 Simón Miranda1,2,3*Jorge Lagrèze1,2Anne-Sophie Knoll1Andrea Angeli1Richard V. Espley3Andrew P. Dare3Mickael Malnoy1Stefan Martens1*
Simón Miranda1,2,3*Jorge Lagrèze1,2Anne-Sophie Knoll1Andrea Angeli1Richard V. Espley3Andrew P. Dare3Mickael Malnoy1Stefan Martens1*- 1Research and Innovation Center, Fondazione Edmund Mach, San Michele all’Adige, Trento, Italy
- 2Center Agriculture Food and Environment (C3A), University of Trento, Trento, Italy
- 3The New Zealand Institute for Plant and Food Research Limited, Auckland, New Zealand
Sieboldin is a specialised secondary metabolite of the group of dihydrochalcones (DHC), found in high concentrations only in some wild Malus species, closely related to the domesticated apple (Malus × domestica L.). To date, the first committed step towards the biosynthesis of sieboldin remains unknown. In this study, we combined transcriptomic analysis and a de novo transcriptome assembly to identify two putative 3-hydroxylases in two wild Malus species (Malus toringo (K. Koch) Carriere syn. sieboldii Rehder, Malus micromalus Makino) whose DHC profile is dominated by sieboldin. We assessed the in vivo activity of putative candidates to produce 3-hydroxyphloretin and sieboldin by de novo production in Saccharomyces cerevisiae. We found that CYP98A proteins of wild Malus accessions (CYP98A195, M. toringo and CYP98A196, M. micromalus) were able to produce 3-hydroxyphloretin, ultimately leading to sieboldin accumulation by co-expression with PGT2. CYP98A197-198 genes of M. × domestica, however, were unable to hydroxylate phloretin in vivo. CYP98A195-196 proteins exerting 3-hydroxylase activity co-localised with an endoplasmic reticulum marker. CYP98A protein model from wild accessions showed mutations in key residues close to the ligand pocket predicted using phloretin for protein docking modelling. These mutations are located within known substrate recognition sites of cytochrome P450s, which could explain the acceptance of phloretin in CYP98A protein of wild accessions. Screening a Malus germplasm collection by HRM marker analysis for CYP98A genes identified three clusters that correspond to the alleles of domesticated and wild species. Moreover, CYP98A isoforms identified in M. toringo and M. micromalus correlate with the accumulation of sieboldin in other wild and hybrid Malus genotypes. Taken together, we provide the first evidence of an enzyme producing sieboldin in vivo that could be involved in the key hydroxylation step towards the synthesis of sieboldin in Malus species.
Introduction
Dihydrochalcones (DHCs) are specialised metabolites with a limited natural distribution, being isolated from some medicinal plants but abundantly accumulated in Malus species (Rivière, 2016). DHCs are categorised as minor flavonoid together with flavanones, flavanonols, chalcones and retrochalcones because of their restricted natural occurrence (Tomás-Barberán and Clifford, 2000). In 1835, the first DHC, phloridzin (phloretin 2′-O-glucoside), was described as an antipyretic substance isolated from bark and root of cultivated apple tree (Gosch et al., 2010b). For a long time, phloridzin was believed to be exclusive to Malus × domestica L, where it can account for up to 10 – 14% of leaf dry weight (Gosch et al., 2010b). Thus, phloridzin has been used as an effective marker to detect food-derived adulteration with apple (Spinelli et al., 2016; Hrubá et al., 2021). However, trilobatin and sieboldin dominate the polyphenolic profile of some wild Malus relatives (Gutierrez et al., 2018b). DHCs have demonstrated a wide range of beneficial effects against cancer, cardiovascular and free radical-involving diseases (Stangl et al., 2005; Dugé de Bernonville et al., 2010; Szliszka et al., 2010; Viet et al., 2013). Some DHCs have also been reported to act as flavour sweeteners and bitterness blockers, such as trilobatin and neohesperidin DHC (Tomás-Barberán et al., 1995; Wang et al., 2020). According to the currently proposed DHC pathway, 3-hydroxyphloretin, the key precursor of sieboldin in Malus species, would also be an intermediate compound towards the biosynthesis of the flavour enhancer neohesperidin DHC (Ibdah et al., 2018). Phloridzin may act as an anti-diabetic compound by blocking sodium-linked glucose transport and renal reabsorption of glucose in kidneys (Ehrenkranz et al., 2005). Despite the protective effects reported in mammal models, little is known about how these metabolites are biosynthesised and what is their function in planta.
Previous studies have suggested a role of DHCs in plant defence during Erwinia amylovora and Venturia inaequalis infection, pathogens causing fire blight and scab disease in apples, respectively. Although the relevance of phloridzin and its aglycone precursor phloretin in the acquisition of pathogen resistance is still controversial, it has been proposed that DHCs accumulate to cope with the oxidative stress during pathogen response (reviewed in Gosch et al., 2010). Moreover, phloridzin and its aglycone precursor phloretin have shown antibacterial and antifungal properties (Lattanzio et al., 2001; Oleszek et al., 2019; Liu et al., 2021). In addition to the most studied DHCs, sieboldin exhibits antibacterial and antifungal activities, suggesting a complementary role to phloretin and phloridzin against E. amylovora and V. inaequalis infection (Gaucher et al., 2013). Furthermore, overexpressing a chalcone 3-hydroxylase from Cosmos sulphureus Cav. in apple led to increased levels of 3-hydroxyphloridzin, correlating with reduced susceptibility to apple scab and fire blight (Hutabarat et al., 2016). Along with the roles of DHCs in biotic stresses, it has been reported that sieboldin could be involved in the tolerance to oxidative stress in Malus leaves (Dugé de Bernonville et al., 2010).
To get a better understanding of the accumulation of DHCs in plants, a number of studies have attempted to elucidate the biosynthetic pathway, providing a valuable resource to assess their physiological roles. The DHC pathway (Supplementary Figure 1) diverts from the main phenylpropanoid pathway from p-coumaroyl-CoA by the action of a postulated double bond reductase. Then, chalcone synthase (CHS) catalyses the condensation of p-dihydrocoumaroyl-CoA to phloretin (Gosch et al., 2010b). Phloretin can be directly glycosylated at position 2′- or 4′ by Ph-2′ or Ph-4′-O-UDP-glycosyltransferases (Supplementary Figure 1) to produce phloridzin or trilobatin, respectively. Several studies have identified 2′-O-UDP-glycosyltransferases involved in the synthesis of phloridzin (PGT1, Jugdé et al., 2008; Gosch et al., 2010a; Gosch et al., 2012; Yahyaa et al., 2016; Zhou et al., 2017). However, the enzyme responsible for the 4′-O-glycosylation of trilobatin was characterised only recently (PGT2, Wang et al., 2020). Sieboldin has been postulated to derive from hydroxylation of phloretin in position 3 of B-ring before being glycosylated (Ibdah et al., 2018; Supplementary Figure 1). Although the glycosylation of 3-hydroxyphloretin can be carried out at least by PGT2 to produce sieboldin in vitro (Wang et al., 2020), the first committed step to 3-hydroxyphloretin has not been yet identified. To date, the only attempt to identify genes involved in this step resulted in the characterisation of two flavonoid 3′-hydroxylases from M. × domestica (F3′HI and F3′HII), which did not accept phloretin and therefore unlikely to be involved in its 3-hydroxylation (Weissensteiner et al., 2021). In this study, we aimed to identify candidate genes that may account for the 3-hydroxylation of phloretin, that could be involved in DHC formation of wild Malus species. Combining transcriptomic, metabolic and functional analyses, our work reports for the first time a 3-hydroxylase of Malus species that convert phloretin to 3-hydroxyphloretin in vivo, the key missing step towards the production of the specialised metabolite sieboldin naturally occurring in some Malus species.
Materials and methods
Plant material
Plants of the Malus germplasm collection of Fondazione Edmund Mach in Trento, Italy (GPS coordinates 46.181848, 11.119849), consisting in three individuals per accession (Supplementary Table 1) planted in 2011 on M9 T337 rootstock and maintained at the germplasm collection, with winter pruning, spring low-titre nitrogen-based fertilisation and phytosanitary treatments during the growing season following the integrated production regulations of the Autonomous Province of Trento were used in this study. RNA-Seq, de novo transcriptome assembly and functional analysis was carried out on three individuals of cultivated apple (M. × domestica cv. ‘Golden Delicious’) and two wild Malus species (M. toringo syn. sieboldii, PI 613858; M. micromalus, PI 594092) Samples were collected from young and old leaves, and five time points of bud development (dormant, half inch, tight cluster, pink, bloom). Fruits were collected at different time points representing the main physiological changes described during fruit development (Janssen et al., 2008): fruit set, and fruit at 35, 60, 87 and 132 days after full bloom (DAFB). At ripening, fruit were split in two enriched fractions of skin and pulp. Sampling was conducted in the three plants representing each accession in the germplasm collection, and each sample comprised at least 10 fruit per time point. All samples were immediately frozen in liquid nitrogen and stored at -80°C until further processing for molecular and metabolic analysis.
RNA extraction, library construction and RNA-Seq analysis
Total RNA was isolated from 100 mg of fresh frozen sample by CTAB extraction and LiCl precipitation, followed by cleaning using Spectrum™ Plant Total RNA kit (Sigma-Aldrich). RNA quality and quantity were assessed before and after DNase treatment by gel electrophoresis and spectrophotometric measurements of absorbance ratios at 260 and 280 (A260/280) or 230 nm (A260/230). RNA samples with A260/280 and A260/230 ratios between 1.8 - 2.0 were considered acceptable and used for downstream applications. We treated 5 µg of total RNA with TURBO™ DNase (Invitrogen). RNA integrity was determined by Agilent 2100 Bioanalyzer, and RIN values > 8.0 were further used. Three cDNA libraries per accession were prepared from total RNA of young leaves, using random primers and the TrueSeq® stranded Illumina kit and then sequenced by HiSeq 2500 Illumina sequencing system to obtain single-stranded 100 bp reads. Quality filtering and adapter trimming was performed by fastp, with a minimum length set at 50 bp. Then, filtered reads were aligned to M. × domestica genome GDDH13 (Daccord et al., 2017) and counted using featureCounts (Liao et al., 2014). Differentially expressed genes (DEGs) were identified by DESeq2 (Love et al., 2014) in R-project environment, and p-values were adjusted using the procedure of Benjamini-Hochberg. Genes with |Log2(Fold change)| > 2 and p-adj < 0.05 were considered as DEGs. Enrichment analysis with Kyoto Encyclopedia of Genes and Genomes (KEGG) was performed by KOBAS v3.0 (http://kobas.cbi.pku.edu.cn/kobas3/).
De novo transcriptome assembly and functional annotation
De novo transcriptome assemblies of the two wild Malus spp. (M. toringo, M. micromalus) were assembled using Trinity software v 2.9.1 using three biological replicates per accession, with a default k-mer size of 25 and minimum contig length of 200. To remove redundant and alternatively spliced transcripts, identical and near-identical contigs were clustered by CD-HIT-EST tool v4.6, with a similarity threshold of 95%. The quality of the assemblies was evaluated by mapping with Bowtie 2 v2.3.2 to compute contig ExN50 statistics. Transcriptome completeness was assessed by BUSCO v5.2.2. Gene open reading frames (ORFs) were predicted by TransDecoder v5.5.0 using the assembled unique transcripts as input. Functional annotation of the assembled transcripts was conducted using Diamond software v2.0.8.0 by protein BLAST against the non-redundant (nr) database of NCBI with an E-value cut-off of 10-5. Orthologs to M. × domestica were predicted by a reciprocal-best hit approach (RBH), with 70% minimum percentage of identity and 50% minimum percentage query coverage.
Gene expression analysis
We used 2 µg of DNase-treated RNA to synthesise single-stranded cDNA using SuperScript™ III Reverse Transcriptase (Invitrogen) and oligo(dT)20 following manufacturer’s instructions. Gene-specific primer sets (Supplementary Table 2) were designed by Sigma OligoArchitecht. Quantitative RT-PCR (RT-qPCR) reactions were run in a C1000 Touch thermal cycler coupled to CFX96 Detection System (Bio-Rad) using 2X SsoFast™ EvaGreen® Supermix (Bio-Rad). Reactions consisted of 10 ng cDNA, 250 nM of forward and reverse primers in a total volume of 12.5 µL, and were run with a standard thermal profile: 98°C, 5 s; 40 cycles of 98°C, 5 s, 60°C, 10 s, 72°C, 10 s; with a final melting step between 65°C and 95°C. Primer efficiency was determined by LinRegPCR v. 7.5 from amplification plots. The quality of PCR reactions was determined by analysis of the dissociation and amplification curves. The constitutively expressed genes ACT2 (actin 2), EF1α (elongation factor 1 alpha) and UBI (ubiquitin 10) were tested as candidate normalizers. Transcript levels of target genes were conducted in three biological replicates and normalised to the expression of EF1α, which was the most stably expressed gene under the tested conditions according to RefFinder (https://www.heartcure.com.au/reffinder/).
Targeted metabolomic profiling
Young leaves of each accession in the Malus germplasm collection and samples from leaves, buds, flowers, and fruits of M. toringo, M. micromalus and M. × domestica cv. ‘Golden Delicious’ were ground and 100 mg fresh weight (FW) was extracted in 5 mL 80% v/v methanol, sonicated for 20 min at 60 Hz in a water bath at 25°C and kept in dark for 48 h, and then filtered through a 0.22 µm PTFE filter and stored at 4°C. Phenolic compounds from yeast cultures were extracted by ethyl acetate. Briefly, 1 mL of culture of three independent clones was extracted twice with 1 volume ethyl acetate followed by centrifugation at 16,000 g for 2 min. The supernatants were collected in 2 mL microcentrifuge tubes and evaporated in an Eppendorf Concentrator Plus ™, by incubation at 25°C for 1 h under negative pressure. Pellets were resuspended in 200 µL 80% v/v methanol and stored at 4°C until further analysis. Targeted profiling of phenolic compounds was conducted using a Waters Acquity UPLC system coupled to a Waters Xevo® TQ-MS mass spectrometer equipped with an electrospray source using multiple reaction monitoring (MRM), as previously described (Vrhovsek et al., 2012). Summary with MRM parameters used for each metabolite is reported in Supplementary Table 3. Data processing was carried out with default settings in Mass Lynx™ and TargetLynx™ softwares (Waters Co., USA). Metabolite concentration automatically determined by TargetLynx™ software using calibration curves of real metabolite standards was normalised according to the fresh weight of plant material (mg/mg FW), or culture volume used for extraction corrected by the dilution factor after resuspension in methanol (mg/L).
Metabolic engineering of Saccharomyces cerevisiae
We used a S. cerevisiae strain derived from S288C (MATα hoΔ0 his3Δ0 leu2Δ0 ura3Δ0), which produces phloretin by simultaneous expression of HaCHS, ScTSC13, At4CL2, AtPAL2, AmC4H, ScCPR1 in a vector conferring prototrophy to uracil (PAR1; Eichenberger et al., 2017). To attempt in vivo conversion of phloretin into 3-hydroxyphloretin in S. cerevisiae, coding sequences of putative 3-hydroxylases were cloned into MCS-2 of pAT425 yeast expression vector (Ishii et al., 2014) by adding SalI and NotI restriction recognition sites through PCR using Phusion Hot Start II DNA Polymerase (ThermoFisher), followed by digestion with FastDigest™ SalI and NotI (ThermoFisher) and ligation using T4 DNA ligase (ThermoFisher), according to manufacturer’s instructions. To achieve conversion of phloretin into sieboldin, the coding sequence of MtorPGT2-2 (GenBank accession number MN381000) was cloned by PCR amplification with primers incorporating FseI and AvrII restriction sites, and digested with FseI and AvrII (New England Biolabs) to be ligated into the MCS-1 of pAT425 vector harbouring putative 3-hydroxylases. Ligation reactions were transformed into chemically competent E. coli OneShot™ Mach1™ cells (ThermoFisher) and plated in LB agar plates (5 g/L yeast extract, 10 g/L tryptone, 5 g/L NaCl, 15 g/L agar) supplemented with carbenicillin 100 µg/mL. Confirmation of correct cloning was assessed by Sanger sequencing. Transformation of PAR1 strain was conducted by a standard lithium acetate method (Gietz and Schiestl, 2007), plated in minimal SC dropout medium without uracil and leucine (SC-U-L) and grown at 30°C for 3 days. Positive clones were inoculated into 50 mL of SC-U-L and incubated at 30°C, 250 rpm for 72 – 96 h. Final OD600 of a 1:10 dilution was measured and 1 mL of three independent clones per construct was sampled for metabolite profiling.
Subcellular localization
Tobacco (Nicotiana tabacum L. cv. ‘Samsun-NN’) was stably transformed with fluorescent markers of plasma membrane (pm-rk, CD3-1007, ABRC) and endoplasmic reticulum (ER-rk, CD3-959, ABRC) fused to mCherry, as previously described (Japelaghi et al., 2018). CYP98A196 was amplified by PCR using specific primers (Supplementary Table 2), cloned into pCR8™/GW/TOPO™ (Invitrogen) and recombined into pEarleyGate 101 (Earley et al., 2006) by Gateway™ LR™ clonase enzyme mix (Invitrogen) according to manufacturer instructions to produce a C-terminal protein fusion to EYFP. Tobacco leaves were agroinfiltrated following a standard protocol for transient expression (Sparkes et al., 2006) with A. tumefaciens C58C1 (pMP90) harbouring the empty vector pEarleyGate101 or the construct CYP98A196-EYFP. Tobacco leaf protoplasts were prepared following the protocol of Hsu et al., 2021. Confocal imaging was performed 72 hours after agroinfiltration and 16 h after protoplast digestion, using a Leica TCS SP8 confocal microscope and Leica Application Suite X software. EYFP and mCherry signals were visualised with excitation at 488 and 554 nm, respectively. Emission was collected between 510 to 540 nm and 590 to 650 nm for EYFP and mCherry. Autofluorescence of chlorophyll was detected between 660 to 730 nm. Z-stacking was performed in 1 to 5 µm steps, in sections ranging 10 to 50 µm. Image processing consisted of merging acquisition channels and quantifying colocalization.
High resolution melting analysis
DNA was extracted from Malus accessions in the germplasm collection of Fondazione Edmund Mach, as previously described (Allen et al., 2006). HRM analysis was performed in a C1000 Touch thermal cycler coupled to CFX96 Detection System (Bio-Rad) using 2X SsoFast™ EvaGreen® Supermix (Bio-Rad). Reactions consisted of 10 ng genomic DNA, 500 nM of specific primers (Supplementary Table 2) in a total volume of 20 µL, and were run with the following thermal profile: 98°C, 2 min; 40 cycles of 98°C, 5 s, 60°C, 10 s; with a melting step between 60°C to 95°C, with acquisitions in 0.1°C inc./sec., 5 sec./step. Normalisation and temperature shift determination of melting curves were determined with default settings in the Precision Melt Analysis™ software (Bio-Rad). Clusters were automatically calculated using a default Tm difference threshold of 0.15 degrees and pre-melt and post-melt regions were optimised to 70.9 - 71.4°C and 78.3 - 78.8°C, respectively.
Protein modelling and phylogenetic analysis
Protein models were obtained using a plant P450 database (PCPD, http://p450.biodesign.ac.cn/). Protein docking was performed using the predicted protein structures obtained in PCPD and phloretin as a ligand in the web-based tool PCPLD.
Publicly available protein sequences of F3′H, F3′5′H and F6H, as well as CYP98A family members were used to build phylogenetic trees with candidate proteins from M. toringo and M. micromalus using MEGA11. Phylogenetic tree was inferred by the maximum likelihood method with 1000 bootstrap replicates and JTT matrix-based model.
Statistical analysis and data visualisation
Statistical differences among tissues and genotypes for metabolite and expression analysis were determined by Statistica software using two-way analysis of variance (ANOVA), with Tukey post-hoc HSD test, using p value < 0.05 as statistically significant. Results were displayed in GraphPad Prism v6.0.
Results
Targeted phenolic metabolomic profiling of domesticated apple (M. × domestica), M. toringo and M. micromalus wild accessions
To get a detailed profiling of phenylpropanoids throughout the development of wild and domesticated Malus species, we sampled plants of M. toringo, M. micromalus and M. × domestica cv. ‘Golden Delicious’, which are represented by three individuals in the germplasm collection of Fondazione Edmund Mach (Italy). Plants were sampled at different bud stages (dormant, half inch, tight cluster, pink and bloom), and across fruit development (fruit set and 35, 60, 87 and 132 DAFB). Young leaves (YL) were collected in the same time points, along with old leaves (OL) at 132 DAFB of fruit development (Supplementary Figure 2). We examined compounds of the DHC pathway based on a targeted metabolomic profiling of phenolic compounds (Vrhovsek et al., 2012). LC-MS/MS analysis shows that DHC profiles in all tissues of wild accessions are dominated by sieboldin and trilobatin (Figures 1D-I), whilst phloridzin is present only in the domesticated apple (Figures 1A-C). Sieboldin is the predominant DHC found in leaves and bud development of wild species, ranging from 3 to 4 mg/g FW in leaves (Figure 1H) and 1 to 4 mg/g FW in buds (Figure 1G). Meanwhile, trilobatin content varies from 0.1 to 2 mg/g FW in buds and leaves (Figures 1D, E), showing significant higher abundance in M. micromalus compared to M. toringo. In buds, trilobatin and sieboldin show a sharp significant increase from dormant to half inch buds, which is maintained until pink flowers, when both DHCs decrease at bloom stage (Figures 1D, G). In leaves, trilobatin and sieboldin content show no statistical difference during the growing season, tending to be stable across the season (Figures 1E, H). The content of sieboldin and trilobatin in whole fruits is 2.5- to 10-fold and 2- to 20-fold lower compared to leaves, respectively, and shows no statistical difference throughout fruit development (Figures 1F, I). Moreover, skin-enriched fraction accounts for the majority of phloridzin, trilobatin and sieboldin in fruits, in both domesticated and wild species (Supplementary Figures 3G-I). In addition, we detected the aglycone phloretin in leaves and buds of all accessions (Supplementary Figures 3D-F), whilst 3-hydroxyphloretin was predominantly found in leaves, followed by buds and fruit set (Supplementary Figures 3A-C).
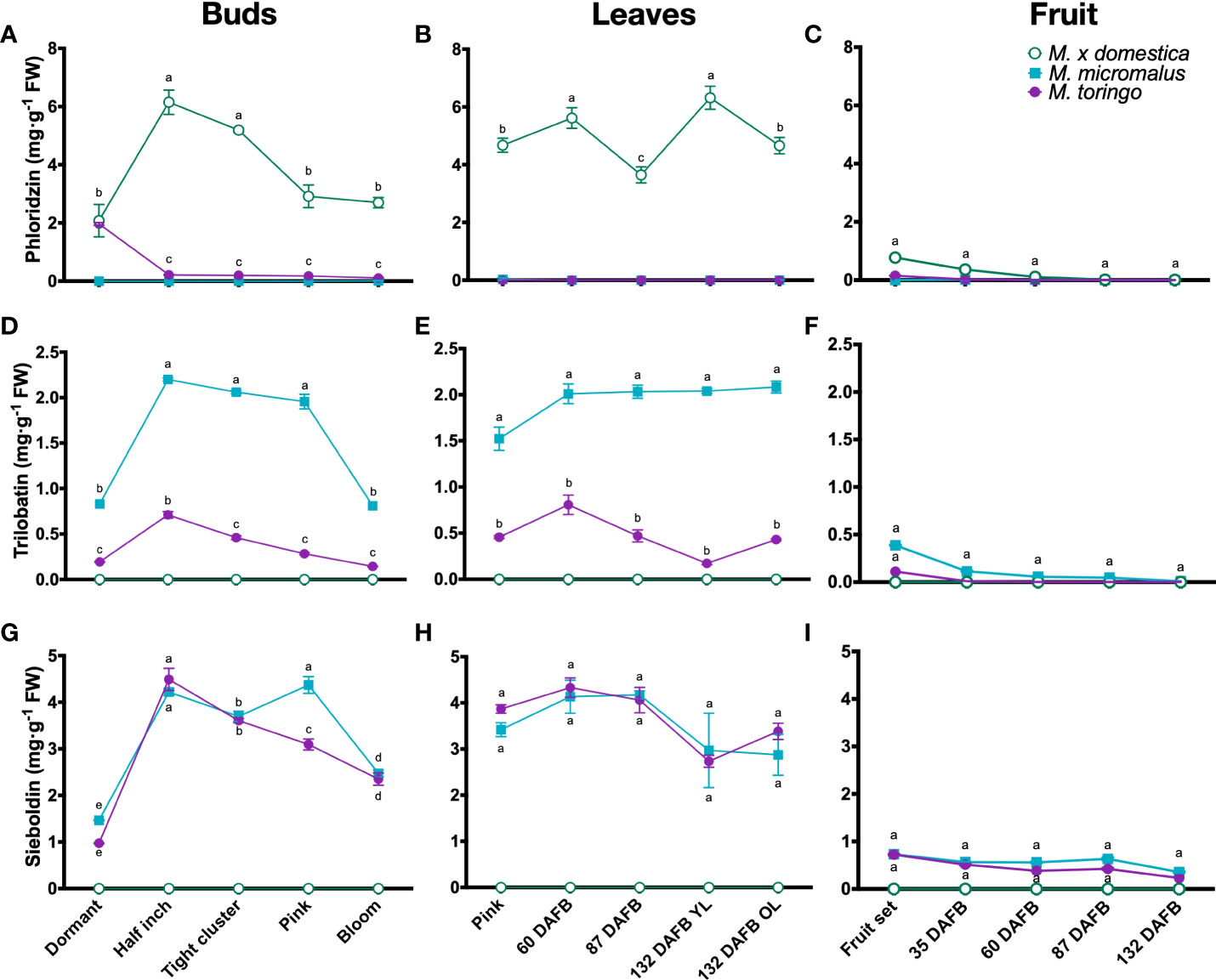
Figure 1 Metabolic profiling of DHCs in wild Malus and domesticated apple. Content of phloridzin (A–C), trilobatin (D–F) and sieboldin (G-I) during bud (A, D, G), young (YL) and old (OL) leaf (B, E, H) and fruit (C, F, I) development of M. toringo, M. micromalus and M. × domestica cv. ‘Golden Delicious’. Fruits were sampled at fruit set and 60, 87, 132 days after full bloom (DAFB). Values are reported in mg·g-1 fresh weight (FW), and represent the mean ± SE of three biological replicates in each sampling point, corresponding to the three individuals representing each accession in the Malus germplasm collection. Letters (a-d) indicate statistical differences (p < 0.05) across genotypes and sampling points as determined by two-way ANOVA and Tukey’s HSD test.
RNA-Seq and de novo assembly of leaf transcriptome of M. toringo and M. micromalus
The transcriptomes of young leaves of M. toringo and M. micromalus were sequenced, as well as the M. × domestica cv. ‘Golden Delicious’. A total of 272,391,814 high-quality 100-bp reads were obtained, with an average of 90,8 million reads per genotype (Supplementary Table 4). GC content of all accessions was 47 – 48%. Between 97.6 – 97.7% and 96.2 – 96.3% bases showed quality scores of Q20 and Q30, respectively. Following adapter trimming and quality filtering, ~98.5% and ~97.1% bases showed Q20 and Q30 scores, indicating that the quality of filtered reads was adequate for further analysis. Aiming to identify differentially expressed genes (DEGs) in wild Malus species compared to domesticated apple, we firstly conducted an RNA-Seq analysis by mapping clean reads by STAR to M. × domestica reference genome GDDH13 (Daccord et al., 2017). Transcriptomes of wild species mapped 71.5% (M. micromalus) and 73.01% (M. toringo) to M. × domestica ‘Golden delicious’ genome, which was similar to the mapping of reads from ‘Golden Delicious’ (72.7%, Supplementary Table 4), whereas unassigned reads due to multiple or ambiguous mapping ranged 24 – 26% and 0.12 – 0.13%, respectively. Mapped reads were counted by featureCounts and identification of DEGs was conducted by DESeq2, considering |fold change| > 2 and p-adj < 0.05. Among the 4,878 and 4,722 genes considered as DEGs in M. micromalus and M. toringo using apple ‘Golden delicious’ reference genome (Supplementary Table 5), we found a common core of 1296 up- and 1,879 down-regulated genes (Figures 2A-C). Gene set enrichment (GSE) analysis using KEGG pathways identified statistically enriched terms in this common set of DEGs, including ‘biosynthesis of secondary metabolites’, ‘ubiquinone and other terpenoid-quinone biosynthesis’ and ‘tyrosine metabolism’ (Figure 2D). Since sieboldin is highly accumulated in M. micromalus and M. toringo leaves, we hypothesised that potential genes involved in its biosynthesis would be among up-regulated DEGs. In order to get the actual coding sequences of up-regulated genes of M. micromalus and M. toringo, we performed a de novo transcriptome assembly. The summary of results is presented in Table 1. Trinity (Grabherr et al., 2013) assembled 139,716 and 132,885 transcripts for M. toringo and M. micromalus, respectively. The expression-informed ExN50 statistic was used as previously described (Hölzer and Marz, 2019), resulting in a transcript length of 2,636 and 2,585 bp in M. toringo and M. micromalus. For each accession, a total of 104,138 and 97,838 unigenes using CD-HIT-EST, and 38,730 and 36,443 open reading frames (ORFs) by TransDecoder were identified. We further evaluated the quality of de novo transcriptome assemblies using BUSCO, a tool that assesses the completeness of transcriptomes by finding a set of near-universal single copy orthologs (Simão et al., 2015). Transcriptomes of Malus species showed 90% of complete (unique and duplicated) ORFs (Table 1). In addition, we used the RBH approach to identify orthologs of M. × domestica ‘Golden Delicious’ in wild accessions, which led to 13,860 and 13,734 orthologs in M. toringo and M. micromalus, respectively. CDS obtained by TransDecoder were aligned against the non-redundant (Nr) database of NCBI, to provide functional annotation that could serve as the basis for retrieving candidate genes to be further analysed (Supplementary Table 6). Combining RNA-Seq results, de novo transcriptomes and functional annotation, we identified two potential 3-hydroxylases for further analyses.
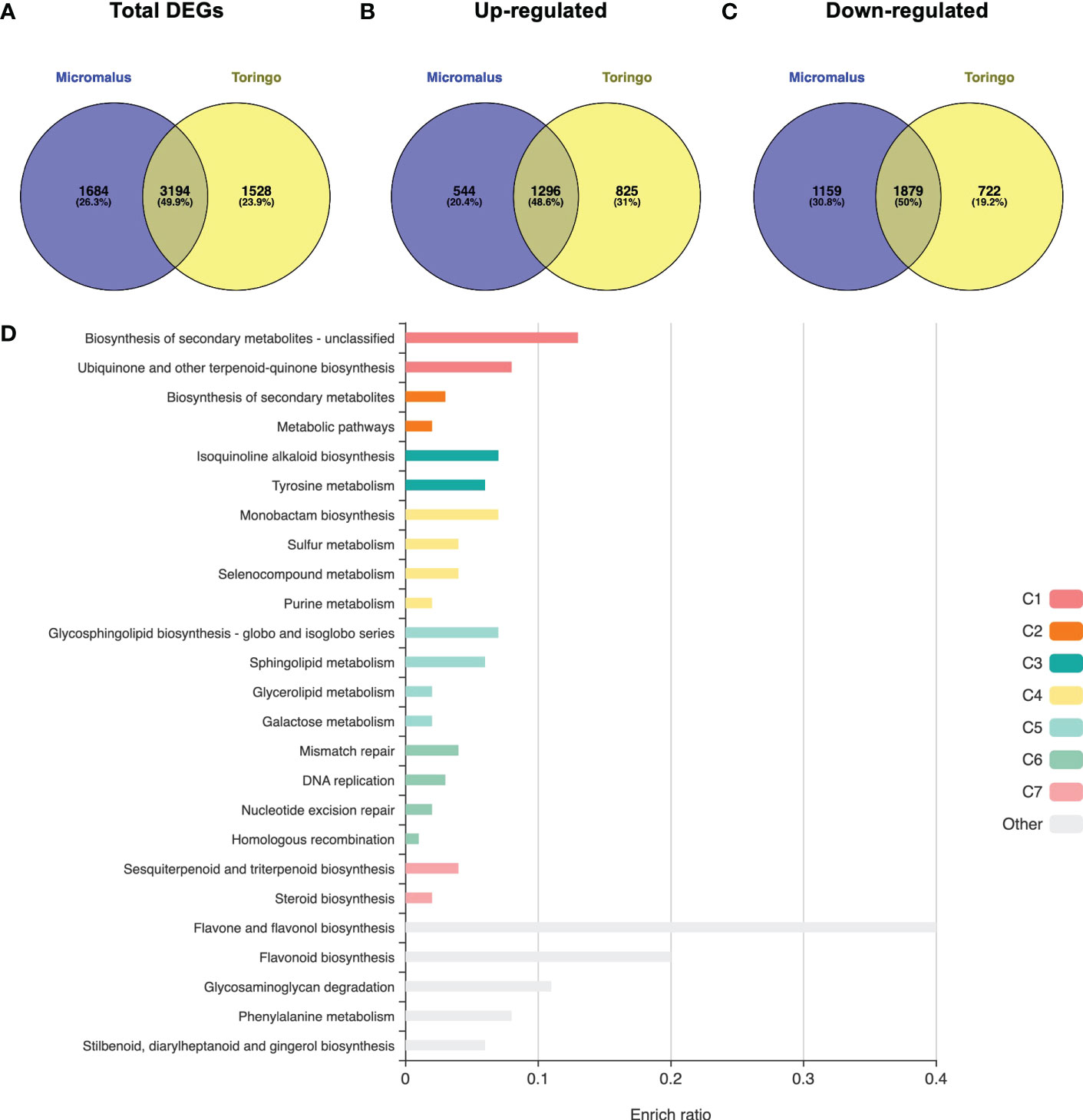
Figure 2 Transcriptomic overview of RNA-Seq results for wild Malus accessions compared to domesticated apple. Venn diagram of total number of DEGs (A), up-regulated (B) and down-regulated (C) DEGs of each Malus genotype. (D) Overrepresentation analysis identified statistically significant KEGG pathways enriched in the common list of up- and down-regulated DEGs of wild Malus species, compared to domesticated apple. Bar size represents the enriched ratio calculated as the number of genes included in the list compared to the background reference Malus × domestica genome.
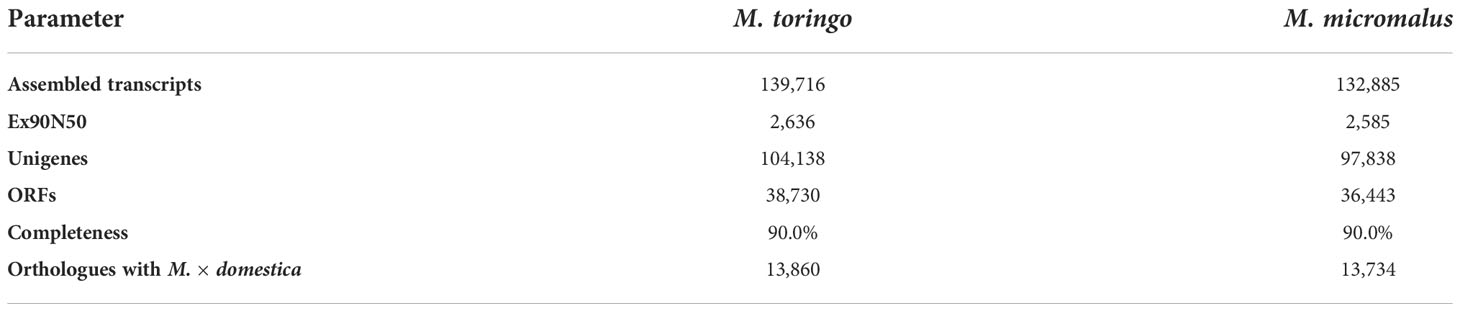
Table 1 Statistics of de novo transcriptome assembly for wild Malus accessions.
In silico analysis and expression analysis by RT-qPCR
We selected genes potentially involved in the DHC pathway of wild Malus species for further expression analysis by RT-qPCR. We found a gene in M. toringo and M. micromalus that was up-regulated 8.6- to 11.3-fold compared to domesticated apple (Supplementary Table 5), annotated as phloretin 4′-O-glycosyltransferase. This gene resulted to encode for PGT2, a glycosyltransferase recently characterised to be involved in trilobatin biosynthesis (Wang et al., 2020). We included CHS, and PAL1 to the expression analysis, considering their role in both flavonoid and DHC pathway (Ibdah et al., 2018). In addition, two genes of M. toringo and M. micromalus were selected as potential 3-hydroxylases to be further evaluated: ‘TRINITY_DN55953_c0_g1_i1.p1’ and ‘TRINITY_DN11639_c0_g1_i2.p1’, annotated as flavonol synthase/flavanone 3-hydroxylase-like. In addition, ‘TRINITY_DN56_c0_g1_i4.p1’ and ‘TRINITY_DN274_c0_g1_i3.p1’ were annotated as cytochrome P450 98A2, and were 36.7- to 36.8-fold up-regulated in M. toringo and M. micromalus compared to the orthologous gene of domesticated apple (MD08G1242900). A second gene in apple genome (MD08G1243000) has a predicted annotation as cytochrome P450 98A2, although it was not identified as the orthologue for ‘TRINITY_DN56_c0_g1_i4.p1’ and ‘TRINITY_DN274_c0_g1_i3.p1’ according to RBH. Among the first 5 best BLAST hits, we found a p-coumaroyl-shikimate 3′-hydroxylase from P. × bretschneideri (99% query cover and 93.36% identity). Cytochrome P450 genes were classified into CYP98 subfamily of cytochrome P450 enzymes (Nelson, 2009) and were assigned the following names: CYP98A195 and CYP98A196 from M. toringo and M. micromalus, respectively; and CYP98A197 (MD08G1242900) and CYP98A198 (MD08G1243000) from M. × domestica. Since cytochrome P450-dependent monooxygenases flavonoid 3′-hydroxylase (F3′H) and chalcone 3-hydroxylase (C3H) are capable of introducing hydroxyl groups in the B ring of flavonoids and DHCs, we aligned the amino acid sequences of the candidate genes of wild Malus with known C3H, F3′H, F6H and F3′5′H of other species and built a phylogenetic tree (Supplementary Figure 4). Proteins of M. toringo and M. micromalus clustered together, apart from F6H, F3′5′H and F3′H groups (Supplementary Figure 4A), suggesting that these genes might diverge in function. The closest members to CYP98A proteins of Malus wild species are CYP98A21 and CYP98A6 from Ammi majus and Lithospermum erythrorhizon, respectively (Supplementary Figure 4B). As a first approach of better characterising these candidate genes, we evaluated their expression profiles by RT-qPCR in selected tissues. The highest expression level of F3′H-like was found at fruit set of M. toringo and M. micromalus, followed by bloom and pink flowers. Leaves showed the lowest expression levels in both wild accessions, although significantly higher compared to M. × domestica (Figure 3A). CYP98A195-196 showed high expression levels in pink and bloom flowers, as well as in young leaves (Figure 3B). CYP98A195-196 transcript was also detected in fruit set of wild species, although significantly lower expression was found compared to leaf and flower tissues (Figure 3B). We also tested a third gene annotated as geraniol 8-hydroxylase-like by RT-qPCR, which showed the highest expression levels in M. × domestica young leaves, fruit set, and flowers at bloom stage (Figure 3C). In pink flowers, geraniol 8-hydroxylase-like showed similar expression levels between M. micromalus and M. × domestica, with statistically lower expression in M. toringo. In addition, the maximum expression for wild accessions was found in fruit set, showing no statistical difference with fruit set of domesticated apple (Figure 3C). The high expression in domestic apple along with higher expression in fruit set compared to the tissues where sieboldin is predominantly accumulated were used as criteria to exclude this third putative hydroxylase for further analyses. Meanwhile, PAL1 was expressed at similar extent in all tissues analysed (Figure 3D). Similarly, CHS was found in all accessions, showing its highest expression in young leaves, followed by pink and blooming flowers (Figure 3E). As in the case of CYP98A195-196, CHS was also expressed in fruit set, with ‘Golden Delicious’ exhibiting a significant higher relative expression compared to wild accessions. PGT2 was highly expressed in M. micromalus in pink flowers and young leaves, whilst M. toringo showed significantly lower expression levels in flower and leaf tissues, although no statistical difference was found at fruit set of both accessions (Figure 3F).

Figure 3 Transcriptional regulation of candidate 3-hydroxylase genes from wild Malus accessions and genes involved in the phenylpropanoid pathway. The expression of F3′H-like (A), CYP98A (B) and geraniol 8-hydroxylase-like (C) genes was assessed in blooming and pink flowers, young leaves and fruit set of M. toringo, M. micromalus and M. × domestica cv. ‘Golden Delicious’ by RT-qPCR. In addition, phenylalanine ammonia lyase (PAL1), chalcone synthase (CHS) and 4′-O-glycosyltransferase (PGT2) genes were assessed. Bars represent mean ± SE for three biological replicates. Letters (a-e) indicate statistical differences (p < 0.05) across genotypes and sampling points as determined by two-way ANOVA and Tukey’s HSD test.
De novo production of sieboldin in Saccharomyces cerevisiae
In order to assess the 3-hydroxylase activity of candidate genes identified by the transcriptomic analysis of wild Malus accessions, we established a heterologous system (summarised in Figure 4A) in a phloretin-producing S. cerevisiae strain by expressing PAL, C4H, 4CL2, CHS and CPR as previously described (Eichenberger et al., 2017). Candidate F3′H-like and CYP98A195-196 genes of M. toringo and M. micromalus were cloned into a high copy yeast expression vector pAT425 and transformed in the phloretin-producing yeast strain. Since pAT425 vector harbours two cloning sites, under the regulation of two different constitutive promoters for yeast expression (Ishii et al., 2014), we initially tested 3-hydroxylase candidate genes under the control of alcohol dehydrogenase (ADH1) and glyceraldehyde-3-phosphate dehydrogenase (TDH3) promoters after 48 h of incubation. Interestingly, 3-hydroxyphloretin was detected only in the phloretin-producing strains expressing the genes CYP98A195 and CYP98A196 of M. toringo and M. micromalus, respectively (Figure 4B). Moreover, expression of CYP98A195-196 under TDH3 promoter led consistently to higher levels of 3-hydroxyphloretin, compared to expression under ADH1 promoter (Figure 4B). To further evaluate the possibility of achieving de novo production of sieboldin in S. cerevisiae using CYP98A196-197 of wild species, we cloned CYP98A197-198 of M. × domestica into the same backbone and co-expressed each CYP98A gene of wild and domesticated Malus accession with the recently characterised 4′-O-glycosyltransferase (PGT2) involved in trilobatin synthesis, which also accepts 3-hydroxyphloretin as substrate in vitro (Wang et al., 2020). To this aim, we cloned PGT2 under ADH1 promoter into the pAT425 vector harbouring the constructs pTDH3::CYP98A195, pTDH3::CYP98A196, pTDH3::CYP98A197 or pTDH3::CYP98A198, and transformed each vector in the phloretin-producing yeast strain followed by incubation for 72 to 96 h. Interestingly, expression of CYP98A genes from M. × domestica did not produce 3-hydroxyphloretin even after 96 h, whereas an increase in 3-hydroxyphloretin was shown in yeast expressing CYP98A195 and CYP98A196 from 72 to 96 h (Figure 4D, Supplementary Figure 5). Furthermore, sieboldin was produced only when CYP98A members from wild accessions were used (Figure 4F, Supplementary Figure 5), leading to levels ranging from 2 - 3 to 3 - 4.5 mg·L-1 after 72 and 96 h, respectively (Figure 4F). As expected, phloretin was accumulated in all yeast strains at similar levels, with the yeast strain transformed with the empty pAT425 vector showing the highest values (Figure 4C). Meanwhile, trilobatin was produced in yeast co-expressing PGT2 with CYP98A genes of wild and domesticated accessions, although higher level of trilobatin was observed in yeast harbouring CYP98A genes of M. × domestica, compared to those expressing CYP98A195 and CYP98A196 (Figure 4E).
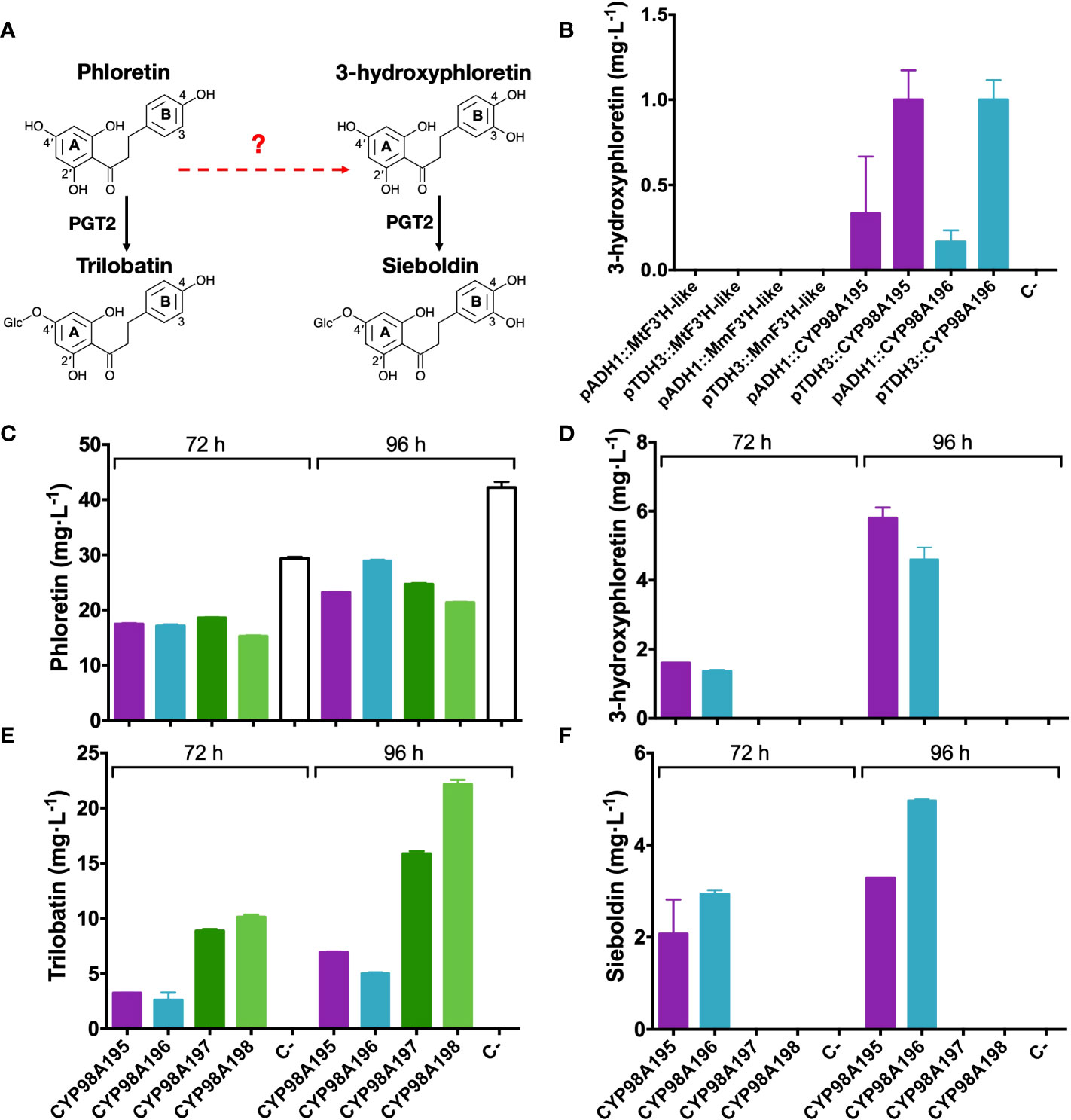
Figure 4 Functional characterisation of 3-hydroxylase candidate genes from wild and domesticated Malus species. (A) Simplified overview of the phenylpropanoid pathway reconstructed in S. cerevisiae, where the characterised enzymatic steps are indicated by solid arrows and 3-hydroxylation reaction potentially catalysed by candidate genes tested is indicated by a red dashed arrow. Relevant carbon residues are indicated by numbers in ring A (2′ and 4′) and B (3 and 4) of DHCs. (B) Detection of 3-hydroxyphloretin after 48 h culture from yeast expressing F3′H-like or CYP98195 and CYP98A196 genes of M. toringo, M. micromalus, respectively or M. × domestica (CYP98A197, CYP98A198) under the control of ADH1 or TDH3 constitutive promoters. In addition, phloretin (C), 3-hydroxyphloretin (D), trilobatin (E) and sieboldin (F) were detected by a targeted UPLC/QqQ/MRM method for phenolic compounds, after incubation for 72 and 96 h of a phloretin-producing S. cerevisiae co-expressing PGT2 and CYP98A genes of wild and domesticated Malus species. Values represent the mean ± SE of extractions from three independent yeast cultures. Negative control (C-) corresponds to the phloretin-producing strain transformed with the empty pAT425 vector.
Assessment of germplasm collection with HRM marker for DHC3H
The orthologous gene of domesticated apple identified in RNA-Seq analysis and functionally characterised corresponds to MD08G1242900 and assigned the name CYP98A197. However, we also identified an adjacent gene in apple annotated as CYP98 A2 in chromosome 8 of the reference ‘Golden Delicious’ genome, corresponding to the gene MD08G1243000 and named CYP98A198. This second gene of domesticated apple shares 95.7% and 93.4% nucleotide and amino acid identity with wild accessions, respectively. Aiming to determine whether SNPs in CYP98A sequences could correlate with the presence or absence of sieboldin, we conducted genetic mapping by high resolution melting (HRM) marker analysis. Using primers flanking a region close to the start codon showing the highest sequence divergence among CYP98A195-198 sequences of wild and domesticated species (Supplementary Figure 6A), HRM marker analysis identified three different clusters in the Malus germplasm accessions with an average confidence level of 98.1% (Supplementary Figures 6B, C, Supplementary Table 7). The three glycosylated DHCs derived from phloretin (phloridzin, trilobatin and sieboldin) found in Malus spp. were determined in young leaves of each genotype. Interestingly, the genotypes evaluated that accumulate sieboldin clustered in group 1 (including M. sargentii, M. toringo, M. micromalus and M. ‘Evereste’), whereas accessions accumulating exclusively phlorizin (including M. baccata, M. × domestica, M. prunifolia, M. robusta, M. halliana, M. × paradisiaca, M. × moerlandsii) categorised in clusters 2 or 3 (Figure 5). M. floribunda and M. × atrosanguinea, as well as some accessions of M. micromalus and M. sargentii accumulate both phloridzin and sieboldin, although they clustered together with accessions where only sieboldin is produced, and HRM marker analysis categorised them in cluster 1 (Figure 5).
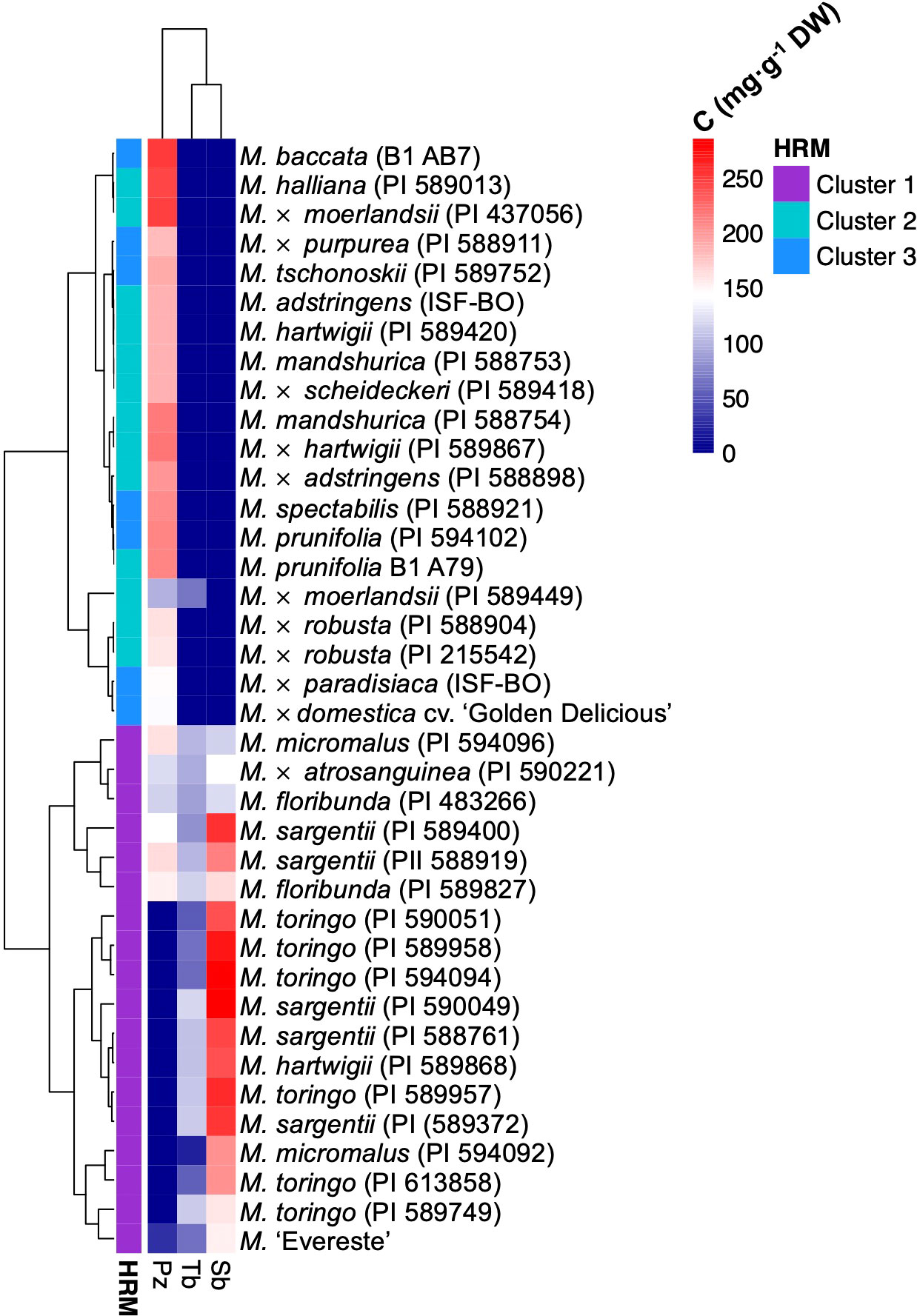
Figure 5 Heatmap clustering Malus germplasm collection by DHC profile and HRM marker analysis for CYP98A195-198 isoforms. Hierarchical heatmap of the concentration of the main DHC forms phloridzin (Pz), trilobatin (Tb) and sieboldin (Sb), for wild and hybrid Malus accessions in a germplasm collection. Gradient of blue and red colours represent a trend of decrease and increase of metabolite, respectively. Metabolite clustering was automatically calculated for genotypes in R-environment. HRM clustering result is displayed at the left of each accession.
Characterisation of DHC3H protein by subcellular localization and protein modelling
Since several steps of the flavonoid pathway occur in the endoplasmic reticulum (Agati et al., 2012), we aimed to determine the subcellular localisation of CYP98A195-196 identified from wild Malus species. We established stable transgenic lines of tobacco overexpressing protein markers of endoplasmic reticulum and plasma membrane fused to mCherry, using ER-rk and PM-rk vectors (Nelson et al., 2007). We selected CYP98A196, given that genes from both wild accessions exhibit 100% amino acid identity. Transient expression of CYP98A196 fused to EYFP in tobacco leaves of transgenic ER-rk and PM-rk lines showed a distinctive pattern (Figure 6A, left and middle panel) that co-localised with the emission of ER-rk marker (Figure 6A, right panel). Fluorescence of CYP98A196-EYFP was distributed in the periphery of epidermal cells showing the characteristic pattern of a network close to the cell borders (Figure 6C, left panel), underneath a more homogeneous fluorescence signal along the surface of cells exhibited by the plasma membrane marker (Figure 6B, middle panel; Figure 6C, right panel). Protoplasts of tobacco leaves were isolated for the same constructs, and CYP98A196-EYFP showed a clear cell peripheral pattern with complete co-localization with ER-rk marker (Figure 6D), similar to the pattern displayed by the marker transformed with the empty vector (Figure 6E). The peripheral localisation of CYP98A196 does not co-localize with the plasma membrane signal, which surrounds the cell in a uniform manner (Figure 6E). The signal of CYP98A196-EYFP surrounds the autofluorescence of chlorophyll, as depicted in Figure 6G
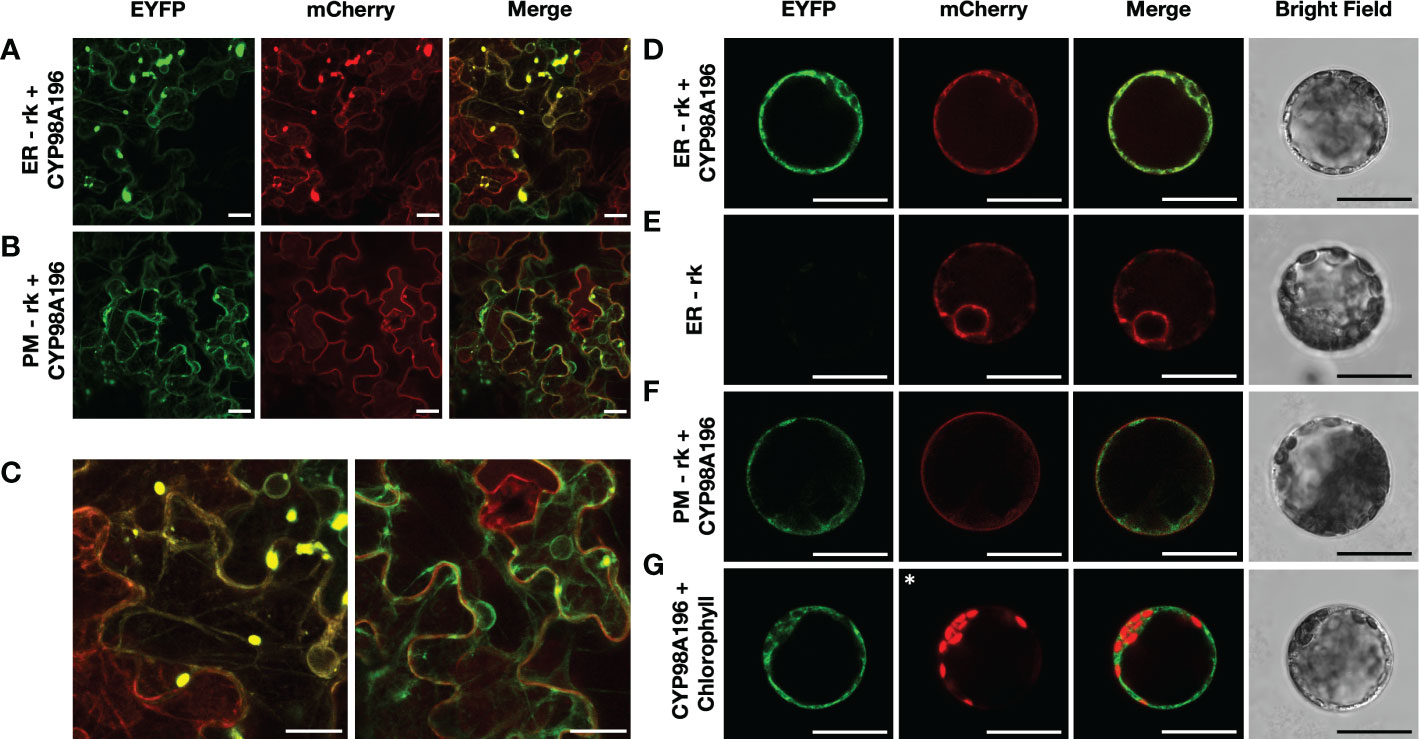
Figure 6 Determination of sub-cellular localisation of CYP98A196. Tobacco epidermal cells were transiently transformed with CYP98A196-EYFP construct by agroinfiltration in a stable transgenic tobacco line expressing the endoplasmic reticulum ER-rk marker (A) or the plasma membrane PM-rk marker (B), both fused to mCherry. Representative results of each construct are presented for EYFP, mCherry and merged visualization, and a zoom of merged fields following co-infiltration of ER-rk and PM-rk with CYP98A196-EYFP are presented in (C), left and right panel, respectively. In addition, protoplasts co-expressing ER-rk marker with CYP98A196-EYFP (D) or empty vector (E), or PM-rk marker with CYP98A196 (F) are presented for EYFP, mCherry, bright field and merged visualization. (G) CYP98A196-EYFP and autofluorescence of chlorophyll instead of mCherry (indicated with an asterisk) collected as described in Materials and Methods. Scale bar: 25 µm.
To examine the structural differences of CYP98A members producing 3-hydroxyphloretin in vivo to those of M. × domestica, we constructed protein models using PCPD server, which combines structural homology and molecular dynamics simulation optimised for plant cytochrome P450s to predict the final protein model (Wang et al., 2021). Then, we carried out a protein-ligand docking by PCPLD using phloretin as potential ligand to predict the position and orientation of phloretin in CYP98A196 protein model. Phloretin is predicted to interact by hydrogen bonds with the residues Trp116 and Met213 and with the heme group. Focusing the attention to residues in the proximity of the ligand pocket predicted, we found that most of the residues within 5 Å were conserved. Five residues, however, were exclusively found in CYP98A196 (Ser104, His212, Met213, Ala302, Thr484). Among them, His212 and Met213 are in close proximity to the A ring of phloretin, with Met213 forming a hydrogen bond with the hydroxyl group at 4′ position of A ring (Figure 7A). In addition, the polar residue Thr302 found in CYP98A197 of M. × domestica has the counterpart with the aliphatic Ala302 in CYP98A196 of M. micromalus, showing physico-chemical conservation with Val302 of CsC3H (Supplementary Figure 7), the only example with reported 3-hydroxylase activity producing 3-hydroxyphloridzin in CsC3H-overexpressing apple lines (Hutabarat et al., 2016). Superimposition of CYP98A196 with the protein model of a CYP98 member coding for C3H that exhibits 3-hydroxyalse activity towards coumaroylshikimate (Schoch et al., 2001) shows that Trp116 of CYP98A196 and Trp112 of C3H are close to the oxygen in the carbonyl group linking A and B ring of phloretin (Figure 7B).
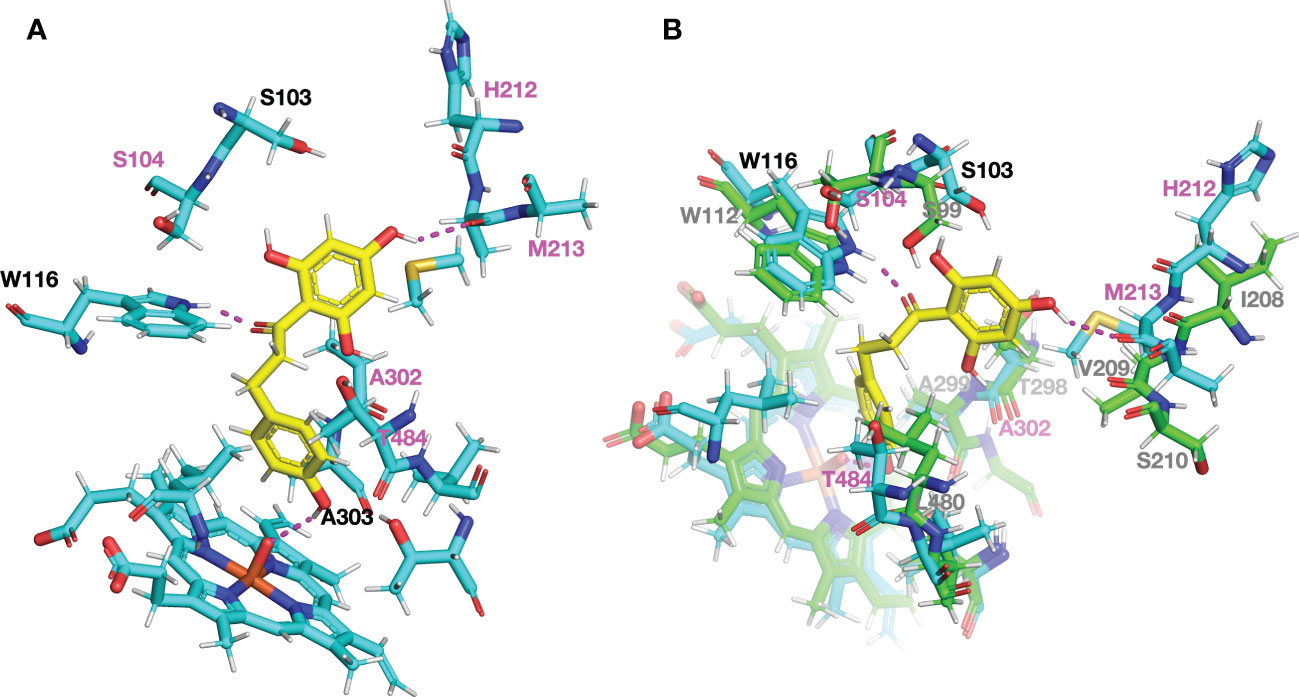
Figure 7 Structural comparison of the ligand pocket predicted for DHC3H model showing 3-hydroxylase activity in vivo. Protein models and docking with phloretin (yellow) were obtained using PCPD and PCPLD, respectively. (A) Amino acid residues found exclusively in CYP98A196 around phloretin within 5 Å are highlighted in magenta (S104, H212, M213, A302, T484), and predicted hydrogen bonds are indicated with dashed lines in magenta. (B) Superimposed models of CYP98A196 with AtCYP98A3, a characterised 3-hydroxylase. Carbon, oxygen, nitrogen and sulphur atoms are shown in light blue, red, blue and orange, respectively.
Discussion
Detailed profiling of dihydrochalcones during plant development in Malus accessions
DHCs are a class of secondary metabolites of the phenylpropanoid pathway, which includes anthocyanins, flavonols and flavan-3-ols with reported benefits for human health (Panche et al., 2016). In contrast to the more common polyphenols, DHCs have a limited natural distribution and the major flavonoid class found in Malus spp. (Rivière, 2016). Phloridzin, the main DHC form of M. × domestica (Gosch et al., 2010b), was thought for long time to be exclusive to apple, leading to propose phloridzin as an authenticity marker for evaluation of apple-derived products in the food industry (Hrubá et al., 2021). Most of the studies have focused on phloridzin, given the protective effects of phloridzin reported in mammal models (Stangl et al., 2005; Szliszka et al., 2010; Viet et al., 2013). However, trilobatin and sieboldin are accumulated in considerable amounts in other Malus species (Gutierrez et al., 2018b). Despite the properties of DHCs as flavour sweeteners and bitterness blockers (Tomás-Barberán et al., 1995; Wang et al., 2020), studies addressing the accumulation patterns and physiological changes of DHCs in planta are still scarce. Histolocalisation showed that DHCs are present in proximity to the vascular system and upper layers of leaves in the ornamental M. cv. ‘Evereste’ (Gaucher et al., 2013). Phloridzin is found in bark (Zhang et al., 2007), root exudates (Hofmann et al., 2009) and leaves of domesticated apple (Petkovsek et al., 2009). In addition, apple cultivars accumulate varying amounts of phloridzin in skin and whole fruit (Łata et al., 2009). Here, we conducted a detailed profiling of DHCs during bud and fruit development, along with a systematic monitoring of foliar DHC content. Polyphenolic profiles of the wild accessions M. toringo and M. micromalus were dominated by the glycosylated DHCs sieboldin, followed by trilobatin, whereas domesticated apple only accumulates phloridzin (Figure 1). Aglycones phloretin and 3-hydroxyphloretin were detected in low concentrations in leaves and fruit set, which may suggest that glycosylation stabilises phloretin and 3-hydroxyphloretin as previously hypothesised (Caputi et al., 2012; Ibdah et al., 2018). We observed a drastic increase of phloridzin, trilobatin and sieboldin in half-inch buds after dormancy (Figures 1A, D, G), reinforcing a previous study that proposed phloridzin as a phase change in apple (Zhang et al., 2007). Foliar content of DHCs was relatively stable across the growing season, showing higher levels of trilobatin in M. micromalus compared to M. toringo (Figures 1D, E), although sieboldin was the predominant DHC found in leaves and exhibited similar levels in both wild accessions (Figure 1H). A previous study showed that sieboldin exerted the highest antioxidant capacity in young leaves, suggesting a role in coping with oxidative stress during development (Dugé de Bernonville et al., 2010). Fruit of wild accessions showed low DHC content compared to leaves and buds, which has been attributed to a dilution effect in domesticated cultivars caused by fruit size (Busatto et al., 2019). In agreement with this hypothesis, we found the highest concentration of phloridzin, trilobatin and sieboldin at fruit set, which constantly decreased until fruit ripening (Figure 1, Supplementary Figure 3). Most of the DHC content in wild and domesticated accessions was attributed to the peel, which in turn has shown positive correlation between DHCs levels in peel and antioxidant capacity (Zhou et al., 2018), further suggesting a role of flavonoids and, among them, DHCs in the response of Malus spp. to oxidative stress faced during the growing season (Tsao et al., 2005).
Identification of candidate genes involved in sieboldin biosynthesis by de novo transcriptome assembly and RNA-Seq analysis
Public genetic resources in Malus species have increased in the last two decades, from the reference genomes for M. × domestica cv. ‘Golden Delicious’ (Velasco et al., 2010; Daccord et al., 2017) and ‘Gala’ (Sun et al., 2020) to sequencing of ‘Gala’ wild progenitors M. sieversii and M. sylvestris (Sun et al., 2020), as well as wild M. baccata and M. prunifolia (Chen et al., 2019; Li et al., 2022). Wild accessions with publicly available genome resources, however, accumulate exclusively (M. sieversii, M. sylvestris and M. baccata) or predominantly (M. prunifolia) phloridzin according to a previous study (Gutierrez et al., 2018b). Therefore, we chose two Malus accessions from the germplasm collection at Fondazione Edmund Mach accumulating sieboldin and trilobatin to initially conduct an RNA-Seq analysis. As a preliminary approach to identify genes that could be involved in sieboldin biosynthesis, we hypothesised that candidate genes of wild accessions should be highly up-regulated compared to the domesticated apple reference genome, provided that reads of wild species align in similar proportion to apple genome. We found that the percentage of mapping was comparable in all genotypes (72.7% in Golden Delicious, 71.5 – 73% for M. toringo and M. micromalus; Supplementary Table 4), indicating that our preliminary approach led to mapping statistics within the acceptable range when conducting RNA-Seq analysis (Conesa et al., 2016). Single-end reads tend to have a slightly lower percentage of uniquely mapped reads (Chhangawala et al., 2015), which may explain the mapping percentage in all genotypes. In addition, multiple mapping was similar across accessions (Supplementary Table 4) that could be explained by recent duplication events during domestication of apple (Velasco et al., 2010; Daccord et al., 2017). Importantly, we identified two candidate genes: F3′H-like, annotated as flavonol synthase/flavanone 3-hydroxylase; and CYP98A195-196, that had also annotation as p-coumaroyl-shikimate 3′-hydroxylase. These genes showed a 4.3 – 4.9-fold (F3′H-like) and 36.8-fold (CYP98A195-196) up-regulation in M. toringo and M. micromalus, respectively (Supplementary Table 5). Cytochrome P450-dependent monooxygenases F3′H and F3′5′H can also introduce hydroxyl groups at the 3′ or 3′ and 5′ position of the B ring of flavonoids, showing differences in substrate specificity and amino acid identity lower than 50% (Seitz et al., 2006). We found that the two candidate genes of wild Malus species clustered (CYP98A195-196) in a phylogenetic tree apart from known F3′H, F3′5′H and F6Hs. To delve into the potential candidates of wild accessions for expression and functional analyses, we did a de novo transcriptome assembly from young leaves, resulting in around 100,000 unigenes and completeness of 90%. These results are in line with previous de novo transcriptomes assembled in other species (Wu et al., 2017; Biswal et al., 2021; Madritsch et al., 2021; Pacheco et al., 2021), where a great proportion of assemblies are short or partial transcripts, as well as pre-mRNA, ncRNA and transcript artifacts (Raghavan et al., 2022). To compensate for the low-expressed and short transcripts that dominate de novo transcriptomes, it has been proposed that expression-informed statistics such as Ex90N50 may reflect better the length of assembled transcripts accounting for 90% of the normalised expression data (Hölzer and Marz, 2019). In our study, Ex90N50 values for M. toringo and M. micromalus were around 2.6 kb, suggesting that half of the transcripts assembled accounting for 90% of the expression data had an acceptable size to retrieve full-length CDS. Prediction of ORFs by TransDecoder resulted in around 36,000 – 39,000 predicted CDS, close to the genes of M. × domestica (Daccord et al., 2017). In addition, we identified several genes involved in the flavonoid pathway, including PAL1, CHS and PGT2. To validate transcriptomic results, we conducted an expression analysis of putative 3-hydroxylases together with genes involved in the phenylpropanoid pathway in selected tissues being the predominant sources of DHCs according to the metabolite profiling. We confirmed that the expression of PAL1 and CHS pattern was similar for all accessions (Figures 3D, E), suggesting that the key steps of the phenylpropanoid pathway are transcriptionally active throughout the developmental stages evaluated. PGT2 expression was detected only in wild accessions, showing its maximum in young leaves and pink flowers of M. micromalus (Figure 3F). Moreover, we analysed three potential 3-hydroxylase candidates, and selected F3′H-like and CYP98A195-196 genes for further analyses, using as criteria the high expression in wild accessions compared to domesticated apple and expression in tissues where DHCs are mainly accumulated.
Functional characterisation of 3-hydroxylase involved in the production of sieboldin in vivo
Several examples utilise de novo transcriptome assemblies for identification of putative genes involved in the naturally-abundant flavonoids (Thole et al., 2019; Biswal et al., 2021), carotenoids (Pacheco et al., 2021) or glucosinolates (Wu et al., 2017). These studies have focused their attention on validating de novo transcriptomes by RT-qPCR to evaluate transcriptional regulation of the genes of interest. To the best of our knowledge, the use of high throughput platforms to attempt the elucidation of genes involved in specialised metabolism of non-model species is still scarce in literature, which counts with examples of GWAS and RNA-Seq in the case of DHCs of apple (Zhou et al., 2017; Kumar et al., 2022). Aiming to provide functional evidence for the involvement of candidate genes in the biosynthesis of the specialised metabolites 3-hydroxyphloretin and sieboldin, we used S. cerevisiae as an heterologous system to test the in vivo activity of candidate P450 monooxygenases, which is a readily suitable model for expressing P450 monooxygenases, because of its endogenous NADPH-cytochrome P450 reductases (CPR) needed for the activity of P450 monooxygenases (Hausjell et al., 2018). Using a yeast strain producing phloretin by metabolic engineering as stablished before (Eichenberger et al., 2017), we expressed candidate genes under the control of two constitutive promoters for protein expression (ADH1, TDH3). Interestingly, we found that only CYP98A195 and CYP98196 were able to convert phloretin into 3-hydroxyphloretin (Figure 4) independently of the promoter used, indicating that CYP98A proteins from wild Malus species exerts a 3-hydroxylase activity in vivo. Phloretin conversion was higher under TDH3 promoter, which is consistent with the higher GFP signal obtained under the control of TDH3 compared to ADH1 promoter (Ishii et al., 2014). We therefore chose the expression under TDH3 promoter to further assess the production of sieboldin by introduction of PGT2, a 4′-O-glycosyltransferase that accepts 3-hydroxyphloretin in vitro (Wang et al., 2020). We demonstrated that CYP98A195 and CYP98A196 co-expressed with PGT2 in the phloretin-producing yeast can convert 3-hydroxyphloretin to its glycosylated form sieboldin, which was not found with the apple proteins of M. × domestica, even after 96 h of incubation (Figure 4). Moreover, we aimed to develop a HRM marker to discriminate different alleles, considering the nucleotide differences found between CYP98A195-196 of wild accessions and M. × domestica CYP98A197-198. Screening a Malus germplasm collection by HRM marker analysis, we recognised three clusters, which are consistent with the nucleotide differences found between CYP98A sequence of M. toringo and M. micromalus and the two genes of M. × domestica (Supplementary Figure 6, Supplementary Table 7). Furthermore, these clusters correlated with the presence or absence of sieboldin (Figure 5), indicating that CYP98A isoforms may be useful to recognise sieboldin-producing Malus accessions. A previous study categorised Malus accessions by the presence of only phloridzin (P), phloridzin and trilobatin (PT), sieboldin and trilobatin (ST) or the three glycosylated DHCs (SPT) (Gutierrez et al., 2018b). In agreement with the accessions of the USDA Geneva Malus Core collection, accessions of M. toringo and M. sargentii of our germplasm collection displayed ST and SPT profiles, respectively, and HRM analysis detected the allele present in M. toringo and M. micromalus. It has been proposed that sieboldin originated from M. toringo, and hybrids of M. toringo with phloridzin-accumulating accessions (M. baccata, M. halliana, M. prunifolia) derives in accessions with ST and SPT profiles (Gutierrez et al., 2018b). The latter progenitors belonged to clusters 2 or 3, meaning that they showed a HRM profile similar to the inactive CYP98A197 and CYP98A198 genes of M. × domestica. Hybrids of M. toringo with phloridzin-accumulating parental accessions evaluated in the germplasm collection (M. floribunda and M. × atrosanguinea) exhibited a SPT profile. Despite the intermediate levels of the three DHCs compared to accessions with P and PT profiles, HRM analysis identified the allele of M. toringo and M. micromalus. Altogether, these results indicate that differences in CYP98A members may be used for screening Malus accessions accumulating exclusively sieboldin or together with other DHCs. A linkage and association analysis of DHCs identified LG7 and LG8 as major determinant loci for trilobatin and sieboldin (Gutierrez et al., 2018a). PGT2 mapped in LG7 of M. × domestica genetic map, corresponding to gene models MD07G1281000 and MD07G1281100 in apple found in chromosome 7 (Wang et al., 2020). Similarly, the genes identified in M. toringo and M. micromalus in our study have their equivalents in chromosome 8 of apple (MD08G1242900 and MD08G1243000), suggesting that CYP98A allele of wild Malus accessions may explain the QTL associated with sieboldin identified in LG8 in Gutierrez et al., 2018a.
To further characterise CYP98A proteins of wild Malus species, we selected CYP98A196 because the amino acid sequence has 100% amino acid identity with CYP98A195 despite sharing 99.94% nucleotide identity, indicating that nucleotide differences correspond to synonymous mutations. It has been proposed that the phenylpropanoid pathway exhibits subcellular organisation, where cytosolic proteins (including PAL and CHS) are associated with other proteins of the pathway anchored to the endoplasmic reticulum (such as C4H and F3′H), forming multienzyme complexes channelling intermediate substrates that compete at different branch points (Winkel-Shirley, 2001). Thus, we assessed the sub-cellular localisation of CYP98A196, and found that it exhibited the characteristic pattern of network around a structure compatible with nucleus. Moreover, CYP98A196 fused to EYFP co-localised with an ER marker created by combining the signal peptide of AtWAK2 at N-terminus and the HDEL ER retention signal at C-terminus of the fluorescent protein (Nelson et al., 2007). The peripheral pattern of CYP98A196-EYFP fusion protein did not co-localised with the plasma membrane marker, surrounding chloroplasts of epidermal cells of tobacco (Figure 6).
CYP98A proteins of wild Malus accessions exhibiting 3-hydroxylase activity share 93.36% - 93.5% amino acid identity with CYP98A197 and CYP98A198 of M. × domestica. Remarkably, these differences are sufficient to produce proteins lacking 3-hydroxylase activity towards phloretin in yeast. Aiming to explore the structural basis that could underlie differences in activity, we conducted a protein model prediction by PCPD, a plant cytochrome P450 database that combines AlphaFold2, molecular dynamics (MD) simulation and ligand docking to score and determine the final model (Wang et al., 2021). AlphaFold has greatly improved the accuracy of protein structure prediction, outperforming standard algorithms (Jumper et al., 2021). Integration of MD-simulation and ligand docking process optimised for cytochrome P450s (PCPLD) resulted in the construction of a broad database specifically designed for simulation of plant cytochrome P450s (Wang et al., 2021). Using PCPD and PCPLD, we found significant differences in DHC3H compared to M. × domestica. Six substrate recognition sites (SRS) have been described in cytochrome P450s (Gotoh, 1992): SRS1 (residues 103 - 126), SRS2 (residues 209 - 216), SRS3 (residues 248 - 255), SRS4 (residues 302 - 320), SRS5 (residues 375 - 385) and SRS6 (residues 485 - 493). Comparing the residues close to the predicted substrate phloretin between DHC3H and M. × domestica, we found that four mutations were located precisely in the substrate recognition sites (Ser104, SRS1; His212, SRS2; Met213, SRS3; Ala302, SRS4), whilst Thr484 was next to the SRS6 (Figure 7, Supplementary Figure 7). Moreover, two hydrogen bonds stabilise the interaction between A ring of phloretin and residues in SRS1 (Trp116, Met216). The former study characterising C3H demonstrated that changes in amino acid residues of SRS1 have an impact on chalcone acceptance (Schlangen et al., 2010). In addition, a previous work demonstrated that substrate specificity and functional differences between F3′H and F3′5′H are determined near the N-terminal (SRS1 - 3) and C-terminal (SRS6) regions, respectively (Seitz et al., 2007). Amongst the vast cytochrome P450 superfamily, nomenclature rules have been stablished to distinguish families (indicated by a number, sharing at least 40% amino acid identity), subfamilies (referred as letters, with at least 55% identity) and isoforms (gene identifiers) based on percentage of amino acid identity (Nelson, 2006). Importantly, several plant cytochrome P450s families have been implicated in the metabolism of specialised metabolites, with reported examples in cyanogenic glucosides, glucosinolates, terpenoids and phenylpropanoids (Kahn et al., 1999; Hamberger and Bak, 2013; Romsuk et al., 2022). Besides CYP71 clan displaying monoterpenoid and sesquiterpenoid diversity and CYP79 members producing amino acid-derived specialised compounds, CYP98A subfamily has been described to transition from general to specialised phenylpropanoid metabolism (Hamberger and Bak, 2013). CYP98A3 and CYP98A22 from Arabidopsis thaliana and Ruta graveolens have been implicated in the 3′-hydroxylation involved in biosynthesis of chlorogenic acid and 3-hydroxylation of lignin monomers (Schoch et al., 2001; Karamat et al., 2012). In addition, CYP98A14 of Coleus blumei is responsible for the 3- and 3′-hydroxylation involved in the biosynthesis of rosmarinic acid (Eberle et al., 2009). Within CYP98A subfamily, a study found that CYP98A family members are highly duplicated, accumulating mutations that have led some members to acquire subsidiary activities involved in the hydroxylation of p-coumaroyltyramine (Morant et al., 2007). Interestingly, gene duplication in the CYP98A8 and CYP98A9 members in Arabidopsis resulted in neofunctionalisation of each member for different substrates, driven by a positive-selective pressure for the acquisition of regulatory elements in the promoter region, transcriptional reprogramming and local nonsynonymous amino acid substitutions (Matsuno et al., 2009). It is likely that amino acid differences in CYP98A195-198 found in Malus species have undergone similar mutations leading to modifying enzymatic activity.
The high expression of DHC3H in the tissues where DHCs are mainly accumulated along with key amino acid features close to the substrate pocket predicted in the protein models of wild accessions may explain the in vivo conversion of phloretin into 3-hydroxyphloretin, which ultimately leads to sieboldin production by co-expression with PGT2. Domesticated apple lines overexpressing CsC3H resulted in the production of 3-hydroxyphloridzin, which correlated with reduced susceptibility to apple scab and fire blight, suggesting a beneficial role of 3-hydroxylated DHCs (Hutabarat et al., 2016). A recent study attempted to discover genes involved in this key step of sieboldin biosynthesis, although the genes F3′HI and F3′HII cloned and characterised from M. × domestica genome did not accept phloretin, leading the authors to conclude that F3′Hs identified were unlikely involved in the 3-hydroxylation towards sieboldin biosynthesis (Weissensteiner et al., 2021). F3′HI and F3′HII, however, accepted naringenin, dihydrokaempferol and kaempferol, indicating a role in the well-studied 3′-hydroxylation of the B ring of flavonoids. The F3′H candidate cloned from wild Malus accessions in our study might share this role, which could explain the lack of 3-hydroxylase activity towards phloretin in S. cerevisiae. Our study describes for the first time an in vivo 3-hydroxylase activity of genes characterised from wild Malus genotypes that accumulate sieboldin as main DHC.
Conclusions
DHCs are specialised secondary metabolites that possess beneficial effects in human health, recently implicated in abiotic and biotic response of plants, as well as plant development. The biosynthesis and physiological regulation of DHCs, however, remains an active field of research. Most of the research has focused on discovering genes involved in phloridzin biosynthesis, the main DHC compound found in M. × domestica. Nevertheless, trilobatin and sieboldin are DHCs that dominate the phenolic profile of some Malus species. Aiming to gain a better understanding of the accumulation of DHCs and their physiological roles in Malus, our study contributes to elucidate the biosynthetic pathway by combining transcriptomic, metabolite and functional analyses. The key 3-hydroxylation converting the precursor phloretin into 3-hydroxyphloretin is the missing step that ultimately leads to sieboldin biosynthesis. Here, we selected candidate genes by RNA-Seq and de novo transcriptome assembly that were functionally assessed by metabolic engineering of S. cerevisiae. CYP98A195-196 from M. toringo and M. micromalus exerted 3-hydroxylase activity in yeast and proved to be an endoplasmic reticulum resident protein. Moreover, protein modelling and ligand docking evidenced key mutations that could explain the acceptance of phloretin to produce 3-hydroxyphloretin, which could be further assessed by site-directed mutagenesis in future studies. Importantly, this study identified genes from wild Malus relatives to domesticated apple that can lead to sieboldin accumulation in vivo, by co-expression with the 4′-O-glycosyltransferase PGT2. In addition, we developed an HRM marker analysis that correlated with the accumulation of sieboldin in wild and hybrid Malus accessions that could be used for selecting plant Malus material. Altogether, our work constitutes the first study reporting a key gene that could account for the missing 3-hydroxylation step involved in the biosynthesis of sieboldin, thus providing the basis for future establishment of genetic engineered lines assessing the physiological roles of this gene in plants.
Data availability statement
The data presented in the study are deposited in the NCBI GenBank repository, accession numbers OP616740 to OP616743, and Sequence Read Archive (SRA) repository, BioProject number PRJNA890040.
Author contributions
SMi and SMa conceived the idea and designed the experiments. AD and RE contributed to the discussion and experiment planning. SMi, SMa, and MM collected plant material. SMi carried out RNA-Seq, gene expression analysis and metabolic profiling. SMi and A-SK carried out yeast in vivo and in vitro assays. AA contributed to LC-MS/MS analysis and visualisation of results. A-SK and SMi were responsible for HRM marker analysis. SMi and JL conducted the de novo transcriptome assembly and functional annotation, as well as agroinfiltration, protoplast preparation and confocal imaging. SMi and SMa wrote the manuscript, with input from all authors. All authors contributed to the article and approved the submitted version.
Funding
The research has been funded by GECT Euregio Tirol–Südtirol–Trentino (IPN55) and the ADP project funded by the Autonomous Province of Trento. Authors SM, RE, and AD were employed by The New Zealand Institute for Plant and Food Research Limited. SMi was partly funded by the same institution in agreement with Fondazione Edmund Mach. The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Acknowledgments
We would like to acknowledge Urska Vhrovsek for technical and scientific support for metabolic profiling at Fondazione Edmund Mach Metabolomics unit. We thank Dr. David Nelson for assigning CYP codes to the cytochrome P450 protein sequences characterised in this study. We would also like to thank Davide Busetti for technical support and maintenance of the Malus germplasm collection at Fondazione Edmund Mach.
Conflict of interest
The author(s) declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1072765/full#supplementary-material
References
Agati, G., Azzarello, E., Pollastri, S., Tattini, M. (2012). Flavonoids as antioxidants in plants: Location and functional significance. Plant Sci. 196, 67–76. doi: 10.1016/j.plantsci.2012.07.014
Allen, G. C., Flores-Vergara, M., Krasynanski, S., Kumar, S., Thompson, ,. W. F. (2006). A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 1, 2320–2325. doi: 10.1038/nprot.2006.384
Biswal, B., Jena, B., Giri, A. K., Acharya, L. (2021). De novo transcriptome and tissue specific expression analysis of genes associated with biosynthesis of secondary metabolites in Operculina turpethum (L.). Sci. Rep. 11, 1–15. doi: 10.1038/s41598-021-01906-y
Busatto, N., Matsumoto, D., Tadiello, A., Vrhovsek, U., Costa, F. (2019). Multifaceted analyses disclose the role of fruit size and skin-russeting in the accumulation pattern of phenolic compounds in apple. PloS One 14, 1–20. doi: 10.1371/journal.pone.0219354
Caputi, L., Malnoy, M., Goremykin, V., Nikiforova, S., Martens, S. (2012). A genome-wide phylogenetic reconstruction of family 1 UDP-glycosyltransferases revealed the expansion of the family during the adaptation of plants to life on land. Plant J. 69, 1030–1042. doi: 10.1111/j.1365-313X.2011.04853.x
Chen, X., Li, S., Zhang, D., Han, M., Jin, X., Zhao, C., et al. (2019). Sequencing of a wild apple (Malus baccata) genome unravels the differences between cultivated and wild apple species regarding disease resistance and cold tolerance. G3 Genes Genomes Genet. 9, 2051–2060. doi: 10.1534/g3.119.400245
Chhangawala, S., Rudy, G., Mason, C. E., Rosenfeld, J. A. (2015). The impact of read length on quantification of differentially expressed genes and splice junction detection. Genome Biol. 16, 1–10. doi: 10.1186/s13059-015-0697-y
Conesa, A., Madrigal, P., Tarazona, S., Gomez-Cabrero, D., Cervera, A., McPherson, A., et al. (2016). A survey of best practices for RNA-seq data analysis. Genome Biol. 17, 1–19. doi: 10.1186/s13059-016-0881-8
Daccord, N., Celton, J. M., Linsmith, G., Becker, C., Choisne, N., Schijlen, E., et al. (2017). High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 49, 1099–1106. doi: 10.1038/ng.3886
Dugé de Bernonville, T., Guyot, S., Paulin, J. P., Gaucher, M., Loufrani, L., Henrion, D., et al. (2010). Dihydrochalcones: Implication in resistance to oxidative stress and bioactivities against advanced glycation end-products and vasoconstriction. Phytochemistry 71, 443–452. doi: 10.1016/j.phytochem.2009.11.004
Earley, K. W., Haag, J. R., Pontes, O., Opper, K., Juehne, T., Song, K., et al. (2006). Gateway-compatible vectors for plant functional genomics and proteomics. Plant J. 45, 616–629. doi: 10.1111/j.1365-313X.2005.02617.x
Eberle, D., Ullmann, P., Werck-Reichhart, D., Petersen, M. (2009). cDNA cloning and functional characterisation of CYP98A14 and NADPH:cytochrome P450 reductase from Coleus blumei involved in rosmarinic acid biosynthesis. Plant Mol. Biol. 69, 239–253. doi: 10.1007/s11103-008-9420-7
Ehrenkranz, J. R. L., Lewis, N. G., Kahn, C. R., Roth, J. (2005). Phlorizin: A review. Diabetes Metab. Res. Rev. 21, 31–38. doi: 10.1002/dmrr.532
Eichenberger, M., Lehka, B. J., Folly, C., Fischer, D., Martens, S., Simón, E., et al. (2017). Metabolic engineering of Saccharomyces cerevisiae for de novo production of dihydrochalcones with known antioxidant, antidiabetic, and sweet tasting properties. Metab. Eng. 39, 80–89. doi: 10.1016/j.ymben.2016.10.019
Gaucher, M., Dugé De Bernonville, T., Lohou, D., Guyot, S., Guillemette, T., Brisset, M. N., et al. (2013). Histolocalization and physico-chemical characterization of dihydrochalcones: Insight into the role of apple major flavonoids. Phytochemistry 90, 78–89. doi: 10.1016/j.phytochem.2013.02.009
Gietz, R. D., Schiestl, R. H. (2007). Quick and easy yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 35–37. doi: 10.1038/nprot.2007.14
Gosch, C., Flachowsky, H., Halbwirth, H., Thill, J., Mjka-Wittmann, R., Treutter, D., et al. (2012). Substrate specificity and contribution of the glycosyltransferase UGT71A15 to phloridzin biosynthesis. Trees - Struct. Funct. 26, 259–271. doi: 10.1007/s00468-011-0669-0
Gosch, C., Halbwirth, H., Schneider, B., Hölscher, D., Stich, K. (2010a). Cloning and heterologous expression of glycosyltransferases from Malus × domestica and Pyrus communis, which convert phloretin to phloretin 2′-O-glucoside (phloridzin). Plant Sci. 178, 299–306. doi: 10.1016/j.plantsci.2009.12.009
Gosch, C., Halbwirth, H., Stich, K. (2010b). Phloridzin: Biosynthesis, distribution and physiological relevance in plants. Phytochemistry 71, 838–843. doi: 10.1016/j.phytochem.2010.03.003
Gotoh, O. (1992). Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J. Biol. Chem. 267, 83–90. doi: 10.1016/s0021-9258(18)48462-1
Grabherr, M. G., Brian, J. H., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2013). Trinity: reconstructing a full-length transcriptome without a genome from RNA-seq data. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883.Trinity
Gutierrez, B. L., Arro, J., Zhong, G. Y., Brown, S. K. (2018a). Linkage and association analysis of dihydrochalcones phloridzin, sieboldin, and trilobatin in Malus. Tree Genet. Genomes 14. doi: 10.1007/s11295-018-1304-7
Gutierrez, B. L., Zhong, G. Y., Brown, S. K. (2018b). Genetic diversity of dihydrochalcone content in malus germplasm. Genet. Resour. Crop Evol. 65, 1485–1502. doi: 10.1007/s10722-018-0632-7
Hamberger, B., Bak, S. (2013). Plant P450s as versatile drivers for evolution of species-specific chemical diversity. Philos. Trans. R. Soc B Biol. Sci. 368. doi: 10.1098/rstb.2012.0426
Hausjell, J., Halbwirth, H., Spadiut, O. (2018). Recombinant production of eukaryotic cytochrome P450s in microbial cell factories. Biosci. Rep. 38, 1–13. doi: 10.1042/BSR20171290
Hofmann, A., Wittenmayer, L., Arnold, G., Schieber, A., Merbach, W. (2009). Root exudation of phloridzin by apple seedlings (Malus × domestica borkh.) with symptoms of apple replant disease. J. Appl. Bot. Food Qual. 82, 193–198.
Hölzer, M., Marz, M. (2019). De novo transcriptome assembly: A comprehensive cross-species comparison of short-read RNA-seq assemblers. Gigascience 8, 1–16. doi: 10.1093/gigascience/giz039
Hrubá, M., Baxant, J., Čížková, H., Smutná, V., Kovařík, F., Ševčík, R., et al. (2021). Phloridzin as a marker for evaluation of fruit product’s authenticity. Czech J. Food Sci. 39, 49–57. doi: 10.17221/239/2020-CJFS
Hsu, C.-T., Lee, W.-C., Cheng, Y.-J., Yuan, Y.-H., Wu, F.-H., Lin, C.-S. (2021). Genome Editing and Protoplast Regeneration to Study Plant–Pathogen Interactions in the Model Plant Nicotiana benthamiana. Front. Genome Ed. 2, 627803. doi: 10.3389/fgeed.2020.627803
Hutabarat, O. S., Flachowsky, H., Regos, I., Miosic, S., Kaufmann, C., Faramarzi, S., et al. (2016). Transgenic apple plants overexpressing the chalcone 3-hydroxylase gene of Cosmos sulphureus show increased levels of 3-hydroxyphloridzin and reduced susceptibility to apple scab and fire blight. Planta 243, 1213–1224. doi: 10.1007/s00425-016-2475-9
Ibdah, M., Martens, S., Gang, D. R. (2018). Biosynthetic pathway and metabolic engineering of plant dihydrochalcones. J. Agric. Food Chem. 66, 2273–2280. doi: 10.1021/acs.jafc.7b04445
Ishii, J., Kondo, T., Makino, H., Ogura, A., Matsuda, F., Kondo, A. (2014). Three gene expression vector sets for concurrently expressing multiple genes in Saccharomyces cerevisiae. FEMS Yeast Res. 14, 399–411. doi: 10.1111/1567-1364.12138
Janssen, B. J., Thodey, K., Schaffer, R. J., Alba, R., Balakrishnan, L., Bishop, R., et al. (2008). Global gene expression analysis of apple fruit development from the floral bud to ripe fruit. BMC Plant Biol. 8, 1–29. doi: 10.1186/1471-2229-8-16
Japelaghi, R. H., Haddad, R., Valizadeh, M., Uliaie, E. D., Javaran, ,. M. J. (2018). High-efficiency agrobacterium-mediated transformation of tobacco (Nicotiana tabacum). J. Plant Mol. Breed 6, 38–50. doi: 10.22058/JPMB.2019.92266.1170
Jugdé, H., Nguy, D., Moller, I., Cooney, J. M., Atkinson, R. G. (2008). Isolation and characterization of a novel glycosyltransferase that converts phloretin to phlorizin, a potent antioxidant in apple. FEBS J. 275, 3804–3814. doi: 10.1111/j.1742-4658.2008.06526.x
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi: 10.1038/s41586-021-03819-2
Kahn, R. A., Fahrendorf, T., Halkier, B. A., Møller, B. L. (1999). Substrate specificity of the cytochrome P450 enzymes CYP79A1 and CYP71E1 involved in the biosynthesis of the cyanogenic glucoside dhurrin in Sorghum bicolor (L.) moench. Arch. Biochem. Biophys. 363, 9–18. doi: 10.1006/abbi.1998.1068
Karamat, F., Olry, A., Doerper, S., Vialart, G., Ullmann, P., Werck-Reichhart, D., et al. (2012). CYP98A22, a phenolic ester 3’-hydroxylase specialized in the synthesis of chlorogenic acid, as a new tool for enhancing the furanocoumarin concentration in ruta graveolens. BMC Plant Biol. 12. doi: 10.1186/1471-2229-12-152
Kumar, S., Molloy, C., Hunt, M., Hong Deng, C., Wiedow, C., Andre, C., et al. (2022). GWAS provides new insights into the genetic mechanisms of phytochemicals production and red skin colour in apple. Hortic. Res. doi: 10.1093/hr/uhac218
Łata, B., Trampczynska, A., Paczesna, J. (2009). Cultivar variation in apple peel and whole fruit phenolic composition. Sci. Hortic. (Amsterdam) 121, 176–181. doi: 10.1016/j.scienta.2009.01.038
Lattanzio, V., Di Venere, D., Linsalata, V., Bertolini, P., Ippolito, A., Salerno, M. (2001). Low temperature metabolism of apple phenolics and quiescence of Phlyctaena vagabunda. J. Agric. Food Chem. 49, 5817–5821. doi: 10.1021/jf010255b
Liao, Y., Smyth, G. K., Shi, W. (2014). FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi: 10.1093/bioinformatics/btt656
Liu, N., Zhang, N., Zhang, S., Zhang, L., Liu, Q. (2021). Phloretin inhibited the pathogenicity and virulence factors against Candida albicans. Bioengineered 12, 2420–2431. doi: 10.1080/21655979.2021.1933824
Li, Z., Wang, L., He, J., Li, X., Hou, N., Guo, J., et al. (2022). Chromosome-scale reference genome provides insights into the genetic origin and grafting-mediated stress tolerance of Malus prunifolia. Plant Biotechnol. J. 20, 1015–1017. doi: 10.1111/pbi.13817
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21. doi: 10.1186/s13059-014-0550-8
Madritsch, S., Burg, A., Sehr, E. M. (2021). Comparing de novo transcriptome assembly tools in di- and autotetraploid non-model plant species. BMC Bioinf. 22, 1–17. doi: 10.1186/s12859-021-04078-8
Matsuno, M., Compagnon, V., Schoch, G. A., Schmitt, M., Debayle, D., Bassard, J. E., et al. (2009). Evolution of a novel phenolic pathway for pollen development. Science 80) 325, 1688–1692. doi: 10.1126/science.1174095
Morant, M., Schoch, G. A., Ullmann, P., Ertunç, T., Little, D., Olsen, C. E., et al. (2007). Catalytic activity, duplication and evolution of the CYP98 cytochrome P450 family in wheat. Plant Mol. Biol. 63, 1–19. doi: 10.1007/s11103-006-9028-8
Nelson, D. R. (2006). “Cytochrome P450 nomenclature 2004,” in Cytochrome P450 protocols. Eds. Phillips, I. R., Shephard, E. A. (Totowa: NJ: Humana Press), 1–10. doi: 10.1385/1-59259-998-2:1
Nelson, D. R. (2009). The cytochrome P450 homepage. Hum. Genomics 4, 59–65. doi: 10.1186/1479-7364-4-1-59
Nelson, B. K., Cai, X., Nebenführ, A. (2007). A multicolored set of in vivo organelle markers for co-localization studies in Arabidopsis and other plants. Plant J. 51, 1126–1136. doi: 10.1111/j.1365-313X.2007.03212.x
Oleszek, M., Pecio, Ł., Kozachok, S., Lachowska-Filipiuk, Ż., Oszust, K., Frąc, M. (2019). Phytochemicals of apple pomace as prospect bio-fungicide agents against mycotoxigenic fungal species–in vitro experiments. Toxins (Basel) 11. doi: 10.3390/toxins11060361
Pacheco, J., Vilanova, S., Grillo-Risco, R., García-García, F., Prohens, J., Gramazio, P. (2021). De novo transcriptome assembly and comprehensive annotation of two tree tomato cultivars (Solanum betaceum cav.) with different fruit color. Horticulturae 7. doi: 10.3390/horticulturae7110431
Panche, A. N., Diwan, A. D., Chandra, S. R. (2016). Flavonoids: An overview. J. Nutr. Sci. 5. doi: 10.1017/jns.2016.41
Petkovsek, M. M., Stampar, F., Veberic, R. (2009). Seasonal changes in phenolic compounds in the leaves of scab-resistant and susceptible apple cultivars. Can. J. Plant Sci. 89, 745–753. doi: 10.4141/CJPS08202
Raghavan, V., Kraft, L., Mesny, F., Rigerte, L. (2022). A simple guide to de novo transcriptome assembly and annotation. Brief Bioinform. 23, 1–30. doi: 10.1093/bib/bbab563
Rivière, C. (2016). Dihydrochalcones: Occurrence in the plant kingdom, chemistry and biological activities. doi: 10.1016/B978-0-444-63932-5.00007-3
Romsuk, J., Yasumoto, S., Seki, H., Fukushima, E. O., Muranaka, T. (2022). Identification of key amino acid residues toward improving the catalytic activity and substrate specificity of plant-derived cytochrome P450 monooxygenases CYP716A subfamily enzyme for triterpenoid production in Saccharomyces cerevisiae. Front. Bioeng. Biotechnol. 10. doi: 10.3389/fbioe.2022.955650
Schlangen, K., Miosic, S., Thill, J., Halbwirth, H. (2010). Cloning, functional expression, and characterization of a chalcone 3-hydroxylase from Cosmos sulphureus. J. Exp. Bot. 61, 3451–3459. doi: 10.1093/jxb/erq169
Schoch, G., Goepfert, S., Morant, M., Hehn, A., Meyer, D., Ullmann, P., et al. (2001). CYP98A3 from Arabidopsis thaliana is a 3′-hydroxylase of phenolic esters, a missing link in the phenylpropanoid pathway. J. Biol. Chem. 276, 36566–36574. doi: 10.1074/jbc.M104047200
Seitz, C., Ameres, S., Forkmann, G. (2007). Identification of the molecular basis for the functional difference between flavonoid 3′-hydroxylase and flavonoid 3′,5′-hydroxylase. FEBS Lett. 581, 3429–3434. doi: 10.1016/j.febslet.2007.06.045
Seitz, C., Eder, C., Deiml, B., Kellner, S., Martens, S., Forkmann, G. (2006). Cloning, functional identification and sequence analysis of flavonoid 3′-hydroxylase and flavonoid 3′,5′-hydroxylase cDNAs reveals independent evolution of flavonoid 3′,5′-hydroxylase in the Asteraceae family. Plant Mol. Biol. 61, 365–381. doi: 10.1007/s11103-006-0012-0
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Sparkes, I. A., Runions, J., Kearns, A., Hawes, C. (2006). Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat. Protoc. 1, 2019–2025. doi: 10.1038/nprot.2006.286
Spinelli, F. R., Dutra, S. V., Carnieli, G., Leonardelli, S., Drehmer, A. P., Vanderlinde, R. (2016). Detection of addition of apple juice in purple grape juice. Food Control 69, 1–4. doi: 10.1016/j.foodcont.2016.04.005
Stangl, V., Lorenz, M., Ludwig, A., Grimbo, N., Guether, C., Sanad, W., et al. (2005). The flavonoid phloretin suppresses stimulated expression of endothelial adhesion molecules and reduces activation of human platelets. J. Nutr. 135, 172–178. doi: 10.1093/jn/135.2.172
Sun, X., Jiao, C., Schwaninger, H., Chao, C. T., Ma, Y., Duan, N., et al. (2020). Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nat. Genet. 52, 1423–1432. doi: 10.1038/s41588-020-00723-9
Szliszka, E., Czuba, Z. P., Mazur, B., Paradysz, A., Krol, W. (2010). Chalcones and dihydrochalcones augment TRAIL-mediated apoptosis in prostate cancer cells. Molecules 15, 5336–5353. doi: 10.3390/molecules15085336
Thole, V., Bassard, J. E., Ramírez-González, R., Trick, M., Ghasemi Afshar, B., Breitel, D., et al. (2019). RNA-Seq, de novo transcriptome assembly and flavonoid gene analysis in 13 wild and cultivated berry fruit species with high content of phenolics. BMC Genomics 20, 1–23. doi: 10.1186/s12864-019-6183-2
Tomás-Barberán, F. A., Borrego, F., Ferreres, F., Lindley, M. G. (1995). Stability of the intense sweetener neohesperidine dihydrochalcone in blackcurrant jams. Food Chem. 52, 263–265. doi: 10.1016/0308-8146(95)92821-Z
Tomás-Barberán, F. A., Clifford, M. N. (2000). Flavanones, chalcones and dihydrochalcones - nature, occurrence and dietary burden. J. Sci. Food Agric. 80, 1073–1080. doi: 10.1002/(SICI)1097-0010(20000515)80:7<1073::AID-JSFA568>3.0.CO;2-B
Tsao, R., Yang, R., Xie, S., Sockovie, E., Khanizadeh, S. (2005). Which polyphenolic compounds contribute to the total antioxidant activities of apple? J. Agric. Food Chem. 53, 4989–4995. doi: 10.1021/jf048289h
Velasco, R., Zharkikh, A., Affourtit, J., Dhingra, A., Cestaro, A., Kalyanaraman, A., et al. (2010). The genome of the domesticated apple (Malus × domestica borkh.). Nat. Genet. 42, 833–839. doi: 10.1038/ng.654
Viet, M. H., Chen, C. Y., Hu, C. K., Chen, Y. R., Li, M. S. (2013). Discovery of dihydrochalcone as potential lead for alzheimer’s disease: in silico and in vitro study. PloS One 8, 4–9. doi: 10.1371/journal.pone.0079151
Vrhovsek, U., Masuero, D., Gasperotti, M., Franceschi, P., Caputi, L., Viola, R., et al. (2012). A versatile targeted metabolomics method for the rapid quantification of multiple classes of phenolics in fruits and beverages. J. Agric. Food Chem. 60, 8831–8840. doi: 10.1021/jf2051569
Wang, H., Wang, Q., Liu, Y., Liao, X., Chu, H., Chang, H., et al. (2021). PCPD: Plant cytochrome P450 database and web-based tools for structural construction and ligand docking. Synth. Syst. Biotechnol. 6, 102–109. doi: 10.1016/j.synbio.2021.04.004
Wang, Y., Yauk, Y. K., Zhao, Q., Hamiaux, C., Xiao, Z., Gunaseelan, K., et al. (2020). Biosynthesis of the dihydrochalcone sweetener trilobatin requires phloretin glycosyltransferase2. Plant Physiol. 184, 738–752. doi: 10.1104/pp.20.00807
Weissensteiner, J., Molitor, C., Marinovic, S., Führer, L., Hassan, S. W., Hutabarat, O. S., et al. (2021). Molecular and enzymatic characterization of flavonoid 3′-hydroxylase of Malus × domestica. Plants 10, 1–11. doi: 10.3390/plants10091956
Winkel-Shirley, B. (2001). Flavonoid biosynthesis. a colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 126, 485–493. doi: 10.1104/pp.126.2.485
Wu, S., Lei, J., Chen, G., Chen, H., Cao, B., Chen, C. (2017). De novo transcriptome assembly of chinese kale and global expression analysis of genes involved in glucosinolate metabolism in multiple tissues. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00092
Yahyaa, M., Davidovich-Rikanati, R., Eyal, Y., Sheachter, A., Marzouk, S., Lewinsohn, E., et al. (2016). Identification and characterization of UDP-glucose:Phloretin 4′-o-glycosyltransferase from Malus × domestica borkh. Phytochemistry 130, 47–55. doi: 10.1016/j.phytochem.2016.06.004
Zhang, X. Z., Zhao, Y. B., Li, C. M., Chen, D. M., Wang, G. P., Chang, R. F., et al. (2007). Potential polyphenol markers of phase change in apple (Malus × domestica). J. Plant Physiol. 164, 574–580. doi: 10.1016/j.jplph.2006.03.011
Zhou, K., Hu, L., Li, P., Gong, X., Ma, F. (2017). Genome-wide identification of glycosyltransferases converting phloretin to phloridzin in Malus species. Plant Sci. 265, 131–145. doi: 10.1016/j.plantsci.2017.10.003
Keywords: dihydrochalcones, 3-hydroxyphloretin, polyphenols, biosynthesis, cytochrome P450, 3-hydroxylation, Malus species
Citation: Miranda S, Lagrèze J, Knoll A-S, Angeli A, Espley RV, Dare AP, Malnoy M and Martens S (2022) De novo transcriptome assembly and functional analysis reveal a dihydrochalcone 3-hydroxylase(DHC3H) of wild Malus species that produces sieboldin in vivo. Front. Plant Sci. 13:1072765. doi: 10.3389/fpls.2022.1072765
Received: 17 October 2022; Accepted: 23 November 2022;
Published: 16 December 2022.
Edited by:
Teresa Docimo, Institute of Bioscience and Bioresources (CNR), ItalyReviewed by:
Lorenzo Caputi, Max Planck Institute for Chemical Ecology, GermanyJIabao Ye, Yangtze University, China
Yi Wang, Beijing Botanical Garden (CAS), China
Yaqiong Wu, Jiangsu Province and Chinese Academy of Sciences, China
Jingwen Zhou, Jiangnan University, China
Copyright © 2022 Miranda, Lagrèze, Knoll, Angeli, Espley, Dare, Malnoy and Martens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simón Miranda, simondavid.mirandachavez@fmach.it; Stefan Martens, stefan.martens@fmach.it