 mice. Briefly, recipient sera were incubated with the donor splenocytes for 20 min at 4°C in the dark. The cells were thoroughly washed and 3 µl of FITC-conjugated anti-mouse Ig (polyclonal; BD biosciences) was added to the samples for a 20 min incubation. The T cells were stained with APC-conjugated anti-CD3 (Clone 145-2C11, BD biosciences) and the B cells were stained with anti-B220 mAb (RA3-6B2; BD biosciences). The samples were analyzed using a BD LSRFortessa X-20 (BD Bioscience, San Jose, CA) and anlayzed using Flow Jo v10.9.0 (Tree Star, San Carlos, CA). Alloantibody production was calculated as median fluorescence intensity fold increase over the negative control (background signal). Non-responders to IPI treatment were defined as mice that demonstrated a 15-fold rise in DSA over background control TFXM during the study period.
Histology and pathological gradings
The grafts from surviving mice were recovered 7 weeks after transplantation. The explanted grafts were bisected and fixed in 10% formalin or frozen. Sections were stained for H&E and whole stained slide were scanned with an Aperio ScanScope XT (Aperio Technologies, Inc., Vista, CA). Images were assessed by a clinical cardiac transplant pathologist (G.G.) using the ImageScope (Aperio Technologies). A determination of AMR, CAV, and acute cellular rejection (ACR) based on visualization of the graft tissue, lymphocytic infiltration, and vasculopathy. A scoring system was developed that assigned scores of either 1 (no evidence of pathology in question), 2 (cannot rule out evidence suspicious for pathology in question), 3 (strong evidence of pathology in question), or 0 (damaged tissue/insufficient tissue for analysis).
Statistics
Experimental results were analyzed by GraphPad Prism software (GraphPad Software 10.0.3, San Diego, CA). All the data are presented as mean with individual values shown in figures. And compared using a non-parametric student's t-test with significance set at p less than 0.05. Survival curves were compared in GraphPad Prism with a Mantel-Cox Log-Rank test.
Results
Post-transplant immunoproteasome inhibitors does not change graft survival but may increase recipient mortality
Human CD52Tg mice received fully MHC mismatched C57BL/6 cardiac allograft. As shown in Figure 1A, heterotopic transplant recipients received peri-transplant alemtuzumab induction, which mediates T cell depletion and promotes long-term graft survival (21). Additional treatment of anti-CD25mAb (PC61 clone) has demonstrated accelerated AMR CAV development (24). This chronic AMR model typically does not promote cessation of heterotopic cardiac allografts (or acute rejection). Similarly, IPI group with additional post-transplant IPI treatment (alemtuzumab/PC61 induction with subsequent IPI 3 times weekly) did not change the graft survival compared to control (alemtuzumab/PC61 induction without maintenance immunosuppression) (Figure 1B). However, it is notable that there were more transplant recipients in the IPI group than the control group that died prior to study completion. One animal from each group died in the first post operative week and were subsequently censored from survival analysis. Of the remaining twenty mice assigned to the IPI group, twelve (60.0%) survived to end point compared to seven of the eight (87.5%) control mice. At the end of the study period, there was no significant difference in survival between the IPI and control groups (p = 0.1503).
Post-transplant immunoproteasome inhibition suppresses post-transplant DSA production
Depletion of regulatory T cells with PC61 can accelerate development of donor-specific alloantibodies (24). As shown in Figure 2, the control group exhibited a gradual elevation of serum DSA. Interestingly, circulating DSA levels were noted to be significantly reduced in the IPI group for both TFXM (14.17 vs. 48.86-fold increase, p = 0.0291; Figure 2A) and BFXM (6.114 vs. 43.53-fold increase, p = 0.0031; Figure 2B) at study endpoint. BFXM was also significantly reduced in the IPI group at 4- and 6-weeks post-transplant (4.022 vs. 10.4, p = 0.0315 and 5.435 vs. 29.40, p = .0172, respectively; Figure 2B). Within the IPI group, five animals were considered non-responders based on DSA elevations as detailed above (Figure 2A).
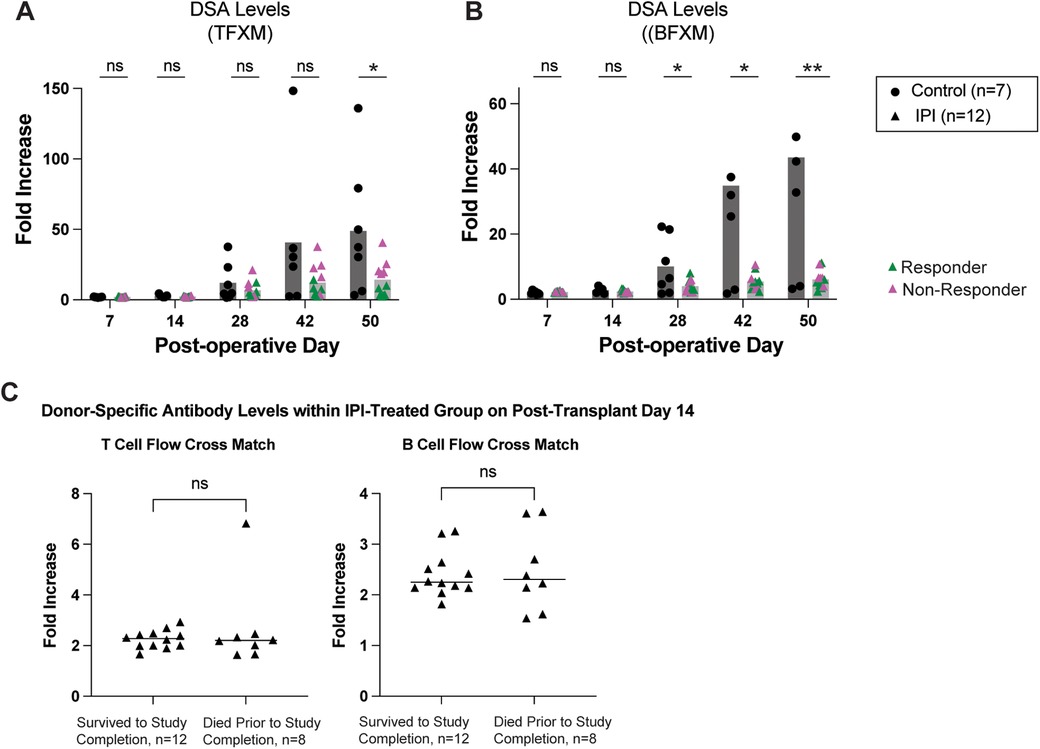
Figure 2. Survival in IPI-treated and control groups. (A) DSA production measured in TFXM. (B) BFXM throughout study period. (C) DSA levels in those that did not survive to study completion and those that died prematurely at day 14 in IPI group. Green triangles represent response to IPI treatment, pink represent nonresponse with DSA elevation. N, numbers indicate biologically independent animals; *p < 0.05; **P < 0.01 using two-tailed parametric paired t-test; NS, indicates no statistical significance.
To assess whether early mortality affected DSA production or vice versa among the 20 IPI-treated mice, we compared DSA levels between mice that died early (n = 8) and those that survived (n = 12) at post-operative day 14—the final timepoint at which sera were collected from all animals (Figure 2C). We detected no significant difference in DSA levels between the two groups as measured in a TFXM (p = 0.3110) or a BFXM (p = 0.5408).
IPI treatment is associated with reduced histologic evidence of AMR
Transplanted cardiac tissue was recovered for histological evaluation in six of seven controls and twelve of twelve IPI-treated animals surviving to study completion. Samples were deemed insufficient for CAV evaluation (score 0) in three of twelve in the IPI group and in two of six control group slides. Samples with insufficient visualization for evaluation were omitted from statistical analysis. Evidence of AMR was more prevalent in the control group than the IPI group (p = 0.0410, Figure 3A). Within the IPI group, evidence of AMR was observed in two of the five IPI-treated recipients deemed IPI-non-responders. There was no difference in incidence of ACR or CAV between control and IPI groups (P = 0.8118 and P = 0.2199, respectively) (Figures 3B,C).
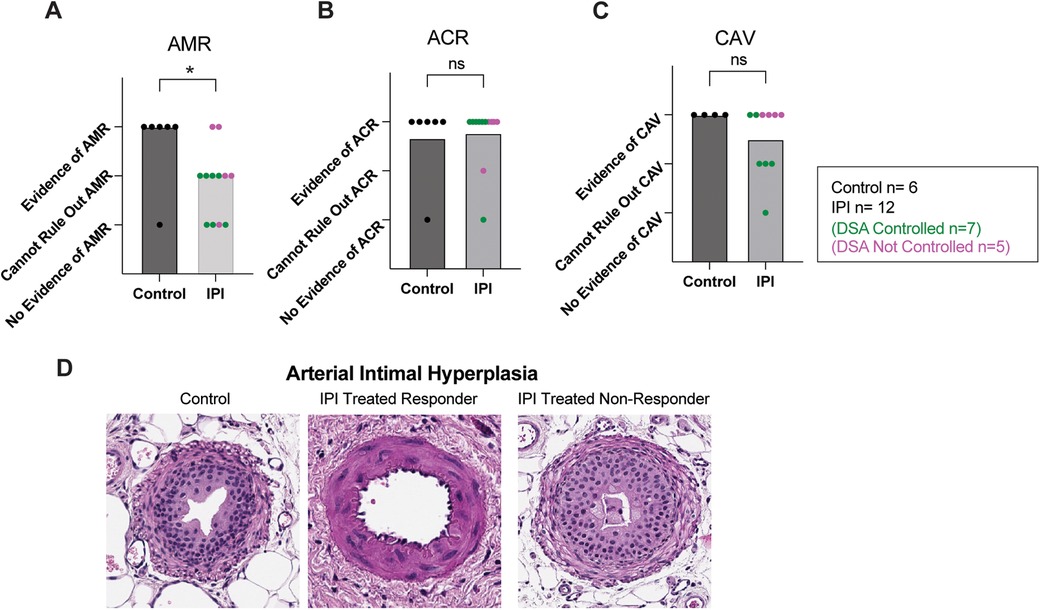
Figure 3. Histologic evidence of graft inflammation, damage, and rejection. (A) AMR scores compared between IPI and control groups, calculated as evidence of AMR = 3, evidence concerning for possible AMR = 2, and no evidence of AMR = 1. (B) ACR scores compared between IPI and control groups, calculated as evidence of ACR = 3, evidence concerning for possible ACR = 2, and no evidence of ACR = 1. (C) CAV scores compared between 9 IPI and 4 control grafts with sufficient vessels recovered for histologic evaluation, calculated as evidence of CAV = 3, evidence concerning for possible CAV = 2, and no evidence of CAV = 1. (D) Representative H&E images of arteries identified within control, IPI-treated responder, and IPI-treated non responder grafts. N, numbers indicate biologically independent animals; *p < 0.05 using two-tailed parametric paired t-test; NS, indicates no statistical significance.
Arterial Intimal hyperplasia was observed in both control and IPI-treated groups. Marked arterial hyperplasia was noted in an IPI-non-responder, while IPI-responders were found to have some non-hyperplastic vessels (Figure 3D).
Discussion
In cardiac transplantation, AMR poses a threat to allograft survival through acceleration of graft dysfunction and development of coronary vasculopathy. While the pathogenesis is multifactorial (25), the development of DSA and subsequent episodes of AMR have been shown to correlate with progression of graft vasculopathy and atherosclerosis (6). Proteasome inhibition has emerged as a prominent strategy to target AMR across the field of transplantation (9, 15, 26). PIs induce apoptosis of antibody-producing plasma cells (27), and are used clinically to reduce DSA in episodes of acute AMR in solid organ transplant patients (28, 29). However, some studies have demonstrated that proteasome inhibition is inconsistently effective in managing AMR clinically (14, 30). Additionally, while plasma cells are particularly susceptible PIs due to high protein turnover rates, proteasomes are ubiquitous organelles, and indiscriminate inhibition leads to off-target effects. Common complications of proteasome inhibitors include cardiotoxicity, nephrotoxicity, anemia, and peripheral neuropathy (31–33). IPIs specifically target subunits of immunoproteasomes in cells of hematopoietic origin and those exposed to pro-inflammatory cytokines (34) instead of targeting constitutively expressed proteasomes in other cells. This selective inhibition has been shown to reduce toxicity profiles without compromising plasma cell depletion activity in multiple myeloma (18) and rat models of kidney transplant (19), however studies have yet to explore the impact of immunoproteasome inhibition on DSA production in heart transplantation and whether attenuating AMR subsequently reduces graft vasculopathy.
We have previously shown that alemtuzumab treatment promoted long-term graft survival in hCD52Tg recipients, however, these recipients developed de novo DSA, allo-B cells, and CAV (21). We also showed that this AMR phenotype can be accelerated or reversed with regulatory T cell depletion or co-stimulation blockade treatment, respectively (35). In the present study, we showed that IPI can attenuate DSA production, and the resultant AMR compared to the control group, but its effectiveness is potentially limited by toxicity and variable treatment responsiveness. Notably, there were three distinct phenotypes observed within the IPI-treated group: IPI response and reduction of DSA, IPI non-response with significant production of DSA, and presumed toxicity-mediated death. Among IPI-treated mice, approximately one-half demonstrated persistently controlled DSA through the study endpoint. These mice had correspondingly reduced incidence of AMR on pathology. CAV was also reduced in the IPI-treated group, however the difference between IPI and control groups was not statistically significant. Notably, some IPI-treated mice did not demonstrate persistently controlled DSA. This differential response has been also observed in multiple PI studies. In cardiac transplant recipients, nonresponse to carfilzomib and bortezomib have been observed clinically (30, 36). The Borteject trial also showed that proteasome inhibition did not perform better than placebo at reducing late antibody mediated rejection in kidney transplant recipients (14). Successful clinical and preclinical studies suggest that proteasome inhibition is most effective when used in combination with other therapies (27, 37, 38). Thus, IPI therapy may be similarly limited as monotherapy in our model. In multiple myeloma, IPI has also been used to enhance efficacy of constitutive proteasome inhibitors through feedback regulation (39). This approach may be possible in the transplant population during acute AMR, but it would increase the risk of toxicity.
A primary goal of immunoproteasome inhibition is reducing toxicity associated with proteasome inhibition through enhanced specificity. Multiple studies have reported improvements in toxicity and adverse effect profiles in vivo and in vitro with IPI compared to PI therapy (10, 20, 32). Recently, IPI demonstrated appropriate safety and tolerability in stage 2 clinical trials for dermatomyositis, despite several participants experiencing significant toxicity (40). Our study found that IPI treatment resulted in a high mortality rate. The premature deaths may be mediated by toxicity, as heterotopic heart transplants are nonfunctional, meaning graft rejection or failure would not affect overall survival. However, the possibility of human error due to more frequent handling cannot be ruled out, as control group animals did not receive placebo injections. The different level of tolerability to IPI may be due to varying levels of immunoproteasome expression in some animals. Post-transplant stress and inflammation could lead to widespread upregulation of immunoproteasomes in the setting of increased secretion of interferon gamma and other inflammatory cytokines (34), which could increase IPI toxicity. The observed heterogenous response to IPI has also been shown in cancer studies as well. Differential expression of proteasome subunits, increased expression of chaperone proteins, and elevated antioxidant concentrations have been shown to be protective against PI-mediated apoptosis (41, 42). Rapid proteasome adaptation (43) may contribute to the unresponsiveness to IPI. Alternating conventional proteasome inhibitors with IPI could help prevent proteasome adaptation and promote more effective plasma cell depletion. Future studies could investigate whether similar signatures predict IPI response in transplant recipients, and whether these could be tested for and utilized clinically.
This study has several limitations. A significant limitation was the unexpected high mortality in the treatment group during the study period. This could skew the recipient demographic, limiting the data to surviving animals. The causes of death in the treatment group were not clarified. Future studies should investigate potential toxicity and mortality and optimize dosing to minimize toxicity while maintaining the efficacy of IPI. Evaluation of CAV, neointimal hyperplasia, and their correlation to AMR was also limited by additional tissue damage. Inflammatory vascular damage in allografts develops through many pathways and is observed clinically with and without AMR (6, 36). While vasculopathy was observed in both IPI and control group grafts, ACR, stress, and inflammation may have contributed to observed inflammation, which may be mitigated with appropriate combination of IPI with other immunomodulatory therapies in the post-transplant period. Additionally, the impact of IPI on immune cell populations has not been fully elucidated. Therefore, future studies should also investigate how prolonged IPI therapy modulates B and T cell subpopulations to better understand its effects on immune responses and antibody production.
In our chronic AMR model, prolonged post-transplant IPI treatment reduced DSA production and AMR development, but it was associated potential toxicity and variable efficacy. The introduction of IPI as a more selective alternative capable of reducing humoral rejection, while expected to reduce toxicity compared to PI, may still cause significant off-target effects. Further characterization of IPI in large animal models, including nonhuman primates, will enhance our understanding of its safety and efficacy. This will improve the utility of IPI in transplantation settings and support its advancement toward clinical application.
Data availability statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Duke University Institutional Animal Care and Use Committee (IACUC). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
AS: Data curation, Formal Analysis, Writing – original draft, Writing – review & editing. IA: Data curation, Formal Analysis, Investigation, Writing – original draft. ID: Data curation, Formal Analysis, Investigation, Writing – original draft. JL: Data curation, Investigation, Writing – review & editing. JY: Investigation, Methodology, Writing – review & editing. RB: Investigation, Methodology, Writing – review & editing. MS: Investigation, Methodology, Writing – review & editing. CG: Methodology, Validation, Writing – review & editing. JW: Writing – review & editing. SK: Investigation, Writing – review & editing. JK: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by American Heart Association (AHA)/Enduring Heart Foundation Research Award (15SDG25710165 to JK), NIH T32 (T32AI141342, to JA) and the ASTS Student Mentorship Grants (to AS and ID).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ACR, Acute cellular rejection; AMR, Antibody-mediated rejection; BFXM, B cell flow crossmatch; CAV, cardiac allograft vasculopathy; DSA, Donor specific antibody; H&E, Hematoxylin and Eosin; IFN-γ, interferon gamma; IPI, immunoproteasome inhibitor; mAb, monoclonal antibody; PI, proteasome inhibitor; TFXM, T cell flow crossmatch; TNF-α, Tumor Necrosis Factor Alpha.
References
1. Wilhelm MJ. Long-term outcome following heart transplantation: current perspective. J Thorac Dis. (2015) 7(3):549–51. doi: 10.3978/j.issn.2072-1439.2015.01.46
2. Ramzy D, Rao V, Brahm J, Miriuka S, Delgado D, Ross HJ. Cardiac allograft vasculopathy: a review. Can J Surg. (2005) 48(4):319–27.16149368
3. Khush KK, Cherikh WS, Chambers DC, Harhay MO, Hayes D Jr., Hsich E, et al. The international thoracic organ transplant registry of the international society for heart and lung transplantation: thirty-sixth adult heart transplantation report—2019; focus theme: donor and recipient size match. J Heart Lung Transplant. (2019) 38(10):1056–66. doi: 10.1016/j.healun.2019.08.004
4. Mehra MR, Raval NY. Metaanalysis of statins and survival in de novo cardiac transplantation. Transplant Proc. (2004) 36(5):1539–41. doi: 10.1016/j.transproceed.2004.05.036
5. Clerkin KJ, Restaino SW, Zorn E, Vasilescu ER, Marboe CC, Mancini DM. The effect of timing and graft dysfunction on survival and cardiac allograft vasculopathy in antibody-mediated rejection. J Heart Lung Transplant. (2016) 35(9):1059–66. doi: 10.1016/j.healun.2016.04.007
6. Loupy A, Toquet C, Rouvier P, Beuscart T, Bories MC, Varnous S, et al. Late failing heart allografts: pathology of cardiac allograft vasculopathy and association with antibody-mediated rejection. Am J Transplant. (2016) 16(1):111–20. doi: 10.1111/ajt.13529
7. Russell ME, Fujita M, Masek MA, Rowan RA, Billingham ME. Cardiac graft vascular disease. Nonselective involvement of large and small vessels. Transplantation. (1993) 56(6):1599–601.8279051
8. Kfoury AG, Hammond ME, Snow GL, Drakos SG, Stehlik J, Fisher PW, et al. Cardiovascular mortality among heart transplant recipients with asymptomatic antibody-mediated or stable mixed cellular and antibody-mediated rejection. J Heart Lung Transplant. (2009) 28(8):781–4. doi: 10.1016/j.healun.2009.04.035
9. Nguyen VP, Kobashigawa JA. Antibody-medicated rejection after heart transplantation: diagnosis and clinical implications. Curr Opin Organ Transplant. (2020) 25(3):248–54. doi: 10.1097/MOT.0000000000000754
10. Neubert K, Meister S, Moser K, Weisel F, Maseda D, Amann K, et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med. (2008) 14(7):748–55. doi: 10.1038/nm1763
11. Kubiczkova L, Pour L, Sedlarikova L, Hajek R, Sevcikova S. Proteasome inhibitors—molecular basis and current perspectives in multiple myeloma. J Cell Mol Med. (2014) 18(6):947–61. doi: 10.1111/jcmm.12279
12. Field-Smith A, Morgan GJ, Davies FE. Bortezomib (velcadetrade mark) in the treatment of multiple myeloma. Ther Clin Risk Manag. (2006) 2(3):271–9. doi: 10.2147/tcrm.2006.2.3.271
13. Li J, Basler M, Alvarez G, Brunner T, Kirk CJ, Groettrup M. Immunoproteasome inhibition prevents chronic antibody-mediated allograft rejection in renal transplantation. Kidney Int. (2018) 93(3):670–80. doi: 10.1016/j.kint.2017.09.023
14. Eskandary F, Regele H, Baumann L, Bond G, Kozakowski N, Wahrmann M, et al. A randomized trial of bortezomib in late antibody-mediated kidney transplant rejection. J Am Soc Nephrol. (2018) 29(2):591–605. doi: 10.1681/ASN.2017070818
15. Everly JJ, Walsh RC, Alloway RR, Woodle ES. Proteasome inhibition for antibody-mediated rejection. Curr Opin Organ Transplant. (2009) 14(6):662–6. doi: 10.1097/MOT.0b013e328330f304
16. McCarthy MK, Weinberg JB. The immunoproteasome and viral infection: a complex regulator of inflammation. Front Microbiol. (2015) 6:21. doi: 10.3389/fmicb.2015.00021
17. Aki M, Shimbara N, Takashina M, Akiyama K, Kagawa S, Tamura T, et al. Interferon-gamma induces different subunit organizations and functional diversity of proteasomes. J Biochem. (1994) 115(2):257–69. doi: 10.1093/oxfordjournals.jbchem.a124327
18. Nair N. Vascular rejection in cardiac allograft vasculopathy: impact on graft survival. Front Cardiovasc Med. (2022) 9:919036. doi: 10.3389/fcvm.2022.919036
19. Li J, Koerner J, Basler M, Brunner T, Kirk CJ, Groettrup M. Immunoproteasome inhibition induces plasma cell apoptosis and preserves kidney allografts by activating the unfolded protein response and suppressing plasma cell survival factors. Kidney Int. (2019) 95(3):611–23. doi: 10.1016/j.kint.2018.10.022
20. Karreci ES, Fan H, Uehara M, Mihali AB, Singh PK, Kurdi AT, et al. Brief treatment with a highly selective immunoproteasome inhibitor promotes long-term cardiac allograft acceptance in mice. Proc Natl Acad Sci U S A. (2016) 113(52):E8425–32. doi: 10.1073/pnas.1618548114
21. Kwun J, Oh BC, Gibby AC, Ruhil R, Lu VT, Kim DW, et al. Patterns of de novo allo B cells and antibody formation in chronic cardiac allograft rejection after alemtuzumab treatment. Am J Transplant. (2012) 12(10):2641–51. doi: 10.1111/j.1600-6143.2012.04181.x
22. Gilliland LK, Walsh LA, Frewin MR, Wise MP, Tone M, Hale G, et al. Elimination of the immunogenicity of therapeutic antibodies. J Immunol. (1999) 162(6):3663–71. doi: 10.4049/jimmunol.162.6.3663
23. Kwun J, Hu H, Schadde E, Roenneburg D, Sullivan KA, DeMartino J, et al. Altered distribution of H60 minor H antigen-specific CD8T cells and attenuated chronic vasculopathy in minor histocompatibility antigen mismatched heart transplantation in Cxcr3-/- mouse recipients. J Immunol. (2007) 179(12):8016–25. doi: 10.4049/jimmunol.179.12.8016
24. Oh B, Yoon J, Farris A 3rd, Kirk A, Knechtle S, Kwun J. Rapamycin interferes with postdepletion regulatory T cell homeostasis and enhances DSA formation corrected by CTLA4-ig. Am J Transplant. (2016) 16(9):2612–23. doi: 10.1111/ajt.13789
25. Kwun J, Knechtle SJ. Overcoming chronic rejection-can it B? Transplantation. (2009) 88(8):955–61. doi: 10.1097/TP.0b013e3181b96646
26. Basler M, Li J, Groettrup M. On the role of the immunoproteasome in transplant rejection. Immunogenetics. (2019) 71(3):263–71. doi: 10.1007/s00251-018-1084-0
27. Woodle ES, Walsh RC, Alloway RR, Girnita A, Brailey P. Proteasome inhibitor therapy for antibody-mediated rejection. Pediatr Transplant. (2011) 15(6):548–56. doi: 10.1111/j.1399-3046.2011.01543.x
28. Velleca A, Shullo MA, Dhital K, Azeka E, Colvin M, DePasquale E, et al. The international society for heart and lung transplantation (ISHLT) guidelines for the care of heart transplant recipients. J Heart Lung Transplant. (2023) 42(5):e1–141. doi: 10.1016/j.healun.2022.10.015
29. Otero J, Albers E, Friedland-Little J, Hong B, Kemna M, Gimferrer I, et al. Use of bortezomib in the treatment of antibody mediated rejection (AMR) in pediatric heart recipients. J Heart Lung Transplant. (2019) 38(4):S468–9. doi: 10.1016/j.healun.2019.01.1192
30. Horn ET, Xu Q, Dibridge JN, Huston JH, Hickey GW, Kaczorowski DJ, et al. Reduction of HLA donor specific antibodies in heart transplant patients treated with proteasome inhibitors for antibody mediated rejection. Clin Transplant. (2023) 37(12):e15132. doi: 10.1111/ctr.15132
31. Fotiou D, Roussou M, Gakiopoulou C, Psimenou E, Gavriatopoulou M, Migkou M, et al. Carfilzomib-associated renal toxicity is common and unpredictable: a comprehensive analysis of 114 multiple myeloma patients. Blood Cancer J. (2020) 10(11):109. doi: 10.1038/s41408-020-00381-4
32. von Brzezinski L, Saring P, Landgraf P, Cammann C, Seifert U, Dieterich DC. Low neurotoxicity of ONX-0914 supports the idea of specific immunoproteasome inhibition as a Side-effect-limiting. Therapeutic Strategy. Eur J Microbiol Immunol (Bp). (2017) 7(3):234–45. doi: 10.1556/1886.2017.00025
33. Lataifeh AR, Nusair A. Fatal pulmonary toxicity due to carfilzomib (kyprolis). J Oncol Pharm Pract. (2016) 22(5):720–4. doi: 10.1177/1078155215588630
34. Tubio-Santamaria N, Ebstein F, Heidel FH, Kruger E. Immunoproteasome function in normal and malignant hematopoiesis. Cells. (2021) 10(7). doi: 10.3390/cells10071577
35. Kwun J, Park J, Yi JS, Farris AB, Kirk AD, Knechtle SJ. IL-21 biased alemtuzumab induced chronic antibody-mediated rejection is reversed by LFA-1 costimulation blockade. Front Immunol. (2018) 9:2323. doi: 10.3389/fimmu.2018.02323
36. Horn ET, Xu Q, Tushak Z, Binko M, Dibridge JN, Huston JH, et al. Significant reduction of donor specific antibodies in heart transplant recipients treated with proteasome inhibitors for antibody mediated rejection. J Heart Lung Transplant. (2022) 41(4):S410–1. doi: 10.1016/j.healun.2022.01.1033
37. Walsh RC, Alloway RR, Girnita AL, Woodle ES. Proteasome inhibitor-based therapy for antibody-mediated rejection. Kidney Int. (2012) 81(11):1067–74. doi: 10.1038/ki.2011.502
38. Kwun J, Burghuber C, Manook M, Ezekian B, Park J, Yoon J, et al. Successful desensitization with proteasome inhibition and costimulation blockade in sensitized nonhuman primates. Blood Adv. (2017) 1(24):2115–9. doi: 10.1182/bloodadvances.2017010991
39. Kegyes D, Gulei D, Drula R, Cenariu D, Tigu B, Dima D, et al. Proteasome inhibition in combination with immunotherapies: state-of-the-art in multiple myeloma. Blood Rev. (2023) 61:101100. doi: 10.1016/j.blre.2023.101100
40. Aggarwal R, Goyal N, Lam D, Ng A, Leff R, Ray K, et al. Zetomipzomib demonstrates favorable long-term safety and tolerability profile without signs of immunosuppression: results from the PRESIDIO study and its open-label extension study in patients with dermatomyositis and polymyositis. American College of Rheumatology Convergence (2023).
41. Mitra AK, Harding T, Mukherjee UK, Jang JS, Li Y, HongZheng R, et al. A gene expression signature distinguishes innate response and resistance to proteasome inhibitors in multiple myeloma. Blood Cancer J. (2017) 7(6):e581. doi: 10.1038/bcj.2017.56
42. McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat. (2008) 11(4–5):164–79. doi: 10.1016/j.drup.2008.08.002
Keywords: immunoproteasome, antibody-mediated rejection, CAV, regulatory T cells, alloantibody
Citation: Schwalb AM, Anwar I, DeLaura I, Ladowski JM, Yoon J, Belloni R, Song M, Glass C, Wang J, Knechtle S and Kwun J (2024) Immunoproteasome inhibition reduces donor specific antibody production and cardiac allograft vasculopathy in a mouse heart transplantation model. Front. Transplant. 3:1494455. doi: 10.3389/frtra.2024.1494455
Received: 10 September 2024; Accepted: 2 December 2024;
Published: 16 December 2024.
Edited by:
Rita Leal, Coimbra Hospital and University Center, PortugalReviewed by:
Paolo De Simone, University of Pisa, ItalyClauda Gidea, Newark Beth Israel Medical Center, United States
Copyright: © 2024 Schwalb, Anwar, DeLaura, Ladowski, Yoon, Belloni, Song, Glass, Wang, Knechtle and Kwun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean Kwun, amVhbi5rd3VuQGR1a2UuZWR1
†These authors have contributed equally to this work