 Zhongyu Zhou1
Zhongyu Zhou1 Jun Qian
Jun Qian Linchun Shi
Linchun Shi Baozhong Duan
Baozhong Duan- 1College of Pharmaceutical Science, Dali University, Dali, China
- 2College of Life Science, Northeast Forestry University, Harbin, China
- 3Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing, China
Isodon rubescens (Hemsley) H. Hara is the source of Donglingcao under the monograph Rabdosiae Rubescentis Herba in Chinese Pharmacopoeia. In the local marketplace, this medicine can be accidentally contaminated, deliberately substituted, or mixed with other related species. The contaminants of herbal products are a threat to consumer safety. Due to the scarcity of genetic information on Isodon plants, more molecular markers are needed to avoid misidentification. In the present study, the complete chloroplast (cp) genome of seven species of Isodon was sequenced, de novo assembled and characterized. The cp genomes of these species universally exhibited a conserved quadripartite structure, i.e., two inverted repeats (IRs) containing most of the ribosomal RNA genes and two unique regions (large single copy and small single copy). Moreover, the genome structure, codon usage, and repeat sequences were highly conserved and showed similarities among the seven species. Five highly variable regions (trnS-GCU-trnT-CGU, atpH-atpI, trnE-UUC-trnT-GGU, ndhC-trnM-CAU, and rps15-ycf1) might be potential molecular markers for identifying I. rubescens and its contaminants. These findings provide valuable information for further species identification, evolution, and phylogenetic research of Isodon.
Introduction
Isodon rubescens (Hemsley) H. Hara belongs to the family Lamiaceae (Liu et al., 2017), which is listed in the Chinese Pharmacopoeia, and the Chinese name is “Donglingcao” (Chinese Pharmacopoeia Commission, 2020). The Rabdosiae Rubescentis Herba has crucial medicinal value in eliminating inflammation, reducing sore throats, and treating malignant tumors (Xue et al., 2020; Guan et al., 2021). The previous survey has revealed that I. rubescens is generally contaminated with common adulterants, such as I. inflexus (Thunb.) Kudô, I. eriocalyx (Dunn) Kudô, I. excisus (Maxim.) Kudô, I. lophanthoides (Buch.-Ham. ex D.Don) H.Hara, I. coetsa (Buch.-Ham. ex D.Don) Kudô, and I. japonicus (Burm.f.) H.Hara (Su et al., 2007; Xia et al., 2013; Ge et al., 2022). These adulterants are usually of poor quality and some might even be toxic (Xia et al., 2013; Duan et al., 2018). As the morphology of these species is similar, interchangeable, and indistinguishable, the identification of these species remains somewhat controversial, which may affect their safety and effectiveness in clinical use (Lazarski et al., 2014; Lin et al., 2019). Therefore, it is imperative to develop a method for accurately identifying I. rubescens and its common adulterants.
With the rapid development of molecular technology in recent years, molecular identification has made significant progress in Chinese medicine, especially molecular markers, which involve sequencing specific sections of the genome to identify differences between individuals of different species or populations (Roman and Houston, 2020). Recent studies have shown high levels of genetic variability within species of Isodon and an associated lack of phylogenetic resolution between different species (Zhong et al., 2010; Chen et al., 2021). Universal DNA markers, such as ITS, psbA-trnH, trnD-trnT, rpl32-trnL, and ETS have been used to identify I. rubescens and its related taxa (Xia et al., 2013; Yu et al., 2014; Chen et al., 2021). Moreover, according to Harris and Klooster (2011), they found that 11 microsatellite loci amplify reliably and are sufficiently variable for studying population genetics in I. rubescens. However, some common adulterants were not included in these studies. Therefore, more scientific and accurate identification methods must be developed. The chloroplast (cp) is an essential organelle that plays a crucial role in plant photosynthesis and biochemical processes (Bendich, 2004). Compared with the gene fragments, the cp genome is relatively conserved and slightly varied (Drouin et al., 2008; Ferrarini et al., 2013), which has been widely used for identifying Paris, Polygonatum, and its contaminants (Kawabe and Miyashita, 2003; Jiang et al., 2022; Wang et al., 2022). Recently, although the complete plastid genomes of I. rubescens has reported by Lian et al. (2022) and Yue et al. (2021), the focus of these papers was to compare the intraspecific variation or characterize one genome information. However, the use of cp genomes for comparing Isodon species with their common adulterants has not been reported.
Our study aims to: (i) contribute new fully-sequenced cp genomes in Isodon and improve the understanding of the overall structure of these genomes, (ii) perform comparative analyses and elucidate the phylogenetic evolution of Isodon cp genomes, and (iii) screen molecular markers to differentiate I. rubescens and its adulterants. In the current work, the complete cp genomes of seven Isodon species were sequenced, de novo assembled, and annotated. These genomes were then used in a comparative analysis of genome structure and evolution relationships. The data acquired in this study increase the genomic resources available for the Isodon genus and provide valuable information support for the phylogenetic analysis and identification of the Isodon genus, as well as safe medical applications of I. rubescens. This study is the first comprehensive research on identifying I. rubescens and its adulterants based on the cp genomes.
Materials and methods
Plant and DNA resources
The fresh, healthy leaves for seven species of I. rubescens and its common adulterants, including I. inflexus, I. eriocalyx, I. excisus, I. lophanthoides, I. coetsa, and I. japonicus, were collected from the Germplasm Resource Garden (Yunnan, China, 24°49′55″N, 102°48′58″E), Kunming Zhifen Biotechnology Co., Ltd. One individual sample of approximately 1.0 g of fresh leaves per plant species was gathered and stored in an ice-filled cooler or refrigerator (4°C) until DNA extractions could be performed. Professor Baozhong Duan authenticated the specimens, and the detailed sample information is available in Supplementary Table S1 and Figure S1. The voucher specimens were deposited in the Dali University herbarium. Genomic DNA was extracted from tissue samples using the Plant Genomic DNA kit (Tiangen, Beijing, China) following the manufacturer’s protocol. The extracted DNA was quantified on high-sensitivity Qubit 4.0 fluorometry (Life Technologies, Inc.), and all PCR products were examined for the presence of amplified products in agarose gels.
DNA sequencing, assembly, and annotation
For sequencing library preparation, we used thirty microlitres of high-quality (>100 ng/μL) DNA per individual. All libraries were sequenced on the Illumina NovaSeq system (Illumina, San Diego, CA). Paired-end sequence reads were trimmed to remove low-quality bases and adapter sequences in the Toolkit_v2.3.3 software. The cp genomes were assembled by GetOrganelle v.1.6.4, exploiting Bowtie2 v.2.4.4, SPAdes v.3.13.0, and Blast v.2.5.0 as dependencies (Jin et al., 2019). After assembly, two online annotation tools, CpGAVAS2 and GeSeq were used to annotate the circular cp genomes (Wick et al., 2015), and the annotated cp genome sequences were submitted to the GenBank database of the National Center for Biotechnology Information (NCBI) (Table 1). Gene maps of the cp genomes were produced with the online IRscope (https://irscope.shinyapps.io/Chloroplot/).
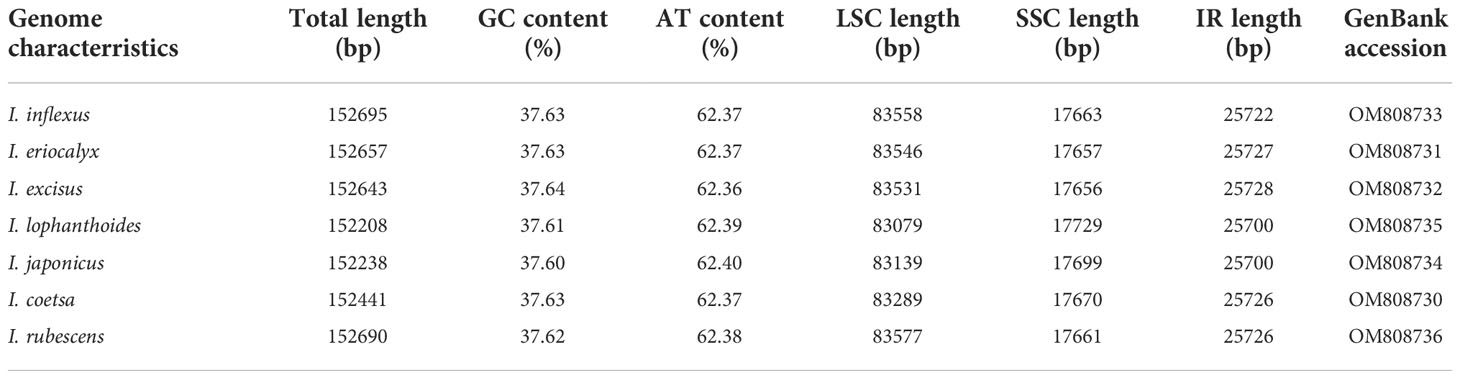
Table 1 Information of Isodon cp genome features.
Repeat analysis
The GC content was analyzed using the Geneious 9.0.2 software (Kearse et al., 2012). Four kinds of the dispersed repeat sequence, including Forward (F), Reverse (R), Palindromic (P), and Complementary (C), were detected using the REPuter program (https://bibiserv.cebitec.uni-bielefeld.de/reputer/) (Kurtz, 2001). The criteria for repeat determination include a minimum repeat size of 20 bp with a similarity between repeat pairs of 90% by putting edit value 3. Furthermore, MISA software (http://pgrc.ipk-gatersleben.de/misa/) was used to evaluate the simple sequence repeats (SSRs) with the parameters of ‘10’ for mono, ‘5’ for di-, ‘4’ for tri-, and ‘3’ for tetra-, penta-, and hexanucleotide motifs (Beier et al., 2017).
Comparative and phylogenetic analyses
Relative synonymous codon usage (RSCU) and codon usage values were analyzed by CodonW v.1.4.2. Moreover, the RSCU values were shown in a heatmap by Tbtools (Chen et al., 2020). The contraction and expansion of IR regions at the junctions were visualized using the online IRscope (https://irscope.shinyapps.io/irapp/) (Amiryousefi et al., 2018). The mVISTA program in Shuffle LAGAN mode was used to compare the cp genomes of seven species of Isodon (Frazer et al., 2004), using the annotation information of I. serra (GenBank NC064127) as a reference. Additionally, the nucleotide variability (Pi) across the cp genome sequences was assessed using DnaSP v.6.12.03, with a window length of 600 sites and a step size of 200 sites (Rozas et al., 2017). A value of Pi higher than 0.014 was recommended as mutational hotspots (Cui et al., 2019a). In phylogenetic analyses, 36 species, including 29 species downloaded from NCBI (Table S2), were used to infer the phylogeny. Simultaneously, two species, Scrophularia dentata (GenBank NC036942) and S. henryi (GenBank NC036943) were used as outgroups (Chase et al., 2016). All these sequences were aligned using MAFFT, and alignments were checked manually. The Maximum-likelihood (ML) tree was reconstructed with an IQtree using default parameters of 1000 iterations, 1000 replications, and best-fit model selection (Nguyen et al., 2015).
Results and discussion
Genome structure
The cp genome size and gene content of seven Isodon species are listed in Table 1. All the genome sizes were similar to that of the reference genome, i.e., around 150 kb. I. inflexu had the largest genome, with a size of 152,701 bp, and I. japonicus had the smallest genome, with a size of 152,208 bp. Moreover, the length of large single copy (LSC) regions ranged from 83,079 bp (I. lophanthoides) to 83,577 bp (I. rubescens), small single copy (SSC) regions ranged from 17,656 bp (I. excisus) to 17,729 bp (I. lophanthoides), and IRa and IRb regions ranged from 25,700 bp (I. lophanthoides and I. japonicus) to 25,728 bp (I. excisus). In addition, the cp genomes of Isodon contained 132 genes, including 88 protein-coding genes, 8 rRNA genes, and 36 tRNA genes, 18 of which are repeated as members of IR regions (Figure 1), which were congruent and largely concordant with recent studies of Isodon (Yue et al., 2021; Lian et al., 2022). It is worth noting that the chIB, chIL, and ycf68 were lost during evolution, typically in most angiosperms (Millen et al., 2001). Meanwhile, the number and types of introns were similar among the seven Isodon species (Table S3). Each of the 18 genes contained one intron, including trnA-UGC (×2), trnI-GAU (×2), rpl2 (×2), and ndhB (×2) were located in the IR, and the genes (trnK-UUU, rps16, trnT-CGU, atpF, rpoC1, trnL-UAA, petB, petD, and rpl16) were located in the LSC, and the ndhA was the only present in the SSC region. Furthermore, ycf3 and clpP have two introns, respectively, consistent with previous genetic studies (Jiang et al., 2022). The GC content ranged from 37.60% to 37.64% and varied among the different regions. The findings were identical to those of other Isodon species (Yue et al., 2021), which is not unexpected, given that the angiosperms possess the highly conserved cp character at the genus level (Meng et al., 2018; Cui et al., 2019b; Shahzadi et al., 2020).
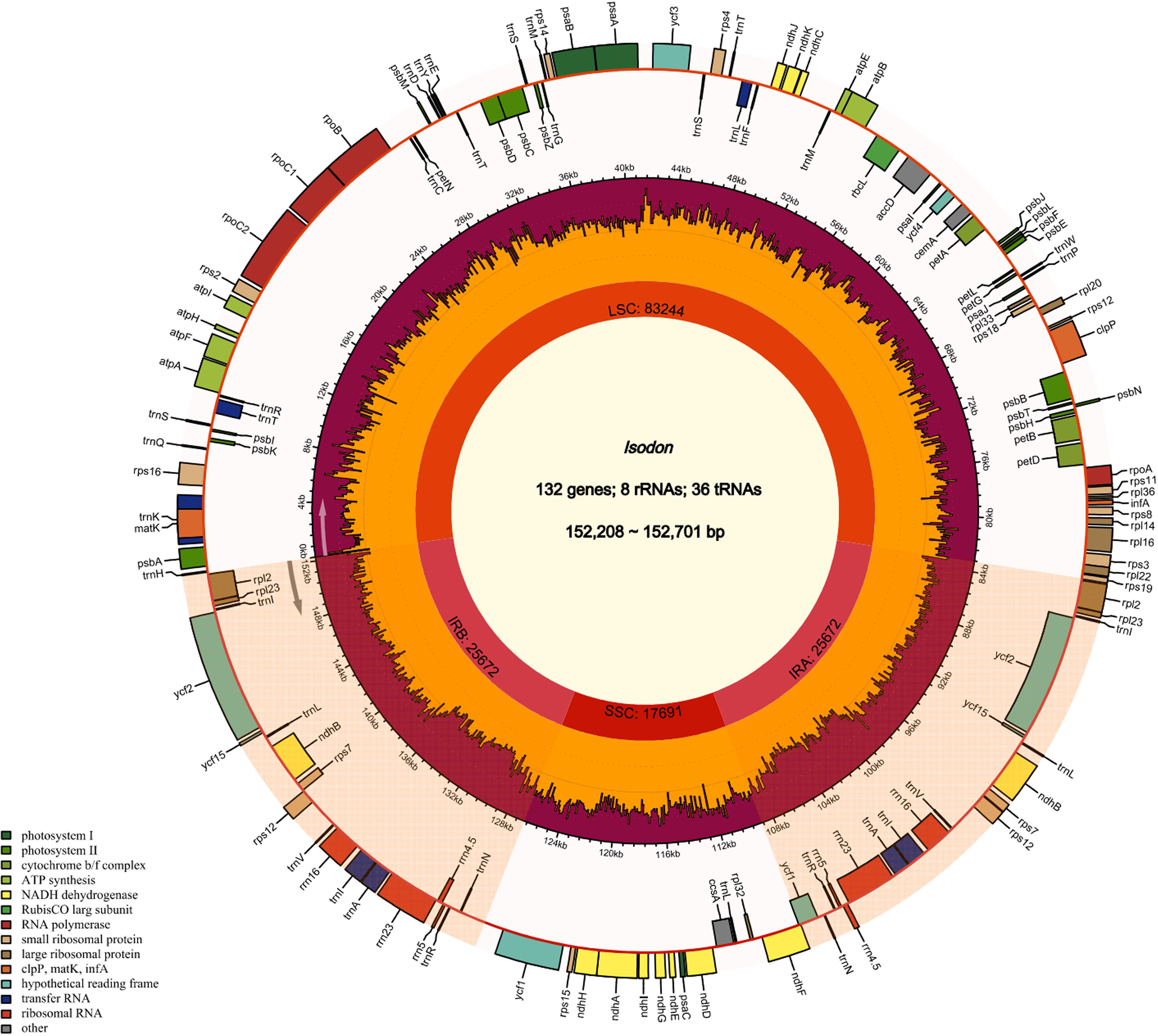
Figure 1 Cp genome map of Isodon.
Codon usage bias of cp genomes
The analyses of relative synonymous codon usage (RSCU) provide information about the encoding frequency of codons for an amino acid. As shown in Table S4, the results of RSCU revealed that the cp protein sequences encoded 21 amino acids, and 30 codons were used frequently in Isodon species, consistent with recent codon usage studies (Lian et al., 2022). Moreover, amino acid frequency analyses confirmed the highest frequency of leucine and isoleucine, whereas cysteine was a rare amino acid (Table S4), which was supported by other researchers based on codon usage bias (Lian et al., 2022). Notably, the use of start codon AUG for methionine and UGG for tryptophan in the Isodon genus showed no codon usage bias, consistent with previous reports of Isodon (Lian et al., 2022). In general, we found high similarities in codon usage and amino acid frequency among the seven species of Isodon. Furthermore, as illustrated in Figure 2, higher RSCU values (≥1) were found for codons with A or T at the 3’ position, which showed high encoding efficacy. Similar findings were reported for codon usage and amino acid frequency in the cp genomes of other angiosperms, which may be attributable to the high overall AT content in the cp genome (Kawabe and Miyashita, 2003; Jiang et al., 2022; Wang et al., 2022). In addition, the codon usage was similar in I. rubescens, I. lophanthoides, and I. japonicaus, whereas the codon usage of I. eriocalyx and I. excisus was relatively close to that of I. inflexus and I. coetsa (Figure 2).
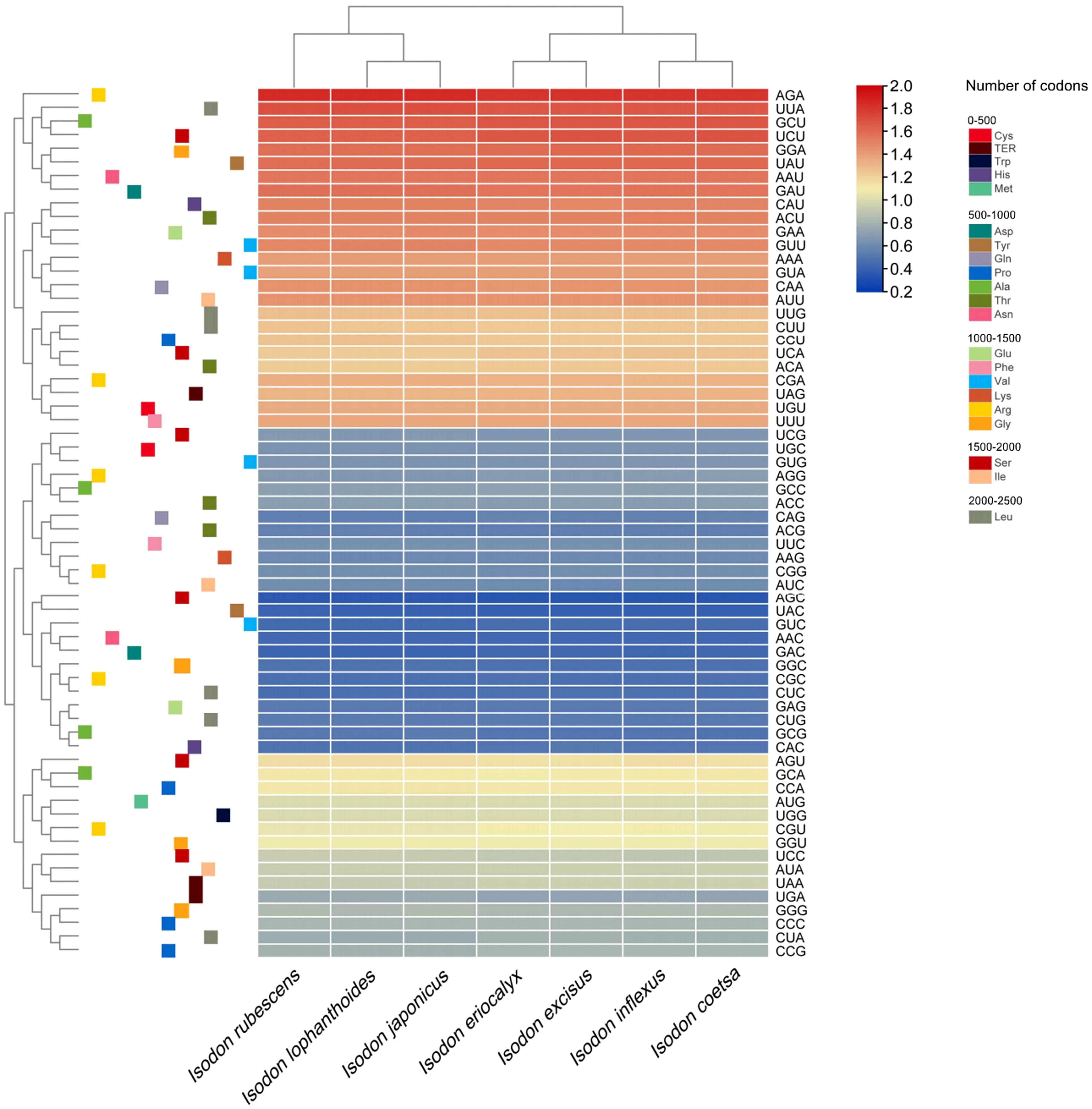
Figure 2 Heat map of the RSCU values among Isodon cp genome.
It was shown that the GC content of synonymous third codons positions (GC3s) was closely related to codon bias, which provided the foundation for assessing the codon usage pattern (Shang et al., 2011). In our study, the values of GC3s ranged from 29.4% to 29.5%, demonstrating that the genus Isodon had a greater preference for the A/U ending codons, which, along with the highly conserved GC content in seven Isodon cp genomes, suggest that natural selection had a profound impact on codon usage patterns (Zhang et al., 2018). In addition, the values for the effective number of codons ranged from 51.73 to 51.81, and both the codon adaptation index and frequency of optimal were less than 0.5. These findings indicated a slight bias of codon usage in the seven Isodon species.
Repeat analysis
Repetitive sequences play a crucial role in the rearrangement and stability of cp genomes (Rahemi et al., 2012; Kumar et al., 2015). A total of 51, 50, 50, 51, 50, 46, and 53 SSRs were identified in I. inflexus, I. eriocalyx, I. excisus, I. lophanthoides, I. japonicus, I. coetsa, and I. rubescens, respectively (Figure 3). More than half of SSRs (62.00% – 69.60%) were mononucleotide A/T motifs, which is consistent with the previous works in the cp genomes of angiosperms (Qian et al., 2013; Liang et al., 2019). The second was dinucleotide (11.65% – 18.00%) with a predominant motif of AT/TA, followed by tetranucleotide repeats (14.00% – 19.61%) with a predominant motif of AAAT/ATTT and AATC/ATTG, trinucleotide (1.89% – 2.17%), pentanucleotide (1.89% – 2.00%) with a predominant motif of AATAT/ATATT, AATAG/ATTCT. Hexanucleotides (1.96% – 4.00%) were absent in the cp genomes of I. eriocalyx, I. coetsa, and I. rubescens. This result was consistent with previous findings that most SSRs include mono- and dinucleotide repeats, while tri-, tetra-, penta-, and hexanucleotide repeat sequences exhibit lower frequencies (Dong et al., 2018).
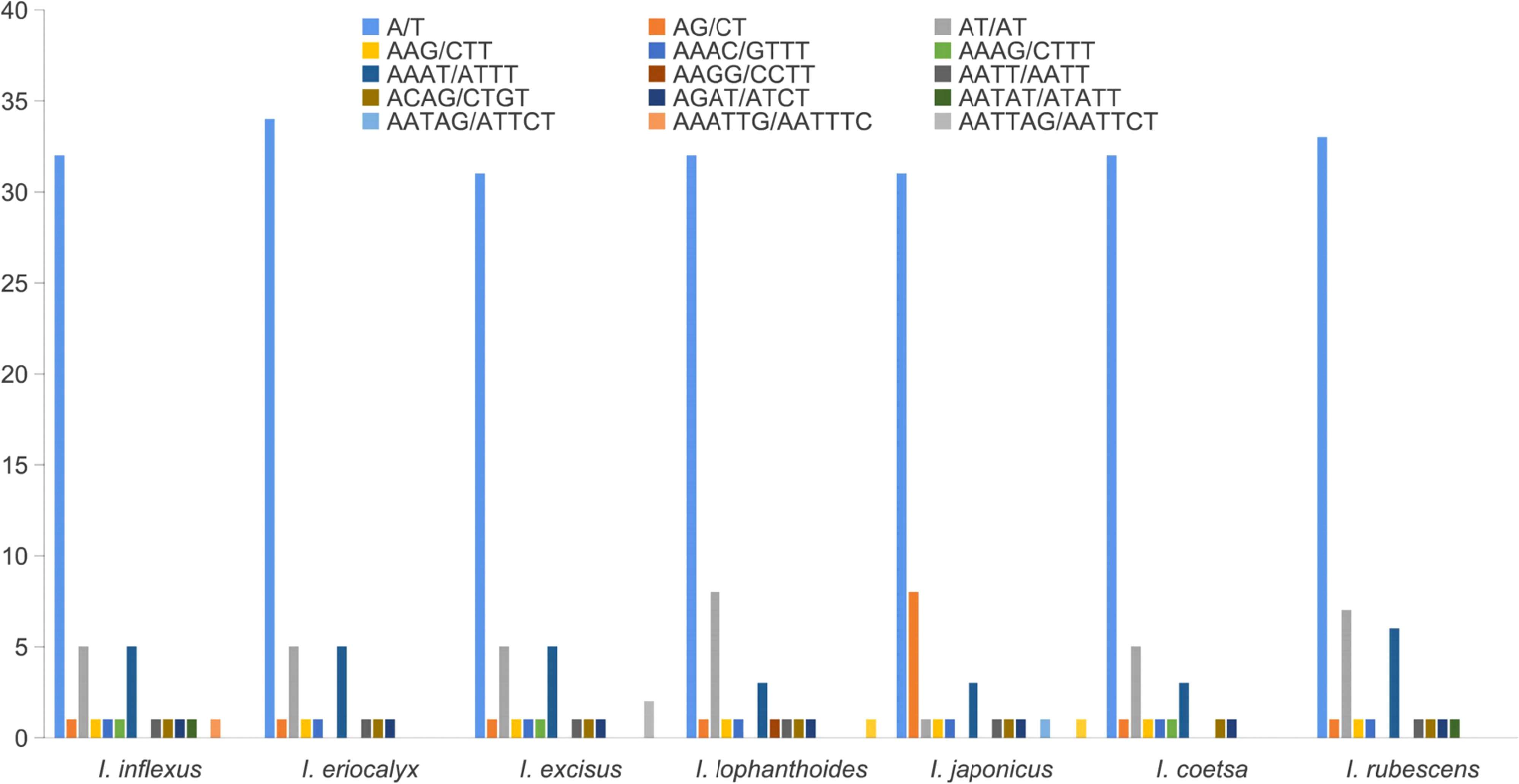
Figure 3 The number and type of SSRs in the cp genome of Isodon.
Moreover, oligonucleotide repeats analysis of four types of repeats in the cp genome, including F, R, P, and C, was performed by REPuter. As illustrated in Figure 4, the number of repeat types varied and presented random permutations among the cp genomes of seven species. The number of repeats varied among these species, but most repeat sequences existed in 20 – 29 bp, which was supported by recent literature (Lian et al., 2022). Meanwhile, the abundance of F and P repeats was higher than that of R and C repeats. A total of 36 F repeats and 47 P repeats were observed in I. rubescens, 38 and 48 in I. inflexus, 34 and 39 in I. eriocalyx, 39 and 48 in I. excisus, 38 and 43 in I. lophanthoides, 39 and 45 in I. japonicus, 42 and 47 in I. coetsa, respectively. These repeats play a crucial role in the generation of substitutions and indels, which makes them important for detecting mutational hotspots (McDonald et al., 2011; Ahmed et al., 2012)
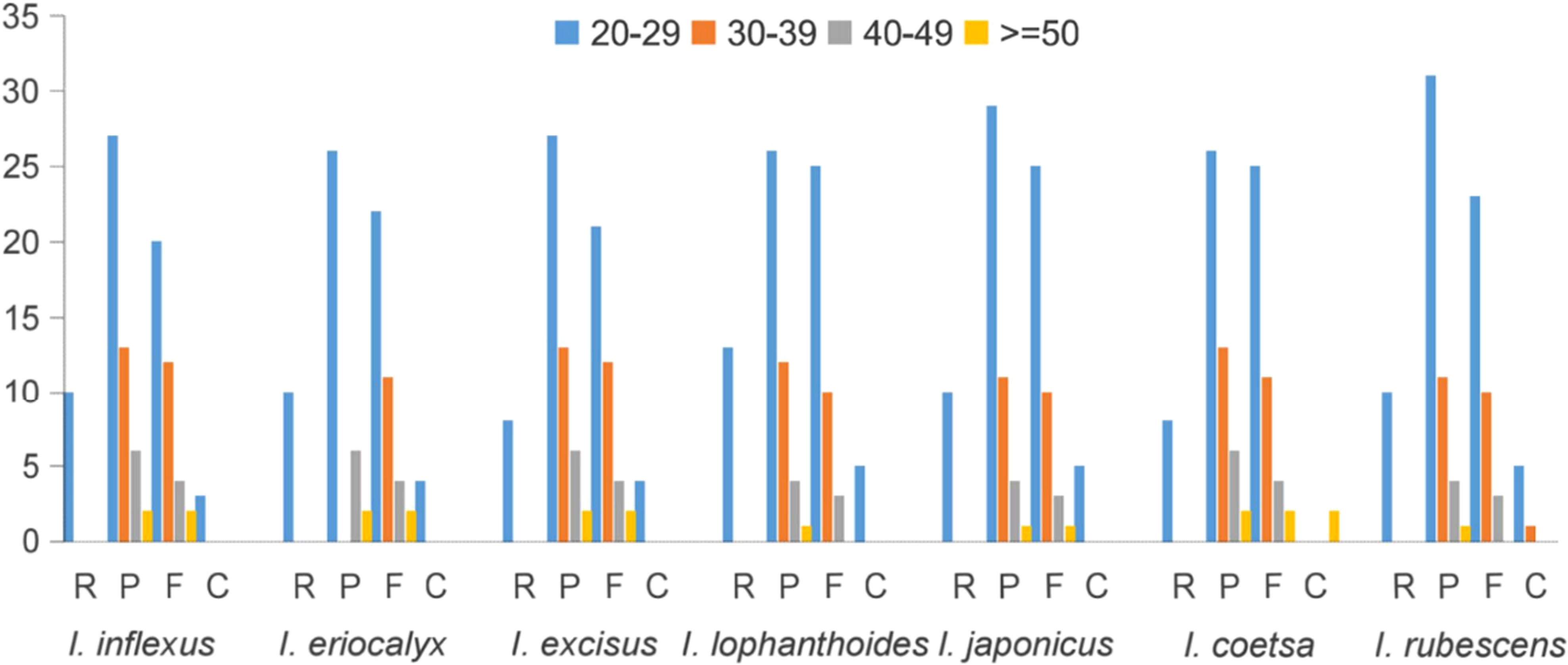
Figure 4 Repeat sequences detected in Isodon cp genome. P, F, C, and R indicate the repeat types: R (Reverse repeats), P (Palindromic repeats), F (Forward repeats), C (Complement repeats).
Inverted repeats
The contraction and expansion of IRs are regarded as crucial evolutionary phenomena that result in the pseudogenes, gene duplication, or the reduction of duplicate genes to a single copy (Abdullah et al., 2020). As illustrated in Figure 5, the rpl22 gene was present in the LSC region, and rpl2 was entirely in the IRb region, consistent with a previous study (Lian et al., 2022). A truncated copy of rps19 gene was observed in all species at the IRb/LSC junction except for I. ternifolious, which starts in LSC and integrates into the IRb regions, while rp12 is exclusively located in the IRb region. In monocotyledons, the rps19 gene is in the IR region (Ahmed et al., 2012; Henriquez et al., 2020), but our findings show that things are different in Lamiaceae. Additionally, the ndhF gene was also found at the junction of IRb/SSC and integrated into the IRb with a size ranging from 42 to 46 kb. At the IRb/LSC junction, another truncated copy of ycf1 gene was observed in all species except for I. ternifolious and I. serra. Contrary to the findings of Henriquez et al. (2020), the de novo assembled genomes of seven Isodon species did not include a ycf pseudogene at the IRa/SSC junction. Neither an internal stop codon nor double peaks were observed in the electropherograms, indicating that the seven Isodon species lack amplified pseudogenes. In addition, psbA and trnH were in the LSC, and rpl2 in the IRa.

Figure 5 Comparisons of the borders of LSC, SSC, and IRa/b regions among the 11 Isodon plastid genomes. The numbers represent the distance between the gene ends and the border sites, and the numbers below represent the length of the LSC, SSC, and IRa/b regions. This Figure is not to scale.
Genome comparison and nucleotide diversity
A comparison of overall sequence variation showed that the cp genome of Isodon is highly conserved, and the coding region is more conserved than non-coding regions. Except for ndhF, ycf1, and ycf2 genes, all protein-coding genes showed a highly conserved character (Figure 6); the intergenic spacers (IGS) with the highest divergence were trnH-GUG-psbA, trnQ-UUG-psbK, trnS-GCU-trnT-CGU, atpH-atpI, trnE-UUC-trnT-GGU, psaA-ycf3, ndhC-trnM-CAU, psbH-petB, and rps15-ycf1, as predicted. In addition, the sliding window analysis revealed that seven regions, including rps16-trnQ-UUG, ndhF, ndhB, ccsA-ndhD, ndhA, ndhH, and ycf1 genes, exhibited higher nucleotide diversity values (> 0.014, Figure 7); the IR regions exhibited lower sequence divergence than LSC and SSC regions, consistent with the previous comparisons of cp genomes (Jiang et al., 2022; Wang et al., 2022). Among these 16 high polymorphic regions, 11 intergenic spacers were found in the trnH-GUG-psbA, trnQ-UUG-psbK, trnS-GCU-trnT-CGU, atpH-atpI, trnE-UUC-trnT-GGU, psaA-ycf3, ndhC-trnM-CAU, psbH-petB, rps15-ycf1, rps16-trnQ-UUG, and ccsA-ndhD. It is noteworthy that the atpH-atpI, rps16-trnQ-UUG, and ndhC-trnM-CAU were also identified as mutational hotspots in a previous study (Lian et al., 2022). IGS was considered one of the evolutionary hotspots that exhibited more significant rates of nucleotide substitutions and indel mutations (Drummond, 2008). Therefore, these IGS might be undergoing more rapid nucleotide substitution at the species level, which could be served as a potential molecular marker for application in phylogenetic analyses of the Isodon genus.
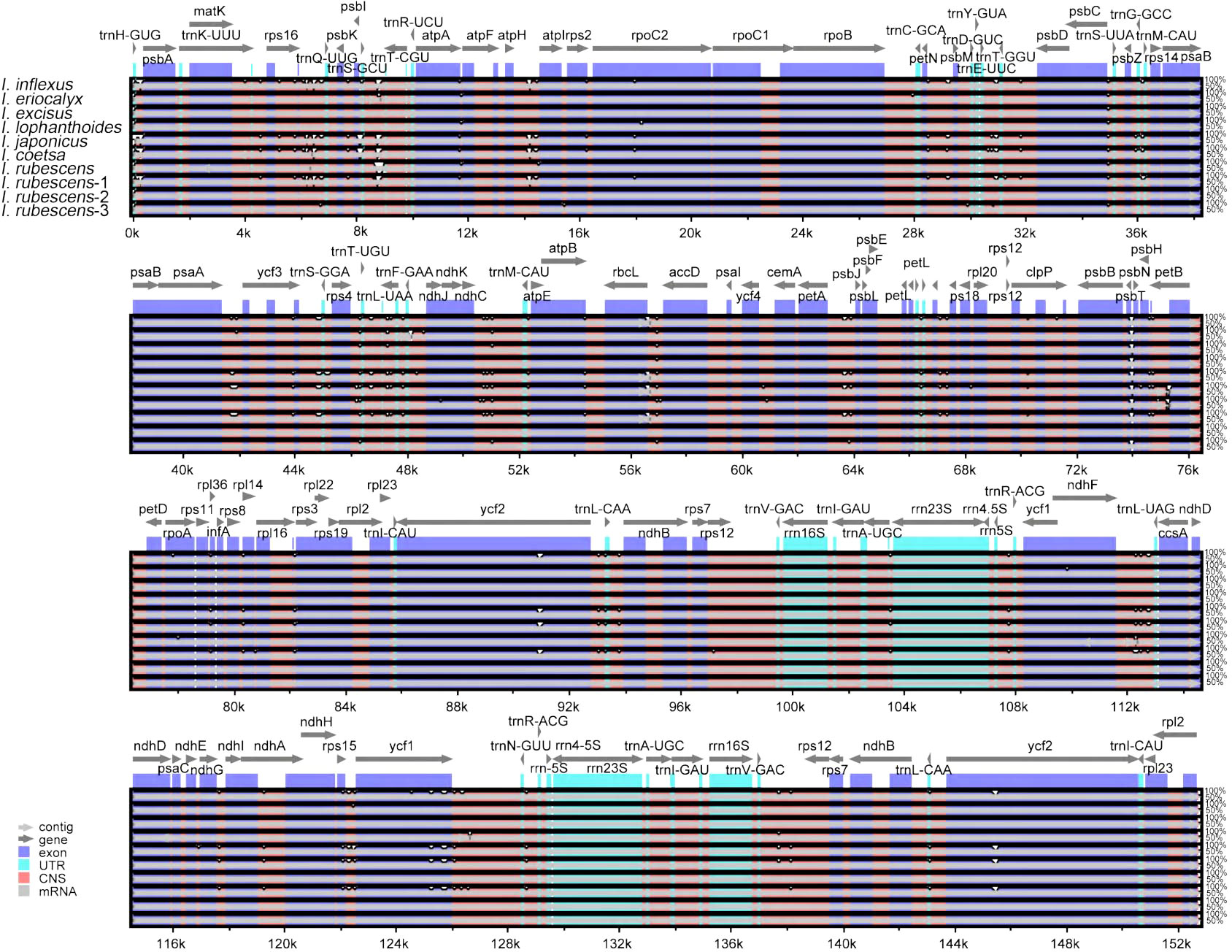
Figure 6 Global comparison of complete genomes of Isodon.
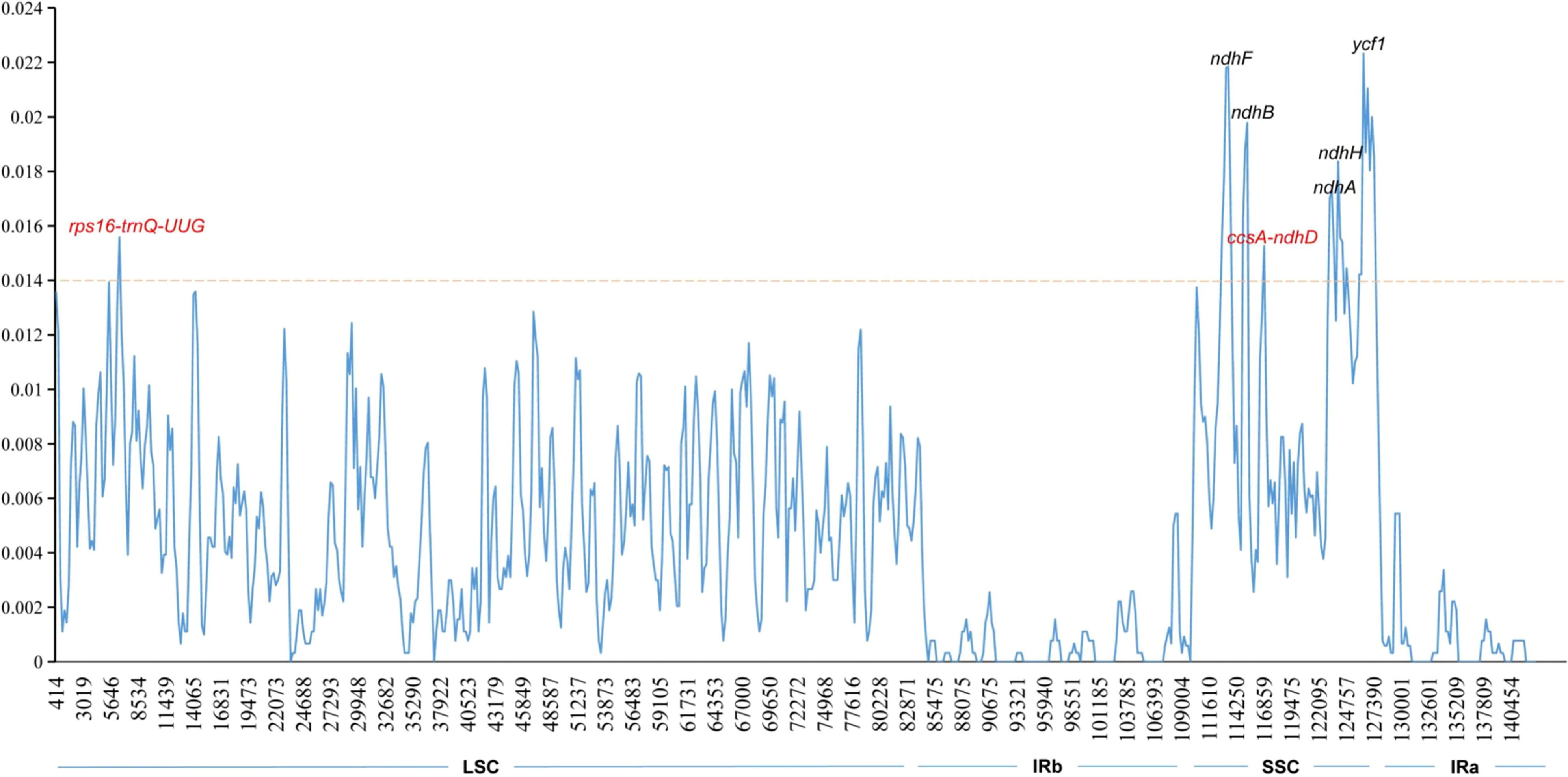
Figure 7 Sliding window analysis of Isodon cp genome. The X-axis represents the midpoint of the window; The Y-axis represents nucleotide diversity values. Window length: 600 bp; step size: 200 bp.
Species authentication analysis based on IGS
Intergenic spacer regions are the most frequently used cp markers for phylogenetic studies at lower taxonomic levels in plants (Shaw et al., 2005), as they are regarded as more variable and could provide more phylogenetically informative characters. To find candidate markers for identifying I. rubescens and its adulterants, the 11 IGSs were extracted using the PhyloSuite v1.2.2 from 14 Isodon species (Zhang et al., 2019). Each of the 11 IGSs was subject to maximum likelihood analyses in IQtree (Nguyen et al., 2015). As illustrated in Figure S2.1-S2.11, five IGSs, including trnS-GCU-trnT-CGU, atpH-atpI, trnE-UUC-trnT-GGU, ndhC-trnM-CAU, and rps15-ycf1 could be distinguished I. rubescens from I. japonicus and I. lophanthoides. Whereas the rest of the Isodon species cannot be distinguished based on these IGSs, bootstrap values for the relationship among these major clades were weak (<70%). These results are partially in line with the previous study, which found that the atpH-atpI and ndhC-trnM-CAU could be potential molecular markers for distinguishing Isodon species (Lian et al., 2022). Moreover, the remaining fragment, including trnS-GCU-trnT-CGU, trnE-UUC-trnT-GGU, and rps15-ycf1 also reported as potential markers for other species identification (Kim et al., 2020; Li et al., 2020; Alzahrani, 2021). Although the previous study has revealed that universal DNA barcodes (e.g., psbA-trnH) could differentiate I. rubescens from their related species (Xia et al., 2013), some common adulterants were not included in this study. Furthermore, the comparative analysis showed that the screened IGSs exhibit higher variability than psbA-trnH. These IGSs could theoretically distinguish the selected 7 species, whereas a much more detailed investigation of identification accuracy and amplification efficiency needs to be accomplished, and more experimental evidence is needed. Moreover, the ML phylogenetic tree was also inferred using a combination of these five IGSs. The results (Figure S3) showed that I. rubescens (GenBank NW018469) and I. rubescens (GenBank NC053708) clustered together as sister group groups and closely related to I. excisus. Simultaneously, I. rubescens (GenBank OM808736) was located in independent branches in the phylogeny, and well-supported sister relationship between I. rubescens and I. japonicus + I. lophanthoides (100% B/S). These results indicated that the combination of five IGSs could effectively discriminate I. rubescens from its common adulterants.
Species identification and phylogenetic analysis
The ML phylogenetic tree was inferred using 36 species, with Scrophularia as the outgroup. As illustrated in Figure 8, on the consensus trees, most nodes were supported with maximum support (100% bootstrap support). Ocimoideae and Lamioideae were sister taxa within the three subfamilies, and Ajugoideae was sister to the clade containing Ocimoideae + Lamioideae. The phylogenetic tree’s crown was occupied by the subfamily Ocimoideae, which included the genera Isodon and Ocimum. These findings confirm the position of Isodon within the Lamiaceae and are consistent with previous phylogenomic studies (Chen et al., 2021; Yue et al., 2021). The genus Isodon is further divided into three clades: (i) clade A, including I. rubescens (GenBank NC053708, GenBank NW018469), I. excise, I. serra, I. nervosus, I. amethystoides, I. coetsa, I. inflexus, I. eriocalyx, and I. rubescens (GenBank NW376483); (ii) clade B, only I. ternifolious; (iii) clade C, including I. japonicas, I. lophanthoides, and I. rubescens (GenBank OM808736). It is worth noting that the I. rubescens (GenBank OM808736) clustered differently from the other three samples (GenBank NC053708, GenBank NW018469, GenBank NW376483) of the same species in the phylogeny, which is consistent with the finding of other researcher based on rps16, trnL-trnF, and ITS sequence (Harris et al., 2012). Moreover, Lian et al. (2022) also found that the samples of I. rubescens from different geographical areas were not recovered as monophyletic and were placed in different branches in the previous report, which is well distinguished by sampling locations, suggesting that the intraspecific diversity was present in I. rubescens. This phenomenon might be explained by the fact that the geographical area of origin might influence the variation of I. rubescens. Another previous study supported the same conclusion; the Artemisia argyi collected from different geographical areas display high intraspecific diversity in the cp genome (Chen et al., 2022). In addition, clade B only includes a species of I. ternifolious, which was supported by the findings of other researchers based on trnD-trnT, psbA-trnH, rpl32-trnL, trnL-trnF, rps16, and nrITS (Zhong et al., 2010; Yu et al., 2014; Chen et al., 2021). Moreover, maximum likelihood analysis demonstrated that I. rubescens (GenBank OM808736) was located in independent branches in the phylogeny and deeply nested within clade C, and the sister relationship between I. rubescens and I. japonicus + I. lophanthoides was highly supported (100% B/S), indicating that the cp genome could discriminate I. rubescens from its common adulterants. The findings supported the results of morphological classification reported by Ge et al. (2022) and Xia et al. (2013).
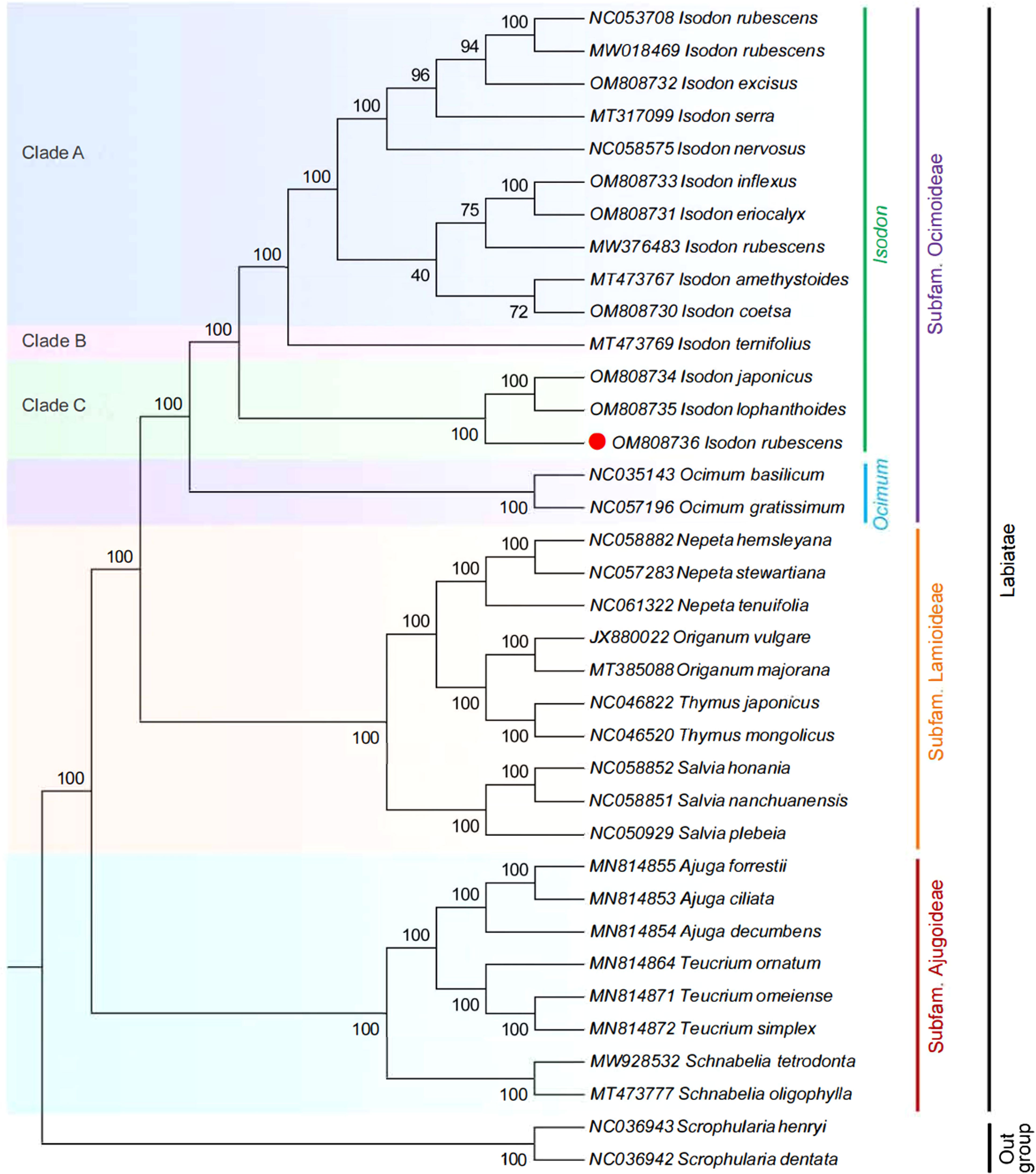
Figure 8 ML phylogenetic tree reconstruction containing the cp genomes of 36 plants. The Sceophularia species were set as the outgroup.
Conclusion
In the current study, the complete cp genome of seven species of Isodon was de novo assembled from Illumina high throughput sequencing reads, and cp genome sequences of I. inflexus, I. eriocalyx, I. excisus, and I. coetsa were reported for the first time. These cp genomes were generally conserved and exhibited similar gene content and genomic structure. Five highly variable cp loci, including trnS-GCU-trnT-CGU, atpH-atpI, trnE-UUC-trnT-GGU, ndhC-trnM-CAU, and rps15-ycf1, were identified, which could serve as potential markers for identifying I. rubescens and its common adulterants. In conclusion, our study provides a powerful tool and valuable scientific reference for the safety and effectiveness of clinical drug use, and it also contributes to the bioprospecting and conservation of Isodon species.
Data availability statement
The data presented in the study are deposited in the GenBank repository, accession numbers were from OM808730 to OM808736.
Author contributions
ZZ, JW, TP, and BD participated in the conception and design of the research. JW, QG, and BD collected the species. JQ, LS, and JD are responsible for analyzing and processing data. ZZ wrote the manuscript. The paper was revised by JQ, LS, and BD. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Yunnan academician expert workstation (202205AF150026, 202105AF150053), the key technology projects in the Yunnan province of China (202002AA100007), and the Yunnan Xingdian talent support plan (YNWR-QNBJ-2020251).
Acknowledgments
We would like to thank Ms. Qingshu Yang for her assistance in obtaining specimens for this study. We also thank Northeast Forestry University and the China Academy of Chinese Medical Sciences for technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1036277/full#supplementary-material
References
Abdullah, Mehmood, F., Shahzadi, I., Waseem, S., Mirza, B., Ahmed, I., et al. (2020). Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): comparative analyses and identification of mutational hotspots. Genomics (San Diego Calif.) 112, 581–591. doi: 10.1016/j.ygeno.2019.04.010
Ahmed, I., Biggs, P. J., Matthews, P. J., Collins, L. J., Hendy, M. D., Lockhart, P. J. (2012). Mutational dynamics of aroid chloroplast genomes. Genome Biol. Evol. 4, 1316–1323. doi: 10.1093/gbe/evs110
Alzahrani, D. A. (2021). Complete chloroplast genome of Abutilon fruticosum: genome structure, comparative and phylogenetic analysis. Plants 10, 270. doi: 10.3390/plants10020270
Amiryousefi, A., Hyvönen, J., Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bendich, A. J. (2004). Circular chloroplast chromosomes: the grand illusion. Plant Cell 16, 1661–1666. doi: 10.1105/tpc.160771
Chase, M. W., Christenhusz, M. J. M., Fay, M. F., Byng, J. W., Judd, W. S., Soltis, D. E., et al. (2016). An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc 181, 1–20. doi: 10.1111/boj.12385
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, Y. P., Huang, C. Z., Zhao, Y., Xiang, C. L. (2021). Molecular and morphological evidence for a new species of Isodon (Lamiaceae) from southern China. Plant Diversity 43, 54–62. doi: 10.1016/j.pld.2020.06.004
Chen, C. J., Miao, Y. H., Luo, D. D., Li, J. X., Wang, Z. X., Luo, M., et al. (2022). Sequence characteristics and phylogenetic analysis of the Artemisia argyi chloroplast genome. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.906725
Chinese Pharmacopoeia Commission (2020). Pharmacopoeia of the peoples republic of China (Beijing: China Med. Sci. Press).
Cui, Y. X., Chen, X. L., Nie, L. P., Sun, W., Hu, H. Y., Lin, Y. L., et al. (2019a). Comparison and phylogenetic analysis of chloroplast genomes of three medicinal and edible Amomum species. Int. J. Mol. Sci. 20, 4040. doi: 10.3390/ijms20164040
Cui, Y. X., Nie, L. P., Sun, W., Xu, Z. C., Wang, Y., Yu, J., et al. (2019b). Comparative and phylogenetic analyses of ginger (Zingiber officinale) in the family zingiberaceae based on the complete chloroplast genome. Plants (Basel) 8, 283–295. doi: 10.3390/plants8080283
Dong, W. L., Wang, R. N., Zhang, N. Y., Fan, W. B., Fang, M. F., Li, Z. H. (2018). Molecular evolution of chloroplast genomes of orchid species: insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 19, 716–736. doi: 10.3390/ijms19030716
Drouin, G., Daoud, H., Xia, J. N. (2008). Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 49, 827–831. doi: 10.1016/j.ympev.2008.09.009
Drummond, C. S. (2008). Diversification of Lupinus (Leguminosae) in the western new world: derived evolution of perennial life history and colonization of montane habitats. Mol. Phylogenet. Evol. 48, 408–421. doi: 10.1016/j.ympev.2008.03.009
Duan, B. Z., Wang, Y. P., Fang, H. L., Xiong, C., Li, X. W., Wang, P., et al. (2018). Authenticity analyses of rhizoma paridis using barcoding coupled with high resolution melting (Bar-HRM) analysis to control its quality for medicinal plant product. Chin. Med-Uk 13, 8–18. doi: 10.1186/s13020-018-0162-4
Ferrarini, M., Moretto, M., Ward, J. A., Šurbanovski, N., Stevanović, V., Giongo, L., et al. (2013). An evaluation of the PacBio RS platform for sequencing and de novo assembly of a chloroplast genome. BMC Genomics 14, 670. doi: 10.1186/1471-2164-14-670
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Ge, J. W., Li, P., Zhang, T. T., Gao, F., Wang, W. Y., Wang, J. N., et al. (2022). Molecular identification of a newly discovered medicinal plant, Isodon rubescens, in Shandong using DNA barcoding. Shandong Sci. 35, 15–20. doi: 10.3976/j.issn.1002-4026.2022.04.003
Guan, Y., Liu, X., Pang, X., Liu, W., Yu, G. X., Li, Y. R., et al. (2021). Recent progress of oridonin and its derivatives for cancer therapy and drug resistance. Med. Chem. Res. 30, 1795–1821. doi: 10.1007/s00044-021-02779-6
Harris, E. S. J., Cao, S. G., Schoville, S. D., Dong, C. M., Wang, W. Q., Jian, Z. Y., et al. (2012). Selection for high oridonin yield in the chinese medicinal plant Isodon (Lamiaceae) using a combined phylogenetics and population genetics approach. PLoS One 7, e50753. doi: 10.1371/journal.pone.0050753
Harris, E. S. J., Klooster, M. R. (2011). Development of microsatellite markers for the medicinal plant Isodon rubescens (Lamiaceae) and related species. Am. J. Bot. 98, e293–e295. doi: 10.3732/ajb.1100190
Henriquez, C. L., Abdullah Ahmed, I., Carlsen, M. M., Zuluaga, A., Croat, T. B., et al. (2020). Evolutionary dynamics of chloroplast genomes in subfamily Aroideae (Araceae). Genomics 112, 2349–2360. doi: 10.1016/j.ygeno.2020.01.006
Jiang, Y., Miao, Y. J., Qian, J., Zheng, Y., Xia, C. L., Yang, Q. S., et al. (2022). Comparative analysis of complete chloroplast genome sequences of five endangered species and new insights into phylogenetic relationships of Paris. Gene 833, 146572. doi: 10.1016/j.gene.2022.146572
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., DePamphilis, C. W., Yi, T. S., et al. (2019). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241–272. doi: 10.1101/256479
Kawabe, A., Miyashita, N. T. (2003). Patterns of codon usage bias in three dicot and four monocot plant species. Genes Genet. Syst. 78, 343–352. doi: 10.1266/ggs.78.343
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kim, G., Lim, C. E., Kim, J., Kim, K., Lee, J. H., Yu, H., et al. (2020). Comparative chloroplast genome analysis of Artemisia (Asteraceae) in East Asia: Insights into evolutionary divergence and phylogenomic implications. BMC Genomics 21, 415–432. doi: 10.1186/s12864-020-06812-7
Kumar, M., Choi, J., Kumari, N., Pareek, A., Kim, S. (2015). Molecular breeding in Brassica for salt tolerance: importance of microsatellite (SSR) markers for molecular breeding in Brassica. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.00688
Kurtz, S. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lazarski, K. E., Moritz, B. J., Thomson, R. J. (2014). The total synthesis of Isodon diterpenes. Angewandte Chemie Int. Edition 53, 10588–10599. doi: 10.1002/anie.201404482
Liang, C. L., Wang, L., Lei, J., Duan, B. Z., Ma, W. S., Xiao, S. M., et al. (2019). A comparative analysis of the chloroplast genomes of four Salvia medicinal plants. Engineering-Prc 5, 907–915. doi: 10.1016/j.eng.2019.01.017
Lian, C. L., Yang, H., Lan, J. X., Zhang, X. Y., Zhang, F., Yang, J. F., et al. (2022). Comparative analysis of chloroplast genomes reveals phylogenetic relationships and intraspecific variation in the medicinal plant Isodon rubescens. PLoS One 17, e266546. doi: 10.1371/journal.pone.0266546
Lin, C. Z., Liu, F. L., Zhang, R. J., Liu, M. T., Zhu, C. C., Zhao, J., et al. (2019). High-performance thin-layer chromatographic fingerprints of triterpenoids for distinguishing between Isodon lophanthoides and Isodon lophanthoides var. gerardianus. J. Aoac Int. 102, 714–719. doi: 10.5740/jaoacint.18-0305
Liu, M., Wang, W. G., Sun, H. D., Pu, J. X. (2017). Diterpenoids from Isodon species: an update. Nat. Prod. Rep. 34, 1090–1140. doi: 10.1039/C7NP00027H
Li, D. M., Ye, Y. J., Xu, Y. C., Liu, J. M., Zhu, G. F. (2020). Complete chloroplast genomes of Zingiber montanum and Zingiber zerumbet: Genome structure, comparative and phylogenetic analyses. PLoS One 15, e236590. doi: 10.1371/journal.pone.0236590
McDonald, M. J., Wang, W., Huang, H., Leu, J. (2011). Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences. PLoS Biol. 9, e1000622. doi: 10.1371/journal.pbio.1000622
Meng, J., Li, X. P., Li, H. T., Yang, J. B., Wang, H. D., He, J. (2018). Comparative analysis of the complete chloroplast genomes of four Aconitum medicinal species. Molecules 23, 1015. doi: 10.3390/molecules23051015
Millen, R. S., Olmstead, R. G., Adams, K. L., Palmer, J. D., Lao, N. T., Heggie, L., et al. (2001). Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 13, 645–658. doi: 10.1105/tpc.13.3.645
Nguyen, L. T., Schmidt, H. A., Haeseler, A. V., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Qian, J., Song, J. Y., Gao, H. H., Zhu, Y. J., Xu, J., Pang, X. H., et al. (2013). The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS One 8, e57607. doi: 10.1371/journal.pone.0057607
Rahemi, A., Fatahi, R., Ebadi, A., Taghavi, T., Hassani, D., Gradziel, T., et al. (2012). Genetic diversity of some wild almonds and related Prunus species revealed by SSR and EST-SSR molecular markers. Plant Syst. Evol. 298, 173–192. doi: 10.1007/s00606-011-0536-x
Roman, M. G., Houston, R. (2020). Investigation of chloroplast regions rps16 and clpP for determination of Cannabis sativa crop type and biogeographical origin. Legal Med-Tokyo 47, 101759. doi: 10.1016/j.legalmed.2020.101759
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Shahzadi, I., Abdullah, Mehmood, F., Ali, Z., Ahmed, I., Mirza, B. (2020). Chloroplast genome sequences of Artemisia maritima and Artemisia absinthium: Comparative analyses, mutational hotspots in genus Artemisia and phylogeny in family asteraceae. Genomics 112, 1454–1463. doi: 10.1016/j.ygeno.2019.08.016
Shang, M. Z., Liu, F., Hua, J. P., Wang, K. B. (2011). Analysis on codon usage of chloroplast genome of Gossypium hirsutum. Sci. Agr. Sin. 44, 245–253. doi: 10.3864/j.issn.0578-1752.2011.02.003
Shaw, J., Lickey, E. B., Beck, J. T., Farmer, S. B., Liu, W., Miller, J., et al. (2005). The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 92, 142–166. doi: 10.3732/ajb.92.1.142
Su, X. H., Dong, C. M., Feng, W. S., Chen, S. Q., Wang, L., Wang, W. L., et al. (2007). Study on the diversity of morphological characteristics of leaves of Rabdosia rubescens(Hemls.) hara. Lishizhen Med. Mat. Med. 18, 2351–2353. doi: 10.3969/j.issn.1008-0805.2007.10.010
Wang, J., Qian, J., Jiang, Y., Chen, X. C., Zheng, B. J., Chen, S. L., et al. (2022). Comparative analysis of chloroplast genome and new insights into phylogenetic relationships of Polygonatum and tribe polygonatea. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.882189
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization ofde novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Xia, Z., Zhang, H. R., Li, H. M., Gao, Z. M. (2013). Molecular authentication and phylogenetic relationship of Isodon rubescens and its relatives. Chin. Trad. Herb. Drugs 44, 2904–2910. doi: 10.7501/j.issn.0253-2670.2013.20.021
Xue, J. T., Yu, S. S., Wang, J. C., Li, C. Y., Yang, M. Y., Shi, Y. L., et al. (2020). Network pharmacological screening of the active ingredients and hypoglycemic effect of Isodon rubescens in the treatment of diabetes. Planta Med. 86, 556–564. doi: 10.1055/a-1147-9196
Yue, Z. Y., Wang, Y., Zhou, B. Z., Wang, H. P. (2021). The complete chloroplast genome of Isodon rubescens, a traditional Chinese herb. Mitochondrial. DNA B Resour. 6, 337–338. doi: 10.1080/23802359.2020.1860704
Yu, X., Maki, M., Drew, B. T., Paton, A. J., Li, H., Zhao, J., et al. (2014). Phylogeny and historical biogeography of Isodon (Lamiaceae): rapid radiation in south-west China and Miocene overland dispersal into Africa. Mol. Phylogenet. Evol. 77, 183–194. doi: 10.1016/j.ympev.2014.04.017
Zhang, Z., Chuang, Y., Huang, N., Mitch, W. A. (2019). Predicting the contribution of chloramines to contaminant decay during Ultraviolet/Hydrogen peroxide advanced oxidation process treatment for potable reuse. Environ. Sci. Technol. 53, 4416–4425. doi: 10.1021/acs.est.8b06894
Zhang, D. S., Hu, P., Liu, T. G., Wang, J., Jiang, S. W., Xu, Q. H., et al. (2018). GC bias lead to increased small amino acids and random coils of proteins in cold-water fishes. BMC Genomics 19, 315–325. doi: 10.1186/s12864-018-4684-z
Keywords: Isodon rubescens, chloroplast genome, species identification, molecular marker, phylogenetic
Citation: Zhou Z, Wang J, Pu T, Dong J, Guan Q, Qian J, Shi L and Duan B (2022) Comparative analysis of medicinal plant Isodon rubescens and its common adulterants based on chloroplast genome sequencing. Front. Plant Sci. 13:1036277. doi: 10.3389/fpls.2022.1036277
Received: 04 September 2022; Accepted: 26 October 2022;
Published: 21 November 2022.
Edited by:
Zemin Ning, Wellcome Sanger Institute (WT), United KingdomReviewed by:
Qingguo Ma, Research Institute of Forestry, Chinese Academy of Forestry, ChinaGang Yao, South China Agricultural University, China
Cornelius Mulili Kyalo, University of Chinese Academy of Sciences, China
Copyright © 2022 Zhou, Wang, Pu, Dong, Guan, Qian, Shi and Duan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baozhong Duan, YnpkdWFuQDEyNi5jb20=; Linchun Shi, bGluY2h1bl9zaGlAMTYzLmNvbQ==