
 J. Anderson Thomson Jr.3,4
J. Anderson Thomson Jr.3,4

- 1 Department of Psychology, Neuroscience and Behaviour, McMaster University, Hamilton, ON, Canada
- 2 Virginia Institute for Psychiatric and Behavioral Genetics, Virginia Commonwealth University, Richmond, VA, USA
- 3 Counseling and Psychological Services, Student Health, University of Virginia, Charlottesville, VA, USA
- 4 Institute of Law, Psychiatry and Public Policy, University of Virginia, Charlottesville, VA, USA
Antidepressant medications are the first-line treatment for people meeting current diagnostic criteria for major depressive disorder. Most antidepressants are designed to perturb the mechanisms that regulate the neurotransmitter serotonin – an evolutionarily ancient biochemical found in plants, animals, and fungi. Many adaptive processes evolved to be regulated by serotonin, including emotion, development, neuronal growth and death, platelet activation and the clotting process, attention, electrolyte balance, and reproduction. It is a principle of evolutionary medicine that the disruption of evolved adaptations will degrade biological functioning. Because serotonin regulates many adaptive processes, antidepressants could have many adverse health effects. For instance, while antidepressants are modestly effective in reducing depressive symptoms, they increase the brain’s susceptibility to future episodes after they have been discontinued. Contrary to a widely held belief in psychiatry, studies that purport to show that antidepressants promote neurogenesis are flawed because they all use a method that cannot, by itself, distinguish between neurogenesis and neuronal death. In fact, antidepressants cause neuronal damage and mature neurons to revert to an immature state, both of which may explain why antidepressants also cause neurons to undergo apoptosis (programmed death). Antidepressants can also cause developmental problems, they have adverse effects on sexual and romantic life, and they increase the risk of hyponatremia (low sodium in the blood plasma), bleeding, stroke, and death in the elderly. Our review supports the conclusion that antidepressants generally do more harm than good by disrupting a number of adaptive processes regulated by serotonin. However, there may be specific conditions for which their use is warranted (e.g., cancer, recovery from stroke). We conclude that altered informed consent practices and greater caution in the prescription of antidepressants are warranted.
Introduction
Serotonin (5-hydroxytryptamine or 5-HT) is an ancient chemical, evolving at least one billion years ago, and it is present in fungi, plants, and animals (Azmitia, 2007). It belongs to a class of biochemicals called monoamines, which also includes norepinephrine (NE), and dopamine (DA). Many adaptive processes evolved to be regulated by serotonin, including cell differentiation, temperature, blood clotting, digestion and gut movement, insulin, electrolyte balance, astrocytic activity, neuronal apoptosis, cerebral blood flow, attention, aggression, mood, reproductive function, and mating behavior (Johnson and Thunhorst, 1997; Azmitia, 2001, 2007, 2010; Jimenez-Trejo et al., 2007; Fonseca et al., 2009; Paulmann et al., 2009).
Drugs affecting the serotonergic system are among the most widely prescribed psychiatric medications. Depression is the most common psychiatric condition for which people seek help (Pincus et al., 1999), and the symptoms are regulated, at least partly, by serotonin and norepinephrine (Heisler et al., 1998; Mayorga et al., 2001; Santarelli et al., 2003; Cryan et al., 2004; Amat et al., 2005, 2006). About 6.6% of adults, or about 13 million of the adult US population, are estimated to have depressive episodes that meet current criteria for major depressive disorder (MDD) within a year’s time (Kessler et al., 2007). Antidepressant medication is the most commonly prescribed treatment for depression (Olfson et al., 2002), and most of these drugs target serotonin and norepinephrine, although dopamine is affected to some degree as well (Stahl, 2008). Commonly prescribed classes of antidepressants are listed in Table 1. Antidepressants may also be prescribed to people with subclinical depression and many other conditions, including dysthymia, bipolar depression, schizoaffective disorder, post psychotic depression, generalized anxiety disorder, panic disorder (with or without agoraphobia), social phobia, substance abuse disorders, anorexia, bulimia, obsessive compulsive disorder, post-traumatic stress disorder, and chronic pain syndromes. Thus, millions of people are prescribed antidepressants and affected by them each year.
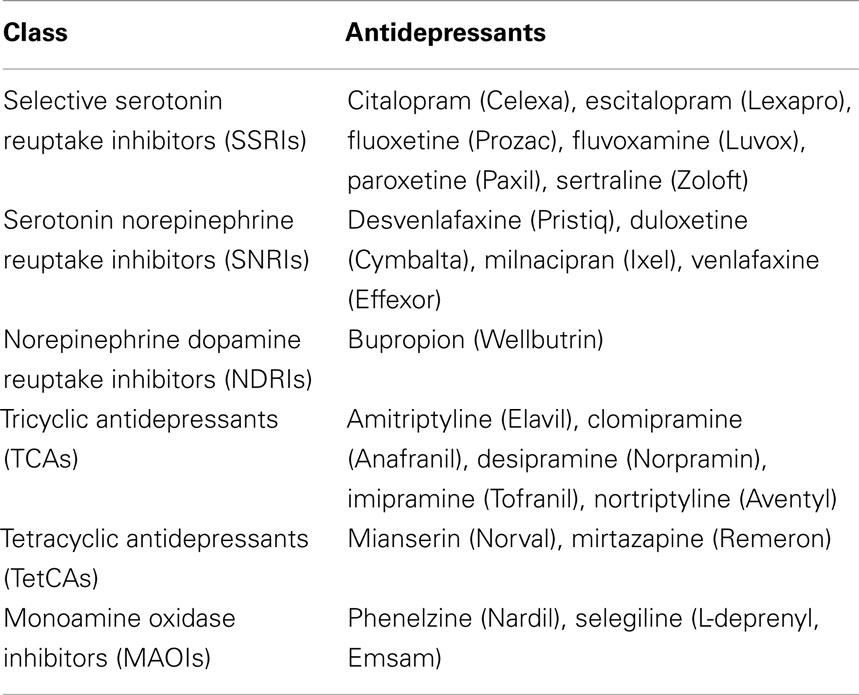
Table 1. Classes of antidepressant drugs along with their chemical and trade names (in parentheses).
The principle of primum non nocere requires physicians to do no harm. However, there is increasing concern that current diagnostic criteria and treatment practices may do more harm than good (Hagen, 2003; Horwitz and Wakefield, 2007; Kirsch et al., 2008; Andrews and Thomson Jr., 2009; Fournier et al., 2010; Wakefield et al., 2010; Andrews et al., 2011; Fava and Offidani, 2011), and these concerns are increasingly expressed in prominent public outlets (Lehrer, 2010; Angell, 2011). Moreover, many of the concerns have been driven by evolutionary conceptions about the nature of disorder (Wakefield, 1992, 1999; Watson and Andrews, 2002; Hagen, 2003; Horwitz and Wakefield, 2007; Andrews and Thomson Jr., 2009; Andrews et al., 2011).
It is a principle of evolutionary medicine that the disruption of adaptive processes will degrade biological functioning (Nesse and Williams, 1994). For instance, a growing body of evidence indicates that fever is an evolutionarily ancient adaptation for coordinating immune responses to infection (Kluger et al., 1997; Hasday et al., 2000; Blatteis, 2003; Appenheimer et al., 2005). Studies in humans have shown that the disruption of fever with antipyretic (fever-reducing) medication has a number of adverse effects on the immune response. In controlled experiments, antipyretic medication lengthens the time it takes the body to clear non-fatal infections such as rhinovirus (common cold) and Varicella zoster (chickenpox; Stanley et al., 1975; Doran et al., 1989; Graham et al., 1990). Acetaminophen also increased the duration of illness in Plasmodium falciparum (malaria) infections in an experimental paradigm (Brandts et al., 1997). In patients with more serious infections, such as bacterial sepsis, correlational studies commonly find fever is associated with increased survival (Bryant et al., 1971; Weinstein et al., 1978, 1983; Mackowiak et al., 1980; Swenson et al., 2007; Rantala et al., 2009). There has been little experimental research on the mortality effects of antipyretic therapy in humans with serious infections. In one experiment, ibuprofen did not affect mortality among patients with bacterial sepsis, but medication was not initiated until the infections had caused organ dysfunction (Bernard et al., 1997). In a preliminary analysis of another experiment where acetaminophen was administered to trauma patients close to the onset of fever, seven out of 44 (16%) who received medication died while only one out of 38 (3%) died in the group receiving no medication (Schulman et al., 2005). The difference was not quite statistically significant (p = 0.06), but the study was halted early. The university’s ethics review board had originally granted a waiver of informed consent based on the assumption that there was minimal risk involved, and the preliminary results strongly suggested that this assumption was erroneous.
Given the broad distribution of serotonin inside and outside of the brain (Berger et al., 2009), and the broad effects of serotonin on adaptive processes, antidepressants could have many adverse health effects. Despite considerable research, there has been little emphasis on the broad serotonergic effects of antidepressants on health in debates about antidepressant use.
In this article, we consider: (1) the broad effects of antidepressants on putative pathological processes thought to be involved in depression; (2) the effects of antidepressants on normal, adaptive processes in the body as a whole; and (3) the issue of whether antidepressants do more harm than good. Ultimately, we come down on the side that the benefits of antidepressants are generally outweighed by their costs (see Table 2), though there may be specific populations where their use is warranted. Notably, our argument does not depend on the efficacy of antidepressants relative to other possible treatments for depression. Rather, it depends on whether it is better for practitioners to refrain from prescribing rather than to start a patient on antidepressants. For this reason, we do not discuss non-pharmacological alternatives, although this is an active area of research. Of course, we do not advocate that practitioners not provide treatment; however, if results of a cost–benefit analysis suggest that providing no medication is a superior choice compared to that of prescribing antidepressants in most situations, then practitioners might be more inclined to employ other treatments with a lower side effect profile.

Table 2. Summary of costly (C) and beneficial (B) effects of antidepressant medications, with some estimate of their effect size or frequency.
Serotonin and Homeostasis
In animals, only about 5% of the body’s serotonin resides in the brain. Most of the body’s serotonin is housed in the gut, with 90% of that stored in enterochromaffin cells (where most of it is synthesized) and the remaining 10% is synthesized by and stored in myenteric interneurons (Gershon, 2004). Enterochromaffin cells also release serotonin into the blood stream, where most of it is taken up by platelets. Serotonin does not pass the adult blood–brain barrier, so central and peripheral sources do not communicate with each other (Hranilovic et al., 2011).
Serotonin is normally under homeostatic control in the brain, the gut, and blood plasma (Hyman and Nestler, 1996; Gershon, 2004; Lesurtel et al., 2008; Keszthelyi et al., 2009; Matondo et al., 2009; Best et al., 2010; Mercado and Kilic, 2010). Homeostasis involves the regulation of an important substance or physiological parameter within a narrow range around an equilibrium. Homeostatic mechanisms are classic examples of evolved adaptations because they maintain important physiological parameters at levels needed for proper biological functioning (Hochochka and Somero, 2002; Woods, 2009), and because they often have complex machinery that could only have evolved by natural selection (Andrews et al., 2011). Minimally, homeostatic mechanisms have a sensor for determining how far the parameter deviates from the equilibrium and feedback mechanisms for bringing the parameter back to equilibrium (Woods, 2009). For instance, the homeostatic control of core body temperature involves neuronal sensors in the preoptic anterior hypothalamus that connect with various efferent pathways to exert feedback and keep temperature at equilibrium (Romanovsky, 2007). Additionally, many homeostatic mechanisms can raise or lower the equilibrium in response to environmental contingencies. Thus, the body often responds to an infection by raising the core body temperature equilibrium – otherwise known as fever (Romanovsky et al., 2005). Feedback mechanisms then maintain core body temperature around this elevated equilibrium.
We now discuss, in turn, how serotonin is homeostatically regulated in the brain, gut, and blood plasma.
Serotonin in the Brain
In the brain, serotonergic neurons originate in the raphe nuclei and project widely to various regions. The dorsal raphe nucleus is the primary source of neurons projecting to forebrain areas. After serotonin is released into the synapse, it is eventually taken back into the presynaptic neuron by serotonin transporter molecules where it is broken down by the enzyme monoamine oxidase A.
Synaptic levels of serotonin are homeostatically regulated, in part, by 5-HT1A autoreceptors located on the somatodendritic regions of serotonergic neurons that control neuronal firing and 5-HT1B autoreceptors located in terminal regions that control the synthesis of serotonin (Best et al., 2010). Thus, when synaptic levels of serotonin are perturbed upwards from their equilibrium level, autoreceptor activity causes a decrease in the firing of serotonergic neurons and a reduction in synthesis, both of which tend to keep synaptic levels relatively stable (Best et al., 2010). Conversely, when synaptic levels are perturbed downwards from equilibrium, firing and synthesis increase to maintain equilibrium levels (Best et al., 2010).
Serotonin in the Gut
Most of the serotonin in the body is produced by enterochromaffin cells that line the digestive tract from the stomach to the colon. Enterochromaffin cells turn over quickly because they form part of the epithelial layer, so neurons cannot form tight junctions with them. Consequently, enterochromaffin cells produce large amounts of serotonin to compensate for the large intervening distance from neurons (Gershon, 2004). However, too much extracellular serotonin in the intestinal lining is harmful and can cause irritable bowel syndrome, characterized by symptoms of pain, diarrhea, constipation, indigestion, bloating, and headache (Gershon, 2004). There are no extracellular enzymes for catabolizing serotonin, and mucosal epithelial cells play an important role in maintaining homeostasis by clearing extracellular serotonin via the same transporter that is expressed in the brain (Gershon and Tack, 2007). These cells then catabolize serotonin by monoamine oxidase or other pathways (Gershon, 2004).
Serotonin in Blood Plasma
The overflow of serotonin produced by enterochromaffin cells is also released into the bloodstream (Gershon and Tack, 2007). Blood plasma levels of serotonin are regulated by platelet cells via the action of the serotonin transporter, which is expressed on the platelet cell membrane (Linder et al., 2007; Mercado and Kilic, 2010).
Transporter molecules are stored in vesicles in the platelet cytoplasm and trafficking between the surface and cytoplasmic stores takes place as a function of plasma levels of serotonin (Mercado and Kilic, 2010). When plasma levels of serotonin are low, a relatively small increase in serotonin triggers a process by which the surface expression of the transporter is increased, promoting the accumulation in platelets and maintaining low plasma levels. Platelets store serotonin in large granules that are crucial to the clotting process (Heger and Collins, 2004), so this homeostatic mechanism for regulating plasma serotonin levels is important for ensuring that clotting mechanisms are primed for the onset of any injuries.
Various cells in the skin (melanocytes, mast cells, Merkel cells) can synthesize and store serotonin (Nordlind et al., 2008). When an injury that punctures the skin takes place, free serotonin levels rapidly increase in the area around the wound (Hernandez-Cueto et al., 2000). A large localized increase in plasma serotonin levels can reverse the surface expression of transporter in platelets (Mercado and Kilic, 2010). Specifically, trafficking reverses direction, reducing transporter expression on the surface, increasing transporter accumulation in cytoplasmic vesicles, and reducing the further uptake of serotonin into the platelet. Moreover, high plasma levels of serotonin initiate a complex process by which the cytoplasmic stores of serotonin trigger the release of procoagulants from the platelet (Mercado and Kilic, 2010), which promotes the clotting process at the site of injury.
The Effects of Antidepressant Medications on Body Systems
According to the most prominent view in medicine and psychiatry, disorders stem from a breakdown or a decline in biological functioning [American Psychiatric Association (APA), 2000b]. Since natural selection is the only natural force capable of generating biological functions, and traits with biological functions are called adaptations, disorders can be understood as involving a breakdown or a decline in the functioning of evolved adaptations (Wakefield, 1992, 1999). In principle, then, interventions that degrade or disrupt the functioning of homeostatic mechanisms can cause disorder.
Antidepressants are most commonly taken in pill form, though there are some that can be taken through a transdermal patch. In either case, they enter the bloodstream, pass through the blood–brain barrier, and affect neuronal functioning. Antidepressants perturb monoamine levels through a variety of mechanisms, the most common of which is by binding to monoamine transporters. In the normally functioning rodent brain, transporter blockade prevents the reuptake of monoamines into the presynaptic neuron, which causes extracellular monoamines to increase from equilibrium levels in forebrain regions within minutes to hours of administration (see Figure 1; Rutter and Auerbach, 1993; Bymaster et al., 2002; Felton et al., 2003). With prolonged antidepressant use, however, the brain’s homeostatic mechanisms buffer this effect by making a number of compensatory changes (Best et al., 2010), including an inhibition of synthesis that causes the entire pool of serotonin in the forebrain (intracellular plus extracellular) to decline (Honig et al., 2009). Consequently, extracellular levels in the forebrain return to equilibrium levels with prolonged treatment (Popa et al., 2010). There are other changes that take place with chronic antidepressant use to maintain homeostasis, including alterations in the density and functioning of serotonin receptors, transporters, and enzymes (Holt and Baker, 1996; Hyman and Nestler, 1996; Adell et al., 2005; Kovacevic et al., 2010).
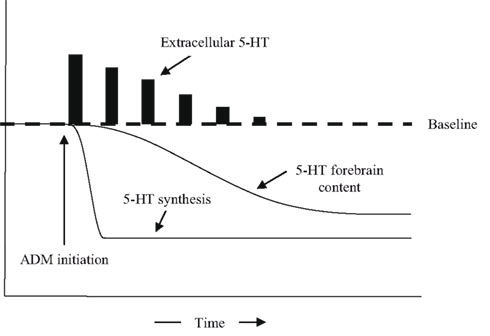
Figure 1. The effects of antidepressant medications on extracellular levels of serotonin, the synthesis of serotonin, and overall forebrain levels of serotonin, as a function of time.
But antidepressants also spread widely throughout the body. Because the serotonin transporter plays a crucial role in the homeostatic regulation of serotonin in the gut and plasma, antidepressants can also influence serotonergic processes in the periphery.
There are a number of ways that antidepressants can disrupt properly functioning homeostatic mechanisms in the brain and periphery and cause disorder. The first way is derived from the fact that it takes several weeks for the decline in synthesis to bring extracellular levels of serotonin in the brain back down to equilibrium levels. During this time, serotonin levels are higher than what they should be. Antidepressants could potentially cause disorder prior to the restoration of equilibrium.
Second, the long-term use of antidepressants can strain homeostatic regulatory mechanisms, because they must make a number of accommodations to restore equilibrium, and this can degrade the functioning of those mechanisms. For instance, it has been suggested that depression could potentially be the result of decline in the functioning of regulatory mechanisms caused by prolonged stress (McEwen, 2000; Ganzel et al., 2010). The same principle suggests that long-term antidepressant use could potentially cause degradation in the functioning of the homeostatic mechanisms controlling serotonin.
Third, antidepressants can trigger disorder when they are discontinued. Even though equilibrium is eventually restored with prolonged antidepressant use, it only does so because the accommodations made by the brain oppose the action of antidepressants (Andrews et al., 2011; Fava and Offidani, 2011). When antidepressants are discontinued, the accommodations made by the brain are unopposed, which should again cause serotonin levels to deviate from equilibrium. Thus, the discontinuation of antidepressants can cause disorder until the brain reverses the accommodations made during treatment.
Finally, antidepressants can cause disorder by effectively disabling a key part of the homeostatic mechanism. For instance, the homeostatic regulation of serotonin in the gut and the blood plasma rely primarily on the serotonin transporter (Gershon, 2004; Mercado and Kilic, 2010). By blocking the transporter, antidepressants interfere with a key part of the homeostatic mechanism such that it is not possible to return the system to the equilibrium.
As we discuss the negative effects of antidepressants on serotonin-regulated processes throughout the body, we sometimes do not know precisely which of these mechanisms is at work or whether other unknown mechanisms are at work. But these mechanisms are proof of the principle that antidepressants can have transient or long-lasting effects that disrupt the proper functioning of homeostatic mechanisms.
Depressed Mood
Research in rodents in which the homeostatic mechanisms regulating serotonin and norepinephrine are temporarily or permanently disabled has provided strong experimental evidence that monoamines play a role in regulating the behavioral symptoms of depression (Heisler et al., 1998; Mayorga et al., 2001; Santarelli et al., 2003; Cryan et al., 2004; Amat et al., 2005, 2006). However, the precise relationship between serotonin and depressive symptoms is a matter of debate (Sapolsky, 2004; Andrews and Thomson Jr., 2009). The oldest neurochemical disorder hypothesis for depression posits that serotonin and other monoamines are depleted – at least in people with severe episodes of depression – and antidepressants correct this imbalance. For the purposes of this paper, we do not need to know the precise relationship. The fact that depressive symptoms are under serotonergic control, and serotonin is under homeostatic control, suggests that the brain could push back against the mood lowering effects of antidepressants during treatment (Andrews et al., 2011).
Modest Efficacy of Antidepressants during Treatment
Antidepressants are widely thought to be effective in reducing symptoms. However, recent research strongly suggests that they are only modestly effective at best. For instance, studies showing a positive effect of antidepressants over placebo have been selectively published (Turner et al., 2008). Turner et al. (2008) applied to the US Food and Drug Administration (FDA) under the Freedom of Information Act (FOIA) to get access to the published and unpublished studies submitted by pharmaceutical companies to get governmental approval for their drugs. They found that 94% of the published studies showed an advantage of antidepressants over placebo. When the published and unpublished studies were considered together, only 51% showed an advantage over placebo.
Kirsch et al. (2008) also applied to the FDA under the FOIA to understand how much more effective antidepressants were at reducing depressive symptoms compared to placebo. Changes in depressive symptoms were measured with the 17-item Hamilton Depression Rating Scale (HDRS; Hamilton, 1960), the most common instrument for assessing the efficacy of antidepressant medications in clinical trials. Scores can range from 0 to 53, and researchers interpret them in a variety of ways (Kearns et al., 1982; Cohn and Wilcox, 1992; Angst et al., 1993). The American Psychiatric Association (APA) (2000a) specifically mentions the scheme used by Kearns et al. (1982), and it is formally used by the National Institute for Clinical Excellence (NICE) in Great Britain (National Institute for Clinical Excellence, 2004) (see Table 3).

Table 3. Interpretation of HDRS score.
Two other things about the HDRS are noteworthy. First, patients with a score of 13 or higher typically meet formal diagnostic criteria for an episode of MDD (Bagby et al., 2004). In other words, many people who meet diagnostic criteria for MDD only have mild/moderate symptoms according to NICE guidelines. Second, NICE guidelines require an antidepressant drug to reduce symptoms by three HDRS points or more than placebo to be considered clinically significant (National Institute for Clinical Excellence, 2004).
On average, Kirsch et al. (2008) found that patients on placebo experienced a 7.8 point reduction in HDRS symptoms while those on antidepressants experienced a 9.6 point reduction. This might seem like both groups improved substantially, but with the exception of one study, patients in all studies were, on average, in the “very severely” depressed range (mean HDRS ≥ 23) at entry. In other words, even with the improvement in symptoms caused by both placebo and antidepressants, most patients would probably still have satisfied diagnostic criteria for MDD. Moreover, on average, antidepressants reduced symptoms by only 1.8 HDRS points more than placebo. While this was a statistically significant difference, this was not sufficient to meet NICE clinical significance guidelines. The difference between antidepressant and placebo did increase with initial HDRS scores, and it reached clinical significance with baseline scores of 28 or higher. However, antidepressants did not reach clinical significance at high HDRS scores because they were increasing in efficacy; rather, it was due to a decline in the efficacy of placebo.
Interpreted straightforwardly, the results suggest that antidepressants do not really have much clinical effect on depressive symptoms, except perhaps in patients in the “very severe” range. The finding that antidepressants have modest symptom reducing effects relative to placebo has been replicated in several other studies (Khan et al., 2002, 2005, 2011; Fournier et al., 2010). The British consider the difference between placebo and antidepressant to be small enough to recommend that antidepressants not be used except in cases of severe depression (Bonin, 2012). Recently, in an interview on the news program 60 Minutes, a representative of the US FDA stated that the symptom reducing effect of antidepressants relative to placebo is “rather small” (Bonin, 2012).
Some have taken the evidence of the limited efficacy of antidepressants as evidence that serotonin is not involved in regulating depressive symptoms. However, limited efficacy is also expected under the hypothesis that the homeostatic mechanisms regulating serotonin are still intact, because the brain is expected to push back against the effects of antidepressants (Andrews et al., 2011).
Effects of Prolonged Antidepressant Treatment
Even among those who respond to antidepressant treatment, longer-term use is associated with a loss of symptom reducing efficacy – sometimes causing a full-blown relapse. This is also consistent with the brain pushing back against the symptom reducing effect of antidepressants. In an early review, studies showed that 9–57% of long-term antidepressant users met formal criteria for a relapse or a recurrence (Byrne and Rothschild, 1998). More recent studies have found similarly high rates of relapse among those who initially remitted on the drug. In one study of fluoxetine, 35.2% met relapse criteria after 6 months of continuous treatment, increasing to 45.9% after 12 months (McGrath et al., 2006). In another study, 68% of patients who initially met remission criteria, and were exposed only to continuous antidepressant treatment, had a relapse over a 2 year period (Bockting et al., 2008). Of course, these studies only report increases in symptoms that meet formal criteria for a relapse. A more general loss of efficacy with prolonged antidepressant use must be substantially higher.
Sequenced Treatment Alternatives to Relieve Depression
The results of the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study are worth detailed description. This study has widely been reported as evidence that antidepressants are effective in the long-term reduction of depressive symptoms, especially if an alternative antidepressant with a broader monoaminergic profile is administered after a narrower one fails (Insel and Wang, 2009). The 3110 patients who met inclusion criteria for STAR*D were put through a maximum of four sequential pharmacological treatments, where a new treatment was initiated conditional on failing to respond to the prior one. The overall remission rate for patients who completed all treatment steps was reported to be 67% (Rush et al., 2006).
However, STAR*D did not include a placebo control group, so any positive responses are not simply attributable to antidepressants – they are attributable to both antidepressants and the placebo effect. Since there was no placebo group, it is not possible to estimate how effective antidepressants were. Moreover, focusing on only those patients who completed all the treatment steps obscures the fact that 93% of the 1518 patients who met criteria for remission in one of the four steps either relapsed during 12 months of treatment or dropped out of the study (Pigott et al., 2010). Even with no placebo control group, this awkward fact also suggests that antidepressants have limited long-term efficacy at best. The 93% relapse/dropout rate was not reported in the primary STAR*D publications. Rather, it was reported by outside researchers who re-analyzed the data (Pigott et al., 2010). These outside researchers have also documented numerous instances of apparent bias in the reporting of results by STAR*D researchers (Pigott et al., 2010).
Increased Risk of Relapse after Antidepressant Discontinuation
When homeostatic mechanisms are perturbed from equilibrium, they produce an opposing force that is proportional to the degree of perturbation. For instance, when you compress a spring from its equilibrium position, it produces an opposing force and the strength of this force increases as you compress it further. Moreover, when you release your hand from the compressed spring, it first overshoots the equilibrium position before returning to equilibrium, and the degree of overshoot is proportional to how much you compressed it.
Antidepressants vary in how much they perturb forebrain monoamine levels, which is analogous to saying that they compress the spring to varying degrees. If the homeostatic mechanisms regulating monoamine levels are properly functioning in most people diagnosed with MDD, then we should see an overshoot in depressive symptoms when they are discontinued. Moreover, the degree of overshoot should be proportional to the strength of the antidepressant.
To test the overshoot prediction, we recently conducted a meta-analysis of studies in which antidepressants are discontinued. Data are lacking on how much antidepressants perturb monoamines in the human brain because it requires invasive techniques, so we examined how much they increase monoamines in a region of the rodent prefrontal cortex implicated in controlling depressive symptoms (Amat et al., 2005). Placebo has no effect on monoamine levels in the rodent brain, but the strongest antidepressants can increase prefrontal monoamine levels by 400% or more (Bymaster et al., 2002). After controlling for covariates, we found strong positive relationships between the risk of relapse after discontinuation and the degree to which the antidepressant used in the study increases serotonin (Figure 2) and norepinephrine (Figure 3). In other words, the more antidepressants perturb monoamine levels in the brain, the more the brain appears to push back, which increases the risk of relapse when the drug is discontinued. This positive relationship also strongly suggests that patients who get better without the use of antidepressants (i.e., when serotonin and norepinephrine levels are not perturbed) will have a lower risk of relapse.
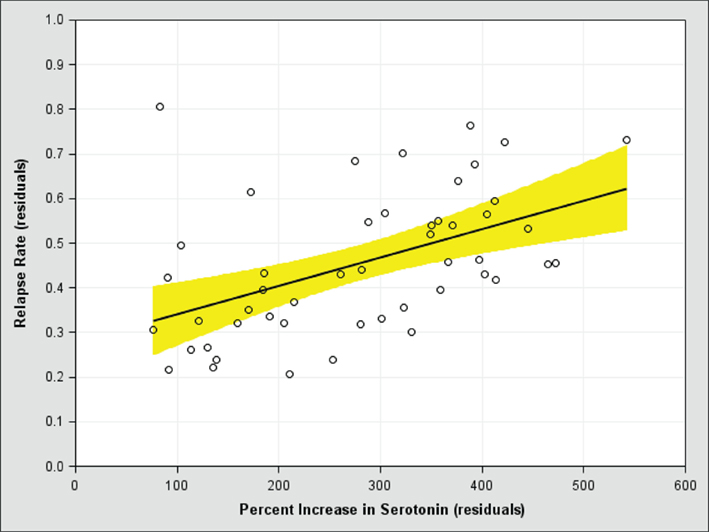
Figure 2. The risk of relapse after antidepressant discontinuation (y-axis) versus the perturbational effect of antidepressants on serotonin in the rodent medial prefrontal cortex (x-axis), after controlling for covariates. A score of 100 on the x-axis means the antidepressant has no effect on serotonin levels.
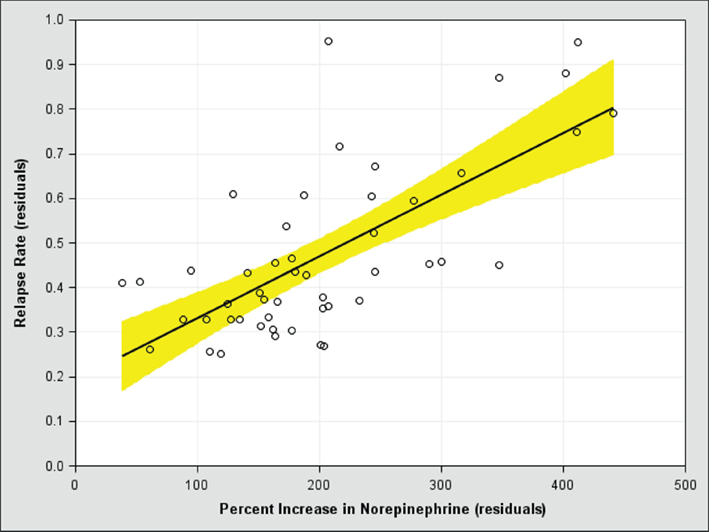
Figure 3. The risk of relapse after antidepressant discontinuation (y-axis) versus the perturbational effect of antidepressants on norepinephrine in the rodent medial prefrontal cortex (x-axis), after controlling for covariates. A score of 100 on the x-axis means the antidepressant has no effect on norepinephrine levels.
Altogether, the results of our meta-analysis further support the hypothesis that the homeostatic mechanisms regulating serotonin and norepinephrine are pushing back against the effects of antidepressants. The fact that this pushback occurs in patients meeting current diagnostic criteria for MDD casts some doubt on the hypothesis that serotonin levels are dysregulated in such patients. This is because the sensor and negative feedback components of the homeostatic mechanisms must be functioning to produce pushback against the effects of antidepressants. However, our results do not rule out some dysfunction in the component that sets the equilibrium.
The results also are inconsistent with hypotheses that antidepressants interrupt stress responses and give the brain a chance to heal so that it can become more resilient to depression (Sapolsky, 2001; Kramer, 2005). Instead, antidepressant use appears to increase susceptibility to depression.
To show how different antidepressants affect the risk of relapse, for this paper we conducted a regression analysis on this dataset including the major antidepressant classes and controlling for the covariates we identified as being important in our prior work. Fluoxetine is unique among the SSRIs in that it increases prefrontal norepinephrine levels through a mechanism other than reuptake blockade (Bymaster et al., 2002), so we separated it from the other SSRIs. The estimated 3-month risk of relapse for patients who had remitted while on placebo was only 21.4%, while the risk after antidepressant discontinuation generally increased with the monoaminergic perturbational effect of the class: 43.3% (SSRI), 47.7% (SNRI), 55.2% (TCA), 61.8% (fluoxetine), and 75.1% (MAOI).
Neuronal Proliferation, Death, and Differentiation
Serotonin is involved in shaping the brain through a number of developmental processes, including cell differentiation, neuronal apoptosis (programmed neuronal death), neurogenesis (the birth and growth of new neurons), and neuroplasticity (Azmitia, 2001). Because of the complex role that serotonin plays in shaping the brain, antidepressants could have complex effects on neuronal functioning.
Many studies have been conducted purporting to show that antidepressants promote neurogenesis (Hanson et al., 2011), and some have argued that this could be a fundamental and beneficial part of the antidepressant response (Santarelli et al., 2003; Duman, 2004; Warner-Schmidt and Duman, 2006; Perera et al., 2011). Many also consider neurogenesis to be useful because it could promote healing of neuronal damage that is purportedly caused by depression (Sapolsky, 2001).
At the same time, the assumption that neurogenesis is a beneficial effect of antidepressants should not be accepted uncritically. Neurogenesis is carefully regulated over the life span because cognitive functioning does not bear a simple relationship to the number of neurons in the brain. In fact, if antidepressants were really effective in promoting the proliferation of new neurons, clinicians would have to weigh any possible utility of antidepressants with the possibility that they could trigger brain tumors (Jackson, 2009). However, there is growing in vitro evidence that antidepressants reduce gliomas and neuroblastomas, and these effects are mediated by neuronal apoptosis (Levkovitz et al., 2005; Cloonan and Williams, 2011). In fact, a recent epidemiological study suggests that prolonged TCA use in humans may protect against gliomas (Walker et al., 2011), although antidepressants may increase the risk of other forms of cancer (Cosgrove et al., 2011). Apoptotic effects are not limited to neoplastic tissue. Antidepressants have been found to cause cell death in non-cancerous hippocampal neurons in vitro (Post et al., 2000; Bartholoma et al., 2002) and in vivo (Sairanen et al., 2005). They have also been found to cause cell death in sperm cells (Tanrikut et al., 2010). In short, there is good evidence from several different lines that antidepressants trigger apoptosis.
It would be extremely odd if antidepressants directly and concomitantly promoted both neurogenesis and neuronal apoptosis. In fact, the evidence of antidepressant triggered neurogenesis is equivocal (Jackson, 2009). At the heart of the matter is the fact that all the studies testing for neurogenesis use a method called 5-bromo-2′-deoxyuridine (BrdU). Other methods may be used as well, but BrdU is nearly always used. BrdU is an analog of the nucleoside thymidine that is incorporated into DNA during synthesis, and the incorporated compound can be detected using immunohistochemistry. Thus, BrdU is a marker of DNA synthesis, which might make it seem to be a good marker of cellular proliferation, since DNA must be synthesized for a cell to divide. However, the interpretation of the BrdU signal is complicated by the fact that BrdU can be incorporated into DNA during processes other than neurogenesis, including DNA repair, abortive cell cycle reentry, and DNA duplication without cellular division (Taupin, 2007). Of particular relevance is the fact that DNA is often synthesized during processes that lead to apoptosis (Taupin, 2007). The interpretational problems with BrdU have led one reviewer to state that BrdU is “one of the most misused techniques in neuroscience” (Taupin, 2007, p. 198). Other commentators have stated that “bromodeoxyuridine (BrdU) labeling is often uncritically accepted as proof of neurogenesis when it may well be attributable to a cell cycle-related cell death” (Herrup et al., 2004, p. 9232).
More recently, researchers have used other methods in conjunction with BrdU to attempt to ascertain the fate of neurons after antidepressant use. Common methods involve assessing neurons for Ki-67 and doublecortin (DCX), both of which are proteins expressed by maturing neurons still going through the cell cycle, and NeuN, which is thought to be a marker of mature neurons that is expressed after doublecortin is downregulated. Positive signals for Ki-67, DCX, or NeuN are frequently interpreted as providing stronger evidence that antidepressants have triggered neuronal growth.
However, a recent study using more sophisticated methodology found no evidence that the antidepressant fluoxetine increased neurogenesis (Kobayashi et al., 2010). But it did find evidence that fluoxetine caused mature neurons to take on immature functional characteristics, including an immature profile of synaptic plasticity and gene expression. Since Ki-67 and DCX are expressed in immature neurons, it is quite possible that dematured neurons increase their expression of Ki-67 and DCX. Importantly, Kobayashi and colleagues found that the dematured neurons still expressed NeuN. Altogether, it is not clear whether any of the methods typically used to assess neuronal growth and maturation are in fact responding only to apoptosis and dematuration.
The dematuration of neurons could be caused by the reduction of serotonin synthesis that occurs when the brain pushes back against the synaptic-enhancing effects of antidepressants (see Figure 1; Honig et al., 2009). Constant serotonergic input is needed to maintain the mature state of neurons (Azmitia, 2001). When serotonin synthesis is inhibited by parachlorophenylalanine, the cytoskeleton disassembles and synapses and dendrites are lost as neuronal processes retract, all of which indicate a return to an immature, undifferentiated state (Chen et al., 1994; Wilson et al., 1998; Azmitia, 2001).
Neuronal dematuration, particularly the retraction of neuronal processes, appears to play a role in triggering apoptosis (Azmitia, 2001), which could explain why antidepressants induce neuronal death. But the functional reason for why “dematuration” triggers apoptosis is not precisely clear. Apoptosis plays a role in removing damaged cells and maintaining homeostasis of differentiated tissues by regulating the equilibrium between the cell death and cell birth (Hengartner, 2000). To maintain tissue homeostasis, inappropriately proliferating cells must be detected and killed. Thus, the body must be able to differentiate between controlled and uncontrolled proliferation to appropriately direct apoptotic processes. Dematuration might be viewed as a signal that the neuron is changing into a malignant proliferative state, making it a target for apoptosis. Similarly, pharmacologically forcing mature neurons to go through a cell cycle causes them to die through apoptotic processes (Herrup et al., 2004), perhaps because the sudden change in proliferative status marks them as possible neoplasms.
Another way antidepressants could target neurons for apoptosis is by directly inflicting structural damage on them, since damaged neurons are often the targets of apoptosis (Cotran et al., 1998; Stergiou and Hengartner, 2004). We are only aware of one study that has tested whether antidepressants cause structural damage to neurons (Kalia et al., 2000). The study authors found that exposure to clinically relevant doses of fluoxetine (11.4 mg/kg, oral) or sertraline (28.6 mg/kg, oral) for only 4 days caused shortened axons, kinks, and swollen nerve terminals in the brains of otherwise healthy rodents. Such morphological changes are often taken as de facto evidence of neuronal damage (Kalia et al., 2000), and similar morphological features are thought to play a role in Parkinson’s disease (Cheng et al., 2010).
Even if they do not directly promote neurogenesis, antidepressants could do so indirectly. When pharmacological interventions trigger neuronal apoptosis, the brain attempts to homeostatically compensate by upregulating neurogenesis. For instance, the induction of neuronal apoptosis in the hippocampus by kainic acid was subsequently followed by an upregulation in neurogenesis in the dentate gyrus and CA3 (Dong et al., 2003). The evidence of neurogenesis was not significant 18 days after the administration of kainic acid, but it was significant after 33 days. One study has shown that fluoxetine increased neuronal progenitor cells, but this was only 30 days after fluoxetine treatment had stopped (Encinas et al., 2006). It seems possible that antidepressants only induce neurogenesis indirectly as a result of the brain attempting to compensate for their apoptotic effects.
In general, neuronal dematuration and damage are likely to interfere with proper brain functioning (Jackson, 2008, 2009). The Parkinsonian-like morphological characteristics caused by antidepressants could explain why antidepressants can induce tardive dyskinesia (involuntary, repetitive body movements; El-Mallakh et al., 2011). In rodents, repeated administration of antidepressants has been found to impair performance on a variety of learning tasks, including passive avoidance, elevated plus maze, and three-panel runway tasks (Ulak et al., 2006; Mutlu et al., 2011). In humans, a recent large epidemiological study has found that prolonged antidepressant use was associated with a 70% increase in the risk of mild cognitive impairment in elderly women and an increased risk of probable dementia (Goveas et al., 2011).
Attention
A common symptom of depression is difficulty concentrating. This is due to the fact that depressed people often have persistent thoughts related to their episode called ruminations that are resistant to distraction and difficult to suppress. These ruminations interfere with the ability to concentrate on other things. In other words, depression clearly affects the allocation of limited attentional resources – particularly working memory (Andrews and Thomson Jr., 2009). These attentional resources are at least partly controlled by serotonin (Carter et al., 2005; Andrews and Thomson Jr., 2009). Consequently, antidepressants could disrupt attentional processes.
One study has shown that the antidepressant sertraline decreases rumination in patients with dysthymia (Kelly et al., 2007). The reduction of rumination is widely thought to be beneficial, however the scientific literature suggests otherwise. Interventions that disrupt rumination (e.g., distraction, thought suppression) do tend to reduce symptoms in the short-term (Morrow and Nolen-Hoeksema, 1990; Nolen-Hoeksema and Morrow, 1993; Vickers and Vogeltanz-Holm, 2003; Park et al., 2004; Andrews et al., 2007), but they tend to increase symptoms in the long-term (Schmaling et al., 2002; Wenzlaff and Luxton, 2003; Hayes et al., 2005), suggesting that the short-term effects are palliative and do not address cause (Andrews and Thomson Jr., 2009). On the other hand, interventions that encourage rumination (e.g., writing about one’s strongest thoughts and feelings related to the episode) have been found to increase insight and shorten the duration of episodes (Hayes et al., 2005, 2007; Gortner et al., 2006; Graf et al., 2008), suggesting that these interventions address cause (Andrews and Thomson Jr., 2009). In other words, the disruption of rumination may not be productive.
In any event, other research shows that antidepressants have negative effects on attention. When administered to non-depressed volunteers for several weeks, they tend to impair cognition (Hindmarch, 2009), particularly on tasks that require highly focused working memory or vigilance (Schmitt et al., 2001, 2002; Riedel et al., 2005).
Of particular interest are experiments showing that antidepressants interfere with driving performance – a task that requires sustained vigilance (Ramaekers et al., 1995; O’Hanlon et al., 1998; Wingen et al., 2005). Recently, these researchers have extended their studies to examine the effects of antidepressants and other prescription drugs on real world driving accidents using a primary care database in the United Kingdom (Gibson et al., 2009). Using the year prior to drug exposure as a baseline [incident rate ratio (IRR) = 1], people who took SSRIs were at a greater risk of a driving accident in the 4-week period before first being prescribed the drug (IRR = 1.7, 95% CI = 1.47–1.99). This suggests that depression, anxiety, or other conditions that lead to antidepressant use are risk factors for accidents. In the first 4-week period of SSRI use, however, the risk of driving accidents returned to baseline (IRR = 0.92, 95% CI = 0.75–1.12). Taken in isolation, this would seem to suggest antidepressants protect against driving accidents. But SSRIs only reduce symptoms after several weeks of continuous use, and this was not a placebo-controlled study, so it is possible that the reduced risk was attributable to other effects of seeking help rather than the SSRI. Indeed, there was a similar reduction in driving accident risk in the 4-week period of first use of other drugs that interfere with attention (benzodiazepines, non-benzodiazepine hypnotics, beta-blockers, opioids, and antihistamines). After 4 weeks of use, however, the risk of driving accidents increased and remained increased for the duration of SSRI treatment (IRR = 1.16, 95% CI = 1.06–1.28). Once SSRI treatment was discontinued, the risk of accidents returned to baseline (IRR = 1.03, 95% CI = 0.92–1.16). Similarly, benzodiazepines, non-benzodiazepine hypnotics, opioids, and antihistamines showed an increased risk of accidents with prolonged use that returned to baseline upon discontinuation. Overall, the pattern suggests that SSRIs – as well as benzodiazepines, hypnotics, opioids, and antihistamines – increase the risk of driving accidents.
Antidepressant use is associated with an increased risk of falling and bone fractures in the elderly (Cumming, 1998). This may also be attributable to the effects of antidepressants on attention, since avoiding falls requires vigilance in the elderly (Holtzer et al., 2007). However, the risk of bone fracture after a fall is probably increased because SSRIs lower bone mineral density (Moret et al., 2009).
Gastrointestinal Function
Antidepressants that block the serotonin transporter can cause long-term increases in serotonin in the gut because transporter uptake is the primary mechanism for clearing serotonin. There are backup transporters that can take up serotonin in the gut, but they are less efficient and do not completely compensate for the loss of function of the serotonin transporter (Gershon and Tack, 2007). As discussed above, elevated levels of serotonin in the intestinal lining are associated with irritable bowel syndrome, characterized by pain, diarrhea, constipation, indigestion, bloating, and headache. Antidepressants that bind to the serotonin transporter could cause similar symptoms. In fact, pain, diarrhea, constipation, indigestion, bloating, and headache are common side effects of antidepressants, with frequencies ranging from 13.8 to 22.9% in one recent study (Zimmerman et al., 2010).
Platelet Activation and the Clotting Process
Serotonin is involved in platelet activation, which is important in the clotting process (Heger and Collins, 2004). Platelets take in serotonin from the plasma via the serotonin transporter, and the serotonin molecules aggregate in the form of dense granules. When these granules are released, they activate processes that cause pro-aggregating proteins to be released from the platelet (Heger and Collins, 2004). Thus, the clotting process requires stores of serotonin in platelets.
Platelet activation also plays a role in atherosclerosis and thrombosis (Gawaz et al., 2005; Weber, 2005). Research on depressed patients has shown that plasma levels of serotonin are reduced, platelet levels are elevated, and pro-aggregating processes are enhanced, possibly increasing the risk of cardiovascular events (Musselman et al., 2000; Bakkaloglu et al., 2008; Flock et al., 2010).
Antidepressants that bind to the serotonin transporter, such as the SSRIs, can affect processes that depend on platelet activation. SSRIs block the uptake of serotonin into the platelet, which causes serum levels of serotonin to increase, platelet levels to decrease, and the inhibition of pro-aggregating processes (Musselman et al., 2000; Bakkaloglu et al., 2008; Flock et al., 2010).
Abnormal Bleeding
Consistent with inhibition of the clotting process, there is growing evidence that SSRIs increase the risk of abnormal bleeding. Antidepressant users are more likely to be hospitalized for abnormal bleeding (Meijer et al., 2004) and lose more blood during elective surgery (van Haelst et al., 2010). A recent population based case-control study in Denmark found that SSRI users are also at modestly increased risk of upper gastrointestinal bleeding, but the risk increases further when SSRIs are used in association with other anti-thrombotic drugs, such as NSAIDs (Table 4; Dall et al., 2009).
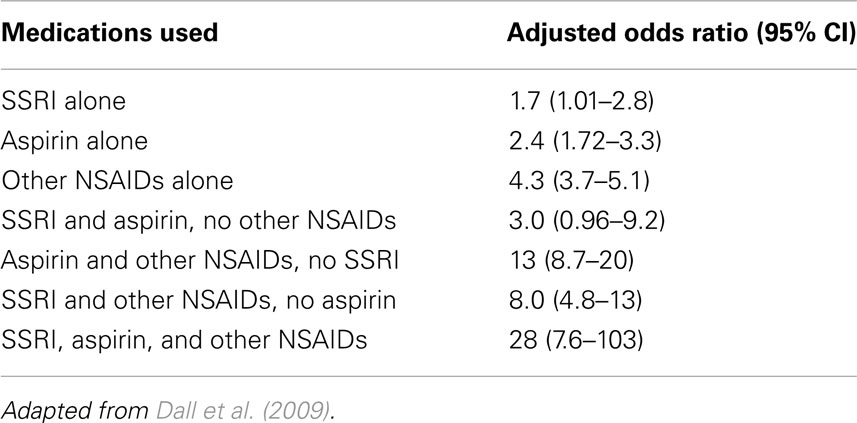
Table 4. Risk of upper gastrointestinal bleeding as a function of current use of SSRIs, aspirin, and other NSAIDs.
Cardiac Events
Depression is associated with an increased risk of cardiovascular disease, particularly myocardial infarction, coronary heart disease, and stroke (Van der Kooy et al., 2007; Pozuelo et al., 2009). As discussed above, these increased risks may be partly due to the effects of serotonin on platelet activation. Researchers have therefore speculated that antidepressants that bind to the serotonin transporter might be useful treatments for cardiac patients.
A recent study suggests that antidepressant use may help protect cardiac patients against cardiac related death while in the hospital (Hata et al., 2011). However, several limitations of the study are worth noting: specifically, the sample size was small, patients were not randomized to the antidepressant treatment, and it was not placebo-controlled. Indeed, the epidemiological literature on antidepressant effects on cardiac events is mixed. TCAs are generally associated with an increase in cardiac events, due to anticholinergic properties, NE reuptake blockade, and other effects (Cohen et al., 1996; Tata et al., 2005). However, studies on SSRIs have produced mixed results, with some showing a protective effect (Sauer et al., 2003; Monster et al., 2004; Schlienger et al., 2004), others showing no effect (Cohen et al., 2000; Meier et al., 2001), and still others showing an increased risk of cardiac events (Tata et al., 2005). Moreover, controlled trials have not, at least thus far, shown that SSRIs or psychotropic treatments have much effect on cardiac events in cardiac patients (Pozuelo et al., 2009).
Cerebrovascular Events
In principle, the anticoagulant properties of SSRIs could reduce occurrences of stroke, which could also increase survival in stroke patients. One widely cited study has explored the mortality effects of antidepressant treatment in the aftermath of stroke (Jorge et al., 2003). One hundred and four stroke patients were randomly assigned to 3 months of antidepressant use (nortriptyline or fluoxetine) or placebo, and then followed up 9 years later. Antidepressant use was associated with a significantly higher 9-year survival rate, which seems to contradict the epidemiological findings discussed above. Unfortunately, the randomization was unsuccessful. The patients in the placebo group were significantly more likely to have diabetes, and they tended to be older, more obese, more likely to have atrial fibrillation, and more likely to have chronic pulmonary obstruction, all of which are serious problems that could have contributed to the higher mortality rate. This study has been criticized for failing to control for these differences and other errors (Sonis, 2004; Ghaemi and Thommi, 2010). If the study authors had re-analyzed the data controlling for these differences, they could have silenced the critics, but the authors declined to do so (Jorge et al., 2004).
Moreover, any beneficial anticoagulant effects may be counter-balanced by negative anticoagulant effects, so the overall effect may be neutral. Indeed, large meta-analyses on the effects of other anticoagulants in stroke patients find that they have no net effect on mortality (Paciaroni et al., 2007; Sandercock et al., 2008). Anticoagulants decrease the risk of recurrent ischemic strokes, but this benefit is offset by the increased risk of intracranial hemorrhages. Similarly, the beneficial effects of anticoagulants in reducing the frequency of pulmonary emboli are counter-balanced by the increased risk of extracranial hemorrhages (Sandercock et al., 2008). Given the strength of this literature, it would be surprising if future research demonstrated an unambiguous positive net effect of antidepressants on mortality in stroke patients.
It has also been argued that antidepressants could promote recovery from stroke (Kramer, 2011). A recent randomized clinical trial in France involved 118 patients with recent ischemic stroke and given early treatment with fluoxetine or placebo. The fluoxetine group showed greater motor recovery at a 90 day follow up (Chollet et al., 2011). Given our discussion above, it is unlikely that fluoxetine promoted recovery due to a direct promotion of neurogenesis. However, the clearance of damaged neurons through apoptosis, the dematuration of adult neurons (which might increase plasticity), and compensatory neurogenesis could have all contributed to greater recovery. Another way in which antidepressants could contribute to motor recovery after stroke is by enhancing extracellular serotonin levels, since serotonin plays a role in repetitive and gross motor activity (Jacobs and Fornal, 1995, 1999). In any event, while promising, the effects of antidepressants on motor recovery after stroke must be confirmed.
Reproductive Function
Serotonin is an important regulator of various aspects of reproduction (Azmitia, 2001). While depression is itself associated with impaired sexual behavior, most antidepressants increase the degree of impaired sexual functioning in men and women, including various aspects of desire, arousal, and orgasm (Serretti and Chiesa, 2009). TCAs and at least some SSRIs have adverse effects on sperm motility, volume, and morphology (Hendrick et al., 2000; Tanrikut et al., 2010). There are also case reports that antidepressants may impair the affective dimensions of romantic love and attachment (Fisher and Thomson, 2006).
Development
Serotonin plays important role in development (Azmitia, 2001), so antidepressants could affect developmental processes. Depression is common among pregnant and lactating mothers, and antidepressants are frequently prescribed (Moret et al., 2009; Ellfolk and Malm, 2010). SSRIs pass through the placental barrier and maternal milk, so antidepressants have the opportunity to affect the development of the fetus and the neonate (Moret et al., 2009; Ellfolk and Malm, 2010). When administered during pregnancy, SSRIs may increase the risk of a preterm delivery and low birth weight, although the results are mixed and inconclusive (Ellfolk and Malm, 2010). Some SSRIs – particularly paroxetine – also appear to be mild fetal teratogens (Bar-Oz et al., 2007; Cole et al., 2007). In one meta-analysis, first-term exposure to paroxetine was associated with an adjusted odds ratio of 1.89 for congenital defects (Cole et al., 2007). SSRI exposure during the third trimester has been associated with medication withdrawal symptoms in the newborn, including abnormal crying, irritability, and convulsions (Moret et al., 2009; Ellfolk and Malm, 2010). Third trimester SSRI exposure has also been associated with an increased risk of persistent pulmonary hypertension in the newborn, which is a serious condition with a mortality rate of about 10% (Ellfolk and Malm, 2010).
Researchers are continuing to investigate the long-term developmental effects of prenatal and neonatal exposure to SSRIs (Ellfolk and Malm, 2010; Homberg et al., 2010). Recently, a study found that fetal exposure to SSRIs, particularly during the first trimester, was associated with an increased risk of developing autism spectrum disorders (Croen et al., 2011).
Electrolyte Homeostasis
Serotonin is important in regulating water balance and electrolyte homeostasis (Johnson and Thunhorst, 1997; Elgot et al., 2009; Fonseca et al., 2009). The body normally keeps serum concentrations of sodium within 135–145 mEq/L, and hyponatremia occurs when levels fall below 130 mEq/L (Moret et al., 2009). Between 120 and 130 mEq/L, hyponatremia can cause nausea, headache, malaise and lethargy, muscle cramps, and disorientation. Lower levels are associated with seizures, coma, respiratory arrest, and death. Although the mechanism is unclear, SSRI use is associated with an increased risk of hyponatremia in the elderly, with studies showing an increased risk ranging from 0.5 to 32% (Jacob and Spinler, 2006). A large epidemiological study in the UK found that the risk of hyponatremia increased by 50% or more for several commonly prescribed antidepressants – citalopram, escitalopram, fluoxetine, and venlafaxine (Coupland et al., 2011). The harmful effects of hyponatremia may be exacerbated in patients who take diuretics and other medications (Moret et al., 2009).
Suicidal Behavior
Depression is a risk factor for suicidal ideation and behavior, which suggests that antidepressants might reduce these risks. However, there is well-known controversy over whether antidepressant use reduces or increases suicidal behavior (Baldessarini et al., 2007; Reeves and Ladner, 2010). The largest, most recent meta-analyses of randomized, placebo-controlled trials generally find a positive association between antidepressant use and suicidal behavior. In one meta-analysis involving 702 trials and over 87,000 patients, SSRI use was associated with an increased risk of suicide attempts (Fergusson et al., 2005). In another meta-analysis of 342 randomized, double-blind, placebo-controlled trials submitted to the FDA involving over 99,000 patients, antidepressant use was associated with an increased risk of suicidal behavior in children, adolescents, and young adults (Stone et al., 2009). But it had no overall effect in adults aged 25–65, and it reduced the risk in the elderly. The reason for the age effect is unknown and worth further investigation. On the other hand, in a recent large epidemiological study in the UK that controlled for many important covariates, including depressive symptoms, patients over 65 years old who were not taking any antidepressant had a 0.25% chance of harming themselves or making a suicide attempt over a 1 year period (Coupland et al., 2011). However, the risk was higher for antidepressant use: 0.43% (TCAs), 0.55% (SSRIs), 1.30% (mirtazapine or venlafaxine). In short, the empirical literature is complex and it is not possible to make any strong conclusions based on it, except that antidepressant-induced suicide is rare (Baldessarini et al., 2007; Reeves and Ladner, 2010). In any event, uncertainty about the effects of antidepressants on suicidal behavior is germane to the costs and benefits of antidepressants.
Other Side Effects
Reviews of other side effects are readily available (Brambilla et al., 2005; Stahl, 2008; Zimmerman et al., 2010), so we do not address them here. We merely note that 50–60% of antidepressant users report experiencing gastrointestinal, sexual, and other significant side effects (Brambilla et al., 2005; Serretti and Chiesa, 2009; Zimmerman et al., 2010), and that these side effects are unpleasant enough that they are one of the most common reasons for discontinuing them (Demyttenaere et al., 2001; Bull et al., 2002).
Do Antidepressants Do More Harm than Good?
It is a principle of evolutionary medicine that the disruption of evolved adaptations will degrade biological functioning. Antidepressants disrupt the functioning of the homeostatic mechanisms that regulate serotonin throughout the body, and our review shows that antidepressants have adverse effects on every major system regulated by serotonin.
Antidepressants increase the brain’s susceptibility to future episodes of depression (Andrews et al., 2011). They cause neuronal damage (Kalia et al., 2000), apoptosis (Post et al., 2000; Bartholoma et al., 2002; Sairanen et al., 2005), and dematuration (Kobayashi et al., 2010). The evidence that antidepressants directly promote neurogenesis is equivocal. It is quite possible that any neurogenesis that does take place is a compensatory response of the brain to antidepressant-induced apoptosis. Antidepressants can also cause problems in early development (Moret et al., 2009; Ellfolk and Malm, 2010), and they have adverse effects on sexual and romantic life (Fisher and Thomson, 2006; Serretti and Chiesa, 2009), and an increased risk of hyponatremia (Jacob and Spinler, 2006), bleeding (Meijer et al., 2004; Dall et al., 2009), and stroke (Smoller et al., 2009; Trifiro et al., 2010).
The primary benefit of antidepressants is thought to be the reduction of depressive symptoms, a view that presupposes depression is the result of brain malfunction. An alternate view is that current diagnostic criteria do not accurately distinguish between true instances of disorder and normal, evolved emotional responses to stressors (Wakefield, 1992, 1999; Watson and Andrews, 2002; Hagen, 2003; Horwitz and Wakefield, 2007; Andrews and Thomson Jr., 2009; Andrews et al., 2011). This latter view suggests that the pharmacological disruption of depressive symptoms could negatively affect the ability to cope with or manage stressors. In any event, we reviewed several findings indicating that the depressed brain pushes back against the symptom reducing effects of antidepressant medications (in patients meeting diagnostic criteria for MDD) in ways indicating that depressive symptoms are under homeostatic control. Thus, even by the debatable metric of symptom reduction, antidepressants are of limited benefit.
The limited symptom reducing effects of antidepressants, and the large number of adverse health effects, suggests that antidepressants could generally do more harm than good – though they may be helpful for certain populations (e.g., those with brain cancers or recovering from stroke). However, it is undoubtedly true that the list of beneficial and harmful effects of antidepressants that we have listed in this review is incomplete. So, while we had difficulty finding strong evidence of beneficial effects, it is possible that antidepressants have a significant beneficial effect not yet identified. Moreover, since the benefits and costs are in different currencies, it is difficult to directly compare them. Finally, the effects differ in their frequency and their effect size, and some of them are rare. In some instances, we simply do not yet have information on effect size or frequency (e.g., antidepressant triggered apoptosis). Each of these issues could make it more difficult to conclude what the overall effect of antidepressants is.
These problems can largely be circumvented by examining the effects of antidepressants on mortality. Mortality data naturally synthesizes the multiple effects of a drug on biological functioning, including those not considered or identified. If the beneficial effects of antidepressants in reducing any harmful symptoms outweigh the detrimental effects in disrupting homeostatic mechanisms, then they should increase survival. Conversely, if the harmful effects of antidepressants outweigh the beneficial effects, then they should decrease survival.
Depression itself is associated with an increased risk of death, primarily through its effects on cardiovascular disease (Van der Kooy et al., 2007; Pozuelo et al., 2009). But does antidepressant use increase or decrease the risk of death? Three recent, large, prospective epidemiological studies have found that, even after controlling for depressive symptoms, antidepressant use is associated with an increased risk of death in the elderly (Smoller et al., 2009; Almeida et al., 2010; Coupland et al., 2011).
The most recent study involved patients in the United Kingdom over 65 years old (average age = 75) who had received a diagnosis of depression between 1996 and 2007 (Coupland et al., 2011). The 1-year risk of death after controlling for a large number of covariates (including the severity of depressive symptoms) was as follows: 7.04% (no antidepressant use), 8.12% (TCAs), 10.61% (SSRIs), and 11.43% (other antidepressants). Put another way, out of 1000 elderly people taking antidepressants, the number of deaths per year caused by antidepressants was estimated to be 10.8 (for TCAs), 35.7 (for SSRIs), and 43.9 (for other antidepressants).
Another study estimated that antidepressants caused the deaths of roughly 5 in 1000 elderly women per year, with no differences in the class of antidepressant (Smoller et al., 2009). The lower estimate in this study could be due to the fact that the women were somewhat younger (age range = 50–79) and perhaps more resilient to the harmful effects of antidepressants.
In comparison, rofecoxib (Vioxx) was pulled off the market amidst evidence that it caused about 7.2 cardiovascular events per 1000 patient-years and a lower risk of death (Bresalier et al., 2005). An important limitation is that the Vioxx studies were randomized, placebo-controlled experiments, while the antidepressant studies were not. But the antidepressant-mortality surveys nevertheless suggest that antidepressants are increasing the risk of death, a point that can only be definitively disproved by conducting controlled experiments on the mortality effects of antidepressants.
Another important limitation of the antidepressant-mortality studies is the fact that they deal exclusively with the elderly. It is possible that young adults who take antidepressants are more resilient to their harmful effects and will experience a much smaller risk of death than the elderly, although this should not simply be assumed. Moreover, many people take antidepressants for years throughout their adult life, and it is unclear what the cumulative impact is over years of use. The cumulative effects of antidepressants on the integrity of the brain and peripheral processes could shorten the lifespan considerably. Current research is unable to rule out this possibility.
Conclusion
We have reviewed a great deal of evidence of the effects of antidepressants on serotonergic processes throughout the body. Some of the effects are widely known, but they have been largely ignored in debates about the utility of antidepressants. Indeed, it is widely believed that antidepressant medications are both safe and effective; however, this belief was formed in the absence of adequate scientific verification. The weight of current evidence suggests that, in general, antidepressants are neither safe nor effective; they appear to do more harm than good.
Further research on their effects is clearly needed. We note a few areas of particular importance. First, after antidepressant discontinuation some people report depressive symptoms that are more severe than the initial symptoms that prompted antidepressant treatment. This should be formally tested, and if confirmed it would provide further support for the homeostatic rebound argument we presented above. Second, more research should be conducted on how best to interpret the BrdU signal in the context of antidepressant use. Does it reflect apoptosis, neurogenesis, or both? Particularly helpful would be studies that examine the temporal relationship between the BrdU signal and independent measures of apoptosis and neurogenesis. Third, more studies should examine the morphological changes in neurons that take place with repeated administration of serotonergic antidepressants (Kalia et al., 2000). Fourth, greater research should be devoted to exploring the possibility that the effects of prolonged antidepressant use on neuronal integrity may cause cognitive decline and dementia (Jackson, 2008; Goveas et al., 2011). Finally, greater research should be devoted to the effects of antidepressants on longevity. The quickest experiments could be done on rodents. That such studies were not done decades ago, before antidepressants were approved for widespread use, is troublesome.
Such research will give us a better picture of the effects of antidepressants on overall functioning. Nevertheless, from a legal, ethical, and public health perspective, it now seems prudent, on the basis of existing evidence, for individual practitioners and professional medical organizations to revise informed consent guidelines and reconsider the status of antidepressants in standards of care for many diagnoses and as the immediate front line treatment for depression. Some patients may be more likely to benefit from antidepressants and less likely to suffer adverse effects, but identifying them will require further research and greater understanding of the etiology of depression.
Patients should be informed that current research suggests that unless they have very severe depression, the symptom reducing effects of antidepressants are modest and are not considered clinically significant. Unless there are rapid-onset adverse side effects, antidepressant therapy usually lasts for months. Patients should be advised that prolonged use might cause mild cognitive impairment and interfere with tasks that require highly focused concentration, such as driving, which may increase the risk of accidents. Patients should also be advised that antidepressants might trigger even more severe depressive episodes when they are discontinued. All patients should be advised of the possible bleeding risks, and physicians should exercise particular caution in prescribing these drugs in conjunction with other diuretic or anti-thrombotic medications. The evidence of harm is strongest in the elderly, who should be advised of the risks of falling, hyponatremia, bleeding, stroke, and death.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Paul W. Andrews and Michael C. Neale were supported, in part, by NIH grant DA-018673 (PI: Michael C. Neale). The authors thank Grace Jackson, Jack Hettema, Reuven Dukas, Irving Kirsch, Steve Hollon, Gautam Ullal and Stefanie Northover for helpful conversations or comments.
Abbreviations
MAOIs, monoamine oxidase inhibitors; MDD, major depressive disorder; NSAIDs, non-steroidal anti-inflammatory drugs; SNRIs, serotonin norepinephrine reuptake inhibitors; SSRIs, selective serotonin reuptake inhibitors; TCAs, tricyclic antidepressants.
References
Adell, A., Castro, E., Celeda, P., Bortolozzi, A., Pazos, A., and Artigas, F. (2005). Strategies for productring faster acting antidepressants. Drug Discov. Today 10, 578–585.
Almeida, O. P., Alfonso, H., Hankey, G. J., and Flicker, L. (2010). Depression, antidepressant use and mortality in later life: the health in men study. PLoS ONE 5, e11266. doi:10.1371/journal.pone.0011266
Amat, J., Baratta, M. V., Paul, E., Bland, S. T., Watkins, L. R., and Maier, S. F. (2005). Medial prefrontal cortex determines how stressor controllability affects behavior and dorsal raphe nucleus. Nat. Neurosci. 8, 365–371.
Amat, J., Paul, E., Zarza, C., Watkins, L. R., and Maier, S. F. (2006). Previous experience with behavioral control over stress blocks the behavioral and dorsal raphe nucleus activating effects of later uncontrollable stress: role of the ventral medial prefrontal cortex. J. Neurosci. 26, 13264–13272.
American Psychiatric Association (APA). (2000a). Handbook of Psychiatric Measures. Washington, DC: American Psychiatric Association.
American Psychiatric Association (APA). (2000b). (DSM-IV-TR) Diagnostic and Statistical Manual of Mental Disorders, 4th Edn, Text Revision Edn. Washington, DC: American Psychiatric Press, Inc.
Andrews, P. W., Aggen, S. H., Miller, G. F., Radi, C., Dencoff, J. E., and Neale, M. C. (2007). The functional design of depression’s influence on attention: a preliminary test of alternative control-process mechanisms. Evol. Psychol. 5, 584–604.
Andrews, P. W., Kornstein, S. G., Halberstadt, L. J., Gardner, C. O., and Neale, M. C. (2011). Blue again: perturbational effects of antidepressants suggest monoaminergic homeostasis in major depression. Front. Psychol. 2:159. doi:10.3389/fpsyg.2011.00159
Andrews, P. W., and Thomson, J. A. Jr. (2009). The bright side of being blue: depression as an adaptation for analyzing complex problems. Psychol. Rev. 116, 620–654.
Angst, J., Delinistula, A., Stabl, M., and Stassen, H. H. (1993). Is a cutoff score a suitable measure of treatment outcome in short-term trials in depression? A methodological meta-analysis. Hum. Psychopharmacol. 8, 311–317.
Appenheimer, M. M., Chen, Q., Girard, R. A., Wang, W. C., and Evans, S. S. (2005). Impact of fever-range thermal stress on lymphocyte-endothelial adhesion and lymphocyte trafficking. Immunol. Invest. 34, 295–323.
Azmitia, E. C. (2001). Modern views on an ancient chemical: serotonin effects on cell proliferation, maturation, and apoptosis. Brain Res. Bull. 56, 413–424.
Azmitia, E. C. (2007). Serotonin and brain: evolution, neuroplasticity, and homeostasis. Int. Rev. Neurobiol. 77, 31–56.
Azmitia, E. C. (2010). “Evolution of serotonin: sunlight to suicide,” in Handbook of the Behavioral Neurobiology of Serotonin, eds C. P. Muller and B. L. Jacobs (Burlington, MA: Academic Press), 3–22.
Bagby, R. M., Ryder, A. G., Schuller, D. R., and Marshall, M. B. (2004). The Hamilton depression rating scale: has the gold standard become a lead weight? Am. J. Psychiatry 161, 2163–2177.
Bakkaloglu, B., Yabanoglu, S., Ozyuksel, B. R., Uaar, G., Ertugrul, A., Demir, B., and Uluğ, B. (2008). Platelet and plasma serotonin levels and platelet monoamine oxidase activity in patients with major depression: effects of sertraline treatment. Turk. J. Biochem. 33, 97–103.
Baldessarini, R. J., Tondo, L., Strombom, I. M., Dominguez, S., Fawcett, J., Licinio, J., Oquendo, M. A., Tollefson, G. D., Valuck, R. J., and Tohen, M. (2007). Ecological studies of antidepressant treatment and suicidal risks. Harv. Rev. Psychiatry 15, 133–145.
Bar-Oz, B., Einarson, T., Einarson, A., Boskovic, R., O’Brien, L., Malm, H., Bérard, A., and Koren, G. (2007). Paroxetine and congenital malformations: meta-analysis and consideration of potential confounding factors. Clin. Ther. 29, 918–926.
Bartholoma, P., Erlandsson, N., Kaufmann, K., Rossler, O. G., Baumann, B., Wirth, T., Giehl, K. M., and Thiel, G. (2002). Neuronal cell death induced by antidepressants: lack of correlation with Egr-1, NF-kappa B and extracellular signal-regulated protein kinase activation. Biochem. Pharmacol. 63, 1507–1516.
Berger, M., Gray, J. A., and Roth, B. L. (2009). The expanded biology of serotonin. Annu. Rev. Med. 60, 355–366.
Bernard, G. R., Wheeler, A. P., Russell, J. A., Schein, R., Summer, W. R., Steinberg, K. P., Fulkerson, W. J., Wright, P. E., Christman, B. W., Dupont, W. D., Higgins, S. B., and Swindell, B. B. (1997). The effects of ibuprofen on the physiology and survival of patients with sepsis. N. Engl. J. Med. 336, 912–918.
Best, J., Nijhout, H. F., and Reed, M. (2010). Serotonin synthesis, release and reuptake in terminals: a mathematical model. Theor. Biol. Med. Model. 7. doi: 10.1186/1742-4682-7-34
Blatteis, C. M. (2003). Fever: pathological or physiological, injurious or beneficial? J. Therm. Biol. 28, 1–13.
Bockting, C. L. H., ten Doesschate, M. C., Spijker, J., Spinhoven, P., Koeter, M. W. J., and Schene, A. H. (2008). Continuation and maintenance use of antidepressants in recurrent depression. Psychother. Psychosom. 77, 17–26.
Bonin, R. (2012). “Treating depression: is there a placebo effect?” in 60 Minutes (New York, NY: CBS Broadcasting, Inc.).
Brambilla, P., Cipriani, A., Hotopf, M., and Barbui, C. (2005). Side-effect profile of fluoxetine in comparison with other SSRIs, tricyclic and newer antidepressants: a meta-analysis of clinical trial data. Pharmacopsychiatry 38, 69–77.
Brandts, C. H., Ndjave, M., Graninger, W., and Kremsner, P. G. (1997). Effect of paracetamol on parasite clearance time in Plasmodium falciparum malaria. Lancet 350, 704–709.
Bresalier, R. S., Sandler, R. S., Quan, H., Bolognese, J. A., Oxenius, B., Horgan, K., Lines, C., Riddell, R., Morton, D., Lanas, A., Konstam, M. A., Baron, J. A., Adenomatous Polyp Prevention on Vioxx (APPROVe) Trial Investigators. (2005). Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N. Engl. J. Med. 352, 1092–1102.
Bryant, R. E., Hood, A. F., Hood, C. E., and Koenig, M. G. (1971). Factors affecting mortality of gram-negative rod bacteremia. Arch. Intern. Med. 127, 120–128.
Bull, S. A., Hunkeler, E. M., Lee, J. Y., Rowland, C. R., Williamson, T. E., Schwab, J. R., and Hurt, S. W. (2002). Discontinuing or switching selective serotonin-reuptake inhibitors. Ann. Pharmacother. 36, 578–584.
Bymaster, F. P., Zhang, W., Carter, P. A., Shaw, J., Chernet, E., Phebus, L., Wong, D. T., and Perry, K. W. (2002). Fluoxetine, but not other selective serotonin uptake inhibitors, increases norepinephrine and dopamine extracellular levels in prefrontal cortex. Psychopharmacology (Berl.) 160, 353–361.
Byrne, S. E., and Rothschild, A. J. (1998). Loss of antidepressant efficacy during maintenance therapy: possible mechanisms and treatments. J. Clin. Psychiatry 59, 279–288.
Carter, O. L., Burr, D. C., Pettigrew, J. D., Wallis, G. M., Hasler, F., and Vollenweider, F. X. (2005). Using psilocybin to investigate the relationship between attention, working memory, and the serotonin 1A and 2A receptors. J. Cogn. Neurosci. 17, 1497–1508.
Chen, L., Hamaguchi, K., Ogawa, M., Hamada, S., and Okado, N. (1994). PCPA reduces both monoaminergic afferents and non-monoaminergic synapses in the cerebral cortex. Neurosci. Res. 19, 111–115.
Cheng, H. C., Ulane, C. M., and Burke, R. E. (2010). Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 67, 715–725.
Chollet, F., Tardy, J., Albucher, J. F., Thalamas, C., Berard, E., Lamy, C., Bejot, Y., Deltour, S., Jaillard, A., Niclot, P., Guillon, B., Moulin, T., Marque, P., Pariente, J., Arnaud, C., and Loubinoux, I. (2011). Fluoxetine for motor recovery after acute ischaemic stroke (FLAME): a randomised placebo-controlled trial. Lancet Neurol. 10, 123–130.
Cloonan, S. M., and Williams, D. C. (2011). The antidepressants maprotiline and fluoxetine induce Type II autophagic cell death in drug-resistant Burkitt’s lymphoma. Int. J. Cancer 128, 1712–1723.
Cohen, H. W., Gibson, G., and Alderman, M. H. (2000). Excess risk of myocardial infarction in patients treated with antidepressant medications: association with use of tricyclic agents. Am. J. Med. 108, 2–8.
Cohen, Z., Bonvento, G., Lacombe, P., and Hamel, E. (1996). Serotonin in the regulation of brain microcirculation. Prog. Neurobiol. 50, 335.
Cohn, J. B., and Wilcox, C. S. (1992). Paroxetine in major depression – a double-blind trial with imipramine and placebo. J. Clin. Psychiatry 53, 52–56.
Cole, J. A., Ephross, S. A., Cosmatos, I. S., and Walker, A. M. (2007). Paroxetine in the first trimester and the prevalence of congenital malformations. Pharmacoepidemiol. Drug Saf. 16, 1075–1085.
Cosgrove, L., Shi, L., Creasey, D. E., naya-McKivergan, M., Myers, J. A., and Huybrechts, K. F. (2011). Antidepressants and breast and ovarian cancer risk: a review of the literature and researchers’ financial associations with industry. PLoS ONE 6, e18210. doi:10.1371/journal.pone.0018210
Cotran, R. S., Kumar, V., Collins, T., and Robbins, S. (1998). Robbins’ Pathologic Basis of Disease. London: W. B. Saunders.
Coupland, C., Dhiman, D., Morriss, R., Arthur, A., Barton, G., and Hippisley-Cox, J. (2011). Antidepressant use and risk of adverse outcomes in older people: population based cohort study. BMJ. 343. doi:10.1136/bmj.d4551
Croen, L. A., Grether, J. K., Yoshida, C. K., Odouli, R., and Hendrick, V. (2011). Antidepressant use during pregnancy and childhood autism spectrum disorders. Arch. Gen. Psychiatry 68, 1104–1112.
Cryan, J. F., O’Leary, O. F., Jin, S. H., Friedland, J. C., Ouyang, M., Hirsch, B. R., Page, M. E., Dalvi, A., Thomas, S. A., and Lucki, I. (2004). Norepinephrine-deficient mice lack responses to antidepressant drugs, including selective serotonin reuptake inhibitors. Proc. Natl. Acad. Sci. U.S.A. 101, 8186–8191.
Cumming, R. G. (1998). Epidemiology of medication-related falls and fractures in the elderly. Drugs Aging 12, 43–53.
Dall, M., De Muckadell, O. B. S., Lassen, A. T., Hansen, J. M., and Hallas, J. (2009). An association between selective serotonin reuptake inhibitor use and serious upper gastrointestinal bleeding. Clin. Gastroenterol. Hepatol. 7, 1314–1321.
Demyttenaere, K., Enzlin, P., Dewe, W., Boulanger, B., De Bie, J., De Troyer, W., and Mesters, P. (2001). Compliance with antidepressants in a primary care setting, 1: beyond lack of efficacy and adverse events. J. Clin. Psychiatry 62, 30–33.
Dong, H. X., Csernansky, C. A., Goico, B., and Csernansky, J. G. (2003). Hippocampal neurogenesis follows kainic acid-induced apoptosis in neonatal rats. J. Neurosci. 23, 1742–1749.
Doran, T. F., Deangelis, C., Baumgardner, R. A., and Mellits, E. D. (1989). Acetaminophen: more harm than good for chickenpox. J. Pediatr. 114, 1045–1048.
Elgot, A., Ahboucha, S., Bouyatas, M. M., Fevre-Montange, M., and Gamrani, H. (2009). Water deprivation affects serotoninergic system and glycoprotein secretion in the sub-commissural organ of a desert rodent Meriones shawi. Neurosci. Lett. 466, 6–10.
Ellfolk, M., and Malm, H. (2010). Risks associated with in utero and lactation exposure to selective serotonin reuptake inhibitors (SSRIs). Reprod. Toxicol. 30, 249–260.
El-Mallakh, R. S., Gao, Y. L., and Roberts, R. J. (2011). Tardive dysphoria: the role of long term antidepressant use in-inducing chronic depression. Med. Hypotheses 76, 769–773.
Encinas, J. M., Vaahtokari, A., and Enikolopov, G. (2006). Fluoxetine targets early progenitor cells in the adult brain. Proc. Natl. Acad. Sci. U.S.A. 103, 8233–8238.
Fava, G. A., and Offidani, E. (2011). The mechanisms of tolerance in antidepressant action. Prog. Neuropsychopharmacol. Biol. Psychiatry 35, 1593–1602.
Felton, T. M., Kang, T. B., Hjorth, S., and Auerbach, S. B. (2003). Effects of selective serotonin and serotonin/noradrenaline reuptake inhibitors on extracellular serotonin in rat diencephalon and frontal cortex. Naunyn Schmiedebergs Arch. Pharmacol. 367, 297–305.
Fergusson, D., Doucette, S., Cranley, K., Glass, K. C., Shapiro, S., Healy, D., Hebert, P., and Hutton, B. (2005). Association between suicide attempts and selective serotonin reuptake inhibitors: systematic review of randomised controlled trials. BMJ 330, 396–399.
Fisher, H. E., and Thomson, J. A. (2006). “Lust, romance, attachment: do the side effects of serotonin-enhancing antidepressants jeopardize romantic love, marriage, and fertility?” in Evolutionary Cognitive Neuroscience, eds S. M. Platek, J. P. Keenan, and T. K. Shackelford (Cambridge, MA: The MIT Press), 245–283.
Flock, A., Zobel, A., Bauriedel, G., Tuleta, I., Hammerstingl, C., Hofels, S., Schuhmacher, A., Maier, W., Nickenig, G., and Skowasch, D. (2010). Antiplatelet effects of antidepressant treatment: a randomized comparison between escitalopram and nortriptyline. Thromb. Res. 126, E83–E87.
Fonseca, F. V., Mecawi, A. S., Araujo, I. G., meida-Pereira, G., magalhaes-Nunes, A. P., Badaue-Passos, D. Jr., and Reis, L. C. (2009). Role of the 5-HT(1A) somatodendritic autoreceptor in the dorsal raphe nucleus on salt satiety signaling in rats. Exp. Neurol. 217, 353–360.
Fournier, J. C., DeRubeis, R. J., Hollon, S. D., Dimidjian, S., Amsterdam, J. D., Shelton, R. C., and Fawcett, J. (2010). Antidepressant drug effects and depression severity: a patient-level meta-analysis. JAMA 303, 47–53.
Ganzel, B. L., Morris, P. A., and Wethington, E. (2010). Allostasis and the human brain: integrating models of stress from the social and life sciences. Psychol. Rev. 117, 134–174.
Gawaz, M., Langer, H., and May, A. E. (2005). Platelets in inflammation and atherogenesis. J. Clin. Invest. 115, 3378–3384.
Gershon, M. D. (2004). Serotonin receptors and transporters: roles in normal and abnormal gastrointestinal motility. Aliment. Pharmacol. Ther. 20, 3–14.
Gershon, M. D., and Tack, J. (2007). The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology 132, 397–414.
Ghaemi, S. N., and Thommi, S. B. (2010). Death by confounding: bias and mortality. Int. J. Clin. Pract. 64, 1009–1014.
Gibson, J. E., Hubbard, R. B., Smith, C. J. P., Tata, L. J., Britton, J. R., and Fogarty, A. W. (2009). Use of self-controlled analytical techniques to assess the association between use of prescription medications and the risk of motor vehicle crashes. Am. J. Epidemiol. 169, 761–768.
Gortner, E. M., Rude, S. S., and Pennebaker, J. W. (2006). Benefits of expressive writing in lowering rumination and depressive symptoms. Behav. Ther. 37, 292–303.
Goveas, J. S., Espeland, M. A., Woods, N. F., Wassertheil-Smoller, S., and Kotchen, J. M. (2011). Depressive symptoms and incidence of mild cognitive impairment and probable eementia in elderly women: the women’s health initiative memory study. J. Am. Geriatr. Soc. 59, 57–66.
Graf, M. C., Gaudiano, B. A., and Geller, P. A. (2008). Written emotional disclosure: a controlled study of the benefits of expressive writing homework in outpatient psychotherapy. Psychother. Res. 18, 389–399.
Graham, N. M. H., Burrell, C. J., Douglas, R. M., Debelle, P., and Davies, L. (1990). Adverse-effects of aspirin, acetaminophen, and ibuprofen on immune function, viral shedding, and clinical status in rhinovirus-infected volunteers. J. Infect. Dis. 162, 1277–1282.
Hagen, E. H. (2003). “The bargaining model of depression,” in Genetic and Cultural Evolution of Cooperation, ed. P. Hammerstein (Cambridge, MA: MIT Press), 95–123.
Hanson, N. D., Owens, M. J., and Nemeroff, C. B. (2011). Depression, antidepressants, and neurogenesis: a critical reappraisal. Neuropsychopharmacology 36, 2589–2602.
Hasday, J. D., Fairchild, K. D., and Shanholtz, C. (2000). The role of fever in the infected host. Microbes Infect. 2, 1891–1904.
Hata, M., Yagi, Y., Sezai, A., Yoshitake, I., Wakui, S., Takasaka, A., Kawachi, H., Murakami, T., Minami, K., and Shiono, M. (2011). Efficacy of prophylactic treatment with selective serotonin reuptake inhibitors for depression after open-heart surgery. Surg. Today 41, 791–794.
Hayes, A. M., Beevers, C. G., Feldman, G. C., Laurenceau, J. P., and Perlman, C. (2005). Avoidance and processing as predictors of symptom change and positive growth in an integrative therapy for depression. Int. J. Behav. Med. 12, 111–122.
Hayes, A. M., Feldman, G. C., Beevers, C. G., Laurenceau, J. P., Cardaciotto, L., and Lewis-Smith, J. (2007). Discontinuities and cognitive changes in an exposure-based cognitive therapy for depression. J. Consult. Clin. Psychol. 75, 409–421.
Heger, C. D., and Collins, R. N. (2004). Platelet activation and “crossover appeal”: Rab and Rho families united by common links to serotonin. Mol. Interv. 4, 79–81.
Heisler, L. K., Chu, H. M., Brennan, T. J., Danao, J. A., Bajwa, P., Parsons, L. H., and Tecott, L. H. (1998). Elevated anxiety and antidepressant-like responses in serotonin 5-HT1A receptor mutant mice. Proc. Natl. Acad. Sci. U.S.A. 95, 15049–15054.
Hendrick, V., Gitlin, M., Altshuler, L., and Korenman, S. (2000). Antidepressant medications, mood and male fertility. Psychoneuroendocrinology 25, 37–51.
Hernandez-Cueto, C., Girela, E., and Sweet, D. J. (2000). Advances in the diagnosis of wound vitality – a review. Am. J. Forensic Med. Pathol. 21, 21–31.
Herrup, K., Neve, R., Ackerman, S. L., and Copani, A. (2004). Divide and die: cell cycle events as triggers of nerve cell death. J. Neurosci. 24, 9232–9239.
Hindmarch, I. (2009). Cognitive toxicity of pharmacotherapeutic agents used in social anxiety disorder. Int. J. Clin. Pract. 63, 1085–1094.
Hochochka, P. W., and Somero, G. N. (2002). Biochemical Adaptation: Mechanism and Process in Physiological Evolution. New York, NY: Oxford University Press.
Holt, A., and Baker, G. B. (1996). Inhibition of rat brain monoamine oxidase enzymes by fluoxetine and norfluoxetine. Naunyn Schmiedebergs Arch. Pharmacol. 354, 17–24.
Holtzer, R., Friedman, R., Lipton, R. B., Katz, M., Xue, X., and Verghese, J. (2007). The relationship between specific cognitive functions and falls in aging. Neuropsychology 21, 540–548.
Homberg, J. R., Schubert, D., and Gaspar, P. (2010). New perspectives on the neurodevelopmental effects of SSRIs. Trends Pharmacol. Sci. 31, 60–65.
Honig, G., Jongsma, M. E., van der Hart, M. C. G., and Tecott, L. H. (2009). Chronic citalopram administration causes a sustained suppression of serotonin synthesis in the mouse forebrain. PLoS ONE 4, e6797. doi:10.1371/journal.pone.0006797
Horwitz, A. V., and Wakefield, J. C. (2007). The Loss of Sadness: How Psychiatry Transformed Normal Sorrow into Depressive Disorder. New York, NY: Oxford University Press.
Hranilovic, D., Blazevic, S., Ivica, N., Cicin-Sain, L., and Oreskovic, D. (2011). The effects of the perinatal treatment with 5-hydroxytryptophan or tranylcypromine on the peripheral and central serotonin homeostasis in adult rats. Neurochem. Int. 59, 202–207.
Hyman, S. E., and Nestler, E. J. (1996). Initiation and adaptation: a paradigm for understanding psychotropic drug action. Am. J. Psychiatry 153, 151–162.
Insel, T. R., and Wang, P. S. (2009). The STAR*D trial: revealing the need for better treatments. Psychiatr. Serv. 60, 1466–1467.
Jacob, S., and Spinler, S. A. (2006). Hyponatremia associated with selective serotonin-reuptake inhibitors in older adults. Ann. Pharmacother. 40, 1618–1622.
Jacobs, B. L., and Fornal, C. A. (1995). Activation of 5-HT neuronal activity during motor behavior. Semin. Neurosci. 7, 401–408.
Jacobs, B. L., and Fornal, C. A. (1999). Activity of serotonergic neurons in behaving animals. Neuropsychopharmacology 21, S9–S15.
Jimenez-Trejo, F., Tapia-Rodriguez, M., Queiroz, D. B. C., Padilla, P., Avellar, M. C. W., Manzano, P. R., Manjarrez-Gutiérrez, G., and Gutiérrez-Ospina, G. (2007). Serotonin concentration, synthesis, cell origin, and targets in the rat caput epididymis during sexual maturation and variations associated with adult mating status: morphological and biochemical studies. J. Androl. 28, 136–149.
Johnson, A. K., and Thunhorst, R. L. (1997). The neuroendocrinology of thirst and salt appetite: visceral sensory signals and mechanisms of central integration. Front. Neuroendocrinol. 18:292–353. doi:10.1006/frne.1997.0153
Jorge, R. E., Robinson, R. G., and Ardnt, S. (2004). Drs. Jorge and Robinson reply. Am. J. Psychiatry 161, 1507–1508.
Jorge, R. E., Robinson, R. G., Arndt, S., and Starkstein, S. (2003). Mortality and poststroke depression: a placebo-controlled trial of antidepressants. Am. J. Psychiatry 160, 1823–1829.
Kalia, M., O’Callaghan, J. P., Miller, D. B., and Kramer, M. (2000). Comparative study of fluoxetine, sibutramine, sertraline and dexfenfluramine on the morphology of serotonergic nerve terminals using serotonin immunohistochemistry. Brain Res. 858, 92–105.
Kearns, N. P., Cruickshank, C. A., Mcguigan, K. J., Riley, S. A., Shaw, S. P., and Snaith, R. P. (1982). A comparison of depression rating scales. Br. J. Psychiatry 141, 45–49.
Kelly, O., Matheson, K., Ravindran, A., Merali, Z., and Anisman, H. (2007). Ruminative-coping among patients with dysthymia before and after pharmacotherapy. Depress. Anxiety 24, 233–243.
Kessler, R. C., Merikangas, K. R., and Wang, P. S. (2007). Prevalence, comorbidity, and service utilization for mood disorders in the United States at the beginning of the twenty-first century. Annu. Rev. Clin. Psychol. 3, 137–158.
Keszthelyi, D., Troost, F. J., and Masclee, A. A. M. (2009). Understanding the role of tryptophan and serotonin metabolism in gastrointestinal function. Neurogastroenterol. Motil. 21, 1239–1249.
Khan, A., Bhat, A., Faucett, J., Kolts, R., and Brown, W. A. (2011). Antidepressant-placebo differences in 16 clinical trials over 10 years at a single site: role of baseline severity. Psychopharmacology (Berl.) 214, 961–965.
Khan, A., Brodhead, A. E., Kolts, R. L., and Brown, W. A. (2005). Severity of depressive symptoms and response to antidepressants and placebo in antidepressant trials. J. Psychiatr. Res. 39, 145–150.
Khan, A., Leventhal, R. M., Khan, S. R., and Brown, W. A. (2002). Severity of depression and response to antidepressants and placebo: an analysis of the Food and Drug Administration database. J. Clin. Psychopharmacol. 22, 40–45.
Kirsch, I., Deacon, B. J., Huedo-Medina, T. B., Scoboria, A., Moore, T. J., and Johnson, B. T. (2008). Initial severity and antidepressant benefits: a meta-analysis of data submitted to the food and drug administration. PloS Med. 5, e45. doi:10.1371/journal.pmed.0050045
Kluger, M. J., Kozak, W., Conn, C. A., Leon, L. R., and Soszynski, D. (1997). “The adaptive value of fever,” in Fever: Basic Mechanisms and Management, 2nd Edn, ed. P. A. Mackowiak (Philadelphia, PA: Lippincott-Raven), 255–266.
Kobayashi, K., Ikeda, Y., Sakai, A., Yamasaki, N., Haneda, E., Miyakawa, T., and Suzuki, H. (2010). Reversal of hippocampal neuronal maturation by serotonergic antidepressants. Proc. Natl. Acad. Sci. U.S.A. 107, 8434–8439.
Kovacevic, T., Skelin, I., and Diksic, M. (2010). Chronic fluoxetine treatment has a larger effect on the density of a serotonin transporter in the Flinders Sensitive Line (FSL) rat model of depression than in normal rats. Synapse 64, 231–240.
Lesurtel, M., Soll, C., Graf, R., and Clavien, P. A. (2008). Role of serotonin in the hepato-gastroIntestinal tract: an old molecule for new perspectives. Cell. Mol. Life Sci. 65, 940–952.
Levkovitz, Y., Gil-Ad, I., Zeldich, E., Dayag, M., and Weizman, A. (2005). Differential induction of apoptosis by antidepressants in glioma and neuroblastoma cell lines – evidence for p-c-Jun, cytochrome c, and caspase-3 involvement. J. Mol. Neurosci. 27, 29–42.
Linder, A. E., Ni, W., Diaz, J. L., Szasz, T., Burnett, R., and Watts, S. W. (2007). Serotonin (5-HT) in veins: not all in vain. J. Pharmacol. Exp. Ther. 323, 415–421.
Mackowiak, P. A., Browne, R. H., Southern, P. M., and Smith, J. W. (1980). Polymicrobial sepsis: an analysis of 184 cases using log-linear models. Am. J. Med. Sci. 280, 73–80.
Matondo, R. B., Punt, C., Homberg, J., Toussaint, M. J. M., Kisjes, R., Korporaal, S. J., Akkerman, J. W., Cuppen, E., and de Bruin, A. (2009). Deletion of the serotonin transporter in rats disturbs serotonin homeostasis without impairing liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G963–G968.
Mayorga, A. J., Dalvi, A., Page, M. E., Zimov-Levinson, S., Hen, R., and Lucki, I. (2001). Antidepressant-like behavioral effects in 5-hydroxytryptamine(1A) and 5-hydroxytryptamine(1B) receptor mutant. J. Pharmacol. Exp. Ther. 298, 1101–1107.
McEwen, B. S. (2000). The neurobiology of stress: from serendipity to clinical relevance. Brain Res. 886, 172–189.
McGrath, P. J., Stewart, J. W., Quitkin, F. M., Ying, C., Alpert, J. E., Nierenberg, A. A., Fava, M., Cheng, J., and Petkova, E. (2006). Predictors of relapse in a prospective study of fluoxetine treatment of major depression. Am. J. Psychiatry 163, 1542–1548.
Meier, C. R., Schlienger, R. G., and Jick, H. (2001). Use of selective serotonin reuptake inhibitors and risk of developing first-time acute myocardial infarction. Br. J. Clin. Pharmacol. 52, 179–184.
Meijer, W. E. E., Heerdink, E. R., Nolen, W. A., Herings, R. M. C., Leufkens, H. G. M., and Egberts, A. C. G. (2004). Association of risk of abnormal bleeding with degree of serotonin reuptake inhibition by antidepressants. Arch. Intern. Med. 164, 2367–2370.
Mercado, C. P., and Kilic, F. (2010). Molecular mechanisms of SERT in platelets: regulation of plasma serotonin levels. Mol. Interv. 10, 231–241.
Monster, T. B. M., Johnsen, S. P., Olsen, M. L., McLaughlin, J. K., and Sorensen, H. T. (2004). Antidepressants and risk of first-time hospitalization for myocardial infarction: a population-based case-control study. Am. J. Med. 117, 732–737.
Moret, C., Isaac, M., and Briley, M. (2009). Problems associated with long-term treatment with selective serotonin reuptake inhibitors. J. Psychopharmacol. 23, 967–974.
Morrow, J., and Nolen-Hoeksema, S. (1990). Effects of responses to depression on the remediation of depressive affect. J. Pers. Soc. Psychol. 58, 519–527.
Musselman, D. L., Marzec, U. M., Manatunga, A., Penna, S., Reemsnyder, A., Knight, B. T., Baron, A., Hanson, S. R., and Nemeroff, C.B. (2000). Platelet reactivity in depressed patients treated with paroxetine: preliminary findings. Arch. Gen. Psychiatry 57, 875–882.
Mutlu, O., Ulak, G., Celikyurt, I. K., Akar, F. Y., and Erden, F. (2011). Effects of citalopram on cognitive performance in passive avoidance, elevated plus maze and three-panel runway tasks in naive rats. Chin. J. Physiol. 54, 36–46.
National Institute for Clinical Excellence. (2004). Depression: Management of Depression in Primary and Secondary Care. London: National Institute for Clinical Excellence.
Nesse, R. M., and Williams, G. C. (1994). Why We Get Sick: The New Science of Darwinian Medicine. New York, NY: Times Books.
Nolen-Hoeksema, S., and Morrow, J. (1993). Effects of rumination and distraction on naturally-occurring depressed mood. Cogn. Emot. 7, 561–570.
Nordlind, K., Azmitia, E. C., and Slominski, A. (2008). The skin as a mirror of the soul: exploring the possible roles of serotonin. Exp. Dermatol. 17, 301–311.
O’Hanlon, J. F., Robbe, H. W. J., Vermeeren, A., Van Leeuwen, C., and Danjou, P. E. (1998). Venlafaxine’s effects on healthy volunteers’ driving, psychomotor, and vigilance performance during 15-day fixed and incremental dosing regimens. J. Clin. Psychopharmacol. 18, 212–221.
Olfson, M., Marcus, S. C., Druss, B., Elinson, L., Tanielian, T., and Pincus, H. A. (2002). National trends in the outpatient treatment of depression. JAMA 287, 203–209.
Paciaroni, M., Agnelli, G., Micheli, S., and Caso, V. (2007). Efficacy and safety of anticoagulant treatment in acute cardioembolic stroke – a meta-analysis of randomized controlled trials. Stroke 38, 423–430.
Park, R. J., Goodyer, I. M., and Teasdale, J. D. (2004). Effects of induced rumination and distraction on mood and overgeneral autobiographical memory in adolescent major depressive disorder and controls. J. Child Psychol. Psychiatry 45, 996–1006.
Paulmann, N., Grohmann, M., Voigt, J. P., Bert, B., Vowinckel, J., Bader, M., Skelin, M., Jevsek, M., Fink, H., Rupnik, M., and Walther, D. J. (2009). Intracellular serotonin modulates insulin secretion from pancreatic beta-cells by protein serotonylation. PLoS Biol. 7, e1000229. doi:10.1371/journal.pbio.1000229
Perera, T. D., Dwork, A. J., Keegan, K. A., Thirumangalakudi, L., Lipira, C. M., Joyce, N., Lange, C., Higley, J. D., Rosoklija, G., Hen, R., Sackeim, H. A., and Coplan, J. D. (2011). Necessity of hippocampal neurogenesis for the therapeutic action of antidepressants in adult nonhuman primates. PLoS ONE 6, e17600. doi:10.1371/journal.pone.0017600
Pigott, H. E., Leventhal, A. M., Alter, G. S., and Boren, J. J. (2010). Efficacy and effectiveness of antidepressants: current status of research. Psychother. Psychosom. 79, 267–279.
Pincus, H. A., Zarin, D. A., Tanielian, T. L., Johnson, J. L., West, J. C., Pettit, A. R., Pettit, A. R., Marcus, S. C., Kessler, R. C., and McIntyre, J. S. (1999). Psychiatric patients and treatments in 1997: findings from the American psychiatric practice research network. Arch. Gen. Psychiatry 56, 441–449.
Popa, D., Cerdan, J., Reperant, C., Guiard, B. P., Guilloux, J. P., David, D. J., and Gardier, A. M. (2010). A longitudinal study of 5-HT outflow during chronic fluoxetine treatment using a new technique of chronic microdialysis in a highly emotional mouse strain. Eur. J. Pharmacol. 628, 83–90.
Post, A., Crochemore, C., Uhr, M., Holsboer, F., and Behl, C. (2000). Differential effects of antidepressants on the viability of clonal hippocampal cells. Biol. Psychiatry 47, 138.
Pozuelo, L., Zhang, J. P., Franco, K., Tesar, G., Penn, M., and Jiang, W. (2009). Depression and heart disease: what do we know, and where are we headed? Cleve. Clin. J. Med. 76, 59–70.
Ramaekers, J. G., Muntjewerff, N. D., and O’Hanlon, J. F. (1995). A comparative-study of acute and subchronic effects of dothiepin, fluoxetine and placebo on psychomotor and actual driving performance. Br. J. Clin. Pharmacol. 39, 397–404.
Rantala, S., Vuopio-Varkila, J., Vuento, R., Huhtala, H., and Syrjanen, J. (2009). Predictors of mortality in beta-hemolytic streptococcal bacteremia: a population-based study. J. Infect. 58, 266–272.
Reeves, R. R., and Ladner, M. E. (2010). Antidepressant-induced suicidality: an update. CNS Neurosci. Ther. 16, 227–234.
Riedel, W. J., Eikmans, K., Heldens, A., and Schmitt, J. A. (2005). Specific serotonergic reuptake inhibition impairs vigilance performance acutely and after subchronic treatment. J. Psychopharmacol. 19, 12–20.
Romanovsky, A. A. (2007). Thermoregulation: some concepts have changed. Functional architecture of the thermoregulatory system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R37–R46.
Romanovsky, A. A., Almeida, M. C., Aronoff, D. M., Ivanov, A. I., Konsman, J. P., Steiner, A. A., and Turek, V. F. (2005). Fever and hypothermia in systemic inflammation: recent discoveries and revisions. Front. Biosci. 10, 2193–2216.
Rush, A. J., Trivedi, M. H., Wisniewski, S. R., Nierenberg, A. A., Stewart, J. W., Warden, D., Niederehe, G., Thase, M. E., Lavori, P. W., Lebowitz, B. D., McGrath, P. J., Rosenbaum, J. F., Sackeim, H. A., Kupfer, D. J., Luther, J., and Fava, M. (2006). Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am. J. Psychiatry 163, 1905–1917.
Rutter, J. J., and Auerbach, S. B. (1993). Acute uptake inhibition increases extracellular serotonin in the rat forebrain. J. Pharmacol. Exp. Ther. 265, 1319–1324.
Sairanen, M., Lucas, G., Ernfors, P., Castren, M., and Castren, E. (2005). Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J. Neurosci. 25, 1089–1094.
Sandercock, P. A. G., Counsell, C., and Kamal, A. K. (2008). Anticoagulants for acute ischaemic stroke. Cochrane Database Syst. Rev. 4, CD000024.
Santarelli, L., Saxe, M., Gross, C., Surget, A., Battaglia, F., Dulawa, S., Weisstaub, N., Lee, J., Duman, R., Arancio, O., Belzung, C., and Hen, R. (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301, 805–809.
Sapolsky, R. M. (2001). Depression, antidepressants, and the shrinking hippocampus. Proc. Natl. Acad. Sci. U.S.A. 98, 12320–12322.
Sauer, W. H., Berlin, J. A., and Kimmel, S. E. (2003). Effect of antidepressants and their relative affinity for the serotonin transporter on the risk of myocardial infarction. Circulation 108, 32–36.
Schlienger, R. G., Fischer, L. M., Jick, H., and Meier, C. R. (2004). Current use of selective serotonin reuptake inhibitors and risk of acute myocardial infarction. Drug Saf. 27, 1157–1165.
Schmaling, K. B., Dimidjian, S., Katon, W., and Sullivan, M. (2002). Response styles among patients with minor depression and dysthymia in primary care. J. Abnorm. Psychol. 111, 350–356.
Schmitt, J. A. J., Kruizinga, M. J., and Riedel, W. J. (2001). Non-serotonergic pharmacological profiles and associated cognitive effects of serotonin reuptake inhibitors. J. Psychopharmacol. 15, 173–179.
Schmitt, J. A. J., Ramaekers, J. G., Kruizinga, M. J., van Boxtel, M. P. J., Vuurman, E. F. P. M., and Riedel, W. J. (2002). Additional dopamine reuptake inhibition attenuates vigilance impairment induced by serotonin reuptake inhibition in man. J. Psychopharmacol. 16, 207–214.
Schulman, C. I., Namias, N., Doherty, J., Manning, R. J., Li, P., Elhaddad, A., Lasko, D., Amortegui, J., Dy, C. J., Dlugasch, L., Baracco, G., and Cohn, S. M. (2005). The effect of antipyretic therapy upon outcomes in critically ill patients: a randomized, prospective study. Surg. Infect. (Larchmt.) 6, 369–375.
Serretti, A., and Chiesa, A. (2009). Treatment-emergent sexual dysfunction related to antidepressants: a meta-analysis. J. Clin. Psychopharmacol. 29, 259–266.
Smoller, J. W., Allison, M., Cochrane, B. B., Curb, J. D., Perlis, R. H., Robinson, J. G., Rosal, M. C., Wenger, N. K., and Wassertheil-Smoller, S. (2009). Antidepressant use and risk of incident cardiovascular morbidity and mortality among postmenopausal women in the Women’s Health Initiative Study. Arch. Intern. Med. 169, 2128–2139.
Stahl, S. (2008). Stahl’s Essential Psychopharmacology: Neuroscientific Basis and Practical Applications, 3rd Edn. Cambridge: Cambridge University Press.
Stanley, E. D., Jackson, G. G., Panusarn, C., and Rubenis, M. (1975). Increased virus shedding with aspirin treatment of rhinovirus infection. JAMA 231, 1248–1251.
Stergiou, L., and Hengartner, M. O. (2004). Death and more: DNA damage response pathways in the nematode C-elegans. Cell Death Differ. 11, 21–28.
Stone, M., Laughren, T., Jones, M. L., Levenson, M., Holland, P. C., Hughes, A., Hammad, T. A., Temple, R., and Rochester, G. (2009). Risk of suicidality in clinical trials of antidepressants in adults: analysis of proprietary data submitted to US Food and Drug Administration. BMJ 339, b2880.
Swenson, B. R., Hedrick, T. L., Popovsky, K., Pruett, T. L., and Sawyer, R. G. (2007). Is fever protective in surgical patients with bloodstream infection? J. Am. Coll. Surg. 204, 815–821.
Tanrikut, C., Feldman, A. S., Altemus, M., Paduch, D. A., and Schlegel, P. N. (2010). Adverse effect of paroxetine on sperm. Fertil. Steril. 94, 1021–1026.
Tata, L. J., West, J., Smith, C., Farrington, P., Card, T., Smeeth, L., and Hubbard, R. (2005). General population based study of the impact of tricyclic and selective serotonin reuptake inhibitor antidepressants on the risk of acute myocardial infarction. Heart 91, 465–471.
Taupin, P. (2007). BrdU immunohistochemistry for studying adult neurogenesis: paradigms, pitfalls, limitations, and validation. Brain Res. Rev. 53, 198–214.
Trifiro, G., Dieleman, J., Sen, E. F., Gambassi, G., and Sturkenboom, M. C. J. M. (2010). Risk of ischemic stroke associated with antidepressant drug use in elderly persons. J. Clin. Psychopharmacol. 30, 252–258.
Turner, E. H., Matthews, A. M., Linardatos, E., Tell, R. A., and Rosenthal, R. (2008). Selective publication of antidepressant trials and its influence on apparent efficacy. N. Engl. J. Med. 358, 252–260.
Ulak, G., Gocmez, S., Erden, F., Tanyeri, P., Utkan, T., Yildiz, F., Mutlu, O., and Gacar, N. (2006). Chronic administration of fluoxetine or venlafaxine induces memory deterioration in an inhibitory avoidance task in rats. Drug Dev. Res. 67, 456–461.
Van der Kooy, K., van Hout, H., Marwijk, H., Marten, H., Stehouwer, C., and Beekman, A. (2007). Depression and the risk for cardiovascular diseases: systematic review and meta analysis. Int. J. Geriatr. Psychiatry 22, 613–626.
van Haelst, I. M. M., Egberts, T. C. G., Doodeman, H. J., Traast, H. S., Burger, B. J., Kalkman, C. J., and van Klei, W. A. (2010). Use of serotonergic antidepressants and bleeding risk in orthopedic patients. Anesthesiology 112, 631–636.
Vickers, K. S., and Vogeltanz-Holm, N. D. (2003). The effects of rumination and distraction tasks on psychophysiological responses and mood in dysphoric and nondysphoric individuals. Cognit. Ther. Res. 27, 331–348.
Wakefield, J. C. (1992). The concept of mental disorder: on the boundary between biological facts and social values. Am. Psychol. 47, 373–388.
Wakefield, J. C. (1999). Evolutionary versus prototype analyses of the concept of disorder. J. Abnorm. Psychol. 108, 374–399.
Wakefield, J. C., Schmitz, M. F., and Baer, J. C. (2010). Does the DSM-IV clinical significance criterion for major depression reduce false positives? Evidence from the National Comorbidity Survey replication. Am. J. Psychiatry 167, 298–304.
Walker, A. J., Card, T., Bates, T. E., and Muir, K. (2011). Tricyclic antidepressants and the incidence of certain cancers: a study using the GPRD. Br. J. Cancer 104, 193–197.
Warner-Schmidt, J. L., and Duman, R. S. (2006). Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment. Hippocampus 16, 239–249.
Watson, P. J., and Andrews, P. W. (2002). Toward a revised evolutionary adaptationist analysis of depression: the social navigation hypothesis. J. Affect. Disord. 72, 1–14.
Weber, C. (2005). Platelets and chemokines in atherosclerosis: partners in crime. Circ. Res. 96, 612–616.
Weinstein, M. P., Iannini, P. B., Stratton, C. W., and Eickhoff, T. C. (1978). Spontaneous bacterial peritonitis: review of 28 cases with emphasis on improved survival and factors influencing prognosis. Am. J. Med. 64, 592–598.
Weinstein, M. P., Murphy, J. R., Reller, L. B., and Lichtenstein, K. A. (1983). The clinical significance of positive blood cultures: a comprehensive analysis of 500 episodes of bacteremia and fungemia in adults. 2. Clinical observations, with special reference to factors influencing prognosis. Rev. Infect. Dis. 5, 54–70.
Wenzlaff, R. M., and Luxton, D. D. (2003). The role of thought suppression in depressive rumination. Cognit. Ther. Res. 27, 293–308.
Wilson, C. C., Faber, K. M., and Haring, J. H. (1998). Serotonin regulates synaptic connections in the dentate molecular layer of adult rats via 5-HT1a receptors: evidence for a glial mechanism. Brain Res. 782, 235–239.
Wingen, M., Bothmer, J., Langer, S., and Ramaekers, J. G. (2005). Actual driving performance and psychomotor function in healthy subjects after acute and subchronic treatment with escitalopram, mirtazapine, and placebo: a crossover trial. J. Clin. Psychiatry 66, 436–443.
Woods, A. H. (2009). “Evolution of homeostatic physiological systems,” in Phenotypic Plasticity of Insects, eds D. Whitman and T. N. Ananthakrishnan (Plymouth: Science Publishers), 655–674.
Keywords: antidepressant medications, depression, mortality, placebo, rebound, risks, safety, side effects
Citation: Andrews PW, Thomson JA Jr, Amstadter A and Neale MC (2012) Primum non nocere: an evolutionary analysis of whether antidepressants do more harm than good. Front. Psychology 3:117. doi: 10.3389/fpsyg.2012.00117
Received: 31 December 2011; Accepted: 30 March 2012;
Published online: 24 April 2012.
Edited by:
Debra Lieberman, University of Miami, USAReviewed by:
Norman Li, Singapore Management University, SingaporeLei Chang, Chinese University of Hong Kong, Hong Kong
Copyright: © 2012 Andrews, Thomson Jr, Amstadter and Neale. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Paul W. Andrews, Department of Psychology, Neuroscience and Behaviour, McMaster University, 1280 Main Street West, Hamilton, ON, Canada L8S 4K1. e-mail:cGFuZHJld3NAbWNtYXN0ZXIuY2E=