
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Microbiomes , 12 September 2023
Sec. Host and Microbe Associations
Volume 2 - 2023 | https://doi.org/10.3389/frmbi.2023.1219960
This article is part of the Research Topic Gut Microbiota and Its Importance on Human Health - the Need for Reliable Measurements to Assess the Microbial Gut Function and Its Correlated Pathologies View all 6 articles
 Nayeli Ortiz-Olvera1†
Nayeli Ortiz-Olvera1† Edith A. Fernández-Figueroa2†
Edith A. Fernández-Figueroa2† Jesús Argueta-Donohué3†
Jesús Argueta-Donohué3† Haydee Miranda-Ortíz4†
Haydee Miranda-Ortíz4† Erika Ruiz-García5,6*†
Erika Ruiz-García5,6*†Despite the numerous fecal microbiota transplantation trials that have been carried out, knowledge about the actual composition of a “healthy microbiota” remains scarce. The aim of this research was to describe the differences in the composition of oral and fecal microbiotas in a super-donor. The microbiota analysis was done using next-generation sequencing of the V3 and V4 regions of the bacterial 16S rRNA gene. The biodiversity found in the mouth was very rich, with 56 species identified, and there was a predominance of the genera Veillonella, Haemophilus, and Streptococcus. It is worth mentioning the presence (2.33%) of Fusobacterium nucleatum in the mouth. In feces, the genera Bacteroides and Faecalibacterium predominated, with the species F. prausnitzii being the most abundant. This analysis shows that the diversity of the microbiota of a super-donor plays a fundamental role in the effectiveness of its product in fecal matter transplantation. This suggests that transplanted gut microorganisms have the ability to maintain or recover health in a dynamic process between the microbiota and the host. Our findings encourage further research which will result in the development of bacterial therapies in infectious and inflammatory diseases.
Despite the numerous fecal microbiome transplantation (FMT) trials that have been carried out, knowledge about the actual composition of a “healthy microbiota” remains scarce. We do not know exactly how transplanted bacteria survive, colonize, and function in the recipient’s gut or, more importantly, what methods can be used to monitor transplanted feces, how to assess whether the product is suitable for transplantation, and whether it contains a “healthy microbiota” (Ianiro et al., 2014; Mukhopadhya et al., 2019).
The availability of novel tools such as next-generation sequencing (NGS) has allowed the identification of genes, and even complete genomes, that have been recovered from complex microbial communities (Human Microbiome Project Consortium, 2012; D’Amore et al., 2016).
Research has shown that some individuals have gut microbiomes with specific characteristics, such as the presence or absence of specific bacteria, phages, and metabolites (unfortunately not fully recognized), which protect the individual donor from the vast majority of gut dysbiosis (Wilson et al., 2019). These individuals are ideal donors and have been called “super-donors” because their fecal microbiota is very suitable for transplantation. However, recent studies have shown that there are some specific characteristics that make the microbiota of super-donors not universally suitable for transplantation (Olesen and Gerardin, 2021).
In this study, the variable regions V3 and V4 of the ribosomal 16S rRNA gene were amplified and sequenced to characterize the oral and fecal microbiota of a “super-donor” whose sample was used in 11 fecal microbiota recipients with Clostridioides difficile infection (8 recurrent and three severe) and who presented a favorable response. All patients presented with remission of diarrhea for a single FMT, motivating us to learn more about the bacterial composition of this “super-donor.”
A single super-donor was recruited and screened following international (American and European) guidelines for fecal microbiota donors (Cammarota et al., 2019). The donor was interviewed about her medical history and lifestyle. Before analyzed the samples, were excluded a history of exposure to infectious agents, drug abuse, and risky social or sexual behavior in the last 6 months.in the donnor. The Rome IV questionnaire was applied to rule out any gut–brain interaction disorder, organic gastrointestinal diseases (celiac disease, inflammatory bowel disease, cancer), metabolic diseases (diabetes mellitus, thyroid pathology, hyperlipidemia), and neurologic diseases. In addition, the super-donor underwent a complete physical examination and laboratory tests. The latter included a complete blood count, and tests for glycemia, electrolytes, inflammatory markers, liver function, and thyroid function. In addition, screening serologies were performed for human immunodeficiency virus (HIV), syphilis, cytomegalovirus, herpes zoster, and hepatitis A, B, and C viruses.
A microbiological stool analysis was performed to rule out pathogenic bacteria (Shigella spp., Salmonella spp., Campylobacter spp., Yersinia spp., and C. difficile); as well as a stool antigen test to rule out Helicobacter pylori. All test results were negative. In addition, no rotavirus or parasite eggs were found in the feces.
The super-donor was a healthy, 26-year-old Mexican woman who was a non-smoker, did not take any medication, and had a body mass index (BMI) of 24.7 kg/m2. From the frequency of food intake, we could see that the average energy intake in 24 h was 1649.8 kcal, which was broken down into carbohydrates 221.3 g, protein 62.8 g, fat 57.6 g, saturated fat 18.72 g, fiber 23.2 g, vitamin A 694.2 mg, vitamin C 156.7 mg, calcium 620.3 mg, iron 9.9 mg, and sodium 2752.5 mg. Energy expenditure in 24 h was calculated at 1576.2 kcal.
A 4-hour fasting oral scraping was performed with a swab; we tried to take a homogeneous sample from the entire mouth. The sample was placed in 1 mL of nuclear lysis solution. In addition, the donor collected the first bowel movement of the day. Samples were frozen until analysis.
DNA extraction from the oral scraping was performed using the Wizard® Genomic DNA Purification Kit (cat. no. A1120; Promega), and DNA from the fecal sample was extracted at room temperature using the QIAamp® PowerFecal® Pro DNA Kit (cat. 51804; QIAGEN®), following the manufacturer’s instructions. Both the DNA samples were quantified and adjusted to 25 ng/μL. The integrity of the DNA was verified in a 1% agarose gel stained with SYBR™ Gold Nucleic Acid Gel Stain (cat. no. S11494; Invitrogen). To create the libraries, 250 ng of DNA was used and the primers reported in the Illumina 16S Metagenomic Sequencing Library Preparation protocol were used to amplify the V3 and V4 regions of bacterial 16S.
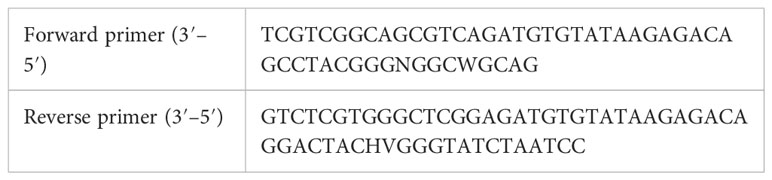
The fragments were amplified using Taq PCR Master mix 2× (cat. no. 1007544; QIAGEN), 1 μL (10 μM) of each primer, DNase- and RNase-free water in a final volume of 50 μL. The PCR assay was as follows: an initial denaturation at 94°C for 3 min, followed by 35 amplification cycles at 94°C for 40 s, 55°C for 2 min, and 72°C for 1 min, and a final extension at 72°C for 10 min. The PCR products were separated on a 1.5% agarose at 100 V for 1 h; the gels were stained as was described. The PCR products were purified using 75 μL of the Agencourt AMPure XP Kit (cat. no A63882; Beckman Coulter™) and eluted in RNase-free water.
Both the samples were sequenced with an Illumina MiSeq platform, 2 × 250, 500 cycles (Illumina Inc.). The reads were processed using DADA2 v1.12 to obtain error-corrected amplicon sequence variant representatives analogous to operational taxonomic units with single-nucleotide resolution (sOTUs). sOTUs annotated as chloroplasts and mitochondria were removed from the analysis.
The sOTUs were annotated with genus-level taxonomy using the Ribosomal Database Project (RDP)-naive Bayesian classifier, as implemented in DADA2, and, if possible, to species level using DADA2, both against the SILVA 16S database, v132.
The resulting OTUs were filtered for possible non-specific amplification using SortMeRNA v2.0 and Infernal v1.1.2. sOTUs with less than one read across all samples were excluded. Multiple sequence alignments of the sOTUs were performed using Infernal v1.1.2. A generalized linear mixed model (GLMM) was calculated for each sOTU to estimate its abundance (i.e., read counts) in relation to predictors. The package glmmTMB v0.1.4 was used to estimate the abundance of each microbe under a zero-inflated Poisson distribution. A multiple hypothesis correction was calculated using the Benjamini–Hochberg test.
The alluvial plots were done in R software (The R Foundation for Statistical Computing, Vienna, Austria) version 4.4.2 and the R packages tidyverse and ggalluvial version 1.3.2 and 0.12.3, respectively, were used. Venn diagrams were created using Microsoft Excel® (Microsoft Corporation, Redmond, WA, USA).
A total of 14,696 and 39,303 sequencing readings of oral and fecal samples were obtained, respectively. The assignment of OTUs for the oral and fecal samples are shown in Table 1 and Figure 1. The general classification of the mouth sample showed that 90.5% of all readings were bacterial, none were Archaea, and 9.5% were not identified at the genus level. The general classification of the fecal sample showed that 92% of all readings were bacterial, none were Archaea, and 8% were not identified at the genus level.
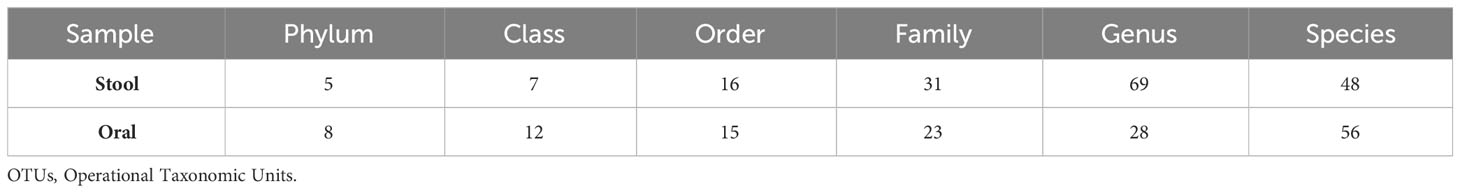
Table 1 Number of OTUs identified in stool and oral cavity in the super-donor.
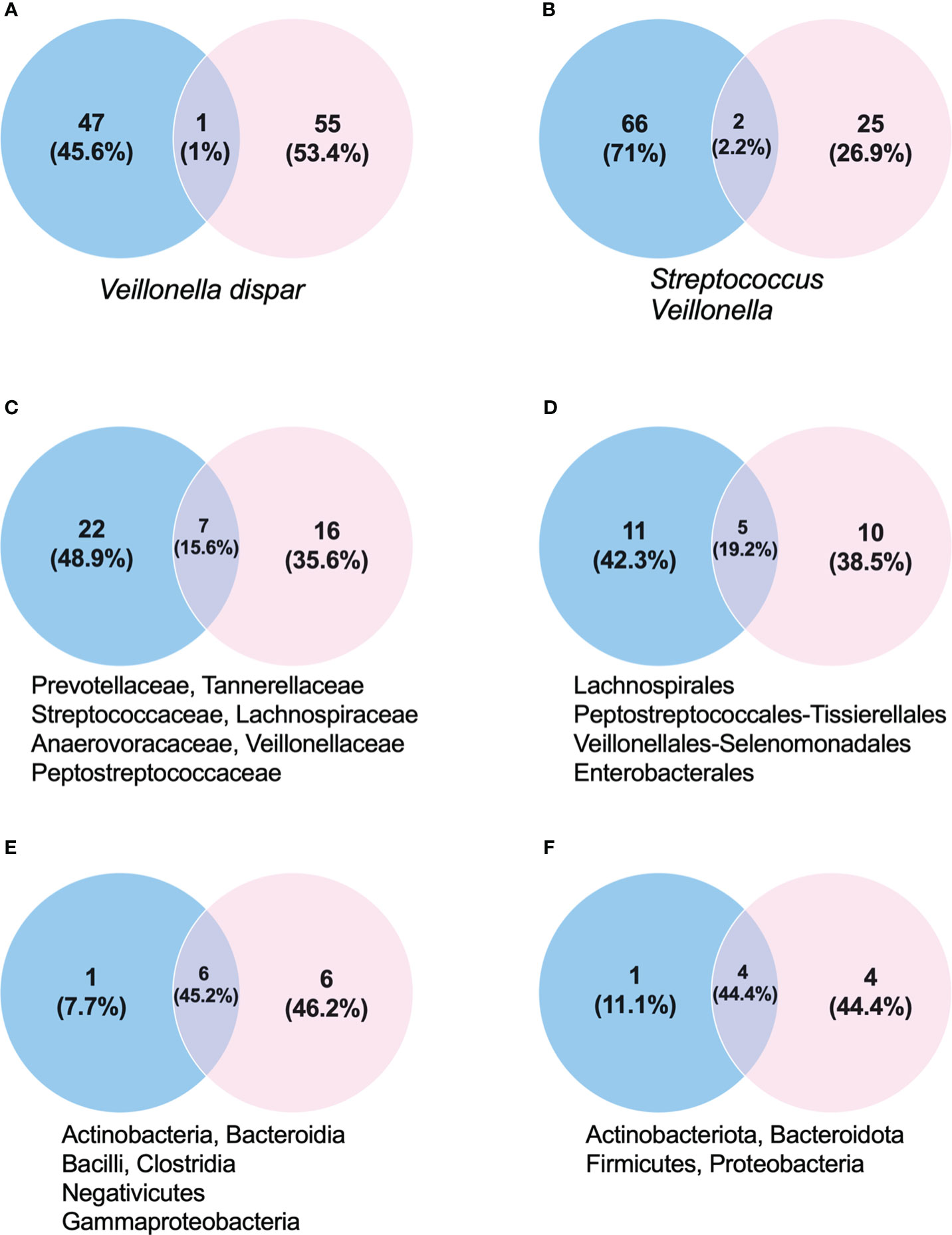
Figure 1 Proportion of taxonomic groups in stool (blue) and mouth (pink) samples. (A) Species. (B) Genus. (C) Families. (D) Order. (E) Class. (F) Phylum. Below each Venn diagram there are the taxonomic classification names shared between the oral and fecal samples.
The biodiversity found in the mouth was very rich, with 56 species identified. Of the eight phyla found, Firmicutes, Proteobacteria, and Bacteroidota predominated. Fusobacteriota, Actinobacteria, and Campylobacterota were also found, but in lower percentages (Figures 1, 2). In the Firmicutes phylum, the predominant class was Negativicutes of the Veillonellales–Selenomonadales order, with a clear predominance of Veillonella parvula at 19.7%; in the Bacilli class, Lactobacillales order of the Streptococcus family, the predominant species were Streptococcus sanguinis at 5.25% and Streptococcus oralis at 4.32% (Figures 1, 2A, B). In the Proteobacteria phylum, the Gammaproteobacteria class, Pasteurellaceae family, Haemophilus genus, Haemophilus haemolyticus species predominated at 11.54%, followed by Haemophilus quentini at 6.14% (Figures 1, 2A, C). On the other hand, in the Bacteroidales order, family Prevotellaceae, the Prevotella genus prevailed with respect to the Bacteroidota phylum, and the species with greater abundance was Prevotella loescheii at 3.95% and Prevotella oris at 1.74%. It is worth mentioning the presence of Fusobacterium nucleatum at 2.33% belonging to the Fusobacteriota phylum (Figures 1, 2A, D).
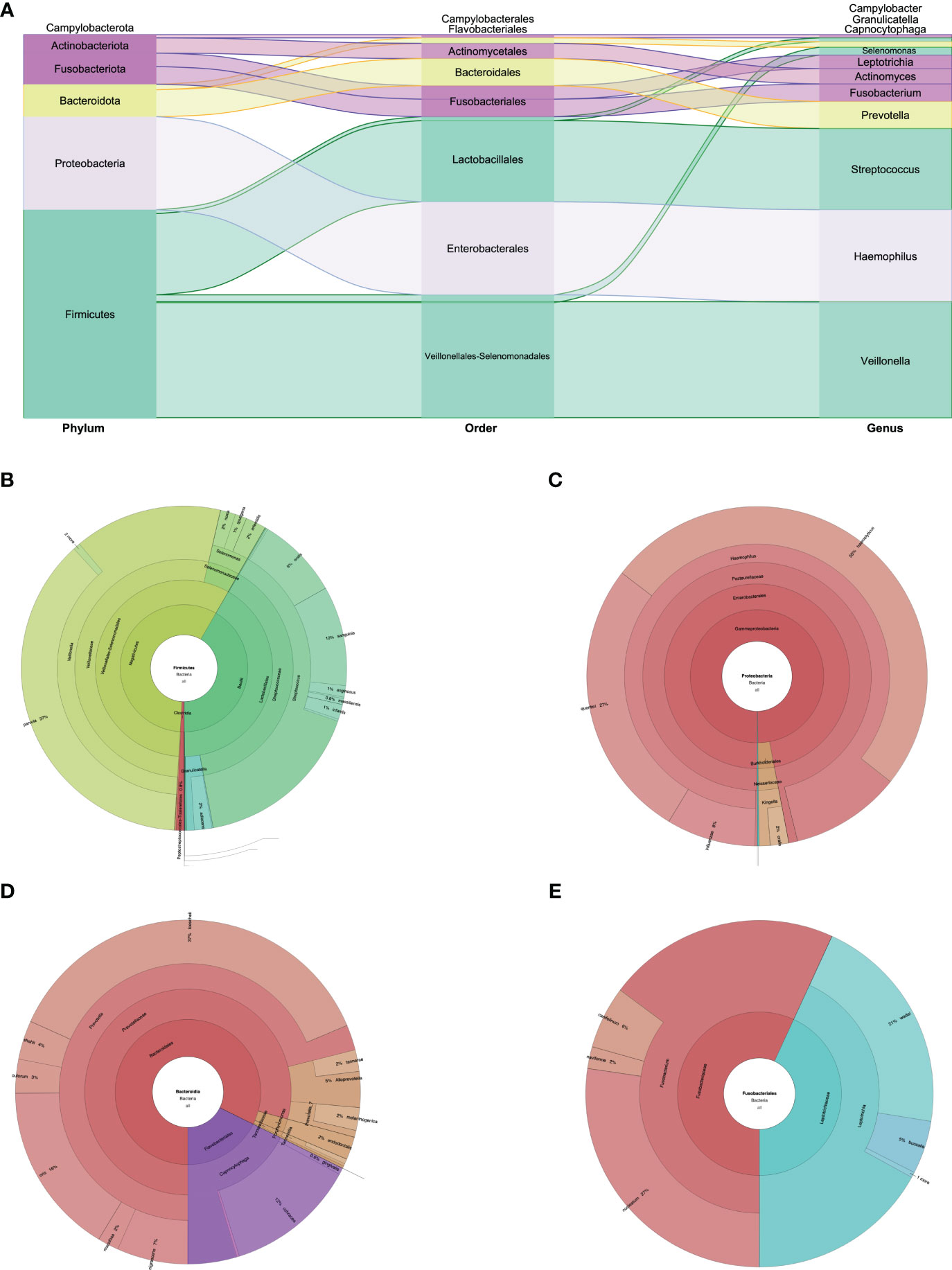
Figure 2 (A) Phylum, order, and genus enrichment in the oral cavity sample of the super-donor. Firmicutes (green), Proteobacteria (lilac), Bacteroidota (yellow), Fusobacteria and Actinobacteria (purple), and Campylobacterota (without color) were the most represented phyla in the oral cavity sample. (B) Taxonomic classification of the Firmicutes phylum. (C) Taxonomic classification of the Proteobacteria phylum. (D) Taxonomic classification of the Bacteroidota phylum. (E) Taxonomic classification of the Fusobacteriota phylum.
In feces we identified 48 species. Firmicutes were the predominant phylum: two classes, Clostridia and Negativicutes, and four families for Clostridia; and the family with greater abundance was Ruminococcaceae, with the Faecalibacterium genus and the Faecalibacterium prausnitzii species at 9.87%. For the Lachnospiraceae family, the predominant genera were Agathobacter at 5% and Lachnospira at 3.2%. We found 2% of the Firmicutes phylum and Christensenellaceae family. In the Negativicutes class we found the Acidaminococcaceae family, with the Phascolarctobacterium genus and the Phascolarctobacterium faecium species at 1.7% (Figures 1, 3A, B). The second phylum with greater abundance was Bacteroidota with two predominant families: Bacteroidaceae and Rikenellaceae. In the Bacteroidaceae and Bacteroides genus, the Bacteroides vulgatus specie predominated with an abundance of 11.8%, followed by Bacteroides stercoris at 4.4% abundance (Figure 3C). Regarding the Rikenellaceae family, the Alistipes genus, with the Alistipes onderdonkii species represented in 1.37% of all species found (Figures 1, 3A, B). The third phylum in order of representation in the fecal sample was Verrucomicrobiota, Verrucomicrobiales order, Akkermansiaceae family, within which the Akkermansia muciniphila species presented an abundance of 1.7%. Other phyla present in a lesser amount were Proteobacteria and Actinobacteriota (Figures 1, 3A).
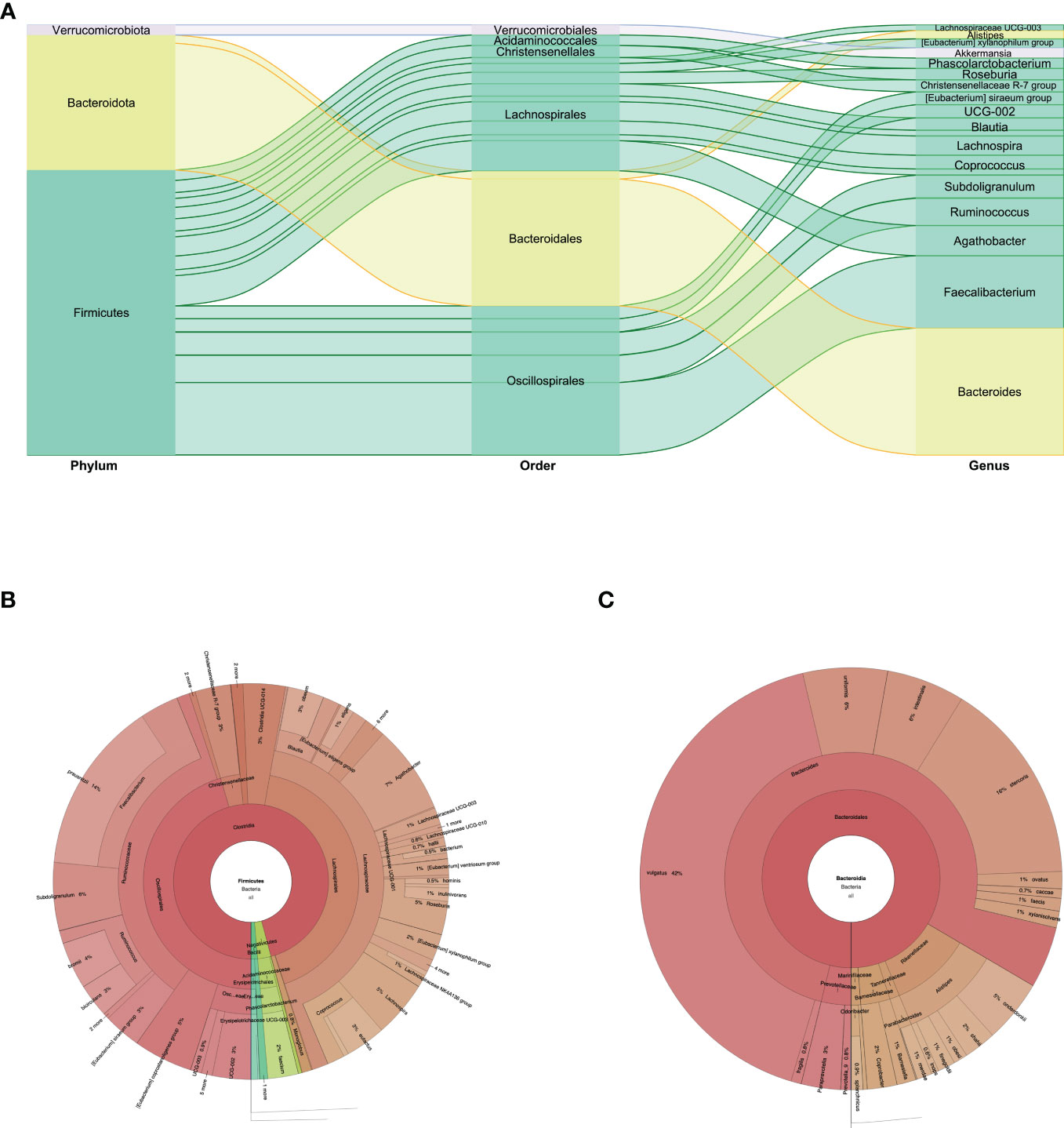
Figure 3 (A) Phylum, order, and genus enrichment in the fecal sample of the super-donor. Firmicutes (green), Bacteroidota (yellow), and Verrucomicrobiota (gray) were the most represented phyla in the fecal sample. (B) Taxonomic classification of the Firmicutes phylum. (C) Taxonomic classification of the Bacteroidota phylum.
Our study aimed to identify and describe the oral and fecal microbiota of a fecal matter super-donor for the treatment of dysbiosis presented in patients with diarrhea associated with C. difficile infection. This super-donor showed a wide diversity in oral and fecal microbiota, with a predominance of Firmicutes and Bacteroidota phyla, but with great variation in families, genera, and species depending on where they were found. We observed that only 2.2% of the microbiota, at the genus level, was similar in both the oral cavity and in the feces (Supplementary 1).
Overall, only 3% of potential candidates to donate product for FMTs are accepted, which shows how exceptional it is to find an ideal donor. Despite the advances in microbiota research, we still do not know what the properties of an “ideal” microbiota are (Kassam et al., 2019). We thus decided to explore the characteristics of our super-donor since all patients who received the product for FMT showed a favorable response to a single transplant, namely stopping the diarrhea.
The sequences obtained in the mouth were representative of the most common species, but were far from describing the total bacterial diversity (Arweiler and Netuschil, 2016). The donor in this study presented predominance in the Veillonellaceae family, with abundance at 27%, and the Veillonella parvula species at 19.7%. The Veillonella parvula species is of interest due to its presence in the supragingival and subgingival dental plaque of the human mouth, where it can cause opportunistic infections. In addition, the Veillonellaceae family is characterized by its mutualistic relationship with early, middle, and late colonizers of the mouth, becoming an important component in oral biofilm ecology (Gronow et al., 2010; Liu et al., 2011).
Another aspect that must be evaluated is the relationship between oral microbiota and other non-oral diseases. In this regard, the best example is the Fusobacteriota phylum, which includes species commonly found in the human oral cavity (Fusobacterium spp.) and in the intestinal tract (Leptotrichia spp.). Fusobacteria are divided into two families: Leptotrichiaceae and Fusobacteriaceae. The latter includes the F. nucleatum species, a multifaceted bacterium involved in various interactions and effects that are sometimes beneficial and sometimes harmful. F. nucleatum was for a long time described as being commonly found in oral microbiota, with integral and beneficial functions in the oral biofilm. However, it is considered an opportunistic pathogen that has the ability to maintain a symbiotic relationship with the host. Recently, a link between the presence of extraoral F. nucleatum and human colorectal cancer has been reported due to the presence of fusobacteria in tumor tissue (Castellarin et al., 2012; Kostic et al., 2012); however, the role of F. nucleatum as part of the cancer-causing microbiota is still being researched (Brennan and Garrett, 2019). It is important to define the causality of F. nucleatum and the diseases to which it has been associated, because this bacterium is ubiquitous in the mouth. However, its presence and abundance in the gut is related to inflammatory bowel disease and colorectal cancer. In our super-donor, we found F. nucleatum only in the mouth, not in feces.
The other family that predominated in this super-donor was Pasteurellaceae, which includes the Haemophilus genus, representing 22% of the oral microbiota with the H. haemolyticus and H. quentini species, and in a lesser amount Haemophilus influenzae, species that are dependent on factors X and V and are positive for urease and some for ornithine decarboxylase. These strains are considered rare and of little clinical significance (Nørskov-Lauritsen, 2014).
The third family that was highly present was Streptococcus, with S. sanguinis and S. oralis species predominating. These species are prominent in the mouth and are generally defined as commensals, but in immunocompromised subjects they are important opportunistic pathogens of systemic infections (Alves et al., 2019).
At the gut level, the composition of the microbiota is determined both by genetic and environmental influences and by diet, through a continuous process that can begin in the maternal uterus (Perez-Muñoz et al., 2017) and fluctuates throughout the life of each individual (Odamaki et al., 2016). In a healthy adult, the bacterial population in the intestine consists predominantly of members of the Firmicutes (60% to 80%) and Bacteroidetes (15% to 30%) (Human Microbiome Project Consortium, 2012) phyla, which are strictly anaerobic, and to a lesser degree Proteobacteria and Actinobacteria phyla. Therefore, our super-donor showed a diversity and distribution of the microbiota in normal feces similar to what is described in healthy subjects, with a predominance of the Firmicutes and Bacteroidota phyla, and a greater abundance of the Bacteroides vulgatus and Faecalibacterium prausnitzii species. The Human Microbiome Project established that Bacteroides are one of the most dominant genera in the healthy human gut and that they play an important role in keeping the gut ecosystem healthy (Human Microbiome Project Consortium, 2012). Patients with intestinal inflammation usually show lower Bacteroides levels (Conte et al., 2006; Zhou and Zhi, 2016). The B. vulgatus species reduces the inflammatory response in the gut, and has an immunoactive function involving the nucleotide-binding oligomerization domain 2 (NOD2) receptor. This receptor plays a critical role in the immune system by acting as a microbial sensor (Nigro et al., 2014). Ramanan et al. showed that B. vulgatus activates the immune response by translocation to the small intestine in a mouse model that lacked functionality in NOD2 (Ramanan et al., 2014). This is relevant because it indicates that the activity of species, such as B. vulgatus, may depend on the genetics of the host; it is also an endogenous modulator in the gut. The interaction of B. vulgatus with other bacteria, such as those of the genus Akkermansia, modulates the intestinal environment and, therefore, the health—or lack thereof—of the host (You et al., 2023). The abundance of B. vulgatus has a direct impact on the synthesis of microbial lipopolysaccharides in the human gut, with a protective effect against atherosclerosis (Yoshida et al., 2018).
The other most abundant bacterial species of human gut microbiota in healthy adults is F. prausnitzii—which in our super-donor accounted for 9.9% of the total bacterial population—as described in the literature (Miquel et al., 2013). This species is an anaerobic, commensal, metabolically active bacterium, and is considered the most important butyrate-producing bacterium in the human colon. To date, it has been considered as a bioindicator of human health since when its population decreases, it favors dysbiosis and other disorders such as inflammatory bowel disease, irritable bowel syndrome, and colorectal cancer, and has even been found in lower levels in dysbiosis due to acute COVID-19 disease, even after recovery (Ferreira-Halder et al., 2017; Baradaran Ghavami et al., 2021; Linares-Garcia et al., 2022). Researchers have also proposed that a decreased abundance of F. prausnitzii could be a predictive biomarker for C. difficile infection in patients with dysbiosis, because patients with this bacterial species have decreased numbers of butyrate-producing bacteria (F. prausnitzii) and increased the lactic acid-producing bacteria (Akkermansia). Even high levels of Akkermansia muciniphila can be a predictive marker for the onset of nosocomial diarrhea and worse prognosis in C. difficile infection. However, it has also been reported that A. muciniphila improves metabolic parameters, such as glucose homeostasis, and reduces inflammation in humans and mice (You et al., 2023). Our donor presented with a high abundance of F. prausnitzii and low abundance of A. muciniphila, similar to what has been described in healthy subjects (Vakili et al., 2020).
Feces donors who are rich in specific members of Clostridium groups IV and XIVa have also been reported to predict a sustained post-FMT response in patients with inflammatory bowel disease (Fuentes et al., 2017). The super-donor in our study also showed a predominance of Clostridia of the orders Lachnospirales and Oscillospirales, with greater abundance of Agathobacter and Lachnospira families. The genera of these families are known to hydrolyze starch and other sugars to produce butyrate and other short-chain fatty acids. All members of Lachnospiraceae are anaerobic, fermenters, and chemoorganotrophic, and some show strong hydrolyzing activity. For example, Blautia and Roseburia of the Lachnospiraceae family represent the genera most involved in the control of intestinal inflammatory processes, atherosclerosis, and maturation of the immune system, demonstrating that the final products of bacterial metabolism (butyrate) mediate these effects (Vacca et al., 2020). It has been reported that the gut microbiome of a super-donor is characterized by enrichment of Ruminococcaceae and Lachnospiraceae families, a characteristic that we observed in our super-donor (Moayyedi et al., 2015). Another interesting finding of this super-donor was the presence of Christsenellaceae, which represented 2% of the microbiotas in her stool. In fact, this family of the Clostridia class was described in 2012. It is widely found in America, Europe, Asia, Africa, and Australia; a greater relative abundance is observed in women, and it is associated with longevity. The relative abundance of Christensenellaceae in the human gut is inversely related to the BMI of the host, with low triglyceride levels, and elevated levels of high-density lipoproteins. In addition, Christensenellaceae is highly inheritable in multiple populations. Further research into the microbial ecology and metabolism of these bacteria should reveal the mechanical foundations of host–health associations, and enable their development as therapeutic agents, such as probiotics (Waters and Ley, 2019).
Because no two gut microbiomes are identical, the definition of a healthy gut microbiome remains undescribed. Despite this, having a stable and diverse gut community is generally correlated with a healthy intestinal state (Human Microbiome Project Consortium, 2012). The microbial diversity of the stool donor has been shown to be one of the most important factors influencing the FMT outcome. Vermeire et al. reported that donors whose products were effective in FMT have significantly greater bacterial richness than those who did not get a response (Vermeire et al., 2016).
The diversity of the microbiota of a super-donor, and the interaction between the different genera and species, plays a fundamental role in the effectiveness of its product in the FMT. This suggests that transplanted gut microorganisms have the ability to maintain or recover health, in a dynamic process between the microbiota and the host. Our findings encourage further research in order to develop bacterial therapies in infectious and inflammatory diseases.
The data presented in the study are deposited in the NCBI repository, accession number PRJNA974497.
The studies involving human participants were reviewed and approved by INCAN/CI/0170/18. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Conception and design of study: NO-O, EF-F, and ER-G. Acquisition of data: EF-F, JA-D, and HM-O. Data analysis and/or interpretation: NO-O, EF-F, JA-D, and HM-O. Drafting of the manuscript and/or critical revision: NO-O, ER-G, and EF-F. Approval of the final version of the manuscript: NO-O, EF-F, JA-D, HM-O, and ER-G. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frmbi.2023.1219960/full#supplementary-material
Alves L. A., de Carli T. R., Harth-Chu E. N., Mariano F. S., Höfling J. F., Stipp R. N., et al. (2019). Oral streptococci show diversity in resistance to complement immunity. J. Med. Microbiol. 68 (4), 600–608. doi: 10.1099/jmm.0.000955
Arweiler N. B., Netuschil L. (2016). The oral microbiota. Adv. Exp. Med. Biol. 902, 45–60. doi: 10.1007/978-3-319-31248-4_4
Baradaran Ghavami S., Pourhamzeh M., Farmani M., Raftar S. K. A., Shahrokh S., Shpichka A., et al. (2021). Cross-talk between immune system and microbiota in COVID-19. Expert Rev Gastroenterol Hepatol 15 (11), 1281–1294. doi: 10.1080/17474124.2021.1991311
Brennan C. A., Garrett W. S. (2019). Fusobacterium nucleatum - symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 17 (3), 156–166. doi: 10.1038/s41579-018-0129-6
Cammarota G., Ianiro G., Kelly C. R., Mullish B. H., Allegretti J. R., Kassam Z., et al. (2019). International consensus conference on stool banking for faecal microbiota transplantation in clinical practice. Gut 68 (12), 2111–2121. doi: 10.1136/gutjnl-2019-319548
Castellarin M., Warren R. L., Freeman J. D., Dreolini L., Krzywinski M., Strauss J., et al. (2012). Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22 (2), 299–306. doi: 10.1101/gr.126516.111
Conte M. P., Schippa S., Zamboni I., Penta M., Chiarini F., Seganti L., et al. (2006). Gut-associated bacterial microbiota in paediatric patients with inflammatory bowel disease. Gut 55 (12), 1760–1767. doi: 10.1136/gut.2005.078824
D’Amore R., Ijaz U. Z., Schirmer M., Kenny J. G., Gregory R., Darby A. C., et al. (2016). A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genomics 17, 55. doi: 10.1186/s12864-015-2194-9
Ferreira-Halder C. V., Faria A. V. S., Andrade S. S. (2017). Action and function of Faecalibacterium prausnitzii in health and disease. Best Pract. Res. Clin. Gastroenterol. 31 (6), 643–648. doi: 10.1016/j.bpg.2017.09.011
Fuentes S., Rossen N. G., van der Spek M. J., Hartman J. H., Huuskonen L., Korpela K., et al. (2017). Microbial shifts and signatures of long-term remission in ulcerative colitis after faecal microbiota transplantation. ISME J. 11 (8), 1877–1889. doi: 10.1038/ismej.2017.44
Gronow S., Welnitz S., Lapidus A., Nolan M., Ivanova N., Glavina Del Rio T., et al. (2010). Complete genome sequence of Veillonella parvula type strain (Te3). Stand Genomic Sci. 2 (1), 57–65. doi: 10.4056/sigs.521107
Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486 (7402), 207–214. doi: 10.1038/nature11234
Ianiro G., Bruno G., Lopetuso L., Beghella F., Laterza L., D'Aversa F., et al. (2014). Role of yeasts in healthy and impaired gut microbiota: the gut mycome. Curr. Pharm. Des. 20 (28), 4565–4593. doi: 10.2174/13816128113196660723
Kassam Z., Dubois N., Ramakrishna B., Ling K., Qazi T., Smith M., et al. (2019). Donor screening for fecal microbiota transplantation. N Engl. J. Med. 381 (21), 2070–2072. doi: 10.1056/NEJMc1913670
Kostic A. D., Gevers D., Pedamallu C. S., Michaud M., Duke F., Earl A. M., et al. (2012). Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 22 (2), 292–298. doi: 10.1101/gr.126573.111
Linares-García L., Cárdenas-Barragán M. E., Hernández-Ceballos W., Pérez-Solano C. S., Morales-Guzmán A. S., Miller D. S., et al. (2022). Bacterial and fungal gut dysbiosis and clostridium difficile in COVID-19: A review. J. Clin. Gastroenterol. 56 (4), 285–298. doi: 10.1097/MCG.0000000000001669
Liu J., Merritt J., Qi F. (2011). Genetic transformation of Veillonella parvula. FEMS Microbiol. Lett. 322 (2), 138–144. doi: 10.1111/j.1574-6968.2011.02344.x
Miquel S., Martín R., Rossi O., Bermúdez-Humarán L. G., Chatel-Guzmán J. M., Sokol H., et al. (2013). Faecalibacterium prausnitzii and human intestinal health. Curr. Opin. Microbiol. 16 (3), 255–261. doi: 10.1016/j.mib.2013.06.003
Moayyedi P., Surette M. G., Kim P. T., Libertucci J., Wolfe M., Onischi C., et al. (2015). Fecal microbiota transplantation induces remission in patients with active ulcerative colitis in a randomized controlled trial. Gastroenterology 149 (1), 102–109.e6. doi: 10.1053/j.gastro.2015.04.001
Mukhopadhya I., Segal J. P., Carding S. R., Hart A. L., Hold G. L. (2019). The gut virome: the ‘missing link’ between gut bacteria and host immunity? Therap Adv. Gastroenterol. 25, 12:1756284819836620. doi: 10.1177/1756284819836620
Nigro G., Rossi R., Commere P.-H., Jay P., Sansonetti P. J. (2014). The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe 15 (6), 792–798. doi: 10.1016/j.chom.2014.05.003
Nørskov-Lauritsen N. (2014). Classification, identification, and clinical significance of Haemophilus and Aggregatibacter species with host specificity for humans. Clin. Microbiol. Rev. 27 (2), 214–240. doi: 10.1128/CMR.00103-13
Odamaki T., Kato K., Sugahara H., Hashikura N., Takahashi S., Xiao J. Z., et al. (2016). Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol. 16, 90. doi: 10.1186/s12866-016-0708-5
Olesen S. W., Gerardin Y. (2021). Re-evaluating the evidence for faecal microbiota transplantation ‘Super-donors’ in inflammatory bowel disease. J. Crohns Colitis 15 (3), 453–461. doi: 10.1093/ecco-jcc/jjaa170
Perez-Muñoz M. E., Arrieta M. C., Ramer-Tait A. E., Walter J. (2017). A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: implications for research on the pioneer infant microbiome. Microbiome 5 (1), 48. doi: 10.1186/s40168-017-0268-4
Ramanan D., San Tang M., Bowcutt R., Cadwell K. (2014). Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity 41 (2), 311–324. doi: 10.1016/j.immuni.2014.06.015
Vacca M., Celano G., Calabrese F. M., Portincasa P., Gobbetti M., De Angelis M. (2020). The controversial role of human gut lachnospiraceae. Microorganisms 8 (4), 573. doi: 10.3390/microorganisms8040573
Vakili B., Fateh A., Asadzadeh Aghdaei H., Sotoodehnejadnematalahi F., Siadat S. D. (2020). Characterization of gut microbiota in hospitalized patients with clostridioides difficile infection. Curr. Microbiol. 77, 1673–1680. doi: 10.1007/s00284-020-01980-x
Vermeire S., Joossens M., Verbeke K., Wang J., Machiels K., Sabino J., et al. (2016). Donor species richness determines faecal microbiota transplantation success in inflammatory bowel disease. J. Crohns Colitis 10 (4), 387–394. doi: 10.1093/ecco-jcc/jjv203
Waters J. L., Ley R. E. (2019). The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. BMC Biol. 17 (1), 83. doi: 10.1186/s12915-019-0699-4
Wilson B. C., Vatanen T., Cutfield W. S., O’Sullivan J. M. (2019). The super-donor phenomenon in fecal microbiota transplantation. Front. Cell Infect. Microbiol. 9, 2. doi: 10.3389/fcimb.2019.00002
Yoshida N., Emoto T., Yamashita T., Watanabe H., Hayashi T., Tabata T., et al. (2018). Bacteroides vulgatus and Bacteroides dorei Reduce Gut Microbial Lipopolysaccharide Production and Inhibit Atherosclerosis. Circulation 138 (22), 2486–2498. doi: 10.1161/CIRCULATIONAHA.118.033714
You H. J., Si J., Kim J., Yoon S., Cha K. H., Yoon H. S., et al. (2023). Bacteroides vulgatus SNUG 40005 restores akkermansia depletion by metabolite modulation. Gastroenterology 164 (1), 103–116. doi: 10.1053/j.gastro.2022.09.040
Keywords: oral microbiota, fecal microbiota, F. prausnitzii, F. nucleatum, C. difficile
Citation: Ortiz-Olvera N, Fernández-Figueroa EA, Argueta-Donohué J, Miranda-Ortíz H and Ruiz-García E (2023) Case Report: Oral and fecal microbiota in a super-donor: the healthy microbiota paradigm for fecal transplantation. Front. Microbiomes 2:1219960. doi: 10.3389/frmbi.2023.1219960
Received: 09 May 2023; Accepted: 17 August 2023;
Published: 12 September 2023.
Edited by:
Lilian Terezinha Costa, Federal University of Rio de Janeiro, BrazilReviewed by:
Shaghayegh Baradaran Ghavami, Shahid Beheshti University of Medical Sciences, IranCopyright © 2023 Ortiz-Olvera, Fernández-Figueroa, Argueta-Donohué, Miranda-Ortíz and Ruiz-García. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erika Ruiz-García, YmV0emFiZTEwMEB5YWhvby5jb20ubXg=
†ORCID: Nayeli Ortiz-Olvera, orcid.org/0000-0002-3406-2918
Edith A. Fernández-Figueroa, orcid.org/0000-0002-5159-8300
Jesús Argueta-Donohué, orcid.org/0000-0003-3668-1066
Haydee Miranda-Ortíz, orcid.org/0000-0003-2275-3727
Erika Ruiz-García, orcid.org/0000-0002-0446-123X
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.