 Bartosz Sekula*
Bartosz Sekula* Zbigniew Dauter
Zbigniew Dauter- Synchrotron Radiation Research Section of Macromolecular Crystallography Laboratory, National Cancer Institute, Argonne, IL, United States
Plants are unique eukaryotes that can produce putrescine (PUT), a basic diamine, from arginine via a three-step pathway. This process starts with arginine decarboxylase that converts arginine to agmatine. Then, the consecutive action of two hydrolytic enzymes, agmatine iminohydrolase (AIH) and N-carbamoylputrescine amidohydrolase, ultimately produces PUT. An alternative route of PUT biosynthesis requires ornithine decarboxylase that catalyzes direct putrescine biosynthesis. However, some plant species lack this enzyme and rely only on agmatine pathway. The scope of this manuscript concerns the structural characterization of AIH from the model legume plant, Medicago truncatula. MtAIH is a homodimer built of two subunits with a characteristic propeller fold, where five αββαβ repeated units are arranged around the fivefold pseudosymmetry axis. Dimeric assembly of this plant AIH, formed by interactions of conserved structural elements from one repeat, is drastically different from that observed in dimeric bacterial AIHs. Additionally, the structural snapshot of MtAIH in complex with 6-aminohexanamide, the reaction product analog, presents the conformation of the enzyme during catalysis. Our structural results show that MtAIH undergoes significant structural rearrangements of the long loop, which closes a tunnel-shaped active site over the course of the catalytic event. This conformational change is also observed in AIH from Arabidopsis thaliana, indicating the importance of the closed conformation of the gate-keeping loop for the catalysis of plant AIHs.
Introduction
Biosynthesis of putrescine (PUT) starts from arginine (ARG) and follows one of the two pathways which comprise agmatine (AGM) or ornithine (ORN) biotransformation (Michael, 2017). The AGM route is important and widely spread among plants, algae, and prokaryotic organisms (Michael, 2016). First, ARG is decarboxylated to AGM by arginine decarboxylase (ADC). The later conversion of AGM to PUT in plants is carried out by agmatine iminohydrolase (AIH) and further by N-carbamoylputrescine amidohydrolase (CPA), an octameric protein with a quaternary structure resembling an incomplete left-handed helix (Sekula et al., 2016). AIH and CPA genes have been acquired by plants through an endosymbiotic gene transfer from the cyanobacterial ancestor of the chloroplast (Illingworth et al., 2003). Actually, plants are unique eukaryotes to biosynthesize PUT via the AGM biotransformation, which makes ADC, AIH, and CPA potential targets for herbicide design (Böger and Sandmann, 1989). In some bacteria, AGM-to-PUT conversion is also catalyzed by agmatine ureohydrolase (agmatinase) (Satishchandran and Boyle, 1986) and most of the cyanobacteria use this enzyme instead of AIH and CPA (Fuell et al., 2010). Some Gram-positive bacteria may also obtain PUT in a catabolic pathway which produces ATP from the carbamoyl phosphate obtained from the AGM-to-PUT transformation (Llacer et al., 2007). The second manner of PUT biosynthesis, the ORN route, is dominant for most eukaryotes, including animals and fungi, and involves ORN decarboxylation catalyzed by ornithine decarboxylase (ODC) (Janowitz et al., 2003). Some plant species like Arabidopsis thaliana and Physcomitrella patens do not have the ODC gene (Hanfrey et al., 2001) and they rely only on the AGM-to-PUT bioconversion. Other plants, which have preserved ODC, may obtain PUT either from AGM or ORN.
PUT is the starting backbone for larger polyamines (PAs) which are produced by specialized aminopropyltransferases, enzymes which use decarboxylated S-adenosylmethionine as a donor of the aminopropyl group. Therefore, the first transfer of the aminopropyl group to PUT, catalyzed by spermidine synthase (SPDS), yields triamine spermidine (SPD). SPD is the substrate for the second transfer which yields symmetrical spermine or unsymmetrical thermospermine. For a long time, it was elusive whether both tetraamines are biosynthesized in plants, but they are actually formed by two distinct proteins, spermine synthase (SPMS) and thermospermine synthase (TSPS). Aminopropyltransferases are distinguished by several structural features that favor each enzyme toward the specific PA production (Sekula and Dauter, 2018).
PAs are essential for the regulation of various physiological processes which secure the proper growth and development of higher plants (Takano et al., 2012; Jimenez-Bremont et al., 2014; Minocha et al., 2014; Tiburcio et al., 2014; Liu et al., 2015). The cationic character of PAs promotes their interactions with anionic proteins and nucleic acids, thus affecting transcription, translation (Gill and Tuteja, 2010; Igarashi and Kashiwagi, 2010; Tiburcio et al., 2014), and the rate of membrane transport (Pottosin et al., 2014; Pottosin and Shabala, 2014). SPD is an important donor of aminobutyl group for the posttranslational modification of the hypusine-containing translation elongation factor eIF5A in eukaryotes and archaea (Prunetti et al., 2016). PAs can also modulate the activity of antioxidant enzymes and thereby influence the concentration of reactive oxygen species (Radhakrishnan and Lee, 2013; Kamiab et al., 2014; Mostofa et al., 2014). PA accumulation is often related with its protective role for the environmental stress conditions and leads to an increase of stress tolerance of the plant (Capell et al., 2004; Alcazar et al., 2010; Wang et al., 2011; Berberich et al., 2015). Meanwhile, defects of PA biosynthesis pathway result in the retardation, sterility, and other developmental pathologies in plants (Hanzawa et al., 2000).
Herein, we describe the structural characterization of AIH from Medicago truncatula (MtAIH), the model legume plant. The enzyme is responsible for the second step of the AGM pathway of PUT biosynthesis, that is the hydrolytic conversion of AGM to N-carbamoylputrescine (NCP) with the release of ammonia. Plant AIHs, as well as CPAs, do not contain chloroplast-targeting peptides and they act in the cytoplasm. This is opposite to the first enzyme of the pathway, ADC, which initializes PUT biosynthesis in plastids (Illingworth et al., 2003). The advantage of AGM production outside plastids could be explained by the availability of AGM in the cytoplasm not only for PUT production but also for the biosynthesis of N-hydroxycinnamoyl conjugates, which may serve as precursors of defensive compounds (Burhenne et al., 2003). AIH belongs to one of the seven types of guanidine-modifying enzymes (GMEs) (Shirai et al., 2006). It is a member of the pentein superfamily that is characterized by the propeller-like arrangement of five repeated motifs that form a narrow channel with a central, negatively charged core (Hartzoulakis et al., 2007). The conserved catalytic triad of GMEs (His, Asp, and Cys) is responsible for a range of activities, which cover transferase and hydrolytic reactions on the guanidine-containing compounds (Hartzoulakis et al., 2007). Although AIHs from various plant species, including corn (Yanagisawa and Suzuki, 1981), soybean (Park and Cho, 1991), and maize (Yanagisawa, 2001) were isolated, there is no published structural characterization of any plant AIH available, except for unpublished reported entries in the Protein Data Bank (PDB) of AIH from A. thaliana (AtAIH, PDB ID 3H7K, 3H7C, 1VKP, Center for Eukaryotic Structural Genomics).
In this work, we present the high-resolution crystal structure of non-liganded MtAIH and the structure with the reaction product analog—6-aminohexanamide (AHX). This, combined with the in-solution small-angle X-ray scattering analysis, provides data for the characterization of this plant AIH and a detailed comparison of plant AIHs (MtAIH and AtAIH) with their bacterial orthologs.
Materials and Methods
Cloning, Overexpression, and Purification of MtAIH
In order to express and purify MtAIH (UniProt ID G7JT50), we used the protocol which was recently successfully applied in the studies of other plant enzymes (Ruszkowski et al., 2018; Sekula et al., 2018). Briefly, the following primers, forward: TACTTCCAATCCAATGCCCATGGCTTTCACATGCCTGCAGAAT and reverse: TTATCCACTTCCAATGTTACTAAATGGCTGGTTGTTGCTGAGTGAT and the cDNA from leaves of M. truncatula as a template were used in a polymerase chain reaction (PCR) prior to obtaining MtAIH open reading frame (MTR_4g112810) with encoded protein starting from codon number 11. The incorporation of MtAIH gene into the pMCSG68 vector (Midwest Center for Structural Genomics) was performed according to the ligase-independent cloning (Kim et al., 2011) protocol. The vector introduces an N-terminal His6-tag followed by the Tobacco Etch Virus (TEV) protease cleavage site to the cloned protein and the Ser-Asn-Ala linker that is not cleaved from the expressed protein. In the next step, the BL21 Gold E. coli competent cells (Agilent Technologies) were transformed with the vector containing the MtAIH gene. The cells were precultured at 37°C in LB medium with the addition of ampicillin (150 μg/ml) overnight. Next, 1.5% v/v of the culture was used as the inoculum of the fresh LB medium with ampicillin. It was cultured at 37°C until OD600 reached a value 1.0. In the next step, the culture was cooled to 10°C for 2 h and then the protein expression was induced with 0.5 mM of isopropyl-β-D-thiogalactopyranoside (IPTG). The protein overexpression was carried out at 18°C for 16 h. Before pelleting the cells in the centrifuge at 3,500 × g for 30 min, the culture was cooled to 4°C. Cell pellets were resuspended in 35 ml of the binding buffer [50 mM HEPES pH 7.4; 500 mM NaCl; 20 mM imidazole; 1 mM tris(2-carboxyethyl)phosphine, TCEP] and frozen at −80°C. Thawed cells were disrupted by sonication in an ice/water bath for 4 min (bursts of 4 s with 26-s intervals). Then, the cellular debris was pelleted by centrifugation at 25,000 × g for 30 min at 4°C.
The first step of MtAIH purification was performed on a column packed with 5 ml of HisTrap HP resin (GE Healthcare) connected to the Vac-Man laboratory vacuum manifold (Promega). The supernatant was applied to the column and washed five times with 40 ml of the binding buffer. The protein elution was performed with 20 ml of the elution buffer (50 mM HEPES pH 7.4; 500 mM NaCl; 400 mM imidazole; 1 mM TCEP). His6-tagged TEV protease (final concentration of 0.1 mg/ml) was used to cleave the His6-tag from MtAIH. This step was simultaneous to the overnight dialysis at 4°C against the dialysis buffer (50 mM HEPES pH 8.0; 500 mM NaCl; 1 mM TCEP). After dialysis, the sample was applied on HisTrap HP resin to remove the cleaved His6-tag and His6-tagged TEV protease. The final step of the purification of MtAIH was size exclusion chromatography on HiLoad Superdex 200 16/60 column (GE Healthcare) connected to an AKTA FPLC system (Amersham Biosciences). The column was equilibrated in 50 mM HEPES pH 7.4, 100 mM KCl, 50 mM NaCl, and 1 mM TCEP.
Crystallization and Data Collection
MtAIH was concentrated with Amicon concentrators (Millipore) to the final concentration of 8 mg/ml, determined by the absorbance measurement at 280 nm with the extinction coefficient of 77,920. The composition of the protein buffer was the same as the buffer used for the size exclusion chromatography. The sample was subjected to crystallization trials with use of Morpheus Screen (Molecular Dimensions) and PEG/Ion Screen (Hampton Research). Unused protein was stored in −80°C in 50-μl aliquots for later use. Crystals of MtAIH were grown in 0.2 M sodium acetate, 20% PEG 3350 at pH 8.0. The MtAIH-AHX complex was obtained by cocrystallization of MtAIH with 10 mM of the ligand in 37th conditions of Morpheus Screen (Molecular Dimensions; 0.12 M Alcohols, 0.1 M Buffer System 1 at pH 6.5, 30% v/v Precipitant Mix 1) diluted with water to 70% of original concentration. Glycerol (25%) was used as a cryoprotectant for the freezing of native crystals. Crystals of MtAIH-AHX were cryoprotected by original 37th conditions of Morpheus Screen. Protein was crystallized by sitting and hanging drop methods.
Diffraction data were collected at SER-CAT 22-ID beamline at the Advanced Photon Source (APS), Argonne National Laboratory, USA. The data were processed with XDS (Kabsch, 2010) and scaled using anisotropic diffraction limits with STARANISO1. The anisotropic cut-off surface for MtAIH data has been determined with best and worst diffraction limits 1.20 and 1.42 Å, respectively. In the case of MtAIH-AHX, the diffraction resolution was truncated between 2.20 and 2.66 Å. Table 1 provides detailed statistics for spherical and anisotropic truncation. Anisotropic data treatment improved the electron density maps of refined structures. Coordinates and structure factors were deposited in the PDB with the following IDs: 6NIB (MtAIH), 6NIC (MtAIH-AHX).
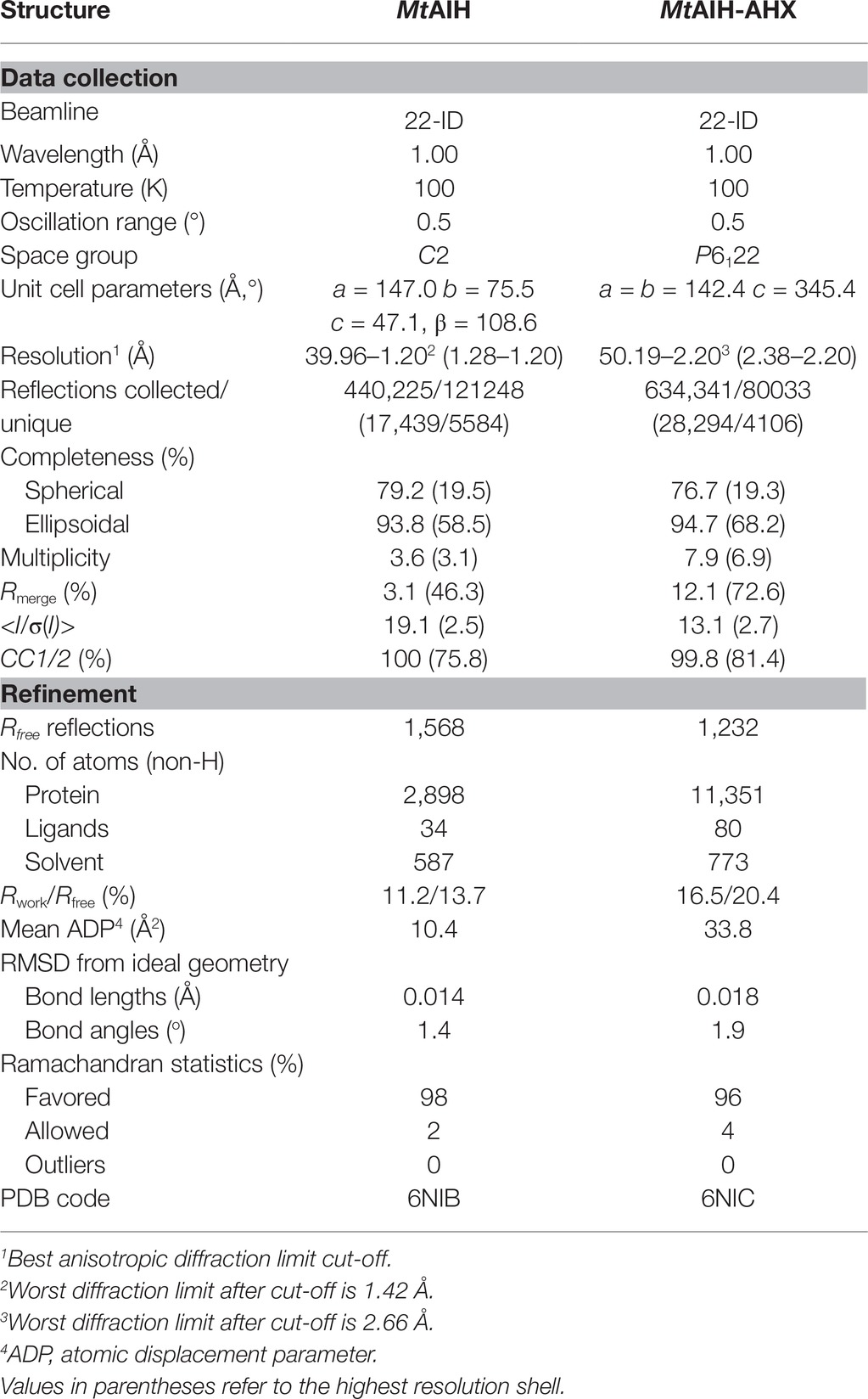
Table 1. Data-collection and refinement statistics.
Structure Determination and Refinement
The structure of MtAIH was solved by molecular replacement in Phaser (McCoy et al., 2007) with the structure of AtAIH (PDB ID 1VKP) as a search model. The initial model was rebuilt in PHENIX AutoBuild (Terwilliger et al., 2008). Then, the structure was subjected to manual and automatic refinement with Coot (Emsley et al., 2010) and Phenix (Adams et al., 2010) with anisotropic B-factors. Refined structure of unliganded MtAIH was used as a model for determination of the structure of MtAIH-AHX that was refined with isotropic B-factors and TLS (Winn et al., 2001, 2003) in Refmac (Murshudov et al., 2011). The refinement was carried out until the Rwork and Rfree values (Brunger, 1992), the geometric parameters, and the overall difference electron density maps were satisfactory. Evaluation of the final structures was performed in PROCHECK (Laskowski et al., 1993) and MolProbity (Chen et al., 2010). The final refinement statistics are given in Table 1.
Small-Angle X-Ray Scattering Data Collection and Analysis
SAXS data were collected from 5.5 mg/ml MtAIH solution at the BioCAT 18-ID beamline (Fischetti et al., 2004) at APS with in-line size exclusion chromatography (SEC-SAXS) to separate sample from aggregates, thus ensuring optimal sample homogeneity. The sample was loaded on a WTC-015S5 column (Wyatt Technologies) connected to an Infinity II HPLC (Agilent Technologies). The sample after the column was sent to the Agilent UV detector, a Multi-Angle Light Scattering (MALS) detector, and a Dynamic Light Scattering (DLS) detector (DAWN Helios II, Wyatt Technologies), and an RI detector (Optilab T-rEX, Wyatt). Molecular weights and hydrodynamic radii were calculated from the MALS and DLS data respectively using the ASTRA 7 software (Wyatt). Afterward, the sample was sent to the SAXS flow cell, a 1.5-mm quartz capillary. Scattering intensity was recorded at 1.03-Å wavelength at room temperature, with 0.5-s exposures every 2 s on a Pilatus3 1M detector (Dectris) placed 3.5 m from the capillary (collected q-range was 0.004–0.4 Å−1). Data reduction and analysis were performed by BioXTAS RAW 1.5.1 (Hopkins et al., 2017). Frames corresponding to the elution peak of the chromatogram were averaged to maximize the signal-to-noise ratio. Several frames immediately proximal to the sample peak (buffer frames) were averaged and subtracted from the sample scattering to obtain the final SAXS curve (Figure 1A). The Rg value calculated from the Guinier (Figure 1B) and distance distribution analysis (Figure 1C) was 30 Å. The calculated maximum dimension of the particle (Dmax) was 96 Å. The qRg limits for further calculations were 0.43–1.29.
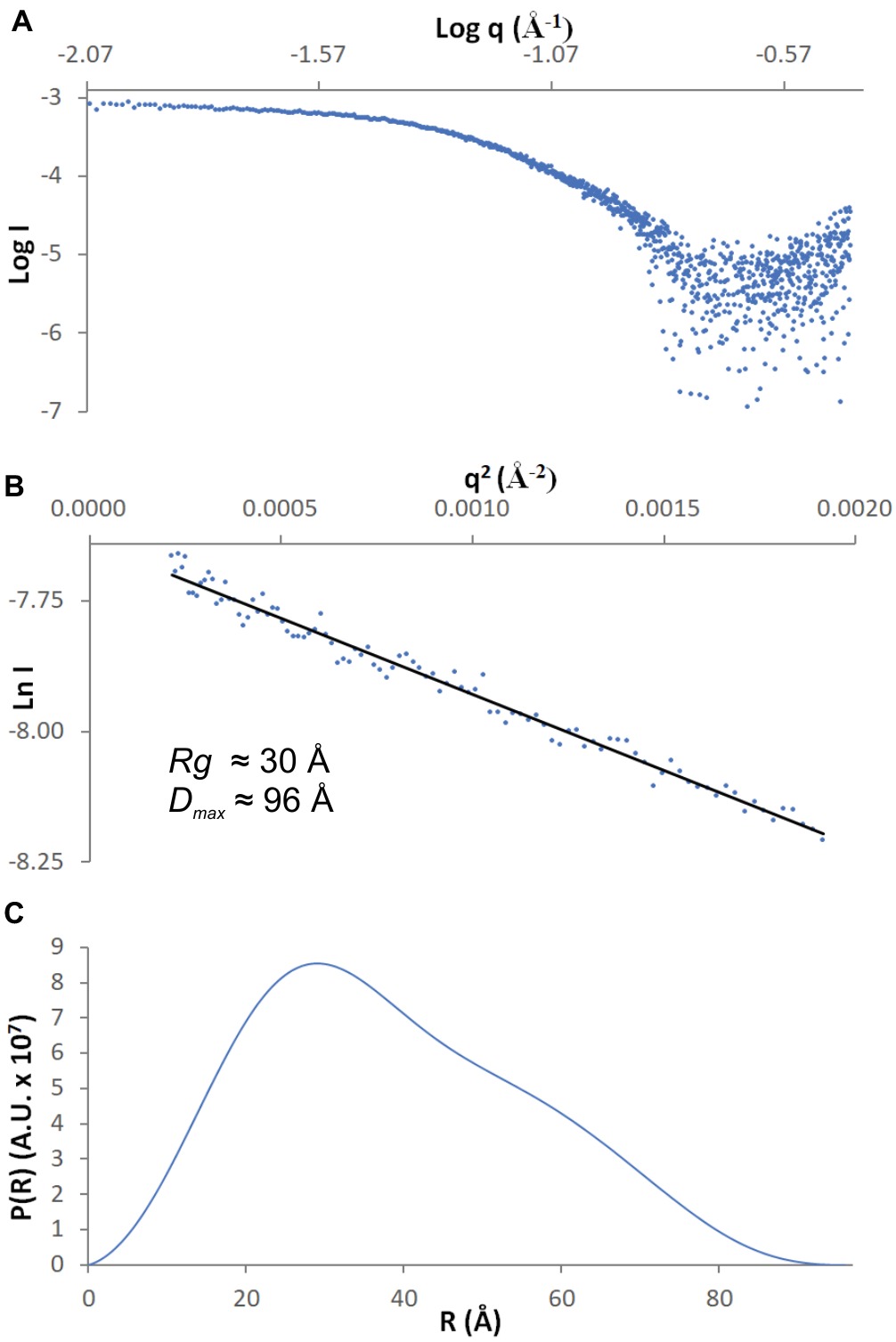
Figure 1. SAXS data. (A) The experimental curve for MtAIH; (B) Guinier plot (blue dots) of the scattering curve with the best fit shown as a black line; (C) Pair-distance distribution function for MtAIH SAXS data.
Ab initio envelopes with the restraint of twofold symmetry were calculated in DAMMIF (Franke and Svergun, 2009), averaged with DAMAVER (Volkov and Svergun, 2003), refined with DAMMIN (Svergun, 1999), and filtered with DAMFILT. SUPCOMB was used for the superposition of the SAXS envelope with the crystallographic dimer of MtAIH. DENSS (Grant, 2018) was used for the calculation of the ab initio electron density maps directly from the SAXS data with no prior information about the symmetry of the molecule.
Other Software Used
Molecular illustrations were created with UCSF Chimera (Pettersen et al., 2004). Ramachandran plot was calculated in Rampage (Lovell et al., 2003). Secondary structure was recognized with ProMotif (Hutchinson and Thornton, 1996) within the PDBsum server (de Beer et al., 2014). Sequence alignments were performed in CLUSTAL W (Thompson et al., 1994) and edited in BioEdit (Hall, 1999).
Results and Discussion
MtAIH Presents the Pentein α/β Propeller Fold
AIHs are assigned by the Structural Classification of Proteins (SCOPe) (Fox et al., 2014) to the porphyromonas-type peptidylarginine deiminase family that is a part of the Superfamily of penteins, characterized by a propeller-like arrangement of five αββαβ units which form a narrow channel in the core (Hartzoulakis et al., 2007). Penteins share a conserved group of residues that recognize the guanidine moiety of the substrate—His, Cys, and two acidic, guanidine-binding residues (usually Asp) which serve to catalyze a range of reactions (Linsky and Fast, 2010). The monomer of MtAIH is no different in this matter, i.e., five motif repeats (I–V) are arranged around fivefold pseudosymmetry axis that is aligned with the catalytic tunnel in the core of the protein (Figures 2A,B). Class, Architecture, Topology, Homology (CATH) server (Sillitoe et al., 2015) matches MtAIH with the L-arginine/glycine amidinotransferase superfamily that belongs to the Class 3 of alpha beta proteins with the architecture of a five-bladed propeller.
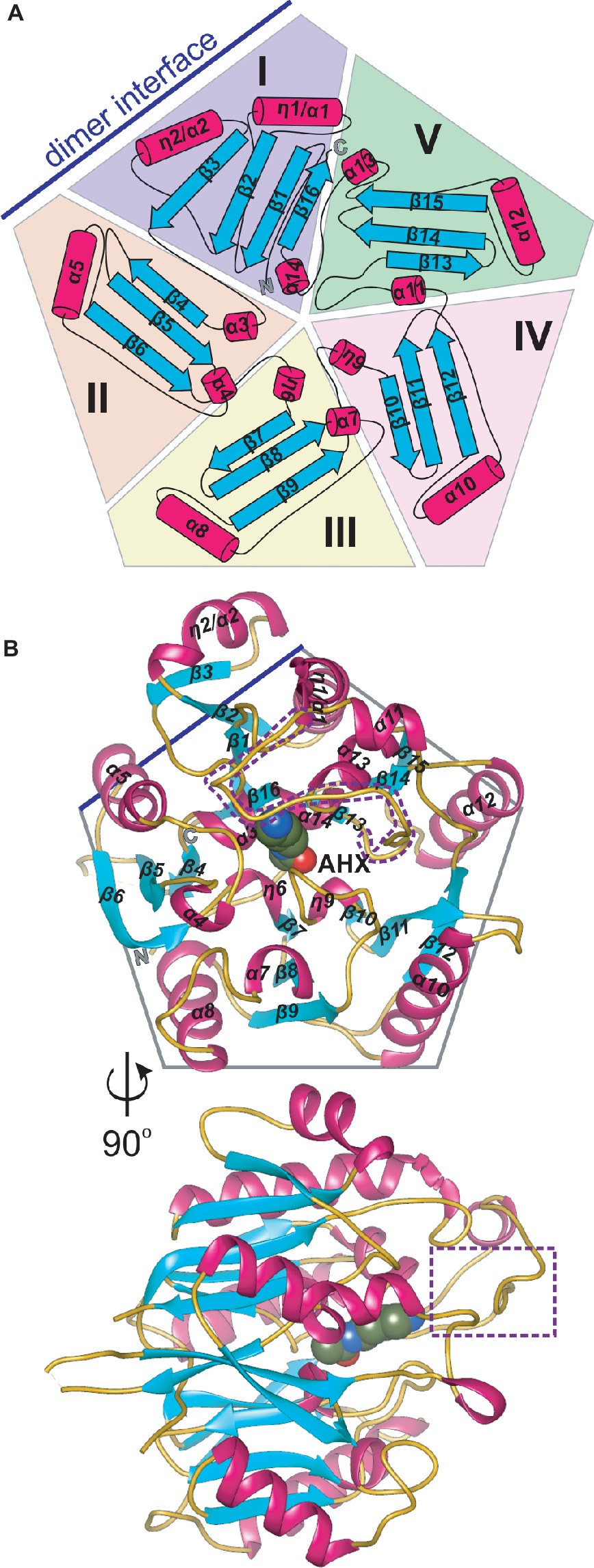
Figure 2. The monomer of MtAIH. (A) Topological diagram of MtAIH with a schematic depiction of the five αββαβ units (I–V) that form internal pseudo fivefold symmetry; secondary structure elements, helices (cylinders) and sheets (arrows) are colored in red and cyan, respectively; blue line indicates the position of the second MtAIH subunit from the dimeric assembly. (B) Ribbon representation of the structure of MtAIH monomer in complex with AHX (shown as spheres); the gate-keeping loop over the active site is marked with a purple dotted line.
The overall globular shape of the MtAIH monomer resembles pentagonal prism, where the longest helices (η1/α1, α1, α7, α9, α11) are positioned in the imaginary vertices of the pentagon with all β-strands running along the direction marked by these helices. Sixteen strands in MtAIH form five β-sheets, one four-stranded, and four with three strands each. All β-sheets are oriented toward the center of the molecule forming five “blades” of the propeller (Figure 2B). Repeat I with its four-stranded β-sheet actually disturbs the overall fivefold pseudosymmetry of the molecule, i.e., it has the additional strand β3 and the helix η2/α2 which are placed outside the pentagonal shape. Moreover, the αββαβ motif of unit I is, in fact, discontinuous and it is fully formed with the complementation of C-termini, more precisely, by α14 and β16 (Figure 2A). In the center of the molecule, four short helices (α3, η6, η9, α14), that directly precede internal strands from repeats I–IV, line the surface of the negatively charged central channel, that is the active site. These core helices are placed on N-terminal sides of the inner strands of β sheets from repeats I–IV. Only the inner strand of repeat V (β13) is not directly preceded by a short helix. Instead, the first helix of this repeat (α11) is actually longer than helices that build the active site and it is placed almost outside of the outline of the protein which precludes it from the interactions with the substrate in the active site. Additionally, α11 is flanked by long coils. One of these coils (residues 291–314) covers the active site entrance and plays a crucial role in the substrate recognition (see below for details). MtAIH has a very high structural similarity to the other plant ortholog, AtAIH (unpublished, PDB ID 3H7K, overall sequence identity is 70%), with the 0.6 Å root mean square deviation of the superposed structures. Both plant AIHs have a similar organization of secondary structure and almost identical architecture of the active site.
MtAIH Forms Symmetric Dimers
The molecular mass of MtAIH calculated from MALS is 81 kDa, which almost ideally agrees with the molecular weight of two monomers (theoretical mass of the MtAIH construct is 41.2 kDa). The dimeric assembly of MtAIH is also shown by SAXS envelope even though the calculated mass of MtAIH from SAXS was underestimated to be ~60 kDa. The envelope with twofold symmetry restraints (Figure 3A) and ab initio electron density map that was calculated with no information about the molecule symmetry (Figure 3B) clearly correspond to the MtAIH dimer in the crystal lattice where two subunits are related by twofold symmetry. It is worth to note that both crystal structures, MtAIH and MtAIH-AHX, present different crystallographic symmetry (see Table 1) with one and four chains in the asymmetric unit, respectively. In the case of the unliganded structure, the dimer is created by a crystallographic twofold axis, while in the MtAIH-AHX complex, there are two almost identical dimers in the asymmetric unit. The comparison of our results with the crystal structure of AtAIH (Figure 3C) clearly shows that AtAIH also forms dimers analogical to MtAIH.
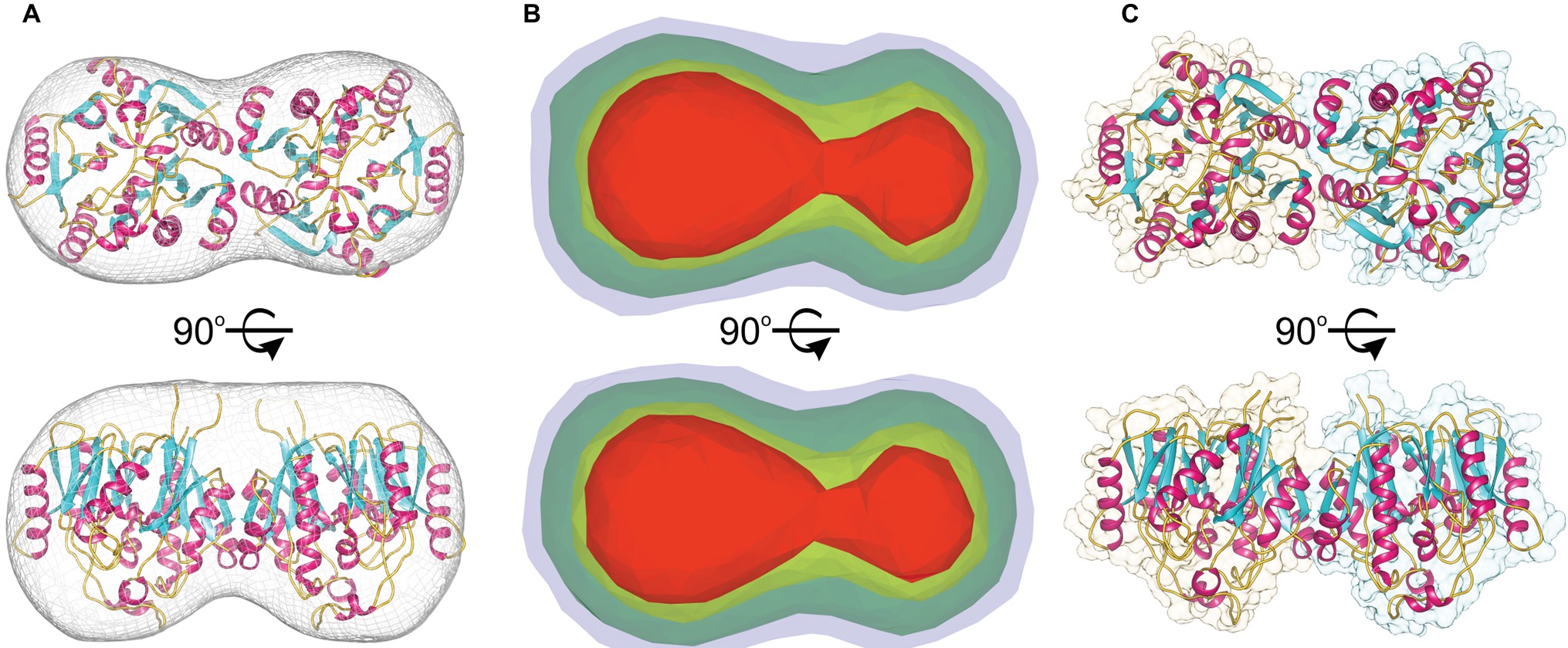
Figure 3. MtAIH dimers. (A) Ab initio SAXS envelope (gray mesh) with the superposed crystallographic dimer of MtAIH; (B) Ab initio electron density maps of MtAIH calculated by DENSS from the SAXS data; contours of the map are as follows: 5σ (red), 3.5σ (yellow), 1.4σ (green), 0.7σ (blue). (C) The shape of the crystallographic dimer in the AtAIH structure (PDB ID 1VKP).
The results are somewhat contrary to the analysis of the MtAIH crystal structure done with the PISA server (Krissinel and Henrick, 2007). It shows that monomer of MtAIH has the surface area ~14,000 A2 with the biggest interface area ~900 Å2 that is shared with the closest monomer in the crystal lattice. It is about 6.5% of the monomer surface and it was estimated by the PISA server to have no role in the complex formation. However, in both MtAIH crystal forms, this interface area between two subunits is preserved and, taking it together with SAXS and MALS results, it is, in fact, responsible for the formation of MtAIH dimers. The intersubunit interactions on this interface involve 25 residues (the same set from both interacting subunits) where about half of them create 20 hydrogen bonds or salt bridges. The interface residues belong to the β2, β3, and η2/α2 from repeat I and α5 which belongs to the repeat II (Figure 2B). Two subunits of AtAIH in the crystal structure (PDB ID 3H7K) share the analogical interface area and the PISA server analysis does not recognize it as a dimer either. Most of the interacting residues are preserved in both plant orthologs, but AtAIH evidently presents fewer interacting residues which altogether form only 14 hydrogen bonds.
A dimeric assembly was independently reported for the other plant AIHs, including AtAIH (Janowitz et al., 2003), AIH from maize (ZmAIH) (Yanagisawa, 2001) and rice (OsAIH) (Mohan Chaudhuri and Ghosh, 1985). The reported exception (Park and Cho, 1991) is a 70-kDa monomeric protein from soybean that was described to have AIH activity. However, the authors did not provide the sequence of the isolated protein. Also, any record classified as AIH matches to the reported description. The sequence alignment (Figure 4) of the dimeric plant AIHs shows that 19 residues (out of the pool of 25 which form the interface in MtAIH) are identical or very similar in all four plant AIHs. A similar extent of conservation applies to polar and hydrophobic residues on the dimer interface. Identical polar positions are: Gln24, Glu58, Thr61, Ser65, Gln68, Arg73, Arg81, Glu84, Ser86 Lys145, Glu150, and Arg151. These residues in MtAIH are involved in 14 hydrogen bonds, that is, 70% of all hydrogen bonds found in the interface analysis.
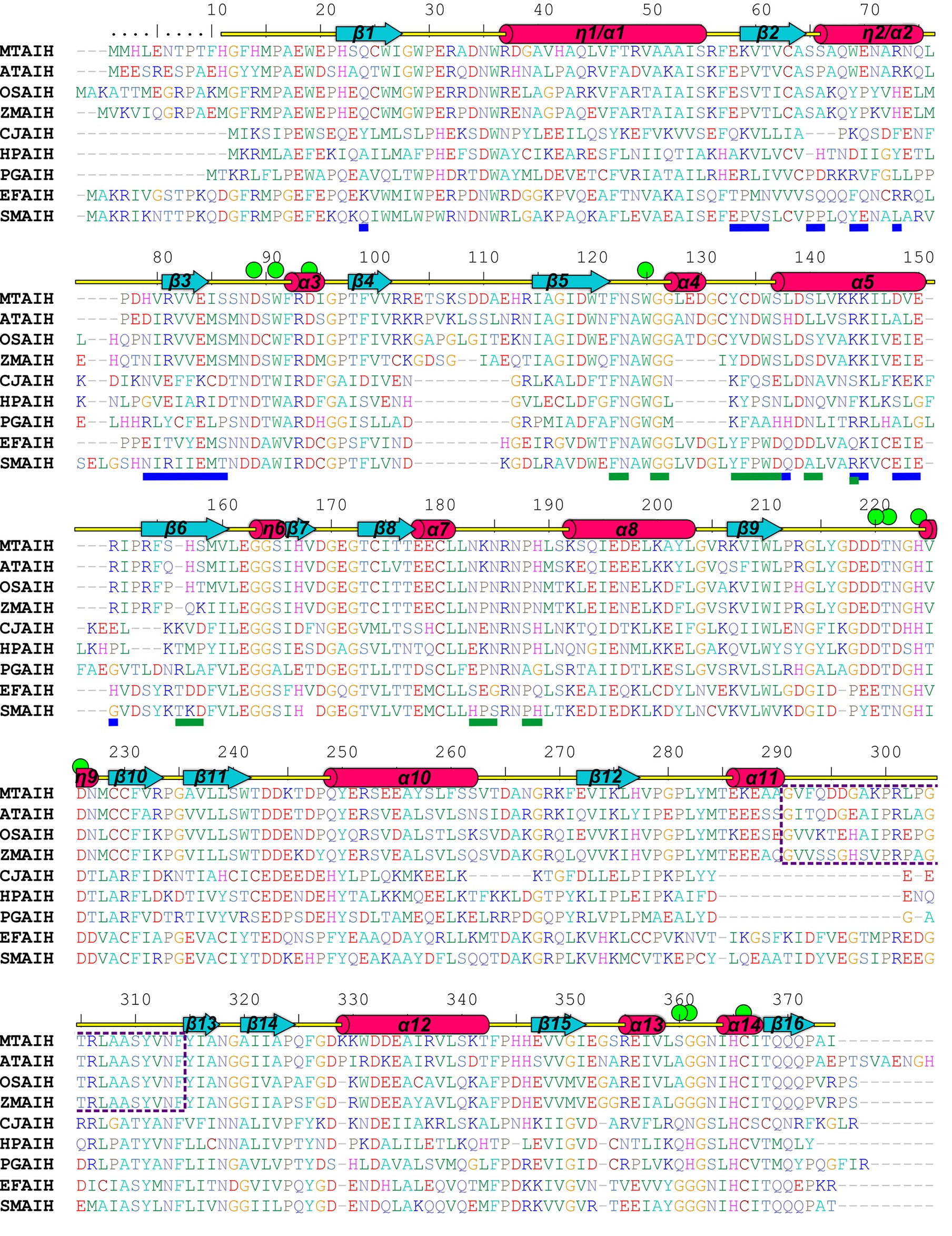
Figure 4. Sequence alignment of selected AIHs. The alignment was made with the following AIH sequences (UniProt accession numbers are given in square brackets, as well as the sequence identity with MtAIH): MtAIH [G7JT50], AtAIH [Q8GWW7, 70% sequence identity], OsAIH [Q01KF3, 65%], ZmAIH [C0PHP8, 64%], CjAIH [Q0P9V0, 26%], HpAIH [O24890, 26%], PgAIH [Q7MXM8, 27%], EfAIH [Q837U5, 44%], SmAIH [Q8DW17, 45%]. Sequence positions above the alignment and annotation of the secondary structure elements (α helices and 310 helices, η, are shown as red cylinders and β strands are shown as cyan arrows) correspond to MtAIH. Residues are color-coded by type. Green circles indicate residues that form the active site or participate in the interactions with the bound substrate. Blue and green lines below the alignment indicate residues that form dimer interface in MtAIH and HpAIH, respectively. The gate-keeping loop over the active site of plant AIHs is marked with a purple dotted line.
Analyzing sequence conservation of all plant AIHs (Figure 5), there is no obvious highly conserved area around the dimer interface that can be distinguished right away. However, when considering the conservation of particular residues involved in the hydrogen bonding between both subunits, most of them stand out as highly conserved (Val60, Arg73, Arg81, Val82, Glu84, Ser86, Lys145), whereas only three are very variable residues (Asp148, Val149, Arg151). Moreover, hydrophobic interactions seem to be important for the dimer formation as well—at the center of the interface between dimer mates there is a patch of apolar residues (Trp69, Val83, Ile85, Val149). All of these residues are apolar in plant AIHs.
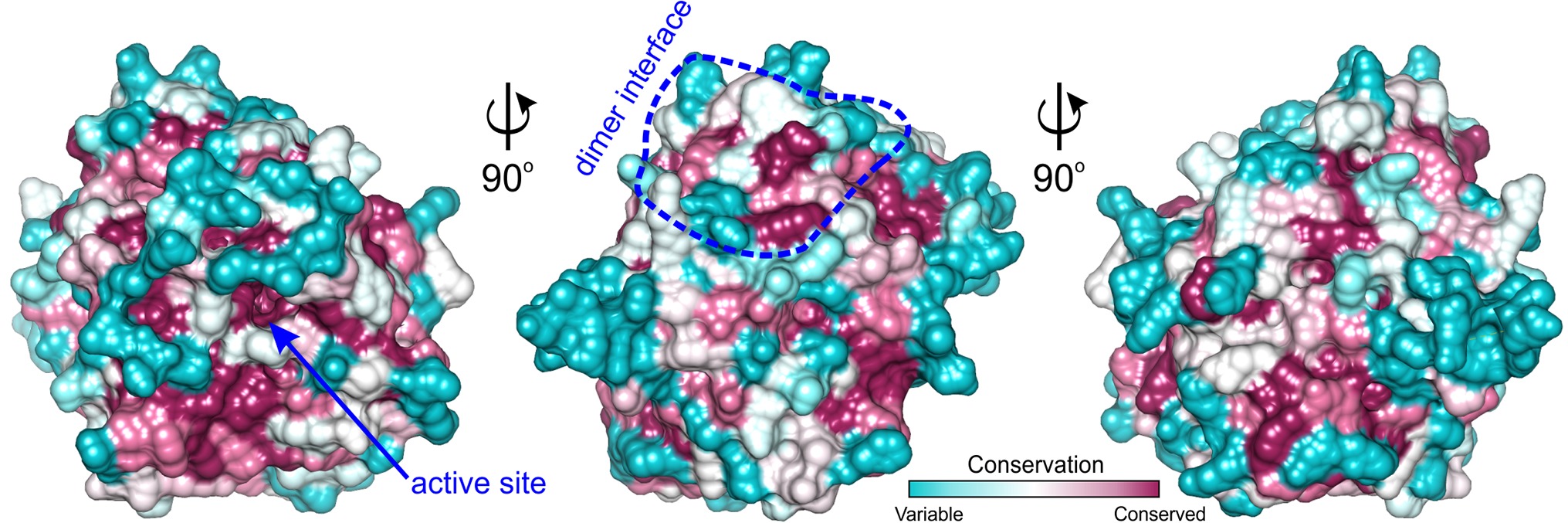
Figure 5. ConSurf analysis for MtAIH. Surface representation of the MtAIH monomer where surface residues are color-coded by the conservation score calculated from the sequence alignment of plant AIHs; the analysis was done in ConSurf (Ashkenazy et al., 2016); dimer interface is circled in blue. The pool of 184 plant AIH sequences was selected from the protein sequences classified to agmatine deiminase family (IPR017754) by InterPro (Finn et al., 2017); clear outliers where the sequence length was shorter than 340 or longer than 420 were manually excluded from the alignment.
Bacterial AIH Analogs With Various Biological Assemblies
In the PDB, there are several structures of bacterial representatives of AIHs that not necessarily form dimers like plant AIHs (Figure 6A). The bacterial AIHs form tetramers, like AIH from Streptococcus mutans (SmAIH, PDB ID 2EWO) and Enterococcus faecalis (EfAIH, PDB ID 2JER) (Llacer et al., 2007) or monomers like AIH from Campylobacter jejuni (CjAIH, PDB ID 6B2W) (Shek et al., 2017). There are also dimeric bacterial AIHs like Helicobacter pylori (HpAIH, PDB ID 3HVM) (Jones et al., 2010a) or Porphyromonas gingivalis (PgAIH, PDB ID 1ZBR). HpAIH and PgAIH both present analogical dimer interface, however, it is different than the dimer interface of plant AIHs (Figure 6B). In these two bacterial dimeric AIHs, the interface residues are from repeats II and III. To be more precise, these residues correspond to the residues from β5, α4, α5, β6, and the loop between α7 and α8 of MtAIH (Figure 4). This dimer interface of both bacterial AIHs is even smaller (~700 Å2, slightly above 5% of the monomer surface) than that of plant AIHs. A closer look at the superposition of MtAIH with bacterial dimeric AIHs (Figure 6B, right panel) reveals the bacterial interface to be placed very close to the region of repeat II that in MtAIH forms short helix α4. In plant AIHs, this region is important for the ligand binding and the conformation of α4 shows that it would create severe steric clashes with the dimer mate. These regions of bacterial dimeric AIHs and CjAIH are five residues shorter. On the other hand, a closer look at the dimer interface of plant AIHs (Figure 6A, left panel) shows the different orientation of two helices in bacterial dimeric AIHs—η2/α2 and α5 with significantly different residues in these helices.
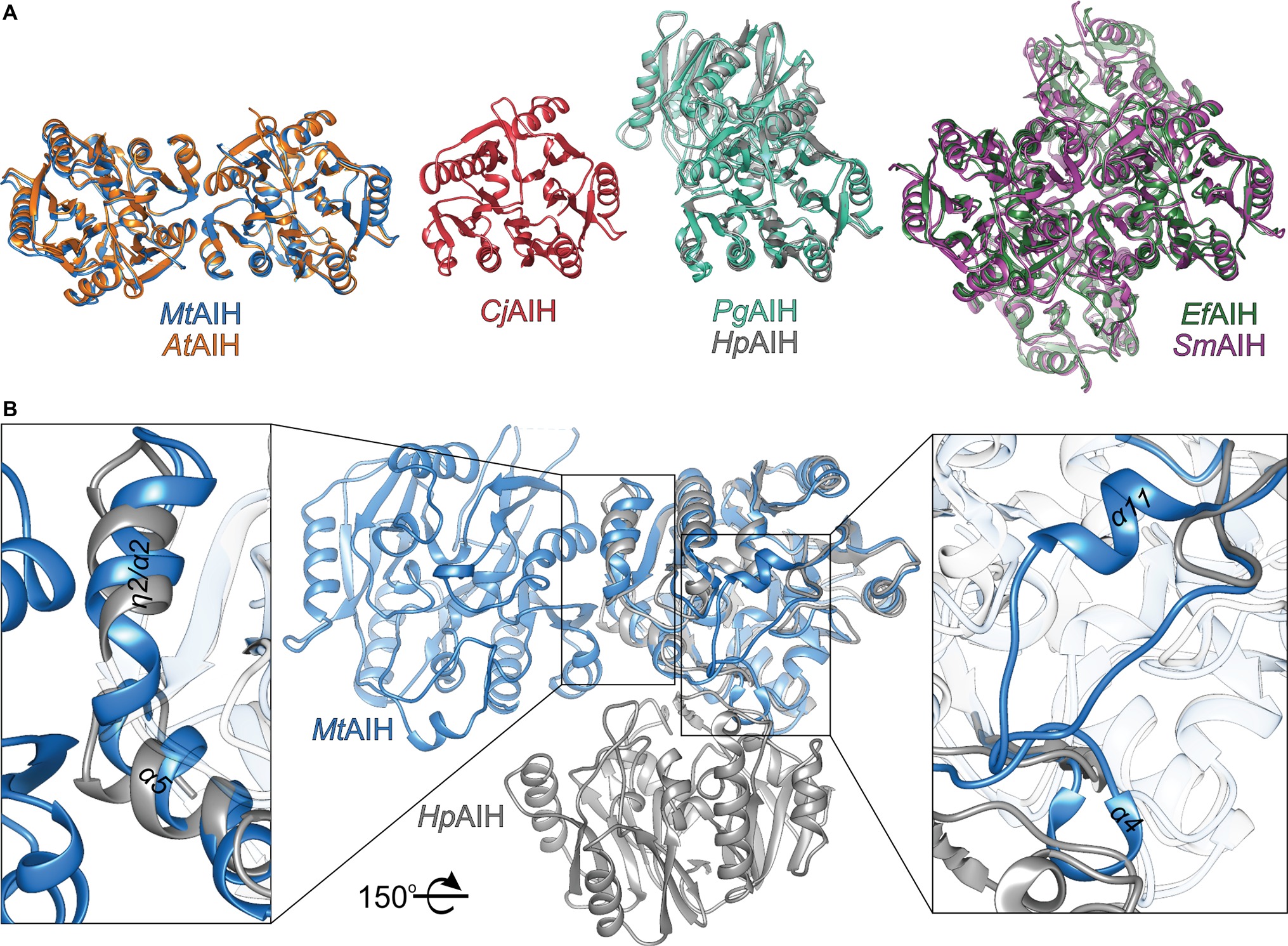
Figure 6. Comparison of AIHs from different species. (A) Various assemblies of AIHs (starting from the left): dimeric plant AIHs (AtAIH, orange, and MtAIH, blue), bacterial monomeric AIH (CjAIH, red), dimeric AIHs (PgAIH, light green, HpAIH, gray), and tetrameric AIHs (EfAIH, dark green, and SmAIH, violet); for clear comparison, orientation of at least one subunit from each superposition is the same as the orientation of MtAIH. (B) Comparison of the secondary structure elements that form dimer interfaces of plant and bacterial AIHs (superposition of MtAIH, blue, with HpAIH, gray). Rectangles indicate zoomed regions; for clarity, part of the chains are semitransparent; the structure is rotated 150° with respect to the orientation in panel A.
Substrate Binding Mode of Plant AIHs
The MtAIH-AHX structure was obtained by cocrystallization and presents the bound AHX in three out of four protein chains that are present in the asymmetric unit. The ligand used for this study structurally differs from the physiological reaction product of MtAIH, NCP, by the methylene which substitutes the secondary amine of NCP (adjacent to the carbamoyl moiety). Therefore, the conformation of the complex is similar to the conformation of the enzyme with the bound product after the reaction, representing a highly probable NCP binding mode (Figure 7A). It is worth to note that the MtAIH-AHX structure shows a somewhat dynamic character where relative conformation of the ligand and surrounding residues (especially close to the terminal amine of AHX) is slightly different in each chain. Residues which are closer to the entrance of the active site have the B factor value significantly higher than the average B factor for the structure. The mean B factor value of the AHX (~35 Å2) is comparable to the structure average (~34 Å2), but it is still higher than the B factor of residues placed deep in the cavity. This can be explained by the nonphysiological character of AHX in comparison to AGM or even NCP; binding of the ligand was enabled due to its high concentration. The other available plant AIH structure which shows the details about the ligand binding mode is the structure of AtAIH with the reaction intermediate (Figure 7B, PDB ID 3H7K, unpublished structure). Therefore, the substrate binding mode of plant AIHs can be described by the analogy of these two AIHs.
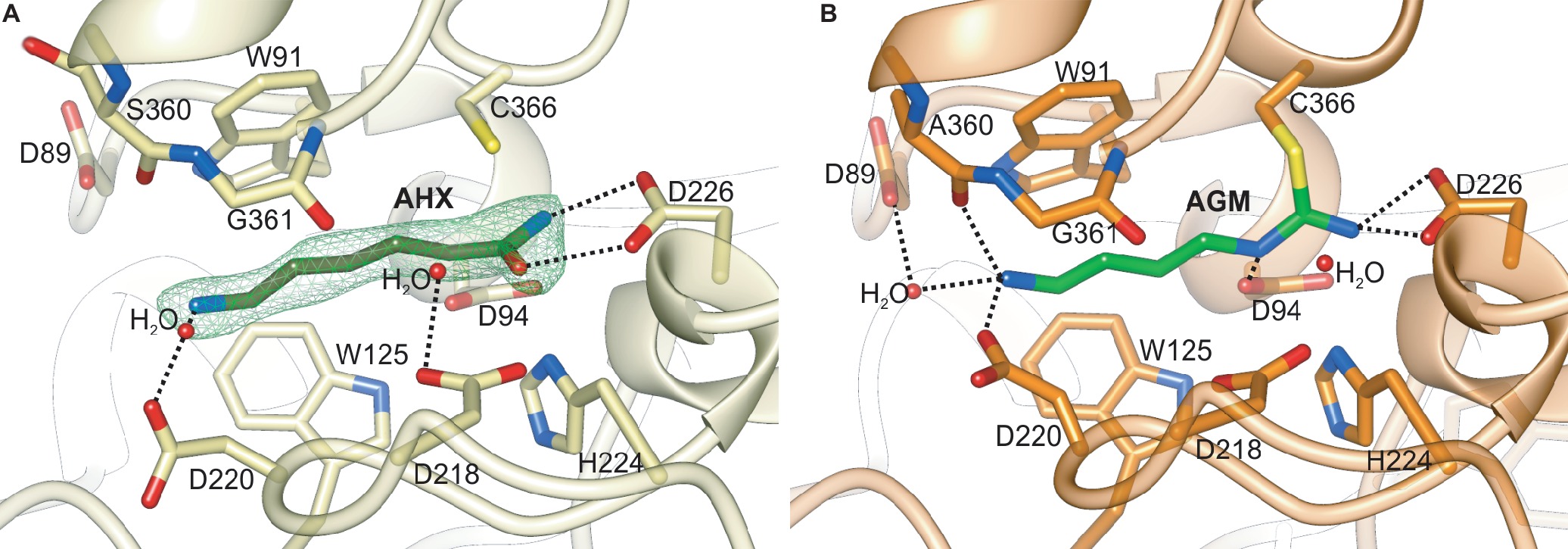
Figure 7. The catalytic site of plant AIHs. (A) Close-up view of the catalytic site of MtAIH with bound AHX (dark green) in the chain C of the MtAIH-AHX structure; green mesh represents Polder omit maps (contoured at 5σ) calculated in Phenix (Liebschner et al., 2017). (B) Imidine covalent intermediate of hydrolyzed AGM (lime green) captured in the crystal structure of AtAIH (PDB ID 3H7H, unpublished structure). Dashed black lines indicate important hydrogen bonding interactions described in the main text.
The active site of MtAIH is formed as a negatively charged channel (Figure 8) that is covered by coil region which links repeat IV and V (residues 291–314) which forms a kind of a lid over the catalytic site (Figure 2B). The active site itself is highly conserved among plant AIHs with the exception of Trp125 which in plant species can also be replaced by Tyr. Most likely, this does not drastically alter the shape and character of the channel. In the case of MtAIH, the channel is formed by side chains of Trp91 and Trp125 where the planes of their indole rings are positioned almost perpendicularly to each other. The character of this region resembles the active site entrance of MtCPA (Sekula et al., 2016), where also Trp residues shape the tunnel which guides to the catalytic Cys residue. In plant AIHs on the other side of the tunnel, there is Gly361 which due to the lack of side chain leaves the necessary void space for the ammonia and water molecules that are important for catalysis (see below for details).
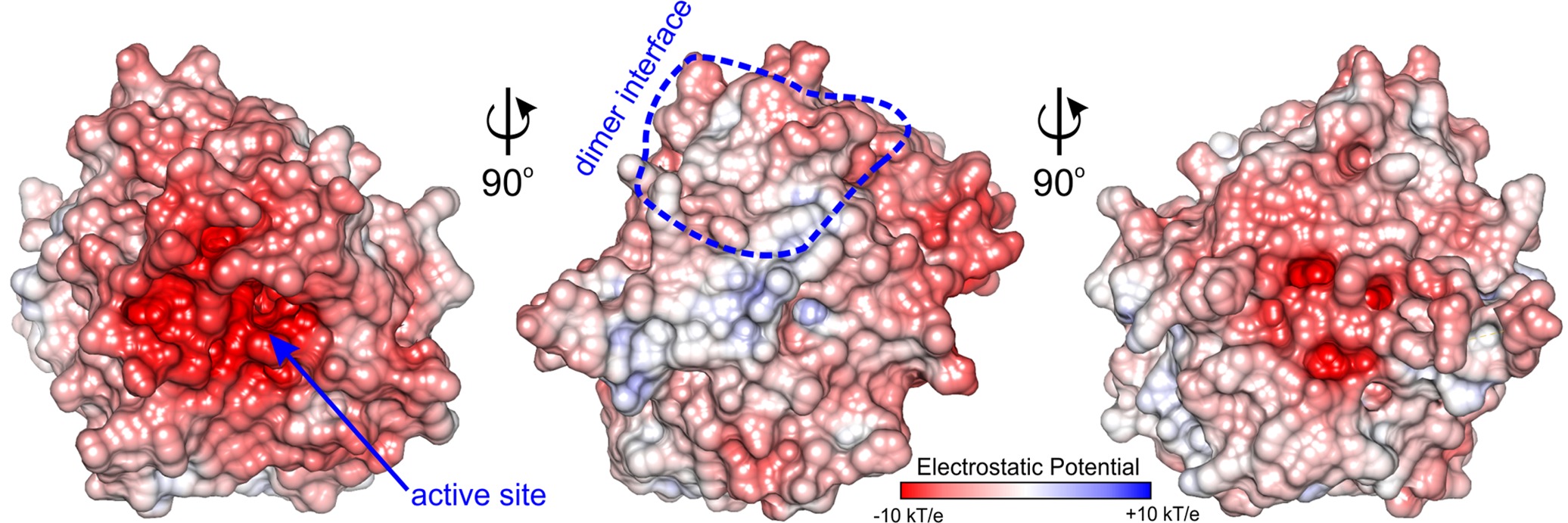
Figure 8. Charge distribution of MtAIH. Surface electrostatic potential mapped on the surface of MtAIH monomer calculated in PDB2PQR and APBS (Baker et al., 2001; Dolinsky et al., 2004); dimer interface is circled in blue.
Generally, GMEs bind their substrates with three different modes—1, 2A, 2B (Shirai et al., 2006). Of course, the bound substrates are structurally very diverse and also the orientation of the guanidine moiety placed in the vicinity of the catalytic triad is not always the same. In mode 1, substrates are bound in a way that their terminal parts (more distant from the catalytic triad) interact with residues from repeat IV and V. This promotes a completely different orientation of guanidine moiety at the bottom of the catalytic channel, which is rotated in comparison to the other two binding modes. The bound ligands in modes 2A and 2B interact with residues from repeat II and III. In the case of plant AIHs, the substrate binding mode corresponds to the mode 2 and is more similar to 2A, where the terminal amine group of AGM interacts with residues from repeat II. Therefore, the guanidine moiety of bound AGM reaches the active site bottom with a catalytic triad (in MtAIH these are Cys366, Asp226, and His224) pointing toward Asp226. Polar residues that interact with the AGM guanidine moiety are Asn94, Asn226, and His224. They are responsible for the positioning of the plane of guanidine moiety very similar to the orientation of amide moiety of AHX (Figure 7A) so that it is susceptible to the nucleophilic attack from sulfur atom of Cys366 that is placed almost ideally on the line normal to the amide plane of AHX. The terminal amine of AGM placed by the entrance of the channel is stabilized by direct H-bonds with Asp220, Ala360, and a water-mediated H-bond with Asp89, analogous to the interactions observed in the AtAIH structure (Figure 7B, PDB ID 3H7K, unpublished structure). Therefore, AGM that binds within the catalytic site of AIH is stabilized by a network of polar interactions involving every heteroatom in the substrate and by a series of hydrophobic interactions between its trimethylene moiety and the hydrophobic residues in the active site channel that connects the entrance with the catalytic site.
Concerted Conformational Rearrangements Upon Ligand Binding
The close vicinity of the MtAIH active site is surrounded by two very flexible regions built mostly by long loops. One is the gate-keeping loop covering the active site (residues 291–314), which is a linker between repeat IV and V (Figure 2B). The other concerns the fragment 122–136 which belongs to the repeat II, where α4 is located. The gate-keeping loop is very variable in plant AIHs except for Arg301 and Arg306. Both regions are disordered in the non-liganded MtAIH structure (Figure 9A), more precisely fragments between Glu129-Cys134 and Pro300-Tyr-293 were excluded from the structure due to the lack of electron density maps that would show their conformation. On the other hand, in the structure of MtAIH-AHX complex, the electron density clearly shows their position (Figure 9B). This feature is also observed in AtAIH (PDB ID 3H7K) with reaction intermediate, where the conformation of these coiled regions is fully modeled. Altogether, when the ligand is bound in the active site, these two regions come close together to form hydrogen bonds: between carbonyl oxygen of Cys132 and amide nitrogen of Lys299, and between backbone nitrogen of Cys134 and carbonyl oxygen of Gly297. Additionally, the guanidine group of Arg301 from the gate-keeping loop creates H-bonds with Asp220 and Asn35 in close vicinity of the AGM binding site. Therefore, this disorder-to-order transition secures the appropriate conformation of the bound substrate before reaction and the opening of the lid loop helps with product release after catalysis. The concerted disorder-to-order transition upon ligand binding was also observed in CjAIH (Shek et al., 2017), however, it concerned different regions. More precisely, in CjAIH, regions that showed conformational change upon substrate binding correspond to residues 122–136 and 212–224 of MtAIH, therefore to the loops which are flanking α4 of repeat II and η9 which links repeats III and IV. The latter fragment in plant AIHs has a different sequence which, together with 18-residues shorter region of 278–314, results in the different recognition of the terminal amine of bound AGM in bacterial AIHs. Moreover, the sequence comparison of the plant AIHs suggests that the concerted conformational change of the gate-keeping coiled regions upon substrate binding can be characteristic for other plant AIHs as well. Likely, this feature can distinguish plant and bacterial orthologs, especially from those which present a shorter loop link between repeat IV and V.
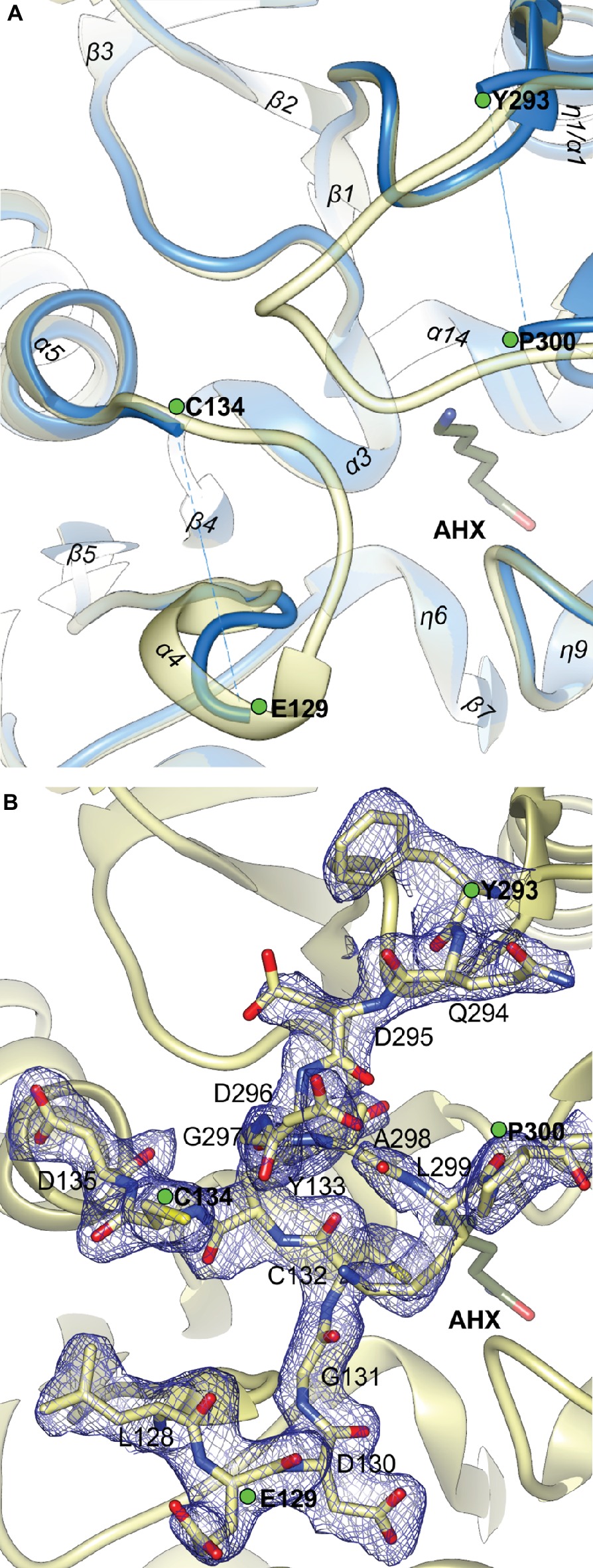
Figure 9. The gate-keeping loop of MtAIH. (A) The superposition of non-liganded MtAIH structure (blue ribbons) with the structure of MtAIH-AHX complex (yellow semitransparent ribbons); green dots depict last visible residues from the loops 122–136 and 291–314 in the non-liganded MtAIH structure. (B) Close-up view of the 2Fo–Fc electron density map contoured at 1σ (blue mesh) for the residues 128–135 and 294–300 in the chain C of MtAIH-AHX structure.
Cys366 Forms a Covalent Intermediate With AGM
The catalytic mechanism of guanidine-modifying enzymes is very similar to the cysteine proteases (Shirai et al., 2006). For AIHs it was structurally studied with bacterial orthologs (Llacer et al., 2007; Jones et al., 2010b) and involves the creation of a thioester covalent intermediate.
The reaction starts after binding of AGM when the gate-keeping loop is closed and the sulfur atom of Cys366 is ready to perform a nucleophilic attack on the central carbon of AGM guanidine moiety to form a tetrahedral covalent adduct. Then, His224 (positioned on the other side of the plane of amide moiety of AHX, Figure 7A) donates the proton to the closest amine of the intermediate, thus acting as a general acid for the reaction. This leads to the break of the adjacent bond and release of ammonia. Subsequently, ammonia is most likely H-bonded with the OD1 of Asp226 and it can be replaced by a water molecule (most likely the one, which is H-bonded with Asp218 in MtAIH-AHX structure, Figure 9A) so the reaction can proceed. This water molecule is presumably moved deeper in the active site to be activated by transferring a proton to the His224/Glu226 charge relay network to form a hydroxide ion, so it can make a nucleophilic attack on the carbon of amidino intermediate to form another tetrahedral intermediate. The most probable position of the hydroxide ion which attacks the central carbon is represented by water in AtAIH structure (Figure 7B). The intermediate collapses to form N-carbamoyl putrescine with a planar ureido carbon. The product of enzymatic action of AIH presents the conformation analogical to that of AHX (with an additional hydrogen bond with Asp94). Finally, the gate-keeping loops can be opened to release the product.
Conclusions
The presented work described MtAIH and compared its crystal structures with the other plant dimeric ortholog, AIH from A. thaliana. We have cross-validated our results with the reports on different plant AIHs highlighting residues that take part in the formation of AIH dimers in plants. These are residues from β2, β3, and η2/α2 from repeat I and α5 from repeat II. Plant AIHs are characterized by a different dimer interface to that observed in dimeric bacterial AIHs.
The crystallographic snapshots of MtAIH together with deposited AtAIH structures showed the detailed conformation of the coiled region that during the catalysis form a lid over the active site of plant AIHs. This loop is responsible for the recognition of the terminal amine of the bound AGM and provides necessary stabilization of the ligand in time of the catalytic event. Interestingly, the structural analysis of plant AIHs showed different disorder-to-order transition of the gate-keeping loops to that observed in bacterial orthologs, which shows that substrate recognition mechanism of plant AIHs differentiates them from bacterial AIH orthologs, especially those which present a shorter loop link between repeat IV and V.
Data Availability
The datasets generated for this study can be found in Protein Data Bank, 6NIB, 6NIC.
Author Contributions
BS planned and performed the experiments, analyzed the results, and wrote the manuscript. ZD analyzed the results and supervised the work.
Funding
This project was supported by the Intramural Research Program of the NCI, Center for Cancer Research. Diffraction data were collected at the Advanced Photon Source (APS), Argonne National Laboratory (ANL) at the SER-CAT beamline 22-ID, supported by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences under Contract W-31-109-Eng-38. SAXS research on the 18-ID BioCAT beamline used resources of APS, a DOE Office of Science User Facility operated by ANL (contract DE-AC02-06CH11357), a project supported by grant 9 P41 GM103622 from the National Institute of General Medical Sciences (NIGMS). Use of the PILATUS 3 1M detector was provided by grant 1S10OD018090-01 from NIGMS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Srinivas Chakravarthy, BioCAT, for the assistance during SAXS experiments.
Abbreviations
ADC, Arginine decarboxylase; AHX, 6-aminohexanamide; AIH, Agmatine iminohydrolase; CPA, N-carbamoylputrescine amidohydrolase; GME, Guanidine-modifying enzyme; NCP, N-carbamoylputrescine; ODC, Ornithine decarboxylase; PA, Polyamine; PUT, Putrescine; SPD, Spermidine; SPM, Spermine.
Footnotes
References
Adams, P. D., Afonine, P. V., Bunkoczi, G., Chen, V. B., Davis, I. W., Echols, N., et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221. doi: 10.1107/S0907444909052925
Alcazar, R., Planas, J., Saxena, T., Zarza, X., Bortolotti, C., Cuevas, J., et al. (2010). Putrescine accumulation confers drought tolerance in transgenic Arabidopsis plants over-expressing the homologous Arginine decarboxylase 2 gene. Plant Physiol. Biochem. 48, 547–552. doi: 10.1016/j.plaphy.2010.02.002
Ashkenazy, H., Abadi, S., Martz, E., Chay, O., Mayrose, I., Pupko, T., et al. (2016). ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350. doi: 10.1093/nar/gkw408
Baker, N. A., Sept, D., Joseph, S., Holst, M. J., and McCammon, J. A. (2001). Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 98, 10037–10041. doi: 10.1073/pnas.181342398
Berberich, T., Sagor, G. H. M., and Kusano, T. (2015). “Polyamines in plant stress response” in Polyamines: A universal molecular nexus for growth, survival, and specialized metabolism. eds. T. Kusano and H. Suzuki (Tokyo, Japan: Springer), 155–168.
Brunger, A. T. (1992). Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475. doi: 10.1038/355472a0
Burhenne, K., Kristensen, B. K., and Rasmussen, S. K. (2003). A new class of N-hydroxycinnamoyltransferases. Purification, cloning, and expression of a barley agmatine coumaroyltransferase (EC 2.3.1.64). J. Biol. Chem. 278, 13919–13927. doi: 10.1074/jbc.M213041200
Capell, T., Bassie, L., and Christou, P. (2004). Modulation of the polyamine biosynthetic pathway in transgenic rice confers tolerance to drought stress. Proc. Natl. Acad. Sci. USA 101, 9909–9914. doi: 10.1073/pnas.0306974101
Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., et al. (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21. doi: 10.1107/S0907444909042073
de Beer, T. A., Berka, K., Thornton, J. M., and Laskowski, R. A. (2014). PDBsum additions. Nucleic Acids Res. 42, D292–D296. doi: 10.1093/nar/gkt940
Dolinsky, T. J., Nielsen, J. E., McCammon, J. A., and Baker, N. A. (2004). PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667. doi: 10.1093/nar/gkh381
Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501. doi: 10.1107/S0907444910007493
Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., et al. (2017). InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199. doi: 10.1093/nar/gkw1107
Fischetti, R., Stepanov, S., Rosenbaum, G., Barrea, R., Black, E., Gore, D., et al. (2004). The BioCAT undulator beamline 18ID: a facility for biological non-crystalline diffraction and X-ray absorption spectroscopy at the advanced photon source. J. Synchrotron Radiat. 11, 399–405. doi: 10.1107/S0909049504016760
Fox, N. K., Brenner, S. E., and Chandonia, J.-M. (2014). SCOPe: structural classification of proteins—extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 42, D304–D309. doi: 10.1093/nar/gkt1240
Franke, D., and Svergun, D. I. (2009). DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346. doi: 10.1107/S0021889809000338
Fuell, C., Elliott, K. A., Hanfrey, C. C., Franceschetti, M., and Michael, A. J. (2010). Polyamine biosynthetic diversity in plants and algae. Plant Physiol. Biochem. 48, 513–520. doi: 10.1016/j.plaphy.2010.02.008
Gill, S. S., and Tuteja, N. (2010). Polyamines and abiotic stress tolerance in plants. Plant Signal. Behav. 5, 26–33. doi: 10.4161/psb.5.1.10291
Grant, T. D. (2018). Ab initio electron density determination directly from solution scattering data. Nat. Methods 15, 191–193. doi: 10.1038/nmeth.4581
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98.
Hanfrey, C., Sommer, S., Mayer, M. J., Burtin, D., and Michael, A. J. (2001). Arabidopsis polyamine biosynthesis: absence of ornithine decarboxylase and the mechanism of arginine decarboxylase activity. Plant J. 27, 551–560. doi: 10.1046/j.1365-313X.2001.01100.x
Hanzawa, Y., Takahashi, T., Michael, A. J., Burtin, D., Long, D., Pineiro, M., et al. (2000). ACAULIS5, an Arabidopsis gene required for stem elongation, encodes a spermine synthase. EMBO J. 19, 4248–4256. doi: 10.1093/emboj/19.16.4248
Hartzoulakis, B., Rossiter, S., Gill, H., O’Hara, B., Steinke, E., Gane, P. J., et al. (2007). Discovery of inhibitors of the pentein superfamily protein dimethylarginine dimethylaminohydrolase (DDAH), by virtual screening and hit analysis. Bioorg. Med. Chem. Lett. 17, 3953–3956. doi: 10.1016/j.bmcl.2007.04.095
Hopkins, J. B., Gillilan, R. E., and Skou, S. (2017). BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Crystallogr. 50, 1545–1553. doi: 10.1107/S1600576717011438
Hutchinson, E. G., and Thornton, J. M. (1996). PROMOTIF—a program to identify and analyze structural motifs in proteins. Protein Sci. 5, 212–220. doi: 10.1002/pro.5560050204
Igarashi, K., and Kashiwagi, K. (2010). Modulation of cellular function by polyamines. Int. J. Biochem. Cell Biol. 42, 39–51. doi: 10.1016/j.biocel.2009.07.009
Illingworth, C., Mayer, M. J., Elliott, K., Hanfrey, C., Walton, N. J., and Michael, A. J. (2003). The diverse bacterial origins of the Arabidopsis polyamine biosynthetic pathway. FEBS Lett. 549, 26–30. doi: 10.1016/S0014-5793(03)00756-7
Janowitz, T., Kneifel, H., and Piotrowski, M. (2003). Identification and characterization of plant agmatine iminohydrolase, the last missing link in polyamine biosynthesis of plants. FEBS Lett. 544, 258–261. doi: 10.1016/S0014-5793(03)00515-5
Jimenez-Bremont, J. F., Marina, M., Guerrero-Gonzalez Mde, L., Rossi, F. R., Sanchez-Rangel, D., Rodriguez-Kessler, M., et al. (2014). Physiological and molecular implications of plant polyamine metabolism during biotic interactions. Front. Plant Sci. 5:95. doi: 10.3389/fpls.2014.00095
Jones, J. E., Causey, C. P., Lovelace, L., Knuckley, B., Flick, H., Lebioda, L., et al. (2010a). Characterization and inactivation of an agmatine deiminase from Helicobacter pylori. Bioorg. Chem. 38, 62–73. doi: 10.1016/j.bioorg.2009.11.004
Jones, J. E., Dreyton, C. J., Flick, H., Causey, C. P., and Thompson, P. R. (2010b). Mechanistic studies of agmatine deiminase from multiple bacterial species. Biochemistry 49, 9413–9423. doi: 10.1021/bi101405y
Kabsch, W. (2010). Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132. doi: 10.1107/S0907444909047337
Kamiab, F., Talaie, A., Khezri, M., and Javanshah, A. (2014). Exogenous application of free polyamines enhance salt tolerance of pistachio (Pistacia vera L.) seedlings. Plant Growth Regul. 72, 257–268. doi: 10.1007/s10725-013-9857-9
Kim, Y., Babnigg, G., Jedrzejczak, R., Eschenfeldt, W. H., Li, H., Maltseva, N., et al. (2011). High-throughput protein purification and quality assessment for crystallization. Methods 55, 12–28. doi: 10.1016/j.ymeth.2011.07.010
Krissinel, E., and Henrick, K. (2007). Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797. doi: 10.1016/j.jmb.2007.05.022
Laskowski, R. A., Macarthur, M. W., Moss, D. S., and Thornton, J. M. (1993). Procheck—a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. doi: 10.1107/S0021889892009944
Liebschner, D., Afonine, P. V., Moriarty, N. W., Poon, B. K., Sobolev, O. V., Terwilliger, T. C., et al. (2017). Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Biol. Crystallogr. 73, 148–157. doi: 10.1107/s2059798316018210
Linsky, T., and Fast, W. (2010). Mechanistic similarity and diversity among the guanidine-modifying members of the pentein superfamily. Biochim. Biophys. Acta 1804, 1943–1953. doi: 10.1016/j.bbapap.2010.07.016
Liu, J. H., Wang, W., Wu, H., Gong, X., and Moriguchi, T. (2015). Polyamines function in stress tolerance: from synthesis to regulation. Front. Plant Sci. 6:827. doi: 10.3389/fpls.2015.00827
Llacer, J. L., Polo, L. M., Tavarez, S., Alarcon, B., Hilario, R., and Rubio, V. (2007). The gene cluster for agmatine catabolism of Enterococcus faecalis: study of recombinant putrescine transcarbamylase and agmatine deiminase and a snapshot of agmatine deiminase catalyzing its reaction. J. Bacteriol. 189, 1254–1265. doi: 10.1128/jb.01216-06
Lovell, S. C., Davis, I. W., Arendall, W. B. III., de Bakker, P. I., Word, J. M., Prisant, M. G., et al. (2003). Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins 50, 437–450. doi: 10.1002/prot.10286
McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C., and Read, R. J. (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. doi: 10.1107/s0021889807021206
Michael, A. J. (2016). Biosynthesis of polyamines and polyamine-containing molecules. Biochem. J. 473, 2315–2329. doi: 10.1042/bcj20160185
Michael, A. J. (2017). Evolution of biosynthetic diversity. Biochem. J. 474, 2277–2299. doi: 10.1042/BCJ20160823
Minocha, R., Majumdar, R., and Minocha, S. C. (2014). Polyamines and abiotic stress in plants: a complex relationship. Front. Plant Sci. 5:175. doi: 10.3389/fpls.2014.00175
Mohan Chaudhuri, M., and Ghosh, B. (1985). Agmatine deiminase in rice seedlings. Phytochemistry 24, 2433–2435. doi: 10.1016/S0031-9422(00)83058-7
Mostofa, M. G., Yoshida, N., and Fujita, M. (2014). Spermidine pretreatment enhances heat tolerance in rice seedlings through modulating antioxidative and glyoxalase systems. Plant Growth Regul. 73, 31–44. doi: 10.1007/s10725-013-9865-9
Murshudov, G. N., Skubak, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., et al. (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367. doi: 10.1107/S0907444911001314
Park, K. H., and Cho, Y. D. (1991). Purification of monomeric agmatine iminohydrolase from soybean. Biochem. Biophys. Res. Commun. 174, 32–36. doi: 10.1016/0006-291X(91)90480-U
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Pottosin, I., and Shabala, S. (2014). Polyamines control of cation transport across plant membranes: implications for ion homeostasis and abiotic stress signaling. Front. Plant Sci. 5:154. doi: 10.3389/fpls.2014.00154
Pottosin, I., Velarde-Buendia, A. M., Bose, J., Fuglsang, A. T., and Shabala, S. (2014). Polyamines cause plasma membrane depolarization, activate Ca2+-, and modulate H+-ATPase pump activity in pea roots. J. Exp. Bot. 65, 2463–2472. doi: 10.1093/jxb/eru133
Prunetti, L., Graf, M., Blaby, I. K., Peil, L., Makkay, A. M., Starosta, A. L., et al. (2016). Deciphering the translation initiation factor 5A modification pathway in halophilic archaea. Archaea 2016:7316725. doi: 10.1155/2016/7316725
Radhakrishnan, R., and Lee, I. J. (2013). Spermine promotes acclimation to osmotic stress by modifying antioxidant, abscisic acid, and jasmonic acid signals in soybean. J. Plant Growth Regul. 32, 22–30. doi: 10.1007/s00344-012-9274-8
Ruszkowski, M., Sekula, B., Ruszkowska, A., and Dauter, Z. (2018). Chloroplastic serine hydroxymethyltransferase from Medicago truncatula: a structural characterization. Front. Plant Sci. 9:584. doi: 10.3389/fpls.2018.00584
Satishchandran, C., and Boyle, S. M. (1986). Purification and properties of agmatine ureohydrolyase, a putrescine biosynthetic enzyme in Escherichia coli. J. Bacteriol. 165, 843–848. doi: 10.1128/jb.165.3.843-848.1986
Sekula, B., and Dauter, Z. (2018). Crystal structure of thermospermine synthase from Medicago truncatula and substrate discriminatory features of plant aminopropyltransferases. Biochem. J. 475, 787–802. doi: 10.1042/bcj20170900
Sekula, B., Ruszkowski, M., and Dauter, Z. (2018). Structural analysis of phosphoserine aminotransferase (Isoform 1) from Arabidopsis thaliana–the enzyme involved in the phosphorylated pathway of serine biosynthesis. Front. Plant Sci. 9:876. doi: 10.3389/fpls.2018.00876
Sekula, B., Ruszkowski, M., Malinska, M., and Dauter, Z. (2016). Structural investigations of N-carbamoylputrescine amidohydrolase from Medicago truncatula: insights into the ultimate step of putrescine biosynthesis in plants. Front. Plant Sci. 7:350. doi: 10.3389/fpls.2016.00350
Shek, R., Dattmore, D. A., Stives, D. P., Jackson, A. L., Chatfield, C. H., Hicks, K. A., et al. (2017). Structural and functional basis for targeting Campylobacter jejuni agmatine deiminase to overcome antibiotic resistance. Biochemistry 56, 6734–6742. doi: 10.1021/acs.biochem.7b00982
Shirai, H., Mokrab, Y., and Mizuguchi, K. (2006). The guanidino-group modifying enzymes: structural basis for their diversity and commonality. Proteins 64, 1010–1023. doi: 10.1002/prot.20863
Sillitoe, I., Lewis, T. E., Cuff, A., Das, S., Ashford, P., Dawson, N. L., et al. (2015). CATH: comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 43, D376–D381. doi: 10.1093/nar/gku947
Svergun, D. I. (1999). Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886. doi: 10.1016/S0006-3495(99)77443-6
Takano, A., Kakehi, J., and Takahashi, T. (2012). Thermospermine is not a minor polyamine in the plant kingdom. Plant Cell Physiol. 53, 606–616. doi: 10.1093/pcp/pcs019
Terwilliger, T. C., Grosse-Kunstleve, R. W., Afonine, P. V., Moriarty, N. W., Zwart, P. H., Hung, L. W., et al. (2008). Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69. doi: 10.1107/S090744490705024X
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Tiburcio, A. F., Altabella, T., Bitrian, M., and Alcazar, R. (2014). The roles of polyamines during the lifespan of plants: from development to stress. Planta 240, 1–18. doi: 10.1007/s00425-014-2055-9
Volkov, V. V., and Svergun, D. I. (2003). Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864. doi: 10.1107/S0021889803000268
Wang, B. Q., Zhang, Q. F., Liu, J. H., and Li, G. H. (2011). Overexpression of PtADC confers enhanced dehydration and drought tolerance in transgenic tobacco and tomato: effect on ROS elimination. Biochem. Biophys. Res. Commun. 413, 10–16. doi: 10.1016/j.bbrc.2011.08.015
Winn, M. D., Isupov, M. N., and Murshudov, G. N. (2001). Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133. doi: 10.1107/S0907444900014736
Winn, M. D., Murshudov, G. N., and Papiz, M. Z. (2003). Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321. doi: 10.1016/S0076-6879(03)74014-2
Yanagisawa, H. (2001). Agmatine deiminase from maize shoots: purification and properties. Phytochemistry 56, 643–647. doi: 10.1016/S0031-9422(00)00491-X
Keywords: polyamine biosynthesis, putrescine, beta/alpha propeller fold, penteins, agmatine deiminase, guanidine-modifying enzymes
Citation: Sekula B and Dauter Z (2019) Structural Study of Agmatine Iminohydrolase from Medicago truncatula, the Second Enzyme of the Agmatine Route of Putrescine Biosynthesis in Plants. Front. Plant Sci. 10:320. doi: 10.3389/fpls.2019.00320
Edited by:
Rubén Alcázar, University of Barcelona, SpainReviewed by:
Jarrod B. French, Stony Brook University, United StatesPedro Carrasco, University of Valencia, Spain
Copyright © 2019 Sekula and Dauter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bartosz Sekula, YmFydG9zei5zZWt1bGFAbmloLmdvdjs=c2VrdWxhLmJhcnRvc3pAZ21haWwuY29t