 Jennifer Malcolm
Jennifer Malcolm Shauna Culshaw
Shauna Culshaw- Oral Sciences, University of Glasgow Dental School, School of Medicine, Dentistry and Nursing, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, United Kingdom
There are well established epidemiological links between rheumatoid arthritis and periodontitis. Recent data have started to shed light on the mechanisms that might underlie the relationship between these two complex diseases. Unravelling the roles of distinct pathways involved in these mechanisms has the potential to yield novel preventative and therapeutic strategies for both diseases. Perhaps most intriguingly, this represents an area where understanding the biology in the oral cavity might reveal fundamental advances in understanding immune regulation and the relationships between the host and microbiome. Here we seek to discuss aspects of the adaptive immune response that might link periodontitis and rheumatoid arthritis.
Introduction
Autoimmune diseases predominantly develop in individuals with genetic susceptibility after exposure to environmental factors; suggesting gene:environment interactions are necessary to drive loss of immune tolerance, and emergence of pathological adaptive autoimmune responses to host tissues. However, it is increasingly clear that autoreactive CD4+ T cell and autoantibody responses can develop consequent to chronic inflammatory or metabolic disturbances, even in the absence of defined genetic susceptibility (1–3). Further, evidence is emerging that environmental exposures and environment: gene interactions driving autoimmunity may be temporally and functionally distinct (4–6). This raises the possibility that adaptive autoreactivity emerges consequent to environmental exposures as a component of chronic inflammation; and favours the progression to autoimmune disease only in genetically susceptible individuals.
Rheumatoid arthritis (RA) is a joint destructive autoimmune disease associated with adaptive immune responses towards post-translationally modified self-proteins. This autoreactivity can be readily detected as autoantibodies reactive with self-proteins containing citrulline modifications, known as ACPA. Importantly, ACPA can emerge asymptomatically in patient sera many years before the onset of joint inflammation, and strongly associate with chronic inflammatory insults at mucosal surfaces, such as smoking and periodontitis (5, 7). In people expressing human leukocyte antigen (HLA) class II alleles containing an amino acid motif known as the shared epitope [HLA-SE, reviewed (8)], this initial ACPA response can evolve and mature; a process associated with the transition to symptomatic joint inflammation. This is supported by recent data revealing that HLA-SE alleles are required for the transition from ACPA positivity to ACPA positive RA (as is widely used in the literature, in the following discussion, ACPA positivity/ACPA positive RA refers to seropositivity) (5, 6). A corollary to these data is that the pathways predisposing to ACPA positivity can occur independently of HLA-SE risk alleles following exposure to relevant environmental insults, and only predispose to autoimmune disease in genetically susceptible individuals. This distinction suggests that autoreactivity secondary to chronic inflammation or infection precipitates autoimmune responses that may either progress, or not progress, to autoimmune disease based on genetic susceptibility.
Here we review recent developments related to the impact of gene: environment interactions in the pathogenesis of rheumatoid arthritis. Using these new developments as a framework, we discuss the implications for understanding mucosal inflammatory disease and risk for RA, using periodontitis as an exemplar. Finally, we review data derived from recent mechanistic studies that offer clues and new hypotheses to determine how aberrant inflammation in the oral cavity predisposes to the development of RA.
The distinction between ACPA positivity and ACPA positive RA
Retrospective serological studies of patients with RA reveal the presence of ACPA many years before the onset of clinically evident joint inflammation (4, 9, 10). Smoking is the strongest environmental risk factor for ACPA positivity and progression to ACPA positive RA (11). In a large Swedish twin study, smoking was associated with ACPA positivity, but APCA positive RA was associated with smoking only in individuals expressing HLA-SE (HLA-shared epitope) risk alleles (12). This observation that smoking, not HLA-SE, confers risk for ACPA positivity, while smoking in an HLA-SE background confers risk for ACPA positive RA was replicated in a meta-analysis (6). Subsequent studies have revealed that ACPA's in ACPA positive RA show somatic hypermutations, as measured by variable-domain glycosylation. This ACAP maturation is dependent on HLA-SE (13). Glycosylation of IgG ACPA is largely absent from ACPA positive individuals who do not progress to RA (14). Notably, somatic hypermutation is a T-cell dependent process—T cells help B cells to generate an array of antibody variants, with selection for those antibodies with highest affinity. Therefore, HLA-SE-restricted antigen-presentation to CD4+ T cells is implicated in the transition from ACPA positivity to ACPA positive RA (5, 13).
Mucosal origins of ACPA
Identification of smoking as the predominant environmental risk factor for ACPA positive RA, together with observations that ACPA emerge in patients sera up to ten years before the development of joint inflammation, have led to the hypothesis that ACPA are triggered at mucosal surfaces subject to chronic inflammatory insults (15, 16). In support of this mucosal origin hypothesis is the finding that diverse mucosal inflammatory diseases are associated with elevated ACPA positivity compared with healthy individuals (Table 1). How ACPA are triggered, and the biological function of ACPA at mucosal surfaces remain unclear. It is possible that ACPA could represent a biomarker for shared inflammatory pathways occurring at distinct mucosal surfaces, in response to diverse environmental exposures.
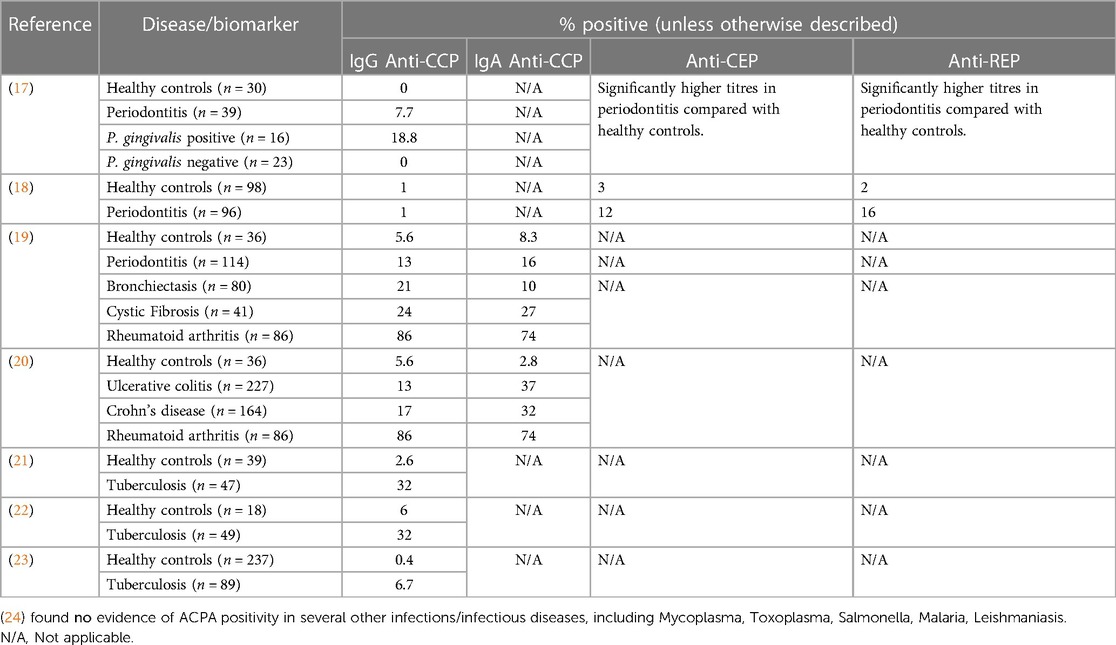
Table 1. Association of ACPA positivity with mucosal exposures in patients without RA.
ACPA positivity in individuals without arthritis is associated with the presence of mucosal inflammatory exposures, including smoking, but also exposure to dysbiotic microbial communities in the gingival and intestinal tissues (Table 1). This suggests that the disease processes that give rise to ACPA, and subsequently predispose to the development of ACPA positive RA in genetically susceptible individuals are active at mucosal surfaces subject to chronic inflammatory insults. In both periodontitis and inflammatory bowel diseases, the homeostatic relationship between host-tissues and the resident polymicrobial communities in health is lost, leading to immune-mediated tissue damage driven by dysregulated host immune responses to dysbiotic polymicrobial communities. Thus, in both diseases, environment and/or genetic factors change the interaction between host-tissues and resident polymicrobial communities, leading to a break down in mucosal barrier function. It has been posited that the mucosal IgA ACPA response may have a protective role in binding and neutralising citrullinated proteins released during NETosis, and that systemic IgG ACPA responses develop consequent to breached mucosal barrier function (15). In support of this hypothesis, it was recently demonstrated that people with both RA and periodontitis can suffer repeated breaches in mucosal barrier function, leading to oral bacteraemia's and changes in circulating monocyte populations. Further, the authors revealed that resident bacteria in the mouth can be externally citrullinated (presumed to be the result of NETosis) and that hypermutated ACPA derived from RA plasmablasts can bind citrullinated oral bacteria (25).
Oral mucosal origins of ACPA
As indicated above, the gingival tissues are a site of potential ACPA induction. Serological studies of patients with periodontitis reveal small, but measurable, serum ACPA (Table 1). Whilst the autoantibody profile in patients with periodontitis is variable, and the titres are relatively low, the disease itself is common, and the autoreactivity is greater than observed in healthy controls. Autoantibodies against both citrullinated (anti-CCP) and native forms of proteins (specific for both host and microbial) have been observed. As such, there is evidence of an adaptive immune response to microbial antigens and antigens that share homology with human proteins, such as α-enolase, and host proteins such as vimentin and fibrinogen (26). It is noteworthy that only very few infectious diseases appear to result in anti-CCP immunity. Auto-antibodies against both native and citrullinated peptides have been documented in patients with TB (with reports varying between around 6% of patients up to 37% of patients). There are small studies reporting anti-citrullinated peptide immunity in leishmaniasis, and some in hepatitis C, Lyme disease, Chagas disease and Yersinia infections. Interestingly, these anti-citrullinated peptide antibodies were not found in malaria, syphilis, salmonella, chlamydia, legionella, streptococcus pyogenes, nor infectious endocarditis. It should be noted that these studies used different assays and so absolute comparisons are challenging. Nonetheless, not all infection appear to carry equal risk for ACPA positivity.
Some studies point to a relationship between ACPA-positive RA and periodontitis associated bacteria (27); other studies show associations specifically with Porphyromonas gingivalis (28–30). P. gingivalis is associated with periodontitis and described as a keystone pathogen capable of orchestrating microbial dysbiosis (31). P. gingivalis expresses a P. gingivalis Peptidyl Arginine Deiminase, (PPAD) that converts arginine residues to citrulline on both microbial and host proteins. Together with the combined expression of P. gingivalis' arginine gingipain, a protease capable of cleaving proteins at arginine peptide bonds, PPAD can generate non-endogenous C-terminal citrullinated peptides. The candidate autoantigens human fibrinogen and α-enolase were proteolytically cleaved and citrullinated following incubation with P. gingivalis (Wegner, 2010). There is a connection between P. gingivalis-mediated citrullination, inflammation in the gingivae and the subsequent generation of ACPA (28–30).
Whether anti-citrullinated autoimmunity can be triggered in the periodontal tissues and progress to joint inflammation consequent to failed mucosal compartmentalisation and evolution of the ACPA response remains to be determined.
Antigen-processing and presentation
Data demonstrating that the periodontal tissues replicate the citrullinome of the arthritic joint (32), that oral bacteria are highly citrullinated (25), and the hypothesis that ACPA might evolve to bind citrullinated proteins at mucosal surfaces are intriguing (15), especially when considered in light of a recent study revealing that citrullination alters antigen-processing, leading to presentation of cryptic epitopes recognised by CD4+ T cells from patients with rheumatoid arthritis (33).
Previous experimental studies have revealed that T cells reactive with immunodominant self-epitopes are rendered tolerant, while T cells potentially reactive with immunorecessive or “cryptic” self-epitopes can escape tolerance (34, 35). During normal physiological self-antigen processing, cryptic epitopes are defined as those that are normally hidden from T cell immunosurveillance because they are not available to bind (or cannot bind) MHC complexes. However, under inflammatory conditions, modification of self-proteins, for example via post-translational modification, can change how self-proteins are processed by altering proteolytic cleavage, leading to the generation of new epitopes, or modified-epitopes with altered affinity for MHC complexes (36). Indeed, oxidative modifications introduced to the host cell proteome during metabolic stress are linked with changes in MHCII antigen-processing (2).
The study by Curran et al, reveal that citrullination of host proteins alters their antigenic processing giving rise to an alternative set of MHC epitopes compared with epitopes presented following processing of unmodified proteins. Since cryptic self-epitopes can have homology with microbial epitopes (37), and T cells reactive with cryptic self-epitopes can be engaged by immunisation with foreign antigen (38), it is conceivable that altered-antigen presentation following processing of modified proteins could underlie hypotheses relevant to bystander activation and molecular mimicry.
Notably, while some cryptic epitopes are implicated in the induction and propagation of pathogenic autoimmunity, others can help to down-regulate pathogenic autoimmune responses (36). In a model of adjuvant-induced arthritis (AA) in the Lewis rat, diversification of the T cell response to cryptic epitopes from mycobacterial 65 kDa heat-shock protein (Bhsp65), which are cross-reactive with a rat heat-shock protein, alleviate the course of AA (39). Furthermore, immunisation of Lewis rats with cryptic peptides derived from Bhsp65, protected mice from induction of AA following immunisation with Mycobacterium tuberculosis H37Ra (37). These data demonstrate that adaptive autoimmune responses can be engaged in both the propagation and regulation of autoimmune disease depending on the nature of the epitopes presented to autoreactive CD4+ T cells. Importantly, while in the Lewis rat induction of AA induces both arthritogenic and regulatory autoimmune responses, in a related strain of rat (Fischer 344) the course of AA can be modulated solely by exposure to environmental factors. Fischer rats raised in a barrier-facility are susceptible to AA, in contrast Fischer rats raised in a conventional facility had a reduced incidence of AA. Notably, naïve Fischer rats raised in the conventional facility demonstrated evidence of prior T cell activation towards regulatory cryptic epitopes derived from mycobacterial Bhsp65. This was attributed to spontaneous priming of T cells against regulatory epitopes of Bhsp65 from molecular homologues derived from microbes present in the conventional facility (40). These data demonstrate that environmental exposures can modulate the course and severity of autoimmune disease by determining whether the outcome of adaptive self-recognition is immunopathogenic or immunoregulatory.
Functional consequences of antigen modification
Post-translational modification (PTM) poses a danger for the development of autoimmunity by changing how proteins are processed for antigen-presentation, potentially giving rise to cryptic (33) and/or modified-epitopes (2, 3) recognised by autoreactive T cells. Whereas cryptic epitopes are anticipated to be recognised as “non-self” and to recruit a different T cell population compared with T cells recruited by dominant self-epitopes, PTM-modified epitopes can be recognised as non-self (2, 3), or as we have shown, can change the interaction between antigen-presenting cells and antigen-specific CD4+ T cells, leading to altered functional responses (41).
CD4+ T cells from OTII mice express a transgenic T cell receptor specific for ovalbumin peptide323–339 (pOVA) and are useful for studying the interaction between antigen-presenting cells and functional outcomes in CD4+ T cells. Using this system, w modified the C-terminal arginine of pOVA to generate C-terminal citrullinated pOVA (pOVA-cit) to show that pOVA-cit changed the interaction between antigen-presenting cells and OTII T cells, leading to changes in T cell function in the context of immune-priming and immune-tolerance. Importantly, we demonstrated that OTII T cells responding to pOVA-cit were less dependent on co-stimulatory checkpoints for robust effector responses, compared with the OTII response to native pOVA. Further, using an oral tolerance model, we demonstrated that immunisation with pOVA-cit was sufficient to breach immune tolerance to native ovalbumin in vivo. This proof-of-concept study reveals that non-endogenous C-terminal citrullination can change the way CD4+ T cells “see” and respond to MHCII antigens.
The OTII TCR recognises a 9 amino acid core epitope (329–337) of pOVA (42). Therefore, the citrullinated residue (339) of pOVA: cit is predicted to be in the peptide-flanking region, and available to interact with the OTII TCR. Modification of flanking regions of MHCII peptides can determine CD4+ T cell functional outcomes, by selecting different populations of CD4+ T cells, or as predicted for pOVA: cit, by modifying the affinity of MHCII: peptide interactions with cognate TCRs (43, 44). Ultimately, whether endogenous or non-endogenous citrullination favours T cell autoimmunity will be dependent upon the T cell repertoire selected in the thymus, and at the site of inflammation.
Discussion
The epidemiological relationship between periodontitis and RA well-established (45), and there is good evidence that periodontitis and infection with P. gingivalis are risk factors for ACPA positivity, at least in some patients. However, it remains to be determined whether ACPA positivity consequent to periodontitis, or P. gingivalis exposure favours transition to ACPA positive RA—and if so, whether this is based on genetic susceptibility, or other factors. Periodontitis is highly prevalent and a significant challenge for affected individuals, and its socioeconomic costs are substantial (46). Understanding the aberrant immune response in periodontitis has potential to improve treatment and prevention. Moreover, we propose that periodontitis could represent a platform to determine the causal pathways underlying the emergence and maturation of ACPA responses predisposing to RA. Broadly, there is an opportunity to understand fundamental and causal pathways of autoimmunity, autoimmune pre-disease, and perhaps the clinically manifest autoimmune disease. There is a significant clinical unmet need for this information. Patients with Rheumatoid arthritis (RA) have benefitted from transformative advances in therapies. RA disease remission (or relatively low disease activity) is achieved for more patients than ever. However, advanced therapies are expensive, can have significant side effects, and are not effective in every patient. Partial response and non-response still represent an unmet need, and therapy is usually lifelong therapy (47). The goal of drug-free remission remains elusive. RA still inflicts significant burdens on individuals and society. Annual direct costs (e.g., medications, medical and other care, adaptations) and indirect costs (e.g., lost productivity) are estimated at USD10,000–30,000 per patient, with some estimates up USD83,000 (48). Of patients treated with anti-TNF over 6 years who were consistently defined as “in remission” 57% of these patients report some compromised physical function (49). A study of 640 patients over 8 years found that 20% of RA patients who were defined as in remission reported Health Assessment Questionnaire (HAQ) scores > 1; indicating moderate to severe disability (50). The HAQ scores were significantly higher in patients with progressing RA who did not achieve remission. However, these and other studies (51) show that a sizable proportion of patients considered to achieve “good” outcomes still suffer. Understanding the biology in the mouth may offer transformative approaches to these significant challenges.
Author contributions
JM: Writing – original draft, Writing – review & editing. SC: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Santambrogio L, Marrack P. The broad spectrum of pathogenic autoreactivity. Nat Rev Immunol. (2023) 23(2):69–70. doi: 10.1038/s41577-022-00812-2
2. Clement CC, Nanaware PP, Yamazaki T, Negroni MP, Ramesh K, Morozova K, et al. Pleiotropic consequences of metabolic stress for the major histocompatibility complex class II molecule antigen processing and presentation machinery. Immunity. (2021) 54(4):721–36. doi: 10.1016/j.immuni.2021.02.019
3. Clement CC, Osan J, Buque A, Nanaware PP, Chang YC, Perino G, et al. PDIA3 epitope-driven immune autoreactivity contributes to hepatic damage in type 2 diabetes. Sci Immunol. (2022) 7(74):eabl3795. doi: 10.1126/sciimmunol.abl3795
4. Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MH, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. (2004) 50(2):380–6. doi: 10.1002/art.20018
5. Scherer HU, van der Woude D, Toes REM. From risk to chronicity: evolution of autoreactive B cell and antibody responses in rheumatoid arthritis. Nat Rev Rheumatol. (2022) 18(7):371–83. doi: 10.1038/s41584-022-00786-4
6. Wouters F, Maurits MP, van Boheemen L, Verstappen M, Mankia K, Matthijssen XME, et al. Determining in which pre-arthritis stage HLA-shared epitope alleles and smoking exert their effect on the development of rheumatoid arthritis. Ann Rheum Dis. (2022) 81(1):48–55. doi: 10.1136/annrheumdis-2021-220546
7. Potempa J, Mydel P, Koziel J. The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat Rev Rheumatol. (2017) 13(10):606–20. doi: 10.1038/nrrheum.2017.132
8. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. (1987) 30(11):1205–13. doi: 10.1002/art.1780301102
9. Kokkonen H, Mullazehi M, Berglin E, Hallmans G, Wadell G, Ronnelid J, et al. Antibodies of IgG, IgA and IgM isotypes against cyclic citrullinated peptide precede the development of rheumatoid arthritis. Arthritis Res Ther. (2011) 13(1):R13. doi: 10.1186/ar3237
10. Arkema EV, Goldstein BL, Robinson W, Sokolove J, Wagner CA, Malspeis S, et al. Anti-citrullinated peptide autoantibodies, human leukocyte antigen shared epitope and risk of future rheumatoid arthritis: a nested case-control study. Arthritis Res Ther. (2013) 15(5):R159. doi: 10.1186/ar4342
11. Klareskog L, Malmstrom V, Lundberg K, Padyukov L, Alfredsson L. Smoking, citrullination and genetic variability in the immunopathogenesis of rheumatoid arthritis. Semin Immunol. (2011) 23(2):92–8. doi: 10.1016/j.smim.2011.01.014
12. Hensvold AH, Magnusson PK, Joshua V, Hansson M, Israelsson L, Ferreira R, et al. Environmental and genetic factors in the development of anticitrullinated protein antibodies (ACPAs) and ACPA-positive rheumatoid arthritis: an epidemiological investigation in twins. Ann Rheum Dis. (2015) 74(2):375–80. doi: 10.1136/annrheumdis-2013-203947
13. Kissel T, van Schie KA, Hafkenscheid L, Lundquist A, Kokkonen H, Wuhrer M, et al. On the presence of HLA-SE alleles and ACPA-IgG variable domain glycosylation in the phase preceding the development of rheumatoid arthritis. Ann Rheum Dis. (2019) 78(12):1616–20. doi: 10.1136/annrheumdis-2019-215698
14. Hafkenscheid L, de Moel E, Smolik I, Tanner S, Meng X, Jansen BC, et al. N-linked glycans in the variable domain of IgG anti-citrullinated protein antibodies predict the development of rheumatoid arthritis. Arthritis Rheumatol. (2019) 71(10):1626–33. doi: 10.1002/art.40920
15. Holers VM, Demoruelle MK, Kuhn KA, Buckner JH, Robinson WH, Okamoto Y, et al. Rheumatoid arthritis and the mucosal origins hypothesis: protection turns to destruction. Nat Rev Rheumatol. (2018) 14(9):542–57. doi: 10.1038/s41584-018-0070-0
16. Catrina AI, Joshua V, Klareskog L, Malmstrom V. Mechanisms involved in triggering rheumatoid arthritis. Immunol Rev. (2016) 269(1):162–74. doi: 10.1111/imr.12379
17. Lappin DF, Apatzidou D, Quirke AM, Oliver-Bell J, Butcher JP, Kinane DF, et al. Influence of periodontal disease, Porphyromonas gingivalis and cigarette smoking on systemic anti-citrullinated peptide antibody titres. J Clin Periodontol. (2013) 40(10):907–15. doi: 10.1111/jcpe.12138
18. de Pablo P, Dietrich T, Chapple IL, Milward M, Chowdhury M, Charles PJ, et al. The autoantibody repertoire in periodontitis: a role in the induction of autoimmunity to citrullinated proteins in rheumatoid arthritis? Ann Rheum Dis. (2014) 73(3):580–6. doi: 10.1136/annrheumdis-2012-202701
19. Janssen KM, de Smit MJ, Brouwer E, de Kok FA, Kraan J, Altenburg J, et al. Rheumatoid arthritis-associated autoantibodies in non-rheumatoid arthritis patients with mucosal inflammation: a case-control study. Arthritis Res Ther. (2015) 17(1):174. doi: 10.1186/s13075-015-0690-6
20. Janssen KMJ, Hop H, Vissink A, Dijkstra G, de Smit MJ, Brouwer E, et al. Levels of anti-citrullinated protein antibodies and rheumatoid factor, including IgA isotypes, and articular manifestations in ulcerative colitis and crohn’s disease. Int J Environ Res Public Health. (2020) 17(21):8054. doi: 10.3390/ijerph17218054
21. Elkayam O, Segal R, Lidgi M, Caspi D. Positive anti-cyclic citrullinated proteins and rheumatoid factor during active lung tuberculosis. Ann Rheum Dis. (2006) 65(8):1110–2. doi: 10.1136/ard.2005.045229
22. Kakumanu P, Yamagata H, Sobel ES, Reeves WH, Chan EK, Satoh M. Patients with pulmonary tuberculosis are frequently positive for anti-cyclic citrullinated peptide antibodies, but their sera also react with unmodified arginine-containing peptide. Arthritis Rheum. (2008) 58(6):1576–81. doi: 10.1002/art.23514
23. Mori S, Naito H, Ohtani S, Yamanaka T, Sugimoto M. Diagnostic utility of anti-cyclic citrullinated peptide antibodies for rheumatoid arthritis in patients with active lung tuberculosis. Clin Rheumatol. (2009) 28(3):277–83. doi: 10.1007/s10067-008-1035-5
24. Schellekens GA, Visser H, de Jong BA, van den Hoogen FH, Hazes JM, Breedveld FC, et al. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. (2000) 43(1):155–63. doi: 10.1002/1529-0131(200001)43:1%3C155::AID-ANR20%3E3.0.CO;2-3
25. Brewer RC, Lanz TV, Hale CR, Sepich-Poore GD, Martino C, Swafford AD, et al. Oral mucosal breaks trigger anti-citrullinated bacterial and human protein antibody responses in rheumatoid arthritis. Sci Transl Med. (2023) 15(684):eabq8476. doi: 10.1126/scitranslmed.abq8476
26. Lundberg K, Wegner N, Yucel-Lindberg T, Venables PJ. Periodontitis in RA-the citrullinated enolase connection. Nat Rev Rheumatol. (2010) 6(12):727–30. doi: 10.1038/nrrheum.2010.139
27. Manoil D, Courvoisier DS, Gilbert B, Moller B, Walker UA, Muehlenen IV, et al. Associations between serum antibodies to periodontal pathogens and preclinical phases of rheumatoid arthritis. Rheumatology (Oxford). (2021) 60(10):4755–64. doi: 10.1093/rheumatology/keab097
28. Sherina N, de Vries C, Kharlamova N, Sippl N, Jiang X, Brynedal B, et al. Antibodies to a citrullinated porphyromonas gingivalis epitope are increased in early rheumatoid arthritis, and can be produced by gingival tissue B cells: implications for a bacterial origin in RA etiology. Front Immunol. (2022) 13:804822. doi: 10.3389/fimmu.2022.804822
29. Oluwagbemigun K, Yucel-Lindberg T, Dietrich T, Tour G, Sherina N, Hansson M, et al. A cross-sectional investigation into the association between Porphyromonas gingivalis and autoantibodies to citrullinated proteins in a German population. Ther Adv Musculoskelet Dis. (2019) 11:1759720X19883152. doi: 10.1177/1759720X19883152
30. Eriksson K, Fei G, Lundmark A, Benchimol D, Lee L, Hu YOO, et al. Periodontal health and oral microbiota in patients with rheumatoid arthritis. J Clin Med. (2019) 8(5):630–48. doi: 10.3390/jcm8050630
31. Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. (2011) 10(5):497–506. doi: 10.1016/j.chom.2011.10.006
32. Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med. (2016) 8(369):369ra176. doi: 10.1126/scitranslmed.aaj1921
33. Curran AM, Girgis AA, Jang Y, Crawford JD, Thomas MA, Kawalerski R, et al. Citrullination modulates antigen processing and presentation by revealing cryptic epitopes in rheumatoid arthritis. Nat Commun. (2023) 14(1):1061. doi: 10.1038/s41467-023-36620-y
34. Cibotti R, Kanellopoulos JM, Cabaniols JP, Halle-Panenko O, Kosmatopoulos K, Sercarz E, et al. Tolerance to a self-protein involves its immunodominant but does not involve its subdominant determinants. Proc Natl Acad Sci USA. (1992) 89(1):416–20. doi: 10.1073/pnas.89.1.416
35. Moudgil KD, Sercarz EE. Dominant determinants in hen eggwhite lysozyme correspond to the cryptic determinants within its self-homologue, mouse lysozyme: implications in shaping of the T cell repertoire and autoimmunity. J Exp Med. (1993) 178(6):2131–8. doi: 10.1084/jem.178.6.2131
36. Moudgil KD, Sercarz EE. Understanding crypticity is the key to revealing the pathogenesis of autoimmunity. Trends Immunol. (2005) 26(7):355–9. doi: 10.1016/j.it.2005.05.007
37. Durai M, Kim HR, Moudgil KD. The regulatory C-terminal determinants within mycobacterial heat shock protein 65 are cryptic and cross-reactive with the dominant self homologs: implications for the pathogenesis of autoimmune arthritis. J Immunol. (2004) 173(1):181–8. doi: 10.4049/jimmunol.173.1.181
38. Moudgil KD, Southwood S, Ametani A, Kim K, Sette A, Sercarz EE. The self-directed T cell repertoire against mouse lysozyme reflects the influence of the hierarchy of its own determinants and can be engaged by a foreign lysozyme. J Immunol. (1999) 163(8):4232–7. doi: 10.4049/jimmunol.163.8.4232
39. Durai M, Gupta RS, Moudgil KD. The T cells specific for the carboxyl-terminal determinants of self (rat) heat-shock protein 65 escape tolerance induction and are involved in regulation of autoimmune arthritis. J Immunol. (2004) 172(5):2795–802. doi: 10.4049/jimmunol.172.5.2795
40. Moudgil KD, Kim E, Yun OJ, Chi HH, Brahn E, Sercarz EE. Environmental modulation of autoimmune arthritis involves the spontaneous microbial induction of T cell responses to regulatory determinants within heat shock protein 65. J Immunol. (2001) 166(6):4237–43. doi: 10.4049/jimmunol.166.6.4237
41. Malcolm J, Mukanthu MN, Brown J, Campbell L, Adrados-Planell A, Butcher JP, et al. C-terminal citrullinated peptide alters antigen-specific APC:T cell interactions leading to breach of immune tolerance. Under Rev J Autoimmun. (2023) 135:102994. doi: 10.1016/j.jaut.2023.102994
42. Robertson JM, Jensen PE, Evavold BD. DO11.10 and OT-II T cells recognize a C-terminal ovalbumin 323–339 epitope. J Immunol. (2000) 164(9):4706–12. doi: 10.4049/jimmunol.164.9.4706
43. Dai YD, Sercarz EE. Antigen processing patterns determine GAD65-specific regulation vs. pathogenesis. Front Biosci (Landmark Ed). (2009) 14(1):344–51. doi: 10.2741/3248
44. Moudgil KD, Sercarz EE, Grewal IS. Modulation of the immunogenicity of antigenic determinants by their flanking residues. Immunol Today. (1998) 19(5):217–20. doi: 10.1016/S0167-5699(97)01233-4
45. Lopez-Oliva I, Malcolm J, Culshaw S. Periodontitis and rheumatoid arthritis-global efforts to untangle two complex diseases. Periodontol 2000. (2024):1–19. doi: 10.1111/prd.12530.
46. The Economist Intelligence Unit. Time to Take Gum Disease Seriously: The Societal and Economic Impact of Periodontitis. (2021).
47. Alivernini S, Firestein GS, McInnes IB. The pathogenesis of rheumatoid arthritis. Immunity. (2022) 55(12):2255–70. doi: 10.1016/j.immuni.2022.11.009
48. Hsieh PH, Wu O, Geue C, McIntosh E, McInnes IB, Siebert S. Economic burden of rheumatoid arthritis: a systematic review of literature in biologic era. Ann Rheum Dis. (2020) 79(6):771–7. doi: 10.1136/annrheumdis-2019-216243
49. Einarsson JT, Geborek P, Saxne T, Kristensen LE, Kapetanovic MC. Sustained remission improves physical function in patients with established rheumatoid arthritis, and should be a treatment goal: a prospective observational cohort study from southern Sweden. J Rheumatol. (2016) 43(6):1017–23. doi: 10.3899/jrheum.150995
50. Svensson B, Andersson M, Forslind K, Ajeganova S, Hafstrom I, group Bs. Persistently active disease is common in patients with rheumatoid arthritis, particularly in women: a long-term inception cohort study. Scand J Rheumatol. (2016) 45(6):448–55. doi: 10.3109/03009742.2016.1147595
Keywords: periodontitis, rheumatoid arthritis, autoimmunity, Porphyromonas gingivalis, autoantibody
Citation: Malcolm J and Culshaw S (2024) Aberrant immunity in the oral cavity—a link with rheumatoid arthritis?. Front. Oral. Health 5:1430886. doi: 10.3389/froh.2024.1430886
Received: 10 May 2024; Accepted: 4 June 2024;
Published: 14 June 2024.
Edited by:
Georgios N. Belibasakis, Karolinska Institutet (KI), SwedenReviewed by:
Daniel Manoil, University of Geneva, Switzerland© 2024 Malcolm and Culshaw. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shauna Culshaw, c2hhdW5hLmN1bHNoYXdAZ2xhc2dvdy5hYy51aw==