 Alla B. Salmina1*
Alla B. Salmina1* Yulia K. Komleva1
Yulia K. Komleva1 István A. Szijártó2
István A. Szijártó2 Yana V. Gorina1
Yana V. Gorina1 Olga L. Lopatina1
Olga L. Lopatina1 Galina E. Gertsog1
Galina E. Gertsog1 Milos R. Filipovic3
Milos R. Filipovic3 Maik Gollasch2*
Maik Gollasch2*- 1Department of Biochemistry, Medical, Pharmaceutical and Toxicological Chemistry, Krasnoyarsk State Medical University named after Prof. V.F. Voino-Yasenetsky, Krasnoyarsk, Russia
- 2Experimental and Clinical Research Center, Charité - University Medicine Berlin and the Max Delbrück Center for Molecular Medicine, Berlin, Germany
- 3Department of Chemistry and Pharmacy, Friedrich-Alexander-University of Erlangen-Nürnberg, Erlangen, Germany
Alzheimer's type of neurodegeneration dramatically affects H2S and NO synthesis and interactions in the brain, which results in dysregulated vasomotor function, brain tissue hypoperfusion and hypoxia, development of perivascular inflammation, promotion of Aβ deposition, and impairment of neurogenesis/angiogenesis. H2S- and NO-signaling pathways have been described to offer protection against Alzheimer's amyloid vasculopathy and neurodegeneration. This review describes recent developments of the increasing relevance of H2S and NO in Alzheimer's disease (AD). More studies are however needed to fully determine their potential use as therapeutic targets in Alzheimer's and other forms of vascular dementia.
Cerebrovascular Dysfunction in AD
Due to the increasing prevalence and high severity, Alzheimer's disease (AD) is one of today's major health challenges. AD typically manifests after the age of 60. As the population ages, this disease impacts a greater percentage of the world-wide population. AD is a progressive neurodegenerative disorder characterized by deposition of amyloid-beta (Aβ) in the brain tissue that leads to cognitive, memory, and behavioral impairments. Several hypotheses were proposed to explain the pathogenesis of AD: Aβ toxicity, cholinergic dysfunction, abnormal phosphorylation of tau protein, development of oxidative stress due to action of endogenous or exogenous pro-oxidants, impairment of neurogenesis, excessive cell death, neuroinflammation etc. (Salmina, 2009).
The following strategies have been proposed for the pharmacotherapy of AD: (1) prevention of accumulation of aberrant proteins (suppression of Àβ production or activation of Àβ clearance); (2) modulation of neuroplasticity; (3) suppression of neuroinflammation; (4) stimulation of neurogenesis and other brain repair mechanisms. However, treatments aimed at reducing Aβ deposits showed no or little success, therefore, alternative approaches appear to be more prospective. In this context, current prevention and treatment strategies are increasingly focused on AD associated vasculopathy.
Cerebrovascular dysfunction is not only a marker of ischemic brain pathology, but also of neurodegenerative disorders such as AD. The vascular hypothesis of AD suggests that neurodegenerative pathology begins with cerebral hypoperfusion and cerebral microvascular abnormalities associated with extensive Aβ deposition and blood-brain barrier disruption. Contribution of cerebrovascular dysfunction to the pathogenesis of AD is evident not only in humans (Saito et al., 2015) but also in experimental models of AD (Han et al., 2015). It is associated with prominent oxidative stress, Aβ-impaired cerebral circulation, and alterations in the neurovascular unit or blood-brain barrier (Han et al., 2008; Bell and Zlokovic, 2009).
Deposition of Aβ is an important mechanism of cerebrovascular dysfunction in AD, and cerebral amyloid angiopathy facilitates progression of AD and cognitive impairment. Cerebral amyloid angiopathy in AD is caused by the accumulation of Aβ in small-sized and medium-sized blood vessels, mostly in arteries (Biffi and Greenberg, 2011). In severe angiopathy, amyloid deposits replace degenerating vessel smooth muscle cells, thus resulting in microhemorrhages, ischemic lesions, and encephalopathies (Yamada, 2015). Accumulation of Aβ leads to extensive neoangiogenesis and hypervascularity associated with abnormal blood-brain barrier leakiness in AD (Biron et al., 2011), but the vessels have smaller diameter suggesting vasomotor dysfunction and/or vascular remodeling in AD (Burke et al., 2014). Mechanisms of cerebral amyloid angiopathy in AD include insufficiency of perivascular drainage of Aβ leading to its accumulation in the vessel wall; development of perivascular inflammation and microhemorrhages, vascular oxidative stress (Park et al., 2011; Hawkes et al., 2014; Boncoraglio et al., 2015). We also found that Aβ peptides elicit a signal transduction pathway in vascular cells, induced by α1-adrenergic receptor activation (Haase et al., 2013). However, it is not clear which molecular events and amyloid toxic action affect the time-course of neurodegeneration. Nevertheless, it is clear that there is a strong correlation between the prevalence of cerebral amyloid angiopathy and age. As such, up to 40% of the elderly population without clinical manifestations of AD demonstrate features of cerebral amyloid angiopathy, and up to 80% of people suffering from AD exhibit signs of cerebral amyloid angiopathy (Jellinger, 2002). Deciphering the molecular mechanisms of cerebral amyloid angiopathy would be beneficial for development of novel therapeutic and diagnostic strategies.
Endothelial cells in the cerebral vasculature may contribute to the formation of amyloid deposits surrounding the cerebral blood vessels. Several recent studies have highlighted that endothelial cells might be the target for the toxic action of heavily aggregated proteins, glia-derived cytokines, and stimuli inducing oxidative and metabolic stress in AD brains (Salmina et al., 2010). Cerebral endothelial cells are in the close connection with pericytes, astrocytes, and neurons in the neurovascular unit, therefore altered paracrine and autocrine interactions of these cells might be critically involved in the development of microvascular abnormalities in AD.
Gaseous Transmitters in the Brain: General Characteristics
Production of gaseous transmitters by mammalian cells has attract much attention in past few years. Hydrogen sulfide (H2S) is now considered the third gaseous transmitter besides nitric oxide (NO) and carbon monoxide (CO) to contribute to the regulation of cardiac function, systemic and pulmonary blood pressure and vasomotor activity, control inflammation, and angiogenesis (Köhn et al., 2012; Lo Faro et al., 2014). Being produced by various cell types, these gaseous transmitters can easily penetrate the plasma membrane thus inducing wide spectrum of signaling cascades in the target cells. In brain, H2S, CO, and NO are released by astroglial cells, neurons, or endothelial cells (Figure 1), and can functionally interact.
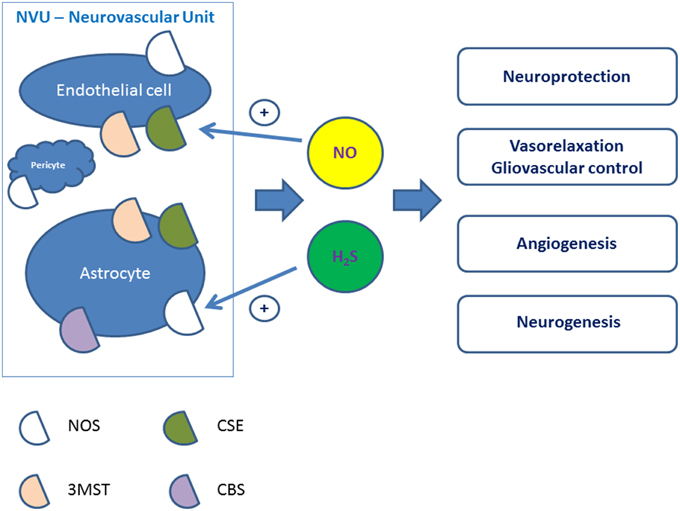
Figure 1. Putative physiological effects of NO and H2S produced in the cells of the neurovascular unit. NVU, neurovascular unit; NOS, nitric oxide synthase; MST, 3-mercaptopyruvate sulfurtransferase; CSE, cystathionine-γ-lyase; CBS, cystathionine-β-synthase.
The action of NO in the brain is extensively studied. NO is now known as a potent vasorelaxant, neurotransmitter, pro-inflammatory, and pro-oxidant molecule (Guix et al., 2005; Kovac et al., 2011; Tabatabaei and Girouard, 2014), even there are some data on antioxidant activity of NO (Hummel et al., 2006). Coordinated expression and activity of nitric oxide synthase (NOS), as either constitutive or inducible isoforms, in endothelial and neuronal cells regulates local NO release to contribute to local neurovascular and metabolic coupling, and neuronal excitability. Molecular targets of NO are cysteine and tyrosine residues in cell proteins that can be nitrosylated or nitrated, respectively (Hess et al., 2005). Although the neurotoxic vs. neuroprotective activity of NO is still a matter of debate, and even dose-dependent effects should be considered (Tripathy et al., 2015), the majority of the data suggests that dysregulated NO production facilitates neurodegeneration (Figure 2; Jullienne and Badaut, 2013).
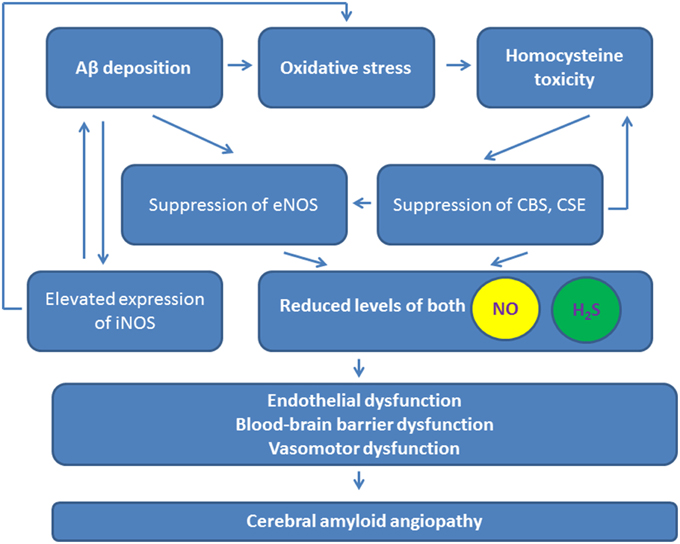
Figure 2. Contribution of NO and H2S to the pathogenesis of cerebral amyloid angiopathy. Purple arrows, reduced levels and actions of NO and H2S. iNOS, inducible nitric oxide synthase; eNOS, endothelial nitric oxide synthase; CSE, cystathionine-γ-lyase; CBS, cystathionine-β-synthase.
Almost similar activities can be attributed to CO produced by heme oxygenase. CO acts as a modulator of cerebral vasomotor function (Parfenova et al., 2012b), but in the contrast to NO which can easily react with superoxide anion to produce peroxynitrite (i.e., in proinflammatory conditions or in reperfusion) with the ultimate pro-oxidant activity, CO exerts anti-oxidant properties (Parfenova et al., 2012a). Molecular targets of CO are potassium (BKCa) channels, guanylyl cyclase, NADPH oxidase, and the heme-containing components of the mitochondria respiratory chain (Parfenova and Leffler, 2008). In general, CO is considered to be a cytoprotective mediator in the brain, and elevation of its level may improve cerebrovascular outcome of brain injury (Liu et al., 2015).
Endogenous H2S acts as a potent regulator of various biological processes mainly related to vasomotor function (Figure 2). H2S regulates intracellular calcium concentrations via L-type calcium channels, T-type calcium channels, sodium/calcium exchangers, transient receptor potential channels, β-adrenergic receptors, and N-methyl-D-aspartate receptors (NMDA) in various cells (Zhang et al., 2015).
Exogenous H2S can produce relaxation of a number of non-cerebral systemic arteries by mechanisms involving opening of adenosine 5′-triphosphate (ATP)-sensitive potassium (KATP) channels, intermediate (IK) and small conductance potassium channels (SK), KCNQ-type voltage-gated potassium (Kv7.x) channels, regulating the Cl−/ transporter, decreasing adenosine triphosphate levels, and/or release of endogenous vasodilatory prostanoids (Köhn et al., 2012). Of note, exogenous H2S can produce vasoconstrictions at low concentrations (< 100 μM) in some vessels, probably via inhibition of cAMP/protein kinase A (PKA) pathway in smooth muscle cells and/or interaction with the endothelial nitric oxide synthase/nitric oxide (eNOS/NO) pathway (Kubo et al., 2007a).
The biological effects of H2S in the central nervous system and cerebral circulation are less clear. Cerebral vessels express H2S-generating enzymes (Chertok and Kotsyuba, 2012), thus, cerebral endothelial cells may use H2S to induce cerebrovascular relaxation to increase local blood flow. H2S produced by astrocytes or endothelial cells within the neurovascular unit may mediate gliovascular control adjusting local blood perfusion to the actual needs in the activated brain area (Figure 1). Biological effects of H2S include modulation of neuronal excitability, regulation of vessel tone, anti-oxidant and anti-inflammatory activity, regulation of vascular tone, angiogenesis, and blood-brain barrier permeability (Geng et al., 2015; Kimura, 2015). In general, H2S demonstrates neuroprotective properties (Zhang and Bian, 2014), but the sensitivity of the cells to H2S depends on developmental status of the cells: differentiated cell have greater sensitivity to H2S compared to progenitor cells (Tsugane et al., 2007).
There is evidence that microvascular function is not controlled by the activity of these three gaseous transmitters independently by themselves, but rather by a complex interaction between NO, CO, and/or H2S gases, which enables a complex pattern of hemodynamic microvascular control in the brain (Dyson et al., 2014) and can affect the function of neuronal cells (Pong and Eldred, 2009). As an example, nitrosothiols are organic compounds or functional groups containing a nitrosogroup attached to the sulfur atom of a thiol. S-Nitrosated proteins (SNOs) serve to transmit nitric oxide (NO) bioactivity in vivo; their reaction with H2S, however, results in the formation of HSNO which can promote further trans-nitrosation of specific protein targets or give NO and nitroxyl (HNO), both of which could have biological activity aimed to promote smooth muscle cell relaxation (Filipovic et al., 2012). Oxidation of H2S could lead to formation of polysulfides, which are very reactive with cell tiols (Greiner et al., 2013; Wedmann et al., 2014). TRPA1 channels are proposed to represent potential targets for the stimulatory effects of polysulfides in the cells (Hatakeyama et al., 2015), and these channels are importantly also affected by lipid peroxidation metabolites and glycolysis by-product methylglyoxal (Eberhardt et al., 2012; Sullivan et al., 2015). Activation of TRPA1 channels enables Ca2+ signal-effector coupling at discrete sites along the endothelium to evoke graded cerebral artery vasodilation (Qian et al., 2013).
H2S-generating Machinery in Brain Cells
H2S is produced from cysteine due to the activity of various enzymes (Figure 1). Within the neurovascular unit, cystine as a cysteine precursor is taken up by astrocytes and then cysteine is released from astroglial cells for neuronal and endothelial needs (Guebel and Torres, 2004). Import of l-cystine into astrocytes and corresponding efflux of l-glutamate is provided by the x(c)—antiporter whose expression is elevated in neuroinflammation or brain hypoxia (Jackman et al., 2010, 2012). Cysteine is further converted into H2S or taurine (with anti-oxidant and anti-inflammatory activities), and is a mainly used for glutathione synthesis.
The endogenous levels of H2S in the brain and various organs were recently re-evaluated and found to be much lower than previously estimated (Ishigami et al., 2009; Wintner et al., 2010). Nevertheless, H2S is generated by two major enzymes, namely cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) using vitamin B6 as cofactor. Some authors believe that CSE plays a major role in generating H2S in cardiovascular system, while CBS is responsible for H2S production in the brain (Wang et al., 2014), i.e., by activated astrocytes due to shift in intracellular pH required for release of H2S from bound sulfur (Ishigami et al., 2009). CSE, but not CBS, was detected in cerebral microvessels, whereas CBS was detected in brain parenchyma (Leffler et al., 2011).
CBS and CSE may also use homocysteine for H2S synthesis (Wang, 2012): initial conversion of homocysteine to cyctathionine followed by conversion to cysteine. When H2S is synthesized from cysteine due to activity of CBS or CSE/3MST, l-serine or pyruvate are the side products, respectively (Olson et al., 2013).
Brain transsulfuration pathway involving cysteine, homocysteine, and cystathionine generates H2S and the atypical amino acid lanthionine (Hensley and Denton, 2015). The latter regulates brain cells autophagy (Harris-White et al., 2015), neuritis outgrowth (Hubbard et al., 2013), and is currently discussed as a candidate for correction of neurological alterations seen in neurodegeneration (Hensley and Denton, 2015).
Increased expression of transsulfuration pathway enzyme CSE is caused by dietary restriction, particularly, by shortage in the sulfur amino acids consumption. As a result, H2S production is elevated and its cytoprotective properties become more evident. It was proposed that dietary restriction-mediated positive effect on longevity might have a relation to permanent H2S-mediated protection from various pathogenic stimuli (Hine et al., 2015).
Since CSE expression was not confirmed in brain cells, CBS is considered to be the major H2S-producing enzyme in the brain. In agreement, astroglial cells express CBS at high level (Wang, 2012). However, H2S was identified in the brain of CBS-knockout mice. This led to the identification of a third H2S-generating pathway, which is regulated by 3-mercaptopyruvate sulfurtransferase (3MST) along with cysteine aminotransferase (CAT; Tanizawa, 2011) in the presence of thioredoxin. Although expression of CBS is mainly attributed to astrocytes (Enokido et al., 2005) and microglia (Du et al., 2014), 3MST is mainly expressed in neurons and endothelial cells (Figure 1). Of note, down-regulation of 3MST expression was detected in astroglial cells after stroke (Zhao et al., 2013). In general, astrocytic production of H2S is almost 10 times higher compared to H2S production in microglial cells (Wang et al., 2014). Endothelial cells expressing 3MST represent also a source for H2S production from cysteine and alpha-ketoglutarate (Shibuya et al., 2009); noteworthy, cerebral endothelial cells may use H2S to induce smooth muscle cell relaxation to increase local blood flow. Therefore, H2S produced by astrocytes or endothelial cells within the neurovascular unit may mediate gliovascular control adjusting local blood perfusion to the actual needs in the activated brain area (Figure 1). Nevertheless, it is still a matter of debate, which enzyme(s) is (are) responsible for brain injury-associated alterations in endogenous H2S production (Zhao et al., 2013). Recently, it was found that target inhibition of different H2S-generating enzymes can be achieved in experimental conditions by pharmacological approach, i.e., with DL-propargylglycine, which is an inhibitor for CSE, aspartate which is an inhibitor for 3MST, or O-(carboxymethyl)hydroxylamine hemihydrochloride, which is an inhibitor for CBS (Jiang et al., 2015). This approach revealed pivotal roles of 3MST and CSE in ischemia-reperfusion-associated blood-brain barrier alterations.
It is generally accepted that H2S and NO interact with each other at the level of expression of the enzymes producing these molecules where mutual down-regulation effect is demonstrated (Rong-na et al., 2011; Figure 2). In macrophages, both NO and H2S irreversibly suppress NOS activity (Heine et al., 2015), thus providing negative feedback control. In contrast, there are some reports on mutually stimulatory activity of these two gases on their production (Kolluru et al., 2013). Therefore, H2S may stimulate NO production in endothelial cells to promote neoangiogenesis (Altaany et al., 2013) (Figure 1). However, some authors report that eNOS can be directly inhibited by H2S (Kubo et al., 2007b), and this effect can underlie neuroprotective effects of H2S in neonatal brain hypoxia (Wang et al., 2013b). Since post-ischemic upregulation of eNOS triggers cerebral angiogenesis accompanied by increased BBB permeability, ischemia/hypoxia-induced vascular plasticity might be regulated by the altering production of NO and H2S in different phases of ischemia and reperfusion in order to provide adequate proangiogenic microenvironment and to prevent dramatic alterations in the BBB structural integrity. Thus, H2S and NO produced within the neurovascular unit may attenuate vasogenic brain edema formation and protect the brain by enhancing cerebral blood flow and neoangiogenesis.
H2S can produce vasorelaxation due to inhibition of RhoA-dependent signaling cascades in SMC (Nalli et al., 2015). The same is true for NO action (Sawada et al., 2001). Taking into consideration that inhibition of RhoA downstream molecular machinery (i.e., Rho-associated coiled-coil forming protein kinases) leads to the upregulation and activation of eNOS (Noma et al., 2012), one can propose that H2S may also exert NOS-stimulatory effects under (patho)physiological conditions (Kram et al., 2013). However, this effect has not been demonstrated in brain cells so far. Similarly, H2S-induced up-regulation of heme oxygenase expression has been observed in various non-cerebral tissues (Zhang et al., 2013; D'Araio et al., 2014; Wang et al., 2015), however, evidence for such an effect in brain cells is missing.
More importantly there is increasing body of evidence that H2S can react directly with NO (Yong et al., 2010; Filipovic et al., 2013; Eberhardt et al., 2014; Dux et al., 2015). Nitroxyl (HNO), as a one-electron reduced sibling of nitric oxide, has been proposed as a main mediator of this direct reaction between NO and H2S. Our research team has shown that nitroxyl, formed in the reaction of H2S and NO, can activate TRPA1 channels by oxidizing critical cysteine residues which leads to Ca2+ influx and subsequent calcitonin gene-related peptide (CGRP) release from sensory nerve ending. This could contribute to systemic blood pressure regulation as well as regulation of cerebral blood flow (Eberhardt et al., 2014). The importance of this H2S+NO/HNO/TRPA1/CGRP signaling cascade has been recently established in the context of meningeal blood flow and pathology of migraines (Dux et al., 2015).
Thus, the cells of the neurovascular unit (endothelial cells, astrocytes) produce NO and H2S whose biological effects support intercellular communications, gliovascular control, cell proliferation and development (Figure 1). Compromised production or action of NO and H2S may be associated with the development of various neurodegenerative and cerebrovascular disorders.
H2S and NO in the Alzheimer's Type of Angiopathy
Vascular Aβ Deposition
Under normal conditions, NO protects endothelial cells and adjusts cerebrovascular function to the actual blood flow needs in active brain regions. NOS activity in endothelial cells prevents APP overexpression and Aβ synthesis (Katusic and Austin, 2014). In AD, Aβ impairs endothelial function due to inhibition of eNOS activity caused by alterations in intracellular calcium homeostasis and protein phosphorylation pattern (Gentile et al., 2004). Deposition of Aβ, thickening and hyalinization of the media of small and medium-size vessels, and apoptosis of endothelial and smooth muscle cells correlate with reduced NOS expression in cerebral vessels (de la Monte et al., 2000). Deficiency of NOS results in increased Aβ production and neuroinflammation development (Austin et al., 2013). These alterations are accompanied with development of oxidative stress (Lamoke et al., 2015). Oxidative stress is a key contributor to development of cerebral amyloid angiopathy, vessel constriction, and cerebral amyloid angiopathy-related microhemorrhages (Han et al., 2015). Thus, permanent inhibition of eNOS in endothelial cells results in vascular dysfunction and impairment of cerebral microcirculation (Lin et al., 2014), and leads to further progression in Aβ tissue deposition. Endothelial eNOS affects transport of Aβ through the blood-brain barrier, therefore, dysregulation of eNOS activity or expression would lead to promotion of amyloid deposition in the brain tissue as it was previously shown in Provias and Jeynes (2014) (Figure 2).
H2S can also inhibit Aβ production in the cells due to suppression of γ-secretase activity (Nagpure and Bian, 2014) and an inhibit Aβ deposition, most likely, due to suppression of fibril formation (Rosario-Alomar et al., 2015). However, eNOS activity can be stimulated by H2S in endothelial cells (Chen et al., 2014) via calcium ions release from intracellular stores (Kida et al., 2013). Thus, cerebral amyloid angiopathy-associated changes in eNOS expression and activity might be linked to dysregulated production of H2S. Indeed, in AD, CBS activity, and H2S production are reduced in the brain (Eto et al., 2002), and plasma H2S levels are negatively correlated with the severity of AD (Giuliani et al., 2013), thus, neuroprotective and angioprotective properties of H2S are reduced (Figure 2). Therefore, it is not surprising that restoration of H2S levels can exhibit beneficial effects in AD (Fan et al., 2013; Kamat et al., 2013). Of note, similar effects have been observed for NO-donors currently tested as novel drug candidates in AD (Chegaev et al., 2015).
Perivascular Inflammation
Cerebral amyloid angiopathy is also associated with development of perivascular inflammation, microglial and astroglial activation, cytokines, and chemokines production. In addition, brain tissue hypoperfusion results in hypoxic alterations overlapping with Aβ-induced neurodegeneration. NO and H2S are well-known to exhibit anti-inflammatory effects, but dysregulated production of these gaseous transmitters at the sites of inflammation may promote degenerative changes. CBS producing H2S is localized to astroglial cell lineage and is up-regulated in reactive astrocytes (Kimura, 2015). Elevated expression of iNOS in brain endothelial cells is a known common feature of Alzheimer's type of vascular pathology associated with hypoxic injury (Sanchez et al., 2012; Figure 2). However, little is known about a putative role of H2S in perivascular inflammation in AD, despite this gaseous substance may exhibit neuroprotective effects due to anti-inflammatory activity (Giuliani et al., 2013). Similar effects have been demonstrated for CO (Cuadrado and Rojo, 2008).
Cerebral Hypoperfusion and Endothelial Dysfunction
Alzheimer's type of neurodegeneration starts from chronic cerebral hypoperfusion. It is interesting to note that metabolism of an allosteric activator of CBS—S-adenosyl-l-methionine (SAM)—is altered in chronic cerebral hypoperfusion (Wu et al., 2014). Thus, it is tempting to speculate that disturbances in SAM cycle would affect CBS activity leading to impaired H2S synthesis in the affected brain regions. Moreover, AD progression in humans is associated with decreased levels of SAM in the cerebrospinal fluid (Linnebank et al., 2010), while SAM has been proved to serve as cognitive-enhancing agent in AD animal model (Montgomery et al., 2014). Previously, such data have been recognized as evidence of altered methylation patterns seen in AD, but probably the summarized picture should include diminished activity of CBS in SAM-deficient brain.
Homocysteine is one of the most important risk factors of AD inducing endothelial dysfunction, angiopathy, and memory deficits. Toxic effects of homocysteine and the product of its spontaneous oxidation, homocysteic acid, are linked to activation of NMDA receptors, induction of intracellular calcium ion imbalance and oxidative stress (Boldyrev, 2009; Boldyrev et al., 2013). Homocysteine metabolism actually links NO and H2S pathways in AD (Figure 2). Both NO and H2S protect vascular and neuronal cells from homocysteine-induced injury (Dayal et al., 2014; Wei et al., 2014). Elevated plasma homocysteine level leads to reduction in nitric oxide bioavailability due to suppression of NOS activity (Lai and Kan, 2015). At the same time, homocysteine can be converted to l-cysteine and H2S due to activity of CBS and CSE, however, toxic concentrations of homocysteine suppress CBS and CSE activities in the brain along with increased expression of NMDA receptors in neuronal cells (Kamat et al., 2015a). These effects are linked to excitotoxicity and blood-brain barrier disruption. Thereby, activity of H2S- and NO-generating enzymes is decreased in AD, and homocysteine-mediated toxicity results in endothelial cell loss and progression of neurodegeneration (Figure 2). Recent findings suggest that exogenous H2S supply may normalize dysregulated expression of NMDA receptors, CBS, and CSE in renovascular diabetic remodeling (Kundu et al., 2015). H2S is able to restore decreased levels of eNOS, CD31, VE-cadherin, and endothelin-1 expression in brain endothelial cells subjected to the toxic action of homocysteine in vitro (Kamat et al., 2015b) and in vivo (Kamat et al., 2013), however whether or not this effect can be reproduced in cerebral amyloid angiopathy remains to be elucidated.
Nitrosation and Sulfhydration of Vessel Proteins in AD
Less attention has been paid to alternative molecular mechanisms of H2S functioning in the central nervous system. By analogy to NO which induces reversible nitrosation of target proteins, H2S may induce reversible protein modification (persulfidation, or alternatively S-sulfhydration; Gadalla and Snyder, 2010). Presumably, CSE (or other enzymes?) generates H2S to persulfidate the targets. Current evidence suggests that protein persulfidation adheres closely to the generally acknowledged paradigm for S-nitrosation by NO. Whereas, nitrosation appears to diminish cysteine reactivity, persulfidation seems to enhance it. Persulfidation may mediate various reported physiological actions of H2S, i.e., relaxation of blood vessels through the endothelial-derived relaxing factor activity of H2S involves opening of ATP–sensitive potassium channels (Yang et al., 2008), which are substrate for posttranslational modifications. Persulfidation and tyrosine nitration have been reported to occur at different subunits of KATPchannels, SUR2B, and Kir6.1, respectively, and pretreatment of the cells with H2S donor NaHS results in the suppression of NO-induced nitration (Kang et al., 2015). KATP channels protect endothelial cells (Chen et al., 2015), control release of NO and eicosanoids further acting on smooth vessel cells to produce vasorelaxation (Minamino and Hori, 2007). Besides that, Kir6.1 is a pore-forming subunit in astroglial plasma membrane KATP channels (Thomzig et al., 2001), which are involved in the intercellular communication within the neurovascular unit (Velasco et al., 2000; Sun and Hu, 2010). Thus, coordinated action of H2S and NO on KATP channels expressed in endothelial, astroglial and vascular smooth muscle cells may be an important regulatory mechanism of cerebral vasomotor activity impaired in AD.
Among the proteins modified by S-nitrosation, ryanodine receptors (RyR) are well-known targets, and NO-mediated S-nitrosation of RyRs mediates calcium release in neuronal cells (Kakizawa et al., 2013). In physiological conditions, RyRs are activated by cyclic adenosine diphosphate-ribose (cADP-ribose), a product of catalytic activity of NAD+-glycohydrolase/CD38 (Higashida et al., 2007). RyR2 and RyR3 are expressed in astrocytes, and RyR-dependent signaling has also been reported in vascular endothelium where three RyR isoforms have been identified. RyR3 are important for astrocytes migration (Matyash et al., 2002). RyR3 appears to be more broadly expressed, with predominance in neurons, and the three subtypes are expressed in large cerebral arteries as well as in the cerebral microcirculation (Dabertrand et al., 2013). Key role of RyR in Aβ production and learning and memory performances has been proposed (Oulès et al., 2012). In AD, there is a dysregulated expression of RyR, particularly in RyR2 splice variants (Bruno et al., 2012) and RyR3 (Liu et al., 2014a). Currently, there are no data on NO-mediated nitrosation or H2S-mediated sulfhudration of RyR in AD. However, the wide spectrum of S-nitrosated proteins seen in AD (Zahid et al., 2014; Zaręba-Kozioł et al., 2014) and very recent data on the inhibitory action of H2S-donor on RyR activity do not rule out this possibility (Tomasova et al., 2015).
Mitochondrial Dysfunction in Endothelial Cells
Brain endothelial cells have much higher density of mitochondria comparing to endothelial cells in other tissues (Oldendorf et al., 1977), and mitochondrial activity is extremely important for the regulation of endothelial cell metabolism (Salmina et al., 2015). Therefore, production of NO and H2S in or their action at mitochondria of endothelial cells should be considered in the context of AD pathogenesis. As expected, mitochondrial failure associated with energy shortage, calcium ion imbalance, and reactive oxygen species overproduction has been repeatedly confirmed in AD. These processes link mitochondrial dysfunction to neurodegeneration and neuroinflammation as well as to the blood-brain barrier dysfunction and impairment of cerebral endothelium repair (Sochocka et al., 2013).
Mitochondria represent the “cross-road” for the gaseous transmitters within the cells. Mitochondrial location was demonstrated for CSE, CBS (due to translocation in stressed cells), 3MST, and NOS. H2S inhibits mitochondrial production of reactive oxygen species, supports oxidative phosphorylation and prevents apoptosis (Guo et al., 2012; Szabo et al., 2014). Dose-dependent effects for H2S activity on mitochondrial function was demonstrated: low concentrations (below 1 μM) stimulate mitochondrial respiration, while high concentrations (higher than 3 μM) inhibit mitochondrial respiration. Such effects of H2S require adequate levels of Krebs cycle activity, therefore a co-ordinatory role of intramitochondrially produced H2S in the connection of citric acid cycle and oxidative phosphorylation has been proposed (Módis et al., 2013). NO is involved in the regulation of multiple aspects of mitochondrial functioning due to its ability to bind heme iron centers, to nitrosylate proteins (i.e., complex I) to inhibit electron flux, and to take part in reactive oxygen species formation (Stefano and Kream, 2015). Activation of mitochondrial KATP channels in cerebral endothelial cells leads to NOS activation, NO production, and vasodilation (Katakam et al., 2013) whereas insulin resistance-associated impairment of KATP channels alters above-mentioned mechanism (Katakam et al., 2009).
In AD, aberrant KATP channels activity and endothelium-mediated vasodilation was reported many years ago (Chi et al., 1999). At present, these data could be interpreted as an indirect effect of NO or H2S insufficiency resulting in mitochondrial impairment. Of note, CO is also known to induce uncoupling of mitochondrial respiration dependent on the activation of mito BKCa channels and inhibition of glycolysis independent of mito BKCa channels (Kaczara et al., 2015). Thus, all three gaseous transmitters demonstrate different behavioral patterns in mitochondria: stimulation or inhibition of mitochondrial respiration by H2S, uncoupling of mitochondrial respiration by CO, and inhibition of respiration by NO.
Mitochondrial biogenesis in endothelial cells is one of the requirements for effective angiogenesis, and is usually associated with the suppression of glycolysis. NO stimulates mitochondrial biogenesis in many cell types (Nisoli et al., 2004; Miller et al., 2013), H2S maintains mitochondrial DNA copy number that is important for mitochondrial biogenesis (Li and Yang, 2015), thereby exogenous H2S supports mitochondrial biogenesis in brain cells subjected to hypoxia/ischemia (Pan et al., 2014). Thus, suppression of mitochondrial biogenesis seen in AD (Rice et al., 2014; Burté et al., 2015) might be, at least partially, caused by reduced levels of endogenous NO and H2S. On the other hand, accumulation of S-nitrosocysteines due to NO-mediated nitrosation of mitochondrial proteins may have direct toxic effect especially under the conditions of oxidative stress: NAD+ depletion, ATP deficit, mitophagy induction in endothelial cells similarly to previously obtained data (Diers et al., 2013). However, it should also be considered that H2S may efficiently suppress homocysteine-induced mitophagy in cerebral endothelial cells (Kamat et al., 2015b), thereby acting also as a functional antagonist of NO.
Aberrant Angiogenesis and Neurogenesis in AD
Compromised angiogenesis takes place in AD, and plays a role in the progression of neurodegeneration. Aβ promotes neoangiogenesis and hypervascularity (Biron et al., 2011) that is supported by the elevated levels of proangiogenic factors—angiogenin and tissue inhibitor of matrix metalloproteinase-4—in the plasma of patients with AD (Qin et al., 2015). However, the present data are controversial since other authors found decreased serum levels of angiogenin in AD (Kim and Kim do, 2012). In both studies, there was a correlation between the levels of angiogenin and the severity of cognitive impairment, probably due to assessment of disease manifestations at different phases of progression. Other studies found that angiogenin stimulates NO synthesis and release from endothelial cells (Trouillon et al., 2010), whereas H2S increases angiogenin expression in endothelial cells (Geng et al., 2015). Thus, it is tempting to speculate that this machinery—[H2S → angiogenin → NO]—is dysregulated in AD, thereby contributing to abnormal angiogenesis.
H2S and NO themselves serve as potent angiogenic molecules acting individually or in the combination with other angiogenic factors to promote endothelial progenitor cells migration, proliferation (Lee et al., 2009; Kimura, 2015; Lu et al., 2015; Figure 1). H2S activates KATP channels that possess angiogenic properties being involved into the cell response to the action of VEGF, and contribute to NO release from endothelial cells (Umaru et al., 2015). The Kir6.1 subunit of KATP channels, which is target for NO-induced nitration (Kang et al., 2015), is required for the angiogenic activity of VEGF (Umaru et al., 2015). Thus, the possibility arises that decreased and/or imbalanced production of H2S and NO observed in AD does diminish the angiogenic potential of factors acting through KATP channels.
Angiogenesis is important not only for reparative processes in adult brain, but also for supporting neuroplasticity. Multiregional dysregulation of neurogenesis, impaired neuronal migration and maturation are common in AD (Hamilton et al., 2010). The vascular microenvironment within the neurogenic niche is integrated by signaling molecules secreted from endothelial cells in the brain vasculature or by direct contact with these cells (Goldberg and Hirschi, 2009). Endothelial-secreted factors from the brain vasculature regulate proliferation, survival/self-renewal, differentiation, and migration of neural stem/progenitor cells within the neurogenic niches. There are many paracrine effectors of the brain vasculature on neurogenesis, such as VEGF, EGF, bFGF, Notch ligands. It is known that H2S directly activates VEGF receptor 2 (VEGFR2), thus providing pro-angiogenic effect (Tao et al., 2013).
Recent finding suggest that constituent release of VEGF from neural stem/progenitor cells highly expressing VEGFR2 in the neurogenic niches supports their functional activity (Ara et al., 2010; Kirby et al., 2015). Therefore, local production of H2S could potentiate these paracrine and autocrine regulatory processes supporting neurogenesis and angiogenesis. H2S and l-cysteine are also known to promote proliferation of neural stem cells (Wang et al., 2013a; Liu et al., 2014b). RyR2 expressed in neural stem/progenitors embryonic cells take part in neurogenesis (Yu et al., 2008). At the same time, endogenous production of NO downstream of RyR activation is required for the positive regulation of proliferation of hippocampal neural progenitor cells derived from embryonic mice (Yoneyama et al., 2011). However, impairment of neural stem cells proliferation due to NO-mediated nitration of the EGF receptor and suppression signaling through the ERK/MAPK pathway was reported (Carreira et al., 2014). Together, targeting H2S and NO as putative local regulators of neurogenesis within the neurogenic niches may represent a novel approach for restoration of brain cell proliferation and differentiation in AD (Figure 2).
Conclusion
Being produced by the cells of the neurovascular unit, H2S, and NO act mainly as functionally additive molecules contributing to the regulation of the local blood flow, maintenance of endothelial integrity, and controlling intercellular communications. Synergistic action of physiological levels of NO and H2S is apparent in their neuroprotective, angiogenesis promoting activity as well as in the gliovascular control providing adequate blood supply to the active brain zones. Therefore, amyloid-induced suppression of NO and H2S production in endothelial and astroglial cells results in impairment of endothelial function and cerebral microcirculation. Also, acting at the same direction, NO, and H2S suppress Aβ production and deposition in cerebral microvessels. However, antagonism in the action of two gaseous transmitters is evident in their ability to modify proteins: NO-induced nitrosation usually suppress the activity of target proteins, whereas H2S-induced sulfhydration leads to activation of target proteins. Contrary to H2S that maintain mitochondrial DNA copy number, NO stimulates mitophagy in endothelial cells. However, it should be noted that data obtained with NO and H2S in complex biological systems should be considered very carefully because of dose-dependent action of both the gaseous mediators on cell metabolism and functioning: the final outcome could be diametrically opposite depending on the actual concentration. That is why our current understandings on the role of NO and H2S in (patho)physiological conditions are very far from the resultant conclusions.
It is clear that Alzheimer's type of neurodegeneration dramatically affects H2S and NO synthesis and their interactions resulting in dysregulated vasomotor function, brain tissue hypoperfusion and hypoxia, development of perivascular inflammation, promotion of Aβ deposition, and impairment of neurogenesis/angiogenesis (Figure 2). Better understanding of key cellular, molecular, and pathobiochemical mechanisms of H2S and NO action will provide new directions for the development of high-performance technologies for neural regeneration and neuroprotection with potential impact in clinical medicine.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
AS, YK, YG, and OL are supported by a grant given by the President Council of the Russian Foundation to support Leading Scientific Teams (project N 1172.2014.7). We also gratefully acknowledge financial support by the Deutsche Akademische Austauschdienst (DAAD) (YK) and Deutsche Forschungsgemeinschaft (DFG) (MG).
References
Altaany, Z., Yang, G., and Wang, R. (2013). Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J. Cell. Mol. Med. 17, 879–888. doi: 10.1111/jcmm.12077
Ara, J., Fekete, S., Zhu, A., and Frank, M. (2010). Characterization of neural stem/progenitor cells expressing VEGF and its receptors in the subventricular zone of newborn piglet brain. Neurochem. Res. 35, 1455–1470. doi: 10.1007/s11064-010-0207-2
Austin, S. A., Santhanam, A. V., Hinton, D. J., Choi, D. S., and Katusic, Z. S. (2013). Endothelial nitric oxide deficiency promotes Alzheimer's disease pathology. J. Neurochem. 127, 691–700. doi: 10.1111/jnc.12334
Bell, R. D., and Zlokovic, B. V. (2009). Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol. 118, 103–113. doi: 10.1007/s00401-009-0522-3
Biffi, A., and Greenberg, S. M. (2011). Cerebral amyloid angiopathy: a systematic review. J. Clin. Neurol. 7, 1–9. doi: 10.3988/jcn.2011.7.1.1
Biron, K. E., Dickstein, D. L., Gopaulm, R., and Jefferies, W. A. (2011). Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer's disease. PLoS ONE 6:e23789. doi: 10.1371/journal.pone.0023789
Boldyrev, A. A. (2009). Molecular mechanisms of homocysteine toxicity. Biochemistry (Mosc). 74, 589–598. doi: 10.1134/S0006297909060017
Boldyrev, A., Bryushkova, E., Mashkina, A., and Vladychenskaya, E. (2013). Why is homocysteine toxic for the nervous and immune systems? Curr. Aging Sci. 6, 29–36. doi: 10.2174/18746098112059990007
Boncoraglio, G. B., Piazza, F., Savoiardo, M., Farina, L., DiFrancesco, J. C., Prioni, S., et al. (2015). Prodromal Alzheimer's disease presenting as cerebral amyloid angiopathy-related inflammation with spontaneous amyloid-related imaging abnormalities and high cerebrospinal fluid anti-Aβ autoantibodies. J. Alzheimers Dis. 45, 363–367. doi: 10.3233/JAD-142376
Bruno, A. M., Huang, J. Y., Bennett, D. A., Marr, R. A., Hastings, M. L., and Stutzmann, G. E. (2012). Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer's disease. Neurobiol. Aging. 33, 1001.e1–1001.e6. doi: 10.1016/j.neurobiolaging.2011.03.011
Burke, M. J., Nelson, L., Slade, J. Y., Oakley, A. E., Khundakar, A. A., and Kalaria, R. N. (2014). Morphometry of the hippocampal microvasculature in post-stroke and age-related dementias. Neuropathol. Appl. Neurobiol. 40, 284–295. doi: 10.1111/nan.12085
Burté, F., Carelli, V., Chinnery, P. F., and Yu-Wai-Man, P. (2015). Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 11, 11–24. doi: 10.1038/nrneurol.2014.228
Carreira, B. P., Morte, M. I., Santos, A. I., Lourenço, A. S., Ambrósio, A. F., Carvalho, C. M., et al. (2014). Nitric oxide from inflammatory origin impairs neural stem cell proliferation by inhibiting epidermal growth factor receptor signaling. Front. Cell. Neurosci. 8:343. doi: 10.3389/fncel.2014.00343
Chegaev, K., Federico, A., Marini, E., Rolando, B., Fruttero, R., Morbin, M., et al. (2015). NO-donor thiacarbocyanines as multifunctional agents for Alzheimer's disease. Bioorg. Med. Chem. 23, 4688–4698. doi: 10.1016/j.bmc.2015.05.050
Chen, P. H., Fu, Y. S., Wang, Y. M., Yang, K. H., Wang, D. L., and Huang, B. (2014). Hydrogen sulfide increases nitric oxide production and subsequent S-nitrosylation in endothelial cells. ScientificWorldJournal 2014:480387. doi: 10.1155/2014/480387
Chen, X., Han, W., Zhang, Y., Cui, W., Pan, Z., Jin, X., et al. (2015). The molecular pathway of ATP-sensitive potassium channel in endothelial cells for mediating arteriole relaxation. Life Sci. 137, 164–169. doi: 10.1016/j.lfs.2015.07.009
Chertok, V. M., and Kotsyuba, A. E. (2012). Distribution of H2S synthesis enzymes in the walls of cerebral arteries in rats. Bull. Exp. Biol. Med. 154, 104–107. doi: 10.1007/s10517-012-1886-2
Chi, X., Sutton, E. T., Thomas, T., and Price, J. M. (1999). The protective effect of K+ channel openers on beta-amyloid induced cerebrovascular endothelial dysfunction. Neurol. Res. 21, 345–351.
Cuadrado, A., and Rojo, A. I. (2008). Heme oxygenase-1 as a therapeutic target in neurodegenerative diseases and brain infections. Curr. Pharm. Des. 14, 429–442. doi: 10.2174/138161208783597407
Dabertrand, F., Nelson, M. T., and Brayden, J. E. (2013). Ryanodine receptors, calcium signaling, and regulation of vascular tone in the cerebral parenchymal microcirculation. Microcirculation 20, 307–316. doi: 10.1111/micc.12027
D'Araio, E., Shaw, N., Millward, A., Demaine, A., Whiteman, M., and Hodgkinson, A. (2014). Hydrogen sulfide induces heme oxygenase-1 in human kidney cells. Acta Diabetol. 51, 155–157. doi: 10.1007/s00592-013-0501-y
Dayal, S., Blokhin, I. O., Erger, R. A., Jensen, M., Arning, E., Stevens, J. W., et al. (2014). Protective vascular and cardiac effects of inducible nitric oxide synthase in mice with hyperhomocysteinemia. PLoS ONE 9:e107734. doi: 10.1371/journal.pone.0107734
de la Monte, S. M., Sohn, Y. K., Etienne, D., Kraft, J., and Wands, J. R. (2000). Role of aberrant nitric oxide synthase-3 expression in cerebrovascular degeneration and vascular-mediated injury in Alzheimer's disease. Ann. N.Y. Acad. Sci. 903, 61–71. doi: 10.1111/j.1749-6632.2000.tb06351.x
Diers, A. R., Broniowska, K. A., and Hogg, N. (2013). Nitrosative stress and redox-cycling agents synergize to cause mitochondrial dysfunction and cell death in endothelial cells. Redox Biol. 1, 1–7. doi: 10.1016/j.redox.2012.11.003
Du, C., Jin, M., Hong, Y., Li, Q., Wang, X. H., Xu, J. M., et al. (2014). Downregulation of cystathionine β-synthase/hydrogen sulfide contributes to rotenone-induced microglia polarization toward M1 type. Biochem. Biophys. Res. Commun. 451, 239–245. doi: 10.1016/j.bbrc.2014.07.107
Dux, M., Will, C., Vogler, B., Filipovic, M. R., and Messlinger, K. (2015). Meningeal blood flow is controlled by H2S-NO crosstalk activating a HNO-TRPA1-CGRP signalling pathway. Br. J. Pharmacol. doi: 10.1111/bph.13164. [Epub ahead of print].
Dyson, R. M., Palliser, H. K., Latter, J. L., Chwatko, G., Glowacki, R., and Wright, I. M. (2014). A role for H2S in the microcirculation of newborns: the major metabolite of H2S (thiosulphate) is increased in preterm infants. PLoS ONE 9:e105085. doi: 10.1371/journal.pone.0105085
Eberhardt, M., Dux, M., Namer, B., Miljkovic, J., Cordasic, N., Will, C., et al. (2014). H2S and NO cooperatively regulate vascular tone by activating a neuroendocrine HNO-TRPA1-CGRP signalling pathway. Nat. Commun. 5, 4381. doi: 10.1038/ncomms5381
Eberhardt, M. J., Filipovic, M. R., Leffler, A., de la Roche, J., Kistner, K., Fischer, M. J., et al. (2012). Methylglyoxal activates nociceptors through transient receptor potential channel A1 (TRPA1): a possible mechanism of metabolic neuropathies. J. Biol. Chem. 287, 28291–28306. doi: 10.1074/jbc.M111.328674
Enokido, Y., Suzuki, E., Iwasawa, K., Namekata, K., Okazawa, H., and Kimura, H. (2005). Cystathionine beta-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J. 19, 1854–1856. doi: 10.1096/fj.05-3724fje
Eto, K., Asada, T., Arima, K., Makifuchi, T., and Kimura, H. (2002). Brain hydrogen sulfide is severely decreased in Alzheimer's disease. Biochem. Biophys. Res. Commun. 293, 1485–1488. doi: 10.1016/S0006-291X(02)00422-9
Fan, H., Guo, Y., Liang, X., Yuan, Y., Qi, X., Wang, M., et al. (2013). Hydrogen sulfide protects against amyloid beta-peptide induced neuronal injury via attenuating inflammatory responses in a rat model. J. Biomed. Res. 27, 296–304. doi: 10.7555/JBR.27.20120100
Filipovic, M. R., Eberhardt, M., Prokopovic, V., Mijuskovic, A., Orescanin-Dusic, Z., Reeh, P., et al. (2013). Beyond H2S and NO interplay: hydrogen sulfide and nitroprusside react directly to give nitroxyl (HNO). A new pharmacological source of HNO. J. Med. Chem. 56, 1499–1508. doi: 10.1021/jm3012036
Filipovic, M. R., Miljkovic, J. Lj., Nauser, T., Royzen, M., Klos, K., Shubina, T., et al. (2012). Chemical characterization of the smallest S-nitrosothiol, HSNO; cellular cross-talk of H2S and S-nitrosothiols. J. Am. Chem. Soc. 134, 12016–12027. doi: 10.1021/ja3009693
Gadalla, M. M., and Snyder, S. H. (2010). Hydrogen sulfide as a gasotransmitter. J. Neurochem. 113, 14–26. doi: 10.1111/j.1471-4159.2010.06580.x
Geng, Y., Li, E., Mu, Q., Zhang, Y., Wei, X., Li, H., et al. (2015). Hydrogen sulfide inhalation decreases early blood-brain barrier permeability and brain edema induced by cardiac arrest and resuscitation. J. Cereb. Blood Flow Metab. 35, 494–500. doi: 10.1038/jcbfm.2014.223
Gentile, M. T., Vecchione, C., Maffei, A., Aretini, A., Marino, G., Poulet, R., et al. (2004). Mechanisms of soluble beta-amyloid impairment of endothelial function. J. Biol. Chem. 279, 48135–48142. doi: 10.1074/jbc.M407358200
Giuliani, D., Ottani, A., Zaffe, D., Galantucci, M., Strinati, F., Lodi, R., et al. (2013). Hydrogen sulfide slows down progression of experimental Alzheimer's disease by targeting multiple pathophysiological mechanisms. Neurobiol. Learn. Mem. 104, 82–91. doi: 10.1016/j.nlm.2013.05.006
Goldberg, J. S., and Hirschi, K. K. (2009). Diverse roles of the vasculature within the neural stem cell niche. Regen. Med. 4, 879–897. doi: 10.2217/rme.09.61
Greiner, R., Pálinkás, Z., Bäsell, K., Becher, D., Antelmann, H., Nagy, P., et al. (2013). Polysulfides link H2S to protein thiol oxidation. Antioxid. Redox Signal. 19, 1749–1765. doi: 10.1089/ars.2012.5041
Guebel, D. V., and Torres, N. V. (2004). Dynamics of sulfur amino acids in mammalian brain: assessment of the astrocytic-neuronal cysteine interaction by a mathematical hybrid model. Biochim. Biophys. Acta 1674, 12–28. doi: 10.1016/j.bbagen.2004.05.005
Guix, F. X., Uribesalgo, I., Coma, M., and Muñoz, F. J. (2005). The physiology and pathophysiology of nitric oxide in the brain. Prog. Neurobiol. 76, 126–152. doi: 10.1016/j.pneurobio.2005.06.001
Guo, W., Kan, J. T., Cheng, Z. Y., Chen, J. F., Shen, Y. Q., Xu, J., et al. (2012). Hydrogen sulfide as an endogenous modulator in mitochondria and mitochondria dysfunction. Oxid. Med. Cell. Longev. 2012:878052. doi: 10.1155/2012/878052
Haase, N., Herse, F., Spallek, B., Haase, H., Morano, I., Qadri, F., et al. (2013). Amyloid-β peptides activate α1-adrenergic cardiovascular receptors. Hypertension 62, 966–972. doi: 10.1161/HYPERTENSIONAHA.113.01348
Hamilton, L. K., Aumont, A., Julien, C., Vadnais, A., Calon, F., and Fernandes, K. J. (2010). Widespread deficits in adult neurogenesis precede plaque and tangle formation in the 3xTg mouse model of Alzheimer's disease. Eur. J. Neurosci. 32, 905–920. doi: 10.1111/j.1460-9568.2010.07379.x
Han, B. H., Zhou, M. L., Abousaleh, F., Brendza, R. P., Dietrich, H. H., Koenigsknecht-Talboo, J., et al. (2008). Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: contribution of soluble and insoluble amyloid-beta peptide, partial restoration via gamma-secretase inhibition. J. Neurosci. 28, 13542–13550. doi: 10.1523/JNEUROSCI.4686-08.2008
Han, B. H., Zhou, M. L., Johnson, A. W., Singh, I., Liao, F., Vellimana, A. K., et al. (2015). Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc. Natl. Acad. Sci. U.S.A. 112, E881–E890. doi: 10.1073/pnas.1414930112
Harris-White, M. E., Ferbas, K. G., Johnson, M. F., Eslami, P., Poteshkina, A., Venkova, K., et al. (2015). A cell-penetrating ester of the neural metabolite lanthionine ketimine stimulates autophagy through the mTORC1 pathway: evidence for a mechanism of action with pharmacological implications for neurodegenerative pathologies. Neurobiol. Dis. doi: 10.1016/j.nbd.2015.03.007. [Epub ahead of print].
Hatakeyama, Y., Takahashi, K., Tominaga, M., Kimura, H., and Ohta, T. (2015). Polysulfide evokes acute pain through the activation of nociceptive TRPA1 in mouse sensory neurons. Mol. Pain. 11:24. doi: 10.1186/s12990-015-0023-4
Hawkes, C. A., Jayakody, N., Johnston, D. A., Bechmann, I., and Carare, R. O. (2014). Failure of perivascular drainage of β-amyloid in cerebral amyloid angiopathy. Brain Pathol. 24, 396–403. doi: 10.1111/bpa.12159
Heine, C. L., Schmidt, R., Geckl, K., Schrammel, A., Gesslbauer, B., Schmidt, K., et al. (2015). Selective irreversible inhibition of neuronal and inducible nitric-oxide synthase in the combined presence of hydrogen sulfide and nitric oxide. J. Biol. Chem. 290, 24932–24944. doi: 10.1074/jbc.m115.660316
Hensley, K., and Denton, T. T. (2015). Alternative functions of the brain transsulfuration pathway represent an underappreciated aspect of brain redox biochemistry with significant potential for therapeutic engagement. Free Radic. Biol. Med. 78, 123–134. doi: 10.1016/j.freeradbiomed.2014.10.581
Hess, D. T., Matsumoto, A., Kim, S. O., Marshall, H. E., and Stamler, J. S. (2005). Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6, 150–166. doi: 10.1038/nrm1569
Higashida, H., Salmina, A. B., Olovyannikova, R. Y., Hashii, M., Yokoyama, S., Koizumi, K., et al. (2007). Cyclic ADP-ribose as a universal calcium signal molecule in the nervous system. Neurochem. Int. 51, 192–199. doi: 10.1016/j.neuint.2007.06.023
Hine, C., Harputlugil, E., Zhang, Y., Ruckenstuhl, C., Lee, B. C., Brace, L., et al. (2015). Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 160, 132–144. doi: 10.1016/j.cell.2014.11.048
Hubbard, C., Benda, E., Hardin, T., Baxter, T., St John, E., O'Brien, S., et al. (2013). Lanthionine ketimine ethyl ester partially rescues neurodevelopmental defects in unc-33 (DPYSL2/CRMP2) mutants. J. Neurosci. Res. 91, 1183–1190. doi: 10.1002/jnr.23239
Hummel, S. G., Fischer, A. J., Martin, S. M., Schafer, F. Q., and Buettner, G. R. (2006). Nitric oxide as a cellular antioxidant: a little goes a long way. Free Radic. Biol. Med. 40, 501–506. doi: 10.1016/j.freeradbiomed.2005.08.047
Ishigami, M., Hiraki, K., Umemura, K., Ogasawara, Y., Ishii, K., and Kimura, H. (2009). A source of hydrogen sulfide and a mechanism of its release in the brain. Antioxid. Redox Signal. 11, 205–214. doi: 10.1089/ars.2008.2132
Jackman, N. A., Melchior, S. E., Hewett, J. A., and Hewett, S. J. (2012). Non-cell autonomous influence of the astrocyte system xc- on hypoglycaemic neuronal cell death. ASN Neuro 4:e00074. doi: 10.1042/ AN20110030
Jackman, N. A., Uliasz, T. F., Hewett, J. A., and Hewett, S. J. (2010). Regulation of system x(c)(-)activity and expression in astrocytes by interleukin-1β: implications for hypoxic neuronal injury. Glia 58, 1806–1815. doi: 10.1002/glia.21050
Jellinger, K. A. (2002). Alzheimer disease and cerebrovascular pathology: an update. J. Neural. Transm. 109, 813–836. doi: 10.1007/s007020200068
Jiang, Z., Li, C., Manuel, M. L., Yuan, S., Kevil, C. G., McCarter, K. D., et al. (2015). Role of hydrogen sulfide in early blood-brain barrier disruption following transient focal cerebral ischemia. PLoS ONE 10:e0117982. doi: 10.1371/journal.pone.0117982
Jullienne, A., and Badaut, J. (2013). Molecular contributions to neurovascular unit dysfunctions after brain injuries: lessons for target-specific drug development. Future Neurol. 8, 677–689. doi: 10.2217/fnl.13.55
Kaczara, P., Motterlini, R., Rosen, G. M., Augustynek, B., Bednarczyk, P., Szewczyk, A., et al. (2015). Carbon monoxide released by CORM-401 uncouples mitochondrial respiration and inhibits glycolysis in endothelial cells: a role for mitoBKCa channels. Biochim. Biophys. Acta 1847, 1297–1309. doi: 10.1016/j.bbabio.2015.07.004
Kakizawa, S., Yamazawa, T., and Iino, M. (2013). Nitric oxide-induced calcium release: activation of type 1 ryanodine receptor by endogenous nitric oxide. Channels (Austin) 7, 1–5. doi: 10.4161/chan.22555
Kamat, P. K., Kalani, A., Givvimani, S., Sathnur, P. B., Tyagi, S. C., and Tyagi, N. (2013). Hydrogen sulfide attenuates neurodegeneration and neurovascular dysfunction induced by intracerebral-administered homocysteine in mice. Neuroscience 252, 302–319. doi: 10.1016/j.neuroscience.2013.07.051
Kamat, P. K., Kalani, A., Tyagi, S. C., and Tyagi, N. (2015b). Hydrogen sulfide epigenetically attenuates homocysteine-induced mitochondrial toxicity mediated through NMDA receptor in mouse brain endothelial (bEnd3) cells. J. Cell. Physiol. 230, 378–394. doi: 10.1002/jcp.24722
Kamat, P. K., Kyles, P., Kalani, A., and Tyagi, N. (2015a). Hydrogen sulfide ameliorates homocysteine-induced Alzheimer's disease-like pathology, blood–brain barrier disruption, and synaptic disorder. Mol. Neurobiol. doi: 10.1007/s12035-015-9212-4. [Epub ahead of print].
Kang, M., Hashimoto, A., Gade, A., and Akbarali, H. I. (2015). Interaction between hydrogen sulfide-induced sulfhydration and tyrosine nitration in the KATP channel complex. Am. J. Physiol. Gastrointest. Liver Physiol. 308, G532–G539. doi: 10.1152/ajpgi.00281.2014
Katakam, P. V., Domoki, F., Snipes, J. A., Busija, A. R., Jarajapu, Y. P., and Busija, D. W. (2009). Impaired mitochondria-dependent vasodilation in cerebral arteries of Zucker obese rats with insulin resistance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R289–R298. doi: 10.1152/ajpregu.90656.2008
Katakam, P. V., Wappler, E. A., Katz, P. S., Rutkai, I., Institoris, A., Domoki, F., et al. (2013). Depolarization of mitochondria in endothelial cells promotes cerebral artery vasodilation by activation of nitric oxide synthase. Arterioscler. Thromb. Vasc. Biol. 33, 752–759. doi: 10.1161/ATVBAHA.112.300560
Katusic, Z. S., and Austin, S. A. (2014). Endothelial nitric oxide: protector of a healthy mind. Eur. Heart J. 35, 888–894. doi: 10.1093/eurheartj/eht544
Kida, M., Sugiyama, T., Yoshimoto, T., and Ogawa, Y. (2013). Hydrogen sulfide increases nitric oxide production with calcium-dependent activation of endothelial nitric oxide synthase in endothelial cells. Eur. J. Pharm. Sci. 48, 211–215. doi: 10.1016/j.ejps.2012.11.001
Kim, Y. N., and Kim do, H. (2012). Decreased serum angiogenin level in Alzheimer's disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 38, 116–120. doi: 10.1016/j.pnpbp.2012.02.010
Kimura, H. (2015). Hydrogen sulfide and polysulfides as signaling molecules. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 91, 131–159. doi: 10.2183/pjab.91.131
Kirby, E. D., Kuwahara, A. A., Messer, R. L., and Wyss-Coray, T. (2015). Adult hippocampal neural stem and progenitor cells regulate the neurogenic niche by secreting VEGF. Proc. Natl. Acad. Sci. U.S.A. 112, 4128–4133. doi: 10.1073/pnas.1422448112
Köhn, C., Dubrovska, G., Huang, Y., and Gollasch, M. (2012). Hydrogen sulfide: potent regulator of vascular tone and stimulator of angiogenesis. Int. J. Biomed. Sci. 8, 81–86.
Kolluru, G. K., Shen, X., and Kevil, C. G. (2013). A tale of two gases: NO and H2S, foes or friends for life? Redox Biol. 1, 313–318. doi: 10.1016/j.redox.2013.05.001
Kovac, A., Erickson, M. A., and Banks, W. A. (2011). Brain microvascular pericytes are immunoactive in culture: cytokine, chemokine, nitric oxide, and LRP-1 expression in response to lipopolysaccharide. J. Neuroinflammation 8:139. doi: 10.1186/1742-2094-8-139
Kram, L., Grambow, E., Mueller-Graf, F., Sorg, H., and Vollmar, B. (2013). The anti-thrombotic effect of hydrogen sulfide is partly mediated by an upregulation of nitric oxide synthases. Thromb. Res. 132, e112–e117. doi: 10.1016/j.thromres.2013.07.010
Kubo, S., Doe, I., Kurokawa, Y., Nishikawa, H., and Kawabata, A. (2007a). Direct inhibition of endothelial nitric oxide synthase by hydrogen sulfide: contribution to dual modulation of vascular tension. Toxicology 232, 138–146. doi: 10.1016/j.tox.2006.12.023
Kubo, S., Kajiwara, M., and Kawabata, A. (2007b). Dual modulation of the tension of isolated gastric artery and gastric mucosal circulation by hydrogen sulfide in rats. Inflammopharmacology 15, 288–292. doi: 10.1007/s10787-007-1590-4
Kundu, S., Pushpakumar, S., and Sen, U. (2015). MMP-9- and NMDA receptor-mediated mechanism of diabetic renovascular remodeling and kidney dysfunction: hydrogen sulfide is a key modulator. Nitric Oxide 46, 172–185. doi: 10.1016/j.niox.2015.02.003
Lai, W. K., and Kan, M. Y. (2015). Homocysteine-Induced Endothelial Dysfunction. Ann. Nutr. Metab. 67, 1–12. doi: 10.1159/000437098
Lamoke, F., Mazzone, V., Persichini, T., Maraschi, A., Harris, M. B., Venema, R. C., et al. (2015). Amyloid β peptide-induced inhibition of endothelial nitric oxide production involves oxidative stress-mediated constitutive eNOS/HSP90 interaction and disruption of agonist-mediated Akt activation. J. Neuroinflammation 12:84. doi: 10.1186/s12974-015-0304-x
Lee, C. Z., Xue, Z., Hao, Q., Yang, G. Y., and Young, W. L. (2009). Nitric oxide in vascular endothelial growth factor-induced focal angiogenesis and matrix metalloproteinase-9 activity in the mouse brain. Stroke 40, 2879–2881. doi: 10.1161/STROKEAHA.109.552059
Leffler, C. W., Parfenova, H., Basuroy, S., Jaggar, J. H., Umstot, E. S., and Fedinec, A. L. (2011). Hydrogen sulfide and cerebral microvascular tone in newborn pigs. Am. J. Physiol. Heart Circ. Physiol. 300, H440–H447. doi: 10.1152/ajpheart.00722.2010
Li, S., and Yang, G. (2015). Hydrogen sulfide maintains mitochondrial DNA replication via demethylation of TFAM. Antioxid. Redox Signal. 23, 630–642. doi: 10.1089/ars.2014.6186
Lin, A. J., Liu, G., Castello, N. A., Yeh, J. J., Rahimian, R., Lee, G., et al. (2014). Optical imaging in an Alzheimer's mouse model reveals amyloid-β-dependent vascular impairment. Neurophotonics 1:011005. doi: 10.1117/1.NPh.1.1.011005
Linnebank, M., Popp, J., Smulders, Y., Smith, D., Semmler, A., Farkas, M., et al. (2010). S-adenosylmethionine is decreased in the cerebrospinal fluid of patients with Alzheimer's disease. Neurodegener. Dis. 7, 373–378. doi: 10.1159/000309657
Liu, D., Wang, Z., Zhan, J., Zhang, Q., Wang, J., Zhang, Q., et al. (2014b). Hydrogen sulfide promotes proliferation and neuronal differentiation of neural stem cells and protects hypoxia-induced decrease in hippocampal neurogenesis. Pharmacol. Biochem. Behav. 116, 55–63. doi: 10.1016/j.pbb.2013.11.009
Liu, J., Fedinec, A. L., Leffler, C. W., and Parfenova, H. (2015). Enteral supplements of a carbon monoxide donor CORM-A1 protect against cerebrovascular dysfunction caused by neonatal seizures. J. Cereb. Blood Flow Metab. 35, 193–199. doi: 10.1038/jcbfm.2014.196
Liu, J., Supnet, C., Sun, S., Zhang, H., Good, L., Popugaeva, E., et al. (2014a). The role of ryanodine receptor type 3 in a mouse model of Alzheimer disease. Channels (Austin) 8, 230–242. doi: 10.4161/chan.27471
Lo Faro, M. L., Fox, B., Whatmore, J. L., Winyard, P. G., and Whiteman, M. (2014). Hydrogen sulfide and nitric oxide interactions in inflammation. Nitric Oxide 41, 38–47. doi: 10.1016/j.niox.2014.05.014
Lu, A., Wang, L., and Qian, L. (2015). The role of eNOS in the migration and proliferation of bone-marrow derived endothelial progenitor cells and in vitro angiogenesis. Cell Biol. Int. 39, 484–490. doi: 10.1002/cbin.10405
Matyash, M., Matyash, V., Nolte, C., Sorrentino, V., and Kettenmann, H. (2002). Requirement of functional ryanodine receptor type 3 for astrocyte migration. FASEB J. 16, 84–86. doi: 10.1096/fj.01-0380fje
Miller, M. W., Knaub, L. A., Olivera-Fragoso, L. F., Keller, A. C., Balasubramaniam, V., Watson, P. A., et al. (2013). Nitric oxide regulates vascular adaptive mitochondrial dynamics. Am. J. Physiol. Heart Circ. Physiol. 304, H1624–H1633. doi: 10.1152/ajpheart.00987.2012
Minamino, T., and Hori, M. (2007). Protecting endothelial function: a novel therapeutic target of ATP-sensitive potassium channel openers. Cardiovasc. Res. 73, 448–449. doi: 10.1016/j.cardiores.2006.11.014
Módis, K., Coletta, C., Erdélyi, K., Papapetropoulos, A., and Szabo, C. (2013). Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 27, 601–611. doi: 10.1096/fj.12-216507
Montgomery, S. E., Sepehry, A. A., Wangsgaard, J. D., and Koenig, J. E. (2014). The effect of S-adenosylmethionine on cognitive performance in mice: an animal model meta-analysis. PLoS ONE 9:e107756. doi: 10.1371/journal.pone.0107756
Nagpure, B. V., and Bian, J. S. (2014). Hydrogen sulfide inhibits A2A adenosine receptor agonist induced β-amyloid production in SH-SY5Y neuroblastoma cells via a cAMP dependent pathway. PLoS ONE 9:e88508. doi: 10.1371/journal.pone.0088508
Nalli, A. D., Rajagopal, S., Mahavadi, S., Grider, J. R., and Murthy, K. S. (2015). Inhibition of RhoA-dependent pathway and contraction by endogenous hydrogen sulfide in rabbit gastric smooth muscle cells. Am. J. Physiol. Cell Physiol. 308, C485–C495. doi: 10.1152/ajpcell.00280.2014
Nisoli, E., Falcone, S., Tonello, C., Cozzi, V., Palomba, L., Fiorani, M., et al. (2004). Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. U.S.A. 101, 16507–16512. doi: 10.1073/pnas.0405432101
Noma, K., Kihara, Y., and Higashi, Y. (2012). Striking crosstalk of ROCK signaling with endothelial function. J. Cardiol. 60, 1–6. doi: 10.1016/j.jjcc.2012.03.005
Oldendorf, W. H., Cornford, M. E., and Brown, W. J. (1977). The large apparent work capability of the blood-brain barrier: a study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann. Neurol. 1, 409–417. doi: 10.1002/ana.410010502
Olson, K. R., Deleon, E. R., Gao, Y., Hurley, K., Sadauskas, V., Batz, C., et al. (2013). Thiosulfate: a readily accessible source of hydrogen sulfide in oxygen sensing. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305, R592–R603. doi: 10.1152/ajpregu.00421.2012
Oulès, B., Del Prete, D., Greco, B., Zhang, X., Lauritzen, I., Sevalle, J., et al. (2012). Ryanodine receptor blockade reduces amyloid-β load and memory impairments in Tg2576 mouse model of Alzheimer disease. J. Neurosci. 32, 11820–11834. doi: 10.1523/JNEUROSCI.0875-12.2012
Pan, H., Xie, X., Chen, D., Zhang, J., Zhou, Y., and Yang, G. (2014). Protective and biogenesis effects of sodium hydrosulfide on brain mitochondria after cardiac arrest and resuscitation. Eur. J. Pharmacol. 741, 74–82. doi: 10.1016/j.ejphar.2014.07.037
Parfenova, H., and Leffler, C. W. (2008). Cerebroprotective functions of HO-2. Curr. Pharm. Des. 14, 443–453. doi: 10.2174/138161208783597380
Parfenova, H., Leffler, C. W., Basuroy, S., Liu, J., and Fedinec, A. L. (2012a). Antioxidant roles of heme oxygenase, carbon monoxide, and bilirubin in cerebral circulation during seizures. J. Cereb. Blood Flow Metab. 32, 1024–1034. doi: 10.1038/jcbfm.2012.13
Parfenova, H., Tcheranova, D., Basuroy, S., Fedinec, A. L., Liu, J., and Leffler, C. W. (2012b). Functional role of astrocyte glutamate receptors and carbon monoxide in cerebral vasodilation response to glutamate. Am. J. Physiol. Heart Circ. Physiol. 302, H2257–H2266. doi: 10.1152/ajpheart.01011.2011
Park, L., Wang, G., Zhou, P., Zhou, J., Pitstick, R., Previti, M. L., et al. (2011). Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-beta. Proc. Natl. Acad. Sci. U.S.A. 108, 5063–5068. doi: 10.1073/pnas.1015413108
Pong, W. W., and Eldred, W. D. (2009). Interactions of the gaseous neuromodulators nitric oxide, carbon monoxide, and hydrogen sulfide in the salamander retina. J. Neurosci. Res. 87, 2356–2364. doi: 10.1002/jnr.22042
Provias, J., and Jeynes, B. (2014). The role of the blood-brain barrier in the pathogenesis of senile plaques in Alzheimer's disease. Int. J. Alzheimers Dis. 2014:191863. doi: 10.1155/2014/191863
Qian, X., Francis, M., Solodushko, V., Earley, S., and Taylor, M. S. (2013). Recruitment of dynamic endothelial Ca2+ signals by the TRPA1 channel activator AITC in rat cerebral arteries. Microcirculation 20, 138–148. doi: 10.1111/micc.12004
Qin, W., Jia, X., Wang, F., Zuo, X., Wu, L., Zhou, A., et al. (2015). Elevated plasma angiogenesis factors in Alzheimer's disease. J. Alzheimers Dis. 45, 245–252. doi: 10.3233/JAD-142409
Rice, A. C., Keeney, P. M., Algarzae, N. K., Ladd, A. C., Thomas, R. R., Bennett, J. P., et al. (2014). Mitochondrial DNA copy numbers in pyramidal neurons are decreased and mitochondrial biogenesis transcriptome signaling is disrupted in Alzheimer's disease hippocampi. J. Alzheimers Dis. 40, 319–330. doi: 10.3233/JAD-131715
Rong-na, L., Xiang-jun, Z., Yu-han, C., Ling-qiao, L., and Gang, H. (2011). Interaction between hydrogen sulfide and nitric oxide on cardiac protection in rats with metabolic syndrome. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 33, 25–32. doi: 10.3881/j.issn.1000-503X.2011.01.007
Rosario-Alomar, M. F., Quiñones-Ruiz, T., Kurouski, D., Sereda, V., Ferreira, E. B., Jesús-Kim, L. D., et al. (2015). Hydrogen sulfide inhibits amyloid formation. J. Phys. Chem. B 119, 1265–1274. doi: 10.1021/jp508471v
Saito, S., Yamamoto, Y., and Ihara, M. (2015). Mild cognitive impairment: at the crossroad of neurodegeneration and vascular dysfunction. Curr. Alzheimer Res. 12, 507–512. doi: 10.2174/1567205012666150530202508
Salmina, A. B. (2009). Neuron-glia interactions as therapeutic targets in neurodegeneration. J. Alzheimers. Dis. 16, 485–502. doi: 10.3233/JAD-2009-0988
Salmina, A. B., Inzhutova, A. I., Malinovskaya, N. A., and Petrova, M. M. (2010). Endothelial dysfunction and repair in Alzheimer-type neurodegeneration: neuronal and glial control. J. Alzheimers Dis. 22, 17–36. doi: 10.3233/JAD-2010-091690
Salmina, A. B., Kuvacheva, N. V., Morgun, A. V., Komleva, Y. K., Pozhilenkova, E. A., Lopatina, O. L., et al. (2015). Glycolysis-mediated control of blood-brain barrier development and function. Int. J. Biochem. Cell Biol. 64, 174–184. doi: 10.1016/j.biocel.2015.04.005
Sanchez, A., Tripathy, D., Yin, X., Desobry, K., Martinez, J., Riley, J., et al. (2012). p38 MAPK: a mediator of hypoxia-induced cerebrovascular inflammation. J. Alzheimers Dis. 32, 587–597. doi: 10.3233/JAD-2012-120829
Sawada, N., Itoh, H., Yamashita, J., Doi, K., Inoue, M., Masatsugu, K., et al. (2001). cGMP-dependent protein kinase phosphorylates and inactivates RhoA. Biochem. Biophys. Res. Commun. 280, 798–805. doi: 10.1006/bbrc.2000.4194
Shibuya, N., Mikami, Y., Kimura, Y., Nagahara, N., and Kimura, H. (2009). Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J. Biochem. 146, 623–626. doi: 10.1093/jb/mvp111
Sochocka, M., Koutsouraki, E. S., Gasiorowski, K., and Leszek, J. (2013). Vascular oxidative stress and mitochondrial failure in the pathobiology of Alzheimer's disease: a new approach to therapy. CNS Neurol. Disord. Drug Targets 12, 870–881. doi: 10.2174/18715273113129990072
Stefano, G. B., and Kream, R. M. (2015). Nitric oxide regulation of mitochondrial processes: commonality in medical disorders. Ann. Transplant. 20, 402–407. doi: 10.12659/AOT.894289
Sullivan, M. N., Gonzales, A. L., Pires, P. W., Bruhl, A., Leo, M. D., Li, W., et al. (2015). Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci. Signal. 8:ra2. doi: 10.1126/scisignal.2005659
Sun, X. L., and Hu, G. (2010). ATP-sensitive potassium channels: a promising target for protecting neurovascular unit function in stroke. Clin. Exp. Pharmacol. Physiol. 37, 243–252. doi: 10.1111/j.1440-1681.2009.05190.x
Szabo, C., Ransy, C., Módis, K., Andriamihaja, M., Murghes, B., Coletta, C., et al. (2014). Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 171, 2099–2122. doi: 10.1111/bph.12369
Tabatabaei, S. N., and Girouard, H. (2014). Nitric oxide and cerebrovascular regulation. Vitam. Horm. 96, 347–385. doi: 10.1016/B978-0-12-800254-4.00014-3
Tanizawa, K. (2011). Production of H2S by 3-mercaptopyruvate sulphurtransferase. J. Biochem. 149, 357–359. doi: 10.1093/jb/mvr018
Tao, B. B., Liu, S. Y., Zhang, C. C., Fu, W., Cai, W. J., Wang, Y., et al. (2013). VEGFR2 functions as an H2S-targeting receptor protein kinase with its novel Cys1045-Cys1024 disulfide bond serving as a specific molecular switch for hydrogen sulfide actions in vascular endothelial cells. Antioxid. Redox. Signal. 19, 448–464. doi: 10.1089/ars.2012.4565
Thomzig, A., Wenzel, M., Karschin, C., Eaton, M. J., Skatchkov, S. N., Karschin, A., et al. (2001). Kir6.1 is the principal pore-forming subunit of astrocyte but not neuronal plasma membrane K-ATP channels. Mol. Cell Neurosci. 18, 671–690. doi: 10.1006/mcne.2001.1048
Tomasova, L., Pavlovicova, M., Malekova, L., Misak, A., Kristek, F., Grman, M., et al. (2015). Effects of AP39, a novel triphenylphosphonium derivatised anethole dithiolethione hydrogen sulfide donor, on rat haemodynamic parameters and chloride and calcium Cav3 and RyR2 channels. Nitric Oxide 46, 131–144. doi: 10.1016/j.niox.2014.12.012
Tripathy, D., Chakraborty, J., and Mohanakumar, K. P. (2015). Antagonistic pleiotropic effects of nitric oxide in the pathophysiology of Parkinson's disease. Free Radic Res. 49, 1129–1139. doi: 10.3109/10715762.2015.1045505
Trouillon, R., Kang, D. K., Park, H., Chang, S. I., and O'Hare, D. (2010). Angiogenin induces nitric oxide synthesis in endothelial cells through PI-3 and Akt kinases. Biochemistry 49, 3282–3288. doi: 10.1021/bi902122w
Tsugane, M., Nagai, Y., Kimura, Y., Oka, J., and Kimura, H. (2007). Differentiated astrocytes acquire sensitivity to hydrogen sulfide that is diminished by the transformation into reactive astrocytes. Antioxid. Redox Signal. 9, 257–269. doi: 10.1089/ars.2007.9.257
Umaru, B., Pyriochou, A., Kotsikoris, V., Papapetropoulos, A., and Topouzis, S. (2015). ATP-sensitive potassium channel activation induces angiogenesis in vitro and in vivo. J. Pharmacol. Exp. Ther. 354, 79–87. doi: 10.1124/jpet.114.222000
Velasco, A., Tabernero, A., Granda, B., and Medina, J. M. (2000). ATP-sensitive potassium channel regulates astrocytic gap junction permeability by a Ca2+-independent mechanism. J. Neurochem. 74, 1249–1256. doi: 10.1046/j.1471-4159.2000.741249.x
Wang, G., Li, W., Chen, Q., Jiang, Y., Lu, X., and Zhao, X. (2015). Hydrogen sulfide accelerates wound healing in diabetic rats. Int. J. Clin. Exp. Pathol. 8, 5097–5104.
Wang, J. F., Li, Y., Song, J. N., and Pang, H. G. (2014). Role of hydrogen sulfide in secondary neuronal injury. Neurochem. Int. 64, 37–47. doi: 10.1016/j.neuint.2013.11.002
Wang, R. (2012). Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 92, 791–896. doi: 10.1152/physrev.00017.2011
Wang, Z., Liu, D. X., Wang, F. W., Zhang, Q., Du, Z. X., Zhan, J. M., et al. (2013a). L-Cysteine promotes the proliferation and differentiation of neural stem cells via the CBS/H2S pathway. Neuroscience 237, 106–117. doi: 10.1016/j.neuroscience.2012.12.057
Wang, Z., Zhan, J., Wang, X., Gu, J., Xie, K., Zhang, Q., et al. (2013b). Sodium hydrosulfide prevents hypoxia-induced behavioral impairment in neonatal mice. Brain Res. 1538, 126–134. doi: 10.1016/j.brainres.2013.09.043
Wedmann, R., Bertlein, S., Macinkovic, I., Böltz, S., Miljkovic, J. Lj., Muñoz, L. E., et al. (2014). Working with “H2S”: facts and apparent artifacts. Nitric Oxide 41, 85–96. doi: 10.1016/j.niox.2014.06.003
Wei, H. J., Xu, J. H., Li, M. H., Tang, J. P., Zou, W., Zhang, P., et al. (2014). Hydrogen sulfide inhibits homocysteine-induced endoplasmic reticulum stress and neuronal apoptosis in rat hippocampus via upregulation of the BDNF-TrkB pathway. Acta Pharmacol. Sin. 35, 707–715. doi: 10.1038/aps.2013.197
Wintner, E. A., Deckwerth, T. L., Langston, W., Bengtsson, A., Leviten, D., Hill, P., et al. (2010). A monobromobimane-based assay to measure the pharmacokinetic profile of reactive sulphide species in blood. Br. J. Pharmacol. 160, 941–957. doi: 10.1111/j.1476-5381.2010.00704.x
Wu, X., Sun, J., Zhang, X., Li, X., Liu, Z., Yang, Q., et al. (2014). Epigenetic signature of chronic cerebral hypoperfusion and beneficial effects of S-adenosylmethionine in rats. Mol. Neurobiol. 50, 839–851. doi: 10.1007/s12035-014-8698-5
Yamada, M. (2015). Cerebral amyloid angiopathy: emerging concepts. J. Stroke 17, 17–30. doi: 10.5853/jos.2015.17.1.17
Yang, G., Wu, L., Jiang, B., Yang, W., Qi, J., Cao, K., et al. (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322, 587–590. doi: 10.1126/science.1162667
Yoneyama, M., Kawada, K., Shiba, T., and Ogita, K. (2011). Endogenous nitric oxide generation linked to ryanodine receptors activates cyclic GMP / protein kinase G pathway for cell proliferation of neural stem/progenitor cells derived from embryonic hippocampus. J. Pharmacol. Sci. 115, 182–195. doi: 10.1254/jphs.10290FP
Yong, Q. C., Hu, L. F., Wang, S., Huang, D., and Bian, J. S. (2010). Hydrogen sulfide interacts with nitric oxide in the heart: possible involvement of nitroxyl. Cardiovasc. Res. 88, 482–491. doi: 10.1093/cvr/cvq248
Yu, H. M., Wen, J., Wang, R., Shen, W. H., Duan, S., and Yang, H. T. (2008). Critical role of type 2 ryanodine receptor in mediating activity-dependent neurogenesis from embryonic stem cells. Cell Calcium 43, 417–431. doi: 10.1016/j.ceca.2007.07.006
Zahid, S., Khan, R., Oellerich, M., Ahmed, N., and Asif, A. R. (2014). Differential S-nitrosylation of proteins in Alzheimer's disease. Neuroscience 256, 126–136. doi: 10.1016/j.neuroscience.2013.10.026
Zaręba-Kozioł, M., Szwajda, A., Dadlez, M., Wysłouch-Cieszyñska, A., and Lalowski, M. (2014). Global analysis of S-nitrosylation sites in the wild type (APP) transgenic mouse brain-clues for synaptic pathology. Mol. Cell Proteomics 13, 2288–2305. doi: 10.1074/mcp.M113.036079
Zhang, C. Y., Li, X. H., Zhang, T., Fu, J., and Cui, X. D. (2013). Hydrogen sulfide upregulates heme oxygenase-1 expression in rats with volume overload-induced heart failure. Biomed. Rep. 1, 454–458. doi: 10.3892/br.2013.87
Zhang, W., Xu, C., Yang, G., Wu, L., and Wang, R. (2015). Interaction of H2S with calcium permeable channels and transporters. Oxid. Med. Cell Longev. 2015:323269. doi: 10.1155/2015/323269
Zhang, X., and Bian, J. S. (2014). Hydrogen sulfide: a neuromodulator and neuroprotectant in the central nervous system. ACS Chem. Neurosci. 5, 876–883. doi: 10.1021/cn500185g
Keywords: H2S, vascular dysfunction, Alzheimer disease (AD), NO, KCNQ channels, obesity
Citation: Salmina AB, Komleva YK, Szijártó IA, Gorina YV, Lopatina OL, Gertsog GE, Filipovic MR and Gollasch M (2015) H2S- and NO-Signaling Pathways in Alzheimer's Amyloid Vasculopathy: Synergism or Antagonism? Front. Physiol. 6:361. doi: 10.3389/fphys.2015.00361
Received: 22 September 2015; Accepted: 16 November 2015;
Published: 11 December 2015.
Edited by:
John D. Imig, Medical College of Wisconsin, USAReviewed by:
Siu-Lung Chan, University of Vermont, USAAdán Dagnino-Acosta, University of Colima, Mexico
Copyright © 2015 Salmina, Komleva, Szijártó, Gorina, Lopatina, Gertsog, Filipovic and Gollasch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alla B. Salmina, YWxsYXNhbG1pbmFAbWFpbC5ydQ==;
Maik Gollasch, bWFpay5nb2xsYXNjaEBjaGFyaXRlLmRl