 Laura Martinez-Mateu
Laura Martinez-Mateu Javier Saiz
Javier Saiz Ademuyiwa S. Aromolaran
Ademuyiwa S. Aromolaran- 1Centro de Investigación e Innovación en Bioingeniería, Universitat Politècnica de València, Valencia, Spain
- 2Cardiac Electrophysiology and Metabolism Research Group, VA New York Harbor Healthcare System, Brooklyn, NY, United States
- 3Department of Cell Biology, State University of New York Downstate Medical Center, Brooklyn, NY, United States
- 4Department of Physiology & Cellular Biophysics, Columbia University, New York, NY, United States
Obesity mechanisms that make atrial tissue vulnerable to arrhythmia are poorly understood. Voltage-dependent potassium (IK, IKur, and IK1) and L-type calcium currents (ICa,L) are electrically relevant and represent key substrates for modulation in obesity. We investigated whether electrical remodeling produced by high-fat diet (HFD) alone or in concert with acute atrial stimulation were different. Electrophysiology was used to assess atrial electrical function after short-term HFD-feeding in guinea pigs. HFD atria displayed spontaneous beats, increased IK (IKr + IKs) and decreased ICa,L densities. Only with pacing did a reduction in IKur and increased IK1 phenotype emerge, leading to a further shortening of action potential duration. Computer modeling studies further indicate that the measured changes in potassium and calcium current densities contribute prominently to shortened atrial action potential duration in human heart. Our data are the first to show that multiple mechanisms (shortened action potential duration, early afterdepolarizations and increased incidence of spontaneous beats) may underlie initiation of supraventricular arrhythmias in obese guinea pig hearts. These results offer different mechanistic insights with implications for obese patients harboring supraventricular arrhythmias.
Introduction
High-fat diet (HFD)-induced obesity is associated with insulin resistance, Type 2 diabetes mellitus (T2DM), and dyslipidemias (Schulze et al., 2016). A recent estimation by the National Institutes of Health (NIH) shows that obesity and its co-morbidities, affects 17% of children and young adults in the United States, while over one-third of adults are overweight and/or obese (Jensen et al., 2014). Moreover, obesity is an independent and key contributor to the expanding prevalence of atrial fibrillation (AF) (Michael et al., 2009; Lau et al., 2012; Abed and Wittert, 2013), a serious condition affecting approximately 2.7 million people in the United States (www.heart.org/en/health-topics/atrial-fibrillation).
In population-based cohort studies, obese individuals showed a 49% increase in vulnerability to AF when compared to non-obese individuals (Wanahita et al., 2008). A third wave of the Nord-Trøndelag Health prospective cohort study (HUNT3), demonstrated that being overweight or obese increased the AF risk by 18 and 59% (Garnvik et al., 2018); while epidemiological studies revealed a 4–5% increased risk of AF for each one unit increase in body mass index (BMI) (De Sensi et al., 2018). Despite the growing knowledge that obesity and AF are serious conditions with significant implications for public health, the molecular mechanisms that underlie atrial remodeling in obesity are poorly understood.
Atrial electrical remodeling due to pathological changes in functional expression of major atrial ion channels is an important signature of AF initiation (Aromolaran and Boutjdir, 2017; Dan and Dobrev, 2018; Heijman et al., 2018; Rahm et al., 2018). The human atrial action potential (AP) is defined by: phase 0 controlled by a large inward sodium current (INa) (Ni et al., 2017; Pezhouman et al., 2018), followed by calcium entry through L-type calcium channels (ICa,L) due to Cav1.2/Cav1.3 channels (Barana et al., 2014); a plateau phase maintained by a balance between inward and outward currents. Repolarization is controlled by fast transient outward potassium currents (Ito), ultra-rapid (IKur), rapid (IKr), and slow (IKs) delayed rectifier currents (Nerbonne and Kass, 2005). The resting membrane potential is determined by the inwardly rectifying potassium currents (IK1) (Ji et al., 2017; Whittaker et al., 2017).
Therefore, altered ion channel function that either increases potassium currents (Aromolaran et al., 2016) or decreases calcium currents (Van Wagoner et al., 1997, 1999; Christ et al., 2004; Mancarella et al., 2008) would be expected to promote an accelerated repolarization process, leading to shortened action potential duration (APD) (Boutjdir et al., 1986; Aromolaran et al., 2016), atrial refractoriness (Boutjdir et al., 1986), leading to ectopic firing (Nattel, 2002; Iwasaki et al., 2011; Czick et al., 2016) and re-entry mechanisms (such as single or multiple wave patterns) (Wakili et al., 2011; Nattel and Dobrev, 2017). To advance the current knowledge of AF mechanisms and development of mechanism-based therapeutic interventions, future studies will require robust approaches including coupling of experimental data with computational modeling (Grandi et al., 2011, 2019; Bai et al., 2018; Martinez-Mateu et al., 2018).
Obesity mechanisms may have a direct impact on the electrical activity of the heart. How the relative functional expression of potassium and calcium channels is altered in HFD-induced obesity remains unknown. It is known that chronically elevated levels of free-fatty acids (FFAs) is associated with obesity-related pathological changes (including insulin-resistance, hyperglycemia, T2DM, and inflammation) (Sonnenberg et al., 2004; Boden et al., 2005; Matafome and Seica, 2017; Sharma et al., 2017). Therefore, hyperlipidemia may represent a common link among obesity and its comorbidities and increased vulnerability to arrhythmias (Chiu et al., 2001; Rennison and Van Wagoner, 2009; Djousse et al., 2013; Fretts et al., 2014).
Furthermore, unlike the monounsaturated oleic acid (OA), saturated free-fatty acids including palmitic acid (PA) (O’Connell et al., 2015; Aromolaran et al., 2016), have been shown to promote adverse ion channel function and are prime candidates for mediating obesity-related adverse electrical remodeling leading to arrhythmias. Therefore, understanding lipotoxicity as a key obesity mechanism is an essential part of the wider effort to understand atrial electrical remodeling in obese patients with arrhythmias.
Unlike rodent models (Matsuura and Ehara, 1997; Nakaya et al., 2000; Killeen et al., 2008a,b), guinea pigs display robust expressions of IKr and IKs (Hume and Uehara, 1985; Aoki et al., 2012; Aromolaran et al., 2014, 2016). To date there have been increasing number of studies utilizing guinea pig atrial myocytes (Baro and Escande, 1989a,b; Inoue et al., 1993a,b; Bunemann et al., 1996; Bosch et al., 2003; Ochi et al., 2006; Zankov et al., 2006; Suzuki et al., 2014), to understand adverse ion channel mechanisms in disease states. Similar to humans, guinea pigs have high low-density lipoprotein/high-density lipoprotein (LDL/HDL) ratios, express key enzymes involved in lipoprotein metabolism (Fernandez et al., 1995; Sharman et al., 2008), and have higher concentrations of hepatic free cholesterol versus esterified cholesterol ratio (Angelin et al., 1992; Roy et al., 2000). These properties in addition to their use as models of arrhythmias (Laurita and Rosenbaum, 1997; O’Hara and Rudy, 2012; Liu et al., 2014; Mickelson and Chandra, 2017; Dey et al., 2018), further underscores the suitability of using obese guinea pigs in our studies.
In the present study we investigated the effect of HFD on the electrophysiological properties of major atrial ion channels, namely: the delayed rectifier potassium current IK (or IKr + IKs), IKur, IK1, and ICa,L currents in guinea pig atrial myocytes and determined its role in atrial arrhythmogenesis. Our data show increased IK and decreased L-type calcium channel densities in atrial myocytes from HFD-fed guinea pigs, whereas IKur and IK1 densities were unchanged. The data suggest that these changes are the cause of atrial arrhythmogenesis and therefore may underlie key electrical events (accelerated repolarization, shortened action potential duration, atrial refractoriness, conduction abnormalities, ectopic firing and single/multiple wave re-entrant mechanisms) that lead to AF onset and/or its maintenance in obesity.
Methods
High-Fat Diet Feeding in Guinea Pig Model
Guinea pigs (male/female; 200–250 g) were purchased from Charles River Laboratories (Wilmington, MA). Guinea pigs were randomly separated into two groups namely: control and overweight/obese. Controls were fed, ad libitum, a low-fat diet (LFD, Research Diets Inc., New Brunswick, NJ USA) containing (in kcal%): 10 fat, 70 carbohydrates, 20 protein, and 2,300 corn starch. HFD groups were fed a palatable high sucrose diet (in which most of the corn starch, was replaced with 1,014 kcal% sucrose), containing 45% of its kcal from fat, 35% from carbohydrates, and 20% from protein. The HFD was calorically rich (4.21 kcal/g versus 3.49 kcal/g for LFD) due to its higher fat content mostly from lard. HFD contained saturated and unsaturated free fatty acids (FFA), which provided 31.4 and 68.6% of the fat-derived calories (Caillier et al., 2012; Patoine et al., 2013). A detailed description and composition of the diets has previously been provided by Drolet’s group (Caillier et al., 2012). Guinea pigs were fed LFD or HFD for a duration of 50 days (~7 weeks), while monitoring temporal changes in weight 2–3 times weekly.
Isolation of Guinea Pig Atrial Myocytes
Guinea pig atrial myocytes were isolated as previously described (Aromolaran et al., 2016; Puckerin et al., 2016). Briefly, adult male and female Hartley guinea pigs were deeply anesthetized with isoflurane in accordance with the guidelines of the Columbia University Animal Care and Use Committee. Excised hearts were Langendorff perfused with Tyrode solution containing (in mM): 118 NaCl, 4.8 KCl, 1 CaCl2, 10 glucose, 1.25 MgSO4, and 1.25 K2HPO4 (pH = 7.4) for 5 min. The heart was then perfused with Ca2+-free Tyrode solution for 10 min before switching to Ca2+-free Tyrode solution containing Collagenase B (final concentration, 0.6 mg/ml; Boehringer Mannheim, Indianapolis, IN) for an additional 10 min. The heart was subsequently perfused with high K solution containing (in mM): 70 KOH, 50 L-glutamic acid, 40 KCl, 20 Taurine, 20 KH2PO4, 3 MgCl2, 10 glucose, 10 HEPES, and 1 EGTA (pH 7.4, with KOH), for 5–10 min. The digested tissue was placed in fresh high K solution, minced into smaller pieces, and then re-suspended several times to dissociate the cells. The cell suspension was filtered through a mesh and allowed to settle for 15–20 min. The pellet was resuspended in M199 media supplemented with 10% fetal bovine serum (FBS) and then plated on laminin-coated coverslips. Media were replaced with M199 containing 1% FBS 1–3 h after initial plating before experiments. All experiments were performed at room temperature (23–25°C).
Generation of hERG Plasmid Constructs
hERG 1a and hERG 1b constructs were a gift from Dr. Gail Robertson (University of Wisconsin). Yellow fluorescent protein (YFP) and cyan fluorescent protein/probe (CFP) were fused in frame to the C-terminus of hERG 1a or 1b using overlap extension PCR. A high-affinity 13 amino acid (WRRYYESSLEPYPD) surface epitope bungarotoxin-binding sequence (BBS) (Aromolaran et al., 2014) was introduced between amino acid residues 177–178 and 517–518, located within the extracellular loop of, respectively, hERG 1a S3-S4 domains using a QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) according to the manufacturer’s instructions. All cDNA plasmids were sequenced to verify that no error was introduced during preparation.
HEK293 Cell Culture and Transfection
Low-passage-number HEK293 cells (ATCC, Manassas, VA, USA) were maintained in DMEM supplemented with 10% FBS and 100 μg ml−1 penicillin-streptomycin at 37°C. In all experiments, cells were transiently transfected with 1 μg T-antigen and a total of 6 μg of the respective DNA plasmids, using the calcium phosphate precipitation method. For flow cytometry assays, hERG channel subunits were transfected in 1:1 (or 3:3 μg, BBS-hERG 1a-YFP + 1b-CFP or hERG 1a-YFP + 1b-CFP) ratio. Cells were cultured in supplemented DMEM at 37°C for 24 h before transfection.
Preparation of Bovine Serum Albumin-Conjugated Free Fatty Acid Solutions
Palmitic acid (PA) and oleic acid (OA) were prepared as previously described (Aromolaran et al., 2016). FA-free (20%) bovine serum albumin (BSA, Roche) was dissolved in DPBS. PA and OA (Sigma-Aldrich, St. Louis, MO, USA) were dissolved in ethanol to produce a stock solution of 0.2 M fatty acid. 20% BSA and 0.2 M FA were then mixed in 20:1 volume ratio. Desired concentrations of PA or OA were prepared from ~10 mM fatty acid stock solutions, in M199 culture media or Tyrode’s solution. The control solution was prepared with BSA, ethanol, and DPBS.
Electrophysiology
Patch clamp experiments in adult guinea pig atrial myocytes were performed at room temperature (20–25°C), using an EPC-10 patch clamp amplifier (HEKA Electronics) controlled by PatchMaster software (HEKA) as described (Puckerin et al., 2016). Briefly, coverslips with myocytes were placed on the stage of an inverted microscope (Eclipse Ti-U, Nikon). The internal solution contained (in mM): 130 KCl, 1 MgCl2, 0.4 GTP, 5 EGTA, 5 K2ATP, and 10 HEPES (pH 7.2). External solution contained (in mM): 137 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES (pH 7.4). Pipette resistance was typically 1.5–2 MΩ and series resistance was compensated 80–90% before each recording. Membrane potentials were corrected for liquid junctional potential. AP’s were recorded from single atrial myocytes in current clamp mode by passing depolarizing currents for 20 ms at subthreshold (1.5×) intensity. Population I–V curves for IK were generated by step depolarizations (−40 to +100 mV), from a holding potential of −50 mV in 10 mV increments for 3 s, followed by a repolarizing step to −50 mV for 1 s, with each pulse repeated every 10 s. The external solution contained nifedipine (5 μM), to block Ca currents. IKur was studied with a protocol consisting of a 300 ms depolarizing pulses from a −40 to +100 mV from a holding potential of −50 mV in 10 mV increments, followed a repolarizing step to −50 mV. IK1 was activated from −40 mV to test potentials ranging from −120 to +10 mV in 10 mV steps for 1 s. IKur and IK1 currents were measured in an external solution contained chromanol 293B (100 μM), E4031 (5 μM), and nifedipine (5 μM) to block IKs, IKr, and ICa,L, respectively. The internal solution for ICa,L recordings contained (in mM): 18 CsCl, 108 Cs-aspartate, 1.2 MgCl2, 11 HEPES, 1 EGTA, 10 glucose, and 2 MgATP (pH = 7.2, adjusted with Tris). External solution contained (in mM): 6 CsCl, 140 NaCl, 10 glucose, 1 MgCl2, 2 CaCl2, and 5 HEPES (pH = 7.4 adjusted with NaOH). The ICa,L was activated by a series of 250-ms depolarization pulses from −90 mV holding potential to test potentials ranging from −40 to +60 mV (10 mV step) at 10-s intervals. Currents were sampled at 20 kHz and filtered at 5 or 10 kHz.
Epicardial Electrograms From Isolated Hearts
LFD- or HFD hearts were Langendorff-perfused with Krebs-Henseleit buffer solution containing the following (in mM): 118 NaCl, 4.7 KCl, 1.2 KH2PO4, 1.2 MgSO4, 1.8 CaCl2, 25 NaHCO3, and 11.1 glucose, (pH 7.4), bubbled with 95% O2–5% CO2 at 35 ± 1°C. An eight-channel amplifier (Powerlab System, AD instruments, Oxfordshire, United Kingdom) was used to record epicardial atrial electrical activity using two chlorinated electrodes that were placed on the heart (Mancarella et al., 2008). A third module was a stimulator that was controlled by a computer which was designed to deliver programmed fast burst stimulation protocol (rectangular pulses 10 ms in duration, 1 V in amplitude with various pulse-pulse duration 23–190 ms), followed by three extra-stimuli S1-S2-S3 delivered at 300 beats/min, with S1 (250 ms for 50 beats); S2 (50 ms for 100 beats); S3 (20 ms for two beats). The stimuli were delivered to the right atrium to induce atrial arrhythmogenesis or arrhythmia.
Quantification of Cell Surface Bungarotoxin-Binding Sequence-hERG-Yellow Fluorescent Protein Channels With Quantum Dots
Relative surface expression of BBS-tagged hERG subunits was quantitatively determined using quantum dot labeling or used in flow cytometry (using a BD LSRII Cell Analyzer) to determine the BBS-tagged hERG 1a-YFP or hERG 1b-YFP surface density as described previously (Aromolaran et al., 2014).
Confocal Imaging
HEK293 cells were imaged using a Zeiss LSM 510 META scanning confocal microscope. CFP and YFP fluorescence signals were captured at, respectively, 458 and 514 nm argon laser lines for excitation and the red fluorescence signals of quantum dot were imaged using the 633 nm helium-neon laser line for excitation. Eight-bit images were obtained using identical laser powers, photomultiplier gain, and pinhole size.
Computer Simulations
We used computer simulations in order to study in human atrial myocytes the effects of the experimentally observed effects of HFD-induced obesity in guinea pig. For this purpose, we simulated the electrical activity of a human atrial myocyte corresponding to a non-obese individual (or LFD) using ionic models described by Courtemanche et al., (1998) and Koivumäki et al. (2011). Then, we modified several ionic currents based on the changes measured in our experiments within the voltage range of the AP, i.e., between −80 and +40 mV (see Table 1). Within this voltage range, for HFD myocytes, IKr and IKs were increased by 100% and ICa,L was decreased by 70%, while for HFD myocytes after pacing only ICaL, IK1 and IKur were modified with respect to LFD myocytes (ICaL and IKur decreased by 40% and IK1 increased by 112%). Other variations experimentally observed were not considered since they were significantly out of the voltage range in which the AP is contained, as shown in Figures 1, 3. As the computational models differentiate between IKr and IKs, same variation was applied to both currents to account for the IK measurements.
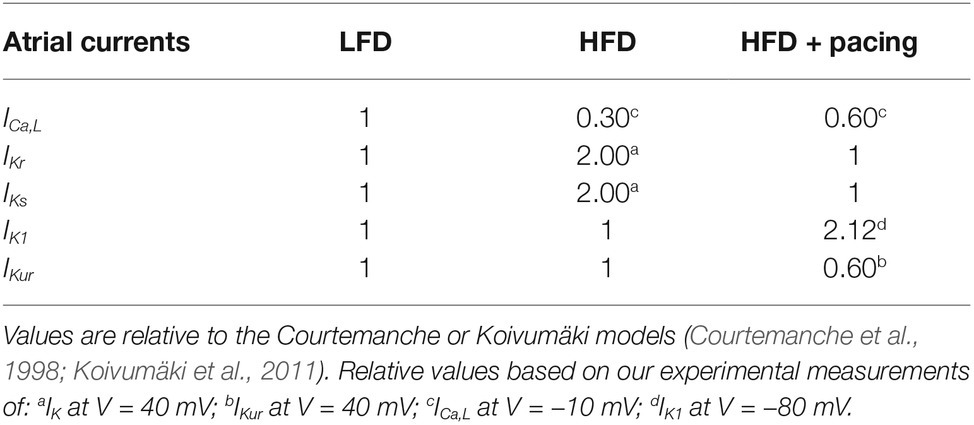
Table 1. Currents modified in the HFD myocyte, with and without pacing, with respect to the low-fat diet myocyte used for human atrial myocyte simulation models.
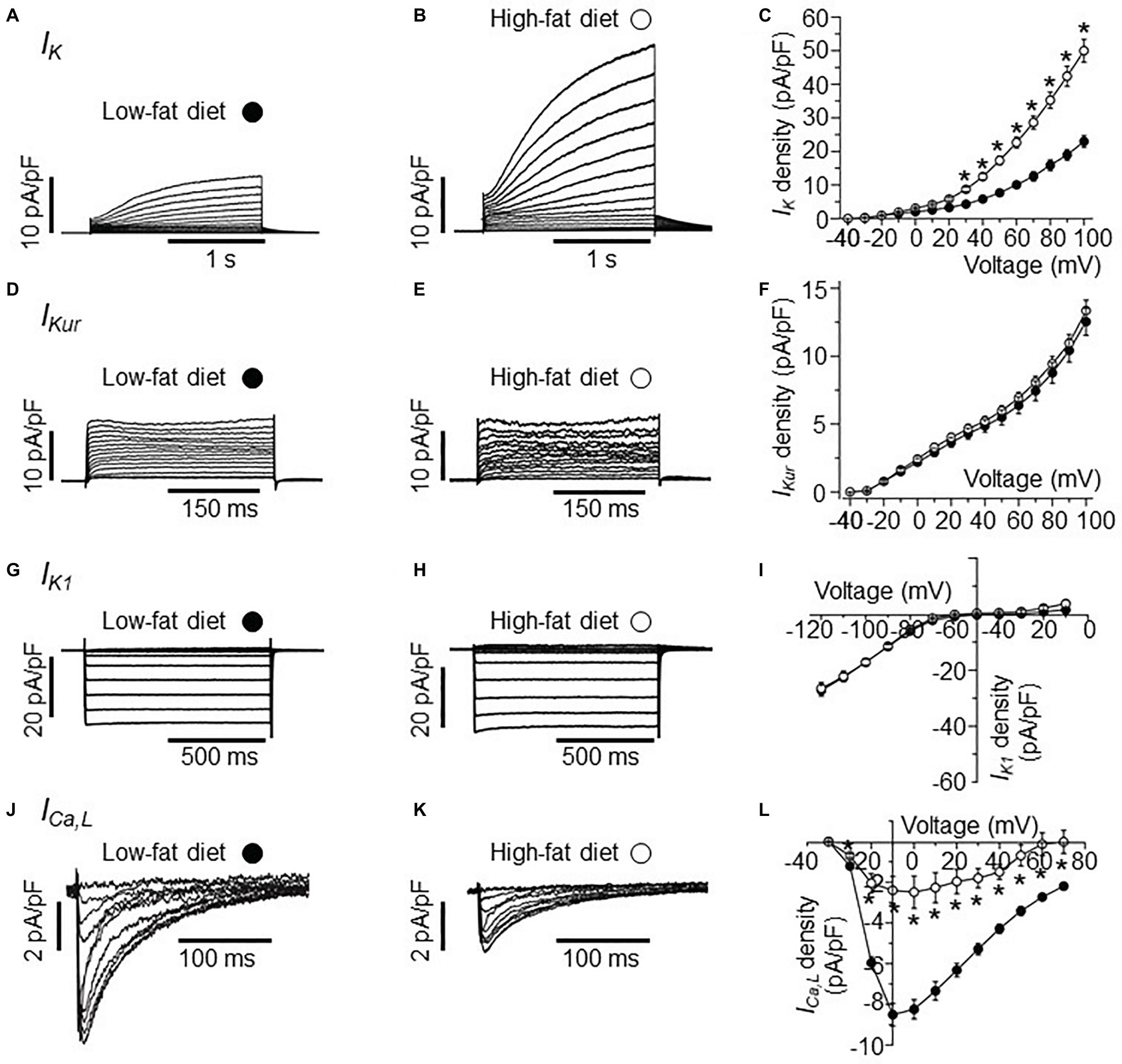
Figure 1. Electrophysiological properties of IK, IKur, IK1, and ICa,L currents in HFD atrial myocytes. (A) Representative IK traces from right atrial myocytes isolated from LFD-fed guinea pigs. (B) Exemplar IK currents measured in HFD atrial myocytes. (C) Population I-V curves for peak IK currents measured in LFD (●, n = 10) or HFD (○, n = 9) atrial myocytes. Exemplar currents and I-V relationships for indicated IKur (D–F), IK1 (G–I), and ICa,L (J–L), same format as (A–C). Data points are means ± S.E.M, n = 5–20. *p < 0.05. Data were generated from three different guinea pig cardiomyocyte preparations.
Data and Statistical Analyses
Electrophysiological data were analyzed off-line using built in functions in Fitmaster (HEKA), and Origin software. Currents are either expressed as current densities (pA/pF). Time constants for current activation, were obtained by fitting the data to a single exponential function. Time constants of rate of IK tail current deactivation, measured at −50 mV was fitted to a single exponential function of the form y = y0 + A1e(−x/t), where t is the time. Data are reported as means ± S.E.M. For all electrophysiology and biochemistry assays, statistical differences were obtained from one-way ANOVA with Bonferroni post hoc analysis or two-tailed unpaired t test for comparisons between groups and considered significant at p < 0.05.
Results
Effects of High-Fat Diet on IK and ICa,L in Guinea Pig Right Atrial Myocytes
In the initial set of experiments, we investigated the effects of HFD on IK current measured in atrial myocytes isolated from HFD-fed guinea pigs compared to LFD-fed controls using whole-cell patch clamp (Figure 1). Compared to IK measured in freshly isolated atrial myocytes from LFD-fed controls (Figure 1A), HFD feeding significantly increased the IK density (Figure 1B) at all potentials positive to +20 mV (Figure 1C). The data is in line with our previous finding that atrial IK density is significantly increased by HFD feeding in guinea pigs (Aromolaran et al., 2016). At +100 mV, IK peak density was increased by 115% (from 23.52 ± 1.32 pA/pF, n = 10, to 50.7 ± 3.45 pA/pF, n = 9, *p < 0.05, Figure 1C, see Table 2).

Table 2. Comparison of averaged ion current densities measured in atrial myocytes isolated from LFD- and HFD-fed guinea pigs after 50 days.
Next, we assessed whether currents generated by KCNA5 and KCNJ5 channel subunits are sensitive to HFD feeding. We measured IKur (KCNA5) and IK1 (KCNJ5) in atrial myocytes. Figure 1D shows IKur current measured in atrial myocytes isolated from LFD-fed guinea pig atria. As demonstrated in Figure 1E, IKur density measured in HFD atria myocytes was not different (p > 0.05) from control at all potentials tested (Figure 1F). At +100 mV, IKur density was 12.71 ± 0.74 pA/pF (n = 18, Figure 1F) and 13.36 ± 0.78 pA/pF (n = 9, Figure 1F, see Table 2) for LFD and HFD, respectively.
A similar picture emerged with IK1 (Figure 1G); at −120 mV IK1 density was −27.25 ± 1.67 pA/pF (n = 20) and −26.3 ± 2.09 pA/pF (n = 11, Table 2) in LFD and HFD atrial myocytes, respectively (Figure 1I). This finding suggests that HFD feeding does not indiscriminately affect all potassium currents.
We next determined whether ICa,L density is affected in response to HFD feeding. Compared to LFD-fed control atrial myocytes (Figure 1J), HFD-fed guinea pigs displayed a significantly blunted ICa,L density (Figure 1K). Atrial myocytes from HFD-fed guinea pigs displayed a significantly reduced ICa,L peak density (at +10 mV) compared with LFD-fed controls (reduced by 69.5% or from −7.88 ± 0.57 pA/pF in LFD (n = 16, Table 2) vs. −2.40 ± 0.68 pA/pF in HFD (n = 5); *p < 0.05, Figure 1L). The pooled current-voltage (I-V) relationship further revealed a depolarization shift (10 mV) in HFD atrial cells (Figure 1L) and supported previous reports (Gaspo et al., 1997; Mancarella et al., 2008; Nattel and Dobrev, 2017) that depression of ICa,L contributes to atrial cell dysfunction leading to vulnerability to AF.
Effects of High-Fat Diet on the Electrical Activity of Atrial Myocytes
To examine the electrical activity of HFD-fed atrial myocytes, we measured atrial action potential, because the dysfunction of IK (or IKr and IKs) (Caballero et al., 2010; Gonzalez de la Fuente et al., 2013; Aromolaran et al., 2014; Puckerin et al., 2016) and ICa,L contribute to arrhythmias (Van Wagoner et al., 1999; Mancarella et al., 2008; Barana et al., 2014), and might trigger arrhythmogenic events in HFD. We used current clamp electrophysiology to assess atrial electrical activity. Figure 2A shows action potential waveforms measured in freshly isolated atrial myocytes obtained from LFD-fed hearts. With HFD feeding (Figure 2B) and exogenous pretreatment (1 h) with palmitic acid (PA) (1 mM, Figure 2C), right atrial myocytes APD was abbreviated in line with our previous findings (Aromolaran et al., 2016). Furthermore, HFD atrial myocytes displayed irregular spontaneous beats with early-afterdepolarization (EAD)-like activity (Figure 2D). Similarly, PA-treated myocytes displayed delayed repolarization with irregular and spontaneous beats (Figure 2E).
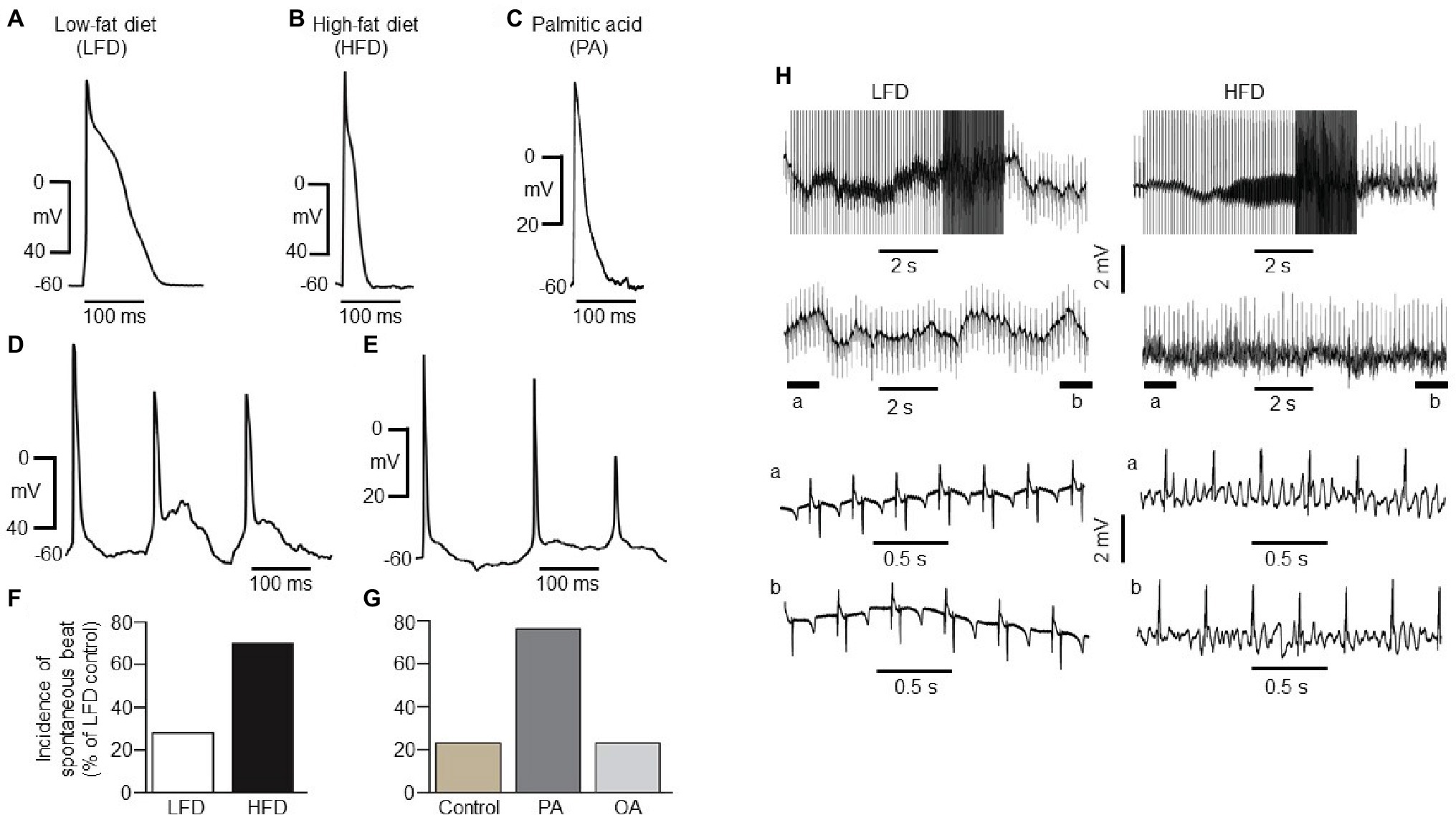
Figure 2. Electrical properties of guinea pig atrial myocytes and ex vivo hearts. (A) Examples of AP waveforms measured in right atrial myocytes isolated from LFD- (A) or HFD-fed (B) guinea pigs, or exposed to exogenous PA (1 mM, ≥1 h, C). HFD and PA pre-treated myocytes had a shortened action potential in line with the previous finding (Aromolaran et al., 2016). (D) Typical spontaneous beats with early delayed afterdepolarizations (EAD)-like activity measured in right atrial myocytes from HFD-fed guinea pigs. (E) Exemplar delayed repolarization with irregular and spontaneous beats in atrial myocytes pretreated with PA. Compared to control myocytes isolated from LFD-fed guinea pigs (F, white column), untreated (G, gold column) or pre-treated with OA (G, light gray column), the incidence of spontaneous beats is significantly greater in HFD (F, black column) and PA-treated myocytes (G, dark gray column). (H) Induction of arrhythmogenesis in right atria of isolated Langendorff perfused hearts from LFD- (left panel) and HFD-fed (right panel) guinea pigs. With rapid right atrial pacing (burst, rectangular pulses 10 ms, 1 V in amplitude with pulse-pulse duration ranging between 20 and 250 ms). Expanded view of atria electrical activities revealed sinus rhythm with LFD (left panel, bottom traces) and uncoordinated atrial activities in HFD-fed guinea pigs (right panel, bottom traces) consistent with AF. Data were generated from six different guinea pig cardiomyocyte preparations.
HFD atrial myocytes [70% or 21 out of 30 cells vs. 6 out of 23 cells (or 26%) for LFD controls, Figure 2F] or PA-treated myocytes [76% or 22 out of 29 cells vs. 3 out of 13 cells (or 26%) for untreated atrial myocytes, Figure 2G] revealed a higher incidence of spontaneous beats. By contrast, 3 out of 15 cells (or 20%) pre-exposed to oleic acid (OA, 1 mM) were arrhythmogenic (Figure 2G). Thus, our data in addition to the findings of Aromolaran and others (Aromolaran et al., 2016) confirm that HFD and PA are pro-arrhythmic while OA is anti-arrhythmic in guinea pigs.
To determine whether HFD hearts will be vulnerable to AF/tachycardia, burst stimulation and extrasystolic beats combined (Mancarella et al., 2008; Diness et al., 2010; Osadchii, 2012; Ashrafi et al., 2016) were delivered in the atria (Mancarella et al., 2008; Diness et al., 2010; Ashrafi et al., 2016) of 50-day-old isolated Langendorff-perfused hearts (Mancarella et al., 2008). We have defined induction of AF as rapid, irregular atrial activity lasting ≥2 s. Compared to LFD-fed hearts (three out of three, Figure 2H, left panel), AF/tachycardia was readily inducible in HFD-fed hearts (three out of three, Figure 2H, right panel), following application of burst stimulation protocol (10 ms and 1 V rectangular pulses, with pulse-pulse durations ranging between 20 and 250 ms). Together, our data demonstrate that increased IK and decreased ICa,L densities predispose to atrial arrhythmogenesis and AF/tachycardia in HFD-fed guinea pigs.
Patch-Clamp Recordings From Atrial Myocytes After Stimulation of the Atrium
Next, we tested the hypothesis that the combination of HFD and pacing might result in further remodeling of the expression of atrial ion channels. Therefore, we studied the electrophysiological properties of IK, IKur, IK1, and ICa,L in atrial myocytes isolated from LFD and HFD-fed guinea pigs after pacing. Whole-cell currents were measured in freshly isolated atrial myocytes within 2–6 h. after stimulations of the atrium (burst stimulation and extrasystolic beats combined). Figures 3A,B shows IK current traces recorded in LFD and HFD atrial myocytes that were subjected to pacing. I-V curves revealed that IK currents remained significantly larger (101.5% increase at +100 mV), when compared to LFD control IK currents (Figure 3C). At +100 mV, averaged IK densities were 23.52 ± 1.32 pA/pF (n = 10) and 47.4 ± 8.38 pA/pF for LFD and HFD (*p < 0.05, Table 3), respectively (Figure 3C). By contrast, we found that IKur and IK1 densities measured in HFD-fed atrial myocytes were altered with pacing. Figure 3D shows control IKur currents measured in LFD atrial myocytes. As illustrated in Figure 3E, IKur density is severely depressed in HFD myocytes subjected to pacing. At +100 mV, IKur density was reduced from 12.71 ± 0.74 pA/pF (n = 18) to 6.80 ± 0.72 pA/pF (n = 6, *p < 0.05) (or by ~49%, Figure 3F; Table 3).
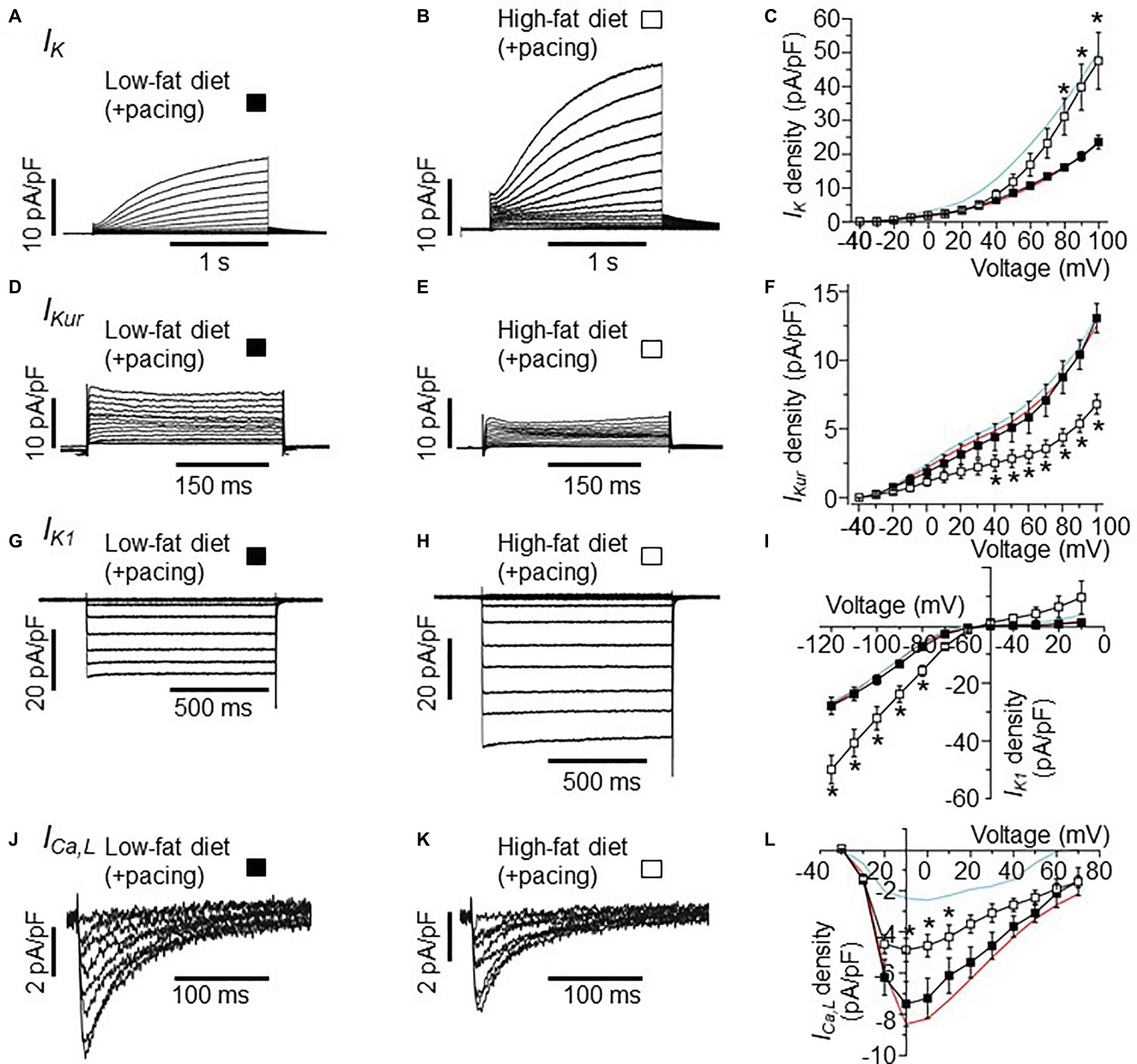
Figure 3. Effects of burst pacing on membrane currents in cardiomyocytes isolated from the atria of LFD and HFD guinea pigs. (A) Selected IK tracings of myocytes from LFD atria after burst pacing. (B) IK currents measured in HFD-fed guinea pigs after burst pacing. (C) Pooled I-V plots for IK density in LFD (■, n = 10) and HFD ( , n = 5) myocytes. Data for IK measured without atria pacing is reproduced for LFD (red trace) and HFD (cyan trace) from Figure 1C. (D–L) Exemplar currents and I-V curves for IKur, IK1, and ICa,L, same format as (A–C). IKur density is reduced (F) after pacing while IK1 density is increased (I). Each data point represents mean ± S.E.M, n = 5–20. *p < 0.05. Data were generated from six different guinea pig cardiomyocyte preparations.
, n = 5) myocytes. Data for IK measured without atria pacing is reproduced for LFD (red trace) and HFD (cyan trace) from Figure 1C. (D–L) Exemplar currents and I-V curves for IKur, IK1, and ICa,L, same format as (A–C). IKur density is reduced (F) after pacing while IK1 density is increased (I). Each data point represents mean ± S.E.M, n = 5–20. *p < 0.05. Data were generated from six different guinea pig cardiomyocyte preparations.

Table 3. Effects of atrial burst pacing on averaged ion current densities measured in myocytes isolated from non-obese and obese guinea pigs after 50 days.
Under similar conditions, IK1 density (Figure 3G) was increased from −27.25 ± 1.67 pA/pF (n = 20) to −49.72 ± 4.95 pA/pF (Figure 3H, or by 82.4%, *p < 0.05) at −120 mV with HFD and pacing (Figure 3I). With ICa,L, the depression in current seen in HFD was not as pronounced with pacing such that the ICa,L density measured at +10 mV in HFD (without pacing) was increased from −2.40 ± 0.68 pA/pF (n = 7) to −4.70 ± 0.68 pA/pF (n = 5, Figures 3J–L; Tables 2, 3) with pacing. Our data suggest that further remodeling of IKur and ICa,L seen with HFD and atrial pacing may be compensatory mechanisms to account for the profound changes in IK and IK1 densities.
Effects of High-Fat Diet and Pacing on the Biophysical Properties of IK
The ability of HFD to increase IK was of particular interest given that an increase in IKs is an important signature of AF (Caballero et al., 2010; Gonzalez de la Fuente et al., 2013; Perez-Hernandez et al., 2016). Therefore, we assessed the biophysical properties (Figure 4) of IK in LFD, HFD, and HFD (with pacing) atrial myocytes. The voltage-dependence of activation of IK, obtained by normalizing the activation curves to the peak current at +100 mV, was not altered in HFD alone myocytes when compared to LFD control myocytes but displayed a slight rightward shift in activation in HFD myocytes subjected to pacing (Figure 4A).
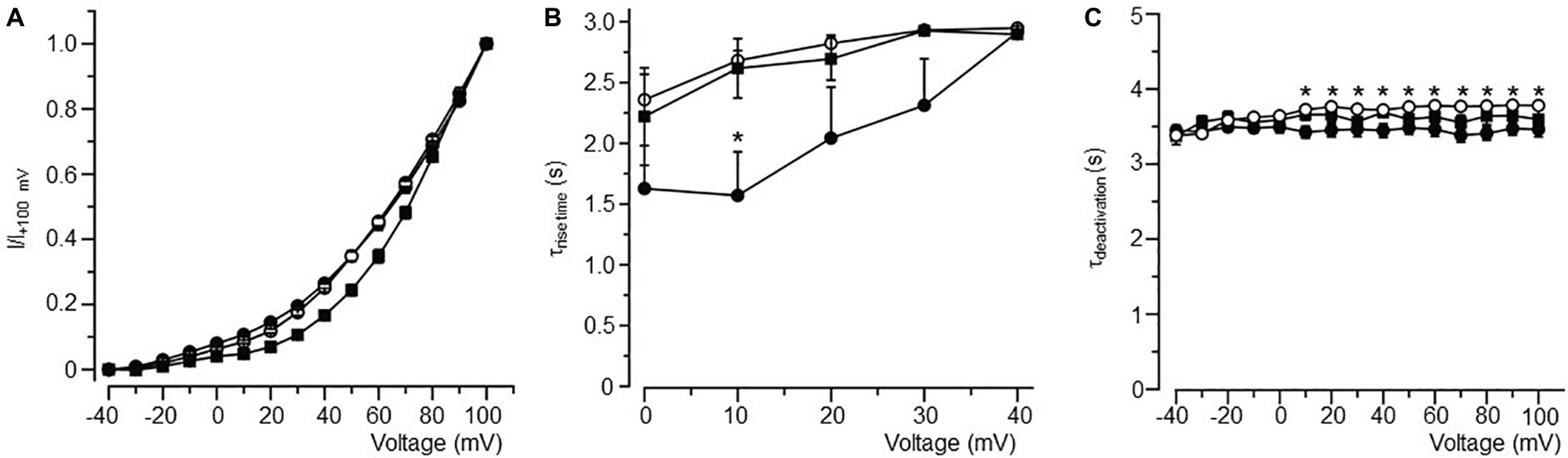
Figure 4. Biophysical properties of IK in atrial myocytes isolated from LFD- and HFD-fed (with and without burst pacing) guinea pigs. (A) Normalized activation curves for IK in LFD (●, n = 10), HFD (○, n = 9), and HFD with pacing (■, n = 5). (B) Relationship between time course (τrise time) of activation and voltage. (C) Plot of the IK tail current deactivation (τdeactivation) relative to voltage. Gating properties of IK are altered with HFD alone or in combination with pacing. *p < 0.05 relative to LFD control myocytes. Unpaired Student’s t-test followed by one-way ANOVA.
IK channels had a slower rise time of activation (τrise, Figure 4B) and deactivation (τdeactivation, Figure 4C) kinetics in HFD myocytes. Averaged τrise was 1.57 ± 0.36 s (n = 10) and 2.68 ± 0.08 s (n = 9, *p < 0.05, Table 4), while τdeactivation was 3.43 ± 0.08 s (n = 10) and 3.72 ± 0.03 s (n = 10, *p < 0.05, Table 4) at +10 mV for LFD and HFD myocytes, respectively. With pacing, τrise is unchanged while τdeactivation is slightly faster (HFD 3.72 ± 0.03 s vs. HFD with pacing 3.65 ± 0.07 s, Figure 4C; Table 4).
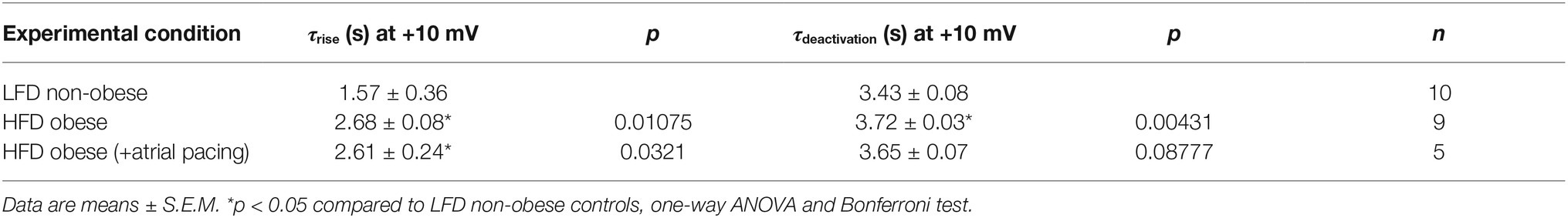
Table 4. Time constants for current activation (τrise) and deactivation (τdeactivation) for IK channels measured in atrial myocytes isolated from LFD non-obese and HFD obese guinea pigs (with and without pacing) after 50 days.
The increase in IK also suggests that HFD may modulate the surface expression of IK channel subunits (KCNQ1 and hERG). We used our bungarotoxin-binding assay (BBS) developed for KCNQ1 subunits (Aromolaran et al., 2014), to test the impact of FFAs elevated in obesity (Aromolaran et al., 2016) on IK channel subunits. First, we validated the feasibility of using this optical assay to assess hERG channel subunits. Figure 5A shows cartoon representation of BBS- and YFP-tagged hERG subunits. Cell surface hERG 1a channel subunits were selectively labeled by exposing BBS-hERG 1a-YFP expressing non-permeabilized HEK293 cells to biotinylated bungarotoxin (BTX-Biotin) followed by streptavidin-conjugated quantum dot-655 (QD655) (Aromolaran et al., 2014). Confocal images of cells expressing BBS-hERG 1a-YFP alone or in combination with hERG 1b-CFP displayed robust red QD655 fluorescence labeling of the cell periphery (Figure 5B).
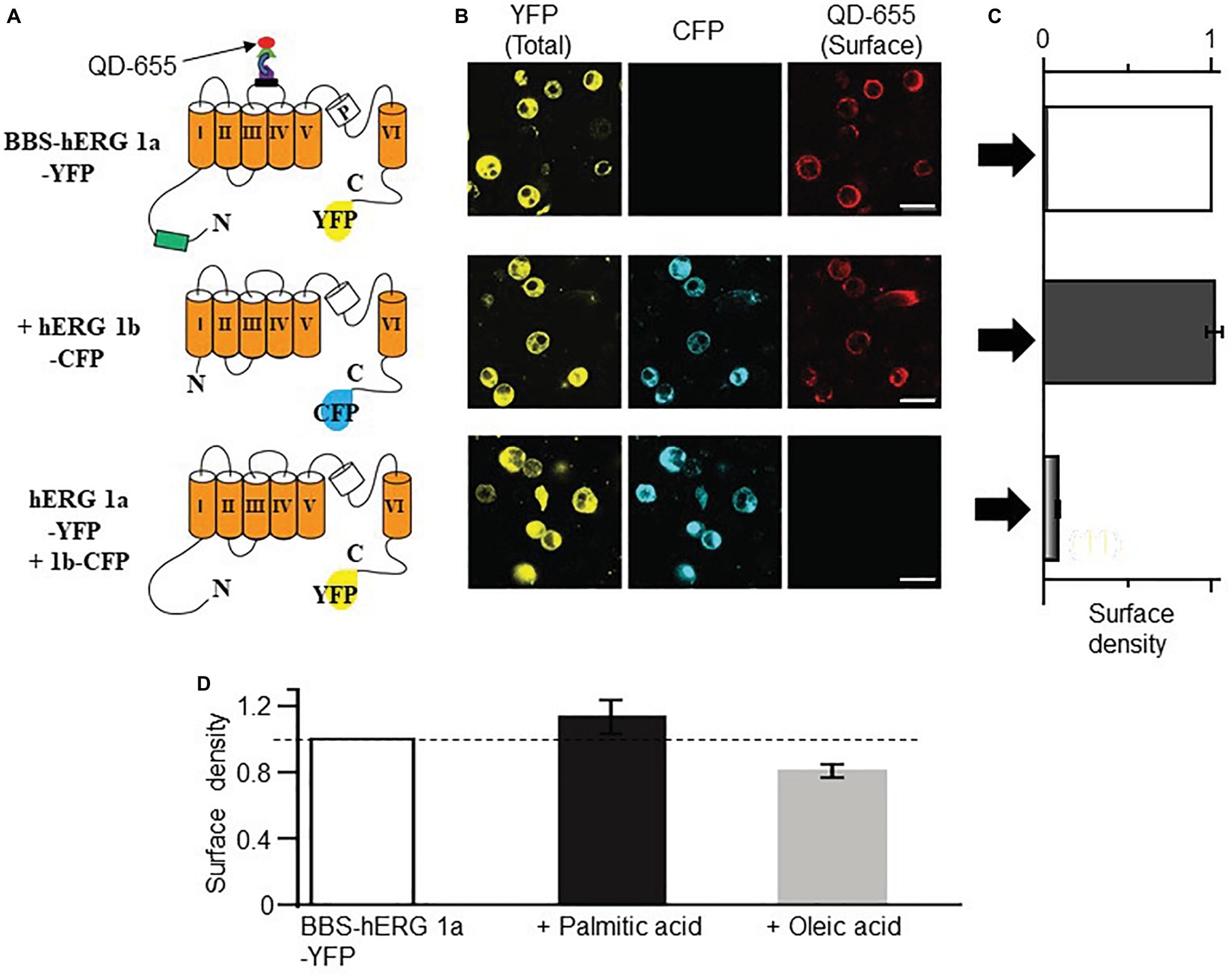
Figure 5. Optical detection of surface hERG subunits with quantum dot in HEK293 cells. (A) Schematic of BBS- and YFP-tagged hERG 1a, CFP-tagged hERG 1b and YFP-tagged hERG 1a lacking the BBS tag. (B) Confocal images of non-permeabilized HEK293 cells expressing BBS-hERG 1a-YFP or hERG 1a-YFP alone and in combination with hERG 1b-CFP, and exposed to QD655 to selectively label surface channels. Panels show hERG channels for YFP (total expression, left panel), CFP (middle panel), and QD655 (surface channels, right panel). Scale bars represent 20 μm. (C) Normalized mean QD655 signals from YFP-positive cells expressing BBS-hERG 1a-YFP alone (white column), BBS-hERG 1a-YFP/hERG 1b-CFP (black column), and hERG 1a-YFP/hERG 1b-CFP (gray column) represent relative surface density. hERG 1a-YFP/hERG 1b-CFP serves as negative control and shows negligible QD655 signal consistent with lack of BBS-tag and therefore negligible channel surface density. (D) Compared to untreated HEK293 cells transiently expressing BBS-HERG 1a-YFP, pretreatment (≥1 h) with PA (1 mM) increased the surface density of hERG channels (~13%), while OA reduced (~20%, light gray column). Dotted line indicates 1. Numbers in parenthesis indicates the number of independent cell culture experiments.
To quantify the QD655 signal, cells were subjected to flow cytometry. The mean QD655 fluorescence of YFP-positive cells was defined as channel surface density or extent of channel surface expression. Measured QD655 signals are normalized to YFP fluorescence (which corresponds to channel protein levels). Quantification of mean QD655 fluorescence indicated that BBS-hERG 1a-YFP alone or when combined with hERG 1b-CFP displayed similar surface density (Figure 5C, 2% increase or from surface density 1.0, n = 12 separate cultures, to 1.02 ± 0.05, 8 separate cultures), in line with published data, indicating hERG 1a traffics independently of hERG 1b to the cell surface in heterologous cells (Phartiyal et al., 2008). Negative controls expressing untagged hERG 1a and 1b displayed no QD655 fluorescence (averaged surface density was 0.18 ± 0.006, five separate cultures), confirming that the red fluorescence signal was specific and only detected the BBS tag, consistent with our previous reports on KCNQ1 trafficking in HEK293 cells and cardiomyocytes (Aromolaran et al., 2014; Kanner et al., 2018).
We used the QD655-labeling approach coupled with flow cytometry to assess the effects of PA (1 mM) and OA (1 mM) on hERG channel surface density as a potential mechanism for increased IK acquired in HFD. Figure 5D shows the effects of PA and OA on the surface density of hERG 1a channels. Relative to control HEK293 cells expressing BBS-hERG channel (from four separate cultures), quantification of mean QD655 fluorescence signal revealed that pretreatment with PA increased the surface density of hERG channels (~13 ± 1%, three separate cultures), whereas OA reduced (~20 ± 0.2%, four separate cultures) the surface density of hERG 1a channels, consistent with our previous report that PA increases hERG currents while OA reduced them in HEK293 cells (Aromolaran et al., 2016). Together, the data suggest that the increase in IK is most likely due to deficits in channel gating and increase in channel trafficking.
Effects of High-Fat Diet and Rapid Atrial Pacing on Human Atrial Electrophysiology Models
To determine whether the effects of HFD-feeding alone or in combination with rapid atrial pacing on whole cell ionic conductance would alter human atrial electrophysiology, we utilized the Courtemanche (Courtemanche et al., 1998) and Koivumäki (Koivumäki et al., 2011) human atrial cardiomyocyte models. Computer simulation (Table 1) results are summarized in Figure 6. As depicted, our simulated human atrial APs (Figure 6A) reveal a strong APD reduction in HFD compared to LFD myocytes, which agrees with our experimental results. APD at 90% repolarization (APD90) was reduced approximately by ~41, ~51, and ~68% in simulated human APs from Courtemanche and Koivumäki models and experimentally recorded guinea pig APs, respectively.
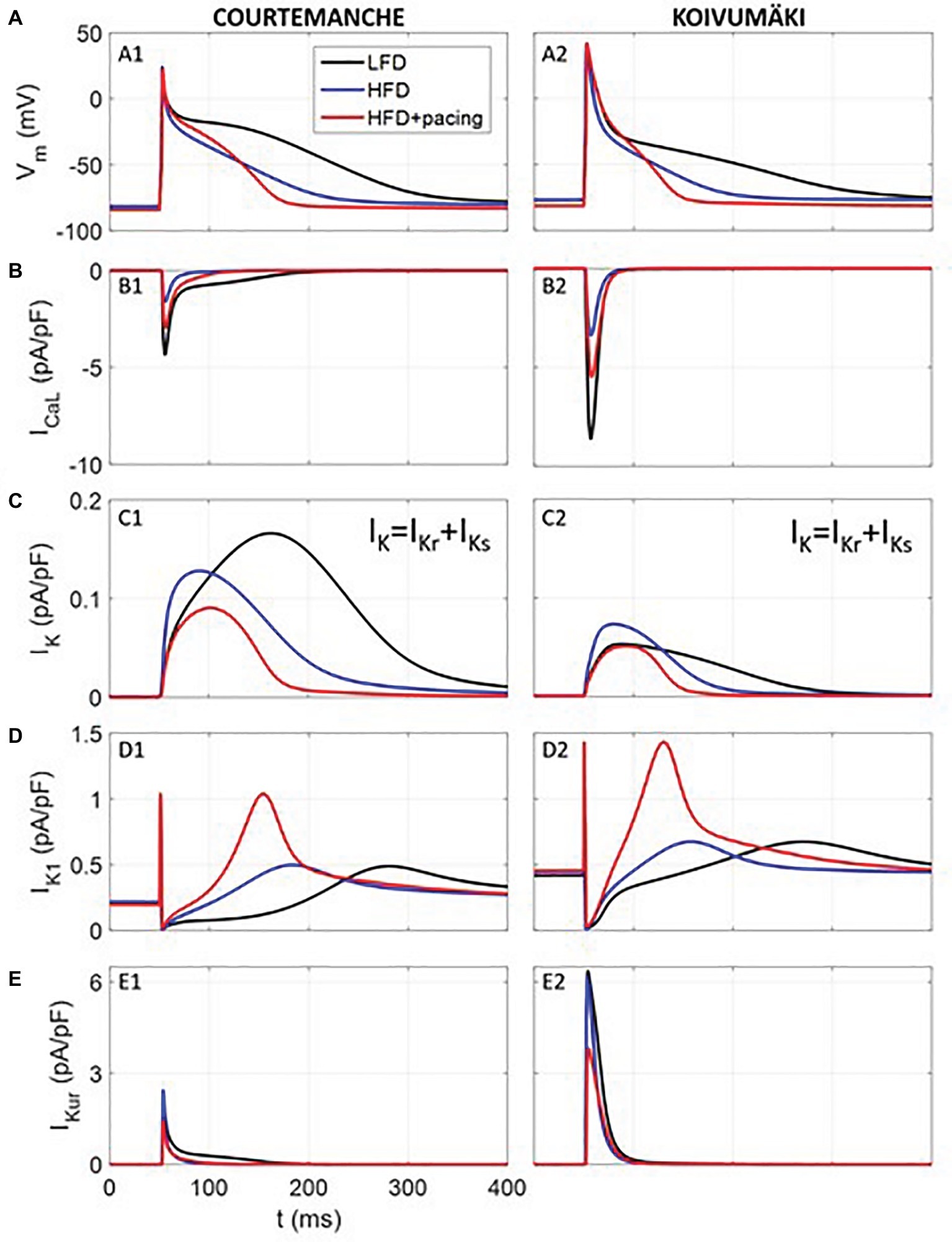
Figure 6. Simulated electrical activity of human atrial myocytes corresponding to LFD and HFD (with and without pacing) individuals. Action potential (A) and ICa,L (B), IK (C), IK1 (D), and IKur (E) current densities. Rapid atrial pacing caused more action potential shortening compared with HFD alone (blue trace), which was intermediate between LFD (black trace) and HFD + pacing (red trace).
As observed in Figure 6, where traces of current densities are shown in panels B-E (IK was composed of IKr and IKs), APD90 reduction in APs from HFD individuals was mainly due to ICa,L current reduction in the first 50 ms of the AP. In fact, when only considering the ICa,L downregulation, APD90 was reduced by 32 and 29% for Courtemanche and Koivumäki models, respectively, which means that the effect of the ICa,L decrease was stronger than the effect of the IK increase in the APD90 shortening. In addition, IKr had a stronger effect than IKs in the APD90 reduction since when considering the ICa,L downregulation together with the IKr upregulation, APD90 shortened by 39% (Courtemanche) and 48% (Koivumäki). In our paced HFD myocytes simulations, APD90 decreased by 55% (Courtemanche) and 62% (Koivumäki) for paced HFD myocytes compared to LFD (by 24% (Courtemanche) and 21% (Koivumäki) if compared to HFD without pacing). As observed in Figure 6, APD90 reduction was due to the downregulated ICa,L and upregulated IK1 currents (when only considering those two currents variations, the APD90 shortened by 60 and 80% for the Courtemanche and Koivumäki models, respectively), while downregulation of IKur current prolonged the APD90. The effect of the IKur downregulation on the APD90 prolongation seems to be more intense for the Koivumäki model. In addition, the maximum AP amplitude of the simulated APs with the Courtemanche and Koivumäki models did not vary significantly in HFD compared to LFD myocytes (5% of increase for experiments), while it did increase by 3% after pacing.
Discussion
In this study, we show that there is a surprising heterogeneity of distinct atrial potassium currents that are sensitive to HFD and underlie the increased susceptibility of obese heart to atrial arrhythmogenic events. Our data further demonstrate that HFD discriminately increases IK density through posttranslational (protein trafficking and channel gating defects) modulation of channel subunits. Only when combined with burst atrial pacing did the altered functional expression of IKur and IK1 phenotypes emerge, manifested as severely reduced IKur density, and a marked increase in IK1 current density. To our knowledge, this is the first study showing that the electrical abnormalities in HFD-fed guinea pig atria included a decrease in ICa,L density. Therefore, we conclude that both increased IK and reduced ICa,L functional expressions are key events that underlie the initial HFD-mediated atrial electrical remodeling and arrhythmogenesis with implications for atrial fibrillation.
Comparison to Previous Studies on High-Fat Diet, K Channels, and Cardiac Electrical Remodeling
In heart, increased IK density (Caballero et al., 2010; Gonzalez de la Fuente et al., 2013; Aromolaran et al., 2016) is an important signature of atrial electrical remodeling and is therefore critically associated with abbreviation of APD and supraventricular arrhythmias (Nattel and Dobrev, 2017). Understanding how HFD affects the functional properties of IK is likely to inform our knowledge regarding vulnerability to atrial arrhythmogenesis in obese patients.
With the exception of our previous report (Aromolaran et al., 2016) on the effect of HFD-induced obesity on IK functional properties, we are not aware of other studies that have assessed modulation by HFD of IK function. Our current findings confirm our previous observations (Aromolaran et al., 2016) and also show that in atrial myocytes from HFD-fed guinea pigs, IK density was approximately two-fold higher, the rise time of activation and deactivation kinetics were slower, and the conductance curve was essentially unchanged when compared with LFD-fed control myocytes.
To further elucidate the molecular mechanisms underlying HFD-mediated effects on IK, we assessed the effects of saturated PA and the monounsaturated OA on the trafficking properties of hERG channel subunits. Previously, we reported that PA likely mediates the effects of obesity on IK in atrial myocytes. In the present study, we discovered that PA increased the surface expression of hERG channel subunits consistent with the notion that the facilitatory effect of HFD on IK involves multiple signaling pathways. These findings may inform experimental strategies for predicting the effects of HFD mechanisms on atrial electrical and cardiac function. Together, our findings provide the new insight that the facilitatory effect of HFD on IK can be attributed to channel gating and trafficking defects.
There have been conflicting data regarding modulation by HFD of atrial specific IKur (or Kv1.5 encoded by KCNA5) (Bhuyan and Seal, 2017) functional expression. Yifan and colleagues (Zhang et al., 2016), demonstrated that the protein expression levels of Kv1.5 was significantly increased in atria of obese mice after 8 weeks. By contrast, Morrow and others (Huang et al., 2013) reported reduced mRNA and protein levels of ventricular Kv1.5 channel subunits (Huang et al., 2013) in HFD-induced obese mice after 20 weeks. The changes reported by Morrow’s group were also associated with ventricular electrical remodeling (including impaired repolarization and QT prolongation) (Huang et al., 2013). Whether changes in Kv1.5 expression translates to altered IKur current density is unknown since electrophysiology measurements were not performed in these studies. Therefore, a definitive role for altered IKur density in HFD-induced cardiac electrical dysfunction requires further investigation.
The differences between our study and these studies may be due to species differences (guinea pig vs. mice) (Zhang et al., 2016), differences in cell-type (atria vs. ventricles) (Huang et al., 2013) or duration of HFD or HFD-induced obesity (7 vs. 20 weeks). Future studies will be needed to further understand the effects of HFD on the spatial selectivity and temporal properties of Kv1.5 transcript and protein levels.
There is currently a lack of clarity about the effects of HFD on IK1 functional expression and atrial arrhythmogenesis. In a HFD obese rat model, Boyet and others (Ashrafi et al., 2016) demonstrated that mRNA levels of Kir2.1 (KCNJ2) and IK1 current density are significantly increased after 8 weeks. However, these changes had negligible effects on ventricular APD (Ashrafi et al., 2016). Similar to IKur, IK1 density is also unchanged in our studies, demonstrating that IKur and IK1 channels are insensitive to HFD in guinea pig atrial myocytes.
With the sole exception of IK, our data revealed that IKur and IK1 current densities are not affected by HFD. This finding is in contrast to the results of Boutjdir and others (Mancarella et al., 2008), which shows that IK density is not altered in an α-1D KO mice model of AF. A similar reduction of ICa,L density with no compensatory changes from other currents has also been described in sinoatrial node cells of the same α-1D calcium channel KO mice (Mangoni et al., 2003). One can speculate that the molecular mechanisms that underlie altered function of distinct atrial ion channels leading to supraventricular arrhythmias may differ depending on the underlying pathology.
Effects of Rapid Pacing on the Atrial Electrophysiology of High-Fat Diet-Fed Guinea Pig
Previous reports have shown that rapid pacing (acute vs. chronic) cause atrial electrophysiological remodeling in humans (Goette et al., 1996; Daoud et al., 1997; Yue et al., 1999; Hadian et al., 2002), and in different animal models of AF (Yu et al., 1998; Tsuchiya et al., 2009; Laszlo et al., 2011). Our data show that acute or short-term rapid pacing increases vulnerability of HFD-fed guinea pigs to atrial arrhythmogenesis compared to LFD-fed controls. Our results also showed that, while the functional expression of IK (increased) and ICa,L (decreased) were essentially unchanged after rapid pacing; IKur density was severely depressed and IK1 current density was increased, in agreement the ability of a brief (10–15 min) episode of rapid atrial pacing to induce cardiac electrical remodeling (Figure 7; Yu et al., 1998; Hadian et al., 2002).
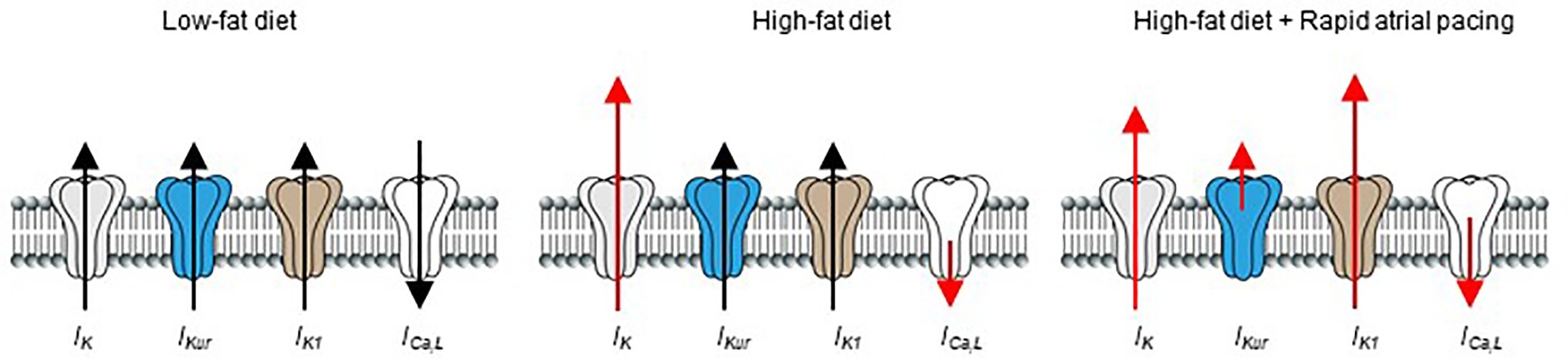
Figure 7. Schematic representation of the effects of HFD and rapid pacing on atrial electrical properties in guinea pig heart. Short-term high-fat diet induced significant electrical remodeling of IK and ICa,L while IKur and IK1 remained unchanged compared to LFD controls. When HFD is combined with pacing IKur is significantly reduced while IK1 is increased in line with their roles in pathogenesis of supraventricular arrhythmias such as atrial fibrillation. The size of red arrows indicates change in the current densities of the ion channels.
Regarding pacing-induced downregulation of IKur and upregulation of IK1 densities, findings have been contradictory. In agreement with our findings, IKur amplitude is significantly reduced in human atrial myocytes at rapid activation rates (Feng et al., 1998; Ehrlich et al., 2008). By contrast, Nattel and colleagues (Yue et al., 1999) did not find any significant changes in the densities of IKur and IK1 in their study. IK1 is also not affected in a rabbit model of rapid atrial pacing (RAP) (Laszlo et al., 2007). In our studies, we utilized 300 beats/min, compared to 400 beats/min and 600 beats/min used in these studies of Nattel and colleagues (Yue et al., 1999) and Bosch’s group (Laszlo et al., 2007). The lower rate in our model combined with a HFD genetic background might cause faster changes in current densities, consistent with the notion that the metabolic state of atria may be an important determinant of the temporal properties and/or sensitivity to pacing.
Alternatively, the different findings may also be due to interspecies differences in the time course of electrical remodeling as previously demonstrated in goat and dog RAP models of AF (Yue et al., 1997, 1999). It was demonstrated that atrial electrical remodeling occurred faster in goat when compared to dog RAP model. Electrical remodeling in a horse model has also been shown to be slower than that in goat or dog (Ausma et al., 2002).
It is reasonable to speculate that the depression of IKur density due to rapid pacing would be expected to prolong APD, and therefore may represent a compensatory mechanism to limit further electrical remodeling and prevent onset of atrial arrhythmogenic events. Our data further suggest that increased IK1 density would be expected to play a critical role in promoting electrical remodeling and atrial arrhythmogenesis leading to the maintenance of supraventricular arrhythmias.
Concerning the extrapolation of our results to humans through computational modeling and simulations, our findings show that remodeling due to obesity might also have proarrhythmic effects. The combination of increased IK and reduced ICa,L in HFD myocytes accelerates repolarization and leads to shorter APDs and shorter refractory period that favor arrhythmogenesis (ectopic and/or reentrant activity). Furthermore, our data suggest that AF mechanisms including the strong APD90 reduction coupled with EAD-like activity and delayed repolarization with irregular and spontaneous beats would explain the higher predisposition of obese individuals to develop atrial arrhythmias (Wanahita et al., 2008). Simulations also suggest that rapid pacing increases vulnerability of obese individuals to arrhythmias, possibly by exacerbating AF mechanisms. This suggestion is in accordance with our experimental results showing that APD90 is shorter with pacing compared to without pacing.
Altogether, we can surmise that in our case, the current variations in humans were similar to those observed in the guinea pig experiments; therefore, the effect of obesity in humans would have a similar behavior to our guinea pig model, as demonstrated by our computer simulations. However, further experiments with human atrial myocytes are needed to validate if variations in the ionic currents are comparable to the ones observed in guinea pig atrial cells.
Study Limitations
This study focused entirely on understanding the effects of HFD feeding on ion channel mechanisms that either initiate or maintain supraventricular arrhythmias. A major limitation of the study is that we did not model the effects of PA and OA on atrial APD, or susceptibility of HFD myocytes to develop EADs, DADs, and spontaneous depolarizations. Our current data, in addition to our previous reports (Aromolaran et al., 2016), strongly suggest that PA will mimic the effects of HFD, while OA will be expected to prevent arrhythmogenic AF mechanisms.
Although our results revealed a significant downregulation of global atrial ICa,L in HFD-fed guinea pigs, it does not differentiate between a role for α-1C and α-1D channels both of which contribute prominently to total ICa,L (Barana et al., 2014). The effects of HFD on the functional expression of ICa,L have been investigated in rat models but with contrasting outcomes. For example, in rats fed a high-fat diet for 15 weeks, it was found that the gene and protein expressions of α-1C calcium channel were unchanged (Lima-Leopoldo et al., 2008; Leopoldo et al., 2011). However, in an unrelated study by the same authors, mRNA expression of α-1C calcium channel is increased at 30 weeks (Lima-Leopoldo et al., 2013). Boyett and others (Ashrafi et al., 2016) also reported increased mRNA levels of ventricular ICa,L after 8 weeks in high fat diet fed rats; while Leopoldo and others (Leopoldo et al., 2011) found no change in protein expression after 15 weeks. In a diet-induced obese Psammomys obesus Gerbil model, mRNA and protein expression levels of the α-1C calcium channel were decreased after 16 weeks (Sahraoui et al., 2016). Furthermore, Gaborit and others (Gaborit et al., 2005) have also shown a significant reduction in the gene expression of α-1C/α-1D in AF patients suggesting a role of both channels in the pathogenesis of AF. Whether or how the functional expression of α-1C and α-1D calcium channels is altered in obese or lipotoxic atria and dyslipidemia is unknown. A previous report by the Boutjdir group (Mancarella et al., 2008) showed that while α-1C mRNA levels were unchanged in α-1D KO mice of AF, ICa,L current density is significantly reduced supporting a role for the α-1D calcium channel in AF. It is intriguing to speculate that similar molecular mechanisms of differential α-1C and α-1D calcium channel expression may underlie the severe reduction of ICa,L and propensity for atrial arrhythmogenesis observed in our model.
Other mechanisms such as a reduced intracellular calcium transient amplitude (Mancarella et al., 2008) may have also contributed to the adverse changes in ICa,L. Therefore, studies that distinguish between the functional expression of Cav1.2/Cav1.3, and altered calcium handling proteins (Aromolaran and Boutjdir, 2017), with implications for reduced ICa,L are likely to provide critical insights for devising targeted therapies in obese patients.
Another limitation of this study is that we did not assess the effects of HFD feeding on the separate components (IKr and IKs) of IK. Differential modulation of IKr and IKs in response to drugs and neurotransmitters has been proposed. For example, Sanguinetti and Jurkiewicz (1991) demonstrated selective inhibition of IKr by the class III antiarrhythmic drug E-4031, while the β-adrenergic agonist isoproterenol facilitated IKs with significant effects on IKr measured in guinea pig ventricular myocytes (Sanguinetti and Jurkiewicz, 1991; Sanguinetti et al., 1991). Furthermore, Matsuura and Ehara (1997) also provided evidence that extracellular ATP discriminately potentiated IKs current density. Therefore, in future studies, it will be important to investigate the selective role of IKr and IKs in obesity-related adverse atrial electrical remodeling that promote reentry mechanisms, incidence of triggered activity, and EADs. Our studies can also be further improved by incorporating comprehensive and robust biophysical analyses (gating mechanisms including activation, inactivation, and deactivation) of remodeled currents (IKr, IKs, IKur, IK1, and ICa,L) investigated in our study and how the pattern of our results may be related to changes in resting membrane potential and contour of the final repolarization phase of the atrial action potential. Work to advance these approaches are currently been conducted in our laboratory.
Although our findings show defects in trafficking and gating mechanisms of IK may underlie adverse remodeling by obesity they do not exclude other mechanisms including increased sympathetic regulation of IK and abnormal transcriptional and posttranslational modifications. It will also be interesting in future studies to examine the contribution of these mechanisms.
Furthermore, the electrophysiological and pharmacological characteristics of guinea pig and human atrial myocytes might substantially differ and therefore, it is important to be cautious when extrapolating results from animal experiments to humans, due to the differences among species (O’Hara and Rudy, 2012). On the other hand, it is well known that the delayed rectifier current in human myocytes, as in other species, is composed of IKr and IKs (Wang et al., 1994). Atrial models available in the literature incorporate the two components and the vast majority present larger IKr than IKs currents. In fact, maximum IKs amplitude is the smallest among all the ionic currents except for the Nygren model (Wilhelms et al., 2012). And even though the maximum IKs current amplitude in the Courtemanche model is larger than in the Koivumäki model, upregulation of this current in the HFD yielded a similar effect in both models. Our data further show that APD90 shortening was mainly due to the contribution of the downregulated ICa,L and upregulated IKr, while IKs slightly affected the APD90. Our findings also suggest that evaluating the obesity-related adverse atrial electrical remodeling observed in the present study at physiological temperatures (37°C versus 20–25°C) may be better predictive of arrhythmogenesis in patients.
Conclusion
Taken altogether, our study is the first to show that HFD negatively modulates IK and ICa,L. Our findings are consistent with the notion that individuals with metabolic disorders may display increased IK and reduced ICa,L with serious implications for cardiac repolarization. Our results predict that: (1) inhibitors of hERG/KCNQ1 channels (Nakaya et al., 2000); (2) activators of Ca channel; or (3) cellular mediators that either limit IKr/IKs channel opening or promote ICa,L opening would be expected to limit abbreviation of atrial APD and therefore vulnerability to supraventricular arrhythmias in patients with metabolic disorders.
By describing the first measurements of the effect of HFD on IK/ICa,L channel function, we reveal that IK/ICa,L channels are sensitive to pathological changes in metabolic substrates and may be involved in the initiation of supraventricular arrhythmias, since both increased IK density and downregulation of ICa,L makes HFD-fed guinea pigs prone to supraventricular arrhythmias. In contrast to observations of increased IK or reduced ICa,L current densities due to remodeling after atrial arrhythmias such as AF has occurred, our data are consistent with the premise that an a priori increase in IK (IKs) (Caballero et al., 2010; Gonzalez de la Fuente et al., 2013) and reduction in ICa,L (Brundel et al., 2001; Barana et al., 2014; Nattel and Harada, 2014) current density may play a key role in adverse atrial electrical mechanisms that underlie initiation of supraventricular arrhythmias underlain by metabolic disorders.
Furthermore, dietary interventions involving: (1) removal of dietary PA; (2) increasing dietary OA; (3) increased exercise training; and (4) multiple combinations of the different interventions may reduce arrhythmias due to obesity. In this context, a previous report by Paulino and others (Paulino et al., 2010) demonstrated that exercise and caloric restriction prevented cardiac defects in obese rats through modulation of calcium handling proteins. Recently, Wang and Fitts (2017, 2018) also demonstrated that upregulation of ventricular KATP channel accelerates cardiac repolarization and shortens APD during stress and exercise in rats. These findings and our data suggest that an exercise protocol that prevents excessive weight gain and therefore pathological accumulation of saturated FFAs will be expected to prevent adverse potassium and calcium channel remodeling in our HFD-induced obese guinea pigs. We are currently investigating modulation by exercise of cardiac potassium channels with implications for supraventricular arrhythmias that are acquired in metabolic disorders.
Collectively, our findings revealed new directions into the dynamic functional interplay between the biophysical properties of potassium and calcium channels in supraventricular tissues, which is likely to inform development of targeted interventions toward the management of atrial arrhythmias including AF. Our data suggest that we may be able to better prevent the initiation of supraventricular arrhythmias in metabolic disorders with more effective ion channel drugs, especially with those that are targeted to IK and ICa,L.
Data Availability Statement
All datasets generated for this study are included in the manuscript/supplementary files.
Ethics Statement
The animal study was reviewed and approved by IACUC, VA Healthcare System, Brooklyn, New York and Columbia University IACUC.
Author Contributions
LM-M designed experiments and finalized manuscript. JS conceived and designed experiments, obtained funding, and finalized manuscript. AA conceived and designed experiments, analyzed results, obtained funding, and wrote the paper.
Funding
This study was supported by an AHA (13SDG16850065 to AA), NIH (R01 HL147044 to AA), and Programa Prometeu de la Conselleria d’Educació, Formació I Ocupació de la Generalitat Valenciana, award number PROMETEU/2016/088.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Kelly Ann Aromolaran (Weill Cornell Medical Center) for her critical review of the manuscript.
Abbreviations
AF, Atrial fibrillation; APD, Action potential duration; FA, Fatty acid; hERG, Human éther-a-go-go-related gene; HFD, High-fat diet; ICa,L, L-type calcium current; IK, Delayed rectifier potassium current; IK1, Inwardly rectifying potassium current; IKr, Rapidly activating delayed rectifier potassium current; IKs, Slowly activating delayed rectifier potassium current; IKur, Ultra-rapid delayed rectifier potassium current; INa, Large inward sodium current; Ito, Fast transient outward potassium current; LFD, Low-fat diet; OA, Oleic acid; PA, Palmitic acid.
References
Abed, H. S., and Wittert, G. A. (2013). Obesity and atrial fibrillation. Obes. Rev. 14, 929–938. doi: 10.1111/obr.12056
Angelin, B., Olivecrona, H., Reihner, E., Rudling, M., Stahlberg, D., Eriksson, M., et al. (1992). Hepatic cholesterol metabolism in estrogen-treated men. Gastroenterology 103, 1657–1663. doi: 10.1016/0016-5085(92)91192-7
Aoki, Y., Hatakeyama, N., Yamamoto, S., Kinoshita, H., Matsuda, N., Hattori, Y., et al. (2012). Role of ion channels in sepsis-induced atrial tachyarrhythmias in guinea pigs. Br. J. Pharmacol. 166, 390–400. doi: 10.1111/j.1476-5381.2011.01769.x
Aromolaran, A. S., and Boutjdir, M. (2017). Cardiac ion channel regulation in obesity and the metabolic syndrome: relevance to long QT syndrome and atrial fibrillation. Front. Physiol. 8, 1–7. doi: 10.3389/fphys.2017.00431
Aromolaran, A. S., Colecraft, H. M., and Boutjdir, M. (2016). High-fat diet-dependent modulation of the delayed rectifier K(+) current in adult guinea pig atrial myocytes. Biochem. Biophys. Res. Commun. 474, 554–559. doi: 10.1016/j.bbrc.2016.04.113
Aromolaran, A. S., Subramanyam, P., Chang, D. D., Kobertz, W. R., and Colecraft, H. M. (2014). LQT1 mutations in KCNQ1 C-terminus assembly domain suppress IKs using different mechanisms. Cardiovasc. Res. 104, 501–511. doi: 10.1093/cvr/cvu231
Ashrafi, R., Yon, M., Pickavance, L., Yanni Gerges, J., Davis, G., Wilding, J., et al. (2016). Altered left ventricular ion channel transcriptome in a high-fat-fed rat model of obesity: insight into obesity-induced arrhythmogenesis. J. Obes. 2016:7127898. doi: 10.1155/2016/7127898
Ausma, J., van-der-Vusse, G., and Allessie, M. (2002). Atrial fibrillation: does ischemia play a role. Einthoven 100, 375–383.
Bai, J., Gladding, P. A., Stiles, M. K., Fedorov, V. V., and Zhao, J. (2018). Ionic and cellular mechanisms underlying TBX5/PITX2 insufficiency-induced atrial fibrillation: insights from mathematical models of human atrial cells. Sci. Rep. 8:15642. doi: 10.1038/s41598-018-33958-y
Barana, A., Matamoros, M., Dolz-Gaiton, P., Perez-Hernandez, M., Amoros, I., Nunez, M., et al. (2014). Chronic atrial fibrillation increases microRNA-21 in human atrial myocytes decreasing L-type calcium current. Circ. Arrhythm. Electrophysiol. 7, 861–868. doi: 10.1161/CIRCEP.114.001709
Baro, I., and Escande, D. (1989a). A Ca2+-activated K+ current in guinea-pig atrial myocytes. Pflugers Arch. 414(Suppl. 1):S168.
Baro, I., and Escande, D. (1989b). A long lasting Ca2+-activated outward current in guinea-pig atrial myocytes. Pflugers Arch. 415, 63–71.
Bhuyan, R., and Seal, A. (2017). Dynamics and modulation studies of human voltage gated Kv1.5 channel. J. Biomol. Struct. Dyn. 35, 380–398. doi: 10.1080/07391102.2016.1144528
Boden, G., She, P., Mozzoli, M., Cheung, P., Gumireddy, K., Reddy, P., et al. (2005). Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-kappaB pathway in rat liver. Diabetes 54, 3458–3465. doi: 10.2337/diabetes.54.12.3458
Bosch, R. F., Schneck, A. C., Csillag, S., Eigenberger, B., Gerlach, U., Brendel, J., et al. (2003). Effects of the chromanol HMR 1556 on potassium currents in atrial myocytes. Naunyn Schmiedeberg’s Arch. Pharmacol. 367, 281–288. doi: 10.1007/s00210-002-0672-5
Boutjdir, M., Le Heuzey, J. Y., Lavergne, T., Chauvaud, S., Guize, L., Carpentier, A., et al. (1986). Inhomogeneity of cellular refractoriness in human atrium: factor of arrhythmia? Pacing Clin. Electrophysiol. 9, 1095–1100.
Brundel, B. J., Van Gelder, I. C., Henning, R. H., Tieleman, R. G., Tuinenburg, A. E., Wietses, M., et al. (2001). Ion channel remodeling is related to intraoperative atrial effective refractory periods in patients with paroxysmal and persistent atrial fibrillation. Circulation 103, 684–690. doi: 10.1161/01.CIR.103.5.684
Bunemann, M., Liliom, K., Brandts, B. K., Pott, L., Tseng, J. L., Desiderio, D. M., et al. (1996). A novel membrane receptor with high affinity for lysosphingomyelin and sphingosine 1-phosphate in atrial myocytes. EMBO J. 15, 5527–5534. doi: 10.1002/j.1460-2075.1996.tb00937.x
Caballero, R., de la Fuente, M. G., Gomez, R., Barana, A., Amoros, I., Dolz-Gaiton, P., et al. (2010). In humans, chronic atrial fibrillation decreases the transient outward current and ultrarapid component of the delayed rectifier current differentially on each atria and increases the slow component of the delayed rectifier current in both. J. Am. Coll. Cardiol. 55, 2346–2354. doi: 10.1016/j.jacc.2010.02.028
Caillier, B., Pilote, S., Patoine, D., Levac, X., Couture, C., Daleau, P., et al. (2012). Metabolic syndrome potentiates the cardiac action potential-prolonging action of drugs: a possible ‘anti-proarrhythmic’ role for amlodipine. Pharmacol. Res. 65, 320–327. doi: 10.1016/j.phrs.2011.11.015
Chiu, H. C., Kovacs, A., Ford, D. A., Hsu, F. F., Garcia, R., Herrero, P., et al. (2001). A novel mouse model of lipotoxic cardiomyopathy. J. Clin. Invest. 107, 813–822. doi: 10.1172/JCI10947
Christ, T., Boknik, P., Wohrl, S., Wettwer, E., Graf, E. M., Bosch, R. F., et al. (2004). L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation 110, 2651–2657. doi: 10.1161/01.CIR.0000145659.80212.6A
Courtemanche, M., Ramirez, R. J., and Nattel, S. (1998). Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. Am. J. Phys. 275, H301–H321. doi: 10.1152/ajpheart.1998.275.1.H301
Czick, M. E., Shapter, C. L., and Silverman, D. I. (2016). Atrial fibrillation: the science behind its Defiance. Aging Dis. 7, 635–656. doi: 10.14336/ad.2016.0211
Dan, G. A., and Dobrev, D. (2018). Antiarrhythmic drugs for atrial fibrillation: imminent impulses are emerging. Int. J. Cardiol. Heart Vasc. 21, 11–15. doi: 10.1016/j.ijcha.2018.08.005
Daoud, E. G., Knight, B. P., Weiss, R., Bahu, M., Paladino, W., Goyal, R., et al. (1997). Effect of verapamil and procainamide on atrial fibrillation-induced electrical remodeling in humans. Circulation 96, 1542–1550. doi: 10.1161/01.CIR.96.5.1542
De Sensi, F., Costantino, S., Limbruno, U., and Paneni, F. (2018). Atrial fibrillation in the cardiometabolic patient. Minerva Med. 157–167. doi: 10.23736/s0026-4806.18.05882-2
Dey, S., DeMazumder, D., Sidor, A., Foster, D. B., and O’Rourke, B. (2018). Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circ. Res. 123, 356–371. doi: 10.1161/CIRCRESAHA.118.312708
Diness, J. G., Sorensen, U. S., Nissen, J. D., Al-Shahib, B., Jespersen, T., Grunnet, M., et al. (2010). Inhibition of small-conductance Ca2+-activated K+ channels terminates and protects against atrial fibrillation. Circ. Arrhythm. Electrophysiol. 3, 380–390. doi: 10.1161/CIRCEP.110.957407
Djousse, L., Benkeser, D., Arnold, A., Kizer, J. R., Zieman, S. J., Lemaitre, R. N., et al. (2013). Plasma free fatty acids and risk of heart failure: the cardiovascular health study. Circ. Heart Fail. 6, 964–969. doi: 10.1161/CIRCHEARTFAILURE.113.000521
Ehrlich, J. R., Ocholla, H., Ziemek, D., Rutten, H., Hohnloser, S. H., and Gogelein, H. (2008). Characterization of human cardiac Kv1.5 inhibition by the novel atrial-selective antiarrhythmic compound AVE1231. J. Cardiovasc. Pharmacol. 51, 380–387. doi: 10.1097/FJC.0b013e3181669030
Feng, J., Yue, L., Wang, Z., and Nattel, S. (1998). Ionic mechanisms of regional action potential heterogeneity in the canine right atrium. Circ. Res. 83, 541–551. doi: 10.1161/01.RES.83.5.541
Fernandez, M. L., Conde, A. K., Ruiz, L. R., Montano, C., Ebner, J., and McNamara, D. J. (1995). Carbohydrate type and amount alter intravascular processing and catabolism of plasma lipoproteins in guinea pigs. Lipids 30, 619–626. doi: 10.1007/BF02536998
Fretts, A. M., Mozaffarian, D., Siscovick, D. S., Djousse, L., Heckbert, S. R., King, I. B., et al. (2014). Plasma phospholipid saturated fatty acids and incident atrial fibrillation: the cardiovascular health study. J. Am. Heart Assoc. 3:e000889. doi: 10.1161/JAHA.114.000889
Gaborit, N., Steenman, M., Lamirault, G., Le Meur, N., Le Bouter, S., Lande, G., et al. (2005). Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillation. Circulation 112, 471–481. doi: 10.1161/CIRCULATIONAHA.104.506857
Garnvik, L. E., Malmo, V., Janszky, I., Wisloff, U., Loennechen, J. P., and Nes, B. M. (2018). Physical activity modifies the risk of atrial fibrillation in obese individuals: the HUNT3 study. Eur. J. Prev. Cardiol. 25, 1646–1652. doi: 10.1177/2047487318784365
Gaspo, R., Bosch, R. F., Talajic, M., and Nattel, S. (1997). Functional mechanisms underlying tachycardia-induced sustained atrial fibrillation in a chronic dog model. Circulation 96, 4027–4035. doi: 10.1161/01.CIR.96.11.4027
Goette, A., Honeycutt, C., and Langberg, J. J. (1996). Electrical remodeling in atrial fibrillation. Time course and mechanisms. Circulation 94, 2968–2974. doi: 10.1161/01.CIR.94.11.2968
Gonzalez de la Fuente, M., Barana, A., Gomez, R., Amoros, I., Dolz-Gaiton, P., Sacristan, S., et al. (2013). Chronic atrial fibrillation up-regulates beta1-adrenoceptors affecting repolarizing currents and action potential duration. Cardiovasc. Res. 97, 379–388. doi: 10.1093/cvr/cvs313
Grandi, E., Dobrev, D., and Heijman, J. (2019). Computational modeling: what does it tell us about atrial fibrillation therapy? Int. J. Cardiol. 287, 155–161. doi: 10.1016/j.ijcard.2019.01.077
Grandi, E., Pandit, S. V., Voigt, N., Workman, A. J., Dobrev, D., Jalife, J., et al. (2011). Human atrial action potential and Ca2+ model: sinus rhythm and chronic atrial fibrillation. Circ. Res. 109, 1055–1066. doi: 10.1161/CIRCRESAHA.111.253955
Hadian, D., Zipes, D. P., Olgin, J. E., and Miller, J. M. (2002). Short-term rapid atrial pacing produces electrical remodeling of sinus node function in humans. J. Cardiovasc. Electrophysiol. 13, 584–586. doi: 10.1046/j.1540-8167.2002.00584.x
Heijman, J., Guichard, J. B., Dobrev, D., and Nattel, S. (2018). Translational challenges in atrial fibrillation. Circ. Res. 122, 752–773. doi: 10.1161/CIRCRESAHA.117.311081
Huang, H., Amin, V., Gurin, M., Wan, E., Thorp, E., Homma, S., et al. (2013). Diet-induced obesity causes long QT and reduces transcription of voltage-gated potassium channels. J. Mol. Cell. Cardiol. 59, 151–158. doi: 10.1016/j.yjmcc.2013.03.007
Hume, J. R., and Uehara, A. (1985). Ionic basis of the different action potential configurations of single guinea-pig atrial and ventricular myocytes. J. Physiol. 368, 525–544. doi: 10.1113/jphysiol.1985.sp015874
Inoue, M., Inoue, D., Ishibashi, K., Sakai, R., Omori, I., Yamahara, Y., et al. (1993b). Effects of pilsicainide on the atrial fibrillation threshold in guinea pig atria. A comparative study with disopyramide, lidocaine and flecainide. Jpn. Heart J. 34, 301–312.
Inoue, D., Shirayama, T., Omori, I., Inoue, M., Sakai, R., Ishibashi, K., et al. (1993a). Electrophysiological effects of flecainide acetate on stretched guinea pig left atrial muscle fibers. Cardiovasc. Drugs Ther. 7, 373–378.
Iwasaki, Y. K., Nishida, K., Kato, T., and Nattel, S. (2011). Atrial fibrillation pathophysiology: implications for management. Circulation 124, 2264–2274. doi: 10.1161/CIRCULATIONAHA.111.019893
Jensen, M. D., Ryan, D. H., Apovian, C. M., Ard, J. D., Comuzzie, A. G., Donato, K. A., et al. (2014). 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association task force on practice guidelines and the Obesity Society. Circulation 129(25 Suppl. 2), S102–S138. doi: 10.1161/01.cir.0000437739.71477.ee
Ji, Y., Varkevisser, R., Opacic, D., Bossu, A., Kuiper, M., Beekman, J. D. M., et al. (2017). The inward rectifier current inhibitor PA-6 terminates atrial fibrillation and does not cause ventricular arrhythmias in goat and dog models. Br. J. Pharmacol. 174, 2576–2590. doi: 10.1111/bph.13869
Kanner, S. A., Jain, A., and Colecraft, H. M. (2018). Development of a high-throughput flow cytometry assay to monitor defective trafficking and rescue of long QT2 mutant hERG channels. Front. Physiol. 9:397. doi: 10.3389/fphys.2018.00397
Killeen, M. J., Sabir, I. N., Grace, A. A., and Huang, C. L. (2008a). Dispersions of repolarization and ventricular arrhythmogenesis: lessons from animal models. Prog. Biophys. Mol. Biol. 98, 219–229. doi: 10.1016/j.pbiomolbio.2008.10.008
Killeen, M. J., Thomas, G., Sabir, I. N., Grace, A. A., and Huang, C. L. (2008b). Mouse models of human arrhythmia syndromes. Acta Physiol (Oxf.) 192, 455–469. doi: 10.1111/j.1748-1716.2007.01822.x
Koivumäki, J. T., Korhonen, T., and Tavi, P. (2011). Impact of sarcoplasmic reticulum calcium release on calcium dynamics and action potential morphology in human atrial myocytes: a computational study. PLoS Comput. Biol. 7:e1001067. doi: 10.1371/journal.pcbi.1001067
Laszlo, R., Schwiebert, M., Menzel, K. A., Eick, C., Schreiner, B., and Schreieck, J. (2011). Effects of dexamethasone and atorvastatin on atrial sodium current and its early tachycardia-induced electrical remodeling in rabbits. Pak. J. Pharm. Sci. 24, 383–387.
Laszlo, R., Winkler, C., Wohrl, S., Laszlo, S., Eick, C., Schreieck, J., et al. (2007). Influence of verapamil on tachycardia-induced alterations of PP1 and PP2A in rabbit atrium. Exp. Clin. Cardiol. 12, 175–178.
Lau, C. P., Tse, H. F., Siu, C. W., and Gbadebo, D. (2012). Atrial electrical and structural remodeling: implications for racial differences in atrial fibrillation. J. Cardiovasc. Electrophysiol. 23(Suppl. 1), S36–S40. doi: 10.1111/jce.12022
Laurita, K. R., and Rosenbaum, D. S. (1997). Implications of ion channel diversity to ventricular repolarization and arrhythmogenesis: insights from high resolution optical mapping. Can. J. Cardiol. 13, 1069–1076.
Leopoldo, A. S., Lima-Leopoldo, A. P., Sugizaki, M. M., do Nascimento, A. F., de Campos, D. H., Luvizotto Rde, A., et al. (2011). Involvement of L-type calcium channel and SERCA2a in myocardial dysfunction induced by obesity. J. Cell. Physiol. 226, 2934–2942. doi: 10.1002/jcp.22643
Lima-Leopoldo, A. P., Leopoldo, A. S., Silva, D. C., Nascimento, A. F., Campos, D. H., Luvizotto Rde, A., et al. (2013). Influence of long-term obesity on myocardial gene expression. Arq. Bras. Cardiol. 100, 229–237. doi: 10.5935/abc.20130045
Lima-Leopoldo, A. P., Sugizaki, M. M., Leopoldo, A. S., Carvalho, R. F., Nogueira, C. R., Nascimento, A. F., et al. (2008). Obesity induces upregulation of genes involved in myocardial Ca2+ handling. Braz. J. Med. Biol. Res. 41, 615–620. doi: 10.1590/S0100-879X2008000700011
Liu, T., Takimoto, E., Dimaano, V. L., DeMazumder, D., Kettlewell, S., Smith, G., et al. (2014). Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a guinea pig model of heart failure. Circ. Res. 115, 44–54. doi: 10.1161/CIRCRESAHA.115.303062
Mancarella, S., Yue, Y., Karnabi, E., Qu, Y., El-Sherif, N., and Boutjdir, M. (2008). Impaired Ca2+ homeostasis is associated with atrial fibrillation in the alpha1D L-type Ca2+ channel KO mouse. Am. J. Physiol. Heart Circ. Physiol. 295, H2017–H2024. doi: 10.1152/ajpheart.00537.2008
Mangoni, M. E., Couette, B., Bourinet, E., Platzer, J., Reimer, D., Striessnig, J., et al. (2003). Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc. Natl. Acad. Sci. USA 100, 5543–5548. doi: 10.1073/pnas.0935295100
Martinez-Mateu, L., Romero, L., Ferrer-Albero, A., Sebastian, R., Rodriguez Matas, J. F., Jalife, J., et al. (2018). Factors affecting basket catheter detection of real and phantom rotors in the atria: a computational study. PLoS Comput. Biol. 14:e1006017. doi: 10.1371/journal.pcbi.1006017
Matafome, P., and Seica, R. (2017). Function and dysfunction of adipose tissue. Adv. Neurobiol. 19, 3–31. doi: 10.1007/978-3-319-63260-5_1
Matsuura, H., and Ehara, T. (1997). Selective enhancement of the slow component of delayed rectifier K+ current in guinea-pig atrial cells by external ATP. J. Physiol. 503, 45–54. doi: 10.1111/j.1469-7793.1997.045bi.x
Michael, G., Xiao, L., Qi, X. Y., Dobrev, D., and Nattel, S. (2009). Remodelling of cardiac repolarization: how homeostatic responses can lead to arrhythmogenesis. Cardiovasc. Res. 81, 491–499. doi: 10.1093/cvr/cvn266
Mickelson, A. V., and Chandra, M. (2017). Hypertrophic cardiomyopathy mutation in cardiac troponin T (R95H) attenuates length-dependent activation in guinea pig cardiac muscle fibers. Am. J. Physiol. Heart Circ. Physiol. 313, H1180–h1189. doi: 10.1152/ajpheart.00369.2017
Nakaya, H., Furusawa, Y., Ogura, T., Tamagawa, M., and Uemura, H. (2000). Inhibitory effects of JTV-519, a novel cardioprotective drug, on potassium currents and experimental atrial fibrillation in guinea-pig hearts. Br. J. Pharmacol. 131, 1363–1372. doi: 10.1038/sj.bjp.0703713
Nattel, S. (2002). New ideas about atrial fibrillation 50 years on. Nature 415, 219–226. doi: 10.1038/415219a
Nattel, S., and Dobrev, D. (2017). Controversies about atrial fibrillation mechanisms: aiming for order in chaos and whether it matters. Circ. Res. 120, 1396–1398. doi: 10.1161/CIRCRESAHA.116.310489
Nattel, S., and Harada, M. (2014). Atrial remodeling and atrial fibrillation: recent advances and translational perspectives. J. Am. Coll. Cardiol. 63, 2335–2345. doi: 10.1016/j.jacc.2014.02.555
Nerbonne, J. M., and Kass, R. S. (2005). Molecular physiology of cardiac repolarization. Physiol. Rev. 85, 1205–1253. doi: 10.1152/physrev.00002.2005
Ni, H., Whittaker, D. G., Wang, W., Giles, W. R., Narayan, S. M., and Zhang, H. (2017). Synergistic anti-arrhythmic effects in human atria with combined use of sodium blockers and acacetin. Front. Physiol. 8:946. doi: 10.3389/fphys.2017.00946
O’Connell, R. P., Musa, H., Gomez, M. S., Avula, U. M., Herron, T. J., Kalifa, J., et al. (2015). Free fatty acid effects on the atrial myocardium: membrane ionic currents are remodeled by the disruption of T-tubular architecture. PLoS One 10:e0133052. doi: 10.1371/journal.pone.0133052
Ochi, R., Momose, Y., Oyama, K., and Giles, W. R. (2006). Sphingosine-1-phosphate effects on guinea pig atrial myocytes: alterations in action potentials and K+ currents. Cardiovasc. Res. 70, 88–96. doi: 10.1016/j.cardiores.2006.01.010
O’Hara, T., and Rudy, Y. (2012). Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. Am. J. Physiol. Heart Circ. Physiol. 302, H1023–H1030. doi: 10.1152/ajpheart.00785.2011
Osadchii, O. E. (2012). Electrophysiological determinants of arrhythmic susceptibility upon endocardial and epicardial pacing in guinea-pig heart. Acta Physiol. 205, 494–506. doi: 10.1111/j.1748-1716.2012.02428.x
Patoine, D., Levac, X., Pilote, S., Drolet, B., and Simard, C. (2013). Decreased CYP3A expression and activity in guinea pig models of diet-induced metabolic syndrome: is fatty liver infiltration involved? Drug Metab. Dispos. 41, 952–957. doi: 10.1124/dmd.112.050641
Paulino, E. C., Ferreira, J. C., Bechara, L. R., Tsutsui, J. M., Mathias, W. Jr., Lima, F. B., et al. (2010). Exercise training and caloric restriction prevent reduction in cardiac Ca2+-handling protein profile in obese rats. Hypertension 56, 629–635. doi: 10.1161/HYPERTENSIONAHA.110.156141
Perez-Hernandez, M., Matamoros, M., Barana, A., Amoros, I., Gomez, R., Nunez, M., et al. (2016). Pitx2c increases in atrial myocytes from chronic atrial fibrillation patients enhancing IKs and decreasing ICa,L. Cardiovasc. Res. 109, 431–441. doi: 10.1093/cvr/cvv280
Pezhouman, A., Cao, H., Fishbein, M. C., Belardinelli, L., Weiss, J. N., and Karagueuzian, H. S. (2018). Atrial fibrillation initiated by early afterdepolarization-mediated triggered activity during acute oxidative stress: efficacy of late sodium current blockade. J. Heart Health 4, 1–17. doi: 10.16966/2379-769x.146
Phartiyal, P., Sale, H., Jones, E. M., and Robertson, G. A. (2008). Endoplasmic reticulum retention and rescue by heteromeric assembly regulate human ERG 1a/1b surface channel composition. J. Biol. Chem. 283, 3702–3707. doi: 10.1074/jbc.M708999200
Puckerin, A., Aromolaran, K. A., Chang, D. D., Zukin, R. S., Colecraft, H. M., Boutjdir, M., et al. (2016). hERG 1a LQT2 C-terminus truncation mutants display hERG 1b-dependent dominant negative mechanisms. Heart Rhythm. 13, 1121–1130. doi: 10.1016/j.hrthm.2016.01.012
Rahm, A. K., Lugenbiel, P., Schweizer, P. A., Katus, H. A., and Thomas, D. (2018). Role of ion channels in heart failure and channelopathies. Biophys. Rev. 10, 1097–1106. doi: 10.1007/s12551-018-0442-3
Rennison, J. H., and Van Wagoner, D. R. (2009). Impact of dietary fatty acids on cardiac arrhythmogenesis. Circ. Arrhythm. Electrophysiol. 2, 460–469. doi: 10.1161/CIRCEP.109.880773
Roy, S., Vega-Lopez, S., and Fernandez, M. L. (2000). Gender and hormonal status affect the hypolipidemic mechanisms of dietary soluble fiber in guinea pigs. J. Nutr. 130, 600–607. doi: 10.1093/jn/130.3.600
Sahraoui, A., Dewachter, C., de Medina, G., Naeije, R., Aouichat Bouguerra, S., and Dewachter, L. (2016). Myocardial structural and biological anomalies induced by high fat diet in Psammomys obesus gerbils. PLoS One 11:e0148117. doi: 10.1371/journal.pone.0148117
Sanguinetti, M. C., and Jurkiewicz, N. K. (1991). Delayed rectifier outward K+ current is composed of two currents in guinea pig atrial cells. Am. J. Phys. 260, H393–H399. doi: 10.1152/ajpheart.1991.260.2.H393
Sanguinetti, M. C., Jurkiewicz, N. K., Scott, A., and Siegl, P. K. (1991). Isoproterenol antagonizes prolongation of refractory period by the class III antiarrhythmic agent E-4031 in guinea pig myocytes. Mechanism of action. Circ. Res. 68, 77–84. doi: 10.1161/01.RES.68.1.77
Schulze, P. C., Drosatos, K., and Goldberg, I. J. (2016). Lipid use and misuse by the heart. Circ. Res. 118, 1736–1751. doi: 10.1161/CIRCRESAHA.116.306842
Sharma, D., Garg, S., Mehndiratta, M., Madhu, S. V., and Puri, D. (2017). Relationship of serum apolipoprotein A-V levels, oxidative stress and inflammatory biomarkers with hypertriglyceridemia in type 2 diabetes mellitus. Int. J. Endocrinol. Metab. 15:e44805. doi: 10.5812/ijem.44805
Sharman, M. J., Fernandez, M. L., Zern, T. L., Torres-Gonzalez, M., Kraemer, W. J., and Volek, J. S. (2008). Replacing dietary carbohydrate with protein and fat decreases the concentrations of small LDL and the inflammatory response induced by atherogenic diets in the guinea pig. J. Nutr. Biochem. 19, 732–738. doi: 10.1016/j.jnutbio.2007.09.008
Sonnenberg, G. E., Krakower, G. R., and Kissebah, A. H. (2004). A novel pathway to the manifestations of metabolic syndrome. Obes. Res. 12, 180–186. doi: 10.1038/oby.2004.24
Suzuki, K., Matsumoto, A., Nishida, H., Reien, Y., Maruyama, H., and Nakaya, H. (2014). Termination of aconitine-induced atrial fibrillation by the KACh-channel blocker tertiapin: underlying electrophysiological mechanism. J. Pharmacol. Sci. 125, 406–414. doi: 10.1254/jphs.14023FP
Tsuchiya, H., Nakazato, Y., Hayashi, H., Suzuki, T., Sekita, G., and Daida, H. (2009). Effects of Bepridil on atrial electrical remodeling in short-term rapid pacing. J. Arrhythm. 25, 89–94. doi: 10.1016/S1880-4276(09)80022-7
Van Wagoner, D. R., Pond, A. L., Lamorgese, M., Rossie, S. S., McCarthy, P. M., and Nerbonne, J. M. (1999). Atrial L-type Ca2+ currents and human atrial fibrillation. Circ. Res. 85, 428–436. doi: 10.1161/01.RES.85.5.428
Van Wagoner, D. R., Pond, A. L., McCarthy, P. M., Trimmer, J. S., and Nerbonne, J. M. (1997). Outward K+ current densities and Kv1.5 expression are reduced in chronic human atrial fibrillation. Circ. Res. 80, 772–781. doi: 10.1161/01.RES.80.6.772
Wakili, R., Voigt, N., Kaab, S., Dobrev, D., and Nattel, S. (2011). Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Invest. 121, 2955–2968. doi: 10.1172/JCI46315
Wanahita, N., Messerli, F. H., Bangalore, S., Gami, A. S., Somers, V. K., and Steinberg, J. S. (2008). Atrial fibrillation and obesity – results of a meta-analysis. Am. Heart J. 155, 310–315. doi: 10.1016/j.ahj.2007.10.004
Wang, Z., Fermini, B., and Nattel, S. (1994). Rapid and slow components of delayed rectifier current in human atrial myocytes. Cardiovasc. Res. 28, 1540–1546. doi: 10.1093/cvr/28.10.1540
Wang, X., and Fitts, R. H. (2017). Ventricular action potential adaptation to regular exercise: role of beta-adrenergic and KATP channel function. J. Appl. Physiol. (1985) 123, 285–296. doi: 10.1152/japplphysiol.00197.2017
Wang, X., and Fitts, R. H. (2018). Effects of regular exercise on ventricular myocyte biomechanics and KATP channel function. Am. J. Physiol. Heart Circ. Physiol. 315, H885–h896. doi: 10.1152/ajpheart.00130.2018
Whittaker, D. G., Ni, H., El Harchi, A., Hancox, J. C., and Zhang, H. (2017). Atrial arrhythmogenicity of KCNJ2 mutations in short QT syndrome: insights from virtual human atria. PLoS Comput. Biol. 13:e1005593. doi: 10.1371/journal.pcbi.1005593
Wilhelms, M., Hettmann, H., Maleckar, M. M., Koivumaki, J. T., Dossel, O., and Seemann, G. (2012). Benchmarking electrophysiological models of human atrial myocytes. Front. Physiol. 3:487. doi: 10.3389/fphys.2012.00487
Yu, W. C., Chen, S. A., Lee, S. H., Tai, C. T., Feng, A. N., Kuo, B. I., et al. (1998). Tachycardia-induced change of atrial refractory period in humans: rate dependency and effects of antiarrhythmic drugs. Circulation 97, 2331–2337. doi: 10.1161/01.CIR.97.23.2331
Yue, L., Feng, J., Gaspo, R., Li, G. R., Wang, Z., and Nattel, S. (1997). Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ. Res. 81, 512–525. doi: 10.1161/01.RES.81.4.512
Yue, L., Melnyk, P., Gaspo, R., Wang, Z., and Nattel, S. (1999). Molecular mechanisms underlying ionic remodeling in a dog model of atrial fibrillation. Circ. Res. 84, 776–784. doi: 10.1161/01.RES.84.7.776
Zankov, D. P., Omatsu-Kanbe, M., Isono, T., Toyoda, F., Ding, W. G., Matsuura, H., et al. (2006). Angiotensin II potentiates the slow component of delayed rectifier K+ current via the AT1 receptor in guinea pig atrial myocytes. Circulation 113, 1278–1286. doi: 10.1161/CIRCULATIONAHA.104.530592
Keywords: high-fat diet, hERG, KCNQ1, cardiomyocytes, guinea pig, atria
Citation: Martinez-Mateu L, Saiz J and Aromolaran AS (2019) Differential Modulation of IK and ICa,L Channels in High-Fat Diet-Induced Obese Guinea Pig Atria. Front. Physiol. 10:1212. doi: 10.3389/fphys.2019.01212
Edited by:
Jichao Zhao, The University of Auckland, New ZealandReviewed by:
Sanjay Ram Kharche, University of Western Ontario, CanadaJieyun Bai, Jinan University, China
Copyright © 2019 Martinez-Mateu, Saiz and Aromolaran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ademuyiwa S. Aromolaran, ademuyiwa.aromolaran@downstate.edu