 Chaojie Zhang1,2,3,4
Chaojie Zhang1,2,3,4 Jia Guo1,2,3,4*
Jia Guo1,2,3,4*- 1Nephrology Research Center, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 2Institute of Nephrology, Zhengzhou University, Zhengzhou, China
- 3Henan Province Research Center for Kidney Disease, Zhengzhou, China
- 4Key Laboratory of Precision Diagnosis and Treatment for Chronic Kidney Disease in Henan Province, Zhengzhou, China
The inward-rectifying potassium channel subunit Kir5.1, encoded by Kcnj16, can form functional heteromeric channels (Kir4.1/5.1 and Kir4.2/5.1) with Kir4.1 (encoded by Kcnj10) or Kir4.2 (encoded by Kcnj15). It is expressed in the kidneys, pancreas, thyroid, brain, and other organs. Although Kir5.1 cannot form functional homomeric channels in most cases, an increasing number of studies in recent years have found that the functions of this subunit should not be underestimated. Kir5.1 can confer intracellular pH sensitivity to Kir4.1/5.1 channels, which can act as extracellular potassium sensors in the renal distal convoluted tubule segment. This segment plays an important role in maintaining potassium and acid-base balances. This review summarizes the various pathophysiological processes involved in Kir5.1 and the expression changes of Kir5.1 as a differentially expressed gene in various cancers, as well as describing several other disease phenotypes caused by Kir5.1 dysfunction.
1 Introduction
Ion channels, a type of transmembrane protein, are key pathways for ions to enter and exit cells. Diseases caused by the dysfunction of ion channels in cell and organelle membranes are called to channelopathies (Kim, 2014), which have been found underlie many human diseases, such as epilepsy, arrhythmia, asthma, and diabetes (Kass, 2005; Ashcroft, 2006; Valverde et al., 2011). Among different kinds of ion channels, potassium channels play a decisive role in various physiological processes such as maintenance of the cell resting membrane potential and regulation of hormone secretion and blood pressure. According to their different structures and functions, potassium ion channels can be mainly divided into five categories: 1) inward rectifiers, Kir; 2) four transmembrane segments-2 pores, K2P; 3) voltage-gated, Kv; 4) the Slo (slowpoke) family; and 5) the Ca2+-activated SK family (small conductance calcium activated potassium channels), SKCa (Figure 1) (Gonzalez et al., 2012).

FIGURE 1. Classification of potassium channels. Abbreviations: Kir, inward rectifiers; K2P, four transmembrane segments with two pores; Kv, voltage-gated; Slo, slowpoke; SKCa, the Ca2+ -activated SK family (small conductance calcium activated potassium channels); ATP, adenosine-triphosphate.
Kir channels are indispensable in maintaining cell resting membrane potential, regulating cell excitability, and regulating systemic electrolyte homeostasis. Inward-rectifying Kir channels exhibit the ability to allow potassium ions to pass through them that increases with hyperpolarization and decreases with depolarization. This characteristic is not an inherent characteristic of channel proteins, but requires the participation of intracellular substances, such as Mg2+ and polyamines (Hibino et al., 2010).17 Kir subunits encoded by KCNJ genes have been identified, and functional Kir channels consist of four subunits in a homologous or heterologous manner (Manis et al., 2021). There are seven Kir channel subfamilies (Kir1.x-Kir7.x) that can be divided into four groups according to their function: 1) classical Kir channels (activated by phosphatidylinositol 4,5-bisphosphate, PtdIns(4,5)P2): Kir2.x; 2) G protein-gated Kir channels (regulated by G protein-coupled receptors): Kir3.x; 3) ATP-sensitive K+ channels (regulated by intracellular nucleotides and by various pharmacological agents): Kir6.x; and 4) K+ transport channels (affected by intracellular pH): Kir1.x, Kir4.x, Kir5.x, and Kir7.x (Hibino et al., 2010).
Among them, Kir5.1, encoded by KCNJ16, is expressed in many organs and tissues, such as the brain, kidney, pancreas, thyroid, and testis (Figure 2) (Liu et al., 2000; Hibino et al., 2004a; Hibino et al., 2004b; Wu et al., 2004). Depending on its location, it performs different functions. Recent studies have demonstrated the important role of Kir5.1 channels in different human organs and diseases; for example, KCNJ16 biallelic mutations can lead to salt wasting, disturbed acid-base homeostasis, and sensorineural deafness, (Welling, 2016; Manis et al., 2020; Schlingmann et al., 2021). With the development of researches on Kir5.1, it has been found that Kir5.1 is closely related to the phenotypes of various diseases, such as participating in the formation of salt-sensitive hypertension, affecting respiratory compensatory ability and reproductive ability (Palygin et al., 2017a; Puissant et al., 2019; Zhao et al., 2021). In addition, KCNJ16 has not only been found to be a differentially expressed gene in a variety of cancers, but also KCNJ16 gene mutation is associated with sudden infant death syndrome, Brugada syndrome, lymphoma and other diseases (Juang et al., 2014; Daino et al., 2019). Even more impressive, Kir5.1 was previously thought to be unable to form functional potassium channels alone. However, studies have found that SQM, the carboxy-terminal amino acid sequence of Kir5.1, can bind to the PZD3-SH3 domain of postsynaptic density protein 95 (PSD-95) to form a functional Kir5.1 channel in brain neurons. As a potential target of the protein kinase A (PKA)-mediated signaling pathway, Kir5.1/PSD-95 channels may play an important functional role in synaptic transmission (Tanemoto et al., 2002).
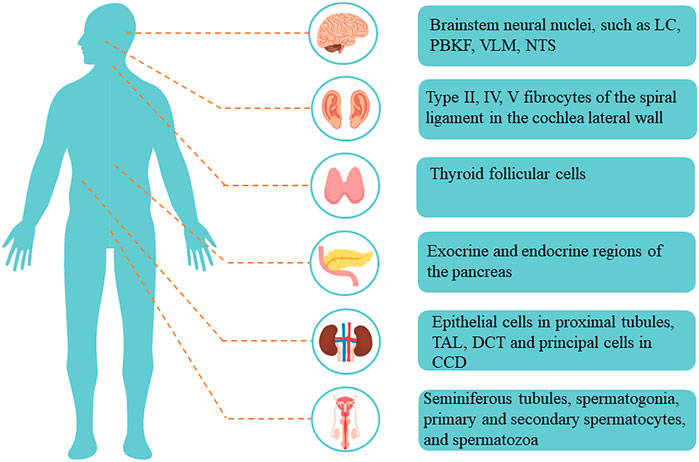
FIGURE 2. Distribution and localization of Kir5.1 in the human body. Abbreviations: LC, locus coeruleus; PBKF, parabrachial, and Kolliker-Fuse; VLM, ventrolateral medullary; NTS, nucleus of solitary tract; TAL, thick ascending limb; DCT, distal convoluted tubule; CCD, cortical collecting duct.
In view of the new findings regarding the role of Kir5.1 in human diseases, we believe that it still has many important functions worth further investigation. This review introduces the inward-rectifying potassium channel Kir5.1 from three aspects: the main physiological characteristics, the pathological processes involved and the connection with various human diseases. Overall, this review broadly summarizes the functions of Kir5.1 reported in existing studies, and provides new ideas for future research exploring the diverse biological functions of Kir5.1 and its possible significance in human diseases (Zhang et al., 2021).
2 Main physiological characteristics of Kir5.1
2.1 Intracellular pH sensitivity of Kir5.1
In 2000, Liu et al. found the new inward-rectifying potassium channel subunit Kir5.1 (encoded by KCNJ16) by searching the Genbank database. It was found that human KCNJ16, localized on chromosome 17q25, and mouse Kcnj16, localized on the distal region of chromosome 11, were highly homologous. Further tissue distribution studies showed that human KCNJ16 was significantly expressed in human renal tubular epithelial cells, pancreatic acinar cells, ductal cells, and the thyroid gland (Liu et al., 2000). Previous studies suggested that Kir5.1 could not form functional homomeric channels, but could form functional heteromeric channels (Kir4.1/5.1 and Kir4.2/5.1) with Kir4.1, encoded by Kcnj10, or Kir4.2, encoded by Kcnj15 (Pessia et al., 2001). By transfecting Kir4.1 alone and co-transfecting Kir4.1 and Kir5.1 in HEK293T cells, researchers found that the involvement of the Kir5.1 subunit conferred intracellular pH (pHi) sensitivity to Kir4.1/5.1 channels. Kir4.1 channel activity remained stable between pHi value was 6–8. When the pHi value was 7, Kir4.1/5.1 channel activity decreased to 20% of that when the pHi value was 7.3, and when the pHi value was 6–6.5, channel activity almost disappeared. In addition, Kir4.1/5.1 could also be activated by intracellular alkalinization. Changing the extracellular pH in this range had no effect on either Kir channel (Tanemoto et al., 2000). However, compared with Kir4.2 channels, the participation of Kir5.1 did not significantly increase the sensitivity of Kir4.2/5.1 to pHi, because of the pH sensing mechanism at the C-terminal of the Kir4.2 subunit making it significantly more sensitive to pHi than the Kir4.1 channel (Pessia et al., 2001). The pHi sensitivity of the Kir4.1/5.1 and Kir4.2/5.1 channels and their localization in renal tubular epithelial cells suggest that they may play an important role in regulating acid-base and electrolyte balances in the kidney.
2.2 Potassium sensor function of Kir5.1
Kir4.1/5.1 is not only the main potassium channel in the basolateral membrane of mouse kidney distal convoluted tubule (DCT) epithelial cells, but is also the extracellular potassium sensor, which determines the potassium conductance of the basolateral membrane, forms the transmembrane potential of tubular epithelial cells, senses the change of extracellular K+ concentration, and changes the membrane potential and intracellular Cl− concentration of tubular epithelial cells (Zhang et al., 2014; Palmer, 2015; Su et al., 2019; Wu et al., 2019). In the renal DCT, NaCl cotransporter (NCC) is mainly responsible for regulating Na+ and Cl− reabsorption and K+ secretion in distal tubules. Kir4.1/5.1 regulates NCC activity through the with-no-lysine kinase (WNK)-STE20/SPS-1-related proline-alanine-rich protein kinase (SPAK) pathway. Finally, it regulates salt reabsorption and K+ secretion in distal tubules and collecting ducts (Terker et al., 2015; Hadchouel et al., 2016; Welling, 2016). For example, in hypokalemia, increased Kir4.1/5.1 activity hyperpolarizes cell membranes, drives intracellular Cl− efflux, causes low Cl− levels in epithelial cells, further activates WNK, activates NCC through the phosphorylation of SPAK, increases sodium reabsorption in the DCT, reduces K+ excretion in distal tubules and collecting ducts, and maintains blood potassium levels. The opposite is true for hyperkalemia (Terker et al., 2015; Welling, 2016). Previous studies have shown that knockout of Kir4.1 can eliminate the effect of dietary potassium on NCC activity (Cuevas et al., 2017; Wang et al., 2018). In addition, Wu et al. found that in wild-type mice a high-potassium diet could inhibit the expression of NCC, whereas a low-potassium diet could promote the expression of NCC. This phenomenon was eliminated in Kir5.1 knockout mice, which had a decreased ability to expel K+ on a high-potassium diet and to retain K+ on a low-potassium diet (Wu et al., 2019). Therefore, knockout of Kir5.1 also abolished the effect of dietary potassium on NCC activity. This suggests that Kir5.1, like Kir4.1, is an indispensable part of the Kir4.1/5.1 channel for its function as a potassium sensor in the DCT. In conclusion, Kir4.1/5.1 regulates NCC activity by sensing changes in extracellular K+ concentration and altering basolateral membrane conductance to ensure that urinary potassium excretion matches dietary potassium intake.
3 Pathophysiological processes related to Kir5.1
3.1 Kir5.1 dysfunction can cause metabolic acidosis and hypokalemia
In the kidney of rats and mice, Kir5.1 is mainly expressed in the basolateral membrane of epithelial cells in the thick ascending limb (TAL), the DCT, and principal cells in the cortical collecting duct (CCD) (Tucker et al., 2000; Lourdel et al., 2002; Lachheb et al., 2008). Kir5.1 is also expressed in proximal tubule (Tucker et al., 2000; Schlingmann et al., 2021). Although there is no research shown that Kir5.1 is localized in the basolateral membrane of the proximal tubule epithelial cells, the expression pattern of Kir5.1 should be consistent throughout the renal tubular segment. Besides, both Kir4.2 and Kir4.1, which bind to Kir5.1 to form functional potassium channels, are localized in the basolateral membrane, and if Kir5.1 is expressed in the apical membrane of the proximal tubule, it is likely to be non-functional (Lourdel et al., 2002; Bignon et al., 2020). Therefore, we suggest that Kir5.1 is mainly expressed in the basolateral membrane of the proximal tubule epithelial cells. In addition, in immunoperoxidase staining of the rat kidney, Kir5.1 expression is also observed in endothelial cells around glomerular capillaries (Tucker et al., 2000).
Previous studies have demonstrated that Kir4.1/5.1 is the main potassium channel in the basolateral membrane of DCT epithelial cells, contributes to K+ circulation with Na+-K+-ATPase, and participates in regulating systemic electrolyte balance and maintaining acid-base homeostasis (Palygin et al., 2017b; Palygin et al., 2018; Lin et al., 2022; Wang and Lin, 2022). Among them, mutations in the KCNJ10 gene encoding the Kir4.1 subunit can cause EAST/SeSAME syndrome (epilepsy, ataxia, sensorineural deafness, tubulopathy/seizures, sensorineural deafness, ataxia, intellectual disability, and electrolyte imbalance), which manifests as epilepsy, ataxia, sensorineural deafness, hypokalemia, and metabolic alkalosis (Bockenhauer et al., 2009; Reichold et al., 2010). In contrast to metabolic alkalosis caused by KCNJ10 gene mutation, mice with a targeted disruption of Kcnj16 and Dahl salt-sensitive (Dahl SS) rats with Kcnj16 gene mutations showed distinct hyperchloremic acidosis. Moreover, urinary ammonium excretion was reduced in mice with targeted disruption of Kcnj16 (Paulais et al., 2011; Puissant et al., 2019). A similar phenomenon has been found in human studies: in 2021, Schlingmann et al. found that eight patients with KCNJ16 biallelic variants at different locations had common clinical features, including salt wasting, acid-base homeostasis disorder, sensorineural deafness, and decreased urinary ammonium excretion, and all but one presented with metabolic acidosis (Schlingmann et al., 2021; Webb et al., 2021). The symptoms of Kir5.1 dysfunction are different from those of Kir4.1 dysfunction, suggesting that Kir5.1 in the kidney is not limited to forming Kir4.1/5.1 channels with Kir4.1 in distal tubules, but also plays other important potential roles in which Kir4.1 is not involved.
In addition, Kir5.1 also plays an essential role in the proximal tubule. The proximal tubule is not only the main reabsorption site of Na+, K+, Cl−, and HCO3−, but is also the main secretion site of H+ and NH4+. To explore the function of Kir5.1 in proximal tubules, Schlingmann et al. further analyzed the expression of Kcnj16, Kcnj10, and Kcnj15 mRNA in the mouse kidney. The results showed that Kcnj16 was widely expressed in renal tubules, whereas Kcnj10 was mostly distributed in the distal tubule, and Kcnj15 was mainly distributed in the proximal tubule (Schlingmann et al., 2021). Interestingly, Kcnj15-deleted mice exhibits hyperchloremic acidosis, decreased bicarbonate reabsorption threshold, and urinary NH4+ excretion dysfunction (Bignon et al., 2020). Moreover, previous studies have shown that Kir4.2 expression is significantly reduced in Kir5.1 knockout mice (Wu et al., 2019). Therefore, Kcnj16 gene mutations may cause the depolarization of proximal tubule epithelial cells through the dysfunction of Kir4.2/5.1 channels and decreased Kir4.2 expression, leading to reduced Na+-HCO3− cotransporter NBCe1 (encoded by SLC4A4) transport in the basolateral membrane. The accumulation of HCO3− in epithelial cells leads to intracellular alkalinization, which can cause abnormal expression levels of glutamine transporters in the basolateral membrane and intracellular ammoniagenic enzymes, and eventually lead to decreased ammonia production (Lee et al., 2018). In Kir5.1 dysfunctional individuals, these mechanisms lead to a phenomenon similar to that observed in Kcnj15-deleted mice, that is, the decreased ability of proximal tubules to reabsorb bicarbonate and secrete NH4+, resulting in metabolic acidosis and decreased urinary ammonium excretion levels, which patients with KCNJ10 mutations do not have (Paulais et al., 2011; Schlingmann et al., 2021). These findings reveal a non-redundant role of Kir5.1 in the proximal tubule. Notably, there is no aminoaciduria or glycosuria in either the Kcnj15 or Kcnj16 gene variants, suggesting that Kir4.2 or Kir5.1 dysfunction caused a phenomenon similar to the permanent forms of familial isolated proximal renal tubular acidosis. Bicarbonate reabsorption disorder and low NH4+ secretion occurred in the absence of extensive proximal tubule reabsorption disorder, suggesting that potassium channels other than Kir4.2 or Kir4.2/5.1 may exist in the proximal tubule to maintain basolateral membrane K+ circulation, thus maintaining normal material reabsorption in the apical membrane of tubular epithelial cells (Bignon et al., 2020; Schlingmann et al., 2021). Interestingly, one patient in this cohort presented with metabolic alkalosis, and further studies revealed that the KCNJ16 mutant in this patient had a strong inhibition of Kir4.1 and milder effect on the Kir4.2/5.1. Therefore, we speculate that whether the KCNJ16 mutant shows metabolic acidosis or metabolic alkalosis depends on whether the effect of the mutant on Kir4.2/5.1 and Kir4.2 in the proximal tubule is dominant or the effect on Kir4.1/5.1 and Kir4.1 in the distal tubule is dominant (Schlingmann et al., 2021).
Another significant manifestation caused by Kir5.1 dysfunction in the kidney is hypokalemia (Paulais et al., 2011; Palygin et al., 2017a; Puissant et al., 2019; Schlingmann et al., 2021). In the body, potassium balance not only requires Na+-K+ pumping and Na+-H+ exchange to change the distribution of intracellular and extracellular fluid, but also requires regulation of potassium excretion by changing the transmembrane potential of renal tubular epithelial cells and the flow rate of distal tubular fluid. Although Kir4.1 and Kir5.1 are both essential for the function of the Kir4.1/5.1 potassium sensor and both cause hypokalemia after knockout, they have different mechanisms (Reichold et al., 2010). KCNJ10 gene mutation can reduce the potassium conductance of the basolateral membrane of DCT epithelial cells, inactivate the WNK–SPAK–NCC pathway, reduce NCC reabsorption, increase the flow rate of distal tubule fluid, quickly remove K+ secreted into the tubule fluid from distal tubules and collecting ducts, and greatly reduce the concentration of K+ in the tubule. The driving force for the secretion of K+ into tubule fluid in tubular epithelial cells increases, leading to a decrease in blood K+ (Zhang et al., 2014). However, in mice with targeted disruption of Kcnj16, owing to the presence of Kir4.1 channels in the basolateral membrane of DCT cells, the activity of NCC after Kir5.1 dysfunction does not decrease, but in fact increases, in response to the increase of Kir4.1 channel conductance (Paulais et al., 2011; Wu et al., 2019). Therefore, the occurrence of hypokalemia in mice with Kir5.1 dysfunction may be related to the widespread distribution of Kir5.1 in renal tubules; Kir4.2/5.1 is expressed in the basolateral membrane of proximal tubular epithelial cells, and Kir4.1/5.1 is expressed in the basolateral membrane of TAL and DCT epithelial cells and CCD principal cells. Kir5.1 dysfunction causes a large range of K+ reabsorption disorders in the basolateral membrane of tubular epithelial cells, resulting in hypokalemia. These studies suggest that Kir5.1 plays an important role in regulating systemic electrolyte homeostasis in the kidney.
3.2 Kir5.1 dysfunction causes loss of salt-sensitive phenotype in Dahl SS rats
Previous studies have found that dietary potassium deficiency can increase blood pressure and salt sensitivity (elevated blood pressure in response to high salt intake) by changing cell membrane voltage, leading to hyperpolarization and stimulating NCC activation (Terker et al., 2015). This is closely related to the potassium sensor function of Kir4.1/5.1. Kir4.1/5.1 can perceive the change of extracellular K+ concentration, change cell membrane potential, and regulate NCC activity to coordinate urinary potassium excretion and dietary potassium intake under normal conditions. When dietary potassium deficiency occurs, the concentration of extracellular K+ decreases, and Kir4.1/5.1 hyperpolarizes the cell membrane and causes NCC activation through the WNK-SPAK-NCC pathway to limit potassium loss, sometimes even at the cost of increasing blood pressure (Terker et al., 2015; Welling, 2016).
Dahl SS rats are a genetically stable and widely used model of hypertension, and their salt sensitivity is characterized by a significant increase in blood pressure after a high-salt diet (Wadei and Textor, 2012). Palygin et al. found that Kir5.1 expression was significantly increased in SS rat kidneys after a high-salt diet, whereas Dahl SS rats with Kcnj16 knockout (SSKcnj16−/−) showed hypotension, and their blood pressure did not increase after a high-salt diet. This suggests that the Kir5.1 channel plays a key role in the development of salt-sensitive hypertension (Palygin et al., 2017a). Thus, targeting Kir5.1 may be an effective way to lower blood pressure during the development of salt-sensitive hypertension.
In addition, SSKcnj16−/− rats also show significant salt consumption, which is not seen in mice with targeted disruption of Kcnj16 (Paulais et al., 2011; Palygin et al., 2017a; Wu et al., 2019). Renal DCT reabsorption not only requires K+ circulation in the basolateral membrane to provide a Na+ concentration gradient favorable for reabsorption in the apical membrane of epithelial cells, but also requires normal NCC activity. This difference in salt consumption may be owing to the different localization of Kir4.1 in the renal tubular epithelial cells of the two knockout animals, although Kir4.1 expression is upregulated in both. In SSKcnj16−/− rats, Kir4.1 is significantly expressed in the cytoplasm, with only a small amount in the basolateral membrane, in contrast to SS rats, where the Kir4.1 channels are located in the basolateral membrane. However, Kir4.1 is significantly upregulated in the basolateral membrane of mice with targeted disruption of Kcnj16 (Paulais et al., 2011; Palygin et al., 2017a). Kir4.1 in the basolateral membrane can maintain K+ circulation and upregulate the expression of NCC in mice with targeted disruption of Kcnj16, thus maintaining normal salt reabsorption. However, in SSKcnj16−/− rats, although the expression of NCC is upregulated, only a small amount of Kir4.1 is present in the basolateral membrane, which is insufficient to maintain normal K+ circulation; this disrupts the Na+ concentration gradient in the apical membrane of epithelial cells, reduces the reabsorption of the DCT, and thus leads to salt consumption (Palygin et al., 2017a). The different localization of Kir4.1 in the kidneys of the two animals with Kir5.1 dysfunction may be owing to their different gene backgrounds (Wu et al., 2019). Furthermore, knockout of Kcnj16 markedly alters the regulation and function of the renin-angiotensin-aldosterone system in Dahl SS rats (Manis et al., 2019). Therefore, as an essential component of potassium sensor and K+ circulation as well as an important regulator of renin-angiotensin-aldosterone system, Kir5.1 plays a key role in the development of salt-sensitive hypertension.
3.3 Kir5.1 dysfunction reduces respiratory compensatory ability against hypercapnia and hypoxia
Arterial CO2 controls respiratory activity in mammals by acting on peripheral respiratory chemoreceptors in the carotid body and aortic body, as well as central respiratory chemoreceptors located in the brainstem. Central respiratory chemoreceptors can drive respiratory activity to be closely linked to arterial pH and CO2 levels, a process thought to be regulated by some PCO2/pH-sensitive channels (Puissant et al., 2019). Studies have found that, in addition to pHi sensitivity, Kir4.1/5.1 channels are also highly sensitive to CO2 (Tucker et al., 2000; Xu et al., 2000; Yang et al., 2000; Zhu et al., 2000). Wu et al. confirmed that Kir5.1 and Kir4.1 were co-expressed in the brainstem neural nuclei involved in the control of respiratory activity, such as the locus coeruleus (LC), parabrachial and Kolliker–Fuse (PBKF) nuclei, the ventrolateral medullary (VLM) area, and the nucleus of the solitary tract (NTS) (Wu et al., 2004). Therefore, the PCO2/pHi sensitive properties and localization of Kir4.1/5.1 channels suggest that they may be involved in the neuronal response to changes in PCO2 and pH in the cellular microenvironment.
The LC is a region in the pons that is chemically sensitive to CO2. Inhibition of Kir4.1/5.1 channels by hypercapnic acidosis can cause depolarization and lead to an increase in the excitability and firing frequency of more than 80% of the LC neurons (Wu et al., 2004; D'Adamo et al., 2011). Although the signaling between LC neurons and the medullary respiratory center remains controversial, increased excitability of LC neurons contributes to the regulation of respiratory activity during hypercapnia. Using mouse brain slices, researchers found that LC neurons in mice lacking Kcnj16 had a reduced increase in firing frequency and a prolonged response time in response to intracellular acidification and hypercapnia compared with wildtype (WT) mice. This suggests that Kir5.1 is an important determinant of PCO2/pHi sensitivity in the LC neurons, is able to link CO2 levels in blood and cerebrospinal fluid to changes in neuronal activity, and may be involved in regulating respiratory responses during hypercapnic acidosis (Wu et al., 2004; D'Adamo et al., 2011).
However, another group of studies suggests otherwise. In 2011, Trapp et al. suggested that Kir5.1 knockout may not directly affect the sensitivity of central and peripheral respiratory chemoreceptors in mice. They used the Kir5.1 knockout mouse (Kir5.1−/−) model to observe changes in the ventilation response of Kir5.1−/− mice at rest and in response to hypoxia and increased ambient CO2. Studies have found that Kir4.1 and Kir5.1 are expressed in both the respiratory center and peripheral carotid body (Wu et al., 2004; Yamamoto et al., 2008), but Kir5.1 knockout has no significant effect on central chemoreceptor sensitivity and carotid body function. The increases of respiratory frequency and respiratory depth in Kir5.1−/− mice during hypercapnia and hypoxia were both lower than those in the WT group, which may be owing to abnormal signal transmission between peripheral chemoreceptors and the respiratory center; however, the mechanism is still unclear (Trapp et al., 2011). In addition, Puissant et al. observed a similar phenotype in SSKcnj16−/− rats, which showed a reduced ventilation response to hypercapnia and hypoxia (Puissant et al., 2019). These results demonstrate the important role of Kir5.1 channels in hypercapnic and hypoxic ventilation responses.
Other studies analyzed the exome data of 155 cases of sudden infant death syndrome (SIDS) and identified the KCNJ16 R137S mutant. It was found that this mutant may cause disorders in signal reception in central respiratory chemoreceptors; thus, KCNJ16 mutation may be a risk factor for SIDS (Neubauer et al., 2022). In conclusion, Kir5.1 knockout reduces the respiratory compensatory ability of the body to hypercapnia and hypoxia, which may be owing to abnormal signal transmission between peripheral respiratory chemoreceptors and central respiratory chemoreceptors or a signal reception disorder of the central chemoreceptors. The specific mechanism needs to be further studied in the future. In addition, the effects of metabolic acidosis caused by proximal tubular dysfunction in Kir5.1 knockout models on respiratory peripheral and central chemoreceptors should also be considered.
4 Research progress of Kir5.1 and human diseases
4.1 Kir5.1 and sensorineural deafness
Endolymph is the extracellular solution in the cochlear canal containing 150 mM K+ that exhibits a potential of approximately +80 mV relative to the adjacent perilymph, which represents the endocochlear potential (EP). The EP is a major driver of auditory signal transduction. External sounds are transmitted through the outer and middle ears, and then stimulate the cochlea of the inner ear. When the sound-driven vibration stimulates the basilar membrane, the hair bundles of hair cells are deflected, thus opening mechano-sensitive channels in the apical membrane of hair cells; then, the K+ in the endolymph flows into and stimulates hair cells. At the same time, the sound-driven vibration is converted into electrochemical signals, which are transmitted along the auditory nerve fibers to the auditory center, finally producing hearing. Subsequently, K+ is secreted by hair cells to the perilymph, further reabsorbed into support cells and spiral ligament fibrocytes, and finally transported to the stria vascularis; from the stria vascularis, K+ is re-secreted to the endolymph to maintain high K+ and EP in the endolymph, thus forming K+ circulation in the cochlea. Interference with any step of the K+ circulation pathway in the cochlea destroys the EP and leads to hearing impairment (Hibino et al., 2004b; Hibino and Kurachi, 2006; Lv et al., 2021).
In rats, Kir4.1 is abundantly expressed in the stria vascularis of the cochlea, whereas Kir5.1 is abundantly expressed in several specific types of fibrocytes (type II, IV, and V fibrocytes) of the spiral ligament in the cochlea lateral wall (Hibino et al., 2004b). Kir4.1 and Kir5.1 have completely different distribution positions in the cochlea, suggesting that they may function as homomeric channels in the cochlea. In addition, the expression of PSD-95 has also been detected in the cochlea. Kir5.1 subunits in fibrocytes are mostly expressed intracellularly, and Kir5.1 can combine with PSD-95 to form functional membrane channels (Tanemoto et al., 2002). Together with Kir4.1, Na+-K+-ATPase, Na+-K+-2Cl−-cotransporter, and other channels, Kir5.1 is involved in forming K+ circulation in the cochlea (Hibino et al., 2004b). Previous studies have demonstrated that mutations in the KCNJ10 gene can cause sensorineural deafness (Bockenhauer et al., 2009; Reichold et al., 2010), but whether Kir5.1 dysfunction can cause hearing impairment is still controversial. Some studies have suggested that Kcnj16 deletion in mice does not cause changes in hearing function, which may be related to compensation through other ion channels, such as KCNMA1, KCNQ4, and KCNE1 (Lv et al., 2021). However, in a group of disease types discovered by Schlingmann et al., eight patients with KCNJ16 biallelic mutations at different positions all developed sensorineural deafness (Schlingmann et al., 2021). Therefore, the function of Kir5.1 in the cochlea and its role in hearing need to be further investigated.
4.2 Kir5.1 and cancer
Studies have found that potassium channels are not only involved in the control of the cell cycle and cell proliferation, but also in the processes of cell adhesion and migration, volume regulation, apoptosis, and angiogenesis, which are closely related to tumor biology, making potassium channels a potential therapeutic target for tumors (Pardo and Stuehmer, 2014; Comes et al., 2015). At present, studies on Kir5.1 and cancer mainly focus on the thyroid, pancreas, and prostate, among others, and the expression changes of Kir5.1 are not consistent in different glands during carcinogenesis (Table 1). In 2014, Ramos et al. found that Kir5.1, Kir4.1, and Kir4.2 were significantly expressed in the thyroid, but the function of Kir5.1 in the thyroid was unclear, although it was suggested to be involved in the transport of thyroglobulin (Ramos et al., 2015). Liu et al. analyzed mRNA datasets from 23 anaplastic thyroid cancer (ATC) samples and 24 normal samples and identified 55 differentially expressed genes (DEGs). Among them, the mRNA expression level of KCNJ16 was low in ATC samples. Most of these DEGs are closely related to biological processes related to tumor progression. Therefore, identification of the key DEGs is of great significance for understanding the molecular mechanism of ATC (Liu et al., 2016). In addition, Kir5.1 is also expressed in both exocrine and endocrine regions of the pancreas (Liu et al., 2000; Pessia et al., 2001). In 2017, from GEO datasets of pancreatic ductal adenocarcinoma (PDAC) and normal pancreatic tissue samples, Jiang et al. found four differentially expressed potassium channels, and KCNJ16 mRNA expression was found to be downregulated in PDAC samples (Jiang et al., 2017). In a 2022 study, RNA transcriptome profiles and clinical data analysis of patients with clear cell renal cell carcinoma (ccRCC) showed that KCNJ16 expression was downregulated in patients with ccRCC, which was believed to be closely related to biological activities such as transmembrane transport and cell structure maintenance. Multiple ion channels are destroyed and altered during the development of ccRCC. Therefore, exploring the alteration of ion channels in the development of ccRCC may facilitate diagnosis and evaluation and inform the corresponding treatment (Zhu et al., 2022).
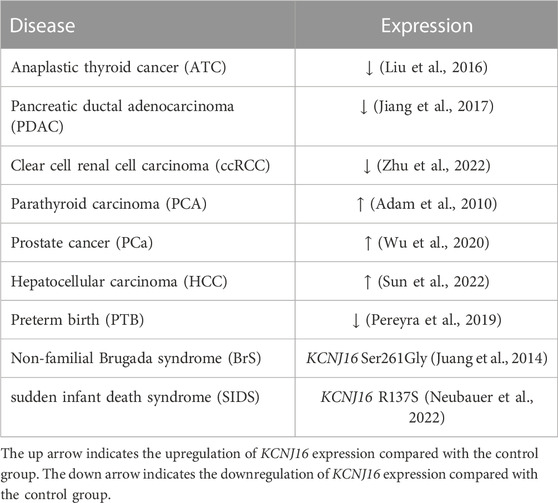
TABLE 1. Expression changes of Kir5.1 as a differentially expressed gene in human diseases.
Abnormally elevated KCNJ16 expression may also lead to carcinogenesis. Through microarray transcriptome analysis of a parathyroid carcinoma (PCA) case and a biopsy from the same patient’s normal parathyroid gland, Adam et al. identified KCNJ16 as one of the DEGs in PCA, and found that KCNJ16 was upregulated in patients with PCA. These DEGs may serve as potential diagnostic markers for PCA (Adam et al., 2010). In addition, RT-qPCR on the mRNA of five prostate cancer cell lines and one normal prostate cell line showed that KCNJ16 was upregulated in prostate cancer cell lines and was a DEG in patients with prostate cancer who had a Gleason score (GS) = 6 and GS ≥ 7. It is helpful to identify patients at high risk of biochemical recurrence after radical prostatectomy (Wu et al., 2020). Liu et al. applied the Significance Analysis of Microarray and Prediction Analysis of Microarray methods of GeneChip to screen out KCNJ16 among 22 risk genes in the liver tissues of patients with chronic hepatitis and hepatocellular carcinoma (HCC) or cirrhosis. Further analysis revealed that KCNJ16 mRNA was significantly upregulated in the HCC group. The detection of DEGs such as KCNJ16 is helpful for the early diagnosis of cancer (Liu et al., 2014; Sun et al., 2022). These results suggest that KCNJ16 can be used in genetic diagnostics for a variety of cancers. Early detection of lesions through genetic screening can provide earlier treatment opportunities and longer survival time for patients.
4.3 Kir5.1 and other diseases
In addition to the above human diseases, the abnormal expression of KCNJ16 is also associated with diseases of other systems, such as the reproductive system, organ development, and the central nervous system. As early as 1999, it was demonstrated that Kir5.1 was expressed in the convoluted seminiferous tubules, spermatogonia, primary and secondary spermatocytes, and spermatozoa of rats (Salvatore et al., 1999). In 2021, Poli et al. used the Kcnj16-deleted mouse model to examine the possible role of Kir5.1 in the reproductive ability of mice. The results of the study found that Kir5.1 knockout mice had smaller testes, an increased proportion of sperm with abnormal morphology, and reduced fertility, compared with WT mice. Therefore, Kir5.1 channels play an important role in the development of mouse testes, the morphology of sperm flagella, and the maintenance of normal reproductive ability (Poli et al., 2021). The cattleyak (CY), a cross breed between cattle and yaks (YKs), has better adaptability to harsh environments, and its performance is much higher than that of YKs, including a larger size, higher quality meat, and higher milk production. However, CY females are fertile, whereas the males are sterile. In a 2020 study, researchers analyzed the epididymis transcriptomes of CY and YK, and found that Kcnj16 was upregulated in CY. Thus, Kcnj16 may be involved in the regulation of fluid and pH balances, and the regulation of luminal pH is important for spermatozoa maturation and storage in the caudal region of the epididymis (Liu et al., 2000; Park et al., 2017). Therefore, the upregulation of Kcnj16 in CY may cause pH and luminal fluid imbalances, and eventually lead to male sterility in CY (Zhao et al., 2021). These studies provide new ideas for the research and diagnosis of human reproductive diseases.
In addition, Brugada syndrome (BrS) is an ion channelopathy associated with sudden cardiac death, and SCN5A is the most common mutated gene, accounting for 20%–25% of patients with BrS (Mizusawa and Wilde, 2012). Disease-targeted multigene sequencing was performed in 15 patients with non-familial BrS without SCN5A variants, and four mutated genes were identified, including KCNJ16. A variety of bioinformatic algorithms have been used for analysis to demonstrate that these mutants are harmful, and the discovery of these genes will help screen patients with non-familial BrS without SCN5A variants (Juang et al., 2014). In addition, through RNA sequencing analysis of the chorioamnion membranes of severe preterm and term fetuses, Pereyra et al. revealed 18 genes were downregulated in preterm infants, including KCNJ16. Abnormal ion channel function may be involved in the pathological process of preterm birth through pathways like Ca2+ signaling, smooth muscle contraction, and inflammatory response. These results help to establish the molecular landscape of preterm infants and improve the understanding of pathological mechanisms in preterm infants (Pereyra et al., 2019).
Moreover, it is known that loss of the transcriptional activation of the potassium channel Kir5.1 by hepatocyte nuclear factor 1 homeobox B drives autosomal-dominant tubulointerstitial kidney disease (Kompatscher et al., 2017). Kcnj16 is frequently mutated in T-cell lymphoma in mice lacking the DNA mismatch repair gene Mlh1, but its function in T-cell lymphoma remains unclear (Daino et al., 2019). In addition, Kir5.1 is also expressed in oligodendrocytes and astrocytes in the central nervous system (Brasko et al., 2017; Patterson et al., 2021), and Kcnj16 knockout produces audiogenic seizures in Dahl SS rats (Manis et al., 2021; Staruschenko et al., 2022). In conclusion, these multiple diseases caused in part or in whole by Kir5.1 dysfunction fully demonstrate its crucial role in the human body, and more studies are needed to explore its potential functions in the future.
5 Conclusion
This review mainly introduces the physiological characteristics of the inward-rectifying potassium channel Kir5.1, the pathophysiological processes related to Kir5.1, and its relationship with human diseases (Table 2). Kir5.1 not only has pHi sensitivity, but also acts as a potassium sensor, allowing its involvement in both the regulation of electrolyte homeostasis and acid-base balance in the kidneys and in the formation of the salt-sensitive phenotype in Dahl SS rats. Kir5.1 is expressed in both central and peripheral respiratory chemoreceptors and can give central respiratory chemoreceptor neurons the ability to sense PCO2 and pHi and regulate respiration by changing their firing frequency. Kir5.1 dysfunction reduces the ability of the body to compensate for hypoxia and hypercapnia. KCNJ16 biallelic mutations can lead to sensorineural deafness, but the role of Kir5.1 in auditory system requires further research. Meanwhile, several genomic analyses have shown that KCNJ16 is a DEG in a variety of cancers, such as ATC, PDAC, and HCC. In addition, potential roles of Kir5.1 in the reproductive system and in organ development have been identified.

TABLE 2. The localization and involvement of Kir5.1 in physiological processes and Kir5.1 dysfunction.
There are several shortcomings in the existing research. In contrast to the large number of studies focused on the kidneys and brain, previous studies on Kir5.1 rarely focus on its functions in the thyroid or pancreas. In 2022, during high-throughput screening of multiple compounds, McClenahan et al. discovered and validated VU6036720, the most potent Kir4.1/5.1 channel-selective inhibitor reported to date. The lack of studies on specific inhibitors of Kir4.1/5.1 channels has limited further investigation into the physiological function of Kir5.1 (McClenahan et al., 2022). In addition, future studies that focus on the regulatory mechanisms of this ion channel will facilitate the potential use of Kir5.1 as a valuable targeted therapy or regulatory strategy, and increase our understanding of this unique Kir channel family member.
Author contributions
CZ: Design and Writing-original draft; JG: Conceptualization and Modification. All authors contributed to and approved the manuscript.
Funding
This work was supported by the Natural Science Foundation of China (grant numbers 1 to JG), and the Science and Technology Research Project of Henan Province (grant numbers YXKC2021034, SBGJ202102148, and ZYQR201912174 to JG).
Acknowledgments
Thanks to Professor Zhangsuo Liu for his suggestions and guidance, as well as the laboratory platform he provided.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adam, M. A., Untch, B. R., and Olson, J. A. (2010). Parathyroid carcinoma: Current understanding and new insights into gene expression and intraoperative parathyroid hormone kinetics. Oncologist 15 (1), 61–72. doi:10.1634/theoncologist.2009-0185
Ashcroft, F. M. (2006). From molecule to malady. Nature 440 (7083), 440–447. doi:10.1038/nature04707
Bignon, Y., Pinelli, L., Frachon, N., Lahuna, O., Figueres, L., Houillier, P., et al. (2020). Defective bicarbonate reabsorption in Kir4.2 potassium channel deficient mice impairs acid-base balance and ammonia excretion. Kidney Int. 97 (2), 304–315. doi:10.1016/j.kint.2019.09.028
Bockenhauer, D., Feather, S., Stanescu, H. C., Bandulik, S., Zdebik, A. A., Reichold, M., et al. (2009). Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N. Engl. J. Med. 360 (19), 1960–1970. doi:10.1056/NEJMoa0810276
Brasko, C., Hawkins, V., De La Rocha, I. C., and Butt, A. M. (2017). Expression of Kir4.1 and Kir5.1 inwardly rectifying potassium channels in oligodendrocytes, the myelinating cells of the CNS. Brain Struct. Funct. 222 (1), 41–59. doi:10.1007/s00429-016-1199-8
Comes, N., Serrano-Albarras, A., Capera, J., Serrano-Novillo, C., Condom, E., Ramon, Y., et al. (2015). Involvement of potassium channels in the progression of cancer to a more malignant phenotype. Biochimica Biophysica Acta-Biomembranes. 1848 (10), 2477–2492. doi:10.1016/j.bbamem.2014.12.008
Cuevas, C. A., Su, X. T., Wang, M. X., Terker, A. S., Lin, D. H., McCormick, J. A., et al. (2017). Potassium sensing by renal distal tubules requires Kir4.1. J. Am. Soc. Nephrol. 28 (6), 1814–1825. doi:10.1681/asn.2016090935
D'Adamo, M. C., Shang, L., Imbrici, P., Brown, S. D. M., Pessia, M., and Tucker, S. J. (2011). Genetic inactivation of Kcnj16 identifies Kir5.1 as an important determinant of neuronal PCO2/pH sensitivity. J. Biol. Chem. 286 (1), 192–198. doi:10.1074/jbc.M110.189290
Daino, K., Ishikawa, A., Suga, T., Amasaki, Y., Kodama, Y., Shang, Y., et al. (2019). Mutational landscape of T-cell lymphoma in mice lacking the DNA mismatch repair gene Mlh1: No synergism with ionizing radiation. Carcinogenesis 40 (2), 216–224. doi:10.1093/carcin/bgz013
Gonzalez, C., Baez-Nieto, D., Valencia, I., Oyarzun, I., Rojas, P., Naranjo, D., et al. (2012). K+ channels: Function-structural overview. Compr. Physiol. 2 (3), 2087–2149. doi:10.1002/cphy.c110047
Hadchouel, J., Ellison, D. H., and Gamba, G. (2016). Regulation of renal electrolyte transport by WNK and SPAK-OSR1 kinases. Annu. Rev. Physiol. 78, 367–389. doi:10.1146/annurev-physiol-021115-105431
Hibino, H., Fujita, A., Iwai, K., Yamada, M., and Kurachi, Y. (2004). Differential assembly of inwardly rectifying K+ channel subunits, Kir4.1 and Kir5.1, in brain astrocytes. J. Biol. Chem. 279 (42), 44065–44073. doi:10.1074/jbc.M405985200
Hibino, H., Higashi-Shingai, K., Fujita, A., Iwai, K., Ishii, M., and Kurachi, Y. (2004). Expression of an inwardly rectifying K+ channel, Kir5.1, in specific types of fibrocytes in the cochlear lateral wall suggests its functional importance in the establishment of endocochlear potential. Eur. J. Neurosci. 19 (1), 76–84. doi:10.1111/j.1460-9568.2004.03092.x
Hibino, H., Inanobe, A., Furutani, K., Murakami, S., Findlay, I., and Kurachi, Y. (2010). Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 90 (1), 291–366. doi:10.1152/physrev.00021.2009
Hibino, H., and Kurachi, Y. (2006). Molecular and physiological bases of the K+ circulation in the mammalian inner ear. Physiology 21, 336–345. doi:10.1152/physiol.00023.2006
Jiang, S., Zhu, L., Yang, J., Hu, L., Gu, J., Xing, X., et al. (2017). Integrated expression profiling of potassium channels identifys KCNN4 as a prognostic biomarker of pancreatic cancer. Biochem. Biophysical Res. Commun. 494 (1-2), 113–119. doi:10.1016/j.bbrc.2017.10.072
Juang, J-M. J., Lu, T-P., Lai, L-C., Ho, C-C., Liu, Y-B., Tsai, C-T., et al. (2014). Disease-Targeted Sequencing of Ion Channel Genes identifies de novo mutations in Patients with Non-Familial Brugada Syndrome. Sci. Rep. 4, 6733. doi:10.1038/srep06733
Kass, R. S. (2005). The channelopathies: Novel insights into molecular and genetic mechanisms of human disease. J. Clin. Invest. 115 (8), 1986–1989. doi:10.1172/jci26011
Kompatscher, A., de Baaij, J. H. F., Aboudehen, K., Hoefnagels, A., Igarashi, P., Bindels, R. J. M., et al. (2017). Loss of transcriptional activation of the potassium channel Kir5.1 by HNF1β drives autosomal dominant tubulointerstitial kidney disease. Kidney Int. 92 (5), 1145–1156. doi:10.1016/j.kint.2017.03.034
Lachheb, S., Cluzeaud, F., Bens, M., Genete, M., Hibino, H., Lourdel, S., et al. (2008). Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. Am. J. Physiology-Renal Physiology 294 (6), F1398–F1407. doi:10.1152/ajprenal.00288.2007
Lee, H. W., Osis, G., Harris, A. N., Fang, L., Romero, M. F., Handlogten, M. E., et al. (2018). NBCe1-A regulates proximal tubule ammonia metabolism under basal conditions and in response to metabolic acidosis. J. Am. Soc. Nephrol. 29 (4), 1182–1197. doi:10.1681/asn.2017080935
Lin, D. H., Duan, X. P., Zheng, J. Y., and Wang, W. H. (2022). Role of inwardly rectifying K+ channel 5.1 (Kir5.1) in the regulation of renal membrane transport. Curr. Opin. Nephrol. Hypertens. 31 (5), 479–485. doi:10.1097/mnh.0000000000000817
Liu, G., Wu, K., and Sheng, Y. (2016). Elucidation of the molecular mechanisms of anaplastic thyroid carcinoma by integrated miRNA and mRNA analysis. Oncol. Rep. 36 (5), 3005–3013. doi:10.3892/or.2016.5064
Liu, J., Zhang, Q., Li, Z., Zhang, Q., Qu, Y., Lu, L., et al. (2014). Predication analysis of microarray data to determine altered gene profiles in liver carcinoma related to HBV-related cirrhosis. Zhonghua gan zang bing za zhi = Zhonghua ganzangbing zazhi = Chin. J. hepatology 22 (8), 625–630. doi:10.3760/cma.j.issn.1007-3418.2014.08.015
Liu, Y., McKenna, E., Figueroa, D. J., Blevins, R., Austin, C. P., Bennett, P. B., et al. (2000). The human inward rectifier K+ channel subunit Kir5.1 (KCNJ16) maps to chromosome 17q25 and is expressed in kidney and pancreas. Cytogenet. Cell Genet. 90 (1-2), 60–63. doi:10.1159/000015662
Lourdel, S., Paulais, M., Cluzeaud, F., Bens, M., Tanemoto, M., Kurachi, Y., et al. (2002). An inward rectifier K+ channel at the basolateral membrane of the mouse distal convoluted tubule: Similarities with kir4-kir5.1 heteromeric channels. J. Physiology-London 538 (2), 391–404. doi:10.1113/jphysiol.2001.012961
Lv, J., Fu, X., Li, Y., Hong, G., Li, P., Lin, J., et al. (2021). Deletion of Kcnj16 in mice does not alter auditory function. Front. Cell Dev. Biol. 9, 630361. doi:10.3389/fcell.2021.630361
Manis, A. D., Hodges, M. R., Staruschenko, A., and Palygin, O. (2020). Expression, localization, and functional properties of inwardly rectifying K+ channels in the kidney. Am. J. Physiology-Renal Physiology 318 (2), F332–F337. doi:10.1152/ajprenal.00523.2019
Manis, A. D., Palygin, O., Isaeva, E., Levchenko, V., LaViolette, P. S., Pavlov, T. S., et al. (2021). Kcnj16 knockout produces audiogenic seizures in the Dahl salt-sensitive rat. Jci Insight 6 (1), e143251. doi:10.1172/jci.insight.143251
Manis, A. D., Palygin, O., Khedr, S., Levchenko, V., Hodges, M. R., and Staruschenko, A. (2019). Relationship between the renin-angiotensin-aldosterone system and renal Kir5.1 channels. Clin. Sci. (Lond) 133 (24), 2449–2461. doi:10.1042/cs20190876
McClenahan, S. J., Kent, C., Kharade, S., Isaeva, E., Williams, J., Han, C., et al. (2022). VU6036720: The first potent and selective in vitro inhibitor of heteromeric Kir4.1/5.1 inward rectifier potassium channels. Mol. Pharmacol. 101, 357–370. doi:10.1124/molpharm.121.000464
Mizusawa, Y., and Wilde, A. A. (2012). Brugada syndrome. Circ. Arrhythm. Electrophysiol. 5 (3), 606–616. doi:10.1161/circep.111.964577
Neubauer, J., Forst, A-L., Warth, R., Both, C. P., Haas, C., and Thomas, J. (2022). Genetic variants in eleven central and peripheral chemoreceptor genes in sudden infant death syndrome. Pediatr. Res. 92, 1026–1033. doi:10.1038/s41390-021-01899-4
Palmer, B. F. (2015). Regulation of potassium homeostasis. Clin. J. Am. Soc. Nephrol. 10 (6), 1050–1060. doi:10.2215/cjn.08580813
Palygin, O., Levchenko, V., Ilatovskaya, D. V., Pavlov, T. S., Pochynyuk, O. M., Jacob, H. J., et al. (2017). Essential role of K(ir)5.1 channels in renal salt handling and blood pressure control. Jci Insight 2 (18), e92331. doi:10.1172/jci.insight.92331
Palygin, O., Pochynyuk, O., and Staruschenko, A. (2018). Distal tubule basolateral potassium channels: Cellular and molecular mechanisms of regulation. Curr. Opin. Nephrol. Hypertens. 27 (5), 373–378. doi:10.1097/mnh.0000000000000437
Palygin, O., Pochynyuk, O., and Staruschenko, A. (2017). Role and mechanisms of regulation of the basolateral K(ir)4.1/K(ir)5.1K(+) channels in the distal tubules. Acta Physiol. 219 (1), 260–273. doi:10.1111/apha.12703
Pardo, L. A., and Stuehmer, W. (2014). The roles of K+ channels in cancer. Nat. Rev. Cancer 14 (1), 39–48. doi:10.1038/nrc3635
Park, Y-J., Battistone, M. A., Kim, B., and Breton, S. (2017). Relative contribution of clear cells and principal cells to luminal pH in the mouse epididymis. Biol. Reproduction 96 (2), 366–375. doi:10.1095/biolreprod.116.144857
Patterson, K. C., Kahanovitch, U., Gonçalves, C. M., Hablitz, J. J., Staruschenko, A., Mulkey, D. K., et al. (2021). Kir 5.1-dependent CO2/H+ -sensitive currents contribute to astrocyte heterogeneity across brain regions. Glia 69 (2), 310–325. doi:10.1002/glia.23898
Paulais, M., Bloch-Faure, M., Picard, N., Jacques, T., Ramakrishnan, S. K., Keck, M., et al. (2011). Renal phenotype in mice lacking the Kir5.1 (Kcnj16) K+ channel subunit contrasts with that observed in SeSAME/EAST syndrome. Proc. Natl. Acad. Sci. U. S. A. 108 (25), 10361–10366. doi:10.1073/pnas.1101400108
Pereyra, S., Sosa, C., Bertoni, B., and Sapiro, R. (2019). Transcriptomic analysis of fetal membranes reveals pathways involved in preterm birth. Bmc Med. Genomics 12, 53. doi:10.1186/s12920-019-0498-3
Pessia, M., Imbrici, P., D'Adamo, M. C., Salvatore, L., and Tucker, S. J. (2001). Differential pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by heteropolymerisation with Kir5.1. J. Physiology-London 532 (2), 359–367. doi:10.1111/j.1469-7793.2001.0359f.x
Poli, G., Hasan, S., Belia, S., Cenciarini, M., Tucker, S. J., Imbrici, P., et al. (2021). Kcnj16 (Kir5.1) gene ablation causes subfertility and increases the prevalence of morphologically abnormal spermatozoa. Int. J. Mol. Sci. 22 (11), 5972. doi:10.3390/ijms22115972
Puissant, M. M., Muere, C., Levchenko, V., Manis, A. D., Martino, P., Forster, H. V., et al. (2019). Genetic mutation of Kcnj16 identifies Kir5.1-containing channels as key regulators of acute and chronic pH homeostasis. Faseb J. 33 (4), 5067–5075. doi:10.1096/fj.201802257R
Ramos, H. E., Dias da Silva, M. R., Carre, A., Silva, J. C., Paninka, R. M., Oliveira, T. L., et al. (2015). Molecular insights into the possible role of Kir4.1 and Kir5.1 in thyroid hormone biosynthesis. Hormone Res. Paediatr. 83 (2), 141–147. doi:10.1159/000369251
Reichold, M., Zdebik, A. A., Lieberer, E., Rapedius, M., Schmidt, K., Bandulik, S., et al. (2010). KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc. Natl. Acad. Sci. U. S. A. 107 (32), 14490–14495. doi:10.1073/pnas.1003072107
Salvatore, L., D'Adamo, M. C., Polishchuk, R., Salmona, M., and Pessia, M. (1999). Localization and age-dependent expression of the inward rectifier K+ channel subunit Kir 5.1 in a mammalian reproductive system. Febs Lett. 449 (2-3), 146–152. doi:10.1016/s0014-5793(99)00420-2
Schlingmann, K. P., Renigunta, A., Hoorn, E. J., Forst, A-L., Renigunta, V., Atanasov, V., et al. (2021). Defects in KCNJ16 cause a novel tubulopathy with hypokalemia, salt wasting, disturbed acid-base homeostasis, and sensorineural deafness. J. Am. Soc. Nephrol. 32 (6), 1498–1512. doi:10.1681/asn.2020111587
Staruschenko, A., Hodges, M. R., and Palygin, O. (2022). Kir5.1 channels: Potential role in epilepsy and seizure disorders. Am. J. Physiol. Cell Physiol. 323 (3), C706–C717. doi:10.1152/ajpcell.00235.2022
Su, X. T., Ellison, D. H., and Wang, W. H. (2019). Kir4.1/Kir5.1 in the DCT plays a role in the regulation of renal K(+) excretion. Am. J. Physiol. Ren. Physiol. 316 (3), F582–F586. doi:10.1152/ajprenal.00412.2018
Sun, Y., Chen, Z. Y., Gan, X., Dai, H., Cai, D., Liu, R. H., et al. (2022). A novel four-gene signature for predicting the prognosis of hepatocellular carcinoma. Scand. J. Gastroenterol. 57, 1227–1237. doi:10.1080/00365521.2022.2069476
Tanemoto, M., Fujita, A., Higashi, K., and Kurachi, Y. (2002). PSD-95 mediates formation of a functional homomeric Kir5.1 channel in the brain. Neuron 34 (3), 387–397. doi:10.1016/s0896-6273(02)00675-x
Tanemoto, M., Kittaka, N., Inanobe, A., and Kurachi, Y. (2000). In vivo formation of a proton-sensitive K+ channel by heteromeric subunit assembly of Kir5.1 with Kir4.1. J. Physiology-London 525 (3), 587–592. doi:10.1111/j.1469-7793.2000.00587.x
Terker, A. S., Zhang, C., McCormick, J. A., Lazelle, R. A., Zhang, C., Meermeier, N. P., et al. (2015). Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 21 (1), 39–50. doi:10.1016/j.cmet.2014.12.006
Trapp, S., Tucker, S. J., and Gourine, A. V. (2011). Respiratory responses to hypercapnia and hypoxia in mice with genetic ablation of Kir5.1 (Kcnj16). Exp. Physiol. 96 (4), 451–459. doi:10.1113/expphysiol.2010.055848
Tucker, S. J., Imbrici, P., Salvatore, L., D'Adamo, M. C., and Pessia, M. (2000). pH dependence of the inwardly rectifying potassium channel, Kir5.1, and localization in renal tubular epithelia. J. Biol. Chem. 275 (22), 16404–16407. doi:10.1074/jbc.C000127200
Valverde, M. A., Cantero-Recasens, G., Garcia-Elias, A., Jung, C., Carreras-Sureda, A., and Vicente, R. (2011). Ion channels in asthma. J. Biol. Chem. 286 (38), 32877–32882. doi:10.1074/jbc.R110.215491
Wadei, H. M., and Textor, S. C. (2012). The role of the kidney in regulating arterial blood pressure. Nat. Rev. Nephrol. 8 (10), 602–609. doi:10.1038/nrneph.2012.191
Wang, M. X., Cuevas, C. A., Su, X. T., Wu, P., Gao, Z. X., Lin, D. H., et al. (2018). Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int. 93 (4), 893–902. doi:10.1016/j.kint.2017.10.023
Wang, W. H., and Lin, D. H. (2022). Inwardly rectifying K(+) channels 4.1 and 5.1 (Kir4.1/Kir5.1) in the renal distal nephron. Am. J. Physiol. Cell Physiol. 323 (2), C277–C288. doi:10.1152/ajpcell.00096.2022
Webb, B. D., Hotchkiss, H., Prasun, P., Gelb, B. D., and Satlin, L. (2021). Biallelic loss-of-function variants in KCNJ16 presenting with hypokalemic metabolic acidosis. Eur. J. Hum. Genet. 29 (10), 1566–1569. doi:10.1038/s41431-021-00883-0
Welling, P. A. (2016). Roles and regulation of renal K channels. Annu. Rev. Physiol. 78, 415–435. doi:10.1146/annurev-physiol-021115-105423
Wu, J., Xu, H., Shen, W., and Jiang, C. (2004). Expression and coexpression of CO(2)-sensitive Kir channels in brainstem neurons of rats. J. Membr. Biol. 197 (3), 179–191. doi:10.1007/s00232-004-0652-4
Wu, P., Gao, Z-X., Zhang, D-D., Su, X-T., Wang, W-H., and Lin, D-H. (2019). Deletion of Kir5.1 impairs renal ability to excrete potassium during increased dietary potassium intake. J. Am. Soc. Nephrol. 30 (8), 1425–1438. doi:10.1681/asn.2019010025
Wu, X., Lv, D., Lei, M., Cai, C., Zhao, Z., Eftekhar, M., et al. (2020). A 10-gene signature as a predictor of biochemical recurrence after radical prostatectomy in patients with prostate cancer and a Gleason score ≥7. Oncol. Lett. 20 (3), 2906–2918. doi:10.3892/ol.2020.11830
Xu, H., Cui, N., Yang, Z., Qu, Z., and Jiang, C. (2000). Modulation of kir4.1 and kir5.1 by hypercapnia and intracellular acidosis. J. Physiol. 524 (3), 725–735. doi:10.1111/j.1469-7793.2000.00725.x
Yamamoto, Y., Ishikawa, R., Omoe, K., and Taniguchi, K. (2008). Expression of inwardly rectifying K+ channels in the carotid body of rat. Histol. Histopathol. 23 (7), 799–806. doi:10.14670/hh-23.799
Yang, Z., Xu, H., Cui, N., Qu, Z., Chanchevalap, S., Shen, W., et al. (2000). Biophysical and molecular mechanisms underlying the modulation of heteromeric Kir4.1-Kir5.1 channels by CO2 and pH. J. Gen. Physiol. 116 (1), 33–45. doi:10.1085/jgp.116.1.33
Zhang, C., Wang, L., Zhang, J., Su, X-T., Lin, D-H., Scholl, U. I., et al. (2014). KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc. Natl. Acad. Sci. U. S. A. 111 (32), 11864–11869. doi:10.1073/pnas.1411705111
Zhang, J., Han, J., Li, L., Zhang, Q., Feng, Y., Jiang, Y., et al. (2021). Inwardly rectifying potassium channel 5.1: Structure, function, and possible roles in diseases. Genes Dis. 8 (3), 272–278. doi:10.1016/j.gendis.2020.03.006
Zhao, W., Ahmed, S., Ahmed, S., Yangliu, Y., Wang, H., and Cai, X. (2021). Analysis of long non-coding RNAs in epididymis of cattleyak associated with male infertility. Theriogenology 160, 61–71. doi:10.1016/j.theriogenology.2020.10.033
Zhu, G., Liu, C., Qu, Z., Chanchevalap, S., Xu, H., and Jiang, C. (2000). CO(2) inhibits specific inward rectifier K(+) channels by decreases in intra- and extracellular pH. J. Cell Physiol. 183 (1), 53–64. doi:10.1002/(SICI)1097-4652(200004)183:1<53::AID-JCP7>3.0.CO;2-R
Keywords: cancer, deafness, Kir5.1, potassium channel, respiration
Citation: Zhang C and Guo J (2023) Diverse functions of the inward-rectifying potassium channel Kir5.1 and its relationship with human diseases. Front. Physiol. 14:1127893. doi: 10.3389/fphys.2023.1127893
Received: 06 January 2023; Accepted: 15 February 2023;
Published: 27 February 2023.
Edited by:
James A. McCormick, Oregon Health and Science University, United StatesReviewed by:
Jerod S. Denton, Vanderbilt University, United StatesDaohong Lin, New York Medical College, United States
Andrew Terker, Vanderbilt University, United States
Copyright © 2023 Zhang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia Guo, ZmNjZ3Vvam5yY0B6enUuZWR1LmNu