 Yongping Li
Yongping Li- 1Key Laboratory of Horticultural Plant Biology (Ministry of Education), College of Horticulture and Forestry Sciences, Huazhong Agricultural University, Wuhan, China
- 2National Key Laboratory of Crop Genetic Improvement, College of Plant Science and Technology, Huazhong Agricultural University, Wuhan, China
- 3Department of Cell Biology and Molecular Genetics, University of Maryland, College Park, College Park, MD, United States
Fragaria vesca, a wild diploid strawberry, has recently emerged as a model for the cultivated strawberry and other members of the Rosaceae. Differentiation and maintenance of meristems largely determines plant architecture, flower development and ultimately fruit yield. However, in strawberry, our knowledge of molecular regulation of meristems in different developmental context is limited. In this study, we hand dissected three types of tissues than contain meristematic tissues corresponding to shoot apical meristem (SAM), flower meristem (FM), and receptacle meristem (REM), in F. vesca for RNA-seq analyses. A total of 3,009 differentially expressed genes (DEGs) were identified through pairwise comparisons. These DEGs were grouped into nine clusters with dynamic and distinct expression patterns. In these nine clusters, 336 transcription factor genes belong to 46 families were identified; some of which were significantly enriched in FM and REM such as the MADS-box family or in REM such as the B3 family. We found conserved and distinctive expression patterns of totally 149 genes whose homologs regulate flowering time or SAM, leaf, and flower development in other plant species. In addition to the ABCE genes in flower development, new MADS box genes were identified to exhibit differential expression in these different tissues. Additionally, the cytokinin and auxin pathway genes also exhibited distinct expression patterns. The Arabidopsis homeobox gene WUSCHEL (WUS), essential for stem cell maintenance, is expressed in organizing center of meristems. The F. vesca homolog FvWUS1 exhibited a broader expression domain in young strawberry flowers than its Arabidopsis counterpart. Altogether, this work provides a valuable data resource for dissecting gene regulatory networks operating in different meristematic tissues in strawberry.
Introduction
Strawberries are herbaceous perennials with unique flower and fruit and distinct plant architectures, such as numerous individual carpels grown on the receptacle of flowers and accessory fruit (Hollender et al., 2012). The cultivated strawberry (Fragaria × ananassa, octoploid) is an important fruit crop worldwide. Together with the cultivated strawberry, there are about 25 recognized Fragaria species in the world, with different ploidies from diploid to decaploid (Staudt, 2009). F. vesca is a diploid progenitor species of F. × ananassa (Edger et al., 2019) and frequently used as a model for cultivated strawberry which is octoploid and a hybrid. One advantage of F. vesca is its high quality genome (Shulaev et al., 2011; Edger et al., 2018). Previously, we profiled the transcriptomes of flower and fruit tissues at different developmental stages in F. vesca (Kang et al., 2013; Hollender et al., 2014). However, the transcriptomes of some important strawberry tissues are still lacking.
Meristems contain stem cells of plants. The shoot apical meristem (SAM) is located on the stem tip, which is further divided into three zones, namely the central zone (CZ), the organizing center (OC), and the peripheral zone (PZ). In strawberry, during vegetative phase, SAM gives rise to new lateral leaf primordia and new lateral branches (branch crown); upon floral induction, SAM becomes inflorescence meristem, which terminates in a floral meristem (FM). As the strawberry inflorescence belongs to dichasial cyme (the primary flower is determinate, and secondary flowers emerge from the base of the peduncle) (Hollender et al., 2012), it is difficult to distinguish floral meristem from inflorescence meristem. In strawberry flower meristem (FM) gives rise to the four whorls of flower organs. Their carpels are not fused and numerous individual carpels emerged from the receptacle sequentially from the base to apex. Thus, the receptacle of a young flower bud maintains its meristematic activity for an extended period of time to continuously give rise to new carpel primordia. Hence, the receptacle is like an FM except that it gives rise to carpels only. For this reason, the developing receptacle is designated as REceptacle Meristem (REM) in this study. Characterization of the REM transcriptome would unveil the underlying regulatory network that specifies its unique development, and facilitates comparative studies with plant species, whose flowers do not have an enlarged receptacle.
Much of our knowledge on the molecular mechanisms underlying SAM and FM development and floral organ identity determination is based on studies of Arabidopsis. The SAM maintenance is regulated by the WUS/CLV signaling module (Schoof et al., 2000; Braybrook and Kuhlemeier, 2010). WUSCHEL (WUS) is a transcription factor expressed in the OC and promotes stem cell fate (Laux et al., 1996; Mayer et al., 1998). CLAVATA3 (CLV3) encodes a small peptide and is a marker gene of the CZ (Fletcher et al., 1999). The WUS protein could move to the CZ to promote the expression of CLV3, while CLV3 and its receptors together inhibit expression of WUS (Brand et al., 2000; Schoof et al., 2000; Yadav et al., 2011; Daum et al., 2014), thus forming a negative feedback loop to maintain the proper size of stem cell population. The WUS/CLV signaling module functions in both SAM and FM to maintain their meristem identity. However, FM eventually terminates after four whorls of floral organs are formed. During flower development, four classes of genes (A, B, C, and E) interact to coordinately specify four floral-organ types (Coen and Meyerowitz, 1991; Krizek and Fletcher, 2005; Causier et al., 2010). Specifically, A (APETALA1/AP1, AP2) and E genes (SEPALLATA/SEP and SEP-like) together specify sepal identity; A, B (AP3, PISTILLATA/PI), and E genes specify petal identity; B, C (AGAMOUS/AG), and E genes specify stamen identity; C and E genes specify carpel identity. Most of the ABCE genes belong to the MADS-box gene family except AP2, which belongs to AP2 family of transcription factors (Parenicova et al., 2003; Arora et al., 2007; Smaczniak et al., 2012).
Thus far, no transcriptome is available for the SAM in strawberry. Previously, we analyzed the transcriptomes of Flower_1–4 (floral stages 1–4) and Receptacle_6–7 (floral stages 6–7) (Hollender et al., 2014), corresponding to FM and REM, respectively. Tissue-specific gene clusters were obtained for the two tissues. However, these tissues were collected by laser capture microdissection (LCM). We found that a lower percentage of raw reads were aligned to the coding sequences for the LCM libraries, that is, 30–40% for LCM and 60–70% for hand-dissected (Hollender et al., 2014). Since LCM and hand-dissection employ very different methods in tissue fixation, sampling, and library construction, a large number of differentially expressed genes are likely caused by technical differences. This is supported by the detection of different expression profiles between LCM and hand-dissection generated from the same tissue (Hollender et al., 2014). Thus, new transcriptomic data using hand-dissected tissues including FM and REM are desirable for comparisons with previously generated data using hand dissection.
In this work, Illumina RNA-seq is utilized to profile hand dissected SAM, FM, and REM tissues to gain insights into genome-wide gene expression dynamics in SAM, FM, and REM of F. vesca. The differentially expressed genes were divided into nine gene clusters with distinct expression patterns. A large number of well-known transcription factors and hormone genes were identified to be highly or specifically expressed in each meristem. One notable finding is that FvWUS1, a homolog of the stem cell maintenance gene WUS in Arabidopsis, is more abundantly expressed in the FM and REM than SAM, which was validated by the FvWUS1pro::GUS reporter in transgenic F. vesca lines as well as RNA in situ hybridization in F. vesca. This expression pattern of WUS in F. vesca is distinctly different from that in Arabidopsis. Together, our results provide a valuable resource for future functional dissection of key genes regulating plant architectures and flower development in strawberry.
Materials and Methods
Plant Materials and Tissue Isolation
The tissues for RNA-seq were dissected manually from YW5AF7, an inbred line of the diploid F. vesca day-neutral variety Yellow Wonder (Slovin et al., 2009), using tweezers under a dissecting microscope. Plants used for tissue isolation were grown in substrate under natural conditions in plastic-covered tunnels in June and July at the Huazhong Agricultural University, when they were flowering. The F. vesca transgenic plants were grown in a growth room under a light intensity of 100 µmol m−2 s−1 with a 16/8 h light/dark photoperiod at 25°C. The samples SAM, FM, and REM were designated as described in Figure 1.
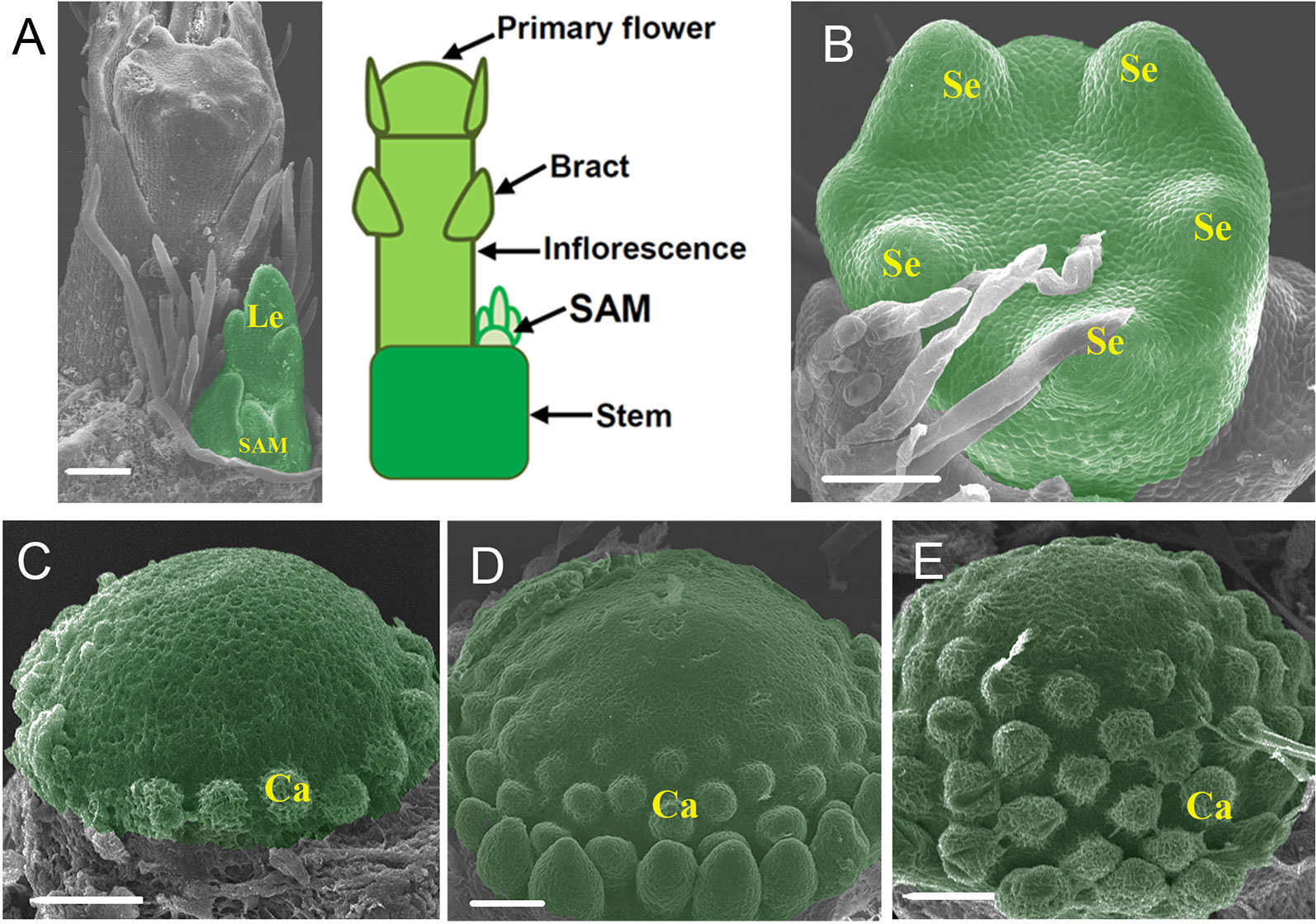
Figure 1 Morphology of the three meristems used for transcriptome analysis. (A) SEM image and diagram showing the tip of a strawberry (F. vesca) shoot with an inflorescence emerging. (B) SEM image of stage 3 flower. The entire flowers were collected as the FM (flower meristem). (C–E) SEM images of the receptacles at stages 5–7, respectively. Sepals, petals, and anthers were removed to expose the receptacles. These receptacles were pooled together as the REM (receptacle meristem). Le, leaf; SAM, shoot apical meristem; Se, sepal; Ca, carpel. The SAM, FM, and REM are false colored green, respectively. Scale Bars = 50 μm.
RNA Extraction and Illumina RNA Sequencing
The tissues were dissected and frozen immediately in liquid N2, and then stored at −80 °C until use. Total RNAs were extracted with the RNeasy Plant Mini Kit (Qiagen, Cat# 74903). Three biological replicates for each tissue type were prepared. Approximately 1 µg of total RNAs per sample was sent to Beijing Genomics Institute (BGI, Wuhan, China) for strand specific library construction and sequencing on Illumina HiSeq2500. In total, 292 million 125 bp paired-end reads were generated. This dataset was deposited at the Sequence Read Archive (SRA) at NCBI (http://www.ncbi.nlm.nih.gov/sra) under the accession number SRP115444.
Gene Expression Analysis
The first 12 nt of each read was trimmed off using fastx_trim in the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). The trimmed reads were mapped against the F. vesca V4 genome with the annotation v4.0.a2 using the program STAR in a 2-pass mode (Dobin et al., 2013; Edger et al., 2018; Li et al., 2019). Only the uniquely mapped reads were retained for further analysis. The aligned reads for each gene were summarized by the program featureCounts (Liao et al., 2014), and TPM (Transcripts Per Million) was used to represent the expression level. The multi-dimensional scaling (MDS) analysis was performed using the log2 transformed TPM of all the expressed genes for each sample with the Euclidean Distance statistical method. Pairwise comparisons were carried out using the R package DESeq2 (Love et al., 2014) to identify the differentially expressed genes (fold change >2 and padj <0.05). All the differentially expressed genes were subjected to K-means clustering with the Euclidean Distance metric in MeV4.8.1 (Saeed et al., 2003). The gene expression plot of each cluster was made using the R package ggplot2.
WGCNA Network Analysis
Gene coexpression networks were constructed using the WGCNA (v1.68) package in R. A total of 97 Illumina RNA-seq libraries generated from 46 tissue types in F. vesca were downloaded to construct the gene coexpression network (Kang et al., 2013; Hollender et al., 2014; Toljamo et al., 2016; Hawkins et al., 2017; Li et al., 2018). A total of 26,192 genes with TPM >2 in at least one of the tissues were used for the WGCNA signed coexpression network analysis (Langfelder and Horvath, 2008), and the average TPM of the three biological replicates was used as the input. Gene modules were obtained using the automatic network construction function blockwiseModules with default settings, except power = 16, minModuleSize = 30, and mergeCutHeight = 0.25. The eigengene value was calculated for each module and used to test the association with each tissue type. The total connectivity and intramodular connectivity (function softConnectivity), kME (for modular membership), and kME-P value were calculated for all the genes, which were clustered into 31 modules. Gene network was visualized using Cytoscape_v3.7.1.
Identification and Phylogenetic Analysis of the MADS-Box Genes in F. vesca
To globally identify the MADS-box genes in F. vesca, the SRF-TF domain (PF00319) was retrieved from the Pfam 27 database (Finn et al., 2013), and the protein sequences were obtained from the V4 genome according to the v4.0.a2 annotation (Edger et al., 2018; Li et al., 2019). The Hidden Markov Model-based HMMER program (version h3.1b2) was used to identify the putative MADS-box proteins with default settings. The full-length protein sequences of the Arabidopsis and strawberry MADS-box genes were aligned by using ClustalW. The unrooted phylogenetic tree was constructed with the MEGA 7 software (Kumar et al., 2016) with the Maximum Likelihood statistical method and bootstrap analysis (1,000 replicates).
Scanning Electron Microscope
Samples were fixed in 2.5% glutaraldehyde at 4°C overnight, treated sequentially with 30%, 50%, 70%, 90% and 100% ethanol for 10 min each, transferred to isoamyl acetate for 20 min, critical point dried, coated with gold for 30 s, and photographed under a scanning electron microscope (JSM-6390LV).
Plasmid Construction
To make the construct FvWUS1pro::GUS, the promoter of FvWUS1 (FvH4_3g04400/gene30464) with a length of 2,076 bp upstream of the translational start codon was isolated by PCR amplification from the genomic DNA of YW5AF7 using the forward primer (5’- AGCGCTGAAGCTTGGCTGCAGAGTCCAAGATCTCTCACTTGCAAG-3’) and the reverse primer (5’-GGACTGACCACCCGGGGATCCTGGTGATGGGCTGAGAGAATGAGA-3’). The fragment was inserted into the binary vector DX2181G (Ye et al., 2012) digested by the PstI/BamHI restriction enzymes using the Gibson cloning method.
Strawberry Transformation
Agrobacterium-mediated strawberry transformation was performed as previously described (Feng et al., 2019). Briefly, leaf strips of the F. vesca variety Hawaii 4 were used as explants, and positive transgenic calli and regenerated plants were screened using 4 mg l−1 hygromycin during transformation.
Gus Staining
The SAM and flower buds at early developmental stages were dissected under a stereomicroscope from five independent T0 transgenic plants. The tissues were stored in cold 90% acetone during dissection and then kept at room temperature for 20 min. Next, acetone was removed and GUS staining solution (1 mM X-glucuronic acid, 0.1 mM EDTA, 0.1% Triton X-100, and 10 mM potassium ferri/ferrocyanide in 100 mM phosphate buffer, pH 7.0) was added to submerge all the materials. After 30 min of vacuum, tissues were incubated overnight at 37°C. The samples were then mounted on clearing solution (chloral hydrate: glycerol: H2O: 8:1:1) for 5 h and observed under differential interference contrast (DIC) optics using a Zeiss Axioscope A1 microscope with a ×0.5 optical adapter. The images were captured and exported using ZEN2.3.
RNA In Situ Hybridization
The SAM and young flower buds of the F. vesca variety YW were fixed in the formaldehyde-acetic acid-ethanol fixative solution and stored at 4°C until use. Floral development stages were designated as previously described (Hollender et al., 2012). For the antisense probe, a 163 bp fragment of FvWUS1 (21–183 bp in the coding sequence) was synthesized and inserted into the pSPT18 vector. The antisense probe was synthesized using the forward primer 5’-CGTTTCACAACTCTCACATGC-3’ and the reverse primer 5’-CATTGGTGATGGGCTGAGA-3’. The M13 fragment was used as the control probe. Synthesis of DIG-labeled RNA probes via in vitro transcription was carried out using the DIG RNA Labeling Kit (SP6/T7) (Roche, Cat# 11175025910). The hybridization signals were detected using the DIG Nucleic Acid Detection Kit (Roche, Cat# 11175041910). Slides were developed in a dark humid container for 24 h and development was stopped and stored with 1×TE. The images were captured using a Zeiss Axioscope A1 microscope with a ×0.5 optical adapter.
Results and Discussion
Morphology of the Three Shoot Meristem Tissues and RNA-Seq Analysis in F. vesca
The shoot apical meristem (SAM) of strawberry is deeply buried by young leaves and surrounded by a young leaf primordium. When flowering, an inflorescence is initiated that occupies a large space on the stem tip and pushes the SAM aside (Figure 1A). In this work, the sample SAM consisted of the typical SAM as well as a subtending leaf primordium and a short stem underneath (Figure 1A). The flower development in F. vesca was divided into 13 stages (Hollender et al., 2012). The sample FM consisted of flower primordia at stages 1 to 4. At stages 1 and 2, the flower primordia look like a dome. At stage 3, five sepal primordia appear (Figure 1B). At stage 4, the flower center is flat, petal primordia start to form, and short trichomes appear on sepals. REM consists of developing receptacles with the carpel primordia emerging from the base to the apex in a spiral pattern at floral stages 5 to 7 (Figures 1C–E). Approximately 70–100 shoot apexes or flower buds were collected by hand dissection to give sufficient quantities of total RNAs for each biological replicate.
RNA-seq was carried out for a total of nine samples (three biological replicates for each meristem type) using the Illumina HiSeq-2500 platform (paired-end, 125bp). The number of raw reads ranged from 29.7 to 34.5 million for each sample, totaling 292.4 million, and an average of 87.7% of the raw reads were mapped to the FvH4 genome with the annotation ver4.0.a2 (Supplementary Table S1) (Li et al., 2019). Genes with a TPM (Transcripts Per Million) value lower than 2 were considered “lowly expressed” and removed from downstream analysis. As a result, 15,106–16,694 genes were expressed among the 34,007 annotated protein-coding genes in each transcriptome. The global relative relationship among the nine samples was examined by multi-dimensional scaling (MDS) plot analysis. In the MDS plot, the three tissue types were separated from each other (Figure 2A), indicating that they are quite distinct. Moreover, the three biological replicates for each tissue were grouped together, suggesting uniform sampling. A Venn diagram was used to reveal the uniquely or commonly expressed genes among the three tissues (Figure 2B). In total, 15,574 genes were commonly expressed, while only 162 genes (FM) to 377 genes (SAM) were specifically expressed in one tissue.
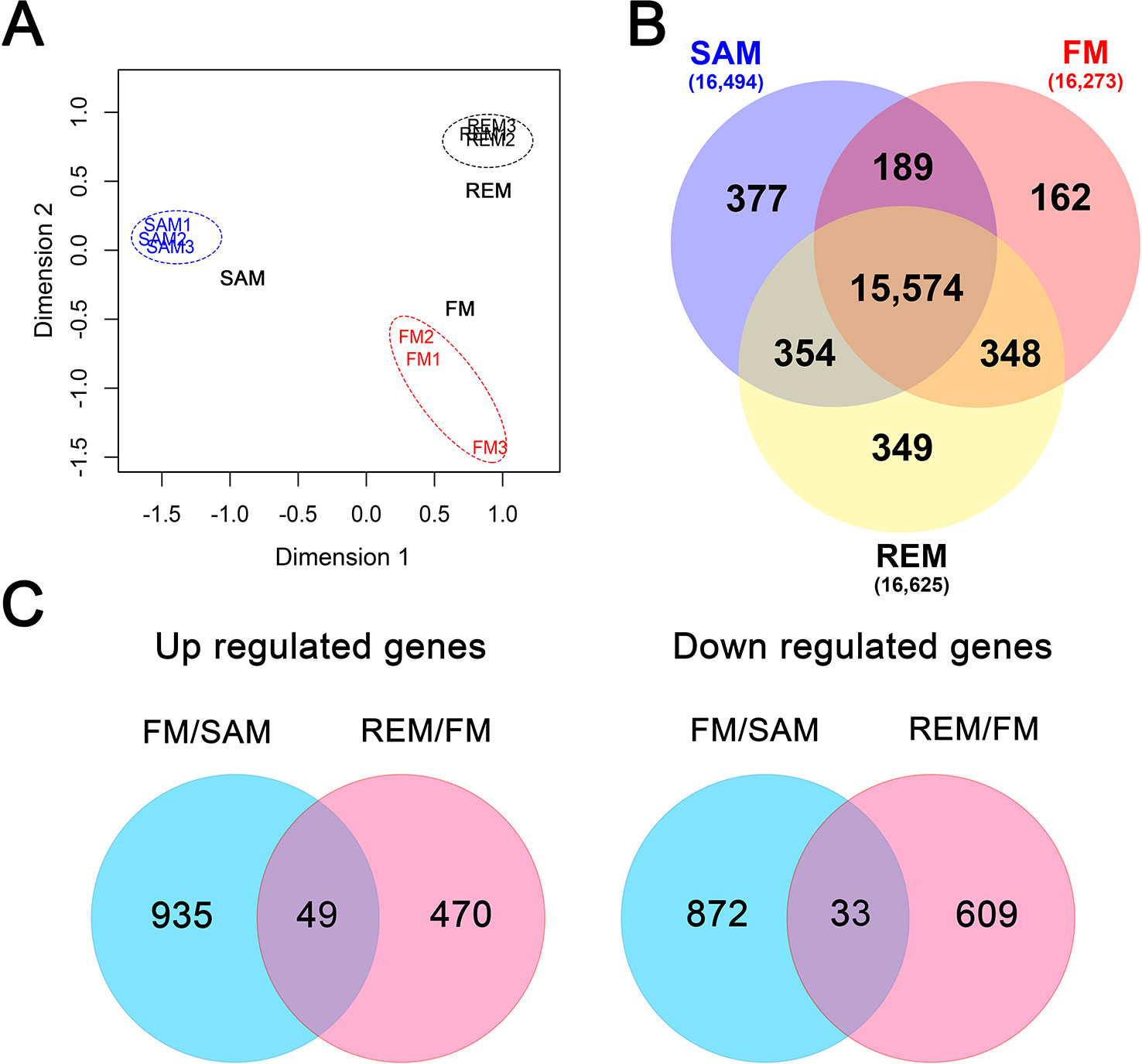
Figure 2 Global analysis of the meristem transcriptomes. (A) MDS plot showing the overall relationship of the three tissue types with three biological replicates. SAM, blue; FM, red; REM, black. Log2-transformed TPM was used with the Euclidean Distance method to measure the pairwise similarity. (B) Venn diagram showing the unique and common genes (TPM > 2) in each tissue. (C) Venn diagram showing the up-regulated and down-regulated genes in the pairwise comparisons (fold change > 2, padj < 0.05).
Differentially Expressed Genes Accompanying Meristem Development
To identify differentially expressed genes (DEGs), pairwise comparisons were carried out between SAM and FM and between FM and REM, respectively, by using the R package DESeq2 (Love et al., 2014). From SAM to FM, 984 genes were significantly up-regulated in FM, while 905 genes were down-regulated in FM (fold change >2, padj ≤0.05) (Figure 2C). In the list of DEGs (Supplementary Data 1), the flower meristem identity genes FvAPETALA1 (FvAP1, FvH4_4g29600) and FvLEAFYa (FvLFYa, FvH4_5g09660) as well as other well-known flower development genes were greatly up-regulated in FM. Gene ontology (GO) analysis revealed that the GO terms of the biosynthetic and metabolic processes were greatly enriched among the up-regulated genes in the FM compared to the SAM (Supplementary Data 1). From FM to REM, 519 and 642 genes were respectively up- and down-regulated in REM (fold change >2, padj ≤0.05) (Figure 2C, Supplementary Data 1). In this comparison, genes involved in the immune responses and transports were significantly enriched in the up-regulated genes. Intriguingly, 49 genes were continuously up-regulated at the two pairwise comparisons, some of which are the homologs of well-studied genes in other plant species, such as the floral meristem identity gene FvLFYa and flower development genes FvAGAMOUS (FvAG, FvH4_3g06720, C gene) and FvSEPALLATA3 (FvSEP3, FvH4_4g23530, E gene) (Mouhu et al., 2013; Hollender et al., 2014). In addition, 33 genes were continuously down-regulated at the two pairwise comparisons, such as FvH4_7g28740 coding for a SOC1-like MADS box transcription factor, the homolog of AGAMOUS-LIKE 42 that promotes floral transition in Arabidopsis (Dorca-Fornell et al., 2011), FvH4_2g21310 and FvH4_3g11120, the homologs of TAWAWA1 regulating the meristem phase change in rice inflorescence (Yoshida et al., 2013).
More DEGs Were Identified Between FM and REM Than the LCM Samples
Previously, we analyzed the transcriptomes of Flower_1–4 and Receptacle_6–7, corresponding to the FM and REM, respectively (Hollender et al., 2014). We wanted to see how the new data is related to the previous LCM data. First, we analyzed the overall relatedness of transcriptomes of the four transcriptome datasets. The MDS plot revealed that Flower_1–4 and Receptacle_6–7 are distantly separated from the three meristem samples generated in this study (Supplementary Figure S1A). Next, we identified the DEGs between Flower_1–4 and Receptacle_6–7 using the same analysis pipeline as described above for FM and REM. Only 200 DEGs were identified between Flower_1–4 and Receptacle_6–7 (Supplementary Data 2), in a sharp contrast to the 1,161 DEGs between FM and REM. Moreover, 54 out of the 131 up-regulated genes and 26 out of the 69 down-regulated genes were not included in the DEGs between FM and REM (Supplementary Figure S1B). Overall, the new transcriptomes generated in this study revealed a higher number of DEGs compared to the previous LCM data. The analyses also suggest that the techniques employed can significantly affect the RNA-seq results.
Dynamic Expression of DEGs During Meristem Development
Combined the three pairwise comparisons (SAM and FM, FM and REM, SAM and REM), a total of 3,009 genes were found to be differentially expressed (fold change >2, padj ≤0.05). These genes were assigned into nine gene clusters with dynamic and distinct expression patterns using the k-means clustering algorithm (Supplementary Data 3). Of these, cluster 4, cluster 2, and cluster 7 contained genes predominantly expressed in the SAM, FM, and REM, respectively (Figure 3A). Cluster 4 contained the greatest number of genes, indicating the uniqueness or higher complexity of SAM. Expression of the cluster 8 genes was gradually increased from SAM to REM. In contrast, cluster 3 genes exhibited an opposing expression trend. In addition, cluster 1 genes were abundantly expressed in both SAM and FM. Cluster 6 genes were abundantly expressed in both FM and REM. Unexpectedly, cluster 5 and 9 genes were abundantly expressed in both SAM and REM. The genes in the nine clusters were also subjected to the GO analysis, respectively (Supplementary Data 3). Four out of the nine clusters (clusters 2, 5, 6, and 7) were found to possess enriched GO terms in the ‘Biological Process’ category.
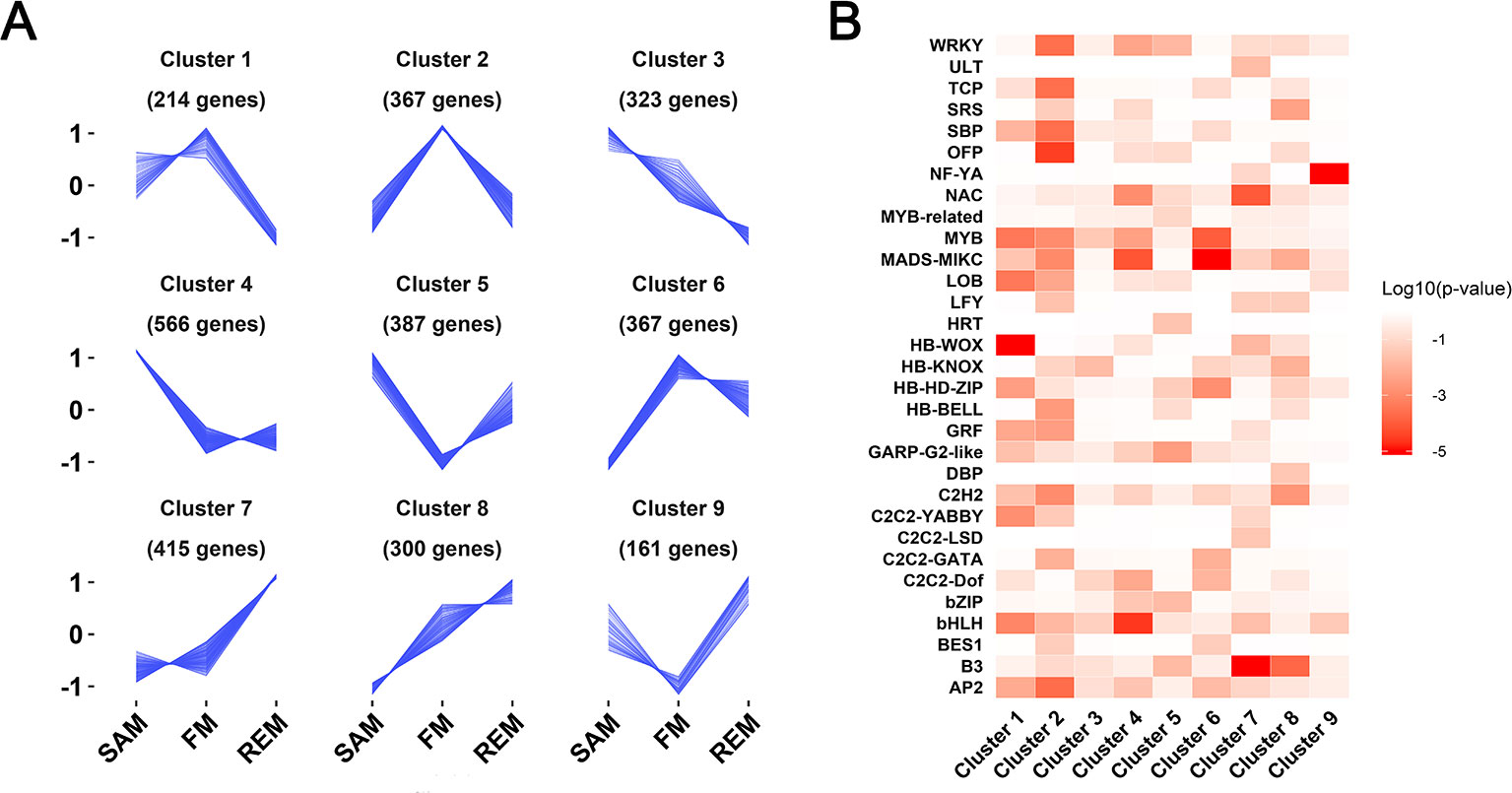
Figure 3 Nine gene clusters with distinct expression patterns. (A) Line plot showing the expression trends of nine gene clusters containing a total of 3,009 differentially expressed genes. Y-axis represents the normalized z-score obtained from the average TPM of three biological replicates. The number of genes in each cluster is indicated. (B) Heatmap showing the enriched transcription factor families in each of the nine gene clusters. Enrichment is indicated by the log10 transformed P values.
Transcription factor genes play essential roles in the regulatory network of meristem maintenance and differentiation (Chen et al., 2018; Tian et al., 2019). Among the 3,009 DEGs that were grouped into nine clusters, 336 genes encode transcription factors belonging to 46 families (Supplementary Data 3). The enriched transcription factor families in each cluster were shown by a heatmap representing the log10 transformed probabilities (Figure 3B). Several clusters possess enriched transcription factor families. For instance, there are four HB-WOX family genes in cluster 1, which are highly expressed in the SAM and FM, but significantly reduced in the REM. Cluster 6 (lowly expressed in the SAM but highly expressed in FM and REM) is enriched with several flower development genes of the MADS-box family, including three B genes (FvAP3, FvH4_1g12260; FvPIa, FvH4_2g27860; FvPIb, FvH4_2g27870) and two E genes (FvSEP4, FvH4_5g13510; FvSEP1-like, FvH4_4g29610). Intriguingly, the B3 family genes (Swaminathan et al., 2008) are preferentially expressed in the REM (cluster 7), which may be involved in receptacle or carpel development. The NF-YA family, encoding a subunit in the heterotrimeric NF-Y transcription factor complex, is enriched in cluster 9, whose functions in the receptacle or carpel development need further investigations.
Examination of Genes in Flowering Time Regulation
Since SEM gives rise to leaves and ultimately flowers, FM and REM give rise to floral organs, we investigated 149 genes whose homologs regulate flowering time or SAM, leaf, and flower development in other plant species (Supplementary Data 4). Firstly, we looked at the flowering time genes with functional studies in strawberry. A key gene regulating flowering time in wild strawberry is FvTFL1, whose mutation caused the day-neutral as it encodes a floral repressor in the photoperiod pathway (Iwata et al., 2012; Koskela et al., 2012). FvTFL1 is predominantly expressed in the shoot apex of short-day F. vesca grown under long days as examined by RNA in situ (Koskela et al., 2012). TFL1 expression is promoted by FvSOC1, the homolog of SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1, which is highly expressed under LD in all tissues except flowers (Mouhu et al., 2013). In contrast, two homologs of FLOWERING LOCUS T, the florigen, are present in F. vesca, called FvFT1 and FvFT2. FvFT1 is only expressed in old leaves, considered as the ortholog of FT, whereas FvFT2 is expressed exclusively in flower buds (Koskela et al., 2012). Expression patterns of these genes were experimentally validated in F. vesca, providing good quality test of our data. Consistent with these results, FvSOC1 and FvTFL1 are highly expressed in the SAM, while FvFT1 is not expressed in any of the three tissues (Figure 4A). In contrast, FvFT2 is expressed in the REM indicating that it may have a different function from FvFT1. FvFT1 and FvTFL1 are members of the phosphatidylethanolamine binding protein (PEBP) family (Karlgren et al., 2011). We further identified other PEBP family members in F. vesca, and found that FvATC and FvBFT are also greatly expressed in the SAM, suggesting potential functions in the control of flowering time. Consistently, ATC is the antiflorigen expressed in old leaves and transported to the shoot apical meristem in Arabidopsis (Huang et al., 2012). NsCET1, the homolog of ATC in tobacco (Nicotiana sylvestris), is demonstrated to move from leaf to the shoot apex via long distance transport of mRNA molecules (Huang et al., 2018). The Arabidopsis BROTHER OF FT AND TFL1 (BFT) has a TFL1-like activity and is expressed in the shoot apical meristem, young leaf and axillary inflorescence meristem (Yoo et al., 2010). These analyses helped identify candidate genes that may participate in flowering time regulation in F. vesca.
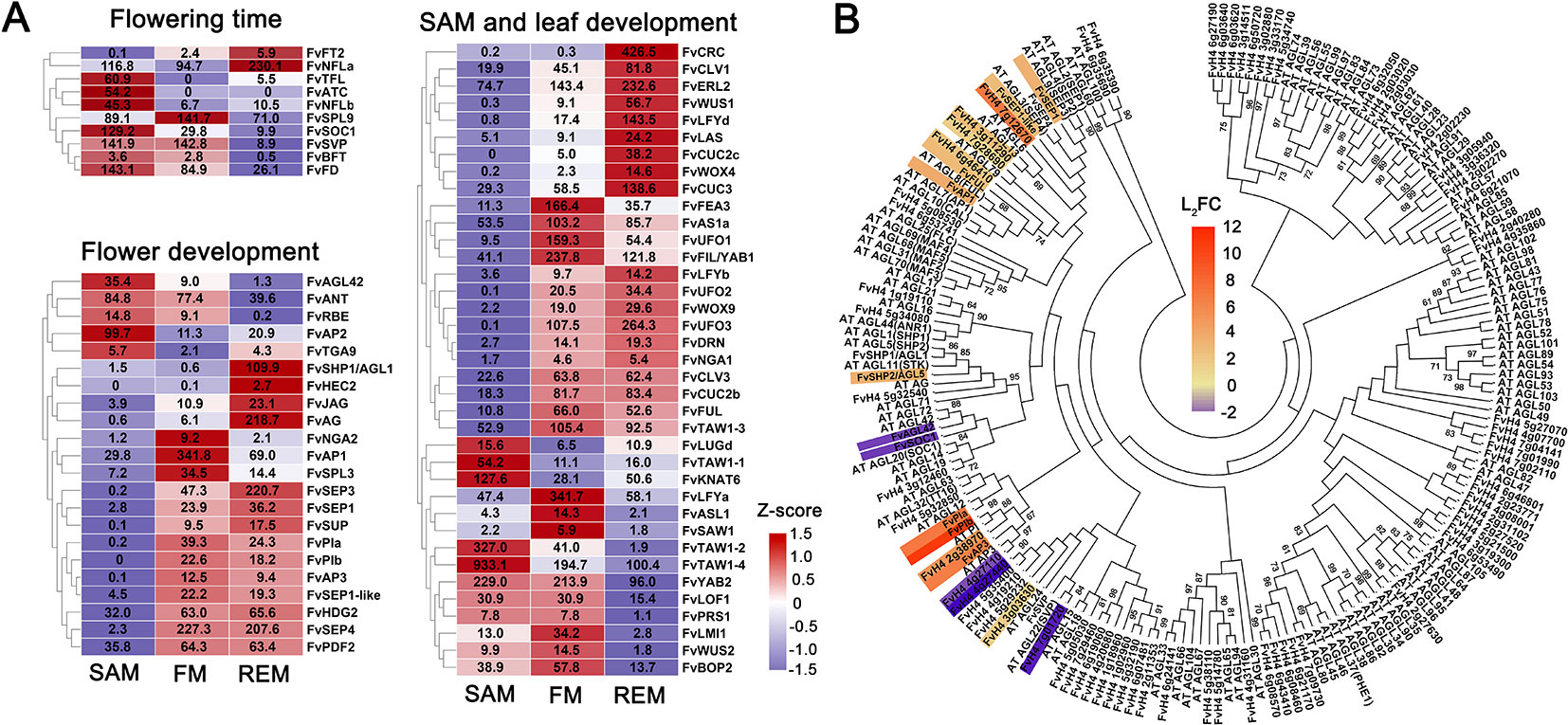
Figure 4 DEGs with known homologs and the MADS-box genes. (A) Heatmap showing the expression patterns of differentially expressed genes regulating the flowering time and development of SAM, leaf, and flower. Z-score obtained from averaged TPM of three biological replicates was used. The numbers in the heatmap indicate the TPM value of each gene in the tissues. (B) Phylogenetic tree showing all the MADS-box genes in F. vesca and Arabidopsis. The tree was generated by the Maximum Likelihood algorithm and bootstrap analysis (1,000 replicates) using MEGA 7. The differentially expressed MADS-box genes between the SAM and FM in our data were indicated with a scaled color (L2FC, log2 fold change). Red indicates up-regulation, and blue indicates down-regulation in the FM.
Analysis of Genes With Roles in SAM and Leaf Development
Our SAM samples consist of both the shoot apical meristem and a young leaf. Thus we expect to see high expression of the key genes regulating the development of both tissues. First, we checked the expression of SAM genes. Among the 149 genes, FvTAW1-2 and FvTAW1-4 are predominantly expressed in the SAM, which are the homologs of TAWAWA1, a regulator of meristem in rice (Yoshida et al., 2013). In F. vesca, there are two close homologs of WUS, FvWUS1 (FvH4_3g04400/gene30464) and FvWUS2 (FvH4_1g11910/gene14621) (Hollender et al., 2014), which share 36% and 38% protein sequence identity with WUS, respectively. Although the identity scores are moderate, they both possess the conserved homeobox (HB) domain and the WUS_box and EAR motifs (Hollender et al., 2014). According to the RNA-seq data, FvWUS1 is barely expressed in the SAM but gradually up-regulated in the FM and REM; in contrast, FvWUS2 is more highly expressed in the SAM and FM compared to REM (Figure 4A). However, the FvWUS2 transcripts are most abundant in anthers (Hollender et al., 2014). It will be interesting to determine if FvWUS1, FvWUS2, or both are the orthologs of Arabidopsis WUS with critical roles in SAM maintenance. In addition, FvCLAVATA1, 2, 3 are abundantly expressed in all three tissue samples, consistent with their roles in Arabidopsis meristems (Supplementary Data 4). The REM in this study was obtained from flowers at stages 6–7, when expression levels of WUS, CLV1, and CLV3 start to decline in Arabidopsis FM (Clark et al., 1997; Schoof et al., 2000; Xu et al., 2018). Therefore, the abundant expression of these genes in strawberry REM may suggest that the strawberry REM needs to maintain its meristematic activity for an extended period of time to ensure continued enlargement of the receptacle leading to the production of more carpels.
Additionally, a number of known genes important for leaf development are also highly expressed in the SAM, which contains of young leaves. For instance, some leaf development genes are specifically expressed in the adaxial or abaxial domain of young leaves to establish the adaxial-abaxial polarity. Several strawberry homologs of the adaxial-abaxial genes are abundantly expressed in the SAM including an HD-ZIPIII transcription factor PHAVOLUTA (FvPHV), abaxially expressed YABBYs (FvYAB2, FvYAB5), and ASYMMETRIC LEAVES1, 2 genes (FvAS1a, FvAS1b, and FvAS2) (McConnell et al., 2001; Lin et al., 2003; Xu et al., 2003; Stahle et al., 2009). Given that the SAM tissue we isolated contains young leaf primordia, it is not surprising that important leaf development genes are also identified.
Expression Patterns of Flower Development Genes
In contrast to SAM, FM and REM do not contain leaf primordia and are producers of floral organs, with REM essentially a late stage FM. Expression patterns of 37 homologs of flower development genes in F. vesca were shown (Figure 4A), including ABCE genes whose expression were previously characterized using LCM-dissected F. vesca tissues (Hollender et al., 2014). Here, we revisited their expression patterns in hand dissected SAM, FM, and REM. Consistently, most of them are highly expressed in the FM and REM (Figure 4A). One exception is FvAP2, which is more highly expressed in the SAM. Previous study in Arabidopsis showed that AP2 is involved in the stem cell maintenance in the SAM (Wurschum et al., 2006), hence FvAP2 might play similar roles in strawberry. Additionally, two floral meristem identity genes, FvLFYa and FvAP1, are greatly induced in FM (Figure 4A), consistent with the expression patterns of their homologs in Arabidopsis (Mandel et al., 1992; Weigel et al., 1992).
Next, we examined candidate floral MADS-box genes that may play important roles in strawberry flower development. Based on the BLAST search for the presence of SRF-TF (PF00319) domain, a total of 84 MADS-box genes were identified in the F. vesca genome version4.0.a2 (Li et al., 2019). Among them, 27 genes are expressed at a level greater than 2 TPM in one of the three meristems, including 12 type I MADS-box genes (M-box only) and 15 type II MADS-box genes (M-box and K-box) (Supplementary Data 4). Moreover, a number of the MADS-box genes are differentially expressed in the FM compared to SAM (Figure 4B). Among the type I genes, the SAM-specific gene FvAGL42 (FvH4_7g28740) is a homolog of Arabidopsis SOC1-like gene AGL42, which promotes Arabidopsis flowering in the shoot apical and axillary meristems through the gibberellins-dependent pathway (Dorca-Fornell et al., 2011). There are several MADS-box genes that are greatly induced in the FM (Figure 4B), whose functions await further investigation.
Expression Patterns of the Hormone Pathway Genes
Cytokinin is a key positive regulatory hormone in meristem maintenance and interacts extensively with the WUS/CLV module at the biosynthesis, degradation, and signaling processes in the SAM of Arabidopsis (Bartrina et al., 2011; Kieber and Schaller, 2014; Lee et al., 2019). We examined expression patterns of the cytokinin pathway genes in strawberry meristems which were previously identified from F. vesca (Supplementary Data 5) (Kang et al., 2013). A total of 19 cytokinin pathway genes are differentially expressed among the three meristem tissues (Figure 5A). For instance, the biosynthetic genes FvLOG3 (LONELY GUY) and FvLOG6, the receptor gene FvAHK4, and four downstream ARR genes are predominantly expressed in the SAM (Figure 5A), indicating a high cytokinin level and active cytokinin signaling in the SAM. In the FM, three CYTOKININ OXIDASE (CKX) genes are highly expressed, suggesting possibly active degradation of cytokinin. Of note, the cytokinin biosynthesis is greatly enhanced in the REM, supported by the up-regulation of four LOG genes, consistent with the rapid growth and enlargement of REM at these stages. These results indicate that cytokinin should also act as a pivotal hormone during the meristem development in strawberry.
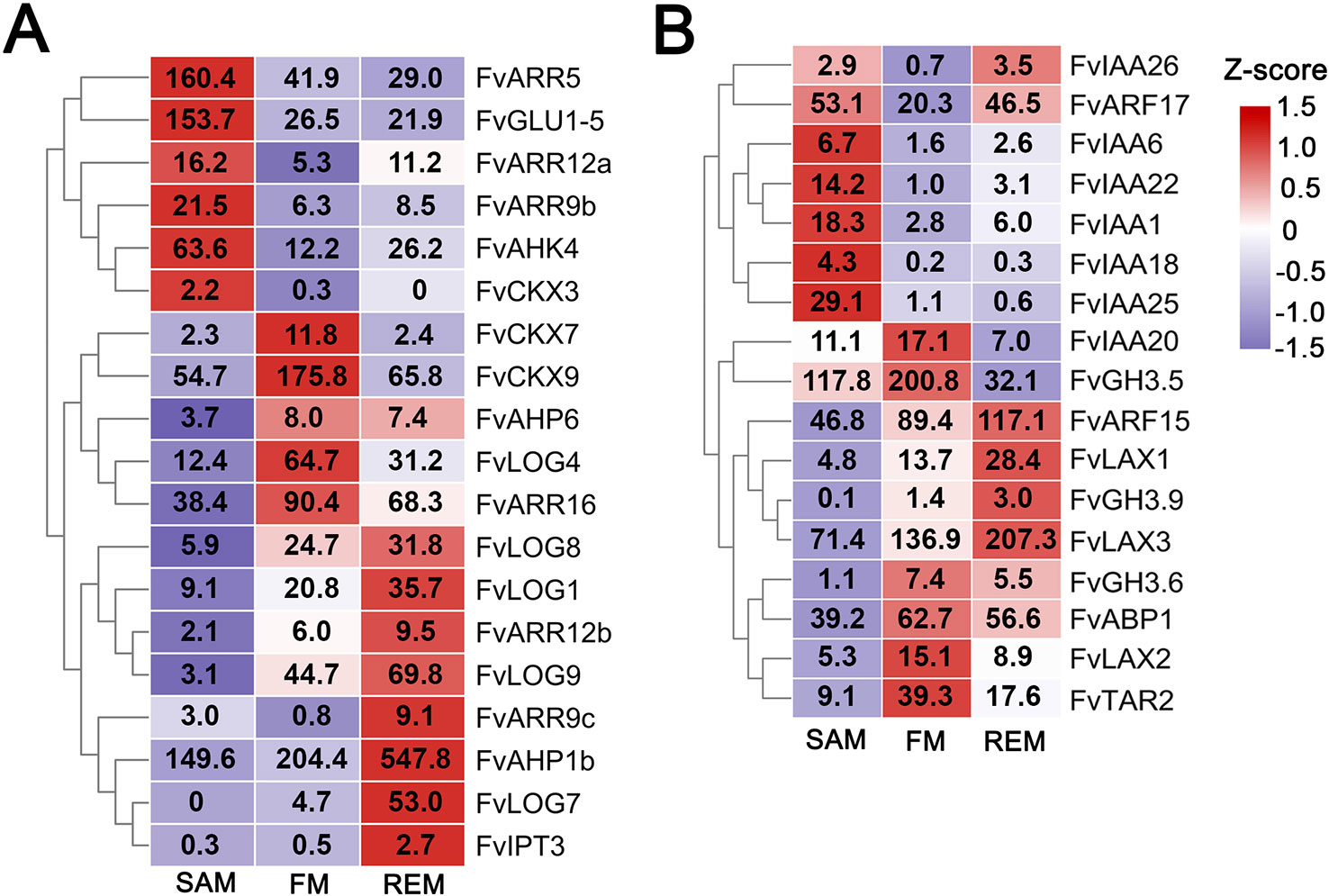
Figure 5 DEGs in the cytokinin and auxin pathways. (A) Heatmap showing the expression patterns of differentially expressed cytokinin pathway genes. (B) Heatmap showing the expression patterns of differentially expressed auxin pathway genes. Z-score obtained from averaged TPM of three biological replicates was used. The numbers in the heatmap indicate the TPM value of each gene in the tissues.
Auxin level and signaling in the PZ of the SAM play important roles in the initiation of leaf and flower primordia and in regulating the phyllotaxis (Sassi and Vernoux, 2013). There are totally 17 auxin pathway genes that were differentially expressed in the three meristems. Noticeably, a number of the AUXIN RESPONSE FACTOR (ARF) and Aux/IAA transcription factor family genes are abundantly expressed in the SAM as well as the other two meristems (Figure 5B, Supplementary Data 5), consistent with the expression study of their Arabidopsis homologs (Vernoux et al., 2011). Especially, FvARF2 (gene08492/FvH4_2g38760), the homolog of MONOPTEROS/ARF5 that helps maintain stem cell homeostasis in the SAM (Vidaurre et al., 2007; Luo et al., 2018), is highly expressed in all three meristems. In addition, three LAX genes, coding for the auxin influx transporters (Peret et al., 2012), are abundantly expressed in the FM and REM, indicating the importance of auxin transport in their development.
The Co-Expression Gene Module Associated With Meristem Tissues
Taking advantage of the RNA-seq data generated from 43 hand dissected tissues in F.vesca (Kang et al., 2013; Hollender et al., 2014; Toljamo et al., 2016; Hawkins et al., 2017) plus three meristem tissues generated here, we constructed a co-expression network using WGCNA (Langfelder and Horvath, 2008). The TPM values of 26,192 genes in these 46 tissues were used for constructing the network, resulting in 31 distinct co-expression modules, each with a different expression profile (Figure 6A). Among these modules, the cyan module contains a total of 347 genes that are abundantly expressed in the three meristem tissues as shown by the module eigengene (Figure 6A; Supplementary Data 6). The gene network of 168 genes with the edge weight higher than 0.2 in the cyan module was visualized by Cytoscape (Figure 6B) (Smoot et al., 2010). We found that the cyan module was enriched with transcription factor genes with roles in meristems as mentioned previously (Figure 4) including FvWUS1, FvLFYs, FvUFOs and FvCUC2s. In addition, some genes constitutively expressed in the three meristem tissues were also identified, such as FvH4_3g43590 that may encode a CLAVATA3/ESR (CLE)-related protein, and the lateral boundary gene FvLOB (FvH4_5g05970). The genes in this cyan module might play roles in the regulation of meristem development.
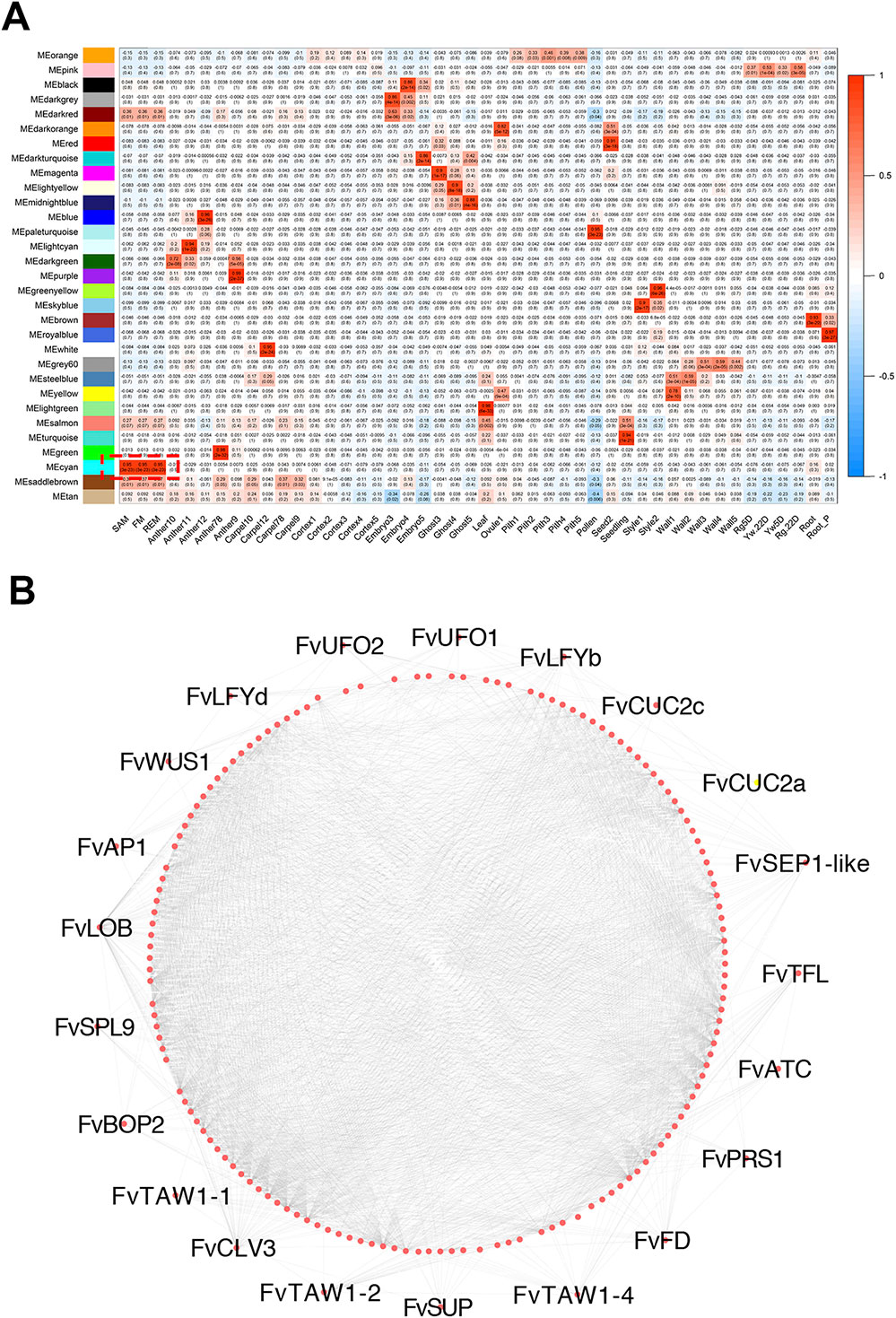
Figure 6 Meristem-associated gene module identified by WGCNA. (A) Heatmap showing module-tissue associations of the gene network. Each row corresponds to a module. Each column corresponds to a specific tissue. The color of each cell at the row-column intersection indicates the eigengene expression value. (B) The correlation network of the meristem-associated cyan module. 168 genes with edge weight higher than 0.2 were visualized by Cytoscape. Each dot represents one gene. Lines represent significantly positive correlation between the connected genes.
Detailed Analysis of the FvWUS1 Expression in Flowers
According to the RNA-seq data, expression of FvWUS1 is gradually increased from the SAM to REM (Figure 4A), which differs from its homologs in other species. To validate this result, the 2,076 bp promoter of FvWUS1 upstream of the translational start codon was isolated and used to drive the β-glucuronidase (GUS) reporter. The FvWUS1pro::GUS construct was stably transformed into the wild type F. vesca variety Hawaii 4. A total of five independent transgenic lines were validated by PCR and then characterized. The SAM and flower buds at stages 1–7 were stained with X-Gluc. In the SAM, no blue color was detected (Figure 7A), in agreement with the very low expression level of FvWUS1 in SAM revealed by the RNA-seq data. In the stages 1–4 flowers, the blue color was detected in the entire FM, but darker in the center region. In stages 6 and 8 flowers, the blue color was present in the young carpels and anthers, which differs dramatically from the restricted expression of WUS in the OC (organizing center) of SAM and FM in Arabidopsis (Mayer et al., 1998).
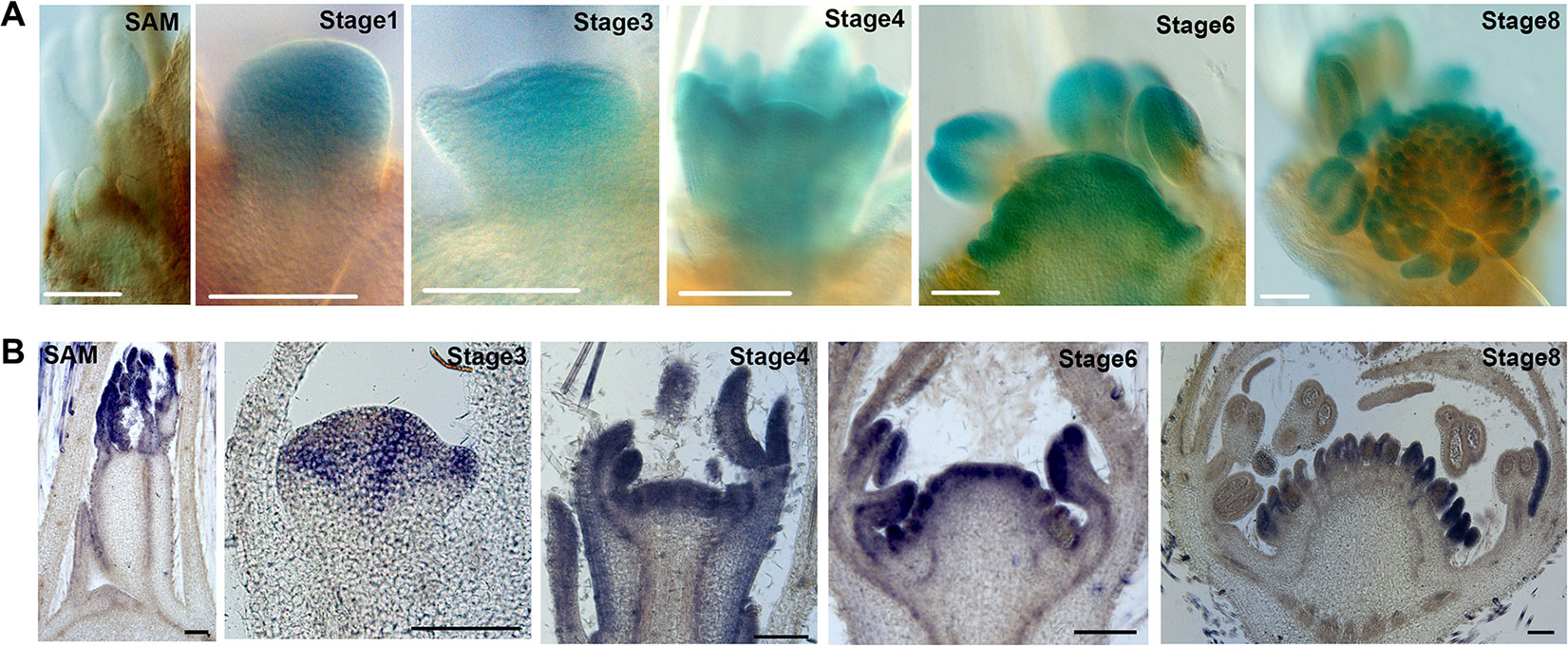
Figure 7 Expression of FvWUS1 in the SAM and young flower buds of F. vesca. (A) GUS expression of the FvWUS1pro::GUS reporter lines in the SAM and flower buds at stages 1, 3, 4, 6, and 8. The SAM and young flower buds were dissected from five independent T0 transgenic lines. (B) Detection of the FvWUS1 transcripts in the SAM with a young leaf and flower buds at stages 3, 4, 6, and 8. 7 μm longitudinal sections of fixed tissues were used. No signal was detected by the control probe. Scale bars: 100 μm.
Since the FvWUS1pro::GUS transgene could miss cis-regulatory element, RNA in situ is more accurate at revealing endogenous FvWUS expression. Therefore, in situ hybridization was conducted in the SAM and young flower buds. Since FvWUS1 and FvWUS2 sequences are highly similar to each other, especially at the 3’ end (Supplementary Figure S2), a 163 bp fragment at the 5’ end was used as an FvWUS1-specific probe. Consistent with the result of FvWUS1pro::GUS reporter expression, FvWUS1 mRNA is detected in the young FM, at the apex of young receptacle, and carpel and stamen; it is not detected in SAM (Figure 7B). Therefore, the expression of FvWUS1 is distinct from WUS expression in Arabidopsis and cucumber (Mayer et al., 1998; Zhao et al., 2018). However, the WUS homologs don’t necessarily have exactly the same expression patterns and functions among different species, such as in rice and maize (Nardmann and Werr, 2006; Suzuki et al., 2019). These results confirmed that FvWUS1 is highly expressed in flower buds at early stages, raising a possibility that FvWUS1 might play a critical role in regulating the number of carpels and ultimately the receptacle fruit size.
Conclusion
In this work, we profiled genome-wide transcriptomic landscape of three different tissues, SAM, FM and REM in the diploid strawberry F. vesca by Illumina RNA-seq. Detailed analysis was performed on the DEGs derived from pairwise comparisons with a focus on homologs of the crucial transcription factor and hormone pathway genes with roles in meristem maintenance, meristem identity, and meristem differentiation. We found that flower and meristem genes, characterized previously in other species, exhibited both conserved and distinctive expression patterns in strawberry. A novel finding is that FvWUS1, a homolog of Arabidopsis WUS, is broadly expressed in young flower meristem, indicating its potential function in floral organ initiation and development. These data provides valuable resources for future functional studies of genes with roles in meristem maintenance and differentiation in strawberry.
Data Availability Statement
The datasets generated for this study can be found in the Sequence Read Archive (SRA) at NCBI (http://www.ncbi.nlm.nih.gov/sra) under the accession number SRP115444.
Author Contributions
CK and ZL conceived and supervised this study. YL analyzed the data. JF, LC, CD, and QG performed the experiments. CK and YL wrote the manuscript. ZL revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (2018YFD1000102) and National Natural Science Foundation of China (31572098, 31772274, and 31822044).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.01624/full#supplementary-material
References
Arora, R., Agarwal, P., Ray, S., Singh, A. K., Singh, V. P., Tyagi, A. K., et al. (2007). MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics 8, 242. doi: 10.1186/1471-2164-8-242
Bartrina, I., Otto, E., Strnad, M., Werner, T., Schmülling, T. (2011). Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and, thus, seed yield in Arabidopsis thaliana. Plant Cell. 23 (1), 69–80. doi: 10.1105/tpc.110.079079
Brand, U., Fletcher, J. C., Hobe, M., Meyerowitz, E. M., Simon, R. (2000). Dependence of stem cell fate in Arabidopsis on a feedback loop regulated by CLV3 activity. Science 289 (5479), 617–619. doi: 10.1126/science.289.5479.617
Braybrook, S. A., Kuhlemeier, C. (2010). How a Plant Builds Leaves. Plant Cell 22 (4), 1006–1018. doi: 10.1105/tpc.110.073924
Causier, B., Schwarz-Sommer, Z., Davies, B. (2010). Floral organ identity: 20 years of ABCs. Semin. Cell Dev. Biol. 21 (1), 73–79. doi: 10.1016/j.semcdb.2009.10.005
Chen, D., Yan, W., Fu, L. Y., Kaufmann, K. (2018). Architecture of gene regulatory networks controlling flower development in Arabidopsis thaliana. Nat. Commun. 9 (1), 4534. doi: 10.1038/s41467-018-06772-3
Clark, S. E., Williams, R. W., Meyerowitz, E. M. (1997). The CLAVATA1 gene encodes a putative receptor kinase that controls shoot and floral meristem size in Arabidopsis. Cell 89 (4), 575–585. doi: 10.1016/s0092-8674(00)80239-1
Coen, E. S., Meyerowitz, E. M. (1991). The war of the whorls: genetic interactions controlling flower development. Nature 353 (6339), 31–37. doi: 10.1038/353031a0
Daum, G., Medzihradszky, A., Suzaki, T., Lohmann, J. U. (2014). A mechanistic framework for noncell autonomous stem cell induction in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 111 (40), 14619–14624. doi: 10.1073/pnas.1406446111
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29 (1), 15–21. doi: 10.1093/bioinformatics/bts635
Dorca-Fornell, C., Gregis, V., Grandi, V., Coupland, G., Colombo, L., Kater, M. M. (2011). The Arabidopsis SOC1-like genes AGL42, AGL71 and AGL72 promote flowering in the shoot apical and axillary meristems. Plant J. 67 (6), 1006–1017. doi: 10.1111/j.1365-313X.2011.04653.x
Edger, P. P., VanBuren, R., Colle, M., Poorten, T. J., Wai, C. M., Niederhuth, C. E., et al. (2018). Single-molecule sequencing and optical mapping yields an improved genome of woodland strawberry (Fragaria vesca) with chromosome-scale contiguity. Gigascience 7 (2), 1–7. doi: 10.1093/gigascience/gix124
Edger, P. P., Poorten, T. J., VanBuren, R., Hardigan, M. A., Colle, M., McKain, M. R., et al. (2019). Origin and evolution of the octoploid strawberry genome. Nat. Genet. 51 (3), 541–547. doi: 10.1038/s41588-019-0356-4
Feng, J., Dai, C., Luo, H., Han, Y., Liu, Z., Kang, C. (2019). Reporter gene expression reveals precise auxin synthesis sites during fruit and root development in wild strawberry. J. Exp. Bot. 70 (2), 563–574. doi: 10.1093/jxb/ery384
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2013). Pfam: the protein families database. Nucleic Acids Res. 42 (D1), D222–D230. doi: 10.1093/nar/gkt1223
Fletcher, J. C., Brand, U., Running, M. P., Simon, R., Meyerowitz, E. M. (1999). Signaling of cell fate decisions by CLAVATA3 in Arabidopsis shoot meristems. Science 283 (5409), 1911–1914. doi: 10.1126/science.283.5409.1911
Hawkins, C., Caruana, J., Li, J., Zawora, C., Darwish, O., Wu, J., et al.(2017). An eFP browser for visualizing strawberry fruit and flower transcriptomes. Hortic. Res. 4, 17029. doi: 10.1038/hortres.2017.29
Hollender, C. A., Geretz, A. C., Slovin, J. P., Liu, Z. (2012). Flower and early fruit development in a diploid strawberry, Fragaria vesca. Planta 235 (6), 1123–1139. doi: 10.1007/s00425-011-1562-1
Hollender, C. A., Kang, C., Darwish, O., Geretz, A., Matthews, B. F., Slovin, J., et al. (2014). Floral transcriptomes in woodland strawberry uncover developing receptacle and anther gene networks. Plant Physiol. 165 (3), 1062–1075. doi: 10.1104/pp.114.237529
Huang, N. C., Jane, W. N., Chen, J., Yu, T. S. (2012). Arabidopsis thaliana CENTRORADIALIS homologue (ATC) acts systemically to inhibit floral initiation in Arabidopsis. Plant J. 72 (2), 175–184. doi: 10.1111/j.1365-313X.2012.05076.x
Huang, N. C., Luo, K. R., Yu, T. S. (2018). Mobility of Antiflorigen and PEBP mRNAs in tomato-tobacco heterografts. Plant Physiol. 178 (2), 783–794. doi: 10.1104/pp.18.00725
Iwata, H., Gaston, A., Remay, A., Thouroude, T., Jeauffre, J., Kawamura, K., et al. (2012). The TFL1 homologue KSN is a regulator of continuous flowering in rose and strawberry. Plant J. 69 (1), 116–125. doi: 10.1111/j.1365-313X.2011.04776.x
Kang, C., Darwish, O., Geretz, A., Shahan, R., Alkharouf, N., Liu, Z. (2013). Genome-scale transcriptomic insights into early-stage fruit development in woodland strawberry Fragaria vesca. Plant Cell 25 (6), 1960–1978. doi: 10.1105/tpc.113.111732
Karlgren, A., Gyllenstrand, N., Kallman, T., Sundstrom, J. F., Moore, D., Lascoux, M., et al. (2011). Evolution of the PEBP gene family in plants: functional diversification in seed plant evolution. Plant Physiol. 156 (4), 1967–1977. doi: 10.1104/pp.111.176206
Kieber, J. J., Schaller, G. E. (2014). Cytokinins. Arabidopsis Book 12, e0168. doi: 10.1199/tab.0168
Koskela, E. A., Mouhu, K., Albani, M. C., Kurokura, T., Rantanen, M., Sargent, D. J., et al. (2012). Mutation in TERMINAL FLOWER1 reverses the photoperiodic requirement for flowering in the wild strawberry Fragaria vesca. Plant Physiol. 159 (3), 1043–1054. doi: 10.1104/pp.112.196659
Krizek, B. A., Fletcher, J. C. (2005). Molecular mechanisms of flower development: an armchair guide. Nat. Rev. Genet. 6 (9), 688–698. doi: 10.1038/nrg1675
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi: 10.1093/molbev/msw054
Langfelder, P., Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 9, 559. doi: 10.1186/1471-2105-9-559
Laux, T., Mayer, K., Berger, J., Jurgens, G. (1996). The WUSCHEL gene is required for shoot and floral meristem integrity in Arabidopsis. Development 122 (1), 87–96. doi: 10.1016/S0070-2153(08)60713-9
Lee, Z. H., Hirakawa, T., Yamaguchi, N., Ito, T. (2019). The roles of plant hormones and their interactions with regulatory genes in determining meristem activity. Int. J. Mol. Sci. 20 (16), 4065. doi: 10.3390/ijms20164065
Li, Y., Wei, W., Feng, J., Luo, H., Pi, M., Liu, Z., et al. (2018). Genome re-annotation of the wild strawberry Fragaria vesca using extensive Illumina- and SMRT-based RNA-seq datasets. DNA Res. 25 (1), 61–70. doi: 10.1093/dnares/dsx038
Li, Y., Pi, M., Gao, Q., Liu, Z., Kang, C. (2019). Updated annotation of the wild strawberry Fragaria vesca V4 genome. Hortic. Res. 6, 61. doi: 10.1038/s41438-019-0142-6
Liao, Y., Smyth, G. K., Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30 (7), 923–930. doi: 10.1093/bioinformatics/btt656
Lin, W. C., Shuai, B., Springer, P. S. (2003). The Arabidopsis LATERAL ORGAN BOUNDARIES-domain gene ASYMMETRIC LEAVES2 functions in the repression of KNOX gene expression and in adaxial-abaxial patterning. Plant Cell 15 (10), 2241–2252. doi: 10.1105/tpc.014969
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. doi: 10.1186/s13059-014-0550-8
Luo, L., Zeng, J., Wu, H., Tian, Z., Zhao, Z. (2018). A molecular framework for auxin-controlled homeostasis of shoot stem cells in arabidopsis. Mol. Plant 11 (7), 899–913. doi: 10.1016/j.molp.2018.04.006
Mandel, M. A., Gustafson-Brown, C., Savidge, B., Yanofsky, M. F. (1992). Molecular characterization of the Arabidopsis floral homeotic gene APETALA1. Nature 360 (6401), 273–277. doi: 10.1038/360273a0
Mayer, K. F., Schoof, H., Haecker, A., Lenhard, M., Jurgens, G., Laux, T. (1998). Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell 95 (6), 805–815. doi: 10.1016/s0092-8674(00)81703-1
McConnell, J. R., Emery, J., Eshed, Y., Bao, N., Bowman, J., Barton, M. K. (2001). Role of PHABULOSA and PHAVOLUTA in determining radial patterning in shoots. Nature 411 (6838), 709–713. doi: 10.1038/35079635
Mouhu, K., Kurokura, T., Koskela, E. A., Albert, V. A., Elomaa, P., Hytonen, T. (2013). The Fragaria vesca Homolog of SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 Represses Flowering and Promotes Vegetative Growth. Plant Cell 25 (9), 3296–3310. doi: 10.1105/tpc.113.115055
Nardmann, J., Werr, W. (2006). The shoot stem cell niche in angiosperms: expression patterns of WUS orthologues in rice and maize imply major modifications in the course of mono- and dicot evolution. Mol. Biol. Evol. 23 (12), 2492–2504. doi: 10.1093/molbev/msl125
Parenicova, L., de Folter, S., Kieffer, M., Horner, D. S., Favalli, C., Busscher, J., et al. (2003). Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell 15 (7), 1538–1551. doi: 10.1105/tpc.011544
Peret, B., Swarup, K., Ferguson, A., Seth, M., Yang, Y., Dhondt, S., et al. (2012). AUX/LAX genes encode a family of auxin influx transporters that perform distinct functions during Arabidopsis development. Plant Cell 24 (7), 2874–2885. doi: 10.1105/tpc.112.097766
Saeed, A. I., Sharov, V., White, J., Li, J., Liang, W., Bhagabati, N., et al. (2003). TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34 (2), 374. doi: 10.2144/03342mt01
Sassi, M., Vernoux, T. (2013). Auxin and self-organization at the shoot apical meristem. J. Exp. Bot. 64 (9), 2579–2592. doi: 10.1093/jxb/ert101
Schoof, H., Lenhard, M., Haecker, A., Mayer, K. F., Jurgens, G., Laux, T. (2000). The stem cell population of Arabidopsis shoot meristems in maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell 100 (6), 635–644. doi: 10.1016/s0092-8674(00)80700-x
Shulaev, V., Sargent, D. J., Crowhurst, R. N., Mockler, T. C., Folkerts, O., Delcher, A. L., et al. (2011). The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 43 (2), 109–116. doi: 10.1038/ng.740
Slovin, J. P., Schmitt, K., Folta, K. M. (2009). An inbred line of the diploid strawberry Fragaria vesca f. semperflorens for genomic and molecular genetic studies in the Rosaceae. Plant Methods 5, 15. doi: 10.1186/1746-4811-5-15
Smaczniak, C., Immink, R. G., Angenent, G. C., Kaufmann, K. (2012). Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development 139 (17), 3081–3098. doi: 10.1242/dev.074674
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P.-L., Ideker, T. (2010). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27 (3), 431–432. doi: 10.1093/bioinformatics/btq675
Stahle, M. I., Kuehlich, J., Staron, L., von Arnim, A. G., Golz, J. F. (2009). YABBYs and the transcriptional corepressors LEUNIG and LEUNIG_HOMOLOG maintain leaf polarity and meristem activity in Arabidopsis. Plant Cell 21 (10), 3105–3118. doi: 10.1105/tpc.109.070458
Staudt, G. (2009). Strawberry biogeography, genetics and systematics. Acta Hortic 842, 71–84. doi: 10.17660/ActaHortic.2009.842.1
Suzuki, C., Tanaka, W., Tsuji, H., Hirano, H. Y. (2019). TILLERS ABSENT1, the WUSCHEL ortholog, is not involved in stem cell maintenance in the shoot apical meristem in rice. Plant Signal Behav. 14 (9), e1640565. doi: 10.1080/15592324.2019.1640565
Swaminathan, K., Peterson, K., Jack, T. (2008). The plant B3 superfamily. Trends Plant Sci. 13 (12), 647–655. doi: 10.1016/j.tplants.2008.09.006
Tian, C., Wang, Y., Yu, H., He, J., Wang, J., Shi, B., et al. (2019). A gene expression map of shoot domains reveals regulatory mechanisms. Nat. Commun. 10 (1), 141. doi: 10.1038/s41467-018-08083-z
Toljamo, A., Blande, D., Karenlampi, S., Kokko, H. (2016). Reprogramming of strawberry (Fragaria vesca) root transcriptome in response to phytophthora cactorum. PloS One 11 (8), e0161078. doi: 10.1371/journal.pone.0161078
Vernoux, T., Brunoud, G., Farcot, E., Morin, V., Van den Daele, H., Legrand, J., et al. (2011). The auxin signalling network translates dynamic input into robust patterning at the shoot apex. Mol. Syst. Biol. 7, 508. doi: 10.1038/msb.2011.39
Vidaurre, D. P., Ploense, S., Krogan, N. T., Berleth, T. (2007). AMP1 and MP antagonistically regulate embryo and meristem development in Arabidopsis. Development 134 (14), 2561–2567. doi: 10.1242/dev.006759
Weigel, D., Alvarez, J., Smyth, D. R., Yanofsky, M. F., Meyerowitz, E. M. (1992). LEAFY controls floral meristem identity in Arabidopsis. Cell 69 (5), 843–859. doi: 10.1016/0092-8674(92)90295-n
Wurschum, T., Gross-Hardt, R., Laux, T. (2006). APETALA2 regulates the stem cell niche in the Arabidopsis shoot meristem. Plant Cell 18 (2), 295–307. doi: 10.1105/tpc.105.038398
Xu, L., Xu, Y., Dong, A., Sun, Y., Pi, L., Xu, Y., et al. (2003). Novel as1 and as2 defects in leaf adaxial-abaxial polarity reveal the requirement for ASYMMETRIC LEAVES1 and 2 and ERECTA functions in specifying leaf adaxial identity. Development 130 (17), 4097–4107. doi: 10.1242/dev.00622
Xu, Y., Prunet, N., Gan, E. S., Wang, Y., Stewart, D., Wellmer, F., et al. (2018). SUPERMAN regulates floral whorl boundaries through control of auxin biosynthesis. EMBO J. 37 (11), e97499. doi: 10.15252/embj.201797499
Yadav, R. K., Perales, M., Gruel, J., Girke, T., Jonsson, H., Reddy, G. V. (2011). WUSCHEL protein movement mediates stem cell homeostasis in the Arabidopsis shoot apex. Genes Dev. 25 (19), 2025–2030. doi: 10.1101/gad.17258511
Ye, R., Zhou, F., Lin, Y. (2012). Two novel positive cis-regulatory elements involved in green tissue-specific promoter activity in rice (Oryza sativa L ssp.). Plant Cell Rep. 31 (7), 1159–1172. doi: 10.1007/s00299-012-1238-8
Yoo, S. J., Chung, K. S., Jung, S. H., Yoo, S. Y., Lee, J. S., Ahn, J. H. (2010). BROTHER OF FT AND TFL1 (BFT) has TFL1-like activity and functions redundantly with TFL1 in inflorescence meristem development in Arabidopsis. Plant J. 63 (2), 241–253. doi: 10.1111/j.1365-313X.2010.04234.x
Yoshida, A., Sasao, M., Yasuno, N., Takagi, K., Daimon, Y., Chen, R., et al. (2013). TAWAWA1, a regulator of rice inflorescence architecture, functions through the suppression of meristem phase transition. Proc. Natl. Acad. Sci. U.S.A. 110 (2), 767–772. doi: 10.1073/pnas.1216151110
Keywords: shoot apical meristem, flower meristem, receptacle meristem, RNA-seq, transcription factor, hormone, FvWUS1, F. vesca
Citation: Li Y, Feng J, Cheng L, Dai C, Gao Q, Liu Z and Kang C (2019) Gene Expression Profiling of the Shoot Meristematic Tissues in Woodland Strawberry Fragaria vesca. Front. Plant Sci. 10:1624. doi: 10.3389/fpls.2019.01624
Received: 03 September 2019; Accepted: 19 November 2019;
Published: 13 December 2019.
Edited by:
Stefan de Folter, Center for Research and Advanced Studies (CINVESTAV), MexicoReviewed by:
Paula Elomaa, University of Helsinki, FinlandChristopher Barbey, University of Florida, United States
Copyright © 2019 Li, Feng, Cheng, Dai, Gao, Liu and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunying Kang, ckang@mail.hzau.edu.cn
†These authors have contributed equally to this work