 Morgan Bennett
Morgan Bennett Tracy E. Hawk
Tracy E. Hawk J. Hollis Rice
J. Hollis Rice Tarek Hewezi
Tarek Hewezi- Department of Plant Sciences, University of Tennessee, Knoxville, TN, United States
A growing body of evidence indicates that epigenetic mechanisms, particularly DNA methylation, play key regulatory roles in plant-nematode interactions. Nevertheless, the transcriptional activity of key genes mediating DNA methylation and active demethylation in the nematode feeding sites remains largely unknown. Here, we profiled the promoter activity of 12 genes involved in maintenance and de novo establishment of DNA methylation and active demethylation in the syncytia and galls induced respectively by the cyst nematode Heterodera schachtii and the root-knot nematode Meloidogyne incognita in Arabidopsis roots. The promoter activity assays revealed that expression of the CG-context methyltransferases is restricted to feeding site formation and development stages. Chromomethylase1 (CMT1), CMT2, and CMT3 and Domains Rearranged Methyltransferase2 (DRM2) and DRM3, which mediate non-CG methylation, showed similar and distinct expression patterns in the syncytia and galls at various time points. Notably, the promoters of various DNA demethylases were more active in galls as compared with the syncytia, particularly during the early stage of infection. Mutants impaired in CG or CHH methylation similarly enhanced plant susceptibility to H. schachtii and M. incognita, whereas mutants impaired in CHG methylation reduced plant susceptibility only to M. incognita. Interestingly, hypermethylated mutants defective in active DNA demethylation exhibited contrasting responses to infection by H. schachtii and M. incognita, a finding most likely associated with differential regulation of defense-related genes in these mutants upon nematode infection. Our results point to methylation-dependent mechanisms regulating plant responses to infection by cyst and root-knot nematodes.
Introduction
DNA methylation and demethylation pathways regulate many aspects of plant development and stress responses through reversible, non-genetic modification of cytosine to 5-methylcytosine (5mC) (Fransz and De Jong, 2002; Henderson and Jacobsen, 2007; Zilberman et al., 2007; Zhang et al., 2010; Matzke and Mosher, 2014; Du et al., 2015; López Sánchez et al., 2016; Hewezi et al., 2018; Hewezi, 2020). In plants, this occurs in the CG, CHG, and CHH sequence contexts (where H is any nucleotide except G) (Lister et al., 2008). The de novo formation of 5mC is catalyzed by DNA methyltransferases DOMAINS REARRANGED METHYLTRANSFERASE2 (DRM2) and DRM3 through the RNA-directed DNA methylation (RdDM) pathway (Cao and Jacobsen, 2002a; Cao and Jacobsen, 2002b; Cao et al., 2003; Henderson et al., 2010). Following DNA replication and cell division, DNA methylation is maintained in a sequence context-specific manner (Saze et al., 2003). CG methylation is maintained mainly by METHYLTRANSFERASE1 (MET1). MET1 paralogs, MET2b and MET3, also contribute to CG maintenance but to a much lesser extent (Kankel et al., 2003; Quadrana et al., 2016). CHROMOMETHYLASE1 (CMT1), CMT2, CMT3, and the RdDM pathway are responsible for maintaining non-CG methylation (Henderson et al., 2010; Law and Jacobsen, 2010; Costa-Nunes et al., 2014; Stroud et al., 2014). 5mC can be actively removed and replaced by cytosine via the base excision repair process mediated by the paralogous DNA demethylases REPRESSOR OF SILENCING 1 (ROS1), DEMETER (DME), DME-LIKE2 (DML2) and DML3 (Choi et al., 2002; Gong et al., 2002; Agius et al., 2006; Morales-Ruiz et al., 2006; Penterman et al., 2007b; Gehring et al., 2009; Zhang and Zhu, 2012). Together, DNA methylation and demethylation fine-tune genome-wide methylation levels and subsequent transcriptional reprogramming (Gong et al., 2002; Penterman et al., 2007a; Penterman et al., 2007b; Zhu et al., 2007; Ortega-Galisteo et al., 2008; Lei et al., 2015; Bennett et al., 2022).
Recent studies have provided intriguing evidence for the involvement of DNA methylation and active demethylation pathways in modulating defense responses against various phytopathogens (Hewezi et al., 2018). Genome-wide methylation profiling of Arabidopsis leaves inoculated with Pseudomonas syringae pv. tomato DC3000 (Pst) revealed widespread changes in plant methylomes, particularly in gene-rich regions (Dowen et al., 2012). Pst-induced DNA methylation changes were associated with differential transcript abundances of a significant number of stress-responsive genes (Dowen et al., 2012). Similarly, infection of canola (Brassica napus) by the fungal pathogen Leptosphaeria maculans triggered differential DNA methylation in the promoters of thousands of genes both in resistant and susceptible cultivars, of which numerous genes are associated with defense responses (Tirnaz et al., 2020). The correlation between genome-wide DNA methylation patterns and gene expression levels were also reported in rice and tobacco in response to infection by rice Black Streaked Dwarf Virus and Cucumber Mosaic Virus, respectively (Wang et al., 2018; Li et al., 2020).
Changes in DNA methylation levels seem to impact plant interactions with fungal pathogens and oomycetes. For example, hypomethylated mutants defective in the establishment of DNA methylation increased plant resistance to Hyaloperonospora arabidopsidis (Hpa), whereas hypermethylated mutants impaired in active DNA demethylation increased plant susceptibility to both Hpa and Fusarium oxysporum (Le et al., 2014; Lopez Sanchez et al., 2016). Gene expression analyses of these mutants provided strong evidence for the involvement of DNA methylation in the regulation of numerous genes with defense- and stress-related functions. Notably, modulation of defense- and stress-responsive genes in hyper- or hypomethylated mutants showed opposite responses to various phytopathogens (López Sánchez et al., 2016).
Cyst nematodes (Globodera and Heterodera spp.) and root-knot nematodes (Meloidogyne spp.) are sedentary obligate biotrophs that infect the root systems of a broad-spectrum of host plants, including the model plant Arabidopsis. Nematode parasitism of host plants is characterized by the formation of syncytia and giant-cells as permanent feeding sites for cyst and root-knot nematodes, respectively. The formation of giant-cells by root-knot nematodes is accompanied by the formation of galls at the site of infection as a result of increasing cell division of neighboring cells. The parasitic second-stage juveniles (J2s) feed from these metabolically hyperactive feeding cells and develop into adult females and complete the life cycle. Several experimental evidences indicate that epigenetic mechanisms, particularly DNA methylation, play important roles in plant-nematode interactions (Hewezi, 2020). For example, mutants partially impaired in non-GC DNA methylation exhibited reduced susceptibility to cyst and root-knot nematodes (Hewezi et al, 2018; Ruiz-Ferrer et al., 2018; Atighi et al., 2021). Genome-wide DNA methylation analysis of soybean roots exposed to soybean cyst nematode (SCN, Heterodera glycines) revealed widespread DNA hypomethylation in the promoter and transcribed regions of a substantial number of genes (Rambani et al., 2015) that were previously reported to be differentially expressed in the syncytium (Ithal et al., 2007; Klink et al., 2009, Klink et al., 2010; Kandoth et al., 2011). Similarly, beet cyst nematode (BCN, Heterodera schachtii) was found to induce considerable increases in hypomethylation levels of protein-coding genes and transposable elements (TEs) (Hewezi et al., 2017; Piya et al., 2017). Further analysis indicated that BCN-induced DNA methylation changes may directly impact the transcript abundance of more than one-fourth of differentially expressed genes in syncytium (Hewezi et al., 2017). Methylome analysis of two near isogenic lines of soybean differing in their response to SCN revealed distinct and specific methylation profiles over protein-coding and miRNA genes as well as TEs (Rambani et al., 2020a; Rambani et al., 2020b). While the susceptible line exhibited global hypomethylation patterns, the resistant line showed global hypermethylation (Rambani et al., 2020a; Rambani et al., 2020b). Global DNA hypomethylation patterns, predominantly in the CHH context, were also observed in early developing galls induced by the root-knot nematode Meloidogyne graminicola in rice (Atighi et al., 2021). In contrast, it has been recently shown that early developing galls formed by Meloidogyne javanica have undergone hypermethylation particularly in the CHG context (Silva et al., 2022).
The biological significance of DNA methylation changes induced by parasitic nematodes was established using various functional assays of overexpression and mutant analyses (Hewezi et al., 2017; Rambani et al., 2020a; Rambani et al., 2020b; Atighi et al., 2021; Silva et al., 2022). However, the transcriptional activity of key genes involved in DNA methylation and active demethylation in nematode feeding sites remains mostly unknown. We addressed this issue by profiling the spatio-temporal expression patterns of genes involved in maintenance and de novo establishment of DNA methylation as well as those involved in active DNA demethylation in the syncytia and galls induced respectively by H. schachtii and M. incognita in Arabidopsis roots. Also, mutants of these genes were assayed for susceptibility to H. schachtii and M. incognita. Our analyses revealed a key role of DNA methylation and active demethylation in mediating plant susceptibility to two evolutionary distant nematode species.
Materials and methods
Plant materials and growth conditions
All transgenic Arabidopsis (Arabidopsis thaliana) lines expressing the GUS reporter gene under the control of various DNA methylation and demethylation related-genes were generated in the Columbia‐0 (Col‐0) background (Bennett et al, 2021). All mutant lines were generated in the Col‐0) background, expect ros1 (CS66099) is in the C24 background. The mutant lines cmt3-7 (CS6365, Lindroth et al., 2001), drm2-2 (CS16386, Cao and Jacobsen, 2002a), and ros1-1 (CS66099, Gong et al., 2002) have been previously characterized. T-DNA insertional mutants for met2 (SALK_102231C and SALK_093835C), met3 (SALK_024049C and SALK_099592C), cmt1 (SALK_138685C and SALK_030404C), cmt2 (SALK_012874C and CS879822), cmt3 (CS6365) drm2 (CS16386 and SALK_129477C), drm3 (SALK_136439C and SALK_024820C), ros1 (CS66099 and SALK_045303C), dml2 (SALK_131712C) and dml3 (SALK_056440C) were obtained from Arabidopsis Biological Resource Center (ABRC) (Supplementary Figure 1A). mRNA expression levels of these genes were quantified in uncharacterized mutant lines and the corresponding wild-type plants using reverse transcription quantitative PCR (RT-qPCR) (Supplementary Figure 1B). Ten-day-old plants grown in plates containing MS medium were used for RNA extraction and quantification of gene expression levels as described below. Length of the main root of 15 two-week-old plants per line was measured, and statistically significant differences from wild-type plants were calculated using t tests (P < 0.05). In addition, shoot and root phenotypes of all mutant lines along with the corresponding wild-type plants were assessed in four-week-old soil-grown plants.
RNA isolation and RT-qPCR
Total RNA was isolated from mutant lines and the corresponding wild-type plants using the method previously described by Verwoerd et al. (1989), and then treated with DNase I (Invitrogen). DNase-treated RNA samples were diluted to a concentration of 50 ng/µL and used as a template for RT-qPCR reactions. The reactions were performed using Verso 1-step RT-qPCR (Thermo Fisher Scientific) in QuantStudio 6 Flex (Applied Biosystems) under the following conditions: 50°C for 15 min, 95°C for 15 min, and 40 cycles of 95°C for 15 s and 55°C for 30 s. Gene expression levels were quantified using 3 biological and 2 technical replicates. Quantification of expression levels was conducted using the 2-ΔΔCT method (Livak and Schmittgen, 2001). Actin8 (AT1G49240) and PROTEIN PHOSPHATASE 2A SUBUNIT A3 (PP2AA3, AT1G13320) were used as internal control to normalize gene expression levels (Piya et al., 2019; Piya et al., 2020; Bennett et al., 2021). ΔCT values determined using Actin8 and PP2AA3 were very similar. Primers used for RT-qPCR assays were provided in Supplementary Table 1.
Histochemical analysis of GUS activity
Seeds of the transgenic GUS reporter lines were sterilized using a 2.8% bleach solution for 5 minutes followed by several washes with sterilized distilled water. The sterilized seeds were randomly planted in culture plates (BD Biosciences) containing modified Knop’s medium solidified with 0.8% Daishin agar (Brunschwig Chemie). Ten-day-old plants were inoculated with about 100 surface-sterilized J2 of Heterodera schachtii or Meloidogyne incognita. Histochemical GUS staining was performed at 3, 7, 10, and 14 days post inoculation (dpi) for H. schachtii and 4, 7, and 14 dpi for M. incognita, according to Jefferson et al. (1987). Samples were incubated with the X-Gluc substrate (5-bromo-4-chloro-3-indolyl-beta-D-glucuronic acid, cyclohexylammonium salt) at 37°C and checked every 30 minutes for colorimetric changes. After staining, the GUS solution was replaced with 70% ethanol to terminate the reaction and samples were stored in ethanol at room temperature. At least two independent transgenic lines were assayed for each reporter line, with at least 24 replicated plants per line. Nematode-inoculated roots were imaged immediately following GUS staining. Images were captured using an EVOS M7000 microscope with a 4x lens for 3 and 4 dpi images and a 2x lens for all other time points.
Nematode susceptibility assays
H. schachtii infection assays were determined in 12-well plates containing modified Knop’s medium (Sijmons et al., 1991) solidified with 0.8% Daishin agar. Seeds of the mutants and corresponding wild type were surface-sterilized and planted in a randomized block design. Ten days after planting, each seedling was inoculated with approximately 200 surface-sterilized J2 of H. schachtii. Following inoculation, the plates were incubated in the dark at 24°C for 3 days to facilitate nematode infection. The plants were then grown in 16-h-light/8-h-dark cycles at 24°C for three weeks before counting the number of J4 females per root system. Two independent experiments were performed with 20 replicates per genotype for each mutant and their corresponding wild-type controls.
For M. incognita infection assays, seeds of the mutants and wild type were surface-sterilized and planted on modified Knop’s-containing plates. Ten-day-old seedlings were then transplanted to pots containing steam‐sterilized sand mixed with top soil at a 3 to 1 ratio and organized in a randomized complete block design. A week after transplantation, each plant was inoculated with approximately 500 J2 M. incognita and grown in 16-h-light/8-h-dark cycles at 26°C. Whole root systems were collected five weeks after inoculation and the number of galls per root system was counted using light microscope. Statistically significant differences between the mutant lines and wild-type controls were calculated using a modified t test in the statistical software package SAS, where P values less than 5% were considered statistically significant.
Results
Promoter activity of DNA methyltransferases and demethylases in the syncytia induced by the beet cyst nematode Heterodera schachtii
Seeds of transgenic Arabidopsis lines (T2 generation) expressing the β-glucuronidase (GUS) reporter, guided by the promoter of 12 different DNA methyltransferase and demethylase genes were planted. Ten‐day‐old seedlings were inoculated with second‐stage juvenile (J2) nematodes of Heterodera schachtii or Meloidogyne incognita. Promoter activity was determined by staining the plants at various time points post inoculation. In response to H. schachtii infection, the major CG methyltransferase MET1 was highly expressed in the syncytia at 3- and 7-day post infection (dpi), as indicated by strong GUS. At later time points, however, no GUS activity directed by MET1 promoter was detected in the syncytium (Figure 1A). The MET1 homolog MET3, whose expression was not detected in roots tissues under non-infected conditions (Bennett et al., 2021), was strongly upregulated in the syncytium at 3 dpi. No activity of MET2b promoter was detected in the syncytium at any time points (Figure 1A). These expression patterns indicate that maintenance of the CG-context methylation in the syncytia is restricted to early stages of syncytium formation and development.
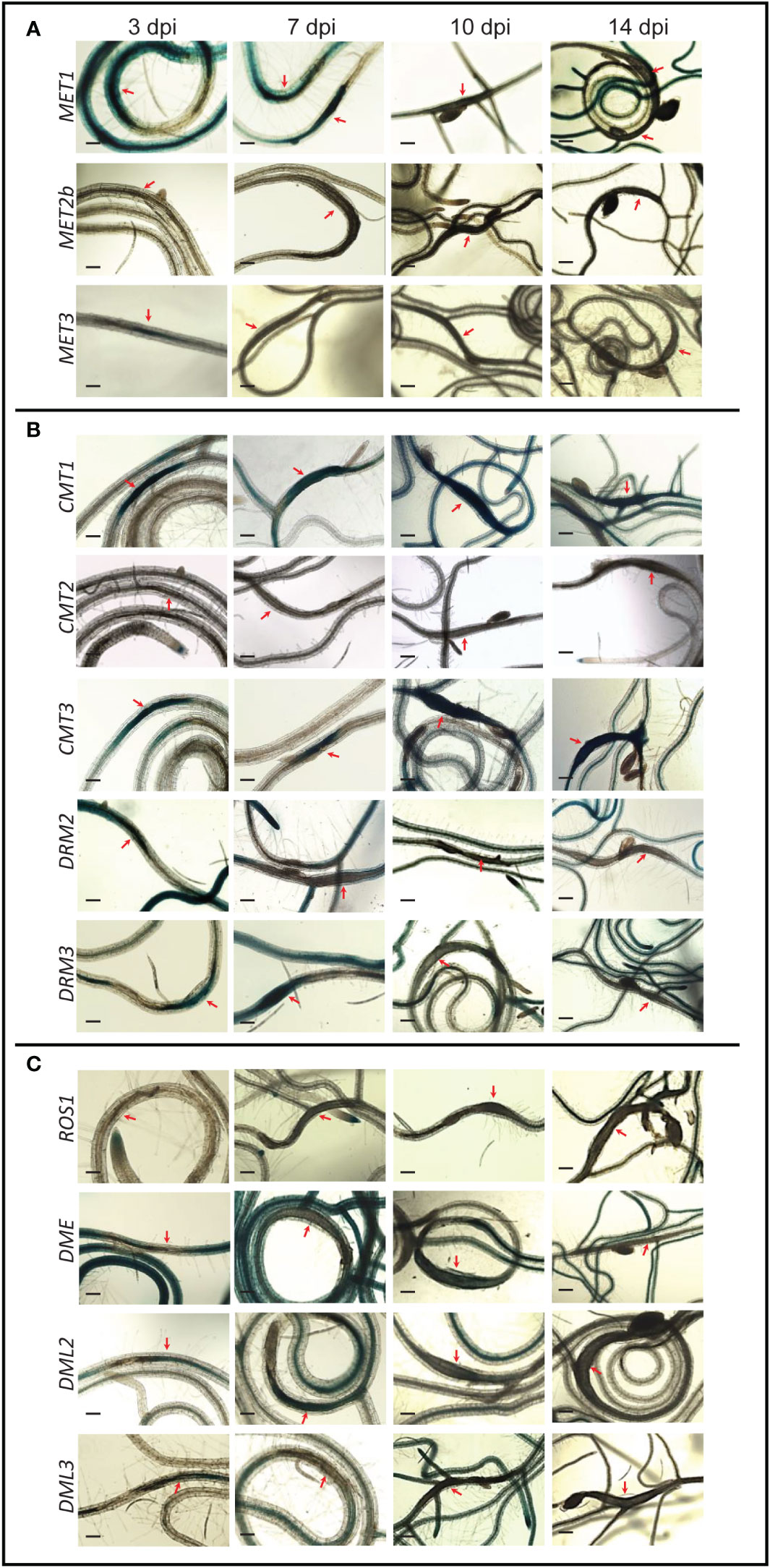
Figure 1 Promoter activity of DNA methyltransferases and demethylases in the syncytia induced by (H) schachtii on Arabidopsis roots. Time-course experiments showing GUS activity controlled by the CG methyltransferases MET1, METb, and MET3 (A), the non-CG methyltransferases CMT1, CMT2, CMT3, DRM2, and DRM3 (B), and the DNA demethylases ROS1, DME, DML2, and DML3 (C) at 3, 7, 10, and 14 dpi. Red arrows point to syncytia. Scale bar = 200 µm.
Promoter activity of CMT3, the main CHG methyltransferase gene, and its homolog CMT1, revealed similar patterns of high expression in the syncytium at all time points (Figure 1B), suggesting that CHG-context methylation is actively maintained in the syncytium during early and late stages of H. schachtii infection. In contrast, the expression of CMT2 was not activated in the syncytium at any time points. The RdDM methyltransferase genes DRM2 and DRM3 showed similar expression patterns of upregulation in the syncytia at 3 dpi and downregulation at 14 dpi (Figure 1B). However, at 7 and 10 dpi DRM2 and DRM3 exhibited opposite expression patterns, i.e., DRM2 was downregulated and DRM3 was upregulated in the syncytia (Figure 1B). These results suggest that de novo DNA methylation is actively established in the syncytia.
The DNA demethylase genes ROS1, DME, and DML3 were downregulated in the syncytia during early and/or late infection stages. Unlike these three DNA demethylases, DML2 was upregulated in the syncytia at 3 and 7 dpi (Figure 1C), suggesting active DNA demethylation in the syncytium is mediated solely through the activity of DML2.
To further confirm promoter activity data, we quantified the expression levels of MET1, MET3, DRM2, ROS1, and DML2 in H. schachtii-infected roots of wild-type Col-0 plants using reverse transcription quantitative PCR (RT-qPCR) at 4 dpi. The expression levels of MET1, DRM2, ROS1, and DML2 were significantly upregulated in infected roots relative to non-infected control plants (Figure S2), confirming the increased promoter activity of these genes upon H. schachtii infection. However, MET3, showed significant downregulation (Supplementary Figure 2) despite its promoter was strongly and specifically upregulated in the syncytium, suggesting opposite regulation of MET3 in roots versus syncytial cells in response to H. schachtii infection.
Mutations of DNA methylation and demethylation-related genes alter plant susceptibility to Heterodera schachtii
We next investigated the impact of mutations in these DNA methylation-related genes on plant response to nematode infection. Homozygous T-DNA insertional mutants for 10 genes were identified and phenotypically analyzed. This included mutants of MET2b (SALK_093835C and SALK_102231C), MET3 (SALK_099592C and SALK_024049C), CMT1 (SALK_138685C and SALK_030404C), CMT2 (SALK_012874C and CS879822), CMT3 (CS6365), DRM2 (CS16386 and SALK_129477C), DRM3 (SALK_024820C and SALK_136439C), ROS1(SALK_045303C and CS66099) DML2 (SALK_131712C), and DML3 (SALK_056440C) (Supplementary Figure 1). Homozygous mutants of MET1 and DME are embryonic lethal or associated with severe developmental irregularities (Choi et al, 2002; Kankel et al., 2003; Saze et al., 2003; Mathieu et al, 2007), and therefore were not included in the susceptibility assays. No severe morphological irregularities in the shoots of mutants assessed in this study were detected when compared with the wild-type plants (Supplementary Figure 3). With the exception of the short root phenotypes observed in the ROS1 mutants (SALK_045303C and CS66099), no noticeable morphological changes in root structure and length were noticed in these mutants as compared with wild-type plants (Supplementary Figure 4).
Seeds of the mutant lines along with the corresponding wild types were planted in 12-well plates and inoculated with freshly hatched J2s of H. schachtii. The numbers of nematode J4 females per root system were counted three weeks after inoculation and used as a measurement of plant susceptibility. As shown in Figure 2A, both mutant alleles of MET2b (SALK_102231C and SALK_093835C) showed susceptibility levels similar to the wild-type Col-0, consistent with the absence of MET2b:GUS activity in the syncytium. In contrast, the MET3 mutant (SALK_024049C) was significantly more susceptible to H. schachtii in comparison with the wild-type Col-0 (Figure 2A), suggesting that CG hypomethylation is associated with increased plant susceptibility to H. schachtii. All mutants of the three CMT genes did not show any significant impact on plant susceptibility (Figures 2B, C). Interestingly, both mutant alleles of DRM2 and DRM3 showed statistically significant increases in plant susceptibility (Figure 2D). These results suggest that non-CG hypomethylation is also associated with increased plant susceptibility to H. schachtii. All tested mutants of the DNA demethylase genes ROS1, DML2, and DML3 were more susceptible as compared with the corresponding wild-type Col-0 or C24 (Figures 2E, F), signifying that inhibition of active DNA demethylation promotes H. schachtii parasitism of Arabidopsis.
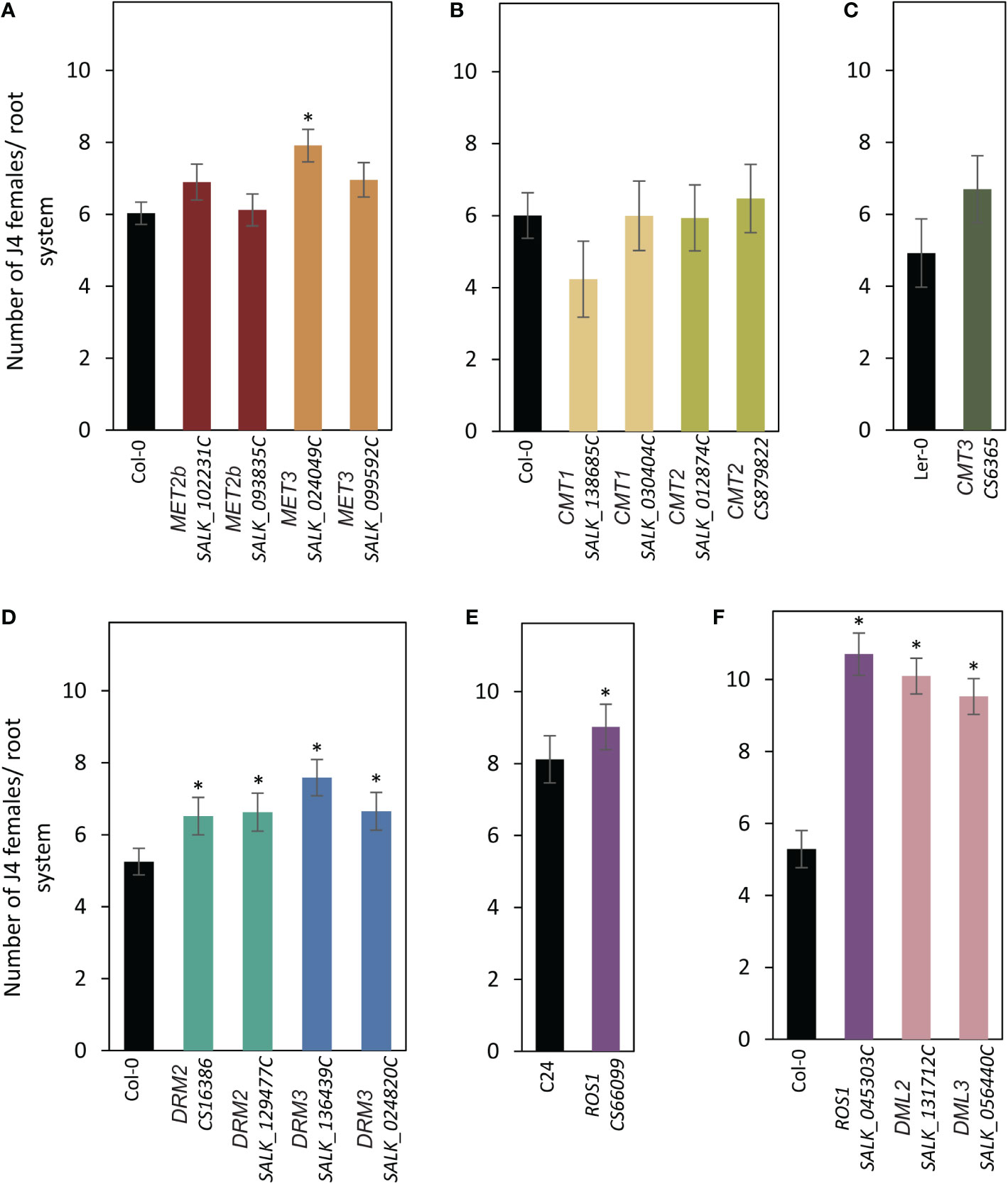
Figure 2 Mutations of DNA methyltransferases and demethylases enhance plant susceptibility to H. schachtii. (A–E): H. schachtii infection assays of mutant alleles of MET2b (SALK_102231C and SALK_093835C), MET3 (SALK_024049C and SALK_099592C) (A), CMT1 (SALK_138685C and SALK_030404C) and CMT2 (SALK_012874C and CS879822) (B), CMT3 (CS6365) (C), DRM2 (CS16386 and SALK_129477C) and DRM3 (SALK_136439C and SALK_024820C) (D), ROS1 (CS66099) (E), ROS1 (SALK_045303C), DML2 (SALK_131712C), and DML3 (SALK_056440C) (F). The number of J4 female nematodes per root system was counted 3 weeks post inoculation. Data are presented as means ± SE (n = 20). Asterisks denote statistically significant differences from the wild-type Col-0, Ler or C24 as determined by t tests (P < 0.05).
Promoter activity of DNA methyltransferases and demethylases in the galls induced by the root-knot nematode Meloidogyne incognita
The CG methyltransferase genes MET1, MET2b, and MET3 were expressed in the galls induced by the root-knot nematode M. incognita at 4 dpi (Figure 3A). At 7 dpi, however, only MET1 showed GUS staining in the galls. At 14 dpi, none of these three genes was activated in the galls, suggesting that CG methylation is maintained in the gall tissues during the early stage of infection.

Figure 3 Promoter activity of DNA methyltransferases and demethylases in the galls induced by M. incognita on Arabidopsis roots. Time-course experiments showing GUS activity controlled by the CG methyltransferases MET1, MET2b, and MET3 (A), the non-CG methyltransferases CMT1, CMT2, CMT3, DRM2, and DRM3 (B), and the DNA demethylases ROS1, DME, DML2, and DML3 (C) in the galls formed by M. incognita at 4, 7, and 14 dpi. Red arrows point to galls. Scale bar = 200 µm.
The CMT gene family members showed varied expression patterns in the galls (Figure 3B). While the promoter activity of CMT1 can be seen in the galls at 4, 7, and 14 dpi, CMT3 promoter was active in the galls only at 7 dpi. In contrast, GUS activity driven by CMT2 promoter was not detected in the galls at any time points (Figure 3B). These data suggest that variable activities of the enzymes mediating CHG methylation occur in the galls induced by M. incognita. DRM2 and DRM3 exhibited high expression in the galls at all time points, implying a role for de novo DNA methylation in establishing gall methylomes during various parasitic stages.
The expression of ROS1, DME, DML2, and DML3 was observed in the galls at 4 dpi (Figure 3C), signifying a role for active DNA demethylation during early stage of M. incognita infection. However, at 7 dpi, only DML2 showed strong GUS staining in the galls (Figure 3C). ROS1 and DML3 showed weak but visible GUS staining at 7 dpi (Figure 3C). At 14 dpi, none of these four DNA demethylase genes showed detectable GUS staining in the galls (Figure 3C), suggesting that active DNA demethylation occurs primarily during the early stage of M. incognita infection.
Furthermore, the expression levels of MET1, MET3, DRM2, ROS1, and DML2 were measured in M. incognita-infected roots of wild-type Col-0 plants at 4 dpi using RT-qPCR to validate the promoter activity of these genes. MET1, MET3, ROS1, and DML2 showed between 2 and 5-fold upregulation in infected roots compared with non-infected roots (Figure S5), consistent with the strong GUS activity of these genes detected after M. incognita infection at this time point.
Mutations of DNA methylation and demethylation-related genes alter plant susceptibility to Meloidogyne incognita
The T-DNA insertional mutants mentioned above were also evaluated for their susceptibility to M. incognita. Three-week-old plants growing in soil were inoculated with freshly hatched J2s of M. incognita, and the number of galls per root system was counted three weeks after inoculation and used to determine susceptibility levels of the mutants in comparison with the corresponding wild-type plants. Of the genes involved in CG methylation, we found that mutant alleles of MET2b (SALK_093835C) and MET3 (SALK_099592C and SALK_024049C) were significantly more susceptible as compared with the wild-type Col-0 plants (Figure 4A). In contrast, mutants of CMT1 (SALK_138685C) or CMT2 (SALK_012874C and CS879822) were statistically less susceptible to M. incognita compared with Col-0 (Figure 4B). Susceptibility level of the CMT3 mutant (CS6365) was comparable to that of wild-type Ler-0 (Figure 4C). Mutant alleles of DRM2 (CS16386 and SALK_129477C) and DRM3 (SALK_136439C) showed statistically significant increases in plant susceptibility (Figure 4D). These results suggest that genes involved in the CG and non-CG methylation may exert opposite effects on plant susceptibility to M. incognita. Interestingly, all examined mutants of the DNA demethylases ROS1 (SALK_045303C and CS66099), DML2 (SALK_131712C), and DML3 (SALK_056440C) were statistically less susceptible to M. incognita as compared with the corresponding wild-type Col-0 or C24 (Figures 4E, F), implying the importance of active DNA demethylation for M. incognita parasitism of Arabidopsis.
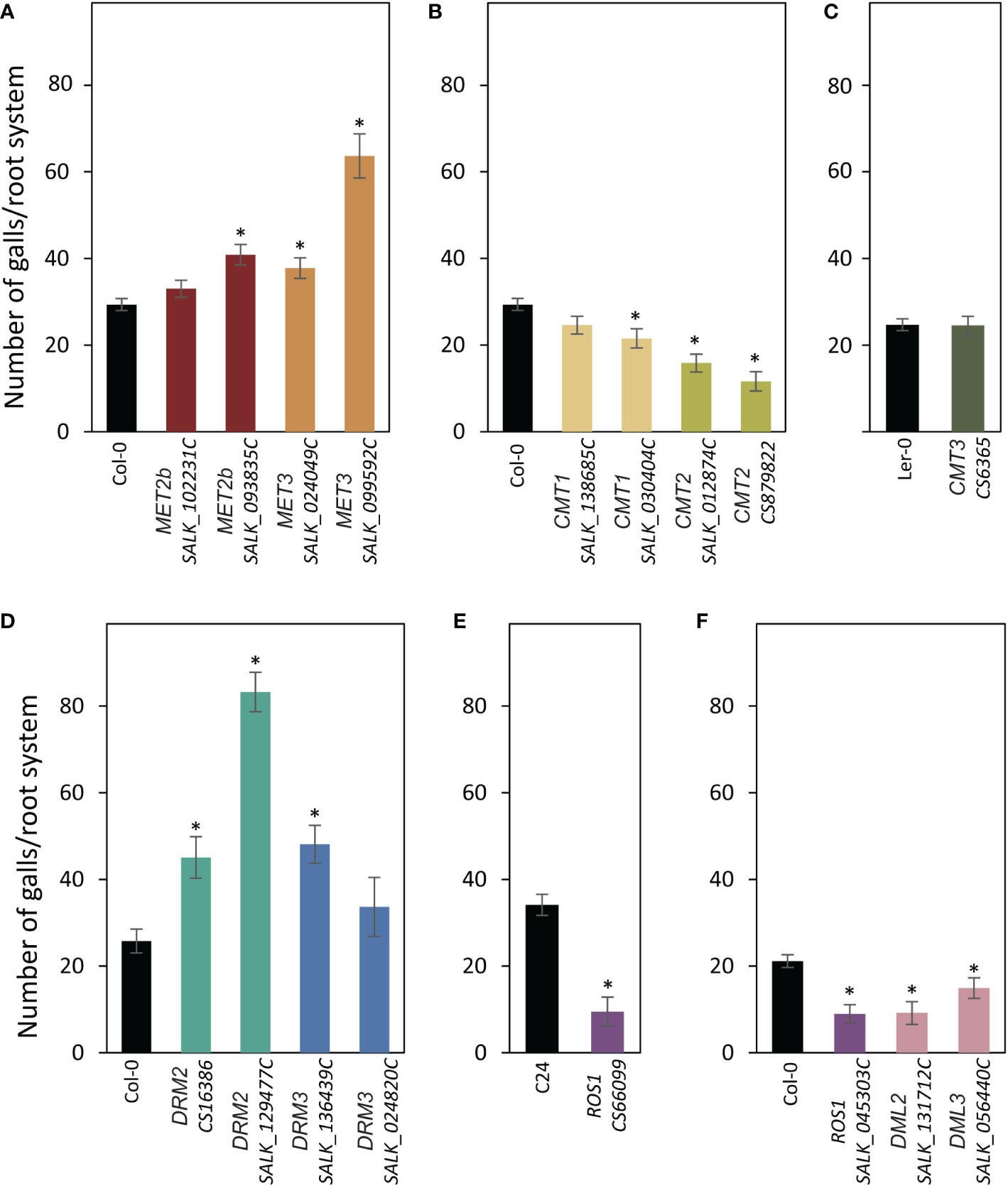
Figure 4 Mutations of DNA methyltransferases and demethylases alter plant susceptibility to M. incognita. (A–E): M. incognita infection assays of mutant alleles of MET2b (SALK_102231C and SALK_093835C), MET3 (SALK_024049C and SALK_099592C) (A), CMT1 (SALK_138685C and SALK_030404C) and CMT2 (SALK_012874C and CS879822) (B), CMT3 (CS6365) (C), DRM2 (CS16386 and SALK_129477C) and DRM3 (SALK_136439C and SALK_024820C) (D), ROS1 (CS66099) (E), ROS1 (SALK_045303C), DML2 (SALK_131712C) and DML3 (SALK_056440C) (F). The number of galls per root system was counted 3 weeks post inoculation. Data are presented as means ± SE (n = 20). Asterisks denote statistically significant differences from the wild-type Col-0, Ler or C24 as determined by t tests (P < 0.05).
Defense response genes are oppositely regulated in DNA demethylase mutants in response to infection by cyst and root-knot nematodes
Several reports have indicated that DNA hypomethylation contributes to the regulatory mechanism of plant-induced defense responses (Yu et al., 2013; Le et al., 2014; Lopez Sanchez et al., 2016). Therefore, we examined the expression levels of pathogenesis related (PR) genes in the DNA demethylase mutants showing contrasting susceptibility phenotypes in response to infection by H. schachtii and M. incognita. We first quantified the expression of PR1, PR5, and PLANT DEFENSIN1.2 (PDF1.2) in the root tissues of ros1 (CS66099 and SALK_045303C), dml2 (SALK_131712C) and dml3 (SALK_056440C) under non-infected conditions. As shown in Figures 5A–C, PR1, PR5 and PDF1.2 expression was comparable to that of the wild-type plants Col-0 or C24.
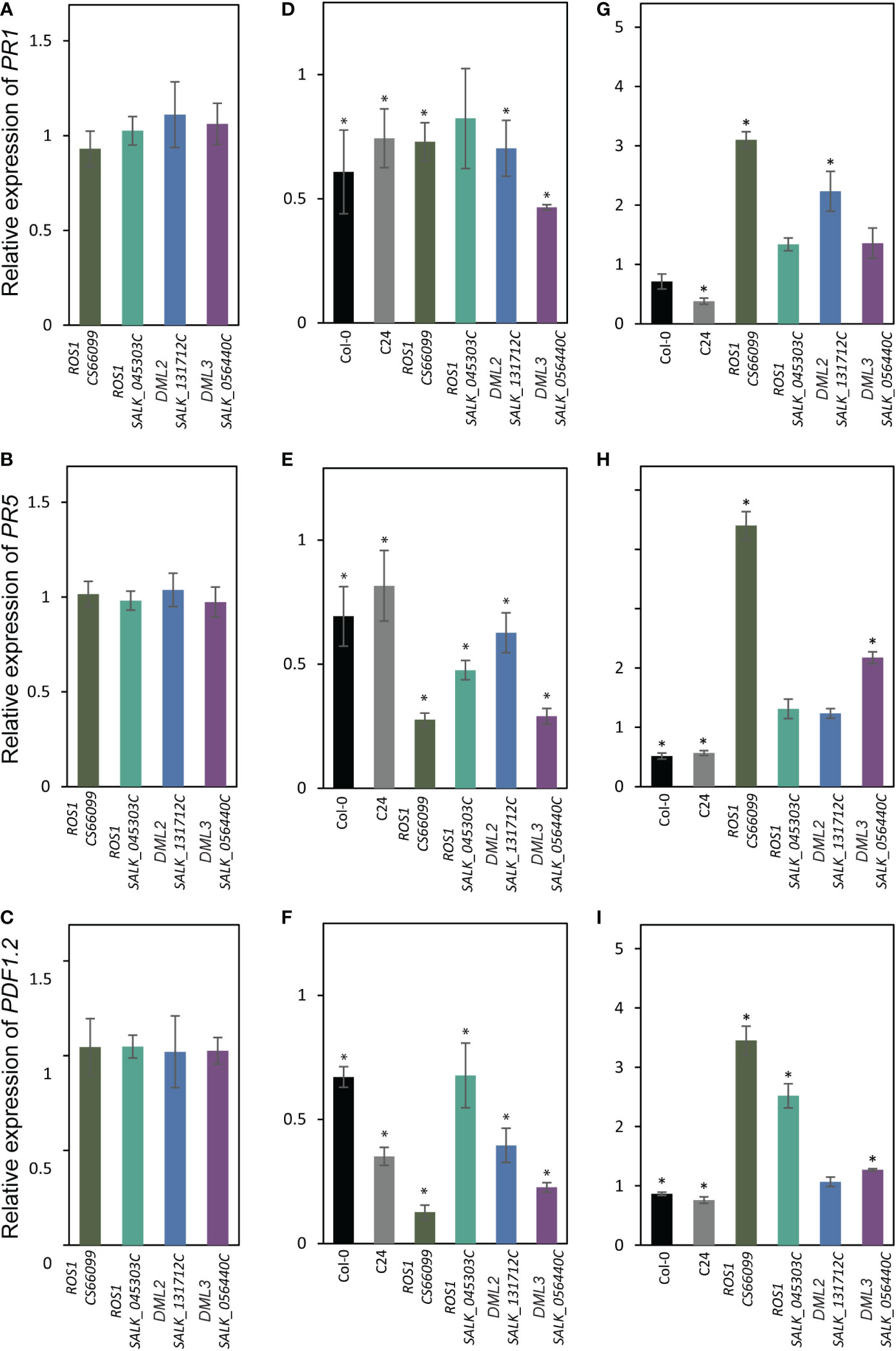
Figure 5 Expression levels of PR genes in DNA demethylase mutants in response to infection by cyst and root-knot nematodes. (A–C): RT-qPCR quantification of the expression of PR1 (A), PR5 ) and PDF1.2 (C) in roots tissues of mutant alleles of ROS1 (CS66099 and SALK_045303C), DML2 (SALK_131712C), and DML3 (SALK_056440C) under non-infected conditions relative to wild-type plants. (D-F): Expression of PR1 (D), PR5 (E) and PDF1.2 (F) in the (H) schachtii-infected roots tissues of wild-type (Col-0 and C24) and mutant alleles of ROS1 (CS66099 and SALK_045303C), DML2 (SALK_131712C), and DML3 (SALK_056440C) relative to non-infected root tissues. (G–I): Expression of PR1 (G), PR5 (H) and PDF1.2 (I) in the M. incognita-infected roots tissues of wild-type (Col-0 and C24) and mutant alleles of ROS1 (CS66099 and SALK_045303C), DML2 (SALK_131712C), and DML3 (SALK_056440C) relative to non-infected root tissues. Relative gene expression levels were obtained from three biological samples and presented as mean ± SE. PP2AA3 and actin8 were used as internal reference genes to normalize gene expression. Asterisks indicate statistically significance differences from control treatments at P < 0.05.
We next quantified the expression of PR1, PR5, and PDF1.2 in the root tissues of these four DNA demethylase mutants and wild-type plants (Col-0 and C24) infected with H. schachtii or M. incognita at 4 dpi. In response to H. schachtii infection, the expression of these genes showed significant downregulation in infected wild-type roots compared with non-infected roots (Figures 5D–F). Similarly, the expression levels of PR1, PR5, and PDF1.2 were significantly downregulated in almost all H. schachtii-infected mutant lines when compared with non-infected mutants (Figures 5D–F). However, the levels of downregulation of PR5 and PDF1.2 were much higher in the infected mutants than the infected wild types (Figures 5E, F).
In response to M. incognita infection, the expression of PR1, PR5, and PDF1.2 was downregulated in the infected wild-type roots versus non-infected roots (Figures 5G–I). In the mutant lines, however, we observed a general trend of upregulation of the PR1, PR5, and PDF1.2 transcripts in M. incognita-infected roots versus non-infected roots (Figures 5G–I). Together, these results suggest that opposite regulation of defense-related genes in the mutant lines in response to infection by H. schachtii and M. incognita may have contributed to the observed opposite susceptibility phenotypes.
Discussion
Our analysis revealed an important role of DNA methylation and active demethylation pathways in mediating Arabidopsis interactions with H. schachtii and M. incognita. Promoter activity and mutant analyses of 12 key genes involved in DNA methylation or active demethylation unveiled important similarity and distinct differences between these two pathosystems in term of expression patterns and plant susceptibility. For example, the promoter activity of MET1 and its homolog MET3 were both detected in the syncytia and galls during the early stage of infection. In contrast, the expression of MET2b was detected only in galls at 4 dpi (Figures 1A, 3A), a finding that is consistent with increased susceptibility of met2b mutant plants (SALK_093835C) to M. incognita but not H. schachtii (Figures 2B, 4B). Strikingly, no such activity of MET3 promoter was detected in the root tissues under non-infected conditions (Bennett et al., 2021), but this promoter was activated specifically in the syncytia and galls only at 3 and 4 dpi, respectively (Figures 1A, 3A). Consistent with this finding, mutant alleles of MET3 were more susceptible to both H. schachtii and M. incognita (Figures 2B, 4B). The finding that MET3 expression is restricted to the developing embryo (Jullien et al., 2012; Bennett et al., 2021) suggests a unique role of the encoding enzyme in reprogramming CG methylation in the syncytia and galls in a way similar to the developing embryos, which undergo extensive DNA methylation changes (Xiao et al., 2006; Gehring et al., 2009; Jullien et al., 2012; Wang et al., 2015; Satyaki and Gehring, 2017).
Notably, none of the examined mutants of the CMT genes were found to alter plant susceptibility to H. schachtii despite the strong activation of CMT1 and CMT3 in the syncytia during both early and late stages of infection. While this finding can be attributed to a possible functional redundancy between CMT1 and CMT3, whose expression was detected in the syncytia (Figure 1B), this explanation doesn’t support the results showing that single mutants of this gene family were altered in their susceptibility to M. incognita (Figures 4B, C). One possible explanation is that global loss of CHG methylation in cmt1 and cmt3 (Stroud et al., 2013) mutants have little or no effect on syncytium formation and function. Alternatively, global decrease in CHG methylation can be compensated with an increase of CG and CHH methylation in the syncytium as recently reported (Rambani et al., 2020b). In support of this hypothesis, the expression of MET1 and MET2b as well as DRM2 and DRM3, which establish CHH methylation de novo, are strongly induced in the syncytia (Figures 1A-B). Although DRMs are established as CHH methyltransferases, they regulate CHG methylation as well (Cao and Jacobsen, 2002b; Stroud et al., 2013).
The intense GUS staining of the DRM2 and DRM3 promoters in the syncytia and galls (Figures 1B, 3B) along with the results showing that mutant alleles of DRM2 and DRM3 were more susceptible to both M. incognita and H. schachtii point to the importance of establishing DNA methylation de novo in syncytial and gall cells as a defense mechanism to control cyst and root-knot nematode infection. Similarly, RdDM-defective mutants were found to be more susceptible to Botrytis cinerea and Plectosphaerella cucumerina (López et al., 2011). More recently, it was shown that tomato DRM5 expression is significantly upregulated in the roots of a resistant tomato cultivar upon infection with a virulent root-knot nematode field population (Leonetti and Molinari, 2020). However, various rice mutants deficient in the RdDM pathway were reported less susceptible to M. graminicola despite the fact that more than 99% of the identified differentially methylated regions in the galls were CHH-hypomethylated (Atighi et al., 2020). Similarly, it has been recently shown that M. javanica reproduction on drm1/drm2 double mutant was greatly reduced (Silva et al., 2022). Together, these findings suggest that the RdDM pathway regulates plant resistance against various phytopathogens (Erdmann and Picard, 2020) including cyst and root-knot nematodes.
It may be important to mention that the differences between the two alleles of MET3 (SALK_099592C and SALK_024049C), CMT1 (SALK_138685C and SALK_030404C), and DRM3 (SALK_136439C and SALK_024820C) in response to infection by H. schachtii or M. incognita are most likely due to difference in gene expression as shown in Supplementary Figure 1B. Similarly, of the two mutant lines of MET2b (SALK_102231 and SALK_093835C) only SALK_093835C exhibited increased susceptibility to M. incognita despite the fact that MET2b is expressed at a similar level in both mutants. The differences between the two mutant alleles could be related to different T-DNA insertion sites (Supplementary Figure 1A), which may impact the function of truncated transcripts.
Remarkably, the promoter activity of ROS1, DME, DML2, and DML3 exhibited opposite expression patterns in the syncytia and galls (Figures 1C, 3C). With the exception of DML2 upregulation in the syncytium at 3 and 7 dpi, the remaining three DNA demethylase genes were either downregulated or not expressed at a detectable level in the syncytia (Figure 1C). In contrast, strong GUS activity was observed for all four demethylase genes in young galls at 4 dpi (Figure 3C). Consistent with the contrasting expression patterns, the mutant alleles of ROS1, DML2, and DML3 showed opposite responses to infection by cyst and root-knot nematodes. Indeed, the DNA demethylase mutants exhibited increased susceptibility to H. schachtii and decreased susceptibility to M. incognita (Figures 2C, 3C). The involvement of active DNA demethylases in disease resistance has been reported in few studies. For example, a loss-of-function mutation in the Arabidopsis ROS1 gene enhanced plant susceptibility to the bacterial pathogen Pseudomonas syringae pv. tomato DC3000 (Pto DC3000) (Yu et al, 2013). In addition, Arabidopsis ros1/dml2/dml3 (rdd) triple mutant displayed enhanced disease susceptibility to the fungal pathogen Fusarium oxysporum (Le et al., 2014). The opposite response of Arabidopsis hypermethylated mutants to various plant pathogens has also been reported. For instance, the hypermethylated mutant ros1 was less resistant to Hpa, but more resistant to the necrotrophic fungi Alternaria brassicicola and Plectosphaerella cucumerina as well as M. javanica (López Sánchez et al., 2016; Silva et al., 2022).
Our gene expression analysis of PR1, PR2, and PDF1.2 pointed to opposite regulation of defense-related genes in the DNA demethylase mutants in response to infection by M. incognita and H. schachtii (Figures 5B, C). This may partially explain the opposite susceptibility phenotypes of these mutants in response to infection by H. schachtii and M. incognita (Figures 2C, 4C), and highlights the complexity of plant defense pathways against plant-parasitic nematodes and their interactions with active demethylation pathway. Our results are consistent with the finding that active DNA demethylases function mainly in regulating the expression of defense- and stress-related genes (Yu et al., 2013; Le et al., 2014; Lopez Sanchez et al., 2016) and point to distinct methylation-dependent mechanisms regulating defense gene expression upon infection by cyst and root-knot nematodes. Basal defense response does not seem to be altered in the demethylase mutants under non-infected conditions because PR1, PR2, and PDF1.2 are similarly expressed in these mutants and wild-type plants (Figures 5A–C). Additionally, it is unlikely that changes in the expression of PR1, PR5, and PDF1.2 are due to changes in methylation status in cis at the promoters of these genes upon nematode infection because nematode-induced potential hypermethylation of these PR genes in four demethylated mutants argues against upregulation of these genes in response to M. incognita infection (Figures 5G–I). Furthermore, PR1, PR2, and PDF1.2 are not subjected to DNA methylation changes upon H. schachtii infection in Col-0 plants (Hewezi et al., 2017). Additionally, it has been shown that the majority of defense-related genes in ros1 mutant are indirectly regulated by DNA methylation (López Sánchez et al., 2016; Halter et al., 2021). Taken together, it is conceivable to suggest that changes in DNA methylation patterns of trans-acting factors directly or indirectly controlling the expression of defense- and stress-related genes in DNA demethylase mutants are the causal factors contributing towards opposite regulation of PR1, PR2, and PDF1.2 upon infection by H. schachtii and M. incognita. Taken together, our results set the stage for future studies to determine the exact mechanism through which DNA methylation and active demethylation of trans-acting factors modulate plant immunity and basal defense response upon infection with parasitic nematodes. This may eventually lead to the development of novel approaches to improve plant resistance to nematode infection.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
TH designed the original research plans. MB performed the majority of the experimental work, analyzed the data, and wrote the first draft. TEH, VL-C, NA, and JR helped MB in data collection and analysis. TH supervised the experimental work, analyzed the data, and wrote the final version. All authors contributed to the article and approved the submitted version.
Funding
University of Tennessee, Institute of Agriculture
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1111623/full#supplementary-material
References
Agius, F., Kapoor, A., Zhu, J.-K. (2006). Role of the arabidopsis DNA glycosylase/lyase ROS1 in active DNA demethylation. Proc. Natl. Acad. Sci. 103, 11796–11801. doi: 10.1073/pnas.0603563103
Atighi, M. R., Verstraeten, B., De Meyer, T., Kyndt, T. (2021). Genome-wide shifts in histone modifications at early stage of rice infection with meloidogyne graminicola. Mol. Plant Pathol. 22, 440–455. doi: 10.1111/nph.16532
Bennett, M., Cleaves, K., Hewezi, T. (2021). Expression patterns of DNA methylation and demethylation genes during plant development and in response to phytohormones. Int. J. Mol. Sci. 22. doi: 10.3390/ijms22189681
Bennett, M., Piya, S., Baum, T. J., Hewezi, T. (2022). miR778 mediates gene expression, histone modification, and DNA methylation during cyst nematode parasitism. Plant Physiol. 189, 2432–2453. doi: 10.1093/plphys/kiac228
Cao, X., Aufsatz, W., Zilberman, D., Mette, M. F., Huang, M. S., Matzke, M., et al. (2003). Role of the DRM and CMT3 methyltransferases in RNA-directed DNA methylation. Curr. Biol. 13, 2212–2217. doi: 10.1016/j.cub.2003.11.052
Cao, X., Jacobsen, S. E. (2002a). Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc. Natl. Acad. Sci. 99 Suppl 4, 16491–16498. doi: 10.1073/pnas.162371599
Cao, X., Jacobsen, S. E. (2002b). Role of the arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol., 12, 1138–1144. doi: 10.1016/s0960-9822(02)00925-9
Choi, Y., Gehring, M., Johnson, L., Hannon, M., Harada, J. J., Goldberg, R. B., et al. (2002). DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in arabidopsis. Cell 110, 33–42. doi: 10.1016/s0092-8674(02)00807-3
Costa-Nunes, P., Kim, J. Y., Hong, E., Pontes, O. (2014). The cytological and molecular role of domains rearranged methyltransferase3 in RNA-dependent DNA methylation of arabidopsis thaliana. BMC Res. Notes 7, 721. doi: 10.1186/1756-0500-7-721
Dowen, R. H., Pelizzola, M., Schmitz, R. J., Lister, R., Dowen, J. M., Nery, J. R., et al. (2012). Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. 109, E2183–E2191. doi: 10.1073/pnas.1209329109
Du, J., Johnson, L. M., Jacobsen, S. E., Patel, D. J. (2015). DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 16, 519–532. doi: 10.1038/nrm4043
Erdmann, R. M., Picard, C. L. (2020). RNA-directed DNA methylation. PloS Genet. 16, e1009034. doi: 10.1371/journal.pgen.1009034
Fransz, P. F., De Jong, J. H. (2002). Chromatin dynamics in plants. Curr. Opin. Plant Biol. 5, 560–567. doi: 10.1016/s1369-5266(02)00298-4
Gehring, M., Bubb, K. L., Henikoff, S. (2009). Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science 324, 1447–1451. doi: 10.1126/science.1171609
Gong, Z., Morales-Ruiz, T., Ariza, R. R., Roldán-Arjona, T., David, L., Zhu, J. K. (2002). ROS1, a repressor of transcriptional gene silencing in arabidopsis, encodes a DNA glycosylase/lyase. Cell 111, 803–814. doi: 10.1016/s0092-8674(02)01133-9
Halter, T., Wang, J., Amesefe, D., Lastrucci, E., Charvin, M., Singla Rastogi, M., et al. (2021). The arabidopsis active demethylase ROS1 cis-regulates defence genes by erasing DNA methylation at promoter-regulatory regions. eLife 10, e62994. doi: 10.7554/eLife.62994
Henderson, I. R., Deleris, A., Wong, W., Zhong, X., Chin, H. G., Horwitz, G. A., et al. (2010). The de novo cytosine methyltransferase DRM2 requires intact UBA domains and a catalytically mutated paralog DRM3 during RNA-directed DNA methylation in arabidopsis thaliana. PloS Genet. 6, e1001182. doi: 10.1371/journal.pgen.1001182
Henderson, I. R., Jacobsen, S. E. (2007). Epigenetic inheritance in plants. Nature 447, 418–424. doi: 10.1038/nature05917
Hewezi, T. (2020). Epigenetic mechanisms in nematode–plant interactions. Annu. Rev. Phytopathol. 58, 119–138. doi: 10.1146/annurev-phyto-010820-012805
Hewezi, T., Lane, T., Piya, S., Rambani, A., Rice, J. H., Staton, M. (2017). Cyst nematode parasitism induces dynamic changes in the root epigenome. Plant Physiol. 174, 405–420. doi: 10.1104/pp.16.01948
Hewezi, T., Pantalone, V., Bennett, M., Neal Stewart, C., Burch-Smith, T. M. (2018). Phytopathogen-induced changes to plant methylomes. Plant Cell Rep. 37, 17–23. doi: 10.1007/s00299-017-2188-y
Ithal, N., Recknor, J., Nettleton, D., Maier, T., Baum, T. J., Mitchum, M. G. (2007). Developmental transcript profiling of cyst nematode feeding cells in soybean roots. Mol. Plant-Microbe Interact. 20, 510–525. doi: 10.1094/MPMI-20-5-0510
Jefferson, R. A., Kavanagh, T. A., Bevan, M. W. (1987). GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 6, 3901–3907. doi: 10.1002/j.1460-2075.1987.tb02730.x
Jullien, P. E., Susaki, D., Yelagandula, R., Higashiyama, T., Berger, F. (2012). DNA methylation dynamics during sexual reproduction in arabidopsis thaliana. Curr. Biol. 22, 1825–1830. doi: 10.1016/j.cub.2012.07.061
Kandoth, P. K., Ithal, N., Recknor, J., Maier, T., Nettleton, D., Baum, T. J., et al. (2011). The soybean Rhg1 locus for resistance to the soybean cyst nematode heterodera glycines regulates the expression of a large number of stress- and defense-related genes in degenerating feeding cells. Plant Physiol. 155, 1960–1975. doi: 10.1104/pp.110.167536
Kankel, M. W., Ramsey, D. E., Stokes, T. L., Flowers, S. K., Haag, J. R., Jeddeloh, J. A., et al. (2003). Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 163, 1109–1122. doi: 10.1093/genetics/163.3.1109
Klink, V. P., Hosseini, P., Matsye, P., Alkharouf, N. W., Matthews, B. F. (2009). A gene expression analysis of syncytia laser microdissected from the roots of the glycine max (soybean) genotype PI 548402 (Peking) undergoing a resistant reaction after infection by heterodera glycines (soybean cyst nematode). Plant Mol. Biol. 71, 525–567. doi: 10.1007/s11103-009-9539-1
Klink, V. P., Hosseini, P., Matsye, P., Alkharouf, N. W., Matthews, B. F. (2010). Syncytium gene expression in Glycine max [PI 88788] roots undergoing a resistant reaction to the parasitic nematode Heterodera glycines. Plant Physiol. and Biochem. 48, 176–193. doi: 10.1016/j.plaphy.2009.12.003
Law, J. A., Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220. doi: 10.1038/nrg2719
Lei, M., Zhang, H., Julian, R., Tang, K., Xie, S., Zhu, J. K. (2015). Regulatory link between DNA methylation and active demethylation in arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 112, 3553–3557. doi: 10.1073/pnas.1502279112
Leonetti, P., Molinari, S. (2020). Epigenetic and metabolic changes in root-knot nematode-plant interactions. Int. J. Mol. Sci. 21, 7759. doi: 10.3390/ijms21207759
Le, T. N., Schumann, U., Smith, N. A., Tiwari, S., Au, P. C., Zhu, Q. H., et al. (2014). DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in arabidopsis. Genome Biol. 15, 458. doi: 10.1186/s13059-014-0458-3
Li, L., He, Y., Zhang, X., Zhang, H., Sun, Z., Li, J., et al. (2020). Alterations of rice (Oryza sativa l.) DNA methylation patterns associated with gene expression in response to rice black streaked dwarf virus. Int. J. Mol. Sci. 21. doi: 10.3390/ijms21165753
Lindroth, A. M., Cao, X., Jackson, J. P., Zilberman, D., Mccallum, C. M., Henikoff, S., et al. (2001). Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 292, 2077–2080. doi: 10.1126/science.1059745
Lister, R., O’malley, R. C., Tonti-Filippini, J., Gregory, B. D., Berry, C. C., Millar, A. H., et al. (2008). Highly integrated single-base resolution maps of the epigenome in arabidopsis. Cell 133, 523–536. doi: 10.1016/j.cell.2008.03.029
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. . Methods 25, 402–408. doi: 10.1006/meth.2001.1262
López, A., Ramírez, V., García-Andrade, J., Flors, V., Vera, P. (2011). The RNA silencing enzyme RNA polymerase v is required for plant immunity. PloS Genet. 7, e1002434. doi: 10.1371/journal.pgen.1002434
López Sánchez, A., Stassen, J. H., Furci, L., Smith, L. M., Ton, J. (2016). The role of DNA (de)methylation in immune responsiveness of Arabidopsis. Plant J. 88, 361–374. doi: 10.1111/tpj.13252
Mathieu, O., Reinders, J., Caikovski, M., Smathajitt, C., Paszkowski, J. (2007). Transgenerational stability of the arabidopsis epigenome is coordinated by CG methylation. Cell 130, 851–862. doi: 10.1016/j.cell.2007.07.007
Matzke, M. A., Mosher, R. A. (2014). RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 15, 394–408. doi: 10.1038/nrg3683
Morales-Ruiz, T., Ortega-Galisteo, A. P., Ponferrada-Marín, M. I., Martínez-Macías, M. I., Ariza, R. R., Roldán-Arjona, T. (2006). DEMETER and REPRESSOR OF SILENCING 1 encode 5-methylcytosine DNA glycosylases. Proc. Natl. Acad. Sci. 103, 6853–6858. doi: 10.1073/pnas.0601109103
Ortega-Galisteo, A. P., Morales-Ruiz, T., Ariza, R. R., Roldán-Arjona, T. (2008). Arabidopsis DEMETER-LIKE proteins DML2 and DML3 are required for appropriate distribution of DNA methylation marks. Plant Mol. Biol. 67, 671–681. doi: 10.1007/s11103-008-9346-0
Penterman, J., Uzawa, R., Fischer, R. L. (2007a). Genetic interactions between DNA demethylation and methylation in arabidopsis. Plant Physiol. 145, 1549–1557. doi: 10.1104/pp.107.107730
Penterman, J., Zilberman, D., Huh, J. H., Ballinger, T., Henikoff, S., Fischer, R. L. (2007b). DNA demethylation in the arabidopsis genome. Proc. Natl. Acad. Sci. 104, 6752–6757. doi: 10.1073/pnas.0701861104
Piya, S., Bennett, M., Rambani, A., Hewezi, T. (2017). Transcriptional activity of transposable elements may contribute to gene expression changes in the syncytium formed by cyst nematode in arabidopsis roots. Plant Signaling Behav. 12, e1362521. doi: 10.1080/15592324.2017.1362521
Piya, S., Binder, B. M., Hewezi, T. (2019). Canonical and noncanonical ethylene signaling pathways that regulate arabidopsis susceptibility to the cyst nematode heterodera schachtii. New Phytol. 221, 946–959. doi: 10.1111/nph.15400
Piya, S., Liu, J., Burch-Smith, T., Baum, T. J., Hewezi, T. (2020). A role for arabidopsis growth-regulating factors 1 and 3 in growth–stress antagonism. J. Exp. Bot. 71, 1402–1417. doi: 10.1093/jxb/erz502
Quadrana, L., Bortolini Silveira, A., Mayhew, G. F., Leblanc, C., Martienssen, R. A., Jeddeloh, J. A., et al. (2016). The arabidopsis thaliana mobilome and its impact at the species level. eLife 5, e15716. doi: 10.7554/eLife.15716
Rambani, A., Hu, Y., Piya, S., Long, M., Rice, J. H., Pantalone, V., et al. (2020a). Identification of differentially methylated miRNA genes during compatible and incompatible interactions between soybean and soybean cyst nematode. Mol. Plant-Microbe Interact. 33, 1340–1352. doi: 10.1094/MPMI-07-20-0196-R
Rambani, A., Pantalone, V., Yang, S., Rice, J. H., Song, Q., Mazarei, M., et al. (2020b). Identification of introduced and stably inherited DNA methylation variants in soybean associated with soybean cyst nematode parasitism. New Phytol. 227, 168–184. doi: 10.1111/nph.16511
Rambani, A., Rice, J. H., Liu, J., Lane, T., Ranjan, P., Mazarei, M., et al. (2015). ). The methylome of soybean roots during the compatible interaction with the soybean cyst nematode. Plant Physiol. 168, 1364–1377. doi: 10.1104/pp.15.00826
Ruiz-Ferrer, V., Cabrera, J., Martinez-Argudo, I., Artaza, H., Fenoll, C., Escobar, C. (2018). Silenced retrotransposons are major rasiRNAs targets in arabidopsis galls induced by meloidogyne javanica. Mol. Plant Pathol. 19, 2431–2445. doi: 10.1111/mpp.12720
Satyaki, P. R., Gehring, M. (2017). DNA methylation and imprinting in plants: machinery and mechanisms. Crit. Rev. Biochem. Mol. Biol. 52, 163–175. doi: 10.1080/10409238.2017.1279119
Saze, H., Scheid, O. M., Paszkowski, J. (2003). Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat. Genet. 34, 65–69. doi: 10.1038/ng1138
Sijmons, P. C., Grundler, F. M. W., Von Mende, N., Burrows, P. R., Wyss, U. (1991). Arabidopsis thaliana as a new model host for plant-parasitic nematodes. Plant J. 1, 245–254. doi: 10.1111/j.1365-313X.1991.00245.x
Silva, A. C., Ruiz-Ferrer, V., Müller, S. Y., Pellegrin, C., Abril-Urías, P., Martínez-Gómez, Á., et al. (2022). The DNA methylation landscape of the root-knot nematode-induced pseudo-organ, the gall, in arabidopsis, is dynamic, contrasting over time, and critically important for successful parasitism. New Phytol. 236, 1888–1907. doi: 10.1111/nph.18395
Stroud, H., Do, T., Du, J., Zhong, X., Feng, S., Johnson, L., et al. (2014). Non-CG methylation patterns shape the epigenetic landscape in arabidopsis. Nat. Struct. Mol. Biol. 21, 64–72. doi: 10.1038/nsmb.2735
Stroud, H., Greenberg, M. V., Feng, S., Bernatavichute, Y. V., Jacobsen, S. E. (2013). Comprehensive analysis of silencing mutants reveals complex regulation of the arabidopsis methylome. Cell 152, 352–364. doi: 10.1016/j.cell.2012.10.054
Tirnaz, S., Merce, C., Bayer, P. E., Severn-Ellis, A. A., Edwards, D., Batley, J. (2020). Effect of leptosphaeria maculans infection on promoter DNA methylation of defence genes in brassica napus. Agronomy 10, 1072. doi: 10.3390/agronomy10081072
Verwoerd, T. C., Dekker, B. M., Hoekema, A. (1989). A small-scale procedure for the rapid isolation of plant RNAs. Nucleic Acids Res. 17, 2362. doi: 10.1093/nar/17.6.2362
Wang, C., Wang, C., Xu, W., Zou, J., Qiu, Y., Kong, J., et al. (2018). Epigenetic changes in the regulation of nicotiana tabacum response to cucumber mosaic virus infection and symptom recovery through single-base resolution methylomes. Viruses 10. doi: 10.3390/v10080402
Wang, P., Xia, H., Zhang, Y., Zhao, S., Zhao, C., Hou, L., et al. (2015). Genome-wide high-resolution mapping of DNA methylation identifies epigenetic variation across embryo and endosperm in maize (Zea may). BMC Genomics 16, 21. doi: 10.1186/s12864-014-1204-7
Xiao, W., Custard, K. D., Brown, R. C., Lemmon, B. E., Harada, J. J., Goldberg, R. B., et al. (2006). DNA methylation is critical for arabidopsis embryogenesis and seed viability. Plant Cell 18, 805–814. doi: 10.1105/tpc.105.038836
Yu, A., Lepère, G., Jay, F., Wang, J., Bapaume, L., Wang, Y., et al. (2013). Dynamics and biological relevance of DNA demethylation in arabidopsis antibacterial defense. Proc. Natl. Acad. Sci. U.S.A. 110, 2389–2394. doi: 10.1073/pnas.1211757110
Zhang, M., Kimatu, J. N., Xu, K., Liu, B. (2010). DNA cytosine methylation in plant development. J. Genet. Genomics 37, 1–12. doi: 10.1016/s1673-8527(09)60020-5
Zhang, H., Zhu, J. K. (2012). Active DNA demethylation in plants and animals. Cold Spring Harb. Symp Quant Biol. 77, 161–173. doi: 10.1101/sqb.2012.77.014936
Zhu, J., Kapoor, A., Sridhar, V. V., Agius, F., Zhu, J. K. (2007). The DNA glycosylase/lyase ROS1 functions in pruning DNA methylation patterns in arabidopsis. Curr. Biol. 17, 54–59. doi: 10.1016/j.cub.2006.10.059
Keywords: Arabidopsis, Heterodera schachtii, Meloidogyne incognita, DNA methylation, active DNA demethylation, promoter activity, syncytium, gall
Citation: Bennett M, Hawk TE, Lopes-Caitar VS, Adams N, Rice JH and Hewezi T (2023) Establishment and maintenance of DNA methylation in nematode feeding sites. Front. Plant Sci. 13:1111623. doi: 10.3389/fpls.2022.1111623
Received: 29 November 2022; Accepted: 07 December 2022;
Published: 10 January 2023.
Edited by:
Chen Jingsheng, Chongqing Three Gorges University, ChinaReviewed by:
Tushar Kanti Dutta, Indian Agricultural Research Institute (ICAR), IndiaZhao Dan, Jilin Agriculture University, China
Copyright © 2023 Bennett, Hawk, Lopes-Caitar, Adams, Rice and Hewezi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tarek Hewezi, dGhld2V6aUB1dGsuZWR1