 Ang Li
Ang Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 22 October 2020
Sec. Striated Muscle Physiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.595800
This article is part of the Research Topic Calcium Homeostasis in Skeletal Muscle Function, Plasticity and Disease View all 14 articles
Mitochondria are both the primary provider of ATP and the pivotal regulator of cell death, which are essential for physiological muscle activities. Ca2+ plays a multifaceted role in mitochondrial function. During muscle contraction, Ca2+ influx into mitochondria activates multiple enzymes related to tricarboxylic acid (TCA) cycle and oxidative phosphorylation, resulting in increased ATP synthesis to meet the energy demand. Pathophysiological conditions such as skeletal muscle denervation or unloading also lead to elevated Ca2+ levels inside mitochondria. However, the outcomes of this steady-state elevation of mitochondrial Ca2+ level include exacerbated reactive oxygen species (ROS) generation, sensitized opening of mitochondrial permeability transition pore (mPTP), induction of programmed cell death, and ultimately muscle atrophy. Previously, both acute and long-term endurance exercises have been reported to activate certain signaling pathways to counteract ROS production. Meanwhile, electrical stimulation is known to help prevent apoptosis and alleviate muscle atrophy in denervated animal models and patients with motor impairment. There are various mechanistic studies that focus on the excitation-transcription coupling framework to understand the beneficial role of exercise and electrical stimulation. Interestingly, a recent study has revealed an unexpected role of rapid mitochondrial Ca2+ transients in keeping mPTP at a closed state with reduced mitochondrial ROS production. This discovery motivated us to contribute this review article to inspire further discussion about the potential mechanisms underlying differential outcomes of physiological mitochondrial Ca2+ transients and pathological mitochondrial Ca2+ elevation in skeletal muscle ROS production.
Skeletal muscle carries out multiple critical functions of human body such as locomotion, metabolism, and thermogenesis (Block, 1994; Qiu et al., 2018). Thus, skeletal muscle atrophy, characterized by loss of muscle mass and strength, could have severe impact on daily living or even become life-threatening (Jackman and Kandarian, 2004). Human and animal studies revealed a variety of etiological factors for skeletal muscle atrophy, including disuse (limb immobilization, unloading) (Jackman and Kandarian, 2004), denervation (spinal motor neuron lesion in patients or surgical transection of motor nerves in animals) (Hoellwarth and Christian Hofer, 2005; Adhihetty et al., 2007), fasting (Qiu et al., 2018), lack of gravity (Fitts et al., 2001), aging (sarcopenia) (Dupont-Versteegden, 2005), cancer (cachexia) (Tisdale, 2010), neuromuscular diseases such as amyotrophic lateral sclerosis (ALS), and spinal muscular atrophy (Fischer et al., 2004; Monani, 2005). Accumulating evidence highlights programmed cell death (apoptosis) as a major cause of muscle fiber loss in skeletal muscle atrophy (Borisov and Carlson, 2000; Tews, 2002; Siu and Alway, 2005; Adhihetty et al., 2007), potentially implicating mitochondrial abnormality as a shared pathological feature underlying muscle atrophy induced by different etiological factors.
Mitochondria take up around 10–15% volume of mammalian skeletal muscle fibers (Eisenberg, 2010), with a certain degree of compositional and function differences observed between the subsarcolemmal, interfibrillar and peri-nuclear subpopulations (Cogswell et al., 1993; Díaz-Vegas et al., 2019). Mitochondria, especially the interfibrillar subpopulation, not only serve as the primary energy provider but also are intimately involved in apoptosis (Adhihetty et al., 2005; Siu and Alway, 2005; Chabi et al., 2008; Wang and Youle, 2009). The connections between mitochondria and apoptosis include:
1. Multiple proapoptotic proteins are located within mitochondria, such as cytochrome c (Cyto c) (Liu et al., 1996), apoptosis-inducing factor (AIF) (Susin et al., 1999), second mitochondria-derived activator of caspase/direct IAP (inhibitor-of apoptosis) binding protein (Smac/Diablo) (Du et al., 2000), Endonuclease G (EndoG) (Li et al., 2001), and high temperature requirement protein A2 (HtraA2) (Hegde et al., 2002).
2. Although healthy mitochondria are not the major contributor to cytosolic ROS in skeletal muscle during contractile activities (Sakellariou et al., 2013; Powers et al., 2016; Henríquez-Olguin et al., 2019), they can significantly contribute to ROS production under pathological conditions (Pottecher et al., 2013; Lejay et al., 2014). Upon overwhelming the cellular antioxidants’ neutralizing capacity, ROS causes oxidative damages to lipids, proteins, and DNA (Bandyopadhyay et al., 1999). Elevated ROS production is frequently observed as an early event of the apoptotic process (Fernandez et al., 2002).
3. Long-term elevation of mitochondrial matrix Ca2+ ([Ca2+]mito) can induce cell apoptosis through increasing the release of proapoptotic proteins and mitochondrial permeability transition pore (mPTP) opening (Haworth and Hunter, 1979; Hirsch et al., 1997; De Giorgi et al., 2002; Borutaite et al., 2003). It is worth noticing that interfibrillar mitochondria are more prone to release proapoptotic factors upon ROS stimulation, potentially due to higher probability of mPTP opening than subsarcolemmal mitochondria (Adhihetty et al., 2005). The detailed mechanisms will be discussed later in this review.
Although long-term [Ca2+]mito elevation is closely associated with excessive ROS production (Adam-Vizi and Starkov, 2010; Peng and Jou, 2010), we observed an interesting phenomenon that mitochondrial Ca2+ transients induced by the electrical stimulation can decrease ROS production in denervated skeletal muscle fibers within a minute (Karam et al., 2017). This phenomenon is different from the excitation–transcription coupling events that help skeletal muscle cope with ROS during exercise or electrical stimulation, which usually occur in the time frame of hours or longer (Tsuboyama-Kasaoka et al., 1998; Cortright et al., 1999; Pilegaard et al., 2003; Egan et al., 2010). Although the underlying molecular mechanism of the instant ROS suppression by the rapid mitochondrial Ca2+ influx remains elusive, through this review we hope to inspire more thoughts and discussion about how Ca2+ temporal profile differentially influences mitochondrial ROS production and cell death.
This review does not intend to comprehensively cover all players involved in mitochondrial Ca2+ handling. However, a brief introduction is needed for meaningful discussions about the connections between [Ca2+]mito, ROS, apoptosis, and muscle atrophy (Figure 1).
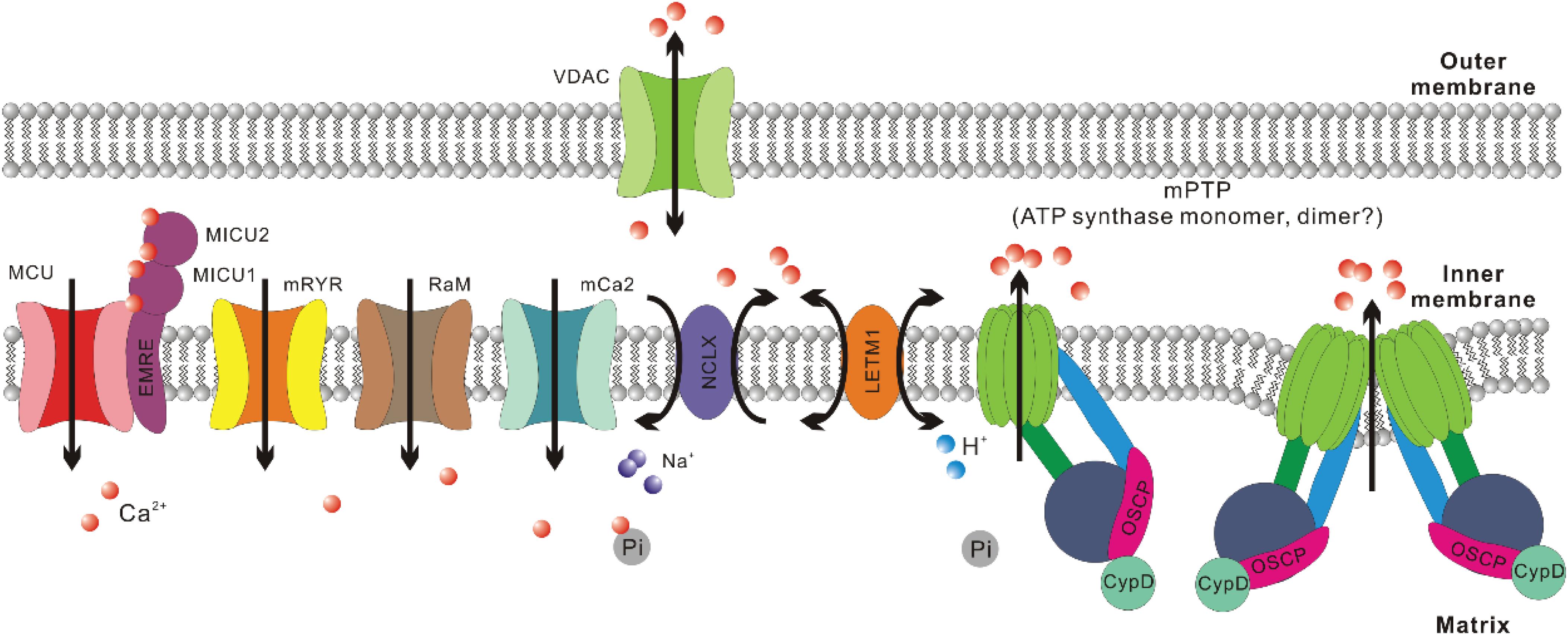
Figure 1. Crucial regulators of Ca2+ homeostasis in mitochondria. For mitochondrial Ca2+ uptake, the major routes include mitochondrial Ca2+ uniporter (MCU), mitochondrial ryanodine receptor (mRYR), rapid mode of mitochondrial Ca2+ uptake (RaM), and mitochondrial calcium channel type 2 (mCa2). The most crucial regulatory subunits of MCU include mitochondrial calcium uptake 1, 2 (MICU1, MICU2) and essential MCU regulator (EMRE). For mitochondrial Ca2+ extrusion, the major routes include Na+/Ca2+/Li+ exchanger (NCLX), mitochondrial H+/Ca2+ exchanger (LETM1), and mitochondrial permeability transition pore (mPTP). Voltage-dependent anion channel (VDAC) is suggested to be the outer mitochondrial membrane (OMM) component of mPTP. Recently, ATP synthase has been confirmed to be the inner mitochondrial membrane (IMM) component of mPTP. There are still debates about whether mPTP is formed by ATP synthase dimer or monomer. Cyclophilin (Cyp) D is a crucial regulator of mPTP opening and interacts with ATP synthase through the oligomycin sensitivity-conferring protein (OSCP) subunit. As to Ca2+ retention in mitochondrial matrix, inorganic phosphate (Pi) helps sequester free Ca2+ in solid precipitates, which could serve as an MCU independent source of mitochondrial Ca2+ upon matrix acidification.
Mitochondria are constantly involved in modulating spatiotemporal profiles of cytosolic Ca2+ ([Ca2+]cyto) in different types of cells under physiological and pathological conditions (Gunter et al., 2004; Szabadkai and Duchen, 2008; Demaurex and Guido, 2017), including cardiac and skeletal muscle (Zhou et al., 1998;Maack et al., 2006; Rizzuto and Pozzan, 2006; Sedova et al., 2006; Csordás and Hajnóczky, 2009; Yi et al., 2011). This is due to their abilities to uptake and extrude Ca2+, as well as retain Ca2+ in their matrix (Szabadkai and Duchen, 2008). Indeed, mitochondrial Ca2+ uptake has been first observed in vivo during skeletal muscle contraction induced by motor nerve stimulation (Rudolf et al., 2004) and later quantified in isolated individual muscle fibers during E-C coupling (Yi et al., 2011; Karam et al., 2017). The Ca2+ uptake routes in mitochondria include mitochondrial Ca2+ uniporter (MCU), mitochondrial ryanodine receptor (mRYR), as well as two other channels with unknown molecular nature: rapid mode of mitochondrial Ca2+ uptake (RaM) and mitochondrial calcium channel type 2 (mCa2) (Buntinas et al., 2001; Kirichok et al., 2004; Altschafl et al., 2007; Michels et al., 2009; Hoppe, 2010). In skeletal muscle, the presence of uptake routes other than MCU still waits to be confirmed. Overexpression of MCU in adult mouse flexor digitorum brevis (FDB) muscle led to notable enhancement in caffeine-induced mitochondrial Ca2+ influx as well as a moderate elevation of the resting [Ca2+]mito level (Mammucari et al., 2015). In contrast, transfection of FDB muscle with short hairpin (sh) RNA against MCU resulted in marked reduction of both resting [Ca2+]mito level and caffeine-induced mitochondrial Ca2+ influx (Mammucari et al., 2015). Furthermore, extensor digitorum longus (EDL) muscle infected by adeno-associated virus (AAV) carrying shRNA against MCU exhibited decreased pyruvate dehydrogenase (PDH) activity (Mammucari et al., 2015), which is known to be dependent on [Ca2+]mito level (Wan et al., 1989; Denton, 2009). Consistently, in skeletal muscles from MCU–/– mice, resting [Ca2+]mito level seems reduced when compared to wild-type controls, while the phosphorylation of PDH, which is negatively correlated with the activity of Ca2+ sensitive phosphatase PDP1, significantly increased (Pan et al., 2013).
The activity of MCU is regulated by MICU1-MICU2 heterodimer, which senses [Ca2+]cyto level (Mallilankaraman et al., 2012; Csordás et al., 2013; Kamer and Mootha, 2014; Patron et al., 2014; Wang et al., 2014). MICU1 is suggested to function as the cooperative activator of MCU, while MICU2 is believed to keep MCU closed at low [Ca2+]cyto level (Patron et al., 2014). Skeletal muscle expresses a unique alternative splice isoform of MICU1, which is more sensitive to Ca2+ than the isoform expressed in other tissues and hence allows activation of MCU at a lower [Ca2+]cyto level (Reane et al., 2016). This is likely an evolutionary adaption to the astounding amount of ATP consumed during muscle contraction since the activities of multiple enzymes involved in ATP synthesis are stimulated by Ca2+ (Wan et al., 1989; Das and Harris, 1990; Murphy et al., 1990; Denton, 2009; Glancy et al., 2013). Indeed, [Ca2+]mito transients in skeletal muscle are larger than those measured in other tissues based on mitoplast patch clamp results (Fieni et al., 2012). Other MCU regulatory subunits include EMRE and MCUb. EMRE helps tether MICU1 and MICU2 to the transmembrane region of MCU, while MCUb is a paralog of MCU that is suggested to negatively regulate MCU complex in a direct manner (Raffaello et al., 2013; Tsai et al., 2016).
The Ca2+ extrusion routes include Na+/Ca2+/Li+ exchanger (NCLX), mPTP and arguably mitochondrial H+/Ca2+ exchanger (LETM1) (Jiang et al., 2009; Hoppe, 2010; Palty et al., 2010; Nowikovsky et al., 2012). NCLX is more intensively expressed in skeletal muscle compared to many other tissues such as cardiac muscle (Palty et al., 2004, 2010). This is accompanied by an ultrafast Ca2+ efflux rate of skeletal muscle mitochondria, which is 2-3 orders faster than cardiac muscle (Rudolf et al., 2004; Palty et al., 2010). mPTP is a nonselective, large conductance megachannel mediating solute (<1.5 kDa) exchange between mitochondrial matrix and the outside milieu (Haworth and Hunter, 1979; Hunter and Haworth, 1979a, b; Kinnally et al., 1992; Szabó and Zoratti, 1992). The opening of mPTP is sensitive to Ca2+, cyclophilin (Cyp) D, oxidizing agents, thiol reagents, depletion of ADP, while its inhibitors include Mg2+, ADP, NADH, antioxidants, and cyclosporin (Cs) A (through interacting with CypD) (Haworth and Hunter, 1979; Hunter and Haworth, 1979a, b; Kinnally et al., 1992; Szabó and Zoratti, 1992). mPTP opening is widely known for its central role in cell death induction under multiple pathological conditions (Bonora et al., 2020). Osmotic influx of water into mitochondrial matrix through these pores results in swollen matrix, dissipated IMM potential and ceased ATP production. ATP dependent ion exchangers/pumps fail to maintain cellular ion homeostasis and finally lead to necrosis (Bonora et al., 2015). Additionally, mPTP opening has also been implied to facilitate the release of intermembrane space factors activating apoptotic pathway (Hirsch et al., 1997; De Giorgi et al., 2002; Borutaite et al., 2003). While the irreversible, high conductance mPTP opening upon [Ca2+]mito overload is detrimental (Ichas et al., 1997; Hoppe, 2010; Karch and Molkentin, 2018), the transient and low conductance mPTP opening is considered a Ca2+ extrusion route that may carry out physiological functions (Ichas and Mazat, 1998; Elrod et al., 2010; Elrod and Molkentin, 2013).
The molecular composition of mPTP has remained elusive for over 60 years (Urbani et al., 2019). Previously mPTP was suggested to form from adenine nucleotide translocator (ANT) or phosphate carrier (PiC) on IMM, as well as voltage-dependent anion channel (VDAC) on OMM (Halestrap and Davidson, 1990; Szabó et al., 1993; Baines, 2009; Halestrap, 2009). However, further researches indicate that they are not the pore forming unit of mPTP, but could be regulatory components (Kokoszka et al., 2004; Krauskopf et al., 2006; Baines et al., 2007). In recent decades, CypD was reported to physically interact with oligomycin sensitivity-conferring protein (OSCP) within ATP synthase (complex V of OXPHOS) (Giorgio et al., 2009) and ATP synthase increases the permeability of IMM to different solutes upon [Ca2+]mito overload (Giorgio et al., 2013, 2017; Alavian et al., 2014). However, the argument about whether it is the monomer or dimer of ATP synthase that carries out the megachannel function has not been settled yet (Mnatsakanyan et al., 2019).
The Ca2+ retention capacity of mitochondrial matrix is believed to heavily rely on the inorganic phosphate (Pi) entered mainly through mitochondrial phosphate carrier (PiC) (Szabadkai and Duchen, 2008; Seifert et al., 2015). Pi can buffer Ca2+ through the formation of osmotically neutral precipitates such as hydroxyapatite and whitlockite (Carafoli, 2010; Chinopoulos and Adam-Vizi, 2010), which theoretically should enable additional Ca2+ uptake through MCU, suppress Ca2+ efflux through NCLX and desensitize mPTP (Zoccarato and Nicholls, 1982; Szabadkai and Duchen, 2008; Seifert et al., 2015). However, these assumptions were challenged as opposite results were observed in PiC knockdown cells and PiC knockout mice (Varanyuwatana and Halestrap, 2012; Kwong et al., 2014). Meanwhile Pi is a long known sensitizer of mPTP (Zoratti and Szabò, 1995). Thus the precise role of the PiC in [Ca2+]mito regulation still waits to be addressed. In addition, Ca–Pi precipitates can dissolve upon acidification and IMM potential disruption, enabling MCU-independent elevation of [Ca2+]mito (Greenawalt et al., 1964; Wolf et al., 2017; Hernansanz-Agustín et al., 2020). For example, in cells experiencing acute hypoxia, complex I of OXPHOS in mitochondria undergoes conformational changes that lead to proton accumulation inside the matrix, dissolution of Ca–Pi precipitates, elevation of [Ca2+]mito and activation of NCLX (Hernansanz-Agustín et al., 2020).
Elevation of [Ca2+]mito level could result in enhanced ROS production through multiple mechanisms (Figure 2):
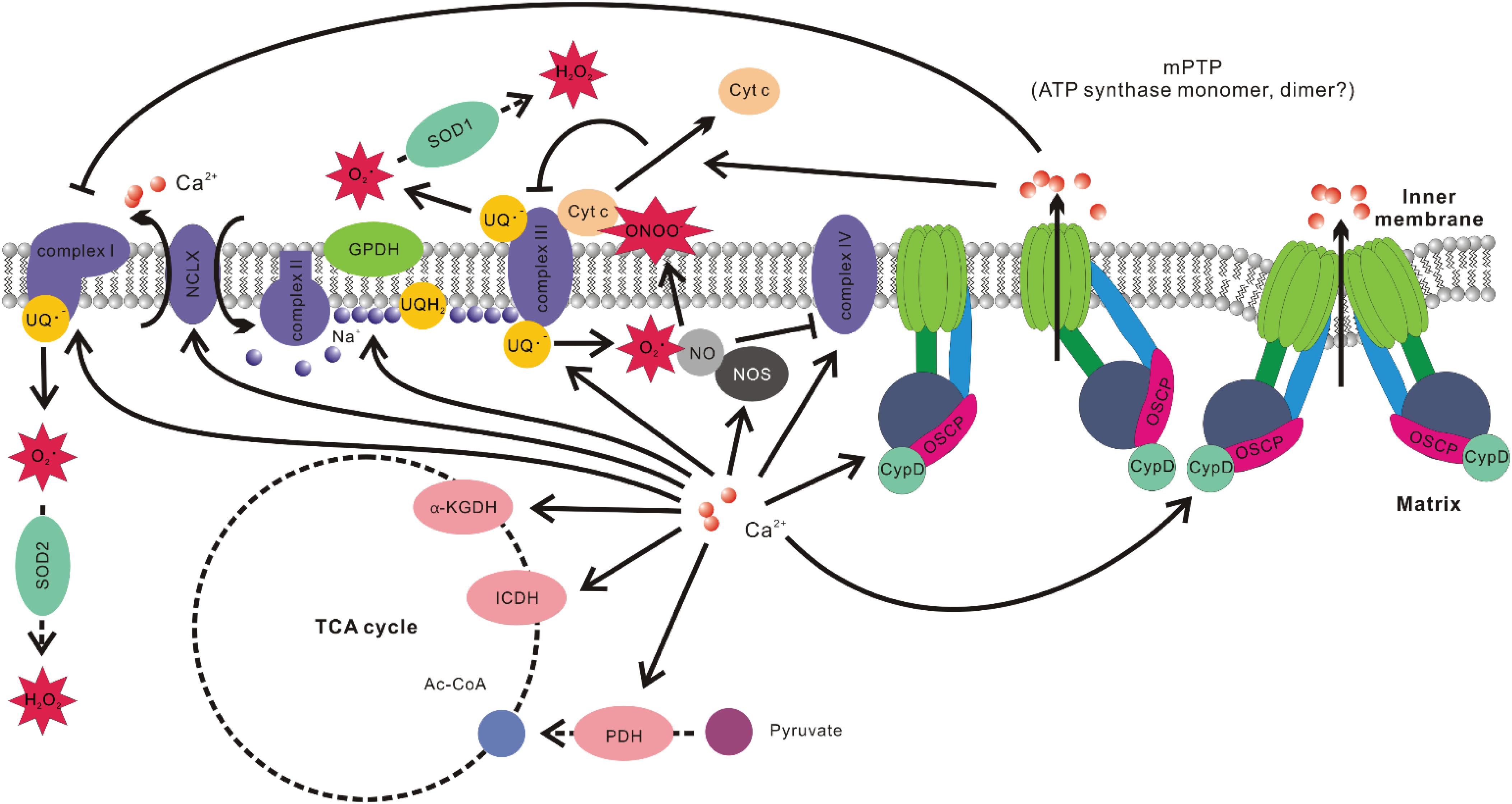
Figure 2. Associations between mitochondrial Ca2+ level and ROS production. Ca2+ promotes the activities of multiple Ca2+ sensitive dehydrogenase such as glycerol phosphate dehydrogenase (GPDH), pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (ICDH), and α-ketoglutarate dehydrogenase (α-KGDH). It also elevates the efficiency of complexes I, III, IV, and V of OXPHOS. Ca2+-induced activation of NCLX results in Na+ influx into the matrix and Na+ interacts with phospholipid in the inner leaflet of IMM, decreases its fluidity, and slows down ubiquinol (UQH2) diffusion. This results in uncoupling of Q cycle and increased ROS production at Qo site in complex III. Ca2+ stimulation of nitric oxide synthase (NOS) increases nitric oxide (NO) production, which inhibits complex IV by competing with O2. NO also readily react with superoxide to form peroxynitrite (ONOO–), which promotes cytochrome c (Cyt c) release. Cyt c release would disrupt the activity of complex III, increase the level of ubisemiquinone (UQ–), which provide one electron to O2 to form superoxide. Ca2+ is also the major stimulant of mPTP opening. The subsequent change of ion strength in mitochondrial intermembrane space disrupts electrostatic interactions between Cyt c and membrane lipid cardiolipin, leading to Cyt c release and hindrance of complex III activity. mPTP opening has also been implicated in inducing conformation changes of complex I, resulting in elevated ROS production.
1. Elevation of [Ca2+]mito stimulates Ca2+ sensitive dehydrogenases, including glycerol phosphate dehydrogenase (GPDH), PDH, isocitrate dehydrogenase (ICDH), and α-ketoglutarate dehydrogenase (α-KGDH) (Wan et al., 1989; Denton, 2009). There is also evidence that Ca2+ increases activities of complexes I, III, IV, and V of OXPHOS (Das and Harris, 1990; Glancy et al., 2013). The faster O2 consumption rate results in increased ROS production under certain circumstances, although the opposite situation also exists (Barja, 2007; Neretti et al., 2009; Silva et al., 2009).
2. Elevation of [Ca2+]mito stimulates mitochondrial nitric oxide synthase (NOS) to produce more nitric oxide (NO). NO can compete with O2 for binding sites on cytochrome c oxidase (complex IV of OXPHOS), which hinders the electron flow and decreases mitochondrial O2 consumption (Giulivi, 2003; Ghafourifar and Cadenas, 2005). The hindrance of the electron flow and the increase of local O2 may boost ROS production (Brookes et al., 2004). On the other hand, NO reacts readily with superoxide and generates peroxynitrite (ONOO–), which is a more potent ROS that causes Cyto c release, lipid peroxidation and oxidative damage to other vulnerable targets (Ghafourifar and Cadenas, 2005).
3. Elevated [Ca2+]mito promotes the opening of mPTP. mPTP opening can lead to changes of ionic strength and hence disrupt the electrostatic interaction between Cyto c and cardiolipin in mitochondrial intermembrane space. Cyto c is required for the activity of ubiquinol-cytochrome c oxidoreductase (complex III of OXPHOS) (Ott et al., 2002). The blockage of complex III activity enhances ROS production by increasing the accumulation of the one-electron donor ubisemiquinone (Turrens et al., 1985; Muller et al., 2002, 2003). mPTP opening also seems to induce conformation changes in NADH-ubiquinone oxidoreductase (complex I of OXPHOS), resulting in increased ROS production (Batandier et al., 2004).
4. [Ca2+]mito overload may stimulate Cyto c release from cardiolipin through competing for cardiolipin binding sites (Grijalba et al., 1999), which affects complex III activity and hence promotes ROS generation.
5. During acute hypoxia, elevation of [Ca2+]mito due to matrix acidification activates NCLX, promoting the import of Na+. Matrix Na+ interacts with phospholipids in the IMM (such as phosphatidylcholine), reducing membrane fluidity, slowing down the diffusion of ubiquinol from glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or complex II to complex III of OXPHOS, resulting in elevated ROS production of complex III at Qo site (Hernansanz-Agustín et al., 2020).
The cytosolic Ca2+ transients are spatiotemporally well-controlled Ca2+ release events from the sarcoplasmic reticulum (SR) responding to the motor nerve activation in a skeletal muscle fiber during excitation contraction (EC)-coupling. Skeletal muscle inactivity including neuromuscular diseases, spinal cord injury, and muscle unloading, etc., could partially or completely disrupt EC-coupling and eliminate cytosolic Ca2+ transients. A pathological hallmark of prolonged muscle inactivity is the enhanced ROS production in muscle fibers (Muller et al., 2007; Powers et al., 2012; Xiao et al., 2018). It is very well established that mitochondria are a major source of ROS production in prolonged muscle inactivity (Powers et al., 2012). Prolonged muscle unloading leads to an elevated resting [Ca2+]cyto (Tischler et al., 1990; Ingalls et al., 1999), which in turn could overload mitochondria, increase the propensity of mPTP opening and stimulate ROS production (Csukly et al., 2006). However, the initial cause of mitochondrial ROS production in inactivated skeletal muscle remains elusive (Hyatt et al., 2019). One question is whether the cessation of physiological Ca2+ transients is an initiating factor for promoting mitochondria ROS production.
Transgenic mouse model expressing a mitochondria-targeted biosensor (mt-cpYFP) allowed a real-time measurement of a ROS-related mitochondrial signal, termed “mitoflash” (Wang et al., 2008; Ding et al., 2015). Although mitoflash activities could be composed of multifaceted signals including matrix alkalization, superoxide, and arguably some other oxidants (Wang et al., 2008, 2016; Schwarzländer et al., 2012; Wei-LaPierre et al., 2013), the mechanism underlying mitoflash events is believed to be linked to transitory opening of mPTP due to the high spatiotemporal correlations between mitoflash events and IMM depolarization (Wang et al., 2008, 2016). Besides transitory opening of mPTP, there are also other potential causes of IMM depolarization during mitoflash events, such as the activation of uncoupling proteins UCP2 and UCP3. However, inhibition of UCP2 by chemical blocker or RNA interference slightly increased, rather than decreased the mitoflash incidence (Wang et al., 2017). Skeletal muscle fibers derived from UCP3 knockout mice exhibited no changes in mitoflash frequency, amplitude or duration compared to wild type controls, while the average area of mitoflash events mildly reduced (McBride et al., 2019). Thus, these functional perturbation experiments do not support the hypothesis of UCP2/3 mediated uncoupling activities as the major contributor of mitoflash signals.
Using this transgenic mouse model, Karam et al detected a fourfold increase of mitoflash activity in skeletal muscle following a short period (24 h) of sciatic nerve transection (Karam et al., 2017). This denervation-induced mitoflash activity could be attenuated by the application of CsA, an established inhibitor of mPTP opening (Giorgio et al., 2018), further suggesting that mitoflash events reflect real-time opening of mPTP in denervated muscle fibers. Consistently, the increased mitoflash activity is associated with an elevated mitochondrial superoxide level reported with MitoSOX Red (Mukhopadhyay et al., 2007) in muscle fibers 24 h after the denervation. Similar results were also reported for muscle fibers derived from the ALS mouse model (SOD1G93A) at a stage before disease symptom onset (Xiao et al., 2018), when motor neuron axon terminal withdrawal (denervation) start to occur in individual muscle fibers (Frey et al., 2000; Fischer et al., 2004).
Due to lack of neuronal impulses, the action potential and physiological [Ca2+]cyto transients are abolished in denervated skeletal muscle. Thus, a hypothesis was proposed that dynamic Ca2+ transients were required to keep mPTP at its closed state and maintain mitochondrial ROS production at the physiological level in skeletal muscle (Karam et al., 2017). Remarkably, when muscle fibers from denervated (24 h) mouse model were exposed to a brief electrical stimulation (40 Hz, 0.5 ms pulses at 8–12 V for a total duration of 350 ms) to restore physiological cytosolic and mitochondrial Ca2+ transients, the area and amplitude of mitoflash events dramatically reduced within a minute to a level comparable to the unstimulated sham muscle fibers (Karam et al., 2017). Importantly, after treating the denervated muscle fibers with Ru360 to block MCU for mitochondrial Ca2+ uptake, electrical stimulation no longer had significant impact on mitoflash activities. Additionally, the levels of mitochondrial superoxide (indicated by MitoSOX Red) also exhibited the same trend of changes under these conditions (Karam et al., 2017). Thus, mitochondrial Ca2+ influxes triggered by physiological cytosolic Ca2+ transients, even brief ones, seems to be capable of inhibiting transitory mPTP opening and ROS generation in mitochondria.
The above results are in line with previous discoveries that electrical stimulations help prevent apoptosis, retard muscle atrophy and improve muscle strength in denervated animal models (Mokrusch et al., 1990; Arakawa et al., 2010; Nakagawa et al., 2017) and patients with motor impairment caused by spinal cord injury or stroke (Dudley et al., 1999; Crameri et al., 2000, 2002; Doucet et al., 2012; Nascimento et al., 2014). However, the studies of the mechanisms underlying these phenomena usually focus on relatively long-term molecular changes, such as gene expression regulation, which usually takes tens of minutes to hours to occur (Voytik et al., 1993; Kostrominova et al., 2005; Arakawa et al., 2010; Peviani et al., 2010; Russo et al., 2010). The events occurred within a minute after electrical stimulation were rarely investigated.
One potential mechanism underlying the role of physiological Ca2+ transients in inhibition of mitochondrial ROS production could be that mitochondrial Ca2+ influxes induced by cytosolic Ca2+ transients suddenly boost the electron flow rate along the respiratory chain, decreasing the reduced state of ROS generators, such as ubisemiquinone generated by complex III of OXPHOS. Meanwhile, the sudden boost of respiratory chain activity also increases O2 consumption rate, decreasing the amount of local O2 available for forming superoxide. These two factors may both contribute to a quick attenuation of superoxide formation. Similar situation also occurs during sudden transition of state 4 respiration to state 3 respiration in isolated mitochondria (Barja, 2007).
Additionally, as illustrated in Figure 3, we propose a second hypothetical mechanism that key mPTP components have two sets of Ca2+ responding structures with different affinities that trigger opposite changes in the propensity of mPTP opening based on the temporal profile of mitochondrial Ca2+ influxes in skeletal muscle fibers. Under this scenario, a steady-state increase of [Ca2+]mito (a likely outcome of steady-state [Ca2+]cyto elevation after denervation or neuronal degenerative disease) promotes mPTP opening through a relatively higher affinity Ca2+ responding structure. The second Ca2+ responding structure with relatively lower Ca2+ binding affinity may respond to the spike of mitochondrial Ca2+ influx following the cytosolic Ca2+ transient during EC-coupling activated by neuronal input or electrical field stimulation and quickly shut down the opening of mPTP in skeletal muscle. More than a coincidence, previous studies identified two binding sites for divalent cations (including Ca2+) on the F1 catalytic domain of ATP synthase. One located in the nucleotide binding pocket within the αβ cleft, the other located in the superficial loop of the β subunit and contains the acidic sequence DELSEED (Hubbard and McHugh, 1996; Giorgio et al., 2017, 2018). The first one is implicated as the “trigger site” of mPTP opening. More specifically, the occupancy of this site by Ca2+ (instead of Mg2+) is proposed to elevate the rigidity of F1 domain and transmit mechanical energy to OSCP, the peripheral stalk and finally to the IMM, leading to the formation of mPTP by ATP synthase dimers (Giorgio et al., 2017, 2018). Interestingly, the physiological role of the other Ca2+ binding site remains unknown. Due to the low affinity nature of this Ca2+ binding site, it may serve as the potential link between mitochondrial Ca2+ transients and quick mPTP shut down. It is also possible that the second Ca2+ responding structure locates on molecules other than ATP synthase, which acts as an accessory safe guard against mPTP opening upon Ca2+ binding during rapid mitochondrial Ca2+ influx. Further structural and functional studies are needed to validate those hypotheses. Nevertheless, the results reported in Karam et al. suggest that the physiological cytosolic and mitochondrial Ca2+ transients induced during EC-coupling are vital to keep mPTP in a closed status in skeletal muscle fibers.
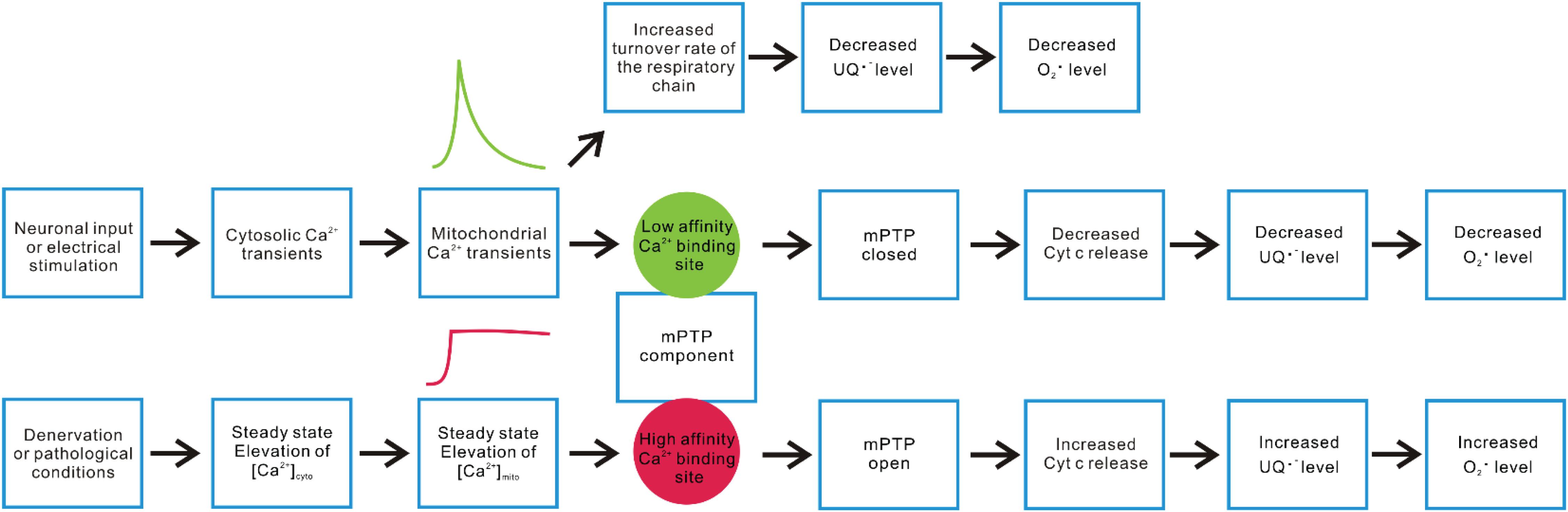
Figure 3. Hypothetical mechanisms underlying differential mitochondrial Ca2+ dynamics and ROS production. Neuronal or electrical stimulation-induced cytosolic Ca2+ transient can result in rapid Ca2+ influx into mitochondria, serving as a stimulant of multiple enzyme complex in OXPHOS that accelerate electron flux along the respiratory chain, leading to decreased accumulation of ubisemiquinone (UQ) and lower O2 partial pressure. Both factors contribute to alleviated superoxide production. On the other hand, we hypothesize that a key mPTP component may have two sets of Ca2+ sensory structures with different Ca2+ affinities, resulting in distinct responses to rapid versus steady-state elevation of [Ca2+]mito. Steady-state elevation of [Ca2+]mito resulting from denervation or other pathological conditions may predominantly trigger the response mediated by a relatively higher affinity structure (such as the Ca2+ binding site within the F1 domain nucleotide binding pockets of ATP synthase), which promotes mPTP opening, enhances Cyt c release, disrupts complex III activity, and increases superoxide production. The rapid mitochondrial Ca2+ transients induced by motor neuron input or electrical stimulation may predominantly activate the response mediated by a relatively lower affinity Ca2+ responding structure, shutting down mPTP and decrease ROS production.
During exercise, muscle contraction dramatically increases ATP turnover rate, which could be more than 100-fold that of the basal rate (Hochachka and McClelland, 1997). To cope with such large demand of ATP, Ca2+ influx into mitochondrial matrix activates multiple enzymes related to TCA cycle and oxidative phosphorylation, elevating the synthesis of ATP in skeletal muscle (Wan et al., 1989; Das and Harris, 1990; Denton, 2009). The high O2 consumption rate and elevated [Ca2+]mito level do not necessarily increase ROS production (Barja, 2007; Silva et al., 2009), which could be at least partially attributable to the exercise-induced signaling pathways, such as AMP-activated protein kinase (AMPK), protein kinase A (PKA), Ca2+/calmodulin-dependent protein kinase (CaMK), mitogen-activated protein kinase (MAPK), protein kinase C (PKC), focal adhesion kinase (FAK), and mammalian target of rapamycin (mTOR), cyclin-dependent kinase (CDK), integrin-linked kinase (ILK), and sirtuin (SIRT) family of protein deacetylases (Sakamoto and Goodyear, 2002; Egan and Zierath, 2013; Hoffman et al., 2015). Although some of these pathways are preferentially induced by acute exercise while the others mediate physiological adaptation to long-term endurance exercise, both of them involve transcriptional regulation and hence belong to excitation-transcription coupling framework (Egan and Zierath, 2013). It is beyond the scope of this review to systematically go through these pathways. But we would like to highlight some of them directly or indirectly involved in ROS regulation (Figure 4A).
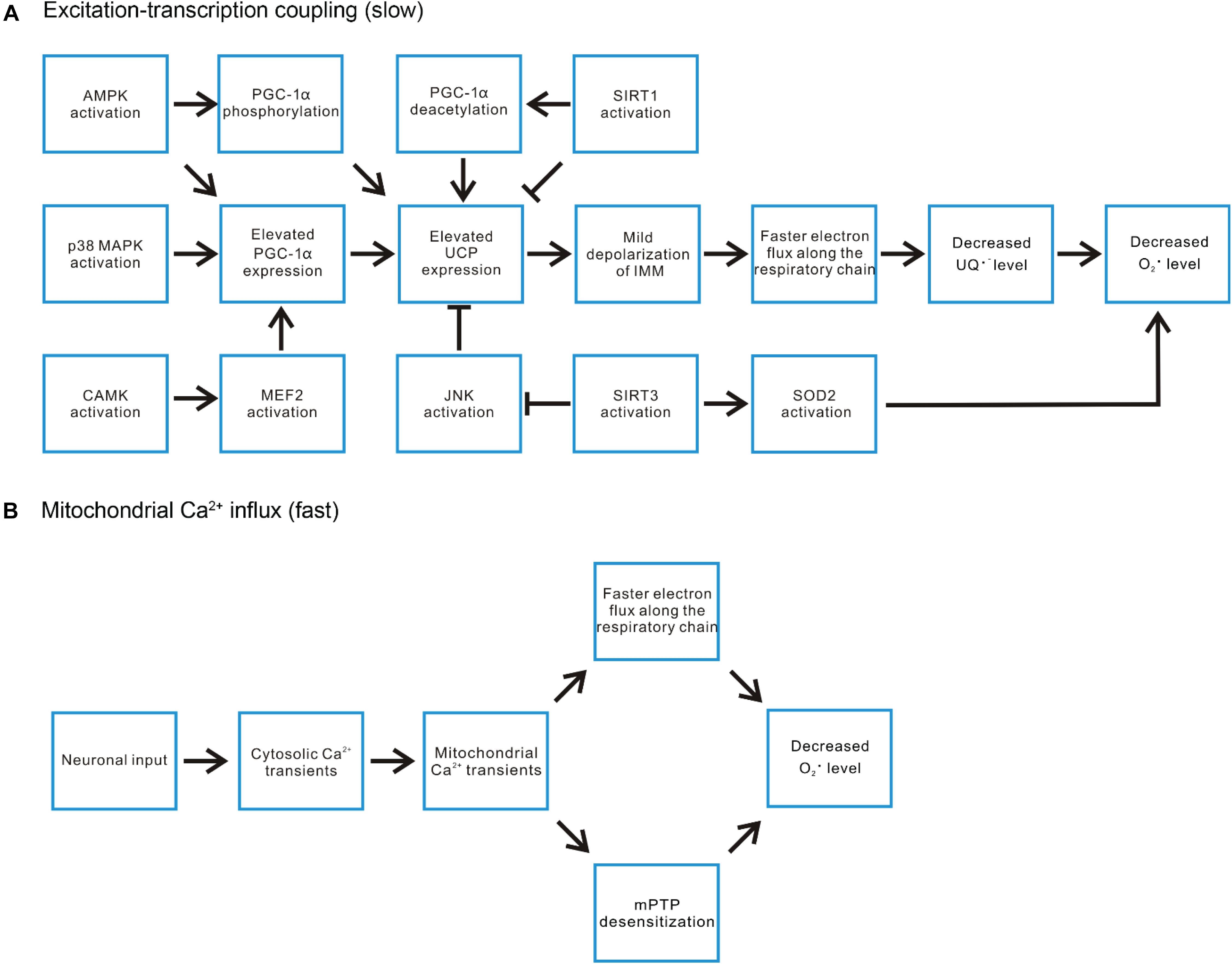
Figure 4. Exercise-induced signaling pathways regulate mitochondrial ROS level. (A) In the aspect of the excitation-transcription events, the ATP deficit during exercise induces AMPK activation, which can both directly phosphorylate and increase the expression of PGC-1α, the pivotal player in countering ROS-related damages. P38 MAPK and certain CAMKs have also been demonstrated to be activated by exercise and induce PGC-1α expression. SIRT1 activation promotes deacetylation of PGC-1α, enhancing its transcriptional activities. On the other hand, SIRT1 represses the expression of uncoupling protein (UCP2, UCP3). UCP induced mild depolarization of IMM accelerates electron flux along the respiratory chain, decreases the level of ubisemiquinone (UQ) and hence reduces superoxide production. Thus, the impact of SIRT1 activation on ROS level is mixed. In contrast, another SIRT family member SIRT3 promotes UCP expression by inhibiting JNK activation. It also promotes superoxide removal through deacetylating SOD2. (B) The rapid mitochondrial Ca2+ flux induced by motor neuron input may decrease ROS production through increasing the turnover rate of the respiratory chain or suppressing mPTP opening.
The three best-characterized MAPK subfamilies are c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and p38 MAPK (Chen et al., 2001). All three subfamilies have been shown to be activated by oxidative stress (Torres and Forman, 2003; Li et al., 2005). Among them, p38 MAPK signaling has been reported to participate in contraction-induced PGC-1α gene expression in skeletal muscle (Akimoto et al., 2004). PGC-1α is a “master regulator” of mitochondrial biogenesis and promotes the expression of OXPHOS enzymes and uncoupling proteins (UCP2 and UCP3) (Wu et al., 1999; Zhou et al., 2000). Uncoupling proteins function to mildly depolarize IMM by increasing proton backflow into the matrix in a fatty acid (FA)-dependent manner (Brand and Esteves, 2005). This process could be described by an FA futile cycling model, in which UCP exports FA– anions into intermembrane space. The anion diffuses away and gets protonated. Then, the protonated FA flip-flops across the membrane to deliver protons electro-neutrally back to the matrix (Jabůrek et al., 1999). UCP2 and UCP3 are not involved in thermogenesis adapting to cold temperature as UCP1 (Brand and Esteves, 2005). Instead, they are believed to serve as a cellular defense mechanism against superoxide formation, which works through accelerating the rates of proton pumping and electron flux along the respiratory chain, decreasing the level of one-electron donors (to O2) generated by complex I and III of OXPHOS (Brand, 2000; Zhou et al., 2000; Echtay et al., 2002; Brand and Esteves, 2005; Ježek et al., 2018). The expression of UCP3 is particularly high in skeletal muscle (Boss et al., 1997; Matsuda et al., 1997; Vidal-Puig et al., 1997; Liu et al., 1998) and is further induced by exercise or fasting (Gong et al., 1997; Tsuboyama-Kasaoka et al., 1998; Cortright et al., 1999). In contrast, denervation of skeletal muscle decreases the expression of UCP3 (Tsuboyama-Kasaoka et al., 1998).
AMPK can also be activated by ROS indirectly through ATP deficit, more specifically the increase of AMP/ATP and creatine/phosphocreatine ratios (Kahn et al., 2005; Irrcher et al., 2009). Acute exercise, due to the increased turnover of ATP, promotes AMPK activation in an intensity dependent manner (Wojtaszewski et al., 2000; Egan et al., 2010). AMPK activation has been report to mediate direct phosphorylation of PGC-1α and transcriptional upregulation of PGC-1α expression (Jäger et al., 2007; Irrcher et al., 2008). Meanwhile acute exercise induced UCP3 expression has also been demonstrated to involve AMPK (Zhou et al., 2000).
CaMKs are implicated in muscle fiber type switch adapting to long-term endurance exercise (Wu et al., 2000). Additionally, they have also been suggested to regulate mitochondrial biogenesis independent of fiber type transformation (Wu et al., 2002). Transgenic mice expressing constitutively active CaMKIV in skeletal muscle exhibited improved resistance to fatigue during repetitive contraction, augmented mitochondrial biogenesis, increased expression of OXPHOS genes (such as subunits of complex I) and PGC-1α (Wu et al., 2002). PGC-1α is a target gene of myocyte-specific enhancer factor 2 (MEF2), one of the transcription factors activated by CaMKIV (Passier et al., 2000). Furthermore, CaMKII has also been suggested to induce PGC-1α expression due to the activation of MEF2 (Liu et al., 2005; Egan and Zierath, 2013).
SIRTs are a family of protein deacetylases sensitive to elevated [NAD+] or NAD+/NADH ratio (Egan and Zierath, 2013). Their activities are closely linked with cellular metabolic status (Schwer and Verdin, 2008). SIRT1 mediated deacetylation is suggested to activate PGC-1α transcriptional activity on other genes (Gerhart-Hines et al., 2007; Gurd, 2011). However, SIRT1 also serves a potent repressor of UCP2 and UCP3 gene expression (Amat et al., 2007, 2009; Schwer and Verdin, 2008). Thus, SIRT1 exerts both positive and negative impact on ROS production. SIRT3 is a major deacetylase for mitochondrial proteins (Jing et al., 2011). In SIRT3 knockout mice, O2 consumption rate decreases while oxidative stress increases, accompanied by enhanced activation of JNK pathway (Jing et al., 2011). Consistently, knocking down SIRT3 in cultured myoblast also led to defective mitochondrial respiration capacity, increased ROS production and JNK pathway activation (Jing et al., 2011). Furthermore, SIRT3 has been shown to deacetylate SOD2, leading to increase in the SOD2 enzymatic activity to convert superoxide into hydrogen peroxide (Tao et al., 2014). Thus, the SIRT3 activity overall helps mitigate ROS accumulation (Tao et al., 2010, 2014).
It is also worth noticing that PGC-1α has an isoform termed PGC-1α4 resulted from alternative promoter usage and splicing (Ruas et al., 2012). This isoform is preferentially induced in mouse and human muscle during resistance exercise training (Ruas et al., 2012). Different from PGC-1α, it does not regulate OXPHOS genes but specifically induces IGF1 and represses myostatin, resulting in muscle hypertrophy (Ruas et al., 2012). Studies have indicated that the induction of PGC-1α4 requires MCU mediated Ca2+ uptake (Mammucari et al., 2015).
Although most studies concerning the beneficiary effect of exercise on ROS control focus on the excitation-transcription coupling framework, there is evidence directly linking acute exercise with improved mitochondrial functions such as decreased susceptibility to mPTP opening and increased O2 respiration rate in cardiac and skeletal muscle (Ascensão et al., 2011; Yoo et al., 2019a, b). These reports are consistent with the mechanisms we proposed underlying the instant decrease of mitochondrial ROS production in denervated skeletal muscle upon electrical stimulation. These two mechanisms may also explain why mitochondria were not the major contributor of ROS during electrical stimulation induced muscle contractions (Sakellariou et al., 2013). Thus, we propose that beside the excitation-transcription coupling framework, the rapid mitochondrial Ca2+ transients generated during exercise serve as a parallel mechanism contributing to prevent excessive ROS production, either through accelerating the turnover rate of the respiratory chain or suppressing mPTP opening (Figure 4B). The rapid mitochondrial Ca2+ influxes could also be one reason why there is no significant mitochondrial contribution to cytosolic ROS level in contractile skeletal muscle fibers.
The elevations of [Ca2+]mito induced by muscle contraction or muscle inactivity share many Ca2+ regulators in common, yet the outcomes are dramatically different in skeletal muscle ROS production. Additionally, electrical stimulation not only rapidly inhibits ROS production in an MCU dependent manner in skeletal muscle, but also prevents apoptosis, retards muscle atrophy in a longer term. These phenomena imply that [Ca2+]mito temporal profile, likely in combination with steady-state [Ca2+]mito level, serves as a toggle switch flipping between the beneficiary versus destructive outcomes. The players downstream of this toggle switch, aside from those relatively slower excitation-transcription coupling events, also include processes that modulate ROS production within the time frame of seconds to minutes. In this review, we proposed two potential mechanisms underlying this rapid process, such as direct regulation of the turnover rate of the respiratory chain and mPTP desensitization. Further experiments are needed to evaluate the validity of these hypotheses.
AL and JZ designed the scheme of this review. All authors contributed to editing the article and approved the submitted version.
Research in our laboratory has been supported by the Muscular Dystrophy Association Grant MDA-4351, NIH AR057404, NIH R01HL138570, NIH R01NS105621, DOD AL170061, the ALS Association (16-IIP-288), Victor E. Speas Foundation, a startup fund from Kansas City University of Medicine and Bioscience, and startup funds from the University of Texas at Arlington to JZ.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AAV, adeno-associated virus; AIF, apoptosis-inducing factor; ALS, amyotrophic lateral sclerosis; AMPK, AMP-activated protein kinase; ANT, adenine nucleotide translocator; CaMK, Ca2+/calmodulin-dependent protein kinase; CDK, cyclin-dependent kinase; CsA, Cyclosporin A; CypD, cyclophilin D; Diablo, direct IAP (inhibitor-of apoptosis) binding protein with low pI; EC-coupling, excitation contraction-coupling; EDL, extensor digitorum longus; EMRE, essential MCU regulator; EndoG, endonuclease G; ERK, extracellular signal-regulated kinase; FAK, focal adhesion kinase; FDB, flexor digitorum brevis; GPDH, glycerol phosphate dehydrogenase; HtraA2, high temperature requirement protein A2; ICDH, isocitrate dehydrogenase; ILK, integrin-linked kinase; IMM, inner mitochondrial membrane; JNK, c-JunN-terminal kinase; MAPK, mitogen-activated protein kinase; mCa2, mitochondrial calcium channel type 2; MCU, mitochondrial Ca2+ uniporter; MICU, mitochondrial Ca2+ uptake; mPTP, mitochondrial permeability transition pore; mTOR, mammalian target of rapamycin; NCLX, Na+/Ca2+/Li+ exchanger; OMM, outer mitochondrial membrane; OSCP, oligomycin sensitivity-conferring protein; OXPHOS, oxidative phosphorylation; PKA, protein kinase A; PKC, protein kinase C; PDH, pyruvate dehydrogenase; RaM, rapid mode of mitochondrial Ca2+ uptake; PiC, phosphate carrier; RaM, rapid mode of mitochondrial Ca2+ uptake; ROS, reactive oxygen species; RYR, ryanodine receptor; SIRT, sirtuin; Smac, second mitochondria-derived activator of caspase; SR, sarcoplasmic reticulum.
Adam-Vizi, V., and Starkov, A. A. (2010). Calcium and mitochondrial reactive oxygen species generation: how to read the facts. J. Alzheimers Dis. 20, S413–S426. doi: 10.3233/JAD-2010-100465
Adhihetty, P. J., Ljubicic, V., Menzies, K. J., and Hood, D. A. (2005). Differential susceptibility of subsarcolemmal and intermyofibrillar mitochondria to apoptotic stimuli. Am. J. Physiol. Cell Physiol. 289, C994–C1001. doi: 10.1152/ajpcell.00031.2005
Adhihetty, P. J., O’leary, M. F., Chabi, B., Wicks, K. L., and Hood, D. A. (2007). Effect of denervation on mitochondrially mediated apoptosis in skeletal muscle. J. Appl. Physiol. 102, 1143–1151. doi: 10.1152/japplphysiol.00768.2006
Akimoto, T., Sorg, B. S., and Yan, Z. (2004). Real-time imaging of peroxisome proliferator-activated receptor-γ coactivator-1α promoter activity in skeletal muscles of living mice. Am. J. Physiol. Cell Physiol. 287, C790–C796. doi: 10.1152/ajpcell.00425.2003
Alavian, K. N., Beutner, G., Lazrove, E., Sacchetti, S., Park, H.-A., Licznerski, P., et al. (2014). An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. U.S.A. 111, 10580–10585. doi: 10.1073/pnas.1401591111
Altschafl, B. A., Beutner, G., Sharma, V. K., Sheu, S.-S., and Valdivia, H. H. (2007). The mitochondrial ryanodine receptor in rat heart: a pharmaco-kinetic profile. Biochim. Biophys. Acta Biomembr. 1768, 1784–1795. doi: 10.1016/j.bbamem.2007.04.011
Amat, R., Planavila, A., Chen, S. L., Iglesias, R., Giralt, M., and Villarroya, F. (2009). SIRT1 controls the transcription of the peroxisome proliferator-activated receptor-γ co-activator-1α (PGC-1α) gene in skeletal muscle through the PGC-1α autoregulatory loop and interaction with MyoD. J. Biol. Chem. 284, 21872–21880. doi: 10.1074/jbc.M109.022749
Amat, R., Solanes, G., Giralt, M., and Villarroya, F. (2007). SIRT1 is involved in glucocorticoid-mediated control of uncoupling protein-3 gene transcription. J. Biol. Chem. 282, 34066–34076. doi: 10.1074/jbc.M707114200
Arakawa, T., Katada, A., Shigyo, H., Kishibe, K., Adachi, M., Nonaka, S., et al. (2010). Electrical stimulation prevents apoptosis in denervated skeletal muscle. NeuroRehabilitation 27, 147–154. doi: 10.3233/NRE-2010-0591
Ascensão, A., Lumini-Oliveira, J., Machado, N. G., Ferreira, R. M., Gonçalves, I. O., Moreira, A. C., et al. (2011). Acute exercise protects against calcium-induced cardiac mitochondrial permeability transition pore opening in doxorubicin-treated rats. Clin. Sci. 120, 37–49. doi: 10.1042/CS20100254
Baines, C. P. (2009). The molecular composition of the mitochondrial permeability transition pore. J. Mol. Cell. Cardiol. 46, 850–857. doi: 10.1016/j.yjmcc.2009.02.007
Baines, C. P., Kaiser, R. A., Sheiko, T., Craigen, W. J., and Molkentin, J. D. (2007). Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 9, 550–555. doi: 10.1038/ncb1575
Bandyopadhyay, U., Das, D., and Banerjee, R. K. (1999). Reactive oxygen species: oxidative damage and pathogenesis. Curr. Sci. 77, 658–666.
Barja, G. (2007). Mitochondrial oxygen consumption and reactive oxygen species production are independently modulated: implications for aging studies. Rejuvenat. Res. 10, 215–224. doi: 10.1089/rej.2006.0516
Batandier, C., Leverve, X., and Fontaine, E. (2004). Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J. Biol. Chem. 279, 17197–17204. doi: 10.1074/jbc.M310329200
Block, B. A. (1994). Thermogenesis in muscle. Annu. Rev. Physiol. 56, 535–577. doi: 10.1146/annurev.ph.56.030194.002535
Bonora, M., Patergnani, S., Ramaccini, D., Morciano, G., Pedriali, G., Kahsay, A. E., et al. (2020). Physiopathology of the permeability transition pore: molecular mechanisms in human pathology. Biomolecules 10:998. doi: 10.3390/biom10070998
Bonora, M., Wieckowski, M. R., Chinopoulos, C., Kepp, O., Kroemer, G., Galluzzi, L., et al. (2015). Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene 34, 1475–1486. doi: 10.1038/onc.2014.96
Borisov, A. B., and Carlson, B. M. (2000). Cell death in denervated skeletal muscle is distinct from classical apoptosis. Anat. Rec. 258, 305–318. doi: 10.1002/(SICI)1097-0185(20000301)258:3<305::AID-AR10>3.0.CO;2-A
Borutaite, V., Jekabsone, A., Morkuniene, R., and Brown, G. C. (2003). Inhibition of mitochondrial permeability transition prevents mitochondrial dysfunction, cytochrome c release and apoptosis induced by heart ischemia. J. Mol. Cell. Cardiol. 35, 357–366. doi: 10.1016/S0022-2828(03)00005-1
Boss, O., Samec, S., Paoloni-Giacobino, A., Rossier, C., Dulloo, A., Seydoux, J., et al. (1997). Uncoupling protein-3: a new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett. 408, 39–42. doi: 10.1016/S0014-5793(97)00384-0
Brand, M. (2000). Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp. Gerontol. 35, 811–820. doi: 10.1016/S0531-5565(00)00135-2
Brand, M. D., and Esteves, T. C. (2005). Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2, 85–93. doi: 10.1016/j.cmet.2005.06.002
Brookes, P. S., Yoon, Y., Robotham, J. L., Anders, M., and Sheu, S.-S. (2004). Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 287, C817–C833. doi: 10.1152/ajpcell.00139.2004
Buntinas, L., Gunter, K. K., Sparagna, G. C., and Gunter, T. E. (2001). The rapid mode of calcium uptake into heart mitochondria (RaM): comparison to RaM in liver mitochondria. Biochim. Biophys. Acta Bioenerg. 1504, 248–261. doi: 10.1016/S0005-2728(00)00254-1
Carafoli, E. (2010). The fateful encounter of mitochondria with calcium: how did it happen? Biochim. Biophys. Acta Bioenerg. 1797, 595–606. doi: 10.1016/j.bbabio.2010.03.024
Chabi, B., Ljubicic, V., Menzies, K. J., Huang, J. H., Saleem, A., and Hood, D. A. (2008). Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging cell 7, 2–12. doi: 10.1111/j.1474-9726.2007.00347.x
Chen, Z., Gibson, T. B., Robinson, F., Silvestro, L., Pearson, G., Xu, B.-E., et al. (2001). MAP kinases. Chem. Rev. 101, 2449–2476. doi: 10.1021/cr000241p
Chinopoulos, C., and Adam-Vizi, V. (2010). Mitochondrial Ca2+ sequestration and precipitation revisited. FEBS J. 277, 3637–3651. doi: 10.1111/j.1742-4658.2010.07755.x
Cogswell, A. M., Stevens, R. J., and Hood, D. A. (1993). Properties of skeletal muscle mitochondria isolated from subsarcolemmal and intermyofibrillar regions. Am. J. Physiol. Cell Physiol. 264, C383–C389. doi: 10.1152/ajpcell.1993.264.2.C383
Cortright, R. N., Zheng, D., Jones, J. P., Fluckey, J. D., Dicarlo, S. E., Grujic, D., et al. (1999). Regulation of skeletal muscle UCP-2 and UCP-3 gene expression by exercise and denervation. Am. J. Physiol. Endocrinol. Metab. 276, E217–E221. doi: 10.1152/ajpendo.1999.276.1.E217
Crameri, R., Weston, A., Climstein, M., Davis, G., and Sutton, J. (2002). Effects of electrical stimulation-induced leg training on skeletal muscle adaptability in spinal cord injury. Scand. J. Med. Sci. Sports 12, 316–322. doi: 10.1034/j.1600-0838.2002.20106.x
Crameri, R. M., Weston, A. R., Rutkowski, S., Middleton, J. W., Davis, G. M., and Sutton, J. R. (2000). Effects of electrical stimulation leg training during the acute phase of spinal cord injury: a pilot study. Eur. J. Appl. Physiol. 83, 409–415. doi: 10.1007/s004210000263
Csordás, G., Golenár, T., Seifert, E. L., Kamer, K. J., Sancak, Y., Perocchi, F., et al. (2013). MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 17, 976–987. doi: 10.1016/j.cmet.2013.04.020
Csordás, G., and Hajnóczky, G. (2009). SR/ER–mitochondrial local communication: calcium and ROS. Biochim. Biophys. Acta Bioenerg. 1787, 1352–1362. doi: 10.1016/j.bbabio.2009.06.004
Csukly, K., Ascah, A., Matas, J., Gardiner, P. F., Fontaine, E., and Burelle, Y. (2006). Muscle denervation promotes opening of the permeability transition pore and increases the expression of cyclophilin D. J. Physiol. 574, 319–327. doi: 10.1113/jphysiol.2006.109702
Das, A. M., and Harris, D. A. (1990). Control of mitochondrial ATP synthase in heart cells: inactive to active transitions caused by beating or positive inotropic agents. Cardiovasc. Res. 24, 411–417. doi: 10.1093/cvr/24.5.411
De Giorgi, F., Lartigue, L., Bauer, M. K., Schubert, A., Grimm, S., Hanson, G. T., et al. (2002). The permeability transition pore signals apoptosis by directing Bax translocation and multimerization. FASEB J. 16, 607–609. doi: 10.1096/fj.01-0269fje
Demaurex, N., and Guido, D. (2017). “The role of mitochondria in the activation/maintenance of SOCE: membrane contact sites as signaling hubs sustaining store-operated Ca 2+ entry,” in Store-Operated Ca2+ Entry (SOCE) Pathways, eds K. Groschner, W. Graier, and C. Romanin (Cham: Springer), 277–296. doi: 10.1007/978-3-319-57732-6_15
Denton, R. M. (2009). Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta Bioenerg. 1787, 1309–1316. doi: 10.1016/j.bbabio.2009.01.005
Díaz-Vegas, A., Eisner, V., and Jaimovich, E. (2019). Skeletal muscle excitation-metabolism coupling. Arch. Biochem. Biophys. 664, 89–94. doi: 10.1016/j.abb.2019.01.037
Ding, Y., Fang, H., Shang, W., Xiao, Y., Sun, T., Hou, N., et al. (2015). Mitoflash altered by metabolic stress in insulin-resistant skeletal muscle. J. Mol. Med. 93, 1119–1130. doi: 10.1007/s00109-015-1278-y
Doucet, B. M., Lam, A., and Griffin, L. (2012). Neuromuscular electrical stimulation for skeletal muscle function. Yale J. Biol. Med. 85:201.
Du, C., Fang, M., Li, Y., Li, L., and Wang, X. (2000). Smac, a mitochondrial protein that promotes cytochrome c–dependent caspase activation by eliminating IAP inhibition. Cell 102, 33–42. doi: 10.1016/S0092-8674(00)00008-8
Dudley, G., Castro, M., Rogers, S., and Apple, D. Jr. (1999). A simple means of increasing muscle size after spinal cord injury: a pilot study. Eur. J. Appl. Physiol. Occup. Physiol. 80, 394–396. doi: 10.1007/s004210050609
Dupont-Versteegden, E. E. (2005). Apoptosis in muscle atrophy: relevance to sarcopenia. Exp. Gerontol. 40, 473–481. doi: 10.1016/j.exger.2005.04.003
Echtay, K. S., Roussel, D., St-Pierre, J., Jekabsons, M. B., Cadenas, S., Stuart, J. A., et al. (2002). Superoxide activates mitochondrial uncoupling proteins. Nature 415, 96–99. doi: 10.1038/415096a
Egan, B., Carson, B. P., Garcia-Roves, P. M., Chibalin, A. V., Sarsfield, F. M., Barron, N., et al. (2010). Exercise intensity-dependent regulation of peroxisome proliferator-activated receptor γ coactivator-1α mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J. Physiol. 588, 1779–1790. doi: 10.1113/jphysiol.2010.188011
Egan, B., and Zierath, J. R. (2013). Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 17, 162–184. doi: 10.1016/j.cmet.2012.12.012
Eisenberg, B. R. (2010). Quantitative ultrastructure of mammalian skeletal muscle. Comp. Physiol. 73–112. doi: 10.1002/cphy.cp100103
Elrod, J. W., and Molkentin, J. D. (2013). Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ. J. 77, 1111–1122. doi: 10.1253/circj.CJ-13-0321
Elrod, J. W., Wong, R., Mishra, S., Vagnozzi, R. J., Sakthievel, B., Goonasekera, S. A., et al. (2010). Cyclophilin D controls mitochondrial pore–dependent Ca 2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 120, 3680–3687. doi: 10.1172/JCI43171
Fernandez, M. G., Troiano, L., Moretti, L., Nasi, M., Pinti, M., Salvioli, S., et al. (2002). Early changes in intramitochondrial cardiolipin distribution during apoptosis. Cell Growth Differ. 13, 449–455.
Fieni, F., Lee, S. B., Jan, Y. N., and Kirichok, Y. (2012). Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun. 3, 1–12. doi: 10.1038/ncomms2325
Fischer, L. R., Culver, D. G., Tennant, P., Davis, A. A., Wang, M., Castellano-Sanchez, A., et al. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240. doi: 10.1016/j.expneurol.2003.10.004
Fitts, R. H., Riley, D. R., and Widrick, J. J. (2001). Functional and structural adaptations of skeletal muscle to microgravity. J. Exp. Biol. 204, 3201–3208.
Frey, D., Schneider, C., Xu, L., Borg, J., Spooren, W., and Caroni, P. (2000). Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J. Neurosci. 20, 2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000
Gerhart-Hines, Z., Rodgers, J. T., Bare, O., Lerin, C., Kim, S. H., Mostoslavsky, R., et al. (2007). Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 26, 1913–1923. doi: 10.1038/sj.emboj.7601633
Ghafourifar, P., and Cadenas, E. (2005). Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci. 26, 190–195. doi: 10.1016/j.tips.2005.02.005
Giorgio, V., Bisetto, E., Soriano, M. E., Dabbeni-Sala, F., Basso, E., Petronilli, V., et al. (2009). Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 284, 33982–33988. doi: 10.1074/jbc.M109.020115
Giorgio, V., Burchell, V., Schiavone, M., Bassot, C., Minervini, G., Petronilli, V., et al. (2017). Ca2+ binding to F-ATP synthase β subunit triggers the mitochondrial permeability transition. EMBO Rep. 18, 1065–1076. doi: 10.15252/embr.201643354
Giorgio, V., Guo, L., Bassot, C., Petronilli, V., and Bernardi, P. (2018). Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 70, 56–63. doi: 10.1016/j.ceca.2017.05.004
Giorgio, V., Von Stockum, S., Antoniel, M., Fabbro, A., Fogolari, F., Forte, M., et al. (2013). Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. U.S.A. 110, 5887–5892. doi: 10.1073/pnas.1217823110
Giulivi, C. (2003). Characterization and function of mitochondrial nitric-oxide synthase. Free Radic. Biol. Med. 34, 397–408. doi: 10.1016/S0891-5849(02)01298-4
Glancy, B., Willis, W. T., Chess, D. J., and Balaban, R. S. (2013). Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 52, 2793–2809. doi: 10.1021/bi3015983
Gong, D.-W., He, Y., Karas, M., and Reitman, M. (1997). Uncoupling protein-3 is a mediator of thermogenesis regulated by thyroid hormone, β3-adrenergic agonists, and leptin. J. Biol. Chem. 272, 24129–24132. doi: 10.1074/jbc.272.39.24129
Greenawalt, J. W., Rossi, C. S., and Lehninger, A. L. (1964). Effect of active accumulation of calcium and phosphate ions on the structure of rat liver mitochondria. J. Cell Biol. 23, 21–38. doi: 10.1083/jcb.23.1.21
Grijalba, M. T., Vercesi, A. E., and Schreier, S. (1999). Ca2+-induced increased lipid packing and domain formation in submitochondrial particles. A possible early step in the mechanism of Ca2+-stimulated generation of reactive oxygen species by the respiratory chain. Biochemistry 38, 13279–13287. doi: 10.1021/bi9828674
Gunter, T. E., Yule, D. I., Gunter, K. K., Eliseev, R. A., and Salter, J. D. (2004). Calcium and mitochondria. FEBS Lett. 567, 96–102. doi: 10.1016/j.febslet.2004.03.071
Gurd, B. J. (2011). Deacetylation of PGC-1α by SIRT1: importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl. Physiol. Nutr. Metab. 36, 589–597. doi: 10.1139/h11-070
Halestrap, A. P. (2009). What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 46, 821–831. doi: 10.1016/j.yjmcc.2009.02.021
Halestrap, A. P., and Davidson, A. M. (1990). Inhibition of Ca2+-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem. J. 268, 153–160. doi: 10.1042/bj2680153
Haworth, R. A., and Hunter, D. R. (1979). The Ca2+-induced membrane transition in mitochondria: II. Nature of the Ca2+ trigger site. Arch. Biochem. Biophys. 195, 460–467. doi: 10.1016/0003-9861(79)90372-2
Hegde, R., Srinivasula, S. M., Zhang, Z., Wassell, R., Mukattash, R., Cilenti, L., et al. (2002). Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 277, 432–438. doi: 10.1074/jbc.M109721200
Henríquez-Olguin, C., Knudsen, J. R., Raun, S. H., Li, Z., Dalbram, E., Treebak, J. T., et al. (2019). Cytosolic ROS production by NADPH oxidase 2 regulates muscle glucose uptake during exercise. Nat. Commun. 10, 1–11. doi: 10.1038/s41467-019-12523-9
Hernansanz-Agustín, P., Choya-Foces, C., Carregal-Romero, S., Ramos, E., Oliva, T., Villa-Piña, T., et al. (2020). Na+ controls hypoxic signalling by the mitochondrial respiratory chain. Nature 586, 287–291. doi: 10.1038/s41586-020-2551-y
Hirsch, T., Marzo, I., and Kroemer, G. (1997). Role of the mitochondrial permeability transition pore in apoptosis. Biosci. Rep. 17, 67–76. doi: 10.1023/A:1027339418683
Hochachka, P., and McClelland, G. (1997). Cellular metabolic homeostasis during large-scale change in ATP turnover rates in muscles. J. Exp. Biol. 200, 381–386.
Hoellwarth, U., and Christian Hofer, D. (2005). Muscle biopsies show that FES of denervated muscles reverses human muscle degeneration from permanent spinal motoneuron lesion. J. Rehabil. Res. Dev. 42(Suppl. 1), 43–53. doi: 10.1682/JRRD.2004.05.0061
Hoffman, N. J., Parker, B. L., Chaudhuri, R., Fisher-Wellman, K. H., Kleinert, M., Humphrey, S. J., et al. (2015). Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise-regulated kinases and AMPK substrates. Cell Metab. 22, 922–935. doi: 10.1016/j.cmet.2015.09.001
Hoppe, U. C. (2010). Mitochondrial calcium channels. FEBS Lett. 584, 1975–1981. doi: 10.1016/j.febslet.2010.04.017
Hubbard, M. J., and McHugh, N. J. (1996). Mitochondrial ATP synthase F1-β-subunit is a calcium-binding protein. FEBS Lett. 391, 323–329. doi: 10.1016/0014-5793(96)00767-3
Hunter, D. R., and Haworth, R. A. (1979a). The Ca2+-induced membrane transition in mitochondria: I. The protective mechanisms. Arch. Biochem. Biophys. 195, 453–459. doi: 10.1016/0003-9861(79)90371-0
Hunter, D. R., and Haworth, R. A. (1979b). The Ca2+-induced membrane transition in mitochondria: III. Transitional Ca2+ release. Arch. Biochem. Biophys. 195, 468–477. doi: 10.1016/0003-9861(79)90373-4
Hyatt, H., Deminice, R., Yoshihara, T., and Powers, S. K. (2019). Mitochondrial dysfunction induces muscle atrophy during prolonged inactivity: a review of the causes and effects. Arch. Biochem. Biophys. 662, 49–60. doi: 10.1016/j.abb.2018.11.005
Ichas, F., Jouaville, L. S., and Mazat, J.-P. (1997). Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 89, 1145–1153. doi: 10.1016/S0092-8674(00)80301-3
Ichas, F., and Mazat, J.-P. (1998). From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low-to high-conductance state. Biochim. Biophys. Acta Bioenerg. 1366, 33–50. doi: 10.1016/S0005-2728(98)00119-4
Ingalls, C. P., Warren, G. L., and Armstrong, R. (1999). Intracellular Ca2+ transients in mouse soleus muscle after hindlimb unloading and reloading. J. Appl. Physiol. 87, 386–390. doi: 10.1152/jappl.1999.87.1.386
Irrcher, I., Ljubicic, V., and Hood, D. A. (2009). Interactions between ROS and AMP kinase activity in the regulation of PGC-1α transcription in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 296, C116–C123. doi: 10.1152/ajpcell.00267.2007
Irrcher, I., Ljubicic, V., Kirwan, A. F., and Hood, D. A. (2008). AMP-activated protein kinase-regulated activation of the PGC-1α promoter in skeletal muscle cells. PLoS One 3:e3614. doi: 10.1371/journal.pone.0003614
Jabůrek, M., VařEcha, M., Gimeno, R. E., Dembski, M., JežEk, P., Zhang, M., et al. (1999). Transport function and regulation of mitochondrial uncoupling proteins 2 and 3. J. Biol. Chem. 274, 26003–26007. doi: 10.1074/jbc.274.37.26003
Jackman, R. W., and Kandarian, S. C. (2004). The molecular basis of skeletal muscle atrophy. Am. J. Physiol. Cell Physiol. 287, C834–C843. doi: 10.1152/ajpcell.00579.2003
Jäger, S., Handschin, C., Pierre, J. S., and Spiegelman, B. M. (2007). AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. U.S.A. 104, 12017–12022. doi: 10.1073/pnas.0705070104
Ježek, P., Holendová, B., Garlid, K. D., and Jabůrek, M. (2018). Mitochondrial uncoupling proteins: subtle regulators of cellular redox signaling. Antioxid. Redox Signal. 29, 667–714. doi: 10.1089/ars.2017.7225
Jiang, D., Zhao, L., and Clapham, D. E. (2009). Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 326, 144–147. doi: 10.1126/science.1175145
Jing, E., Emanuelli, B., Hirschey, M. D., Boucher, J., Lee, K. Y., Lombard, D., et al. (2011). Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. U.S.A. 108, 14608–14613. doi: 10.1073/pnas.1111308108
Kahn, B. B., Alquier, T., Carling, D., and Hardie, D. G. (2005). AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1, 15–25. doi: 10.1016/j.cmet.2004.12.003
Kamer, K. J., and Mootha, V. K. (2014). MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. 15, 299–307. doi: 10.1002/embr.201337946
Karam, C., Yi, J., Xiao, Y., Dhakal, K., Zhang, L., Li, X., et al. (2017). Absence of physiological Ca 2+ transients is an initial trigger for mitochondrial dysfunction in skeletal muscle following denervation. Skeletal Muscle 7:6. doi: 10.1186/s13395-017-0123-0
Karch, J., and Molkentin, J. D. (2018). Identity of the elusive mitochondrial permeability transition pore: what it might be, what it was, and what it still could be. Curr. Opin. Physiol. 3, 57–62. doi: 10.1016/j.cophys.2018.03.001
Kinnally, K. W., Antonenko, Y. N., and Zorov, D. B. (1992). Modulation of inner mitochondrial membrane channel activity. J. Bioenerg. Biomembr. 24, 99–110. doi: 10.1007/BF00769536
Kirichok, Y., Krapivinsky, G., and Clapham, D. E. (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427:360. doi: 10.1038/nature02246
Kokoszka, J. E., Waymire, K. G., Levy, S. E., Sligh, J. E., Cai, J., Jones, D. P., et al. (2004). The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465. doi: 10.1038/nature02229
Kostrominova, T. Y., Dow, D. E., Dennis, R. G., Miller, R. A., and Faulkner, J. A. (2005). Comparison of gene expression of 2-mo denervated, 2-mo stimulated-denervated, and control rat skeletal muscles. Physiol. Genomics 22, 227–243. doi: 10.1152/physiolgenomics.00210.2004
Krauskopf, A., Eriksson, O., Craigen, W. J., Forte, M. A., and Bernardi, P. (2006). Properties of the permeability transition in VDAC1−/− mitochondria. Biochim. Biophys. Acta Bioenerg. 1757, 590–595. doi: 10.1016/j.bbabio.2006.02.007
Kwong, J., Davis, J., Baines, C., Sargent, M., Karch, J., Wang, X., et al. (2014). Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 21, 1209–1217. doi: 10.1038/cdd.2014.36
Lejay, A., Meyer, A., Schlagowski, A.-I., Charles, A.-L., Singh, F., Bouitbir, J., et al. (2014). Mitochondria: mitochondrial participation in ischemia–reperfusion injury in skeletal muscle. Int. J. Biochem. Cell Biol. 50, 101–105. doi: 10.1016/j.biocel.2014.02.013
Li, L. Y., Luo, X., and Wang, X. (2001). Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412, 95–99. doi: 10.1038/35083620
Li, Y.-P., Chen, Y., John, J., Moylan, J., Jin, B., Mann, D. L., et al. (2005). TNF-α acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 19, 362–370. doi: 10.1096/fj.04-2364com
Liu, Q., Bai, C., Chen, F., Wang, R., Macdonald, T., Gu, M., et al. (1998). Uncoupling protein-3: a muscle-specific gene upregulated by leptin in ob/ob mice. Gene 207, 1–7. doi: 10.1016/S0378-1119(97)00596-9
Liu, X., Kim, C. N., Yang, J., Jemmerson, R., and Wang, X. (1996). Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157. doi: 10.1016/S0092-8674(00)80085-9
Liu, Y., Randall, W. R., and Schneider, M. F. (2005). Activity-dependent and-independent nuclear fluxes of HDAC4 mediated by different kinases in adult skeletal muscle. J. Cell Biol. 168, 887–897. doi: 10.1083/jcb.200408128
Maack, C., Cortassa, S., Aon, M. A., Ganesan, A. N., Liu, T., and O’rourke, B. (2006). Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 99, 172–182. doi: 10.1161/01.RES.0000232546.92777.05
Mallilankaraman, K., Doonan, P., Cárdenas, C., Chandramoorthy, H. C., Müller, M., Miller, R., et al. (2012). MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 151, 630–644. doi: 10.1016/j.cell.2012.10.011
Mammucari, C., Gherardi, G., Zamparo, I., Raffaello, A., Boncompagni, S., Chemello, F., et al. (2015). The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep. 10, 1269–1279. doi: 10.1016/j.celrep.2015.01.056
Matsuda, J., Hosoda, K., Itoh, H., Son, C., Doi, K., Tanaka, T., et al. (1997). Cloning of rat uncoupling protein-3 and uncoupling protein-2 cDNAs: their gene expression in rats fed high-fat diet. FEBS Lett. 418, 200–204. doi: 10.1016/S0014-5793(97)01381-1
McBride, S., Wei-Lapierre, L., Mcmurray, F., Macfarlane, M., Qiu, X., Patten, D., et al. (2019). Skeletal muscle mitoflashes, pH, and the role of uncoupling protein-3. Arch. Biochem. Biophys. 663, 239–248. doi: 10.1016/j.abb.2019.01.018
Michels, G., Khan, I. F., Endres-Becker, J., Rottlaender, D., Herzig, S., Ruhparwar, A., et al. (2009). Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels. Circulation 119:2435. doi: 10.1161/CIRCULATIONAHA.108.835389
Mnatsakanyan, N., Llaguno, M. C., Yang, Y., Yan, Y., Weber, J., Sigworth, F. J., et al. (2019). A mitochondrial megachannel resides in monomeric F 1 FO ATP synthase. Nat. Commun. 10, 1–11. doi: 10.1038/s41467-019-13766-2
Mokrusch, T., Engelhardt, A., Eichhorn, K.-F., Prischenk, G., Prischenk, H., Sack, G., et al. (1990). Effects of long-impulse electrical stimulation on atrophy and fibre type composition of chronically denervated fast rabbit muscle. J. Neurol. 237, 29–34. doi: 10.1007/BF00319664
Monani, U. R. (2005). Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron 48, 885–895. doi: 10.1016/j.neuron.2005.12.001
Mukhopadhyay, P., Rajesh, M., Yoshihiro, K., Haskó, G., and Pacher, P. (2007). Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem. Biophys. Res. Commun. 358, 203–208. doi: 10.1016/j.bbrc.2007.04.106
Muller, F., Crofts, A. R., and Kramer, D. M. (2002). Multiple Q-cycle bypass reactions at the Qo site of the cytochrome bc 1 complex. Biochemistry 41, 7866–7874. doi: 10.1021/bi025581e
Muller, F. L., Roberts, A. G., Bowman, M. K., and Kramer, D. M. (2003). Architecture of the Qo site of the cytochrome bc 1 complex probed by superoxide production. Biochemistry 42, 6493–6499. doi: 10.1021/bi0342160
Muller, F. L., Song, W., Jang, Y. C., Liu, Y., Sabia, M., Richardson, A., et al. (2007). Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1159–R1168. doi: 10.1152/ajpregu.00767.2006
Murphy, A. N., Kelleher, J., and Fiskum, G. (1990). Submicromolar Ca2+ regulates phosphorylating respiration by normal rat liver and AS-30D hepatoma mitochondria by different mechanisms. J. Biol. Chem. 265, 10527–10534.
Nakagawa, K., Tamaki, H., Hayao, K., Yotani, K., Ogita, F., Yamamoto, N., et al. (2017). Electrical stimulation of denervated rat skeletal muscle retards capillary and muscle loss in early stages of disuse atrophy. Biomed Res. Int. 2017:5695217. doi: 10.1155/2017/5695217
Nascimento, L. R., Michaelsen, S. M., Ada, L., Polese, J. C., and Teixeira-Salmela, L. F. (2014). Cyclical electrical stimulation increases strength and improves activity after stroke: a systematic review. J. Physiother. 60, 22–30. doi: 10.1016/j.jphys.2013.12.002
Neretti, N., Wang, P.-Y., Brodsky, A. S., Nyguyen, H. H., White, K. P., Rogina, B., et al. (2009). Long-lived Indy induces reduced mitochondrial reactive oxygen species production and oxidative damage. Proc. Natl. Acad. Sci. U.S.A. 106, 2277–2282. doi: 10.1073/pnas.0812484106
Nowikovsky, K., Pozzan, T., Rizzuto, R., Scorrano, L., and Bernardi, P. (2012). The pathophysiology of LETM1. J. Gen. Physiol. 139, 445–454. doi: 10.1085/jgp.201110757
Ott, M., Robertson, J. D., Gogvadze, V., Zhivotovsky, B., and Orrenius, S. (2002). Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. U.S.A. 99, 1259–1263. doi: 10.1073/pnas.241655498
Palty, R., Ohana, E., Hershfinkel, M., Volokita, M., Elgazar, V., Beharier, O., et al. (2004). Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J. Biol. Chem. 279, 25234–25240. doi: 10.1074/jbc.M401229200
Palty, R., Silverman, W. F., Hershfinkel, M., Caporale, T., Sensi, S. L., Parnis, J., et al. (2010). NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. U.S.A. 107, 436–441. doi: 10.1073/pnas.0908099107
Pan, X., Liu, J., Nguyen, T., Liu, C., Sun, J., Teng, Y., et al. (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 15, 1464–1472. doi: 10.1038/ncb2868
Passier, R., Zeng, H., Frey, N., Naya, F. J., Nicol, R. L., Mckinsey, T. A., et al. (2000). CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J. Clin. Invest. 105, 1395–1406. doi: 10.1172/JCI8551
Patron, M., Checchetto, V., Raffaello, A., Teardo, E., Reane, D. V., Mantoan, M., et al. (2014). MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 53, 726–737. doi: 10.1016/j.molcel.2014.01.013
Peng, T. I., and Jou, M. J. (2010). Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 1201, 183–188. doi: 10.1111/j.1749-6632.2010.05634.x
Peviani, S. M., Russo, T. L., Durigan, J. L., Vieira, B. S., Pinheiro, C. M., Galassi, M. S., et al. (2010). Stretching and electrical stimulation regulate the metalloproteinase-2 in rat denervated skeletal muscle. Neurol. Res. 32, 891–896. doi: 10.1179/174313209X459093
Pilegaard, H., Saltin, B., and Neufer, P. D. (2003). Exercise induces transient transcriptional activation of the PGC-1α gene in human skeletal muscle. J. Physiol. 546, 851–858. doi: 10.1113/jphysiol.2002.034850
Pottecher, J., Guillot, M., Belaidi, E., Charles, A.-L., Lejay, A., Gharib, A., et al. (2013). Cyclosporine A normalizes mitochondrial coupling, reactive oxygen species production, and inflammation and partially restores skeletal muscle maximal oxidative capacity in experimental aortic cross-clamping. J. Vasc. Surg. 57, 1100–1108.e2. doi: 10.1016/j.jvs.2012.09.020
Powers, S. K., Radak, Z., and Ji, L. L. (2016). Exercise-induced oxidative stress: past, present and future. J. Physiol. 594, 5081–5092. doi: 10.1113/JP270646
Powers, S. K., Wiggs, M. P., Duarte, J. A., Zergeroglu, A. M., and Demirel, H. A. (2012). Mitochondrial signaling contributes to disuse muscle atrophy. Am. J. Physiol. Endocrinol. Metab. 303, E31–E39. doi: 10.1152/ajpendo.00609.2011
Qiu, J., Fang, Q., Xu, T., Wu, C., Xu, L., Wang, L., et al. (2018). Mechanistic role of reactive oxygen species and therapeutic potential of antioxidants in denervation-or fasting-induced skeletal muscle atrophy. Front. Physiol. 9:215. doi: 10.3389/fphys.2018.00215
Raffaello, A., De Stefani, D., Sabbadin, D., Teardo, E., Merli, G., Picard, A., et al. (2013). The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 32, 2362–2376. doi: 10.1038/emboj.2013.157
Reane, D. V., Vallese, F., Checchetto, V., Acquasaliente, L., Butera, G., De Filippis, V., et al. (2016). A MICU1 splice variant confers high sensitivity to the mitochondrial Ca2+ uptake machinery of skeletal muscle. Mol. Cell 64, 760–773. doi: 10.1016/j.molcel.2016.10.001
Rizzuto, R., and Pozzan, T. (2006). Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol. Rev. 86, 369–408. doi: 10.1152/physrev.00004.2005
Ruas, J. L., White, J. P., Rao, R. R., Kleiner, S., Brannan, K. T., Harrison, B. C., et al. (2012). A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 151, 1319–1331. doi: 10.1016/j.cell.2012.10.050
Rudolf, R. D., Mongillo, M., Magalhães, P. J., and Pozzan, T. (2004). In vivo monitoring of Ca2+ uptake into mitochondria of mouse skeletal muscle during contraction. J. Cell Biol. 166, 527–536. doi: 10.1083/jcb.200403102
Russo, T. L., Peviani, S. M., Durigan, J. L., Gigo-Benato, D., Delfino, G. B., and Salvini, T. F. (2010). Stretching and electrical stimulation reduce the accumulation of MyoD, myostatin and atrogin-1 in denervated rat skeletal muscle. J. Muscle Res. Cell Motil. 31, 45–57. doi: 10.1007/s10974-010-9203-z
Sakamoto, K., and Goodyear, L. J. (2002). Invited review: intracellular signaling in contracting skeletal muscle. J. Appl. Physiol. 93, 369–383. doi: 10.1152/japplphysiol.00167.2002
Sakellariou, G. K., Vasilaki, A., Palomero, J., Kayani, A., Zibrik, L., Mcardle, A., et al. (2013). Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase (s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid. Redox Signal. 18, 603–621. doi: 10.1089/ars.2012.4623
Schwarzländer, M., Murphy, M. P., Duchen, M. R., Logan, D. C., Fricker, M. D., Halestrap, A. P., et al. (2012). Mitochondrial ‘flashes’: a radical concept repHined. Trends Cell Biol. 22, 503–508. doi: 10.1016/j.tcb.2012.07.007
Schwer, B., and Verdin, E. (2008). Conserved metabolic regulatory functions of sirtuins. Cell Metab. 7, 104–112. doi: 10.1016/j.cmet.2007.11.006
Sedova, M., Dedkova, E. N., and Blatter, L. A. (2006). Integration of rapid cytosolic Ca2+ signals by mitochondria in cat ventricular myocytes. Am. J. Physiol. Cell Physiol. 291, C840–C850. doi: 10.1152/ajpcell.00619.2005
Seifert, E. L., Ligeti, E., Mayr, J. A., Sondheimer, N., and Hajnóczky, G. (2015). The mitochondrial phosphate carrier: role in oxidative metabolism, calcium handling and mitochondrial disease. Biochem. Biophys. Res. Commun. 464, 369–375. doi: 10.1016/j.bbrc.2015.06.031
Silva, L. A., Pinho, C. A., Scarabelot, K. S., Fraga, D. B., Volpato, A. M., Boeck, C. R., et al. (2009). Physical exercise increases mitochondrial function and reduces oxidative damage in skeletal muscle. Eur. J. Appl. Physiol. 105, 861–867. doi: 10.1007/s00421-008-0971-8
Siu, P. M., and Alway, S. E. (2005). Mitochondria-associated apoptotic signalling in denervated rat skeletal muscle. J. Physiol. 565, 309–323. doi: 10.1113/jphysiol.2004.081083
Susin, S. A., Lorenzo, H. K., Zamzami, N., Marzo, I., Snow, B. E., Brothers, G. M., et al. (1999). Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397, 441–446. doi: 10.1038/17135
Szabadkai, G., and Duchen, M. R. (2008). Mitochondria: the hub of cellular Ca2+ signaling. Physiology 23, 84–94. doi: 10.1152/physiol.00046.2007
Szabó, I., Pinto, V. D., and Zoratti, M. (1993). The mitochondrial permeability transition pore may comprise VDAC molecules: II. The electrophysiological properties of VDAC are compatible with those of the mitochondrial megachannel. FEBS Lett. 330, 206–210. doi: 10.1016/0014-5793(93)80274-X
Szabó, I., and Zoratti, M. (1992). The mitochondrial megachannel is the permeability transition pore. J. Bioenerg. Biomembr. 24, 111–117. doi: 10.1007/BF00769537
Tao, R., Coleman, M. C., Pennington, J. D., Ozden, O., Park, S.-H., Jiang, H., et al. (2010). Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 40, 893–904. doi: 10.1016/j.molcel.2010.12.013
Tao, R., Vassilopoulos, A., Parisiadou, L., Yan, Y., and Gius, D. (2014). Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid. Redox Signal. 20, 1646–1654. doi: 10.1089/ars.2013.5482
Tews, D. (2002). Apoptosis and muscle fibre loss in neuromuscular disorders. Neuromuscul. Disord. 12, 613–622. doi: 10.1016/S0960-8966(02)00030-5
Tischler, M. E., Rosenberg, S., Satarug, S., Henriksen, E. J., Kirby, C. R., Tome, M., et al. (1990). Different mechanisms of increased proteolysis in atrophy induced by denervation or unweighting of rat soleus muscle. Metabolism 39, 756–763. doi: 10.1016/0026-0495(90)90113-Q
Tisdale, M. J. (2010). Cancer cachexia. Curr. Opin. Gastroenterol. 26, 146–151. doi: 10.1097/MOG.0b013e3283347e77
Torres, M., and Forman, H. J. (2003). Redox signaling and the MAP kinase pathways. Biofactors 17, 287–296. doi: 10.1002/biof.5520170128
Tsai, M.-F., Phillips, C. B., Ranaghan, M., Tsai, C.-W., Wu, Y., Williams, C., et al. (2016). Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. eLife 5:e15545. doi: 10.7554/eLife.15545.019
Tsuboyama-Kasaoka, N., Tsunoda, N., Maruyama, K., Takahashi, M., Kim, H., Ikemoto, S., et al. (1998). Up-regulation of uncoupling protein 3 (UCP3) mRNA by exercise training and down-regulation of UCP3 by denervation in skeletal muscles. Biochem. Biophys. Res. Commun. 247, 498–503. doi: 10.1006/bbrc.1998.8818
Turrens, J. F., Alexandre, A., and Lehninger, A. L. (1985). Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 237, 408–414. doi: 10.1016/0003-9861(85)90293-0
Urbani, A., Giorgio, V., Carrer, A., Franchin, C., Arrigoni, G., Jiko, C., et al. (2019). Purified F-ATP synthase forms a Ca 2+-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat. Commun. 10, 1–11. doi: 10.1038/s41467-019-12331-1
Varanyuwatana, P., and Halestrap, A. P. (2012). The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion 12, 120–125. doi: 10.1016/j.mito.2011.04.006
Vidal-Puig, A., Solanes, G., Grujic, D., Flier, J. S., and Lowell, B. B. (1997). UCP3: an uncoupling protein homologue expressed preferentially and abundantly in skeletal muscle and brown adipose tissue. Biochem. Biophys. Res. Commun. 235, 79–82. doi: 10.1006/bbrc.1997.6740
Voytik, S. L., Przyborski, M., Badylak, S. F., and Konieczny, S. F. (1993). Differential expression of muscle regulatory factor genes in normal and denervated adult rat hindlimb muscles. Dev. Dyn. 198, 214–224. doi: 10.1002/aja.1001980307
Wan, B., Lanoue, K., Cheung, J., and Scaduto, R. (1989). Regulation of citric acid cycle by calcium. J. Biol. Chem. 264, 13430–13439.
Wang, C., and Youle, R. J. (2009). The role of mitochondria in apoptosis. Annu. Rev. Genet. 43, 95–118. doi: 10.1146/annurev-genet-102108-134850
Wang, L., Yang, X., Li, S., Wang, Z., Liu, Y., Feng, J., et al. (2014). Structural and mechanistic insights into MICU1 regulation of mitochondrial calcium uptake. EMBO J. 33, 594–604. doi: 10.1002/embj.201386523
Wang, W., Fang, H., Groom, L., Cheng, A., Zhang, W., Liu, J., et al. (2008). Superoxide flashes in single mitochondria. Cell 134, 279–290. doi: 10.1016/j.cell.2008.06.017
Wang, W., Zhang, H., and Cheng, H. (2016). Mitochondrial flashes: from indicator characterization to in vivo imaging. Methods 109, 12–20. doi: 10.1016/j.ymeth.2016.06.004
Wang, X., Zhang, X., Wu, D., Huang, Z., Hou, T., Jian, C., et al. (2017). Mitochondrial flashes regulate ATP homeostasis in the heart. eLife 6:e23908. doi: 10.7554/eLife.23908
Wei-LaPierre, L., Gong, G., Gerstner, B. J., Ducreux, S., Yule, D. I., Pouvreau, S., et al. (2013). Respective contribution of mitochondrial superoxide and pH to mitochondria-targeted circularly permuted yellow fluorescent protein (mt-cpYFP) flash activity. J. Biol. Chem. 288, 10567–10577. doi: 10.1074/jbc.M113.455709
Wojtaszewski, J. F., Nielsen, P., Hansen, B. F., Richter, E. A., and Kiens, B. (2000). Isoform-specific and exercise intensity-dependent activation of 5′-AMP-activated protein kinase in human skeletal muscle. J. Physiol. 528, 221–226. doi: 10.1111/j.1469-7793.2000.t01-1-00221.x
Wolf, S. G., Mutsafi, Y., Dadosh, T., Ilani, T., Lansky, Z., Horowitz, B., et al. (2017). 3D visualization of mitochondrial solid-phase calcium stores in whole cells. eLife 6:e29929. doi: 10.7554/eLife.29929.029
Wu, H., Kanatous, S. B., Thurmond, F. A., Gallardo, T., Isotani, E., Bassel-Duby, R., et al. (2002). Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science 296, 349–352. doi: 10.1093/emboj/19.9.1963
Wu, H., Naya, F. J., Mckinsey, T. A., Mercer, B., Shelton, J. M., Chin, E. R., et al. (2000). MEF2 responds to multiple calcium-regulated signals in the control of skeletal muscle fiber type. EMBO J. 19, 1963–1973. doi: 10.1016/S0092-8674(00)80611-X
Wu, Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., et al. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124.
Xiao, Y., Karam, C., Yi, J., Zhang, L., Li, X., Yoon, D., et al. (2018). ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharmacol. Res. 138, 25–36.
Yi, J., Ma, C., Li, Y., Weisleder, N., Ríos, E., Ma, J., et al. (2011). Mitochondrial calcium uptake regulates rapid calcium transients in skeletal muscle during excitation-contraction (EC) coupling. J. Biol. Chem. 286, 32436–32443.
Yoo, S.-Z., No, M.-H., Heo, J.-W., Chang, E., Park, D.-H., Kang, J.-H., et al. (2019a). Effects of a single bout of exercise on mitochondria-mediated apoptotic signaling in rat cardiac and skeletal muscles. J. Exer. Rehabil. 15:512.
Yoo, S.-Z., No, M.-H., Heo, J.-W., Park, D.-H., Kang, J.-H., Kim, J.-H., et al. (2019b). Effects of acute exercise on mitochondrial function, dynamics, and mitophagy in rat cardiac and skeletal muscles. Int. Neurourol. J. 23:S22.
Zhou, M., Lin, B.-Z., Coughlin, S., Vallega, G., and Pilch, P. F. (2000). UCP-3 expression in skeletal muscle: effects of exercise, hypoxia, and AMP-activated protein kinase. Am. J. Physiol. Endocrinol. Metab. 279, E622–E629.
Zhou, Z., Matlib, M. A., and Bers, D. M. (1998). Cytosolic and mitochondrial Ca2+ signals in patch clamped mammalian ventricular myocytes. J. Physiol. 507, 379–403.
Zoccarato, F., and Nicholls, D. (1982). The role of phosphate in the regulation of the independent calcium-efflux pathway of liver mitochondria. Eur. J. Biochem. 127, 333–338.
Keywords: skeletal muscle, mitochondrial ROS, mitoflash, mitochondrial Ca2+ homeostasis, transitory mPTP opening, electric field stimulation
Citation: Li A, Yi J, Li X and Zhou J (2020) Physiological Ca2+ Transients Versus Pathological Steady-State Ca2+ Elevation, Who Flips the ROS Coin in Skeletal Muscle Mitochondria. Front. Physiol. 11:595800. doi: 10.3389/fphys.2020.595800
Received: 17 August 2020; Accepted: 05 October 2020;
Published: 22 October 2020.
Edited by:
Enrique Jaimovich, University of Chile, ChileReviewed by:
Takahiko Shimizu, National Center for Geriatrics and Gerontology (NCGG), JapanCopyright © 2020 Li, Yi, Li and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ang Li, ang.li3@uta.edu; Jingsong Zhou, jingsong.zhou@uta.edu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.